
 Christopher T. D. Price1
Christopher T. D. Price1
- 1 Department of Microbiology and Immunology, University of Louisville School of Medicine, Louisville, KY, USA
- 2 Division of Infectious Diseases, Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA
To identify factors contributing to the ability of tubercle bacilli to grow in the lung during active infection, we analyzed RNA expression patterns in bacteria present in patient sputum. Prominent among bacterial transcripts identified were those encoding secreted peptides of the Esat-6 subfamily that includes EsxK and EsxL (Rv1197 and Rv1198). H37Rv esxKL and esxJI transcripts were differentially expressed under different growth conditions, and disruption of these genes altered growth phase kinetics in typical laboratory batch broth cultures. These growth defects, including the reduced intracellular growth of an ΔesxKL mutant in primary human macrophages, were reversed by either low multiplicity co-infection or co-culture with wild-type bacteria, demonstrating the ability of the secreted factors to rescue isogenic mutants. Complementing either only esxL or esxI alone (Rv1198 or Rv1037c) also reduced observed growth defects, indicating these genes encode factors capable of contributing to growth. Our studies indicate that the Mycobacterium tuberculosis Mtb9.9 family secreted factors EsxL and EsxI can act in trans to modulate growth of intracellular bacteria, and are highly expressed during active human lung infection.
Introduction
Mycobacterium tuberculosis exists largely within a natural reservoir of latent infection estimated to comprise almost one-third of the world’s population. These infections are often asymptomatic, as an effective immune response is typically able to restrict bacterial growth, frequently localizing bacilli within nodules or granulomas in the lung. As the host’s immune status eventually declines with age or other compromise, bacteria resume replication, and can grow to high titers in a poorly defined extracellular niche on the interior walls of further-remodeled lung tissue cavities (Grosset, 2003; Hunter, 2011). Transmission to new hosts occurs primarily by way of droplet nuclei generated when high numbers of bacteria are aerosolized as cavities progress to merge with bronchial airways (Mastorides et al., 1999). Although traditionally viewed as a resident of alveolar macrophages, M. tuberculosis possess a relatively large genetic capacity (Bishai, 1998), reflecting an ability to adapt to several different intracellular and extracellular environments (Grosset, 2003; Rohde et al., 2007). These include those in a wide variety of host tissues, and at least temporarily in sputum and desiccated airborne droplet nuclei.
Specifically how M. tuberculosis adapts to these alternate environments encountered during infection remains poorly characterized. Survival and growth within different niches would require activating alternate metabolic pathways, and the elaboration of additional bacterial factors, including those needed to defend against and alter host immune responses. Previous studies have attempted to directly identify some of these by analyzing bacterial gene expression following phagocytosis by resting or activated murine and human macrophages (Graham and Clark-Curtiss, 1999; Schnappinger et al., 2003; Homolka et al., 2010), and within inherently heterogeneous bacterial populations present in resected lung tissues (Timm et al., 2003; Rachman et al., 2006) and sputum (Garton et al., 2008) and in different animal models of disease (Talaat et al., 2007; Kesavan et al., 2009). We therefore sought to identify important features exhibited by tubercle bacilli in the cavitary stage of active human disease by examining bacteria recovered directly from the lungs of untreated patients in quick-frozen sputum specimens.
Analysis of RNA expression patterns in these bacteria indicated that transcripts encoding an Esat-6 subfamily of small secreted proteins (Alderson et al., 2000; Pallen, 2002; Jones et al., 2010; Sutcliffe, 2011) are among the most abundantly expressed mRNAs. The H37Rv genome contains five nearly identical pairs of co-linear open reading frames (ORFs) designated esxKL, esxJI, esxMN, esxPO, and esxWV. These ORFs show little sequence similarity to esxBA (Cfp-10–Esat-6), other than encoding two short ∼100 residue peptides with the 5′ ORF encoding a variant carboxyl terminal “QILSS” motif and the 3′ encoding the Mtb9.9 family of secreted T-cell antigens. All contain a central “WXG100” esx family structural motif, and are thought to encode effectors of an uncharacterized ESX-5 transport system. Further examination of the contributions of these small secreted peptides indicated they are required for normal microbial growth, and likely to have a role in bacterial multiplication during active infection.
Materials and Methods
See Section “Supplementary Materials and Methods” in Appendix for additional details.
Mycobacterial Cultures
Mycobacterium tuberculosis H37Rv (ATCC no. 25618) was obtained from the American Type Culture Collection and grown in Middlebrook 7H9 OADC and 0.05% Tween-80 at 37°C either on a rotary shaker or as set cultures for use as frozen inocula. Frozen stocks were used as inoculums for infections as previously described (Price et al., 2008).
Isolation of RNA from Sputum Samples, Broth-Grown Bacteria, Infected Cells, and Tissues
Tuberculosis patient sputum specimens were obtained by consent from previously untreated and symptomatic walk in patients at the Metro Health Department (Nashville, TN, USA) and the Louisville Metro Department of Public Health and Wellness. Independent un-induced specimens for the study were produced by patients into 50 ml tubes on ice and frozen at −20°C. Frozen sputum samples from the first six smear positive cases in 50 ml tubes were thawed on ice and homogenized by vortexing with 4-mm glass beads and 10% of N-acetyl-L-cysteine solution as previously described (Desjardin et al., 1996). Following centrifugation at 8500 × g for 10 min, pellets were resuspended in 4 ml of TRIZOL (Invitrogen, Carlsbad, CA, USA) at 70°C and 300 μl of phenol reagent was added. The mixture was transferred to a tube containing 0.1 mm zirconia silica beads (Biospec) and processed in a Savant FP-120 homogenizer as previously described (Graham and Clark-Curtiss, 1999). Following additional organic extraction and ethanol precipitations, RNA pellets were washed with 70% ethanol and resuspended in distilled water. RNA from mid-log phase broth cultures and infected monolayers was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacture’s instructions with minor modifications. M. tuberculosis infected C57BL6 mouse lung tissue specimens (∼0.1 g) were obtained at 21 days in the active infection model previously described (Shoen et al., 2004). All normal blood and sputum specimens were obtained with donor consent as approved by the UofL IRB, as was use of animal tissue by the UofL IACUC.
To prepare total RNAs, lysates in Qiagen lysis buffer on ice were briefly heated at 70°C for 1 min and then homogenized with zirconia beads in screw-cap vials with a Savant FP-120 instrument prior to column purification (Graham and Clark-Curtiss, 1999). RNAs so obtained were then treated with DNAse I (Ambion, Austin, TX, USA) as described by the enzyme manufacturer, and stored at –80°C in aliquots as ethanol precipitates.
cDNA Synthesis and Quantitative PCR (RT-qPCR)
RNA was converted to first-strand cDNA with reverse transcriptase Superscript III (Invitrogen) according to manufacture’s instructions using random non-amer primers (Price et al., 2008). For selective capture of transcribed sequences (SCOTS; Graham and Clark-Curtiss, 1999), we used previously described primer for broth RNA (5′-GCCGGAGCTCTGCAGAATTCNNNNNNNNN-3′) and sputum-derived RNA (5′-GACACTCTCGAGACATCACCGGTACCNNNNNNNNN-3′) For quantitative PCR measurements, primers used specifically for measuring sigA and QILSS–Mtb9.9 mRNA levels were chosen using Primer3 software (see Materials and Methods in Appendix) and tested for optimal PCR efficiency using genomic DNA template. Fluorescence-monitored PCR was carried out with an Opticon detection system (MJ Research, Waltham, MA, USA), using Sybr green as the indicator. The major housekeeping sigma factor gene sigA was used as an internal control for the normalization of transcripts as H37Rv lacks the regulation of sigA expression seen in many clinical strains (Manganelli et al., 1999). A “no-reverse transcriptase” control was then used to confirm absence of genomic DNA. Change in steady-state mRNA levels were calculated and statistically analyzed by comparing expression level of sigA and target RNAs in different growth conditions using the REST software tool and a threshold of P-value of <0.05 (Pfaffl et al., 2002).
Selective Capture of Transcribed Sequences
Mycobacterium tuberculosis cDNAs from total amplified cDNAs from sputum specimens were selectively captured by hybridization to denatured sonicated photobiotinylated M. tuberculosis genomic DNA which has been prehybridized with blocking M. tuberculosis ribosomal DNA (rDNA) fragments as previously described (Graham and Clark-Curtiss, 1999). Microbial cDNA–genomic DNA hybrids were then collected by binding to streptavidin-coated beads, and bacterial cDNAs were eluted by alkaline denaturation. Eluted bacterial cDNAs were then amplified by PCR. After three rounds of selective capture, bacterial cDNAs were used as template to prepare radiolabeled probes.
Analysis of M. tuberculosis in vivo Expression by Genome Array Hybridization
Mycobacterium tuberculosis genomic arrays on nylon from prototype and limited production runs (Sigma-Genosys, Panorama Gene Arrays) were used as previously described (Graham et al., 2002). Random primed radiolabeled first-strand cDNA was prepared from four mid-log cultures using 10–20 μg of DNAse-treated total RNA template by incorporation of alpha P[33] deoxycytidine triphosphate (ICN Biomedicals, Irvine, CA, USA) with Superscript III (Invitrogen). For SCOTS, radiolabeled probes were made from 2 μg of captured amplified third round cDNA using random non-amer primers and 50 μCi P[33] dCTP with the modification of including cold dCTP at 1/100 the level of the other three dNTP’s. Nylon membranes were hybridized for 24 h, washed, and exposed to a Fuji phosphorimager plate overnight prior to scanning with a Fuji BAS FLA-2000 (Fuji Scientific Imaging). Scanned images were quantified with ArrayVision software (Imaging Research, ON, Canada) and data analyzed with BRB Array Tools (Simon et al., 2007) extension for Microsoft Excel. Local background subtraction was used correct regional background variations within individual patterns from single-channel data on nylon macro arrays. As SCOTS is intended as a qualitative screening tool, we determined if cDNAs for mRNAs were detected or not detected by comparison of levels for background based on designated blank spots on arrays. Those features showing hybridization levels greater than threefold over the average level for blank array features was considered as detected expression of the corresponding mRNA. Similarly for mid-log broth cultures, we considered the average hybridization value for blanks on all four arrays relative to the average values for hybridization signals for ORFs obtained with four cDNAs from four independent cultures. The hybridization data from these experiments is available at https://louisville.edu/medschool/microbiology/faculty/full-time-faculty/james-e-graham-ph-d.
Identification of Specific esx mRNAs by Targeted Selective Hybridization
Homologous segment of the esxJI genomic DNA was PCR-amplified with (F) primer 5′-CGTGTGATGCGGACAAAG-3′ and (R) primer 5′-ATCTTCCCAGTTCAGCAC-3′ from H37Rv genomic DNA. Three rounds of SCOTS captured cDNA from sputum specimens 8 and 9 were then subjected to three rounds of selective enrichment by hybridizing to biotinylated amplified homologous segment of esxJI. After selective enrichment cDNAs were cloned in pCR2.1-Topo vector (Invitrogen). Individual plasmid clones were characterized by sequencing insert cDNA fragments.
Allele Inactivation of esxKL and esxJI
An H37Rv ΔesxKL, esxJIi interrupted, and ΔesxJI mutants were generated by specialized transduction method (Bardarov et al., 2002) with the previously described modifications (Price et al., 2008). Deletion of the entire gene was confirmed by PCR, and lack of mRNA signal in cDNA prepared from the mutant strain by RT-qPCR. Complement strains were generated by cloning esxK or esxL for ΔesxKL mutant, and esxI for esxJIi interrupted mutant, downstream of constitutive hsp60 promoter in the single-copy integrating vector pLou3.
Primary Human Macrophages and THP-1 Cell Infections
Monocyte derived macrophages (PBMCs) were obtained from two healthy donors as previously described (Schlesinger and Horwitz, 1991; Graham et al., 2002; Price et al., 2008). Briefly, mononuclear cells were isolated by Ficoll (Sigma) gradients and total mononuclear cells incubated in Teflon wells for 5 days in RPMI media (GIBCO). Adherent macrophages were then selected by attachment in 24-well plate at a cell density of 5 × 105 cells and incubated for 5 days for further in vitro maturation prior to infection. Human leukemia macrophage cell line THP-1 was grown in RPMI 1640 to density of 1–2 × 106 cells per ml in 75 cm2 tissue culture flasks. As described by Theus et al. (2004), 3 days prior to infections cells were seeded at 5 × 105 cells per wells in 24-well plate with of 100 nM PMA and 150 units of human IFN-g. Bacterial strains were opsonized with native FBS (final concentration of 10% v/v) and then diluted in RPMI supplemented with 2% v/v FBS for inocula. Monolayers were infected overnight in RPMI-2% FBS lacking PMA and INF-g, and washed 3× prior to adding fresh media, which was changed as 48 h. Colony-forming units (CFU) counts were obtained by lysing triplicate wells at each time point with 1% Triton-X 100.
Surface Labeling of Mycobacteria with Fluorescein-Tagged Succinimidyl Ester
H37Rv and ΔesxKL mutant were labeled with fluorescein-tagged succinimidyl ester (Alexa fluor 488 and 594 from Molecular Probes and Invitrogen) as previously described (Beatty and Russell, 2000). Briefly, bacteria were washed and added to buffer containing 1 mM fluorescein succinimidyl ester. Following 1.5 h incubation at 37°C, bacteria, was washed as inocula (prepared as described above) for macrophage infections. For infections, 5 × 105 cells adhering to circular glass cover slips in 24-well culture plates were infected at MOI of 1:1 with mixture consisting of 50:50 of fluorescent labeled wild-type and ΔesxKL mutant bacteria. At 24, 48, and 72 h post-infections cells were washed three times with HANKS buffer and were fixed at 37°C in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min. Following washing with PBS cover slips were mounted on slide with the Prolong Antifade kit (Invitrogen). Labeled mycobacteria in infected macrophages were visualized directly using an Olympus F500 laser scanning confocal microscope.
esxK and esxL Interaction by Two-Hybrid System
The BacterioMatch Two-Hybrid Vector system (Stratagene) was used according to manufacturer’s instructions. Briefly, the BacterioMatch two-hybrid vectors pBT bait containing the λcI domain and pTRG target containing alpha-RNAP domain, were used as starting plasmids for cloning esxK and esxL genes respectively. The plasmids pBT–esxK and pTRG–esxL were then used for cotransformation of Escherichia coli XL1-Blue cells containing the reporter cassette. The transformed cells were plated on selective media containing 5 mM 3-amino-1,2,4-triazole and incubated at 37°C for 24 h.
Results
Analysis of Sputum Bacteria RNA
Mycobacterium tuberculosis gene expression during active lung infection was investigated by SCOTS and genomic array hybridization (Graham et al., 2002) by collecting sputum bacteria from consenting patients directly into 50 ml centrifuge tubes on ice. Originally developed to characterize RNA expression in M. tuberculosis growing in primary human macrophages, SCOTS uses hybridization with biotinylated bacterial genomic DNA to obtain the microbial component of total cDNA prepared from specimens containing low numbers of bacteria (Graham and Clark-Curtiss, 1999; Graham et al., 2002). A limited number of un-induced sputum specimens were obtained from patients prior to initiating therapy as described in Section “Materials and Methods.” Radiolabeled probes prepared from total normalized bacterial cDNA obtained by three rounds of SCOTS were hybridized with M. tuberculosis H37Rv genomic arrays on nylon. Complex hybridization patterns were observed for five sputum specimens (Figure 1), while a sixth showed very few hybridizations (not shown), and was removed from our analysis of the remaining five specimen cDNAs. Analyses of 16S rRNA copy number by reverse transcription and quantitative PCR (RT-qPCR) indicated an estimated 103–105 genome equivalents per specimen; it is therefore likely that some specimens contained bacteria at levels where variable detection of RNAs occurred at threshold levels (see Figure 1 and below).
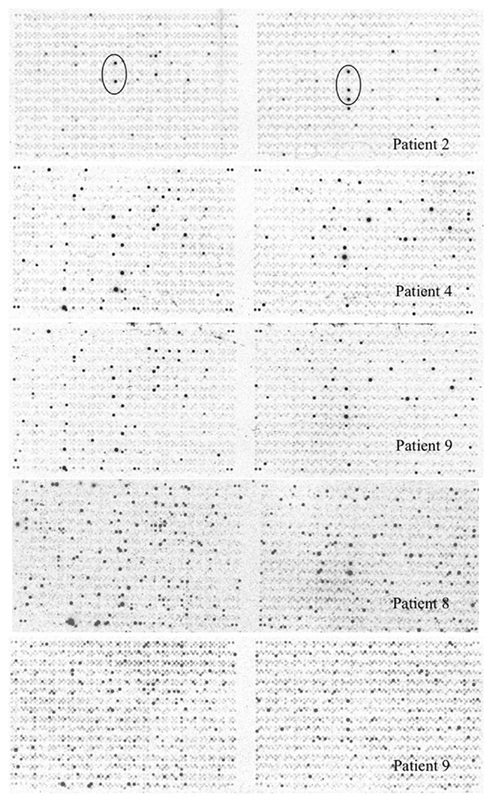
Figure 1. Mycobacterium tuberculosis genomic array hybridized with radiolabeled cDNAs obtained by SCOTS from sputum bacteria. Array patterns obtained by hybridizing cDNAs obtained by three rounds of SCOTS from bacteria in five un-induced sputum specimens as described in Section “Materials and Methods” are shown. Array features showing the highest levels of hybridization for patient 2 (circled) were readily detected for all specimens, and correspond to ORFs Rv1197 (esxK) and Rv1198 (esxL) on the left, and Rv2346c (esxP), Rv2347c (esxO), and Rv1047 on the right.
In order to test the reproducibility of SCOTS and array hybridization at the lowest levels of bacteria present in our set of sputum specimens (i.e., 103 bacteria), we simulated these analyses using a mixture of bacterial RNA derived from broth-grown bacterial cultures (0.01 ng) and 5 μg of HeLa cell RNA. Our results indicated that only hybridization signals in the upper half of the range of relative intensities among those apparent (threefold above background pixel intensity) were consistently detected in all three parallel control samples (data not shown). These experiments indicated that only abundant RNAs were reliably detected in our patient specimens. We therefore focused on identifying highly expressed RNAs in sputum bacteria, including identifying those most likely differentially expressed relative to those at mid-logarithmic phase in standard laboratory broth cultures. (Table 1 and Table S1 in Supplementary Material). Identification of abundant transcripts has been previously described as equally likely to identify important characteristics of different bacteria, and their growth within different environments (Sidders et al., 2007). A simple visual inspection of all six array hybridization patterns readily indicated a group of features hybridizing to all specimen cDNAs. These were nearly identical paired ORFs esxKL and esxPO (Figure 1). A more thorough image analysis indicated a group of 121 array features showing hybridization with SCOTS cDNAs from either all specimens, or four of five specimens (Table S1 in Supplementary Material). Those showing the highest levels of hybridization included the QILSS–Mtb9.9 paired ORFs noted above, and at lower levels esxB (encoding Cfp-10), and esxF. Among these readily detected RNAs we also found a much smaller subset of 29 array features (Table 1) that also consistently showed hybridization intensities close to local background levels (<3×) when first-strand cDNAs from mid-logarithmic phase cultures were used as array probes (data not shown). As shown in Table 1, these included genes potentially unique to M. tuberculosis annotated as “hypothetical,” those with potential roles in dipeptide, asparagine, phosphate, and sulfate transport, in riboflavin and trehalose synthesis, and notably the glyoxylate shunt (aceA), and transcriptional regulation (sigM and Rv0195).
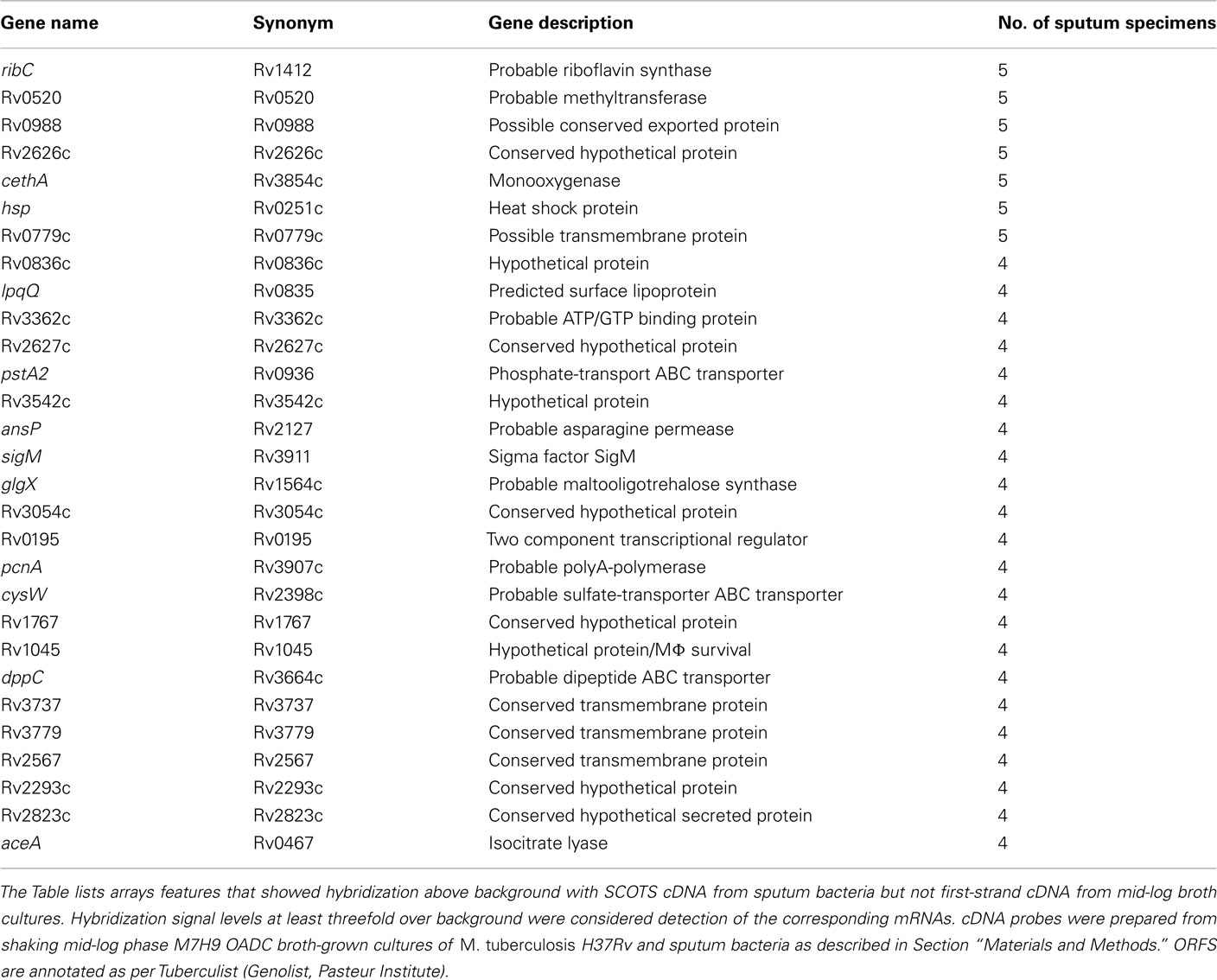
Table 1. Mycobacterium tuberculosi s ORFs corresponding to mRNAs detected either in four or five sputum specimens and potentially expressed in response to growth in the lung.
Identification of RNAs Annotated as esx Highly Expressed in Sputum Bacteria
Visually apparent array hybridization patterns among SCOTS cDNAs from sputum specimens (Figure 1) included array features corresponding to two nearly identical pairs of ORFs annotated as “esx” or esat-6 like genes in the H37Rv genome (as per Tuberculist, Lew et al., 2011). These ORFs in fact show only 10–30% amino acid similarity to Cfp-10 and Esat-6 (Louise et al., 2001), and have also been described as a “QILSS family” (Cole et al., 1998; Sutcliffe, 2011) based on encoding a shared carboxyl terminal motif in the 5′ ORFs, or “Mtb9.9 family” based on of protective CD4+ T-cell antigens encoded by the 3′ ORFs (Alderson et al., 2000). The 5′ and 3′ ORFs among these five pairs encode proteins with greater than 90% amino acid sequence similarity, and except for esxPO (Rv2396–Rv2397), are located just downstream of regions encoding paired PE and PPE gene family members (Cole et al., 1998; Bitter et al., 2009). Only one of these pairs, esxMN (Rv1792–Rv1793) lies within a region encoding a putative esx-type secretion system (Gey Van Pittius et al., 2001), designated ESX-5 (Bitter et al., 2009). This locus may be involved in secretion of QILLS–Mtb9.9 factors, which has been shown to be independent of apparatus encoded at ESX-1 (Champion et al., 2006).
As array features for QILLS–Mtb9.9 family ORFs were likely to have cross-hybridized cDNAs, we extended the SCOTS approach to capture and clone the specific relevant cDNA for RNAs highly expressed in sputum bacteria. A PCR-amplified biotinylated DNA fragment corresponding to the randomly selected EsxJI coding region was used to hybridize relevant cDNAs from normalized cDNA already obtained by SCOTS from sputum specimens 8 and 9 as described in Section “Materials and Methods.” Cloning of cDNAs captured in an E. coli plasmid and sequencing as described indicated that the most abundant cDNAs in the bacteria in both of these specimens were likely those for the esxKL gene pair (Table 2), while those for esxPO were also frequently identified. As these were also the esx transcripts most readily detected by array hybridization in the patient specimen 2 (that showing the least amount of hybridization, Figure 1), we do not think that expected biases in plasmid cloning of cDNAs prevented us from distinguishing among gene family transcripts by this novel approach. PCR analysis of cDNA prepared from H37Rv grown in laboratory cultures with primers spanning intergenic and ORF junctions in the esxKL coding regions indicated that ORFs Rv1197 and Rv1198 and upstream Rv1196 (PPE-18) and Rv1195 (PE-13) can be transcribed in a single polycistronic mRNA (data not shown). Although Goldstone et al. (2009) showed that expression of an upstream transcript encoding PE-13 and PPE-18 can also be independently regulated, analysis by RT-qPCR indicated that mRNA steady-state levels for corresponding upstream PE and PPE ORFs were reduced at least fivefold in both ΔesxKL and esxJIi mutants (data not shown).

Table 2. Mycobacterium tuberculosis esx transcripts identified by novel targeted SCOTS in patient specimens 8 and 9.
esxKL and esxJI mRNAs are Differentially Expressed
Duplication and conservation of nearly identical protein coding capacity among the five QILLS–Mtb9.9 allele pairs (esxJI, esxKL, esxMN, esxPO, and esxWV) suggests that the encoded products are important contributors for M. tuberculosis colonization of normal ecological niches (Cole et al., 1998). In additional to increasing gene copy number, retaining duplications with minimal variation may also reflect capacity for differential expression under different growth conditions (Kumar et al., 2005). We therefore determined steady-state mRNA levels for these gene pairs in M. tuberculosis H37Rv growing at mid-log in standard media by targeting the different intergenic regions with RT-qPCR primers. All transcripts were expressed at different steady-sate levels during growth in broth culture (Table 3), ranging from about fourfold lower (for esxJI) to eightfold higher (for esxPO) than a sigA reference transcript (Manganelli et al., 1999). Bacteria growing in THP-1 macrophages at 48 h post-infection, and in C57BL6 mouse lungs 21 day following high dose infections showed greatly elevated esxJI and reduced esxKL transcript levels relative to bacteria at mid-log in broth cultures (Table 4). As esxJI mRNA levels were at dramatically higher levels (46- to 48-fold) at 21 days post-infection in bacteria present in mouse lung tissues, we attempted to determine levels in bacteria present in a few additional sputum specimens and a single human patient tissue specimen directly by RT-qPCR, but were only able to consistently measure abundant rRNA levels. Taken together, our data demonstrate the differential regulation of esxJI and esxKL mRNA expression in environments including those encountered during infection.
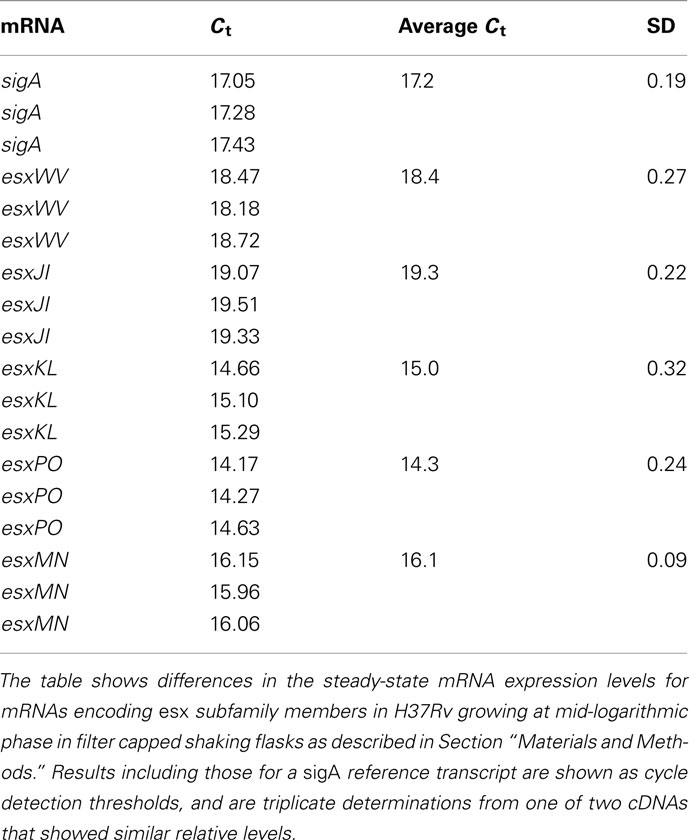
Table 3. QILSS–Mtb9.9 family mRNA are expressed at different levels.
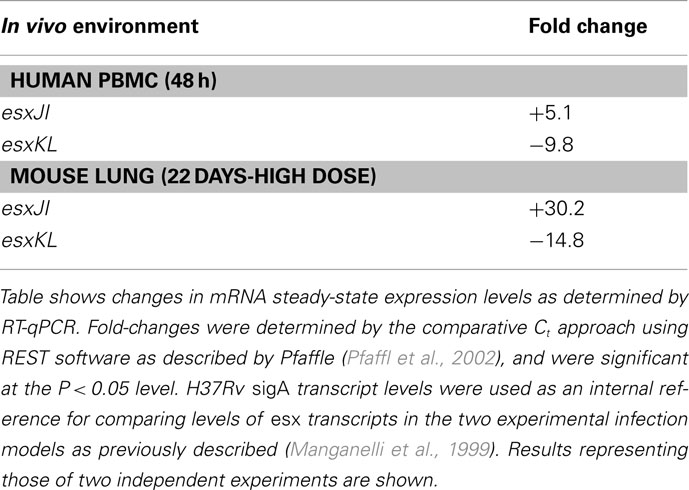
Table 4. esxJI and esxKL mRNAs are differentially expressed in different in vivo environments.
esxKL Contributes to the Growth of M. tuberculosis in Macrophages
As we identified esxKL as highly expressed by M. tuberculosis during active human disease, we sought to investigate contributions of the encoded factors to growth in a cell culture model of infection (Theus et al., 2004; Price et al., 2008). Human monocytic THP-1 cell monolayers were infected at a low MOI of 1: 1 either with H37Rv, an H37Rv ΔesxKL deletion mutant, or esxK or esxL complemented isogenic strains (see Materials and Methods). Mycobacterial growth was monitored as CFU in lysates at 24 and 96 h post-infection. As shown in Figure 2, the ability of the ΔesxKL mutant to colonize these cells was impaired (P < 0.0001 at 4 days) relative to the wild-type. Complementation of the ΔesxKL mutant with esxL, but not esxK, restored intracellular growth (Figure 2B). Additional similar experiments with experimental infection of primary single-donor PBMC as previously described (Graham and Clark-Curtiss, 1999; Price et al., 2008) confirmed these results (Figure 2C). These comparisons of isogenic recombinant strains indicated that the partial but significant intracellular growth defect seen in this model was due to a lack of esxL expression. Further analyses, as describe below, indicated that this defect was also apparent in replication rate during growth in laboratory broth (but not plate) media.
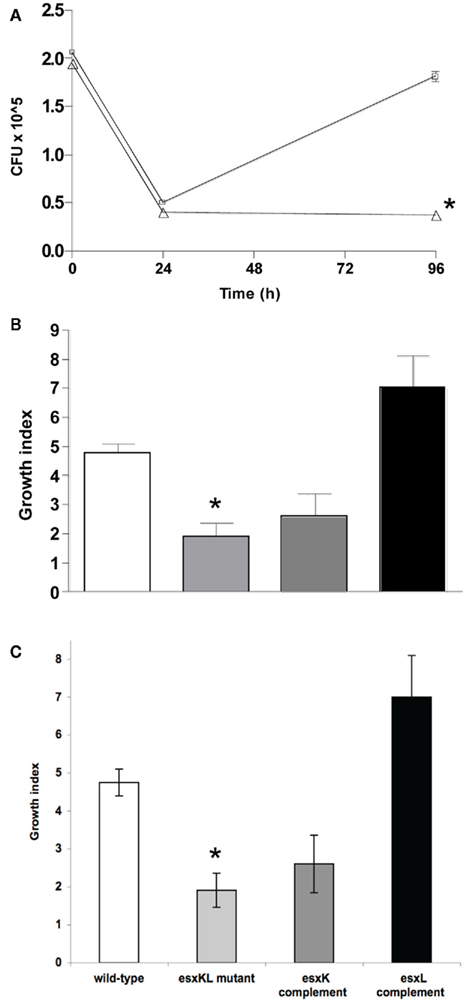
Figure 2. ΔesxKL mutant is defective in intracellular growth. (A) THP-1 cells were infected with wild-type (open squares) or ΔesxKL mutant (open triangles) at a MOI of 1: l. Data points indicate the mean number of CFUs for three independently infected wells and error bars indicate SD from means. Intracellular growth of the ΔesxKL mutant was significantly reduced in three independent experiments (P < 0.005)* at 96 h as determined by unpaired t-test. (B) THP-1 cells were infected at MOI of 1:1 and a growth index as CFU at 96 h relative to CFU at 24 h was determined for wild-type (open bar), ΔesxKL (light gray bar), esxK complemented (dark gray bar), and esxL complemented (black bar) strains. Reduced intracellular growth of ΔesxKL (P < 0.005)* was reversed by restoring only esxL. Data shown is a representative of two independent experiments. (C) The ΔesxKL mutant showed this growth defect in similar experiments with a primary single-donor PBMC infection model, and restoring esxL allowed normal intracellular growth.
EsxK and EsxL Do not Interact in an E. coli Two-hybrid System
Mycobacterium tuberculosis Cfp-10 and Esat-6 proteins show only limited similarity to EsxK and EsxL, but are encoded within related chromosomal regions (Gey Van Pittius et al., 2001) and interact to form a 1:1 dimer (Renshaw et al., 2002). This dimerization is an essential aspect of their function (Renshaw et al., 2005). We therefore determined if we could demonstrate an EsxK–EsxL interaction in an E. coli two-hybrid system (Dove et al., 1997). Although all controls gave appropriate results, no colonies were obtained on selective media when E. coli was co-transformed with pBT–EsxK and pTRG–EsxL fusion plasmids. Western blot analyses confirmed soluble cytoplasmic expression of both EsxK and EsxL fusions protein partners (data not shown). We were therefore unable to demonstrate this QILLS–Mtb9.9 family pair interaction. Mahmood et al. (2011) have recently shown interaction of the most similar pair, EsxWV (Rv3619c–Rv3620c), but with an approximate 10-fold lower Kd than the Cfp-10–Esat-6-pair. If EsxK and EsxL do interact, we were unable to show it with this assay, and as described above, and complementation of esxL alone in the ΔesxKL mutant reversed an intracellular growth defect (Figure 2).
Co-Infection with Wild-Type Restores Growth of a ΔesxKL Mutant
Previous studies have shown that esxL is exported, and found in M. tuberculosis H37Rv culture supernatants (Mattow et al., 2003; Champion et al., 2006). To determine if the esxL encoded secreted protein factor produced by wild-type bacteria could rescue the partial growth defect of the ΔesxKL mutant, THP-1 macrophages were co-infected with equal numbers of H37Rv and ΔesxKL mutant at an MOI of 1:1. Intracellular growth of H37Rv and the ΔesxKL mutant sub-populations and controls were determined by plating cell lysates on different selective agars and determining CFUs (Figure 3A). Even with this relatively insensitive approach, it was clear that co-infection with H37Rv fully restored growth of the ΔesxKL mutant (P < 0.0001) as determine by total lysate CFUs. We then co-infected cells with ΔesxKL, and decreasing numbers of H37Rv. ΔesxKL CFUs in monolayer lysates declined with decreased H37Rv co-Infection levels (Figure 3B). Similar experiments with human primary PBMCs confirmed that wild-type co-infection at low MOI was able to restore the ability of the mutant population to replicate in the same monolayer (data not shown). These results indicated that wild-type bacteria confer normal growth either directly via by providing a trans-acting EsxL secreted protein in the monolayer, or by inhabiting shared cells or phagosomes and modulating a common intracellular environment.
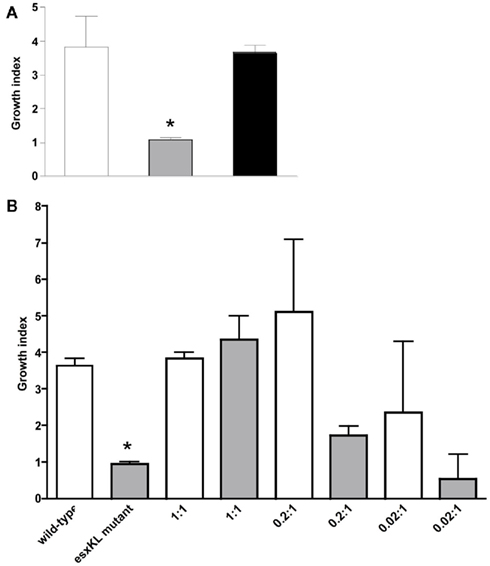
Figure 3. Co-infection with wild-type allows intracellular growth of the ΔesxKL mutant population. (A) THP-1 monolayers were infected with wild-type or ΔesxKL mutant or 1:1 ratio of wild-type and ΔesxKL mutant strains at a MOI of 1:1. CFUs were determined at 24 and 96 h post-infection. Growth indices are shown for wild-type (open bar), ΔesxKL (gray bar), and ΔesxKL sub-population in co-infection (black bar). Reduced intracellular growth of ΔesxKL mutant (P < 0.005)* was rescued by co-infection with wild-type bacteria. A representative of three independent experiments is shown (B) THP-1 monolayers were co-infected with fixed number of ΔesxKL and varying concentration of wild-type H37Rv. Growth index at 96 h post-infection for wild-type (open bar) and ΔesxKL (gray bar) in each co-infection well was determined. Data from a single experiment is shown.
To distinguish between these possibilities, we examined the distribution of differentially labeled bacteria in these low multiplicity infections by fluorescent microscopy. THP-1 macrophages were inoculated with equal numbers of surface labeled wild-type (green dye) and ΔesxKL mutant (red dye) at MOI of 1:1 as described in the Section “Materials and Methods.” At 24, 48, and 72 h post-infection, infected cells were examined by confocal microscopy. As shown in Figure 4, few if any wild-type and ΔesxKL mutant bacteria resided within in the same cells during the course of these infections. Our data are therefore consistent with a secreted EsxL factor produced by wild-type bacteria able to restore growth to bacteria lacking this factor in independently infected cells.
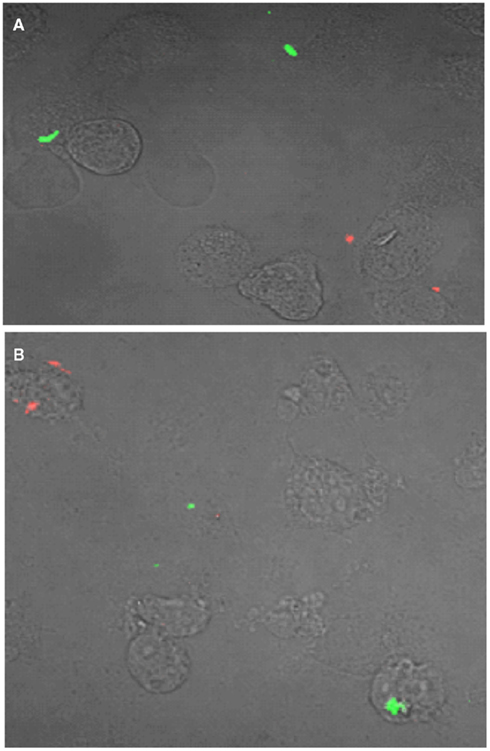
Figure 4. esxKL mutant and wild-type bacteria do not inhabit the same cells during low MOI co-infection. A representative confocal microscopy image of THP-1 cells simultaneously infected with equal proportion of fluorescently labeled wild-type (red) and ΔesxKL mutant (green) at MOI of 1:1 is shown. Cells were infected and fixed at (A) 24 h, and (B) 72 h post-infection as described in Section “Materials and Methods.”
QILLS–Mtb9.9 Mutants Have Growth Defects in Laboratory Cultures
While characterizing the contribution of esxKL to the ability of M. tuberculosis to colonize human cell line macrophages, we also initiated experiments to compare the role of esxJI. esxJI was chosen among the four other QILLS pairs because of its different mRNA expression pattern (Table 4), and otherwise highly similar protein product. Although EsxI and EsxL differ by only two amino acids, both differences are predicted to alter alpha-helical content of the amino terminal halves of the proteins (Figure 5). In an extended effort to generate a ΔesxJI mutant, we were consistently unable to recover hygromycin resistant transformants following allele exchange approach successful in obtaining ΔesxKL strain pairs (see Materials and Methods). We were eventually able to obtain the desired ΔesxJI knockout by allowing transformants plates to incubate for over 2 months (rather than 3 weeks, not shown), but our efforts were then inhibited by the inability of this extremely slow growing ΔesxJI knockout mutant to grow in broth cultures. As an alternate strategy, we instead replaced the intergenic spacer region between esxJ and esxI with a selectable hygromycin resistance marker leaving the two ORFs intact. This “esxJI-interrupted” (esxJIi) mutant formed colonies with a more limited delay (1 week) and was of interest in exhibiting several different growth defects in broth cultures. esxJIi showed an extended lag period prior to replicating at a reduced rate, and then reached stationary phase at a lower density compared to both wild-type and ΔesxKL mutant bacteria (Figure 6A). These growth defects could also be reversed by co-culturing with wild-type bacteria (Figure 6B) or by complementing only the esxI region with a single-copy integrating vector (Figure 6A).
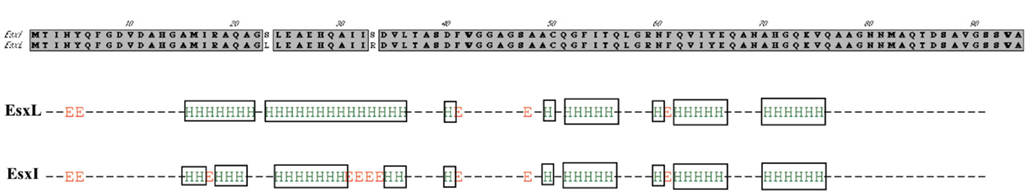
Figure 5. Secondary structure prediction for M. tuberculosis EsxL (Rv1198) and EsxI (Rv1037c). The figures depicts the difference in the alpha-helical secondary structures of EsxL and EsxI based on analysis with nnpredict (Kneller et al., 1990).
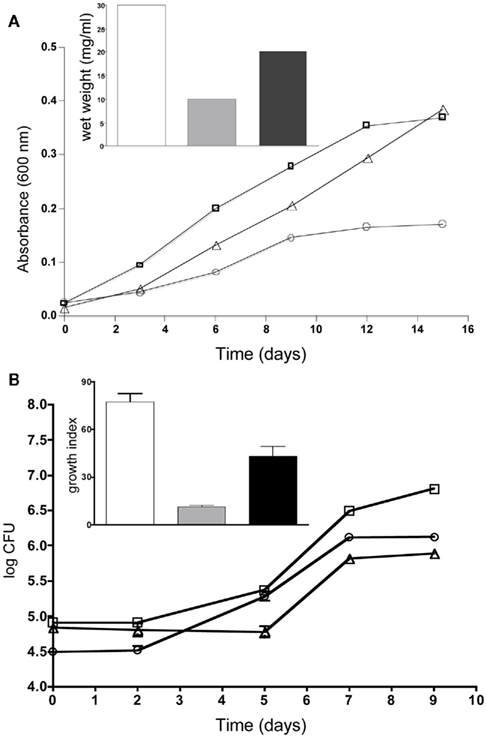
Figure 6. esxJIi mutation reduces growth in cultures. (A) Cultures were grown in 7H9 broth supplemented with 0.05% Tween and OADC and optical density measurements taken at 600 nm. Growth of wild-type is indicated with squares, esxJIi mutant with circles, and esxI complemented strain with triangles. The inset shows wet weight measurements of wild-type (bar), esxJIi (gray bar), and esxI complemented strain (black bar) at 15 days of growth. A representative of three independent experiments is shown. (B) Co-culturing with wild-type bacteria rescues growth of esxJIi mutant. Growth of H37Rv (squares), the esxJIi mutant (triangles), and the esxJIi mutant in co-culture (closed circles) were determined by enumerating CFUs. Plate counts were in triplicate and averaged. Inset shows growth indices for wild-type (bar), esxJIi mutant (gray bar), and esxJIi mutant in co-culture (black bar) calculated as CFU at 9 days relative day 0. Data shown is a representative of three independent experiments where the wild-type and mutant were grown both independently and in co-culture, and showed this difference in growth of the mutant.
As esxI was clearly important for the growth of M. tuberculosis in the absence of any host, we re-examined our ΔesxKL mutant more closely to determine if we had previously failed to detect any similar in vitro growth defects. When comparing H37Rv, ΔesxKL knockout, and esxL complemented strains we were able to observe that the ΔesxKL mutant was growing at a slower growth rate compared to wild-type and complemented strains (Figure 7). Unlike either esxJI mutant, ΔesxKL grew normally on plates (not shown), and unlike esxJIi, to normal stationary population density and without an extended lag in broth cultures (Figure 7). As both mutants showed growth defects in the absence of host cells, our experiments indicate the EsxI and EsxL secreted factors make basic contributions to growth, rather than being specifically involved in colonizing host macrophages.
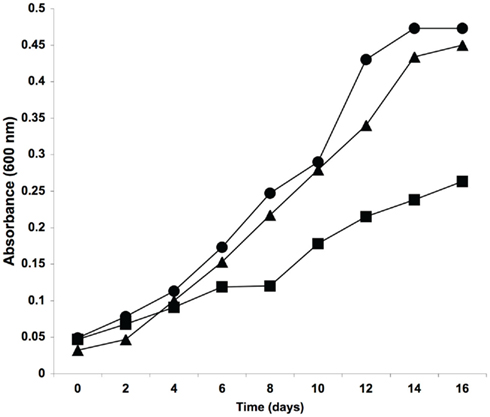
Figure 7. Reduced in vitro growth of ΔesxKL mutant. Cultures were grown in M7H9 broth supplemented with 0.05% Tween and OADC and optical density measurements taken at 600 nm. Circles indicate optical density for wild-type, squares for ΔesxKL mutant, and triangles for esxL complemented mutant strain. The reduced growth rate of the mutant was restored to wild-type levels by complementing with esxL. A representative of three experiments is shown.
Discussion
As active human infections proceed from only a few bacteria surviving aerosol transmission in droplet nuclei to initiate growth within resting alveolar macrophages, and eventually to replication to high titers on the inner walls of pulmonary cavities, tubercle bacilli inhabit quite different in vivo environments. Colonization of these multiple natural ecological niches requires coordinated expression of different sets of genes, including not only those encoding factors that directly influence hosts, but also those that govern both basic and alternative microbial metabolism. Central to the multiple strategies used by tubercle bacilli to evade host immune responses is a need for temporal regulation of bacterial growth and replication among microbes residing in cells and tissues. In both latent and active human disease, the dynamic results of temporal immune responses remodel lung tissue into novel structures that are the hallmark of the disease, potentially benefiting both host and pathogen (Russell, 2007). Among multiple and heterogeneous bacterial populations found in these lesions in human pulmonary infections, individual local races may be won and lost, even at different sites within the same lung. We remain largely unable to answer central questions of how and why the balance between bacterial replication and effective cell-mediated immunity can readily tip in one direction or another. The ability of bacteria to grow within these niches must therefore be viewed as a critical aspect of successful colonization, encompassing fundamental aspects of the ability to both provoke and avoid immune responses, and thereby ultimately determine disease progress and outcome.
Bacteria are traditionally viewed as autonomous free-living single-celled organisms capable of initiating growth in the absence of others and without need of exogenous signals or growth factors typical of eukaryotic cells. Over the last decade, roles for intercellular signaling molecules have become well defined, particularly the serine lactones produced by Gram-negative organisms that modulate density dependent gene expression (Shank and Kolter, 2009), and peptides that regulate genetic exchange and production of adherence factors and exoenzymes in Gram positive bacteria (Thoendel and Horswill, 2010). Previous studies have so far identified only one such putative signal molecule produced by M. tuberculosis, that designated “Rfp” or resuscitation-promoting-factor (Kana et al., 2008). The five members of this family present in H37Rv were identified based on sequence similarity to a Micrococcus luteus protein with a role in emergence from long-term starvation in laboratory cultures. In M. tuberculosis these were also originally described as autocrine growth factors (Mukamolova et al., 2002). However, determinations of the crystal structures of the M. tuberculosis Rfps indicate that they are likely instead actually peptidoglycan hydrolases that remodel bacterial cell walls, thereby allowing new synthesis involved in growth, septation, and cell division (Kana and Mizrahi, 2010). This emphasizes the potential for secreted proteins recognized as acting as “growth factors” to be other than signal molecules per se. Rather they may be diffusible enzymes with mechanistic roles in growth that may or may not be expressed in response to other well-described types of signal molecules.
We do not yet know how EsxI and EsxL contribute to the growth of M. tuberculosis, nor how EsxL secreted by wild-type cells is able to rescue the intracellular growth phenotype of ΔesxKL mutant bacteria growing in different cells in the same monolayer. We believe that there are several likely scenarios. First, even the smallest bacterial proteins would require an active process to exit and enter host cells. It has recently been described by J. S. Schorey’s laboratory that experimentally infected murine cell line macrophages release exosomes containing secreted M. tuberculosis factors. Treatment of cells with bacterial culture filtrate proteins also causes cells to take up and release these microbial factors as membrane enclosed exosomes capable of delivering contents into other cells (Giri et al., 2010). Cells that are not infected are thereby altered, including becoming attenuated in responding to IFN-gamma, and thereby more permissive from intracellular growth of M. tuberculosis (Singh et al., 2011). As macrophage phagosomes containing M. tuberculosis remain in an early endocytic pathway capable of exchanging cellular material with the host cell and the environment (Beatty et al., 2001), secreted EsxK may be able to leave and enter phagosomes of infected host cells through an exocytic pathway. We do not yet know if this is can account for the nearly complete mutant rescue we see with low MOI co-infection in our human primary PBMC infection model.
Alternately, in any cell culture infection model only a percentage of host cells are infected (i.e., 10–30% at the 1:1 MOI used here), and some of these cells get higher number of bacteria (6–10 or more as determined by acid fast staining and microscopy (data not shown). Some cells are therefore likely to lyse early in the experimental time course, and release both bacteria and secreted factors. Uptake of EsxL could therefore also occur by normal macrophage pinocytosis, and complement mutant bacteria through normal endocytoxic vesicle trafficking. Alternately, Renshaw et al. (2005) showed that the Cfp-10–Esat-6 dimer binds specifically to human monocytic cells. NMR solution structure modeling indicated that the complex is unlikely to have a pore forming (cytolytic) or catalytic role, and a requirement for a specific carboxyl terminal domain of Cfp-10 in the interaction is consistent with a host receptor interaction. The authors propose that this may exploit a host cell signaling interaction and alter host cell behavior in a way favorable to the pathogen. Subsequently Pathak et al. (2007) showed that Cfp-10–Esat-6 directly binds Toll-like receptor 2, and thereby inhibits macrophage inflammatory signaling pathways. We do not yet know if these properties are shared by other the Esat-6 subfamily members who contributions to bacterial growth we describe.
Determining the specific mechanisms by which these less well-described Cfp-10–Esat-6 like QILLS–Mtb9.9 secreted factors are able to influence bacterial growth in the absence of host cells, and then during intracellular growth and growth in the lung during active disease will be an important goal for future research. The ability of M. tuberculosis to grow slowly, and to modulate its growth during infection is undoubtedly a key factor in its ability to frequently avoid a sterilizing host immune response. Further defining the contributions of this family of secreted factors to that ability will be important in better understanding this common and deadly disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Public Health Service grant RO1A148635 from the National Institutes of Allergy and Infectious Disease to James E. Graham. The authors thank Michael Cynamon at SUNY Upstate Medical University for providing mouse lung tissue specimens. We thank Linda Reeves-Hammock in Nashville and Forest Arnold, MD in Louisville for their assistance with identifying patients and obtaining patient sputum specimens.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Cellular_and_Infection_Microbiology_-_closed_section/10.3389/fmicb.2011.00266/abstract
Table S1. Highly expressed mRNAs in sputum bacteria. Table shows a list of H37Rv array features that were detected as expressed by hybridizing SCOTS cDNAs probes from bacteria in at least four or five patient sputum specimens. ORFs highlighted in green are those whose mRNAs were not detected (3× over background) by conventional first-strand cDNA probes from H37Rv in standard mid-log shaking broth cultures. Those in blue encode QILLS–Mtb9.9 family pairs. ORFS are annotated as per Tuberculist (Lew et al., 2011).
References
Alderson, M. R., Bement, T., Day, C. H., Zhu, L., Molesh, D., Skeiky, Y. A., Coler, R., Lewinsohn, D. M., Reed, S. G., and Dillon, D. C. (2000). Expression cloning of an immunodominant family of Mycobacterium tuberculosis antigens using human CD4(+) T cells. J. Exp. Med. 191, 551–560.
Bardarov, S., Bardarov, S. Jr., Pavelka, M. S. Jr., Sambandamurthy, V., Larsen, M., Tufariello, J., Chan, J., Hatfull, G., and Jacobs, W. R. Jr. (2002). Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148, 3007–3017.
Beatty, W. L., and Russell, D. G. (2000). Identification of mycobacterial surface proteins released into subcellular compartments of infected macrophages. Infect. Immun. 68, 6997–7002.
Beatty, W. L., Ullrich, H. J., and Russell, D. G. (2001). Mycobacterial surface moieties are released from infected macrophages by a constitutive exocytic event. Eur. J. Cell Biol. 80, 31–40.
Bishai, W. (1998). The Mycobacterium tuberculosis genomic sequence: anatomy of a master adaptor. Trends Microbiol. 6, 464–465.
Bitter, W., Houben, E. N., Bottai, D., Brodin, P., Brown, E. J., Cox, J. S., Derbyshire, K., Fortune, S. M., Gao, L. Y., Liu, J., Gey van Pittius, N. C., Pym, A. S., Rubin, E. J., Sherman, D. R., Cole, S. T., and Brosch, R. (2009). Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 5, e1000507. doi:10.1371/journal.ppat.1000507
Champion, P. A., Stanley, S. A., Champion, M. M., Brown, E. J., and Cox, J. S. (2006). C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 313, 1632–1636.
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., Gordon, S. V., Eiglmeier, K., Gas, S., Barry, C. E. III, Tekaia, F., Badcock, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R., Devlin, K., Feltwell, T., Gentles, S., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Krogh, A., McLean, J., Moule, S., Murphy, L., Oliver, K., Osborne, J., Quail, M. A., Rajandream, M. A., Rogers, J., Rutter, S., Seeger, K., Skelton, J., Squares, R., Squares, S., Sulston, J. E., Taylor, K., Whitehead, S., and Barrell, B. G. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544.
Desjardin, L. E., Perkins, M. D., Teixeira, L., Cave, M. D., and Eisenach, K. D. (1996). Alkaline decontamination of sputum specimens adversely affects stability of mycobacterial mRNA. J. Clin. Microbiol. 34, 2435–2439.
Dove, S. L., Joung, J. K., and Hochschild, A. (1997). Activation of prokaryotic transcription through arbitrary protein-protein contacts. Nature 386, 627–630.
Garton, N. J., Waddell, S. J., Sherratt, A. L., Lee, S. M., Smith, R. J., Senner, C., Hinds, J., Rajakumar, K., Adegbola, R. A., Besra, G. S., Butcher, P. D., and Barer, M. R. (2008). Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 5, e75. doi:10.1371/journal.pmed.0050075
Gey Van Pittius, N. C., Gamieldien, J., Hide, W., Brown, G. D., Siezen, R. J., and Beyers, A. D. (2001). The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G + C Gram-positive bacteria. Genome Biol. 2, 44.
Giri, P. K., Kruh, N. A., Dobos, K. M., and Schorey, J. S. (2010). Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis-infected and culture filtrate protein-treated macrophages. Proteomics 10, 3190–3202.
Goldstone, R. M., Goonesekera, S. D., Bloom, B. R., and Sampson, S. L. (2009). The transcriptional regulator Rv0485 modulates the expression of a pe and ppe gene pair and is required for Mycobacterium tuberculosis virulence. Infect. Immun. 77, 4654–4667.
Graham, J. E., and Clark-Curtiss, J. E. (1999). Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc. Natl. Acad. Sci. U.S.A. 96, 11554–11559.
Graham, J. E., Peek, R. M. Jr., Krishna, U., and Cover, T. L. (2002). Global analysis of Helicobacter pylori gene expression in human gastric mucosa. Gastroenterology 123, 1637–1648.
Grosset, J. (2003). Mycobacterium tuberculosis in the extracellular compartment: an underestimated adversary. Antimicrob. Agents Chemother. 47, 833–836.
Homolka, S., Niemann, S., Russell, D. G., and Rohde, K. H. (2010). Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog. 6, e1000988. doi:10.1371/journal.ppat.1000988
Hunter, R. L. (2011). Pathology of post primary tuberculosis of the lung: an illustrated critical review. Tuberculosis (Edinb) 91, 497–509.
Jones, G. J., Gordon, S. V., Hewinson, R. G., and Vordermeier, H. M. (2010). Screening of predicted secreted antigens from Mycobacterium bovis reveals the immunodominance of the ESAT-6 protein family. Infect. Immun. 78, 1326–1332.
Kana, B. D., Gordhan, B. G., Downing, K. J., Sung, N., Vostroktunova, G., Machowski, E. E., Tsenova, L., Young, M., Kaprelyants, A., Kaplan, G., and Mizrahi, V. (2008). The resuscitation-promoting factors of Mycobacterium tuberculosis are required for virulence and resuscitation from dormancy but are collectively dispensable for growth in vitro. Mol. Microbiol. 67, 672–684.
Kana, B. D., and Mizrahi, V. (2010). Resuscitation-promoting factors as lytic enzymes for bacterial growth and signaling. FEMS Immunol. Med. Microbiol. 58, 39–50.
Kesavan, A. K., Brooks, M., Tufariello, J., Chan, J., and Manabe, Y. C. (2009). Tuberculosis genes expressed during persistence and reactivation in the resistant rabbit model. Tuberculosis (Edinb) 89, 17–21.
Kneller, D. G., Cohen, F. E., and Langridge, R. (1990). Improvements in protein secondary structure prediction by an enhanced neural network. J. Mol. Biol. 214, 171–182.
Kumar, A., Chandolia, A., Chaudhry, U., Brahmachari, V., and Bose, M. (2005). Comparison of mammalian cell entry operons of mycobacteria: in silico analysis and expression profiling. FEMS Immunol. Med. Microbiol. 43, 185–195.
Lew, J. M., Kapopoulou, A., Jones, L. M., and Cole, S. T. (2011). TubercuList – 10 years after. Tuberculosis (Edinb) 91, 1–7.
Louise, R., Skjot, V., Agger, E. M., and Andersen, P. (2001). Antigen discovery and tuberculosis vaccine development in the post-genomic era. Scand. J. Infect. Dis. 33, 643–647.
Mahmood, A., Srivastava, S., Tripathi, S., Ansari, M. A., Owais, M., and Arora, A. (2011). Molecular characterization of secretory proteins Rv3619c and Rv3620c from Mycobacterium tuberculosis H37Rv. FEBS J. 278, 341–353.
Manganelli, R., Dubnau, E., Tyagi, S., Kramer, F. R., and Smith, I. (1999). Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol. Microbiol. 31, 715–724.
Mastorides, S. M., Oehler, R. L., Greene, J. N., Sinnott, J. T., Kranik, M., and Sandin, R. L. (1999). The detection of airborne Mycobacterium tuberculosis using micropore membrane air sampling and polymerase chain reaction. Chest 115, 19–25.
Mattow, J., Schaible, U. E., Schmidt, F., Hagens, K., Siejak, F., Brestrich, G., Haeselbarth, G., Muller, E. C., Jungblut, P. R., and Kaufmann, S. H. (2003). Comparative proteome analysis of culture supernatant proteins from virulent Mycobacterium tuberculosis H37Rv and attenuated M. bovis BCG Copenhagen. Electrophoresis 24, 3405–3420.
Mukamolova, G. V., Turapov, O. A., Young, D. I., Kaprelyants, A. S., Kell, D. B., and Young, M. (2002). A family of autocrine growth factors in Mycobacterium tuberculosis. Mol. Microbiol. 46, 623–635.
Pallen, M. J. (2002). The ESAT-6/WXG100 superfamily – and a new Gram-positive secretion system? Trends Microbiol. 10, 209–212.
Pathak, S. K., Basu, S., Basu, K. K., Banerjee, A., Pathak, S., Bhattacharyya, A., Kaisho, T., Kundu, M., and Basu, J. (2007). Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat. Immunol. 8, 610–618.
Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002). Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36.
Price, C. T., Bukka, A., Cynamon, M., and Graham, J. E. (2008). Glycine betaine uptake by the ProXVWZ ABC transporter contributes to the ability of Mycobacterium tuberculosis to initiate growth in human macrophages. J. Bacteriol. 190, 3955–3961.
Rachman, H., Strong, M., Ulrichs, T., Grode, L., Schuchhardt, J., Mollenkopf, H., Kosmiadi, G. A., Eisenberg, D., and Kaufmann, S. H. (2006). Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 74, 1233–1242.
Renshaw, P. S., Lightbody, K. L., Veverka, V., Muskett, F. W., Kelly, G., Frenkiel, T. A., Gordon, S. V., Hewinson, R. G., Burke, B., Norman, J., Williamson, R. A., and Carr, M. D. (2005). Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 24, 2491–2498.
Renshaw, P. S., Panagiotidou, P., Whelan, A., Gordon, S. V., Hewinson, R. G., Williamson, R. A., and Carr, M. D. (2002). Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J. Biol. Chem. 277, 21598–21603.
Rohde, K., Yates, R. M., Purdy, G. E., and Russell, D. G. (2007). Mycobacterium tuberculosis and the environment within the phagosome. Immunol. Rev. 219, 37–54.
Schlesinger, L. S., and Horwitz, M. A. (1991). Phagocytosis of Mycobacterium leprae by human monocyte-derived macrophages is mediated by complement receptors CR1 (CD35), CR3 (CD11b/CD18), and CR4 (CD11c/CD18) and IFN-gamma activation inhibits complement receptor function and phagocytosis of this bacterium. J. Immunol. 147, 1983–1994.
Schnappinger, D., Ehrt, S., Voskuil, M. I., Liu, Y., Mangan, J. A., Monahan, I. M., Dolganov, G., Efron, B., Butcher, P. D., Nathan, C., and Schoolnik, G. K. (2003). Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J. Exp. Med. 198, 693–704.
Shank, E. A., and Kolter, R. (2009). New developments in microbial interspecies signaling. Curr. Opin. Microbiol. 12, 205–214.
Shoen, C. M., DeStefano, M. S., Sklaney, M. R., Monica, B. J., Slee, A. M., and Cynamon, M. H. (2004). Short-course treatment regimen to identify potential antituberculous agents in a murine model of tuberculosis. J. Antimicrob. Chemother. 53, 641–645.
Sidders, B., Withers, M., Kendall, S. L., Bacon, J., Waddell, S. J., Hinds, J., Golby, P., Movahedzadeh, F., Cox, R. A., Frita, R., Ten Bokum, A. M., Wernisch, L., and Stoker, N. G. (2007). Quantification of global transcription patterns in prokaryotes using spotted microarrays. Genome Biol. 8, R265.
Simon, R., Lam, A., Li, M. C., Ngan, M., Menenzes, S., and Zhao, Y. (2007). Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 3, 11–17.
Singh, P. P., LeMaire, C., Tan, J. C., Zeng, E., and Schorey, J. S. (2011). Exosomes released from M. tuberculosis infected cells can suppress IFN-gamma mediated activation of naive macrophages. PLoS ONE 6, e18564. doi:10.1371/journal.pone.0018564
Sutcliffe, I. C. (2011). New insights into the distribution of WXG100 protein secretion systems. Antonie Van Leeuwenhoek 99, 127–131.
Talaat, A. M., Ward, S. K., Wu, C. W., Rondon, E., Tavano, C., Bannantine, J. P., Lyons, R., and Johnston, S. A. (2007). Mycobacterial bacilli are metabolically active during chronic tuberculosis in murine lungs: insights from genome-wide transcriptional profiling. J. Bacteriol. 189, 4265–4274.
Theus, S. A., Cave, M. D., and Eisenach, K. D. (2004). Activated THP-1 cells: an attractive model for the assessment of intracellular growth rates of Mycobacterium tuberculosis isolates. Infect. Immun. 72, 1169–1173.
Thoendel, M., and Horswill, A. R. (2010). Biosynthesis of peptide signals in Gram-positive bacteria. Adv. Appl. Microbiol. 71, 91–112.
Timm, J., Post, F. A., Bekker, L. G., Walther, G. B., Wainwright, H. C., Manganelli, R., Chan, W. T., Tsenova, L., Gold, B., Smith, I., Kaplan, G., and McKinney, J. D. (2003). Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. U.S.A. 100, 14321–14326.
Appendix
Supplementary materials and methods
Mycobacterial Cultures
Mycobacterium tuberculosis H37Rv (ATCC no. 25618) was obtained from the American Type Culture Collection and grown in Middlebrook 7H9 broth supplemented with OADC enrichment [Oleic acid, albumin(bovine, fraction V,) dextrose, catalase (DIFCO)] and 0.05% Tween-80 at 37°C on a rotary shaker. Aliquots (1 ml) of logarithmic phase bacteria (grown as set cultures at 37°C) were frozen at −80°C. Frozen stocks were used as inoculums for infections by adding 0.1 mm zirconia silica beads (Biospec, Barltesville, OK, USA) and incubating for 48 h at 37°C prior to vortexing and allowing to settle for 1 h prior to withdrawing the upper aqueous phase containing uniform single-cell suspensions (Price et al., 2008).
Isolation of RNA from Sputum Samples, Broth-grown Bacteria, Infected Cells, and Tissues
Frozen sputum samples in 50 ml tubes were thawed on ice and homogenized by vortexing with 4-mm glass beads and 10% of N-acetyl-L-cysteine solution as previously described (Desjardin et al., 1996). The homogenate was then centrifuged at 8500 × g for 10 min. The pellet was resuspended in 4 ml of TRIZOL (InVitrogen, Carlsbad, CA, USA) at 70°C and 300 μl of phenol reagent was added. The mixture was transferred to a tube containing 0.1 mm zirconia silica beads (Biospec) and processed in a Savant FP-120 homogenizer at a setting of 6.5 for 20 s four times and allowed to stand for 5 min on ice. Tubes were centrifuged at 500 × g for 3 min to pellet the glass beads, and the entire aqueous phase was transferred to a new tube and subjected to two rounds of organic extraction. About 200 μl of chloroform was then added to the tube, which was vortexed for 1 min, and then centrifuged at 13,000 × g for 5 min. The aqueous phase portion was removed and subjected to re-extraction with acid phenol–chloroform and then with chloroform–isoamyl alcohol (24:1) and precipitated with an equal volume of isopropanol in presence of 20 μg of glycogen (Ambion, Austin, TX, USA). RNA pellets were washed with 70% ethanol and resuspended in distilled water. Tubes were centrifuged and entire aqueous phase was subjected to two rounds of organic extraction and precipitation at the end of each extraction.
Total RNA form broth-grown mid-log phase bacterial cultures and infected primary human monocyte derived macrophages or THP-1 cells were isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacture’s instructions with modifications as described below. M. tuberculosis infected C57BL6 mouse lung tissue specimens (∼0.1 g) were obtained at 21 days in the active infection model previously described (Shoen et al., 2004). Corresponding lysates and tissues in Qiagen lysis buffer on ice were briefly heated at 70°C for 1 min and then homogenized with zirconia beads in screw-cap vials with a Savant FP-120 instrument prior to column purification (Graham and Clark-Curtiss, 1999). RNA’s so obtained were then treated with DNAse I (Ambion, Austin, TX, USA) as described by the enzyme manufacturer, and stored at –80°C in aliquots as ethanol precipitates.
cDNA synthesis
RNA isolated from broth-grown bacilli, infected macrophages, infected mouse lung tissue specimens or sputum specimens was converted to first-strand cDNA with reverse transcriptase Superscript III (Invitrogen) according to manufacture’s instructions. For quantitative PCR measurements fist-strand cDNA was synthesized using 5 μg of total RNA with random nonamers (NEB, Ipswitch, MA, USA). For SCOTS analysis, a primer with defined 5′ sequence and nine random 3′ nucleotides were used for both first- and second-strand synthesis as previously described (Froussard, 1992). Primers used for cDNA synthesis from broth RNA (5′-GCCGGAGCTCTGCAGAATTCNNNNNNNNN-3′) and sputum-derived RNA (5′-GACACTCTCGAGACATCACCGGTACCNNNNNNNNN-3′) differ in their terminal sequences. Second-strand synthesis was carried out with Klenow fragment of DNA polymerase I (Promega, Madison, WI, USA) following denaturation and reannealing of first-strand cDNA with the primers. Double-strand cDNAs were purified with QIAquick PCR purification columns (Qiagen, Germantown, MD, USA) and PCR amplification was carried out using appropriate primers lacking the 3′-terminal random nucleotides that were used in cDNA synthesis.
Selective Capture of Transcribed Sequences
Mycobacterium tuberculosis cDNAs from total amplified cDNAs from sputum specimens were selectively captured by hybridization to denatured sonicated photobiotinylated M. tuberculosis genomic DNA which has been prehybridized with blocking M. tuberculosis ribosomal DNA (rDNA) fragments as previously described (Graham and Clark-Curtiss, 1999). Microbial cDNA–genomic DNA hybrids were then collected by binding to streptavidin-coated beads, and bacterial cDNAs were eluted by alkaline denaturation. Eluted bacterial cDNAs were then amplified by PCR. After three rounds of selective capture, bacterial cDNAs were used as template to prepare radiolabeled probes.
Analysis of M. tuberculosis in vivo expression by genome array hybridization
Mycobacterium tuberculosis genomic arrays on nylon from prototype and limited production runs (Sigma-Genosys, Panorama Gene Arrays) were used as previously described (Graham et al., 2002). These arrays consist of PCR-amplified and size verified fragments corresponding to the 3875 ORFs of the M. tuberculosis H37Rv genome. Quadruplicate cultures for H37Rv were grown independently for mid-logarithmic phase RNA. Random primed radiolabeled first-strand cDNA was prepared from 10 to 20 μg of DNase-treated total RNA template by incorporation alpha P[33] deoxy cytidine triphosphate (ICN Biomedicals, Irvine, CA, USA) with Superscript III (Invitrogen). To analyze the composition of M. tuberculosis cDNAs obtained by SCOTS, radiolabeled probes were made from 2 μg of captured amplified cDNA using random non-amer primers and 50 μCi P[33] dCTP with the modification of including cold dCTP at 1/100 the level of the other three dNTP’s. Unincorporated nucleotides were removed by Sephadex G50 column chromatography then probes were denatured with 0.2 M NaOH and incubation at 55°C for 15 min. Radiolabeled cDNA were added to the roller containing 10 ml of hybridization solution (Fawcett et al., 2000). Nylon membranes were hybridized for 24 h, washed, covered in a single layer of plastic film (Sara Wrap, 3 M) and exposed to a Fuji phosphorimager plate overnight prior to scanning with a Fuji BAS FLA-2000 (Fuji Scientific Imaging). Quadruplicate scanned images were quantified with ArrayVision software (Imaging Research, ON, Canada) and data analyzed with BRB Array Tools (Simon et al., 2007) extension for Microsoft Excel. Local background subtraction was used correct regional background variations within individual patterns from single-channel data on nylon macro arrays. As SCOTS is intended as a qualitative screening tool, we determined if cDNAs for mRNAs were detected or not detected by comparison of levels for background based on designated blank spots on arrays. Those features showing hybridization levels greater than threefold over the average level for blank array features was considered as detected expression of the corresponding mRNA. Similarly for mid-log broth cultures we considered the average hybridization value for blanks on all four arrays relative to the average values for hybridization signals for ORFs obtained with four cDNAs from four independent cultures.
Identification of esx paralogs by targeted selective hybridization
Homologous segment of the esxJI genomic DNA was PCR-amplified with (F) primer 5′-CGTGTGATGCGGACAAAG-3′ and (R) primer 5′-ATCTTCCCAGTTCAGCAC-3′ from H37Rv genomic DNA. Three rounds of SCOTS captured cDNA from sputum specimens 8 and 9 were then subjected to three rounds of selective enrichment by hybridizing to biotinylated amplified homologous segment of esxJI. After selective enrichment cDNA’s were cloned in pCR2.1-Topo vector (InVitrogen). Individual plasmid clones were characterized by sequencing insert cDNA fragments.
Quantitative PCR (RT-qPCR) analysis
For quantitative PCR measurements, primers used specifically for measuring sigA and QILSS–Mtb9.9 mRNA levels were chosen using Primer3 software and tested for optimal PCR efficiency using genomic DNA template. As esx parlaogs share high sequence similarity, intergenic regions were targeted and mRNA levels measured for relevant transcripts. Primers used were: sigA (F)-GACGAAGACCACGAAGACC, sigA (R)-CATCCCAGACGAAATCACC, esxKL (F)-AATGAACCCCTGGCAGGCCG, esxKL (R)-CACAATACTTTTACAAGCGA, esxOP (F)-GATGAACTCCTGGCAAGCCA, esxOP (R)-CACAATACTTTTACAAGCGA, esxIJ (F)-CTCCTGAGTCGTCGGGCCGG, esxIJ (R)-GGGCGTCCGCGCAAAACATC, esxVW (F)-CTCCTGAGTCGTCGGGCCGG.
esxVW (R)-GGGCGTCCGCGCAAAACATT, esxMN (F)-AATGTGAGAAAGCGTACGCA, esxMN (R)-CCGCGCAAAACATTTCCGGT. For fluorescence PCR reaction mixture appropriate dilution of the cDNA was mixed with primers (250 nM final concentrations) and 10 μl of 2× Dynamo SYBR green qPCR kit (Finnzymes, Espoo, Finland). The PCR reaction was monitored using an Opticon detection system (MJ research, Waltham, MA, USA). The major housekeeping sigma factor gene sigA was used as an internal control for the normalization of transcripts as H37Rv lacks the regulation of sigA expression seen in many clinical strains (Manganelli et al., 1999). A “no-reverse transcriptase” control was then used to confirm absence of genomic DNA. Change in steady-state mRNA levels were calculated and statistically analyzed by comparing expression level of sigA and target RNAs in different growth conditions using the REST software tool and a threshold of P-value of <0.05 (Pfaffl et al., 2002).
Allele inactivation of esxKL and esxJI
An H37Rv ΔesxKL, esxJ-Hyg-I interrupted, and ΔesxJI mutants were generated by specialized transduction method (Bardarov et al., 2002) with some minor modifications (Price et al., 2008). Briefly, 700 base pair regions immediately up and down stream of the targeted region were cloned into up and down stream of hygromycin resistance gene in plasmid pYUB854. Specialized transducing phage were generated by in vitro Lambda packaging (Gigapack XL, Stratagene, La Jolla, CA, USA) of ligation reaction of PacI digested fragments of circularized phAE87 vector and PYUB854 containing 700 bp up down regions. After transducing in E. coli HB101, hygromycin resistant transductants were screened for large desired product. Phasmid DNA was pooled from desired transductants and electroporated into Mycobacterium smegmatis mc2 155 and plates were incubated at 30°C to generate mycobacteriophage plaques. High titer mycobacteriophage preparation were propagated in M. smegmatis and analyzed for temperature sensitivity at 37°C before transducing into H37Rv at MOI 10:1. Mutants were selected on 7H9 agar supplemented with hygromycin. Deletion of the entire gene was confirmed by PCR, and lack of mRNA signal in cDNA prepared from the mutant strain by RT-qPCR. Complement strains were generated by cloning esxK or esxL for ΔesxKL mutant, and esxI for esxJ-Hyg-I interrupted mutant, downstream of constitutive hsp60 promoter in vectors pLou3kan (kanamycin resistance marker), and pL0u3apr (apramycin resistance marker) respectively. These vectors contain the L5 mycobacteriophage attP and integrate the genes at single-copy at bacterial chromosomal attB site.
Primary Human Macrophages and THP-1 Cell Infections
Monocyte derived macrophages were obtained from two healthy donors as previously described (Schlesinger and Horwitz, 1991). Briefly, mononuclear cells were isolated by Ficoll (Sigma) gradients according to the manufacturer’s instructions. Mononuclear cells were cultivated in Teflon wells for 5 days in RPMI media (GIBCO) supplemented with 10% heat inactivated fetal bovine serum (FBS), 4 mM L-glutamine, 25 mM HEPES, non-essential amino acids, and 5 μg/ml vancomycin at 37°C in a 5% CO2 humidified incubator. Adherent macrophages were scraped and plated in 24-well plate at a cell density of 5 × 105 cells in RPMI media and incubated for 5 days for further in vitro maturation prior to infection. Human leukemia macrophage cell line THP-1 were grown in RPMI 1640 to density of 1–2 × 106 cells per ml in 75 cm2 tissue culture flasks. Three days prior to infection with M. tuberculosis, THP-1 cells were cultured in 24-well plate at density of 5 × 105 cells per well in presence of 100 nM phorbol 12-myristate 13-acetate (PMA) and 150 units of human recombinant IFN-gamma (Theus et al., 2004). Bacterial strains were mixed with native FBS (final concentration of 10% v/v) and were incubated for 30 min. Opsonized bacteria were then diluted in RPMI supplemented with 2% v/v FBS to obtain required multiplicity of infection of 1:1. Monolayers were infected with bacteria for overnight and then washed three times with HANKS buffer and fresh RPMI-2% FBS lack PMA and INF-g media was added to monolayers, which changes as 48 h. CFU counts were performed from triplicates wells at each time point by lysis of monolayers with 1% Triton-X 100.
Surface Labeling of Mycobacteria with Fluorescein-tagged Succinimidyl Ester
H37Rv and ΔesxKL mutant were labeled with fluorescein-tagged succinimidyl ester (Alexa fluor 488 and 594 from Molecular Probes and Invitrogen) as previously described (Beatty and Russell, 2000). Briefly, bacteria were washed twice with phosphate-buffered saline (PBS) – 0.5% Tween-80 – 0.2 M sodium bicarbonate (pH 8.8) and resuspended in same buffer containing 1 mM fluorescein succinimidyl ester. Following 1.5 h incubation at 37°C, bacteria was washed three times with PBS–Tween and then used as inoculum (prepared as described above) for macrophage infections. For macrophage infections, 5 × 105 cells were made to adhere to circular glass coverslips in 24-well culture plates and cells were infected at MOI of 1:1 with mixture consisting of 50:50 of fluorescent labeled wild-type and ΔesxKL mutant bacteria. At 24, 48, and 72 h post-infections cells were washed three times with HANKS buffer and were fixed at 37°C in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30. Following washing with PBS coverslips were mounted on slide with the Prolong Antifade kit (InVitrogen). Labeled mycobacteria in infected macrophages were visualized directly using a Olympus F500 laser scanning confocal microscope.
EsxK and EsxL interaction by two-hybrid system
The BacterioMatch Two-Hybrid Vector system (Stratagene) was used according to manufacturer’s instructions. Briefly, the BacterioMatch two-hybrid vectors pBT bait containing the λcI domain and pTRG target containing alpha-RNAP domain, were used as starting plasmids for cloning esxK and esxL genes respectively. The plasmids pBT–esxK and pTRG–esxL were then used for cotransformation of E. coli XL1-Blue cells containing the reporter cassette. The transformed cells were plated on selective media containing 5 mM 3-amino-1,2,4-triazole and incubated at 37°C for 24 h.
References
Fawcett, P., Eichenberger, P., Losick, R., and Youngman, P. (2000). The transcriptional profile of early to middle sporulation in Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 97, 8063–8068.
Keywords: QILLS-paired secreted factors, WGX100, ESX-5, esxO, esxN, esxV
Citation: Bukka A, Price CTD, Kernodle DS and Graham JE (2012) Mycobacterium tuberculosis RNA expression patterns in sputum bacteria indicate secreted Esx factors contributing to growth are highly expressed in active disease. Front. Microbio. 2:266. doi: 10.3389/fmicb.2011.00266
Received: 23 June 2011;
Accepted: 15 December 2011;
Published online: 10 January 2012.
Edited by:
Adel M. Talaat, University of Wisconsin Madison, USAReviewed by:
Srinand Sreevastan, University of Minnesota, USAMartin S. Pavelka, University of Rochester, USA
Xin Li, The Ohio State University, USA
Copyright: © 2012 Bukka, Price, Kernodle and Graham. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: James E. Graham, Department of Microbiology and Immunology, University of Louisville School of Medicine, Health Sciences Center A405, 319 Abraham Flexner Way, Louisville, KY 40202, USA. e-mail:ai5ncmFoYW1AbG91aXN2aWxsZS5lZHU=