 Xia Ding1,2
Xia Ding1,2 Xiao-Jue Peng1Bin-Song Jin1Ming Xiao3Jia-Kuan Chen1,4
Xiao-Jue Peng1Bin-Song Jin1Ming Xiao3Jia-Kuan Chen1,4 Bo Li1,4Chang-Ming Fang4
Bo Li1,4Chang-Ming Fang4 Ming Nie1,4*
Ming Nie1,4*- 1School of Life Sciences and Institute of Life Science, Nanchang University, Nanchang, China
- 2School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing, China
- 3College of Life and Environment Science, Shanghai Normal University, Shanghai, China
- 4Ministry of Education Key Lab for Biodiversity Science and Ecological Engineering, The Institute of Biodiversity Science, Fudan University, Shanghai, China
The spatial distributions of bacterial communities may be driven by multiple environmental factors. Thus, understanding the relationships between bacterial distribution and environmental factors is critical for understanding wetland stability and the functioning of freshwater lakes. However, little research on the bacterial communities in deep sediment layers exists. In this study, thirty clone libraries of 16S rRNA were constructed from a beach wetland of the Poyang Lake along both horizontal (distance to the water-land junction) and vertical (sediment depth) gradients to assess the effects of sediment properties on bacterial community structure and diversity. Our results showed that bacterial diversity increased along the horizontal gradient and decreased along the vertical gradient. The heterogeneous sediment properties along gradients substantially affected the dominant bacterial groups at the phylum and species levels. For example, the NH+4 concentration decreased with increasing depth, which was positively correlated with the relative abundance of Alphaproteobacteria. The changes in bacterial diversity and dominant bacterial groups showed that the top layer had a different bacterial community structure than the deeper layers. Principal component analysis revealed that both gradients, not each gradient independently, contributed to the shift in the bacterial community structure. A multiple linear regression model explained the changes in bacterial diversity and richness along the depth and distance gradients. Overall, our results suggest that spatial gradients associated with sediment properties shaped the bacterial communities in the Poyang Lake beach wetland.
Introduction
Wetland ecosystems are considered the most biologically diverse ecosystems (Iasur-Kruh et al., 2009; Wang et al., 2012). A beach wetland is a landform along the edge of a body of water and is an interface between the land and water. Bacteria are ubiquitous and play key roles in ecosystem functioning, including cycling of the majority of biologically active elements (Woese, 1990; Woese et al., 1990; Gucht et al., 2007; Newton et al., 2011). However, systematic exploration of geographic bacterial patterns through the simultaneous consideration of contemporary environmental variations and stereoscopic spatial distribution (distance and depth) is largely lacking, resulting in a poor understanding of how environmental factors shape bacterial communities in beach wetlands of lake ecosystems (Yannarell and Triplett, 2005; Córdova-Kreylos et al., 2006; Zhou et al., 2008).
Recent studies demonstrate that bacterial communities in lake wetland ecosystems are strongly correlated with a multitude of environmental factors over horizontal gradients ranging from hundreds of kilometers to centimeters (Terrados et al., 1999; Yannarell and Triplett, 2004; Crump et al., 2007). Several studies suggest that bacterial distributions may be spatially predictable rather than random (Ettema and Wardle, 2002). Differences in environmental factors along sediment horizontal gradients largely determine bacterial composition and diversity, such as water content (Drenovsky et al., 2004; Badin et al., 2011), C and N availability (Cookson et al., 2008; Moseman-Valtierra et al., 2010; Mackelprang et al., 2011; Lin et al., 2012), temperature (Hall et al., 2008; Redmond and Valentine, 2011), pH (Lindström et al., 2005), and sediment structure characteristics (Liu et al., 2011). Despite the importance of sediment bacteria in biogeochemical cycling, the bacterial communities in the deeper layers are not well studied (Haglund et al., 2003; Luna et al., 2004); the bacterial communities of the sediment surface layers have been far better studied than those of the deeper layers (Liao et al., 2009; Schauer et al., 2010). Expanding our knowledge of bacterial diversity and distribution from the surface to deeper sediment layers will improve our understanding of biodiversity and functioning of beach sediment.
In this study, we evaluated the spatial distribution of bacterial communities along gradients of both sediment depth and distance to the water-land junction in the Poyang Lake wetland (Figure 1) (Jiangxi Province, mid-China), the largest freshwater lake in China. The aim of this study was to determine whether main bacterial communities are regularly distributed along vertical and horizontal gradients and what environmental factors affect the spatial distributions of bacterial communities. The spatial distribution of the bacterial communities was determined by constructing clone libraries of 16S rRNA and analyzing the associations between the different communities. We hypothesized that different geochemical parameters along vertical and horizontal gradients affect specific bacterial groups in beach sediments.
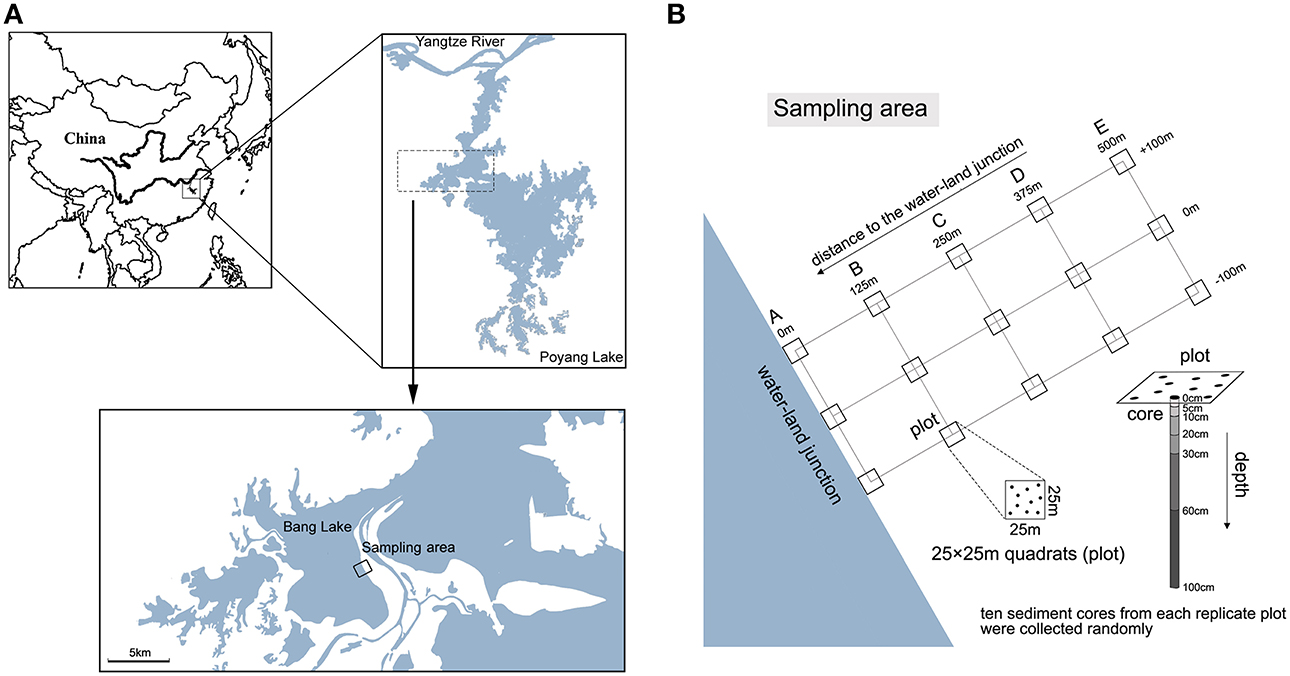
Figure 1. Maps of sampling stations. (A) The sampling sites in the Bang Lake of the Poyang Lake wetland. (B) Core sediment samples were collected from a range of beach wetland locations along gradients of depth and distance to the water-land junction. We sampled 5 sediment locations (A, B, C, D and E), which were 0, 125, 250, 375, and 500 m from the water-land junction, respectively. Three 25 × 25 m quadrats (plots) that were 100 m apart were established in each location. From each replicate plot, 10 sediment cores (diameter 2.5 cm) were collected and pooled.
Methods
Study Location and Sampling
Poyang Lake is located in the northern part of the Jiangxi Province and at the southern bank of the middle reaches of the Yangtze River (29°07′N, 115°59′E) (Figure 1A) (Liu et al., 2011). The lake covers 3283 km2 during the rainy season. The mean annual temperature and precipitation are 17°C and 1636 mm, respectively. The Poyang Lake wetland has a high fluctuation of seasonal water levels between the flood and dry seasons (Xu et al., 2014). In the dry season, the surface area of the lake shrinks to less than one-tenth of the area in the flood season. We sampled beach sediments between the dry and flood seasons, because this stage could capture the major characteristics of beach wetland and obtain great differences in geochemical parameters (e.g., water content) along the spatial gradients. In July 2011, sediment samples were collected from a range of beach wetland locations along gradients of depth and distance to the water-land junction on the shore of Bang Lake, which is an isolated sub-lake located in the core area of Poyang Lake. We chose this lake for avoiding disturbances from agricultural and grazing activities. We sampled 5 sediment locations (A, B, C, D and E), located 0, 125, 250, 375, and 500 m from the water-land junction, respectively. Three 25 × 25 m quadrats (plots) that were 100 m apart were established in each location. From each replicate plot, 10 sediment cores were collected using side opening steel tubes (length 1.2 m, diameter 2.5 cm). Next, the samples were pooled and sieved (mesh size < 2 mm) to remove stones and plant materials. At each location, we sampled 6 depth strata per transect: 0–5, 5–10, 10–20, 20–30, 30–60, and 60–100 cm (Figure 1B). Samples for sediment chemical analysis were stored at 4°C, and samples for clone library analysis were stored at −20°C. All samples were processed within 4 h of collection. Detailed descriptions of the sampling coordinates, sediment chemical properties, sediment depth, and distance to the water-land junction are listed in Table S1.
Sediment Physicochemical Analyses
To understand physiochemical effects from the spatial distances, water content (WC), sediment bulk density (SBD), sediment organic carbon (SOC), and inorganic nitrogen (NH+4, NO−3) were determined. These sediment physicochemical variables were shown to have low co-correlations with each other because the average correlation coefficient r value is 0.27 and only 4 of 15 correlations between these variables are significant (P < 0.05) (Table S2). WC was determined by weighing a sediment sample before and after drying at 105°C for 24–48 h to a constant weight. SBD was determined by oven-drying sediment cores of a fixed volume (Cui et al., 2012). Sediment pH was measured on sediment slurry at a 2.5:1 water: sediment ratio using a glass electrode (Meng et al., 2012). SOC was measured with a TOC analyzer (Analytikjena HT1300, Germany) after removing sediment carbonates using 1 M HCl. Inorganic nitrogen (NH+4, NO−3) was extracted and measured using 2 M KCl and a discrete auto analyzer (Smartchem 200, Westco, France).
DNA Extraction, Clone Library Construction and Sequencing
The total genomic DNA of sediment samples was extracted from 0.5 g (fresh weight) of the sediment sample with the Fast DNA Spin kit for sediments (Qbiogene, Irvine, CA) according to the manufacturer's instructions. After extraction, the DNA samples were immediately frozen at −80°C for further analysis.
The template DNA isolated from the subsamples of each location was pooled so that each subsample was equally represented. The pooled DNA (20 ng for each sample) was analyzed using PCR (predenaturation step of 5 min at 95°C followed by 30 cycles of 1 min at 94°C, 30 s at 53°C and 2 min at 72°C, followed by a final elongation step of 72°C for 15 min), with the bacteria-specific primers 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 1492R (5′-TAC GGY TAC CTT GTT ACG ACT T-3′) (Lane, 1991; Meng et al., 2012). The PCR products were purified using the QIAquick PCR Purification Kit (Qiagen, Germany) and quantified using Nano Drop ND-3000 (Nano-Drop Technologies). The PCR products were subsequently cloned into the pMD18-T vector system (TaKaRa, Japan) and transformed into Escherichia coli Top 10. A total of 2520 recombinant clones were individually chosen from the 30 clone libraries, and partial 16S rRNA gene sequences were determined using a BigDye Terminator V3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and an ABI3730 PRISM Genetic Analyzer (Applied Biosystems).
Sequence Processing and Operational Taxonomic Unit (OTU) Clustering
A total of 2380 sequences from 30 clone libraries (each clone library had 73 to 86 sequences) were retained and deposited into the GenBank database under the accession numbers (KJ013600–KJ015979).
The Ribosomal Database Project (RDP; http://rdp.cme.msu.edu/) classifier was used to assign 16S rRNA gene sequences (Maidak et al., 2001). The sequences were screened and sorted for chimeras within Mothur using the chimera.uchime command (http://www.mothur.org/wiki/Main_Page). The OTU clustering was performed by setting a 0.03 distance limit (equivalent to 97% similarity) using the Mothur program (Schloss et al., 2009).
Statistical Methods for Community Analyses
Canonical Correspondence Analysis (CCA) and Principal Component Analysis (PCA) were used to identify the most important abiotic factors to the bacterial community composition. This analysis was performed using a Multivariate Statistical Package (MVSP) (Kovach Computing, Anglesey, United Kingdom). The relationships between the relative abundance of bacterial species and the taxonomic diversity for groups with shared physico-chemical features were tested with linear regression analyses using SigmaStat 3.5/SigmaPlot 10.0 (SysStat Software Inc., CA).
Results
Sediment Physico-Chemical Characteristics
A total of 30 samples were collected from 5 different locations along a distance gradient, and samples from each of the 6 different depths were collected at each location. The major geographical and physiochemical characteristics of the lake sediments are summarized in Table S1. Across the sampling sites, the WC varied from 32.2 to 23.2%, and the SOC varied from 25.09 to 6.03 g·kg−1. The WC and SOC were highly correlated with the geographic distance to the water-land junction, with both WC (P < 0.001) and SOC (P < 0.05) decreasing as the distance to the water-land junction increased. The SBD varied from 0.27 to 0.62 g·cm−3, and the NH+4 concentration ranged from 14.92 to 107.86 mg·kg−1. Both the SBD and NH+4 were significantly correlated with geographic depth (all P < 0.001). The SBD increased with increasing depth, and NH+4 concentration decreased with increasing depth (Table S2). No significant spatial differences were observed in the sediment pH, which varied from 6.4 to 7.9.
Distribution of Taxa and Phylotypes
The clone libraries were built with careful consideration of sequence quality to ensure significant clone coverage. Across all sediment samples, we obtained a total of 2380 sequences, with 480–510 sequences per location (mean = 500) (Table S3). We successfully classified 79.7% of the obtained sequences.
The dominant phyla (relative abundance >5%) across all locations were Proteobacteria (32.61%), Actinobacteria (14.82%), Acidobacteria (10.38%), Chloroflexi (9.78%), and Firmicutes (5.89%). These phyla accounted for more than 73.48% of the collected bacterial sequences (Figure 2). Spirochaetes, Nitrospira, Armatimonadetes, WS3, Chlorobi, Planctomycetes, Cyanobacteria/Chloroplast, Bacteroidetes, TM7, Verrucomicrobia, and Gemmatimonadetes were present in most of the sediment samples with low relative abundance (<5%) (Figure 2, Table S4). The RDP database revealed the recovery of 21 phyla from lake epilimnia, with 5 of the phyla frequently commonly recovered (Proteobacteria, particularly Betaproteobacteria, with 4300 and 2600 sequences, respectively; Actinobacteria with 3000 sequences; Bacteroidetes with 1900 sequences; Cyanobacteria with 800 sequences; and Verrucomicrobia with 300 sequences) (Newton et al., 2011). However, both the phyla Cyanobacteria and Bacteroidetes were present in low relative abundance in Poyang Lake sediment.
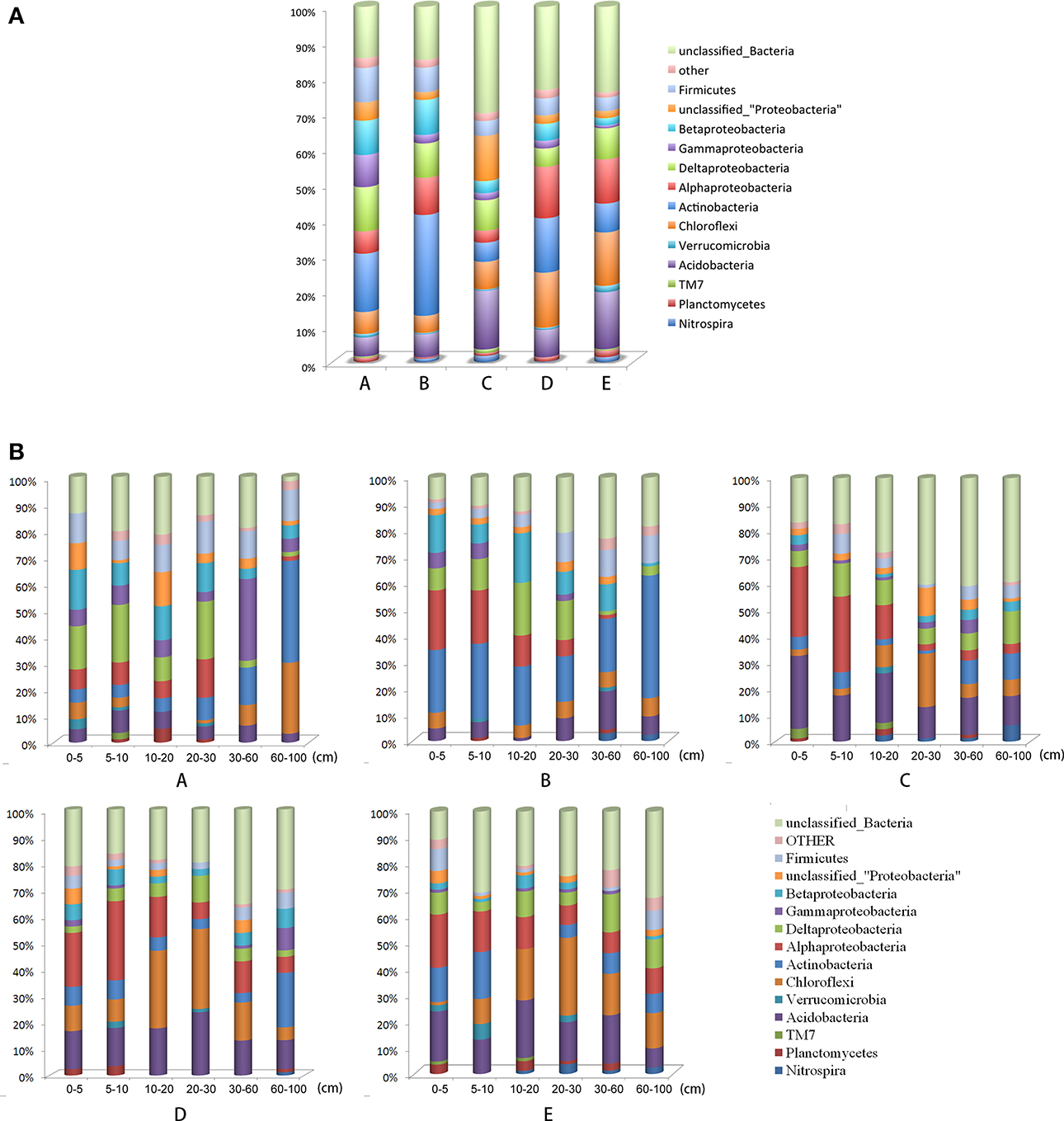
Figure 2. Relative abundances of the dominant bacterial phyla in the sampling locations. Relative abundances were calculated by the proportional frequencies of the DNA sequences that could be classified at the phylum level. (A) A, B, C, D, and E were five sediment locations, located 0, 125, 250, 375, and 500 m from the water-land junction, respectively. (B) At each location (A, B, C, D, and E), there were 6 depth strata sampled per transect: 0–5, 5–10, 10–20, 20–30, 30–60, and 60–100 cm.
The different taxonomic levels of the bacteria were shown to heterogeneously distribute along horizontal and vertical gradients. The decrease in bacterial relative abundance as distance from the water-land junctions increased was particularly sharp for Firmicutes, Deltaproteobacteria, and Gammaproteobacteria (Figure 2). However, the Alphaproteobacteria, Chloroflexi, and Acidobacteria relative abundance increased along the distance gradient (Figure 2). Alphaproteobacteria and Deltaproteobacteria were more abundant at depths less than 30 cm (Table S4).
The heterogeneous distribution along horizontal and vertical gradients was observed more clearly when the bacterial communities were classified at the genus level. Acidobacteria_Gp6, Acidobacteria_Gp2, Acidobacteria_Gp1, Arthrobacter, Pseudolabrys, Thermosporothrix, and Ktedonobacter were detected in nearly all the samples (Figure 3). Othergenera were found in a portion of the samples. Specifically, Acidobacteria_Gp3, TM7_genera_incertae_sedis and Singulisphaera were mainly distributed in surface sediments, whereas Nitrospira was mainly distributed in submerged sediments. Methylocystis, TM7_genera_incertae_sedis, and Acinetobacter were abundant in the water-land junctions, whereas Rhodoplanes, Acidobacteria_Gp7, and Armatimonadetes_gp4 were mainly distributed far from the water-land junctions (Figure 3).
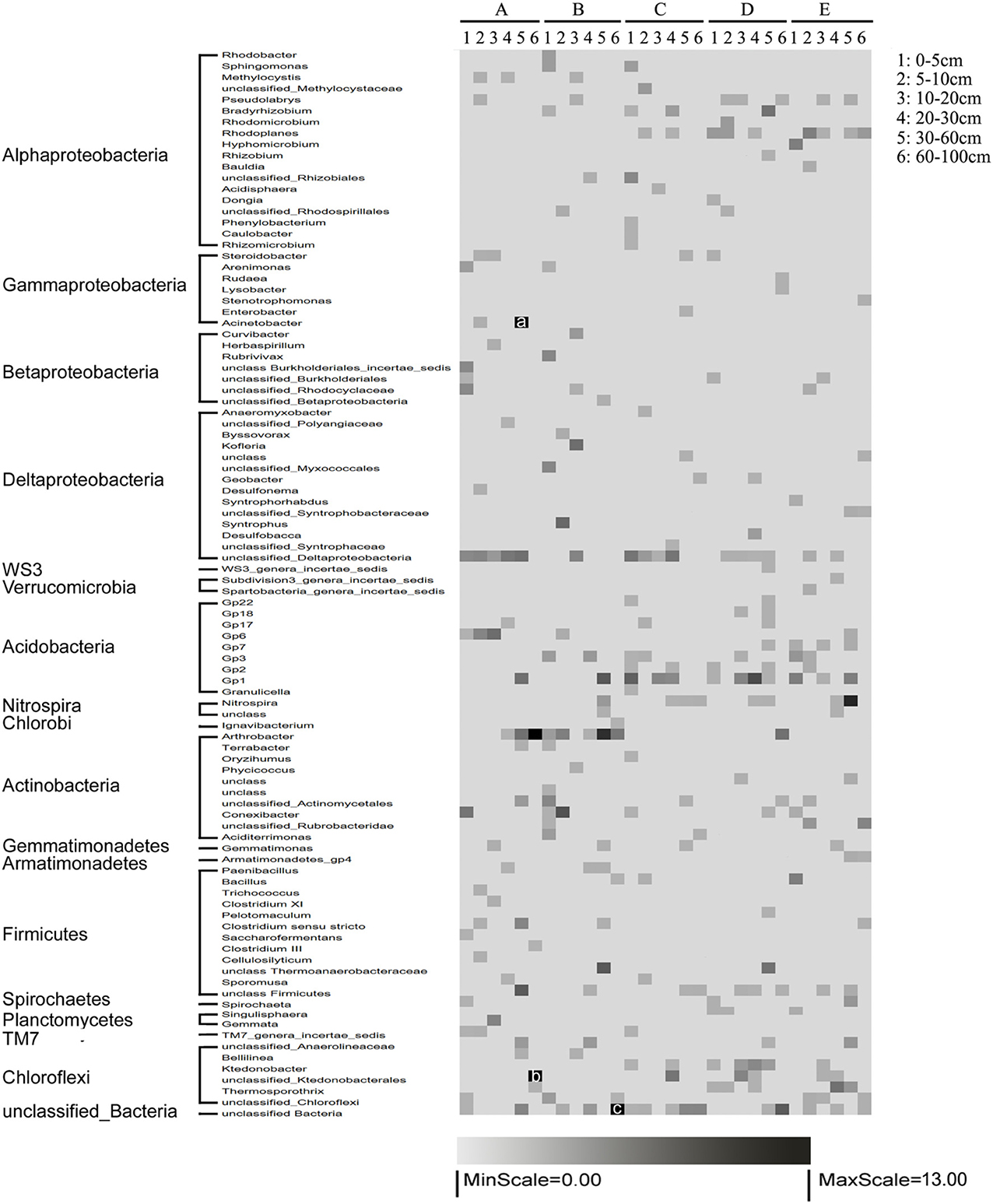
Figure 3. Relative abundance distribution of the dominant operational taxonomic units (OTUs) (97% similarity). A, B, C, D, and E represent the five sediment locations, which were 0, 125, 250, 375, and 500 m from the water-land junction, respectively. Numbers 1, 2, 3, 4, 5, and 6 represent the six depth strata sampled per transect: 0–5, 5–10, 10–20, 20–30, 30–60, and 60–100 cm. The relative abundances of each OTU were normalized to have a mean of 0 and a standard deviation of 100%. The percent abundances of dominant genera in each location are indicated by shading: percentages close to 13 are indicated in black, and those close to 0 are in gray. The actual percentages of a, b, and c are 24, 14, and 19%, respectively.
Relationship between Bacterial Community Structure and Sediment Variables
Canonical correspondence analyses (CCA) were performed to examine the relationship between bacterial community structure and geochemistry. The results indicated that sediment chemical properties and geographical characteristics have different effects on sediment bacteria (Figure 4, Tables S5–S7). Axis 1 and axis 2 were interpreted as the distance to the water-land junction gradient and depth, respectively. The small angles between the WC, SOC and NO−3 vectors along the distance to the water-land junction indicated strong correlations among these variables. Similarly, the small angles between the pH, SBD and NH+4 vectors along the depth indicated strong correlations between these variables. The WC, SOC, and NO−3 showed strong positive correlations with axis 1 and positive correlations with axis 2. The SBD and pH showed strong negative correlations with axis 2 and positive correlations with axis 1. NH+4 showed a strong negative correlation with both the first and second axes. Among the sediment chemical properties, the WC, SBD, NH+4, and SOC were the most important factors in determining the bacterial community structure (Figure 4A). In addition, Planctomycetes, Alphaproteobacteria, Verrucomicrobia, and Nitrospira were significantly associated with NH+4, SBD, and depth. Furthermore, Betaproteobacteria, Gammaproteobacteria, Acidobacteria, and Firmicute were highly associated with the WC, SOC, and distance to the water-land junction (Figure 4B).
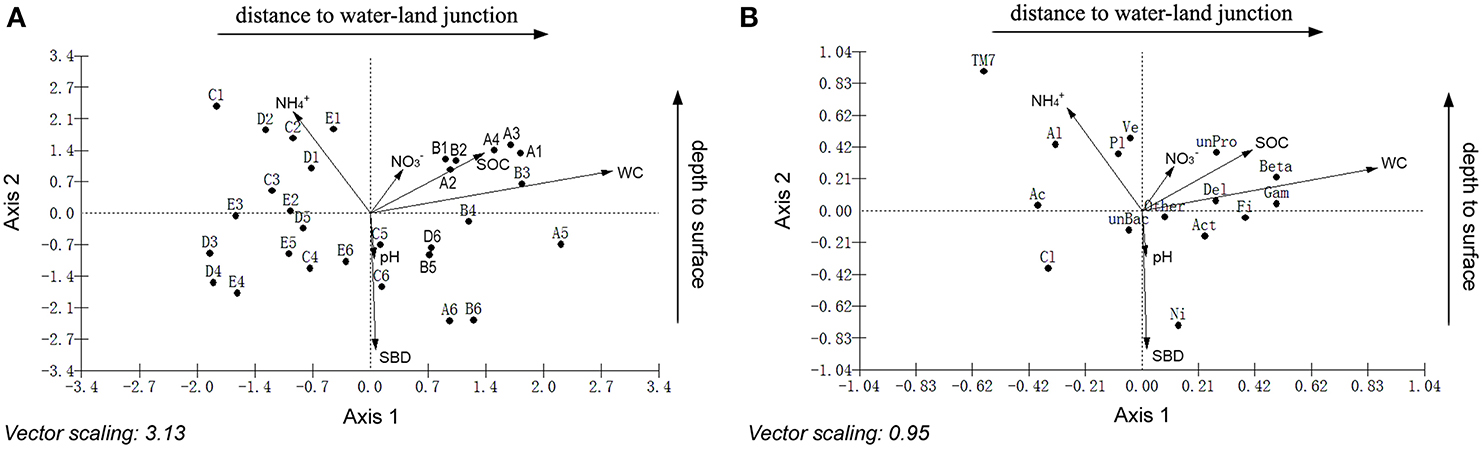
Figure 4. Canonical correspondence analysis (CCA) showing the relationship between the sampling locations and sediment variables (A), and the relationship between the relative abundances of the dominant bacterial phyla and sediment variables (B). The direction of an arrow indicates the steepest increase in the variable, and the length indicates the strength relative to the other variables. The filled circle in (A) represents the sampling locations. A, B, C, D, and E were five sediment locations, which were 0, 125, 250, 375, and 500 m from the water-land junction, respectively. At each location, there were six depth strata sampled per transect: 0–5, 5–10, 10–20, 20–30, 30–60, and 60–100 cm, which were indicated by numbers 1, 2, 3, 4, 5, and 6, respectively. The filled circle in (B) represents the diversity of the bacteria. Bacterial group abbreviations are Pro, Proteobacteria; Al, Alphaproteobacteria; Beta, Betaproteobacteria; Gama, Gammaproteobacteria; Del, Deltaproteobacteria; Act, Actinobacteria; Ac, Acidobacteria; Cl, Chloroflexi; Fi, Firmicutes; Ni, Nitrospira; Pl, Planctomycetes; TM7; Ve, Verrucomicrobia. WC, water content; SBD, sediment bulk density; SOC, soil organic carbon.
We also preformed linear regression analyses to elucidate the relationships between bacterial relative abundances and sediment variables. The relative abundances of dominant bacterial phyla (Proteobacteria, Alphaproteobacteria, Nitrospira, Betaproteobacteria, Acidobacteria, and Firmicute) were significantly correlated with the sampling depth and the distance to the water-land junction (all P < 0.005) (Figure S1, Table S2). The sediment chemical properties (WC, SBD, and NH+4) were also significantly correlated with the distributions of these bacteria across sediment depth and sediment distance to the water-land junction (Figure S2, Table S2). These results were consistent with our above-mentioned CCA observations: sediment chemical properties were divided into two different “factors” determined by sediment depth and distance to the water-land junction (Table S2, Figures 3, 5).
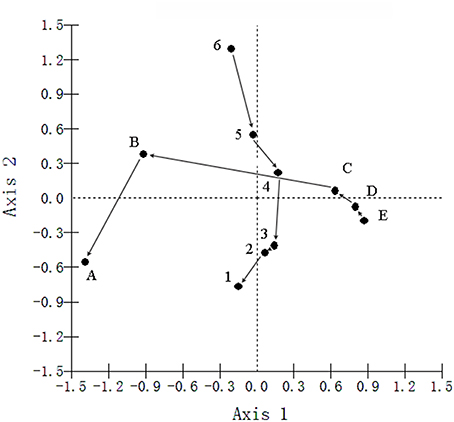
Figure 5. Principal Component Analyses (PCA) of the bacterial communities, with symbols coded by depths or sites. A, B, C, D, and E were the five sediment locations, which were 0, 125, 250, 375, and 500 m from the water-land junction, respectively. At each location, there were six depth strata sampled per transect: 0–5, 5–10, 10–20, 20–30, 30–60, and 60–100 cm, which were indicated by numbers 1, 2, 3, 4, 5, and 6, respectively.
Changes in Bacterial Diversity Along the Depth and Distance
Gradients
Furthermore, PCA was performed to test whether there was a difference between the depth and distance by clustering the samples according to depth regardless of site and clustering the samples according to site (regardless of depth) (Figure 5 and Table S8). The PCA biplot clearly revealed that the bacterial communities were shaped by both depth and distance. It was notable that Axis 1 changed more dramatically within the sites of A, B and C than in the sites of C, D and E (Figure 5).
Discussion
Sediment characteristics of the Poyang Lake varied in space. Our samples were collected between the dry and flood seasons and therefore do not take into account the seasonal variability of the system. Nevertheless, although the results only provide a “snap-shot” of how spatial gradients shaped the bacterial communities, they suggest that multiple environmental factors along spatial gradients can strongly mediate beach bacterial communities in a subtropical freshwater wetland region of China.
The clone library analysis used here was able to target the dominant bacteria. However, the averaged 80 clones per sample could lead to miss some groups of important bacteria, resulting in underestimation of bacterial diversity. Our study suggests that Proteobacteria, Acidobacteria, and Actinobacteria were dominant prokaryotes. These bacteria have been documented as numerically important components in a geographically wide range of freshwater lake habitats, including lakes in North America (Newton et al., 2011), Europe (Glockner et al., 2000), Africa (Humbert et al., 2009), Asia (Wu et al., 2006), and Antarctica (Pearce et al., 2003). But our results suggest that bacterial community composition and diversity were driven by sediment properties (e.g., the WC and SOC), which differed over the depth and distance to the water-land junction spatial gradients. By analyzing the spatial distribution of bacterial communities across a small spatial scale (500 m), our results also indicated that sediment chemical properties were divided into two different “factors.” Spatial gradients in association with varying sediment properties drove bacterial community composition. In addition, our results suggest that different geochemical parameters along vertical and horizontal gradients can affect specific bacterial groups in sediments.
In this study, the WC and nutrient availability (e.g., SOC) significantly increased with increasing distance to the water-land junction. Sediment moisture along the distance to water-land junction exerted a selective pressure on the bacterial community. Sediments with a high of WC harbor the less Acidobacteria and Chloroflexi, and more Betaproteobacteria and Firmicuteacross locations (Figure 4, Figure S2). Nutrient availability along distance gradient is another important factor that influences the bacterial community (Logue and Lindström, 2008). The relative abundance of Actinobacteria decreased as increased SOC (Figure 4). Increased nutrient concentrations could select against the freshwater lake Actinobacteria (Haukka et al., 2006). Organic C in sediment primarily originates from living organisms, such as phytoplankton, plant tissue, and fish (Donohue and Garcia Molinos, 2009). In addition, our results indicated that SOC showed correlations with the abundance of Betaproteobacteria (Figure 4). Several studies also suggested that Betaproteobacteria growth is closely associated with the sediment nutrients (Chen and Chiu, 2000; Lin et al., 2010). The freshwater lake Betaproteobacteria is fast growing and nutrient loving (Newton et al., 2011), which was highly associated with SOC (Figure 4, Figure S1).
The majority of the Alphaproteobacteria belonged to Rhizobiales, which were more abundant in the surface layer sediment samples with low SBD (Figure 3). The SBD varied as the water content of the sediment changed due to deposition and subsequent compaction. SBD generally increases with depth and time as pore water is expelled from the sediment and transported to the surface (Boroujeni et al., 2009). In addition, the Rhizobiales are controller at the hub of the ecosystem N cycle, and often facilitate atmospheric N fixation by plants (Im et al., 2006; Yarwood et al., 2009). NH+4 is mainly produced by aerobic degradation of organic-bound N, which is abundant in upper sediment (Peter et al., 1998). It was not surprising to observe that NH+4 along the depth gradient showed significant correlations with the abundance of Alphaproteobacteria. Taken together, bacterial community composition and diversity were driven by sediment properties, which were differed in the spatial scales (the depth and distance to the water-land junction). O2 is another important regulator of bacterial community structure composition and functioning. For example, O2 availability influences oxidation–reduction reactions in different types of wetland soils (D'angelo and Reddy, 1999). However, we failed to measure O2 availability because of the damage of O2 electrode in the field.
Overall, the sediment physiochemical characteristics had a significant effect on the diversity of beach bacterial communities along spatial gradients in the subtropical freshwater wetland at Poyang Lake. This study would improve our understanding of bacterial diversity in wetland ecosystems.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Laura Nelson for language editing. The work was supported by the National Basic Research Program of China grant 2010CB950602, the National Natural Science Foundation of China grant (31160019, 31160270), the China Postdoctoral Science Foundation grant 201104552, and the Key Laboratory of Poyang Lake Environment and Resource Utilization, Ministry of Education grant Z04996.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00129/abstract
Figure S1. Relationships between the relative abundances of the dominant bacterial phyla and distance and depth. Distance: distance to the water-land junction, depth: sediment depth to surface.
Figure S2. Correlations between the relative abundances of the dominant bacterial phyla and the sediment physiochemical characteristics. WC, water content; SBD, sediment bulk density; SOC, soil organic carbon.
Table S1. All 30 soil physicochemical data.
Table S2. Correlation coefficients between all soil physicochemical variables.
Table S3. Analyses of the 30 bacterial clone libraries.
Table S4. Relative abundance distribution of operational taxonomic units (OTU) (97% similarity).
Table S5. Correlations between the function matrix and the first two axes of the CCA microbial community ordination scores based on intraset scores.
Table S6. Relative abundance at phylum level (CCA).
Table S7. Geochemical characteristics (CCA).
Table S8. Correlations between the function matrix and the first two axes of the PCA microbial community ordination scores based on intraset scores.
References
Badin, A. L., Monier, A., Volatier, L., Geremia, R. A., Delolme, C., and Bedell, J. P. (2011). Structural stability, microbial biomass and community composition of sediments affected by the hydric dynamics of an urban stormwater infiltration basin. Dynamics of physical and microbial characteristics of stormwater sediment. Microb. Ecol. 61, 885–897. doi: 10.1007/s00248-011-9829-4
Boroujeni, H. S., Fathi-Moghadam, M., and Shafaei-Bejestan, M. (2009). Investigation on bulk density of deposited sediments in dez reservoir. Trends Appl. Sci. Res. 4, 148–157. doi: 10.3923/tasr.2009.148.157
Chen, J.-S., and Chiu, C.-Y. (2000). Effect of topography on the composition of soil organic substances in a perhumid subtropical montance forest ecosystem in Taiwan. Geoderma 96, 19–30. doi: 10.1016/S0016-7061(99)00092-0
Cookson, W. R., O'donnell, A. J., Grant, C. D., Grierson, P. F., and Murphy, D. V. (2008). Impact of ecosystem management on microbial community level physiological profiles of postmining forest rehabilitation. Microb. Ecol. 55, 321–332. doi: 10.1007/s00248-007-9278-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Córdova-Kreylos, A. L., Cao, Y., Green, P. G., Hwang, H.-M., Kuivila, K. M., Lamontagne, M. G., et al. (2006). Diversity, composition, and geographical distribution of microbial communities in California salt marsh sediments. Appl. Environ. Microbiol. 72, 3357–3366. doi: 10.1128/AEM.72.5.3357-3366.2006
Crump, R. C., Adams, H. E., Hobbie, J. E., and Kling, G. W. (2007). Biogeography of bacterioplankton in lakes and streams of an Arctic tundra catchment. Ecology 88, 1365–1378. doi: 10.1890/06-0387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cui, J., Meng, H., Nie, M., Chen, X., Li, Z., Bu, N., et al. (2012). Bacterial succession during 500 years of soil development. Ecol. Res. 27, 793–807. doi: 10.1007/s11284-012-0955-3
D'angelo, E. M., and Reddy, K. R. (1999). Regulators of heterotrophic microbial potentials in wetland soils. Soil Biol. Biochem. 31, 815–830. doi: 10.1016/S0038-0717(98)00181-3
Donohue, I., and Garcia Molinos, J. (2009). Impacts of increased sediment loads on the ecology of lakes. Biol. Rev. Camb. Philos. Soc. 84, 517–531. doi: 10.1111/j.1469-185X.2009.00081.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drenovsky, R. E., Vo, D., Graham, K. J., and Scow, K. M. (2004). Soil water content and organic carbon availability are major determinants of soil microbial community composition. Microb. Ecol. 48, 424–430. doi: 10.1007/s00248-003-1063-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ettema, C. H., and Wardle, D. A. (2002). Spatial soil ecology. Trends Ecol. Evol. 17, 177–183. doi: 10.1016/S0169-5347(02)02496-5
Glockner, F. O., Zaichikov, E., Belkova, N., Denissova, L., Pernthaler, J., Pernthaler, A., et al. (2000). Comparative 16S rRNA analysis of lake bacterioplankton reveals globally distributed phylogenetic clusters including an abundant group of actinobacteria. Appl. Environ. Microbiol. 66, 5053–5065. doi: 10.1128/AEM.66.11.5053-5065.2000
Gucht, K. V. D., Cottenie, K., Muylaert, K., Vloemans, N., Cousin, S., Declerck, S., et al. (2007). The power of species sorting: local factors drive bacterial community composition over a wide range of spatial scales. Proc. Natl. Acad. Sci. U.S.A. 104, 20404–20409. doi: 10.1073/pnas.0707200104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haglund, A. L., Lantz, P., Tornblom, E., and Tranvik, L. (2003). Depth distribution of active bacteria and bacterial activity in lake sediment. FEMS Microbiol. Ecol. 46, 31–38. doi: 10.1016/S0168-6496(03)00190-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hall, J. R., Mitchell, K. R., Jackson-Weaver, O., Kooser, A. S., Cron, B. R., Crossey, L. J., et al. (2008). Molecular characterization of the diversity and distribution of a thermal spring microbial community by using rRNA and metabolic genes. Appl. Environ. Microbiol. 74, 4910–4922. doi: 10.1128/AEM.00233-08
Haukka, K., Kolmonen, E., Hyder, R., Hietala, J., Vakkilainen, K., Kairesalo, T., et al. (2006). Effect of nutrient loading on bacterioplankton community composition in lake mesocosms. Microb. Ecol. 51, 137–146. doi: 10.1007/s00248-005-0049-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Humbert, J. F., Dorigo, U., Cecchi, P., Le Berre, B., Debroas, D., and Bouvy, M. (2009). Comparison of the structure and composition of bacterial communities from temperate and tropical freshwater ecosystems. Environ. Microbiol. 11, 2339–2350. doi: 10.1111/j.1462-2920.2009.01960.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iasur-Kruh, L., Hadar, Y., Milstein, D., Gasith, A., and Minz, D. (2009). Microbial population and activity in wetland microcosms constructed for improving treated municipal wastewater. Microb. Ecol. 59, 700–709. doi: 10.1007/s00248-009-9611-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Im, W. T., Kim, S. H., Kim, M. K., Ten, L. N., and Lee, S. T. (2006). Pleomorphomonas koreensis sp. nov., a nitrogen-fixing species in the order Rhizobiales. Int. J. Syst. Evol. Microbiol. 56, 1663–1666. doi: 10.1099/ijs.0.63499-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lane, D. J. (1991). “16S/23 S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Chichester: Wiley), 115–175.
Liao, L., Xu, X. W., Wang, C. S., Zhang, D. S., and Wu, M. (2009). Bacterial and archaeal communities in the surface sediment from the northern slope of the South China Sea. J. Zhejiang Univ. Sci. B 10, 890–901. doi: 10.1631/jzus.B0920181
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, X., Green, S., Tfaily, M. M., Prakash, O., Konstantinidis, K. T., Corbett, J. E., et al. (2012). Microbial community structure and activity linked to contrasting biogeochemical gradients in bog and fen environments of the Glacial Lake Agassiz Peatland. Appl. Environ. Microbiol. 78, 7023–7031. doi: 10.1128/AEM.01750-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lin, Y. T., Huang, Y. J., Tang, S. L., Whitman, W. B., Coleman, D. C., and Chiu, C. Y. (2010). Bacterial community diversity in undisturbed perhumid montane forest soils in Taiwan. Microb. Ecol. 59, 369–378. doi: 10.1007/s00248-009-9574-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lindström, E. S., Agterveld, M. P. K.-V., and Zwart, G. (2005). Distribution of typical freshwater bacterial groups is associated with pH, temperature and lake water retention time. Appl. Environ. Microbiol. 71, 8201–8206. doi: 10.1128/AEM.71.12.8201–8206.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, D., Ding, W., Jia, Z., and Cai, Z. (2011). The impact of dissolved organic carbon on the spatial variability of methanogenic archaea communities in natural wetland ecosystems across China. Appl. Microbiol. Biotechnol. 96, 253–263. doi: 10.1007/s00253-011-3842-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Logue, J. B., and Lindström, E. S. (2008). Biogeography of bacterioplankton in inland waters. Freshw. Rev. 1, 99–114. doi: 10.1608/FRJ-1.1.9
Luna, G. M., Dell'anno, A., Giuliano, L., and Danovaro, R. (2004). Bacterial diversity in deep Mediterranean sediments: relationship with the active bacterial fraction and substrate availability. Environ. Microbiol. 6, 745–753. doi: 10.1111/j.1462-2920.2004.00611.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mackelprang, R., Waldrop, M. P., Deangelis, K. M., David, M. M., Chavarria, K. L., Blazewicz, S. J., et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480, 368–371. doi: 10.1038/nature10576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maidak, B. L., Cole, J. R., Lilburn, T. G., Parker, C. T. Jr., Saxman, P. R., Farris, R. J., et al. (2001). The RDP-II (Ribosomal Database Project). Nucleic. Acids. Res. 29, 173–174. doi: 10.1093/nar/29.1.173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meng, H., Li, K., Nie, M., Wan, J. R., Quan, Z. X., Fang, C. M., et al. (2012). Responses of bacterial and fungal communities to an elevation gradient in a subtropical montane forest of China. Appl. Microbiol. Biotechnol. 97, 2219–2230. doi: 10.1007/s00253-012-4063-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moseman-Valtierra, S. M., Armaiz-Nolla, K., and Levin, L. A. (2010). Wetland response to sedimentation and nitrogen loading: diversification and inhibition of nitrogen-fixing microbes. Ecol. Appl. 20, 1556–1568. doi: 10.1890/08-1881.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Newton, R. J., Jones, S. E., Eiler, A., Mcmahon, K. D., and Bertilsson, S. (2011). A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. 75, 14–49. doi: 10.1128/MMBR.00028-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pearce, D. A., Gast, C. J., Lawley, B., and Ellis-Evans, J. C. (2003). Bacterioplankton community diversity in a maritime Antarctic lake, determined by culture-dependent and culture-independent techniques. FEMS Microbiol. Ecol. 45, 59–70. doi: 10.1016/S0168-6496(03)00110-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peter, B., Risgaard-Petersen, N., and Rysgaard, S. (1998). Interpretation of measured concentration profiles in sediment pore water. Limnol. Oceanogr. 43, 1500–1510. doi: 10.4319/lo.1998.43.7.1500
Redmond, M. C., and Valentine, D. L. (2011). Natural gas and temperature structured a microbial community response to the Deepwater Horizon oil spill. Proc. Natl. Acad. Sci. U.S.A. 109, 20292–20297. doi: 10.1073/pnas.1108756108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schauer, R., Bienhold, C., Ramette, A., and Harder, J. (2010). Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J. 4, 159–170. doi: 10.1038/ismej.2009.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Terrados, J., Duarte, C. M., Kamp-Nielsen, L., Agawin, N. S. R., Gacia, E., Lacap, D., et al. (1999). Are seagrass growth and survival constrained by the reducing conditions of the sediment? Aquat. Bot. 65, 175–197. doi: 10.1016/S0304-3770(99)00039-X
Wang, Y., Sheng, H. F., He, Y., Wu, J. Y., Jiang, Y. X., Tam, N. F., et al. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 78, 8264–8271. doi: 10.1128/AEM.01821-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woese, C. R. (1990). Evolutionary questions: the “progenote.” Science 247, 789. doi: 10.1126/science.2305249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woese, C. R., Kandler, O., and Wheelis, M. L. (1990). Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. U.S.A. 87, 4576–4579. doi: 10.1073/pnas.87.12.4576.
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, Q. L., Zwart, G., Schauer, M., Kamst-Van Agterveld, M. P., and Hahn, M. W. (2006). Bacterioplankton community composition along a salinity gradient of sixteen high-mountain lakes located on the Tibetan Plateau, China. Appl. Environ. Microbiol. 72, 5478–5485. doi: 10.1128/AEM.00767-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xu, X., Zhao, W., Xiao, M., Huang, J., Fang, C., Li, B., et al. (2014). Snails promote methane release from a freshwater lake ecosystem. Front. Environ. Sci. 2:12. doi: 10.3389/fenvs.2014.00012
Yannarell, A. C., and Triplett, E. W. (2004). Within- and between-lake variability in the composition of bacterioplankton communities: investigations using multiple spatial scales. Appl. Environ. Microbiol. 70, 214–223. doi: 10.1128/AEM.70.1.214-223.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yannarell, A. C., and Triplett, E. W. (2005). Geographic and environmental sources of variation in lake bacterial community composition. Appl. Environ. Microbiol. 71, 227–239. doi: 10.1128/AEM.71.1.227-239.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yarwood, S. A., Myrold, D. D., and Hogberg, M. N. (2009). Termination of belowground C allocation by trees alters soil fungal and bacterial communities in a boreal forest. FEMS Microbiol. Ecol. 70, 151–162. doi: 10.1111/j.1574-6941.2009.00733.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, J., Kang, S., Schadt, C. W., Charles, T., and Garten, J. (2008). Spatial scaling of functional gene diversity across various microbial taxa. Proc. Natl. Acad. Sci. U.S.A. 105, 7768–7773. doi: 10.1073/pnas.0709016105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: bacteria, community structure, freshwater lake wetland, Poyang Lake, spatial distribution, 16S rRNA
Citation: Ding X, Peng X-J, Jin B-S, Xiao M, Chen J-K, Li B, Fang C-M and Nie M (2015) Spatial distribution of bacterial communities driven by multiple environmental factors in a beach wetland of the largest freshwater lake in China. Front. Microbiol. 6:129. doi: 10.3389/fmicb.2015.00129
Received: 17 October 2014; Accepted: 03 February 2015;
Published online: 26 February 2015.
Edited by:
Télesphore Sime-Ngando, Centre National de la Recherche, FranceReviewed by:
Ludwig Jardillier, Université Paris-Sud, FranceEmma Jane Rochelle-Newall, Institut de Recherche pour le Développement, France
Copyright © 2015 Ding, Peng, Jin, Xiao, Chen, Li, Fang and Nie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Nie, Ministry of Education Key Lab for Biodiversity Science and Ecological Engineering, The Institute of Biodiversity Science, Fudan University, #2005 Songhu Road, Shanghai 200438, China e-mail:bW5pZUBhYmRuLmFjLnVr