 Chenhong Luo Fumi Goshima Maki Kamakura Yoshifumi Mutoh Seiko Iwata Hiroshi Kimura* Yukihiro Nishiyama
Chenhong Luo Fumi Goshima Maki Kamakura Yoshifumi Mutoh Seiko Iwata Hiroshi Kimura* Yukihiro Nishiyama- Department of Virology, Nagoya University Graduate School of Medicine, Nagoya, Japan
Genital herpes is an intractable disease caused mainly by herpes simplex virus (HSV) type 2 (HSV-2), and is a major concern in public health. A previous infection with HSV type 1 (HSV-1) enhances protection against primary HSV-2 infection to some extent. In this study, we evaluated the ability of HF10, a naturally occurring replication-competent HSV-1 mutant, to protect against genital infection in mice caused by HSV-2. Subcutaneous inoculation of HF10-immunized mice against lethal infection by HSV-2, and attenuated the development of genital ulcer diseases. Immunization with HF10 inhibited HSV-2 replication in the mouse vagina, reduced local inflammation, controlled emergence of neurological dysfunctions of HSV-2 infection, and increased survival. In HF10-immunized mice, we observed rapid and increased production of interferon-γ in the vagina in response to HSV-2 infection, and numerous CD4+ and a few CD8+ T cells localized to the infective focus. CD4+ T cells invaded the mucosal subepithelial lamina propria. Thus, the protective effect of HF10 was related to induction of cellular immunity, mediated primarily by Th1 CD4+ cells. These data indicate that the live attenuated HSV-1 mutant strain HF10 is a promising candidate antigen for a vaccine against genital herpes caused by HSV-2.
Introduction
Herpes simplex virus (HSV) type 1 (HSV-1) and type 2 (HSV-2) belong to the alphaherpesvirus family. HSV-1 and HSV-2 have 50% DNA sequence homology (Kieff et al., 1972). Generally, HSV-1 infects via the oral route, whereas HSV-2 infects via the genital tract. Both exert neurotropic effects and spread to the nervous system (Corey and Spear, 1986; Whitley and Roizman, 2001). HSV-2 is the main causative agent of genital herpes worldwide (Tao et al., 2000). Epidemiological investigations have indicated that the prevalence of HSV-2 in the general population of the USA ranges from 10 to 60%, and genital herpes is one of the most common sexually transmitted diseases (Malvy et al., 2005; Xu et al., 2006). After primary infection via the genital tract, the virus establishes latency within the lumbosacral ganglions, and establishes a state of lifelong infection. Subsequently, the latent virus reactivates intermittently resulting in recurrent disease (Miller et al., 1998; Stanberry et al., 2000). In addition, genital herpes is linked to an increased susceptibility to sexually acquiring and transmitting human immunodeficiency virus (HIV; Freeman et al., 2006; Kapiga et al., 2007), which is not markedly reduced by HSV antiviral therapy (Celum et al., 2008; Watson-Jones et al., 2008). A vaccine would provide a more effective means of preventing or limiting infection, and would greatly relieve the social and economic burden of HSV-2 infection. In developed countries, while childhood acquisition of HSV-1 has decreased, HSV-2 seroprevalence has increased, suggesting the possible protective effect of HSV-1 against HSV-2 infection (Xu et al., 2006). HSV-1 has also become a major causative agent of primary genital herpes in developed countries (Lafferty et al., 2000; Nieuwenhuis et al., 2006).
In the past, efforts to develop an HSV vaccine have included development of inactivated whole-virus vaccines, subunit glycoprotein preparations, DNA plasmids, and attenuated replication-competent viruses. These candidate vaccines were unsuccessful in clinical trials (Stanberry, 2004). The most successful vaccine in human trials was a subunit glycoprotein vaccine that included HSV-2 gD, a major viral envelope antigen, as an immunogen with alum and 2-o-deacylated monophosphoryl lipid A as adjuvants (Bernstein et al., 2005). Although the vaccine appeared safe and effective against genital herpes in guinea pigs, it failed to provide sufficient protection against primary infection in a clinical trial (Stanberry et al., 2002). Furthermore, immunization with HSV-2 gD subunit did not reduce the rate at which women acquired HSV-2 genital herpes (Belshe et al., 2012). Therefore, new strategies for developing HSV vaccines are required. Meanwhile, there are established live vaccines for other alphaherpesviruses, e.g., a modified live virus vaccine that prevents pseudorabies virus infection (PRV/Marker Gold®) in pigs is commercially available (Swenson et al., 1993a,1993b; Van de Walle et al., 2003), and an attenuated live varicella-zoster virus vaccine that prevents chicken pox and shingles (Arvin and Gershon, 1996; Oxman et al., 2005) is in widespread use. Killed viral vaccines have proven to be inferior to live vaccines, in terms of naturally acquired immunity, preventing infection or re-infection, and producing durable immunity. The use of a live HSV vaccine risks inducing latency or reactivation (Cappel, 1976).
HF10 is a spontaneously occurring HSV-1 mutant that lacks functional expression of UL43, UL49.5, UL55, UL56, and latency-associated transcripts (Ushijima et al., 2007). We have demonstrated that HF10 can be attenuated, and that it does not cause any neurotropic effects in mice. Intranasal vaccination of mice with HF10 conferred significant protection against lethal challenge with HSV-1 and HSV-2 (Mori et al., 2005). Thus, HF10 is a promising live attenuated HSV vaccine candidate. It is also a well-known oncolytic virus for cancer therapy (Fujimoto et al., 2006; Kimata et al., 2006; Nakao et al., 2007). In this study, we used HF10 as a live attenuated vaccine, and evaluated the immune response generated and protective effect against HSV-2 genital infection in mice. Subcutaneous inoculation of HF10-immunized mice from lethal infection by HSV-2, and attenuated the development of genital ulcer diseases. Furthermore, we observed inhibition of virus replication and production of interferon-γ (IFN-γ) by splenocytes in response to HSV-2 antigens in the serum of immunized mice. HF10 also induced rapid accumulation of CD4+ and CD8+ cells in the infective focus, and protected mice against HSV-2 genital disease via induction of a cellular immune response.
Materials and Methods
Viruses, Cells, and Antibodies
Vero cells (African green monkey kidney epithelial cells) were grown in Eagle’s minimal essential medium (MEM) supplemented with 10% calf serum. The HSV-1 mutant HF10, the wild-type HSV-1 strains KH7 and KOS, and wild-type HSV-2 strain 186 were titrated in Vero cells. HF10 virus was inactivated by exposure to ultraviolet (UV) light for 30 min using a GL15 UV (Mitsubishi/Osram, Kakegawa, Japan). UV-inactivated virus was not infectious when inoculated into Vero cells. NIH3T3 cells (mouse embryonic fibroblast cell line derived from BALB/c) were grown in Dulbecco’s modified Eagle’s medium containing 10% calf serum. NIH3T3 cells were infected with HSV-2 strain 186 at a multiplicity of infection (MOI) of 3 in the presence of ganciclovir (GCV; 10 μg/ml) or cycloheximide (CHX; 20 μg/ml) for 8 h, and then harvested for stimulating splenocytes. Anti-HSV-1 polyclonal rabbit antibody was purchased from Dako (Glostrup, Denmark). Anti-HSV-2 antibody was acquired from a mixture of anti-HSV-2 UL17, UL42, UL46, UL48, and US11 antibodies generated in our laboratory by immunizing rabbits (Goshima et al., 2000; Kato et al., 2000; Koshizuka et al., 2001). Anti-mouse CD4 antibody and fluorescein isothiocyanate (FITC)-labeled anti-mouse CD8 antibody were purchased from Chemicon International (Temecula, CA, USA) and Thermo Scientific (Rockford, IL, USA), respectively. DRAQ5 (Biostatus Limited, Shepshed, UK) was used to stain cell nuclei. Anti-mouse IgG-conjugated FITC and anti-rabbit IgG-conjugated tetramethylrhodamine-5-(6)-isothiocyanate (TRITC) were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Mouse Strains, Immunization, and Challenge
BALB/c and BALB/c nude mice were obtained from SLC (Hamamatsu, Japan). Six-week-old BALB/c mice were immunized subcutaneously in the rear flank once with 100 μl phosphate-buffered saline (PBS) containing 1 ×106 plaque-forming units (PFUs) of HF10, 1 × 107 PFUs of UV-treated HF10, or PBS only. Mice were challenged 4 weeks or 4 months after immunization, and 7 days prior to challenge they were subcutaneously injected in the neck ruff with 3 mg Depo-Provera (Sigma-Aldrich). For intravaginal challenge, mice were inoculated with 5 × 105 PFUs of HSV-2 strain 186 (approximately 15 × LD50) using a pipette.
For the safety study, 5 × 105 PFUs of HF10 or KH7 were subcutaneously inoculated in the flank of 6-week-old BALB/c nude mice. On days 1 and 5 after infection, mice were sacrificed, and skin samples were harvested for histological and immunohistochemical studies to detect HSV-1 antigens.
All experiments were approved by the University Committee and conducted in accordance with the Guidelines for Animal Experimentation of Nagoya University.
Clinical Observations
Mice were observed daily for signs of genital lesions. The severity of disease was scored as follows: 0, no sign; 1, slight genital erythema and edema; 2, moderate genital inflammation; 3, purulent genital lesions and paralysis; and 4, death.
Evaluation of Acute Infection
Vaginal tracts of mice were washed with 200 μl MEM containing 5% newborn calf serum for 1–5 days after challenge. These were stored at -80°C for virus titration and IFN-γ assays. Viral titers were determined using a standard plaque assay. IFN-γ concentration was determined using a Quantikine Immunoassay kit (R&D Systems, Minneapolis, MN, USA) in an enzyme-linked immunosorbent assay (ELISA).
Neutralizing Antibody Assay
Four weeks after immunization, blood samples were collected via laparotomy from the abdominal aortic arch. After incubation at 37°C for 30 min, blood samples were centrifuged at 3000 rpm for 10 min and serum was collected. To estimate neutralization titers, diluted sera were added to 100 PFUs of HSV-1 strains HF10 and KOS or HSV-2 strain 186, incubated for 30 min at 37°C, and the remaining infectious virus was detected on duplicate Vero cell monolayers.
Immunofluorescent Staining of Vaginal Tissues
Mice were deeply anesthetized with ketamine and xylazine and their vaginas were excised. To examine the distribution of CD4+ and CD8+ cells, frozen sections were stained with a variety of antibodies. In brief, 8 μm frozen sections were blocked with PBS/2% fet al calf serum (FCS), reacted with mouse CD4 monoclonal antibody for 30 min, and stained with anti-mouse IgG-conjugated FITC antibody for 30 min or FITC-labeled anti-mouse CD8 monoclonal antibody for 30 min at 37°C. Slides were then washed with PBS/2% FCS three times, fixed with 4% paraformaldehyde for 15 min, and treated with 0.1% Triton X-100 for 10 min at room temperature. Next, these slides were stained with polyclonal rabbit HSV-2 antibody (described in Section “Viruses, Cells, and Antibodies”) for 30 min at 37°C, washed with PBS, and then treated with a secondary antibody (anti-rabbit IgG-conjugated TRITC) for 30 min at 37°C. Stained slides were washed, incubated with DRAQ5, and mounted with Fluoromount Plus (Diagnostic Biosystems, Pleasanton, CA, USA). Finally, slides were visualized using an LSM 510 laser-scanning confocal microscope (Carl Zeiss, Jena, Germany).
Evaluation of Cellular Immunity
Mice were deeply anesthetized with ketamine and xylazine, and their spleens were excised. Tissues were crushed through a 100-μm nylon cell strainer (BD Biosciences, Franklin Lake, NJ, USA). Erythrocytes were depleted using lysis buffer (BD Biosciences), and spleen cells were suspended in RPMI-1640 medium containing 10% FCS. Spleen cells were plated at 1 × 107 cells/well (2 ml) for stimulation by HSV-2-infected NIH3T3 cells. NIH3T3 cells (2 × 106 cells/35 mm dish) were infected with HSV-2 strain 186 at an MOI of 3 for 3 h. Spleen cells were also plated at 1 × 106 cells/well (500 μl) for stimulation by NIH3T3 cells expressing HSV-2 viral antigens. To produce viral antigens, UL46 and US6 genes and ICP0 cDNA from HSV-2 strain 186 were amplified by polymerase chain reaction and cloned into pcDNA 3.1(+) expression vectors (Invitrogen, Carlsbad, CA, USA). Each plasmid (1.5 μg) was transfected into 1 × 106 NIH3T3 cells/35 mm dish with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, and incubated for 18 h. Plasmid-transfected or HSV-2-infected cells were frozen, thawed, and added to dishes containing splenocytes acquired as described above. Splenocytes were stimulated with these viral antigens at 37°C for 3 h and the medium was collected at 5 and 20 h to quantify IFN-γ concentrations.
Statistics
The statistical significance of differences in disease scores and viral titers on individual days was determined using a Student’s t-test. Survival rates were estimated by the Kaplan–Meier method, and statistical significances were determined by the log-rank test. Viral titers, disease scores, and IFN-γ concentrations in genital washes were expressed as means ± SE. Statistical analysis was performed using SPSS 1.1J software (SPSS Inc., Chicago, IL, USA). Differences with a P-value of <0.05 were considered statistically significant.
Results
Clearance of HF10 After Subcutaneous Inoculation into BALB/c Nude Mice
To confirm the low virulence of HF10, we subcutaneously inoculated HF10 or wild-type KH7 into eight BALB/c nude mice and compared their virulence. No HF10-inoculated nude mice developed zoster or died. In contrast, KH7 infection caused severe zoster formation and death in all mice. HF10 was detected 1 day after inoculation but cleared by day 5 (Figure 1). Conversely, KH7-infected cells were still detectable on day 5. These results confirm the low virulence of HF10 (Mori et al., 2005).
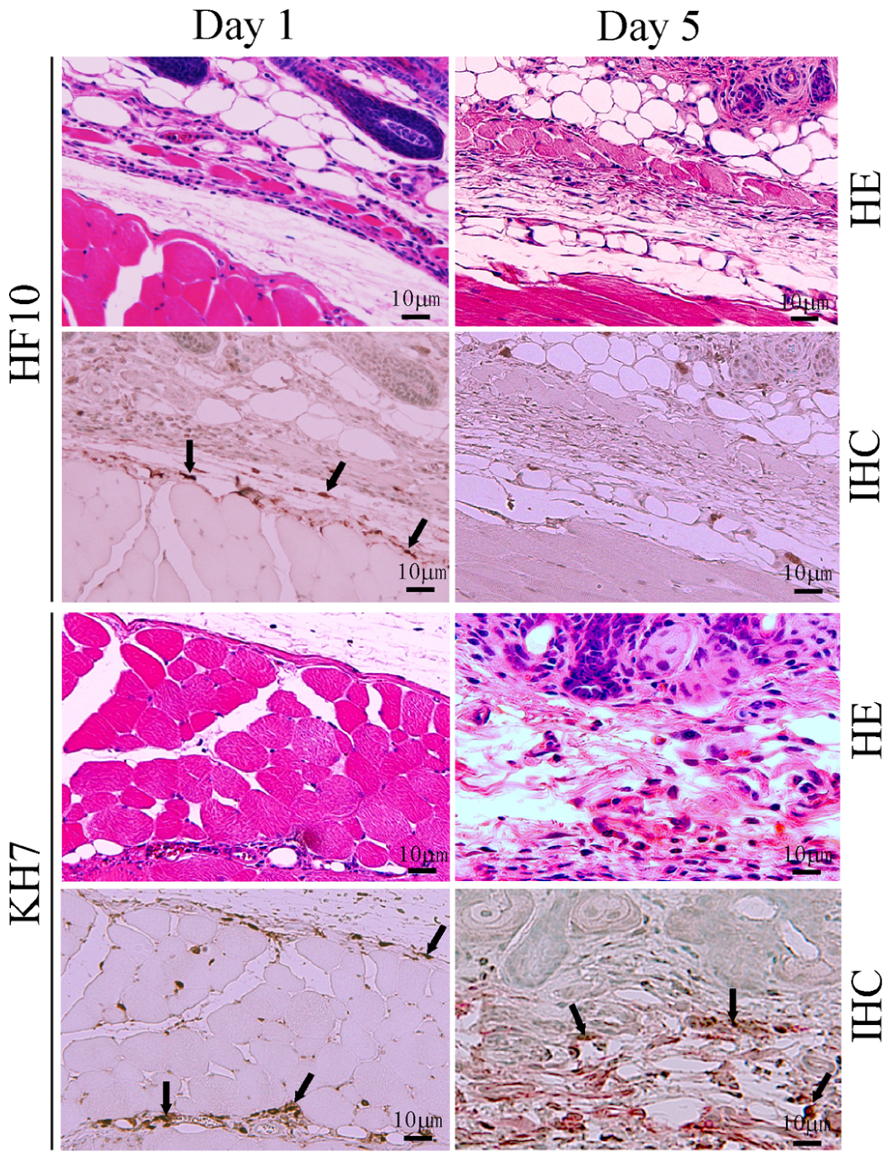
FIGURE 1. Safety study of HF10 in a nude mouse model. HSV-1 mutant HF10 or HSV-1 wild-type KH7 was subcutaneously inoculated into BALB/c nude mice. Skin samples were obtained at days 1 and 5 after infection and evaluated by hematoxylin and eosin (HE) staining and immunohistochemistry (IHC) to detect HSV-1 antigens. Arrows indicate HSV-1-infected cells.
Immunization with HF10 Protects Mice Against HSV-2 Genital Disease
To determine the efficacy of HF10 as a vaccine, we subcutaneously immunized either BALB/c mice with HF10 or UV-inactivated HF10, or performed mock immunization. After 1 month, each group was challenged by intravaginal inoculation of wild-type HSV-2 stain 186. Mice immunized with HF10 had significantly lower titers of virus shedding in the vaginal mucosa 1 day after challenge (Figure 2A). The shedding of strain 186 in HF10-immunized mice was diminished by day 5 after challenge. In control mice, the viral titer decreased by day 3 and then increased (Figure 2A). Viral titers on day 5 were significantly lower in HF10-immunized mice than in unimmunized mice and UV-inactivated HF10-immunized mice. Immunization with HF10 protected mice from development of local genital symptoms and these mice exhibited no signs of systemic disease (Figure 2B); the survival rate was 83.3% (Figure 2C). In contrast, all mice in the control group developed severe genital symptoms and hind paralysis (Figure 2B). Eventually, all succumbed and died within 10 days of challenge (Figure 2C). Genital symptoms and paralysis in mice immunized with UV-inactivated HF10 were slightly weaker than those in unimmunized mice, albeit without statistical significance (Figure 2B). The survival rate of UV-inactivated HF10-immunized mice was 40% (Figure 2C). Thus, immunization with UV-inactivated HF10 partially protected mice from HSV-2 genital disease. We then confirmed the persistent efficacy of HF10 immunization against vaginal infection by HSV-2. Four months after immunization, mice immunized with HF10 were also protected against genital disease to the same level as mice challenged after 1 month (data not shown).
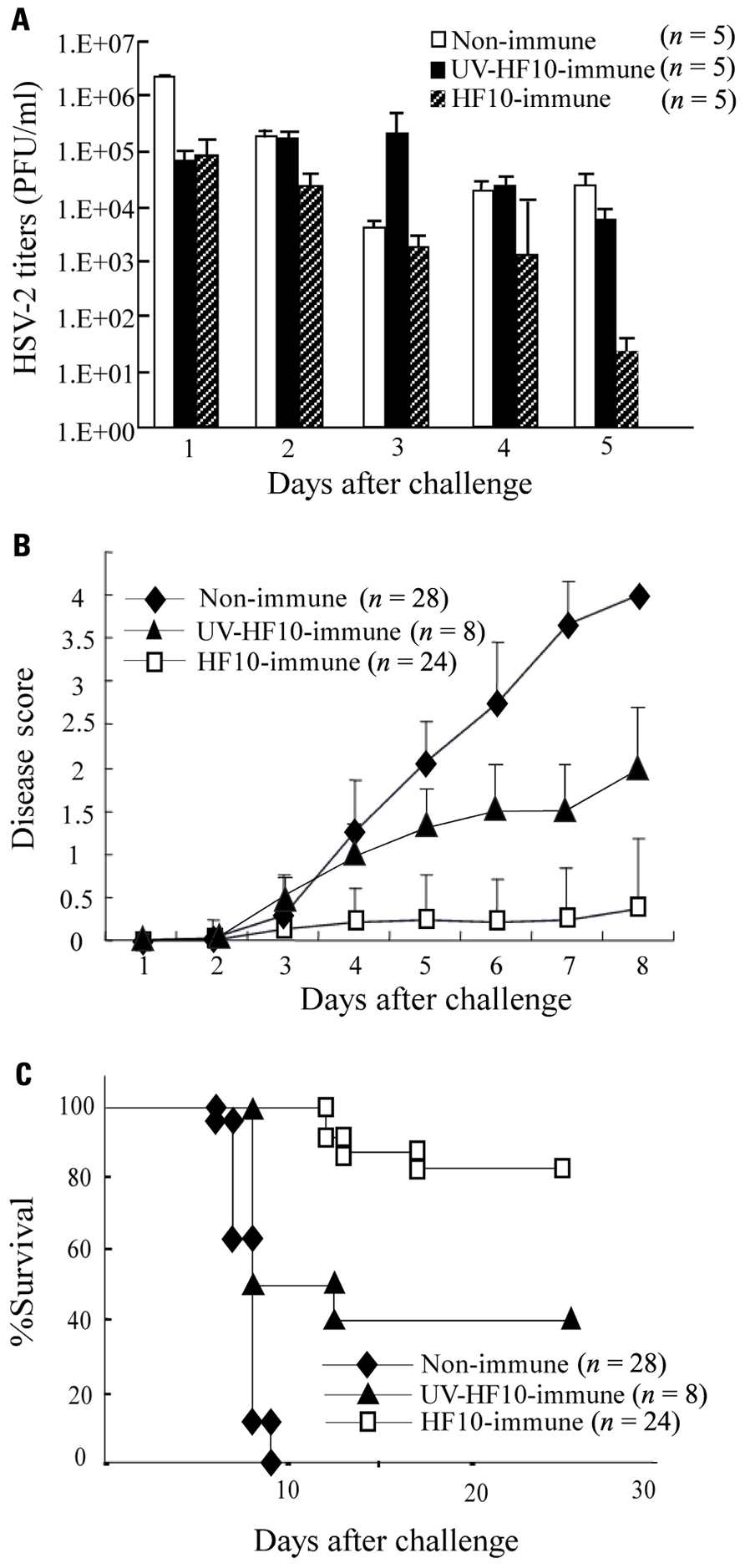
FIGURE 2. Immunization of mice with HF10 followed by lethal HSV-2 challenge via vaginal inoculation. One month after immunization with HF10, 5 × 105 PFUs of wild-type HSV-2 strain 186 were inoculated into mice vagina. Unimmunized mice and UV-inactivated HF10-immunized mice were used as controls. (A) Replication of strain 186 in the genital mucosa was determined by viral titrations of vaginal washes (200 μl). Viral titers were significantly lower in HF10-immunized mice on day 5 than in both unimmunized mice (P < 0.05) and UV-inactivated HF10-immunized mice (P < 0.05). (B) Clinical symptoms were monitored and scored for 8 days. HF10-immunized mice had lower disease scores than unimmunized mice (on day 8, P < 0.0001). Disease scores were not significantly different between UV-inactivated HF10-immunized mice and unimmunized mice (P = 0.065). (C) Survival curve derived by the Kaplan–Meier method. HF10-immunized mice survived longer than unimmunized mice and UV-inactivated HF10 mice (on day 28, P < 0.0001 and P = 0.001, respectively).
Histological examinations of vaginal tissues were performed (Figure 3). In unimmunized mice, HSV-2 antigens were present in mucosal epithelial cells and subepithelial lamina propria of vaginal tissue at days 1 and 4 after challenge, and mucosa in the infective focus dropped out from the epithelium 6 days after challenge. In contrast, in HF10-immunized mice, HSV-2 antigen staining was restricted to the mucosal surface at days 1 and 4 and was undetectable 6 days after challenge. These results indicate that mice immunized with HF10 were protected against severe genital disease caused by HSV-2.
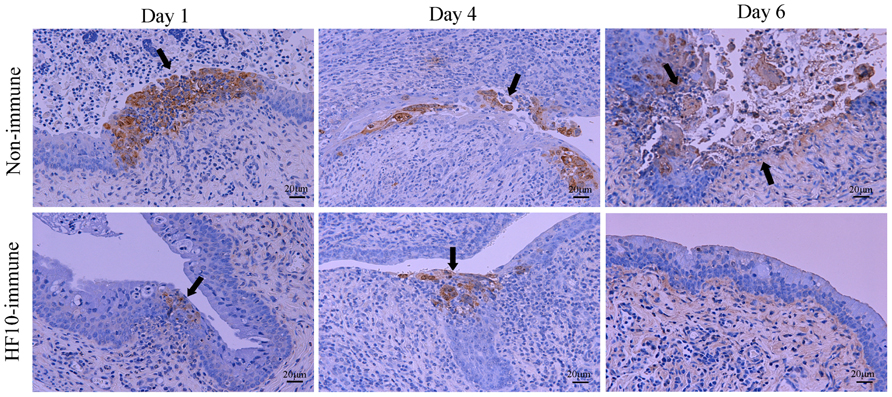
FIGURE 3. Immunohistochemical evaluation after HSV-2 challenge. After inoculation of wild-type HSV-2 strain 186 into HF10-immunized mice or unimmunized mice vagina; vaginal mucosal lesions were excised at days 1, 4, and 6 after challenge, and HSV-2 antigens were stained. Arrows indicate HSV-2-infected cells.
Immune Responses Against HSV-2 Strain 186 in HF10-Immunized Mice
To determine neutralizing antibody titers against HSV-2 strain 186 in HF10-immunized mice, we collected serum 1 month after HF10 immunization and investigated its ability to neutralize HSV-1 strains HF10 and KOS and HSV-2 strain 186 based as determined by reductions in plaque formation (Figure 4A). Serum inhibited HF10 plaque formation at a dilution of 1:128 and KOS plaque formation at a dilution of 1:64. The titers producing a 50% reduction in plaque formation by HF10 and KOS were between 16 and 32. Serum had little effect on plaque formation by HSV-2 strain 186 at lower concentrations, but at a dilution of 1:2 caused a 40% reduction. Plaque formation by UV-inactivated HF10-immunized mouse serum was lower than that of HF10-immunized mice (Figure 4B). To evaluate cellular immune responses, we stimulated spleen cells from each immunized or unimmunized mouse with 186-infected NIH3T3 cells and examined IFN-γ production kinetics. IFN-γ accumulated in the medium of splenocytes from HF10-immunized mice at both 5 and 20 h after stimulation, although IFN-γ levels were similar to those produced by UV-inactivated HF10-immunized mice (Figure 4C). We then investigated IFN-γ concentrations in vaginal washes after challenge (Figure 4D). Although IFN-γ production was recognized in mouse vaginas immunized with UV-inactivated HF10, the quantity was not statistically significantly different from that produced by unimmunized mice. In contrast, IFN-γ concentration in HF10-immunized mice was significantly higher than that in unimmunized mice.
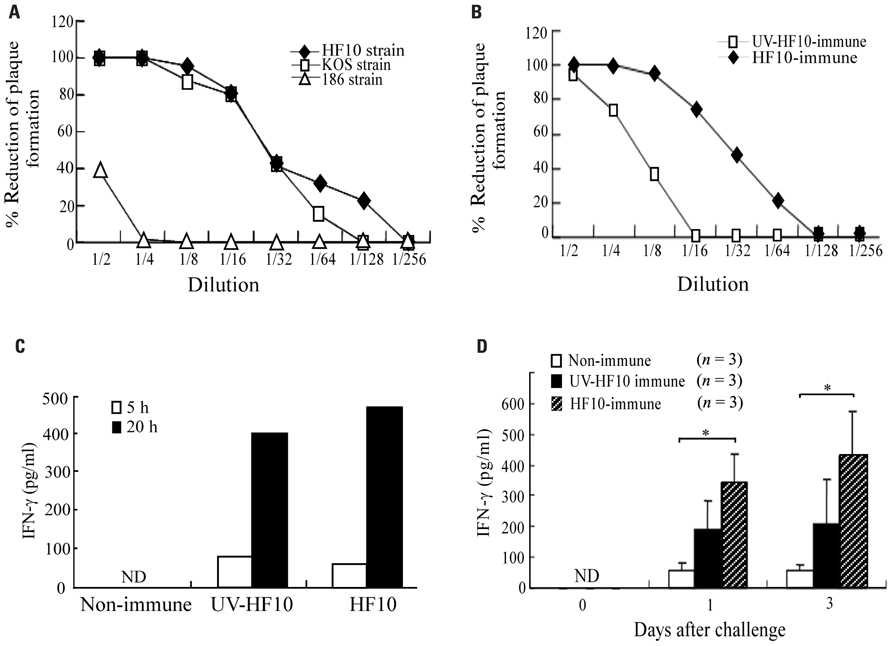
FIGURE 4. Immune responses of HF10-immunized mice. (A) Serum was obtained from HF10-immunized mice (n = 3), and its neutralizing ability against HSV-1 strains (HF10 and KOS) and an HSV-2 strain (186) was investigated by the reduction in plaque formation. (B) Mice were immunized with UV-inactivated HF10 or HF10. After 4 weeks, serum (n = 3) collected and neutralizing ability against HF10 was assayed by the reduction in plaque formation. (C) IFN-γ produced by splenocytes stimulated with HSV-2 strain 186-infected NIH3T3 cells. Splenocytes (1 × 107 cells/dish) from unimmunized (n = 3), UV-inactivated HF10-immunized (n = 3), or HF10-immunized (n = 3) mice were incubated with 186-infected NIH3T3 cells (1 × 107 cells/dish) for 5 and 20 h, and the supernatants were collected. Supernatants from three mice were combined and assayed for IFN-γ concentrations using an ELISA. (D) IFN-γ levels in mice vagina after HSV-2 challenge. Genital tracts of unimmunized, UV-inactivated HF10-immunized, and HF10-immunized mice were washed 0, 1, and 3 days after challenge with strain 186, and the washes were assayed for IFN-γ concentrations by ELISA. *P < 0.05 between unimmunized mice and HF10-immunized mice. ND; not detected.
To confirm that memory cells of HF10-immunized mice cross-reacted with HSV-2 proteins, we expressed HSV-2 ICP0, UL46, and gD in NIH3T3 cells (Muller et al., 2009), used them to stimulate splenocytes, and quantified IFN-γ concentration in the medium. Splenocytes from HF10-immunized mice produced considerable amounts of IFN-γ in response to gD-, ICP0-, and UL46-expressing NIH3T3 cells (Figure 5). To confirm that protein synthesis was necessary, we stimulated splenocytes with 186-infected NIH3T3 cells treated with ganciclovir or cycloheximide. Splenocytes stimulated with ganciclovir-treated cells produced high levels of IFN-γ (474 pg/ml) at 20 h after stimulation compared to the production by cycloheximide-treated cells (82 pg/ml), indicating the requirement for protein synthesis (Figure 5). There was little or no production of IFN-γ by splenocytes from unimmunized mice subjected to identical protein stimulation (Figure 5).
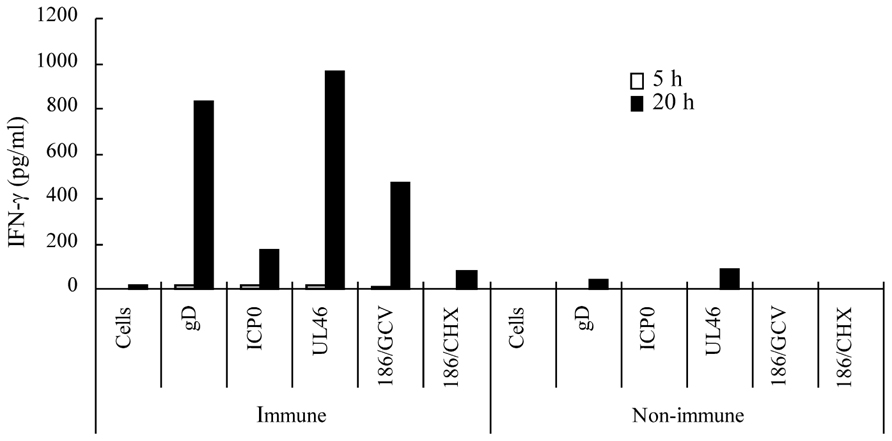
FIGURE 5. Cellular immune responses of HF10-immunized mice to HSV-2 proteins. Splenocytes (1 × 106 cells) from HF10-immunized or unimmunized mice were collected and incubated at 37°C for 3 h. NIH3T3 cells (1 × 106 cells) transfected with expression vectors for HSV-2 gD, ICP0, and UL46 were added into dishes to stimulate splenocytes. To confirm protein synthesis, 186-infected NIH3T3 cells were treated with either 10 μg/ml ganciclovir (186/GCV) or 20 μg/ml cycloheximide (186/CHX) for 8 h and used for stimulation. The media were collected at 5 and 20 h after addition and assayed for IFN-γ concentrations by ELISA. “Cells” denote uninfected NIH3T3 cells used as a negative control.
Furthermore, to investigate cellular responses to HSV-2 genital infection, we performed immunohistochemical studies in mouse vaginas on days 1 and 3 after challenge (Figure 6). In HF10-immunized mice, CD4+ cells localized to the infective focus and invaded the mucosal subepithelial lamina propria on both 1 and 3 days after challenge (Figure 6A). CD8+ cells were only detected 1 day after challenge (Figure 6B). In unimmunized mice, there were no detectable CD4+ and CD8+ cells 1 day after challenge (Figures 6A, B), but there were a few CD4+ cells in the infective focus 3 days after challenge.
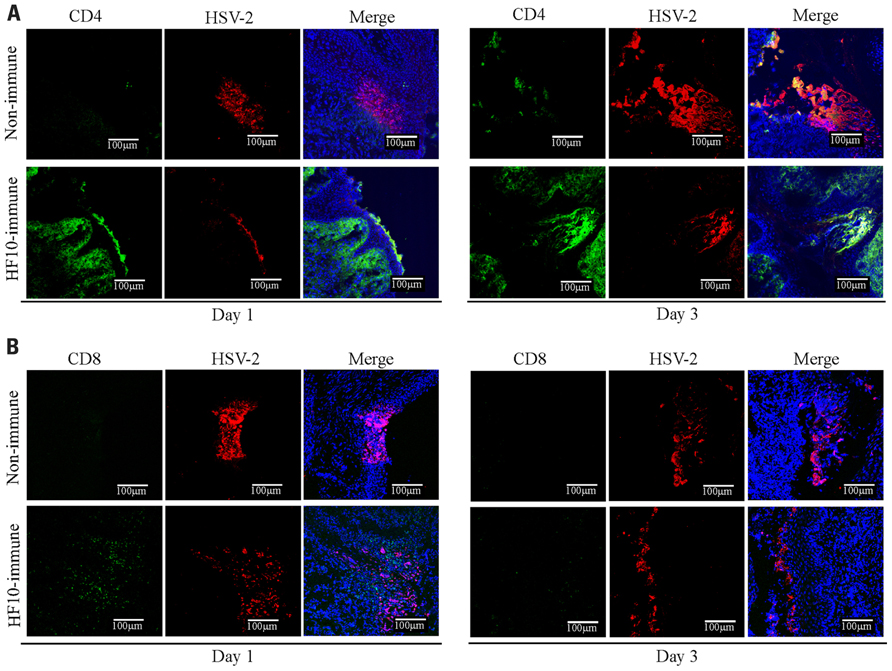
FIGURE 6. Accumulation of CD4+ and CD8+ T cells in the infection focus of vagina after HSV-2 challenge. Unimmunized and HF10-immunized mice were challenged with HSV-2 by intravaginal infection, and vagina was excised 1 or 3 days later. Frozen sections were stained with anti-CD4 (green), anti-CD8 (green), and anti-HSV-2 antibodies (red). Cell nuclei were counterstained with DRAQ5 (blue). Images were captured with confocal microscopy at ×200 magnification. (A) Anti-CD4. (B) Anti-CD8.
Discussion
Genital herpes is an intractable disease of major public health importance. It causes significant morbidity and psychosocial distress and increases the risk of HIV transmission (Freeman et al., 2006; Kapiga et al., 2007). Previous HSV-1 infection provides protection against primary genital HSV-2 infection and its severity, to some extent (Mertz et al., 1992; Bryson et al., 1993). Therefore, immunization with HSV-1 may be useful for preventing infection with or disease caused by HSV-2.
In this study, we evaluated the ability of the spontaneously occurring HSV type 1 mutant HF10 to serve as a vaccine against HSV-2-mediated genital disease. The safety of the vaccine must be considered because HF10 is a replication-competent virus. We determined the complete DNA sequence of HF10 and found that the virus lacks functional expression of UL43, UL49.5, UL55, UL56, and latency-associated transcripts (Ushijima et al., 2007). In addition, HF10 exhibits a relatively high divergence in proteins compared to HSV-1 strain 17. All of these changes occurred spontaneously. UL56 associates with the kinesin motor protein KIF1A, and its absence reduces the neuroinvasiveness of HSV (Rosen-Wolff et al., 1991; Berkowitz et al., 1994; Koshizuka et al., 2002). The LAT promoter region is also reported to be associated with neurovirulence (Jones et al., 2005; Peng et al., 2005). We previously found that HF10 lacks neuroinvasiveness and is at least 10,000-fold less virulent than wild-type HSV-1 in mouse models (Nishiyama et al., 1991; Jiang et al., 1995; Mori et al., 2005). To confirm the safety of HF10, we subcutaneously inoculated HF10 into BALB/c nude mice. HF10 was cleared from the skin by day 5, and no nude mouse developed zoster or died. Using clinical trials of HF10 as cancer virotherapy, we have been evaluating the safety of HF10 through various approaches in preclinical tests (Fujimoto et al., 2006; Kimata et al., 2006; Nakao et al., 2007). Recently, the US Food and Drug Administration approved a phase I clinical trial of HF10 against refractory head and neck cancer (Clinical Trials Gov, Identifier: NCT01017185). Considering the results collectively, we believe that HF10 is a safe vaccine candidate. One striking advantage of HF10 is that it is a naturally occurring HSV-1 mutant and is not genetically engineered. Therefore, in terms of ethical aspects, it is not necessary to consider the safety of foreign gene expression.
Our results indicate that immunization with HF10 protected mice from HSV-2 primary genital infection. Immunization inhibited viral replication in the vagina, reduced local inflammation, controlled emergence of neurological manifestations of HSV-2 infection, and increased survival. We speculate that HF10 can induce adaptive immunity against HSV-2 strain 186. However, serum from the HF10-immunized mice elicited few neutralizing effects against HSV-2. Therefore, humoral immunity does not play a key role in the protection against HSV-2 genital disease. In previous studies, the transfer of serum from HSV-immunized mice to unimmunized mice did not reduce HSV replication in the vaginal mucosa, but rather protected the nervous system and prevented lethality (McDermott et al., 1990; Eis-Hubinger et al., 1993). Mucosal immunity against vaginal HSV-2 infection does not depend on IgA. Regarding cellular immunity, Th1 CD4+ cell help is required for the entry of CD8+ T cells into the genital mucosa during the effector phase of the immune response (Nakanishi et al., 2009). Furthermore, CD4+ effector T cells themselves play a direct antiviral role. In the mouse vaginal infection model, Th1 CD4+ cells enter the infected vagina and produce high levels of IFN-γ, which blocks viral replication (Iijima et al., 2008). Conversely, CD8+ T cells are important for preventing reactivation of latent HSV in neurons (Zhu et al., 2007). Thus, T cell-mediated immunity plays an important role in the prevention of HSV genital disease. Our results indicate that HF10-immunized mice rapidly produced high levels of IFN-γ in their vagina in response to vaginal infection by HSV-2. Furthermore, the vaginal sections of HF10-immunized mice revealed many CD4+ cells concentrated in the infective focus and invading the mucosal subepithelial lamina propria 1 and 3 days after challenge. CD8+ cells were detected only 1 day after challenge. Taking our data and those of previous reports into consideration, protection of HF10-immunized mice against HSV-2 genital disease appears to be associated with cellular immunity mediated mainly by Th1 CD4+ T cells.
In this study, the serum of UV-inactivated HF10-immunized mice had a lower neutralizing titer against HF10 than did that of live HF10-immunized mice. In mice immunized with UV-irradiated HF10, IFN-γ concentration in vaginal washes after HSV-2 challenge were not significantly increased compared to those of unimmunized mice. As a result, UV-inactivated HF10 immunization could not completely protect mice against HSV-2 challenge. These results indicate that UV-inactivated HF10 immunization induced weaker acquired immunity, in accordance with the belief that killed viral vaccines are inferior to live vaccines.
Recently, it has been reported that replication-defective HSV-1 that include the CMV promoter driving the HSV-1 gD expression cassette (CJ9-gD) effectively protects guinea pigs against HSV-2 genital disease by inducing strong cross-reactive immunity (Brans et al., 2008). In mice, CJ9-gD induces strong and persistent humoral and Th1-associated cellular immunity against both HSV-1 and HSV-2 (Brans et al., 2009; Lu et al., 2009). This indicates not only that HSV-1 induces cellular immune responses against HSV-2 but also that gD is a key antigen for immunity. Meanwhile, tegument proteins encoded by UL46, UL47, and UL49 also induce a Th1 type response to HSV-2 infection in BALB/c mice (Muller et al., 2009). In this study, splenocytes from HF10-immunized mice produced IFN-γ in response to gD-, ICP0-, and UL46-expressing NIH3T3 cells, indicating that memory T cells responding to epitopes of HSV-2 gD, ICP0, and a tegument protein encoded by UL46 can be induced in HF10-immunized mice. Taken together with the IFN-γ production in the vagina, HF10 inoculation could induce Th1 type cellular immunity and protect against severe HSV-2 infection. Due to the high amino acid sequence homology between gD-1 and gD-2 (Lasky and Dowbenko, 1984), the memory T cell response to gD may play an important role in the mechanism underlying protection against HSV-2 infection by HF10 inoculation.
In summary, we demonstrated that immunization with HF10, a non-engineered, naturally occurring HSV-1 mutant, protected mice against severe genital disease caused by HSV-2. Immunization with HF10 inhibited HSV-2 replication in the vagina, reduced local inflammation, blocked neuroinvasiveness, and increased survival. The protective effect was related to the induction of cellular immunity mediated mainly by Th1 CD4+ T cells. These results indicate that HF10 is a promising candidate antigen for inclusion in a vaccine against both HSV-1 and HSV-2 infection. Moreover, our data support the hypothesis that previous infection with HSV-1 provides a degree of protection against primary genital HSV-2 infection and attenuates its severity.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This study was supported by a grant from Ministry of Education, Culture, Sports and Technology, Japan (22390088).
References
Arvin, A. M., and Gershon, A. A. (1996). Live attenuated varicella vaccine. Annu. Rev. Microbiol. 50, 59–100.
Belshe, R. B., Leone, P. A., Bernstein, D. I., Wald, A., Levin, M. J., Stapleton, J. T., Gorfinkel, I., Morrow, R. L., Ewell, M. G., Stokes-Riner, A., Dubin, G., Heineman, T. C., Schulte, J. M., Deal, C. D., and Herpevac Trial for Women. (2012). Efficacy results of a trial of a herpes simplex vaccine. N. Engl. J. Med. 366, 34–43.
Berkowitz, C., Moyal, M., Rosen-Wolff, A., Darai, G., and Becker, Y. (1994). Herpes simplex virus type 1 (HSV-1) UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch. Virol. 134, 73–83.
Bernstein, D. I., Aoki, F. Y., Tyring, S. K., Stanberry, L. R., St-Pierre, C., Shafran, S. D., Leroux-Roels, G., Van Herck, K., Bollaerts, A., Dubin, G., and GlaxoSmithKline Herpes Vaccine Study Group. (2005). Safety and immunogenicity of glycoprotein D-adjuvant genital herpes vaccine. Clin. Infect. Dis. 40, 1271–1281.
Brans, R., Akhrameyeva, N. V., and Yao, F. (2009). Prevention of genital herpes simplex virus type 1 and 2 disease in mice immunized with a gD-expressing dominant-negative recombinant HSV-1. J. Invest. Dermatol. 129, 2470–2479.
Brans, R., Eriksson, E., and Yao, F. (2008). Immunization with a dominant-negative recombinant HSV type 1 protects against HSV-1 skin disease in guinea pigs. J. Invest. Dermatol. 128, 2825–2832.
Bryson, Y., Dillon, M., Bernstein, D. I., Radolf, J., Zakowski, P., and Garratty, E. (1993). Risk of acquisition of genital herpes simplex virus type 2 in sex partners of persons with genital herpes: a prospective couple study. J. Infect. Dis. 167, 942–946.
Cappel, R. (1976). Comparison of the humoral and cellular immune response after immunization with live, UV inactivated herpes simplex virus and a subunit vaccine and efficacy of these immunizations. Arch. Virol. 52, 29–35.
Celum, C., Wald, A., Hughes, J., Sanchez, J., Reid, S., Delany-Moretlwe, S., Cowan, F., Casapia, M., Ortiz, A., Fuchs, J., Buchbinder, S., Koblin, B., Zwerski, S., Rose, S., Wang, J., Corey, L., and HPTN 039 Protocol Team. (2008). Effect of aciclovir on HIV-1 acquisition in herpes simplex virus 2 seropositive women and men who have sex with men: a randomised, double-blind, placebo-controlled trial. Lancet 371, 2109–2119.
Corey, L., and Spear, P. G. (1986). Infections with herpes simplex viruses (2). N. Engl. J. Med. 314, 749–757.
Eis-Hubinger, A. M., Schmidt, D. S., and Schneweis, K. E. (1993). Anti-glycoprotein B monoclonal antibody protects T cell-depleted mice against herpes simplex virus infection by inhibition of virus replication at the inoculated mucous membranes. J. Gen. Virol. 74(Pt 3), 379–385.
Freeman, E. E., Weiss, H. A., Glynn, J. R., Cross, P. L., Whitworth, J. A., and Hayes, R. J. (2006). Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20, 73–83.
Fujimoto, Y., Mizuno, T., Sugiura, S., Goshima, F., Kohno, S., Nakashima, T., and Nishiyama, Y. (2006). Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol. 126, 1115–1117.
Goshima, F., Watanabe, D., Takakuwa, H., Wada, K., Daikoku, T., Yamada, M., and Nishiyama, Y. (2000). Herpes simplex virus UL17 protein is associated with B capsids and colocalizes with ICP35 and VP5 in infected cells. Arch. Virol. 145, 417–426.
Iijima, N., Linehan, M. M., Zamora, M., Butkus, D., Dunn, R., Kehry, M. R., Laufer, T. M., and Iwasaki, A. (2008). Dendritic cells and B cells maximize mucosal Th1 memory response to herpes simplex virus. J. Exp. Med. 205, 3041–3052.
Jiang, Y. M., Daikoku, T., Yamamoto, M., Morishima, T., and Nishiyama, Y. (1995). Growth and cytopathogenicity of herpes simplex virus in a macrophage cell line, RAW264: a good indicator of intraperitoneal pathogenicity. Microbiol. Immunol. 39, 905–909.
Jones, C., Inman, M., Peng, W., Henderson, G., Doster, A., Perng, G. C., and Angeletti, A. K. (2005). The herpes simplex virus type 1 locus that encodes the latency-associated transcript enhances the frequency of encephalitis in male BALB/c mice. J. Virol. 79, 14465–14469.
Kapiga, S. H., Sam, N. E., Bang, H., Ni, Q., Ao, T. T., Kiwelu, I., Chiduo, S., Ndibe, U., Seage, G. III, Coplan, P., Shao, J., Rosenberg, Z. F., and Essex, M. (2007). The role of herpes simplex virus type 2 and other genital infections in the acquisition of HIV-1 among high-risk women in northern Tanzania. J. Infect. Dis. 195, 1260–1269.
Kato, K., Daikoku, T., Goshima, F., Kume, H., Yamaki, K., and Nishiyama, Y. (2000). Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145, 2149–2162.
Kieff, E., Hoyer, B., Bachenheimer, S., and Roizman, B. (1972). Genetic relatedness of type 1 and type 2 herpes simplex viruses. J. Virol. 9, 738–745.
Kimata, H., Imai, T., Kikumori, T., Teshigahara, O., Nagasaka, T., Goshima, F., Nishiyama, Y., and Nakao, A. (2006). Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann. Surg. Oncol. 13, 1078–1084.
Koshizuka, T., Goshima, F., Takakuwa, H., Nozawa, N., Daikoku, T., Koiwai, O., and Nishiyama, Y. (2002). Identification and characterization of the UL56 gene product of herpes simplex virus type 2. J. Virol. 76, 6718–6728.
Koshizuka, T., Takakuwa, H., Goshima, F., Murata, T., and Nishiyama, Y. (2001). The US11 gene product of herpes simplex virus has intercellular trafficking activity. Biochem. Biophys. Res. Commun. 288, 597–602.
Lafferty, W. E., Downey, L., Celum, C., and Wald, A. (2000). Herpes simplex virus type 1 as a cause of genital herpes: impact on surveillance and prevention. J. Infect. Dis. 181, 1454–1457.
Lasky, L. A., and Dowbenko, D. J. (1984). DBA sequence analysis of the type-common glycoprotein-D genes of herpes simplex virus types 1 and 2. DNA 3, 23–29.
Lu, Z., Brans, R., Akhrameyeva, N. V., Murakami, N., Xu, X., and Yao, F. (2009). High-level expression of glycoprotein D by a dominant-negative HSV-1 virus augments its efficacy as a vaccine against HSV-1 infection. J. Invest. Dermatol. 129, 1174–1184.
Malvy, D., Halioua, B., Lancon, F., Rezvani, A., Bertrais, S., Chanzy, B., Daniloski, M., Ezzedine, K., Malkin, J. E., Morand, P., De Labareyre, C., Hercberg, S., and El Hasnaoui, A. (2005). Epidemiology of genital herpes simplex virus infections in a community-based sample in France: results of the HERPIMAX study. Sex. Transm. Dis. 32, 499–505.
McDermott, M. R., Brais, L. J., and Evelegh, M. J. (1990). Mucosal and systemic antiviral antibodies in mice inoculated intravaginally with herpes simplex virus type 2. J. Gen. Virol. 71(Pt 7), 1497–1504.
Mertz, G. J., Benedetti, J., Ashley, R., Selke, S. A., and Corey, L. (1992). Risk factors for the sexual transmission of genital herpes. Ann. Intern. Med. 116, 197–202.
Miller, C. S., Danaher, R. J., and Jacob, R. J. (1998). Molecular aspects of herpes simplex virus I latency, reactivation, and recurrence. Crit. Rev. Oral Biol. Med. 9, 541–562.
Mori, I., Liu, B., Goshima, F., Ito, H., Koide, N., Yoshida, T., Yokochi, T., Kimura, Y., and Nishiyama, Y. (2005). HF10, an attenuated herpes simplex virus (HSV) type 1 clone, lacks neuroinvasiveness and protects mice against lethal challenge with HSV types 1 and 2. Microbes Infect. 7, 1492–1500.
Muller, W. J., Dong, L., Vilalta, A., Byrd, B., Wilhelm, K. M., McClurkan, C. L., Margalith, M., Liu, C., Kaslow, D., Sidney, J., Sette, A., and Koelle, D. M. (2009). Herpes simplex virus type 2 tegument proteins contain subdominant T-cell epitopes detectable in BALB/c mice after DNA immunization and infection. J. Gen. Virol. 90(Pt 5), 1153–1163.
Nakanishi, Y., Lu, B., Gerard, C., and Iwasaki, A. (2009). CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462, 510–513.
Nakao, A., Takeda, S., Shimoyama, S., Kasuya, H., Kimata, H., Teshigahara, O., Sawaki, M., Kikumori, T., Kodera, Y., Nagasaka, T., Goshima, F., Nishiyama, Y., and Imai, T. (2007). Clinical experiment of mutant herpes simplex virus HF10 therapy for cancer. Curr. Cancer Drug Targets 7, 169–174.
Nieuwenhuis, R. F., van Doornum, G. J., Mulder, P. G., Neumann, H. A., and van der Meijden, W. I. (2006). Importance of herpes simplex virus type-1 (HSV-1) in primary genital herpes. Acta Derm. Venereol. 86, 129–134.
Nishiyama, Y., Kimura, H., and Daikoku, T. (1991). Complementary lethal invasion of the central nervous system by nonneuroinvasive herpes simplex virus types 1 and 2. J. Virol. 65, 4520–4524.
Oxman, M. N., Levin, M. J., Johnson, G. R., Schmader, K. E., Straus, S. E., Gelb, L. D., Arbeit, R. D., Simberkoff, M. S., Gershon, A. A., Davis, L. E., Weinberg, A., Boardman, K. D., Williams, H. M., Zhang, J. H., Peduzzi, P. N., Beisel, C. E., Morrison, V. A., Guatelli, J. C., Brooks, P. A., Kauffman, C. A., Pachucki, C. T., Neuzil, K. M., Betts, R. F., Wright, P. F., Griffin, M. R., Brunell, P., Soto, N. E., Marques, A. R., Keay, S. K., Goodman, R. P., Cotton, D. J., Gnann, J.W. Jr., Loutit, J., Holodniy, M., Keitel, W. A., Crawford, G. E., Yeh, S. S., Lobo, Z., Toney, J. F., Greenberg, R. N., Keller, P. M., Harbecke, R., Hayward, A. R., Irwin, M. R., Kyriakides, T. C., Chan, C. Y., Chan, I. S., Wang, W. W., Annunziato, P. W., Silber, J. L., and Shingles Prevention Study Group. (2005). A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 352, 2271–2284.
Peng, W., Henderson, G., Inman, M., BenMohamed, L., Perng, G. C., Wechsler, S. L., and Jones, C. (2005). The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal Ganglia of acutely infected mice. J. Virol. 79, 6162–6171.
Rosen-Wolff, A., Lamade, W., Berkowitz, C., Becker, Y., and Darai, G. (1991). Elimination of UL56 gene by insertion of LacZ cassette between nucleotide position 116030 to 121753 of the herpes simplex virus type 1 genome abrogates intraperitoneal pathogenicity in tree shrews and mice. Virus Res. 20, 205–221.
Stanberry, L. R. (2004). Clinical trials of prophylactic and therapeutic herpes simplex virus vaccines. Herpes 11(Suppl. 3), 161A–169A.
Stanberry, L. R., Cunningham, A. L., Mindel, A., Scott, L. L., Spruance, S. L., Aoki, F. Y., and Lacey, C. J. (2000). Prospects for control of herpes simplex virus disease through immunization. Clin. Infect. Dis. 30, 549–566.
Stanberry, L. R., Spruance, S. L., Cunningham, A. L., Bernstein, D. I., Mindel, A., Sacks, S., Tyring, S., Aoki, F. Y., Slaoui, M., Denis, M., Vandepapeliere, P., Dubin, G., and GlaxoSmithKline Herpes Vaccine Efficacy Study Group. (2002). Glycoprotein-D-adjuvant vaccine to prevent genital herpes. N. Engl. J. Med. 347, 1652–1661.
Swenson, S. L., McMillen, J., and Hill, H. T. (1993a). Diagnostic compatibility of a thymidine kinase, inverted repeat, gI, and gpX modified live gene-deleted PRV vaccine with three differential ELISAs. J. Vet. Diagn. Invest. 5, 347–350.
Swenson, S. L., McMillen, J., and Hill, H. T. (1993b). Evaluation of the safety and efficacy of a thymidine kinase, inverted repeat, gI, and gpX gene-deleted pseudorabies vaccine. J. Vet. Diagn. Invest. 5, 341–346.
Tao, G., Kassler, W. J., and Rein, D. B. (2000). Medical care expenditures for genital herpes in the United States. Sex. Transm. Dis. 27, 32–38.
Ushijima, Y., Luo, C., Goshima, F., Yamauchi, Y., Kimura, H., and Nishiyama, Y. (2007). Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect. 9, 142–149.
Van de Walle, G. R., Favoreel, H. W., Nauwynck, H. J., and Pensaert, M. B. (2003). Antibody-induced internalization of viral glycoproteins and gE-gI Fc receptor activity protect pseudorabies virus-infected monocytes from efficient complement-mediated lysis. J. Gen. Virol. 84(Pt 4), 939–948.
Watson-Jones, D., Weiss, H. A., Rusizoka, M., Changalucha, J., Baisley, K., Mugeye, K., Tanton, C., Ross, D., Everett, D., Clayton, T., Balira, R., Knight, L., Hambleton, I., Le Goff, J., Belec, L., Hayes, R., HSV Trial Team, and Steering and Data Monitoring Committees. (2008). Effect of herpes simplex suppression on incidence of HIV among women in Tanzania. N. Engl. J. Med. 358, 1560–1571.
Xu, F., Sternberg, M. R., Kottiri, B. J., McQuillan, G. M., Lee, F. K., Nahmias, A. J., Berman, S. M., and Markowitz, L. E. (2006). Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296, 964–973.
Keywords: genital herpes, live attenuated vaccine, HSV-1, HSV-2
Citation: Luo C, Goshima F, Kamakura M, Mutoh Y, Iwata S, Kimura H and Nishiyama Y (2012) Immunization with a highly attenuated replication-competent herpes simplex virus type 1 mutant, HF10, protects mice from genital disease caused by herpes simplex virus type 2. Front. Microbio. 3:158. doi: 10.3389/fmicb.2012.00158
Received: 09 February 2012; Paper pending published: 07 March 2012;
Accepted: 09 April 2012; Published online: 30 April 2012.
Edited by:
Tatsuya Tsurumi, Aichi Cancer Center, JapanReviewed by:
Tohru Daikoku, University of Toyama, JapanYoshitaka Sato, Kobe University School of Medicine, Japan
Tatsuo Suzutani, Fukushima Medical University School of Medicine, Japan
Copyright: © 2012 Luo, Goshima, Kamakura, Mutoh, Iwata, Kimura and Nishiyama. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Hiroshi Kimura, Department of Virology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, Japan. e-mail: hkimura@med.nagoya-u.ac.jp