 Markus Woegerbauer
Markus Woegerbauer Melanie Kuffner1
Melanie Kuffner1 Sara Domingues
Sara Domingues Kaare M. Nielsen
Kaare M. Nielsen- 1Integrative Risk Assessment – Data – Statistics, GMO Risk Assessment, Austrian Agency for Health and Food Safety, Vienna, Austria
- 2Faculty of Pharmacy and Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 3Department of Pharmacy, University of Tromsø, Tromsø, Norway
- 4Genøk-Center for Biosafety Tromsø, Tromsø, Norway
Intragenic recombination leading to mosaic gene formation is known to alter resistance profiles for particular genes and bacterial species. Few studies have examined to what extent aminoglycoside resistance genes undergo intragenic recombination. We screened the GenBank database for mosaic gene formation in homologs of the aph(3′)-IIa (nptII) gene. APH(3′)-IIa inactivates important aminoglycoside antibiotics. The gene is widely used as a selectable marker in biotechnology and enters the environment via laboratory discharges and the release of transgenic organisms. Such releases may provide opportunities for recombination in competent environmental bacteria. The retrieved GenBank sequences were grouped in three datasets comprising river water samples, duck pathogens and full-length variants from various bacterial genomes and plasmids. Analysis for recombination in these datasets was performed with the Recombination Detection Program (RDP4), and the Genetic Algorithm for Recombination Detection (GARD). From a total of 89 homologous sequences, 83% showed 99–100% sequence identity with aph(3′)-IIa originally described as part of transposon Tn5. Fifty one were unique sequence variants eligible for recombination analysis. Only a single recombination event was identified with high confidence and indicated the involvement of aph(3′)-IIa in the formation of a mosaic gene located on a plasmid of environmental origin in the multi-resistant isolate Pseudomonas aeruginosa PA96. The available data suggest that aph(3′)-IIa is not an archetypical mosaic gene as the divergence between the described sequence variants and the number of detectable recombination events is low. This is in contrast to the numerous mosaic alleles reported for certain penicillin or tetracycline resistance determinants.
Introduction
Mosaic genes are genetic units consisting of DNA segments of different phylogenetic origin leading to sequence patterns which may confer novel phenotypic properties (Smith, 1992; Dowson et al., 1997; Boc and Makarenkov, 2011). The within gene (i.e., intragenic) recombination of DNA fragments increases the genetic plasticity of bacterial genomes and contributes to evolution and adaptability to new environmental conditions (Hanage et al., 2006). The process of mosaic gene formation primarily relies on the uptake of free DNA from the environment by competent bacteria via natural genetic transformation and subsequent integration of the incoming DNA fragment into the bacterial genome through homologous recombination (Smith et al., 1991). The efficiency of DNA segment integration is dependent on sequence similarity between the involved DNA strands. The frequency of homologous recombination decreases in a log-linear relationship with increasing sequence divergence between donor and recipient DNA to the point where it falls below the limit of detection—which is usually the case when pairwise sequence identity drops below 70% (Dowson et al., 1997; Fraser et al., 2007). This stringent similarity requirement may be circumvented by homology-directed illegitimate recombination, a mechanism where the integration of non-homologous DNA fragments is facilitated by the presence of a short homologous anchor sequence in the donor molecule and a region of microhomology on the opposite terminus of the incoming DNA with the target sequence (de Vries and Wackernagel, 2002; Prudhomme et al., 2002); or by double-illegitimate recombination, which is independent of any homology (Hulter and Wackernagel, 2008).
Genetic recombination inducing mosaic patterns in antibiotic resistance genes in bacterial pathogens results in therapy failure in clinical settings (Spratt, 1994; Heinemann and Traavik, 2004). Bacteria capable of lateral transfer of resistance gene fragments have the opportunity to evade selection pressure in response to alternating antibiotic therapy by acquiring new or modifying existing housekeeping genes and/or resistance determinants (Spratt, 1994). A prominent example is the mosaic pattern formation occurring in penicillin binding protein genes in Streptococcus pneumoniae (e.g., pbp2b) and Neisseria spp. (e.g., penA) and in tetracycline resistance determinants [e.g., tet(M), tet(O), tet(W)] in various animal and human pathogens (Spratt et al., 1989; Dowson et al., 1994; Patterson et al., 2007). These mosaic genes confer increased antibiotic resistance to the host bacterium and impact human health by increasing the morbidity and mortality rates of infectious diseases and by amplifying the financial burden of public health systems (Doern et al., 2001; Heinemann and Traavik, 2004; Bush et al., 2011).
An analysis of a potential contribution of the aminoglycoside resistance gene aph(3′)-IIa to the mosaic gene formation and the variability of aph(3′)-II-homologs is of relevance because this resistance gene is one of the most frequently applied selectable markers in genetic engineering and plant gene technology (Miki and McHugh, 2004; Shakya et al., 2011). Due to such technology applications this resistance gene is shed into the environment. Corresponding DNA fragments may additionally undergo chemical modifications when present as free extracellular DNA in the environment (Pontiroli et al., 2007; Chen et al., 2012). A recombination of anthropogenically released aph(3′)-IIa fragments with endogenous aph(3′)-IIa homologs present in competent environmental bacteria may lead to the formation of mosaic phosphotransferases with an altered antibiotic inactivation spectrum.
The enzyme APH(3′)-IIa inactivates the critically important aminoglycoside antibiotics neomycin and kanamycin as well as paromomycin, butirosin, gentamicin B, and ribostamycin (Shaw et al., 1993; WHO, 2012). Amikacin, a crucial second-line antibiotic used exclusively in humans, was shown to be phosphorylated to some extent only under in vitro conditions (Perlin and Lerner, 1986).
There is currently no experimental evidence available to support or disprove the hypothesis that antibiotic marker genes like aph(3′)-IIa may be involved in the formation of mosaic resistance genes. But powerful bioinformatic tools have now become available that allow in silico analysis of lateral intragenic gene transfer events (Boc et al., 2010; Martin et al., 2010; Boc and Makarenkov, 2011; Le et al., 2014).
To determine whether the genetic variability of aph(3′)-IIa like alleles available in GenBank has arisen from mosaic formation we performed a detailed in silico screening for intragenic recombination events in aph(3′)-IIa sequences utilizing phylogeny- and non-phylogeny-based algorithms of the Recombination Detection Program (RDP4) software package and the Genetic Algorithm for Recombination Detection (GARD) (Kosakovsky Pond et al., 2006a; Martin, 2010).
Materials and Methods
Collection of Sequence Data
The aph(3′)-IIa gene from the Escherichia coli transposon Tn5 (Accession number V00618, positions 151–945; 795 nts) was used as query sequence. This reference sequence termed for clarification “EcoAph3IIa” was searched against the bacterial non-redundant nucleotide collection (http://www.ncbi.nlm.nih.gov/nuccore/) and the database of reference genomic sequences (http://www.ncbi.nlm.nih.gov/refseq/). The discontiguous megablast algorithm was used with default settings except for 250 hits to be displayed (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). Vectors, artificial sequences and models were excluded from the search. The search was carried out on September 22nd, 2014.
Sequence Alignments
Sequences producing BLAST matches were downloaded from GenBank, spanning the complete open reading frame when available. Multiple sequence alignments were prepared using the ClustalW algorithm implemented in Bioedit (http://www.mbio.ncsu.edu/bioedit/bioedit.html) (Hall, 2007). The sequence identity matrix option of Bioedit was used to determine the pairwise sequence identity between each sequence and the reference sequence aph(3′)-IIa (EcoAph3IIa). The sequence difference count matrix option of Bioedit was used to determine pairwise nucleotide differences among all aligned sequences.
Selection of Sequence Sets for Recombination Analysis
All sequences sharing more than 60% sequence identity with the reference sequence EcoAph3IIa across their entire length were considered as “aph(3′)-IIa homologs.” Sequences with less than 60% sequence identity were considered as non-homologous. This distinction was based on the observation that aph(3′)-IIb (X90856) and aph(3′)-IIc (HQ424460), the closest described relatives of aph(3′)-IIa among aminoglycoside 3′-O-phosphotransferases (Ramirez and Tolmasky, 2010) share nearly 60% sequence identity with aph(3′)-IIa.
From the bulk of homologs collected from GenBank (Table 1), three sequence datasets were selected for recombination analysis:
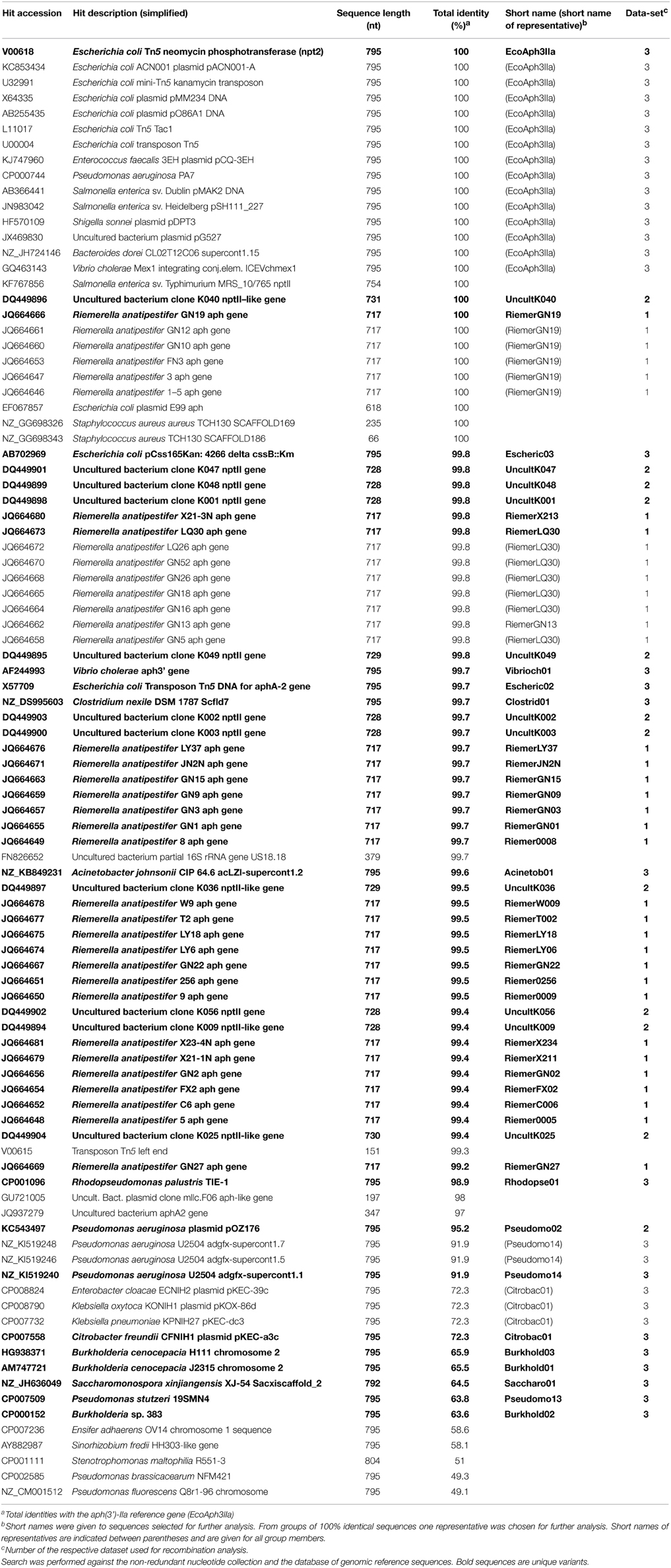
Table 1. The highest scoring BLAST hits for the aph(3′)-IIa gene from E. coli transposon Tn5.
Dataset 1: 36 partial sequences from the Riemerella anatipestifer isolate collection, representing the intra-species variation of aph(3′)-IIa homologs in a pathogen species residing in ducks (yellow bars in Figure S1).
Dataset 2: 11 partial sequences from river water, representing the variation of aph(3′)-IIa homologs occurring in bacterial species recovered from a defined natural aquatic environment (green bars, Figure S1).
Dataset 3: 34 full length aph(3′)-IIa homologs comprising the reference gene EcoAph3IIa and 33 sequences from various bacterial genomes and plasmids. This dataset represented the entire variation of aph(3′)-IIa genes known to date (i.e., as officially deposited in GenBank per September 22nd, 2014 (red, dark blue and light blue bars, Figure S1).
Each dataset was separately aligned with ClustalW and de-replicated to retain one representative sequence per variant. Pairwise differences among all variants were determined to allow selection of sequence subsets for improved recombination detection according to the recommendations of the instruction manual of RDP4 (Martin, 2010). It is indicated that RDP4 is unlikely to detect recombination between extremely similar sequences. The presence of multiple nearly identical sequences in a dataset unnecessarily increases the number of pairwise comparisons and the severity of multiple comparison correction and, thus, reduces sensitivity. The following formula was used for calculating the ratio between the number of sequences (X), length (L) and the minimum required pairwise distances (Y) in the dataset for sequences still eligible for recombination analysis by RDP4: Y = (2 × ln 4X) / L (Martin, 2010). On the other hand, highly divergent sequences increase the risk of false positives as they may cause misalignments and introduce an excess of variable sites into the alignment. Therefore, sequences sharing less than 70% sequence identities have to be handled with caution (Martin, 2010).
Detection of Recombination Events in Aligned Sequence Datasets
Recombination events in multiple sequence alignments were determined using the Recombination Detection Program Beta 4.36 package (RDP4). Seven of the recombination signal detection algorithms available as modules in RDP4 were employed: RDP (Martin, 2010), BootScan (Martin et al., 2005), MaxChi (Smith, 1992), Chimera (Posada and Crandall, 2001), GeneConv (Padidam et al., 1999), SiScan (Gibbs et al., 2000), and 3Seq (Boni et al., 2007). In the general settings for the RDP4 recombination detection procedure, the highest acceptable p-value was set to 0.05, the Bonferroni method was selected to correct for multiple comparisons and the entire process was run in permutational mode with 100 permutations. For the remaining parameters in the general RDP4 options defaults were retained. These defaults involved running PhylPro (Weiller, 1998) and LARD (Holmes et al., 1999) as secondary detection methods. Default settings were also retained for the options in the individual detection modules, except for MaxChi, where the specific window size was set to “variable.” These settings and analysis modules were chosen in accordance with common practice in literature (Keymer and Boehm, 2011; Smith et al., 2012; Thomas et al., 2012; Alvarez-Perez et al., 2013; Freel et al., 2013; Hester et al., 2013; Altamia et al., 2014; Duron, 2014).
Recombination breakpoints in multiple sequence alignments were confirmed with GARD (Kosakovsky Pond et al., 2006b) available at the datamonkey server (http://www.datamonkey.org/) using default values.
Results
Genetic Diversity of Aph(3′)-IIa Homolog Sequences in GenBank
The GenBank database was BLAST-searched for sequences similar to the aph(3′)-IIa gene from the transposon Tn5 of E. coli (EcoAph3IIa). In total 227 hits were obtained. Table 1 summarizes the 94 highest scoring hits, and Figure S1 shows the regions of aph(3′)-IIa matched by these hits.
Eighty nine sequences showed sequence identities of 63–100% with EcoAph3IIa and were considered as aph(3′)-IIa homologs. Their bacterial carriers were of animal (40 isolates), human (28 isolates) and genuine environmental origin (21 isolates) (Figure 1). The large majority originated from avian hosts. Non-vertebrate samples were retrieved from such diverse environments as river water, soil, pig manure, activated sludge, marine sediments, and household installations (Figure 1). Most of the animal bacteria were pathogens (43%) but only a minimal fraction of the environmental isolates could be identified as causative agents for diseases (1%) (Figure 2). The aph(3′)-IIa gene sequence variant carriers comprised the following bacterial taxonomic classes: Actinobacteria, Alphaproteobacteria, Bacilli, Bacteroidia, Betaproteobacteria, Clostridia, Flavobacteria, and Gammaproteobacteria (Figure S2). Complete aph(3′)-IIa homologs had a length of 792–795 nts and discontiguous megablast produced alignment matches of 627–795 bp with the reference gene. Of these 89 homologs 26 were perfect 100% matches and 48 showed over 99% sequence identity with the reference sequence. The 99–100% BLAST matches included two sets of partial sequences originating from bacterial population surveys specifically targeting aph(3′)-IIa diversity: 36 sequences from isolates of the avian pathogen R. anatipestifer collected from diseased ducks (Yang et al., 2012), and 11 sequences from a cultivation independent monitoring of aph(3′)-IIa in Canadian river water samples (Zhu, 2007). As these sequences had been produced by PCR amplification with primers binding within the aph(3′)-IIa gene, sequence information was missing at their ends. Fifteen perfect (100%) and six nearly perfect (>99%) matches over the full gene length were detected in plasmid and genome sequences of bacteria phylogenetically as divergent as E. coli, Bacteroides dorei, and Clostridium nexile. The remaining six 99–100% matches represented gene fragments (66–754 nts; Table 1). Fifteen sequences were found to share 63–99% sequence identity with the reference sequence. These included two short sequence fragments from PCR-based studies on antibiotic resistance genes in water (JQ937279) and activated sludge (GU721005) and 13 complete genes from genomes and plasmids of Pseudomonas aeruginosa, Enterobacter cloacae, Citrobacter freundii, Klebsiella pneumoniae, Klebsiella oxytoca, Saccharomonospora xinjiangensis, Pseudomonas stutzeri, and Burkholderia spp. isolates (Table 1).
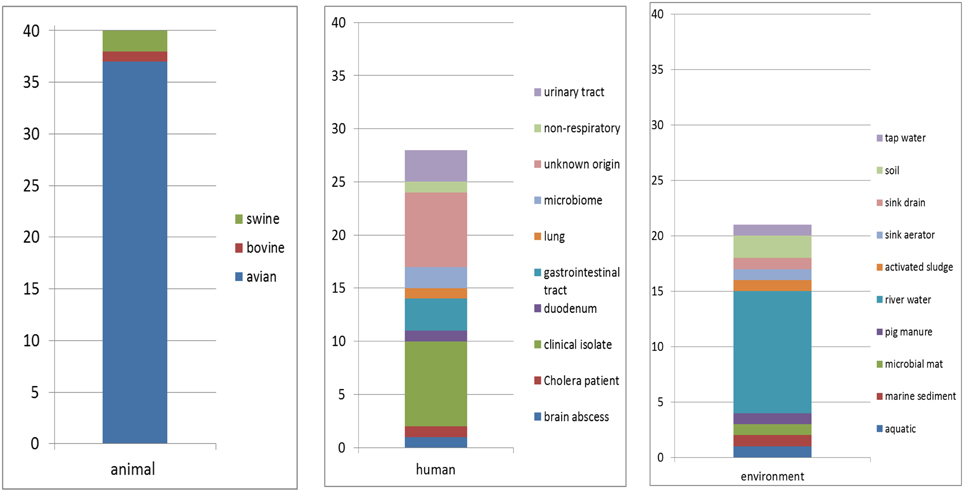
Figure 1. Hosts and habitats of bacterial carriers of aph(3′)-IIa sequence variants. Numbers of isolates are indicated.
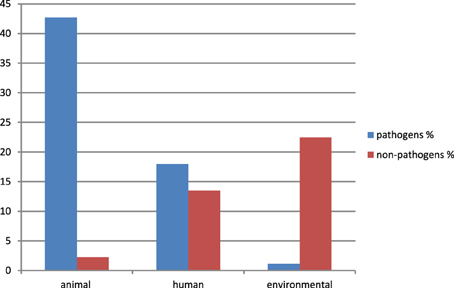
Figure 2. Relative abundance and origin of bacterial isolates carrying aph(3′)-IIa variants. Only isolates explicitly classified as “pathogen” in the GenBank entry or in one of its associated publications or showing a clear history as causative agents for disease as described in Murray et al. (1999), were considered as pathogens. All other isolates were identified as “non-pathogens” (including species characterized as opportunistic pathogens causing rare disease only in immunocompromised patients and “uncultured bacteria” without any additional information available). Data were calculated for a total of 89 isolates (=100%).
The remaining 136 hits shared only 44–59% sequence identity with EcoAph3IIa and, thus, were not considered as aph(3′)-IIa homologs. They included the aph(3′)-IIc gene of Stenotrophomonas maltophilia (HQ424460) and the aph(3′)-IIb (X90856) gene of P. aeruginosa (data not shown). The last sequence match presented in Table 1 and Figure S1 is an open reading frame of a S. maltophilia strain (CP001111) with 97% sequence identity to aph(3′)-IIc. These different aph genes varied in open reading frame length between 783 and 813 nts and produced discontiguous megablast matches spanning 50–370 bp between positions 360 and 720 of aph(3′)-IIa. The region between positions 360 and 720 of the aph(3′)-IIa gene contains two functional domains, known as motif1 and motif2, that are conserved across different clades of the aph gene family (Shaw et al., 1993).
Sequence Variation and Recombination Analysis in Aph(3′)-IIa Homologs from Riemerella anatipestifer Isolates (Dataset 1)
Of the 36 sequences from R. anatipestifer isolates, 25 were unique variants. One unique representative was selected from each group of identical sequences. The most frequent variant (RiemerGN19) was identical with the aph(3′)-IIa reference gene from the E. coli transposon Tn5. The sequences contained parts of the PCR primers used by the survey authors (Yang et al., 2012). After removal of the uninformative primer regions, a 686 nts gene segment, spanning aph(3′)-IIa between position 85 and 770 remained for recombination analysis. In total there were 45 polymorphic sites in the sequence alignment. Pairwise nucleotide differences ranged between 1 and 9 nucleotides. RDP4 analysis of the entire 25 sequence set did not reveal recombination signals. The analysis was repeated with a subset comprising the four most divergent sequences (RiemerX234, RiemerX211, RiemerFX02, RiemerC006). This subset corresponded to the recommendations of the RDP4 developers (Martin, 2010) regarding the relation between number, length and minimum divergence of the sequences. However, no recombination event was detected in this subset.
Sequence Variation and Recombination Analysis in Aph(3′)-IIa Homologs from River Water (Dataset 2)
All of the 11 aph(3′)-IIa sequences extracted from river water were unique variants. Sequence UncultK40 was identical with the aph(3′)-IIa reference gene (EcoAph3IIa). After removal of PCR primer binding sites, a 688 nts gene segment, spanning aph(3′)-IIa between position 27 and 714 remained for recombination analysis. Pairwise nucleotide differences ranged between 1 and 9 nucleotides. RDP4 did not detect recombination events neither in the complete set of 11 sequences, nor in the alignment of the three most divergent sequences (Uncultk56, UncultK009, UncultK025).
Sequence Variation and Recombination Analysis in Full Length Aph(3′)-IIa Homologs from Various Bacterial Genomes and Plasmids (Dataset 3)
Of the 34 available full length homologs originating from various bacterial chromosomes and plasmids 15 were unique variants. The original aph(3′)-IIa gene (EcoAph3IIa) was representative for 15 sequences producing perfect BLAST matches. The three sequences from isolate P. aeruginosa U2504 were identical, and one was retained as representative (Pseudomo14). The aph(3′)-IIa homologs detected in plasmids of E. cloacae, K. oxytoca, K. pneumoniae and C. freundii were identical, and the sequence from Citrobacter was retained as representative for further analysis (Citrobac01). The 15 unique sequences comprised 795 nts, except for Sacharo01, which was one nucleotide triplet shorter. Pairwise sequence differences varied between 1 and 324 nucleotides. Seven recombination detection methods in RDP4 detected a single recombination event in this dataset (Table 2). The results suggested that Pseudomo02 was a mosaic of Pseudomo14 and a sequence highly similar to the reference sequence EcoAph3IIa (Figure 3). The seven methods congruently identified the exchange of a fragment in the region between alignment positions 100 and 500. Figure 3 visualizes the recombination event and highlights the recombination breakpoints at positions 224 and 484, which were proposed congruently by three different methods (Table 2). Analysis of a 5 sequence subset including only sequences with the recommended level of pairwise nucleotide differences (8–239 nts, for explanations see Materials and Methods) confirmed the results obtained with the complete 15 sequence dataset (Table 2). For further confirmation the 15 sequence set was analyzed with GARD. GARD analysis detected a single significant recombination breakpoint signal at position 198 (Table 3). Upon analysis of the five sequence subset, GARD produced several statistically non-significant breakpoint signals, including one at position 482.
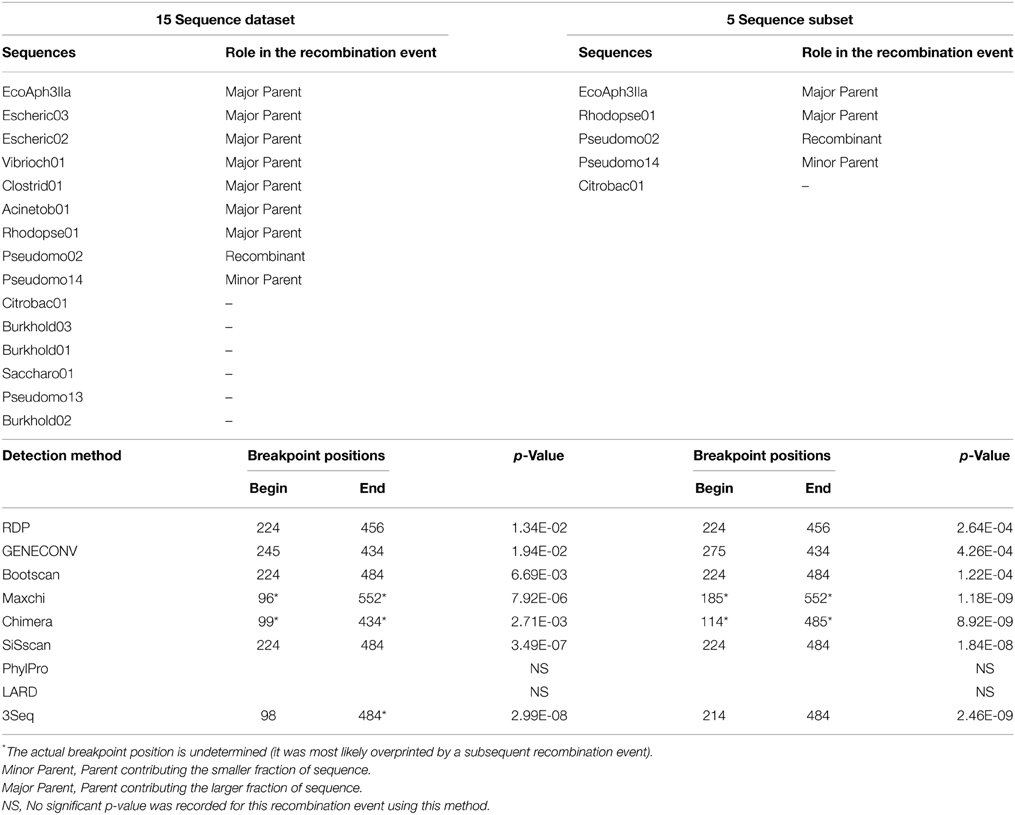
Table 2. Confirmation of the recombination event in dataset 3 (15 full length aph(3′)-IIa homologs from various bacterial species) and in a subset of 5 sequences with GARD.
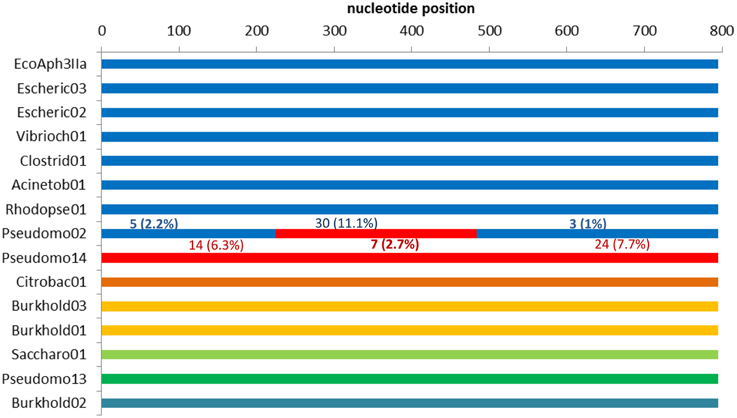
Figure 3. Detection of a recombination event in dataset 3 (15 full length aph(3′)-IIa homologs) by RDP4. The colors indicate sequence similarity and the likely origins of the segments in the recombinant sequence. The upper seven sequences (depicted in blue) are close relatives but not identical. For each segment of the recombinant (Pseudomo02), the number of sites different from those in the corresponding regions of the proposed parents is indicated: differences to EcoAph3IIa are depicted in blue above the segment, differences to Pseudomo14 are given in red below the segment.
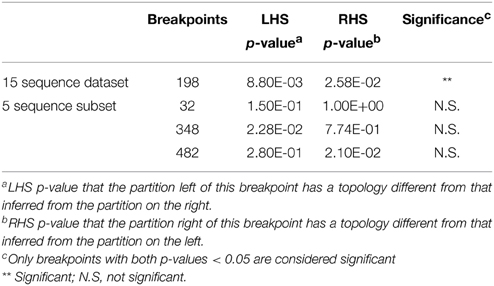
Table 3. Confirmation of the recombination event in dataset 3 (15 full length aph(3′)-IIa homologs from various bacterial species) and in a subset of 5 sequences with GARD.
Discussion
Sequence analysis of antibiotic resistance genes coding for penicillin binding proteins or for tetracycline resistance determinants has revealed horizontal gene transfer events leading to mosaic gene formation (Dowson et al., 1994; Patterson et al., 2007). The aim of this work was to elucidate whether intragenic recombination also occurs in natural homologs of aph(3′)-IIa aminoglycoside resistance genes. To determine the natural variability of aph(3′)-IIa the GenBank database was screened for aph(3′)-IIa variants. The hits were subsequently analyzed for intragenic recombination signals with the RDP4 software package and the web-based tool GARD.
The analysis of the recombination potential of aph(3′)-IIa is of biological relevance because this resistance determinant is inactivating important aminoglycoside antibiotics like kanamycin and neomycin which are vital antimicrobial agents for veterinary purposes and in special cases for human therapeutic applications (WHO, 2012). Additionally, Aph(3′)-IIa was shown in vitro to be capable of extending its antibiotic inactivation spectrum to amikacin—an essential agent for the treatment of severe systemic infections caused by Gram negative bacteria and a crucial second-line antibiotic for combatting multidrug-resistant tuberculosis (Durante-Mangoni et al., 2009; WHO, 2011)–due to an exchange of a single amino acid (Kocabiyik and Perlin, 1992). Although a high-level aph(3′)-IIa-induced amikacin resistant phenotype was only demonstrated so far for an E. coli mutant laboratory strain that showed a reduced aminoglycoside uptake combined with a resistance gene amplification (Perlin and Lerner, 1986) these observations are indicative for a significant effect of aph(3′)-IIa sequence variability on the antibiotic resistance profile of this aminoglycoside phosphotransferase. Nevertheless, we are only aware of two studies dealing explicitly with aph(3′)-IIa sequence variations, both failing to provide a connection between genotype and antibiotic resistance phenotype or induced minimum inhibitory concentrations (MIC) (Zhu, 2007; Yang et al., 2012).
There are only a few studies available on the prevalence of aph(3′)-IIa. Shaw et al. reported 2.5% of all isolates resistant to kanamycin as carriers of aph(3′)-IIa (Shaw et al., 1993). Most of the remaining papers suggested a low abundance of this resistance determinant in natural habitats: aph(3′)-IIa was only rarely detected in bacterial isolates of human (Peirano et al., 2006; Woegerbauer et al., 2014) or environmental origin or in total soil DNA preparations (Leff et al., 1993; Smalla et al., 1993; Ma et al., 2011) or there was evidence of large seasonal fluctuations especially in river waters (Zhu, 2007). These findings indicate that i) bacterial aph(3′)-IIa carrier strains are available providing recombination partners for this resistance determinant and that ii) an artificial exposure of bacterial populations with aph(3′)-IIa copies from anthropogenic sources like laboratory waste discharges or antibiotic resistance marker gene carrying transgenic organisms–eventually in combination with aminoglycoside containing effluents or manure - may increase the likelihood for genetic recombination (Chee-Sanford et al., 2009; Chen et al., 2012).
BLAST search of GenBank revealed only a limited number of aph(3′)-IIa variants with sequence identities between 60 and 99%. This is in contrast to the many mosaic genes coding for penicillin binding proteins or tetracycline resistance determinants for which homologs with a continuous spectrum of sequence identity between 80 and 99% have been identified (Spratt, 1994; Oggioni et al., 1996; Hakenbeck, 2000; Hollingshead et al., 2000; Johansen et al., 2001; Prudhomme et al., 2002; Nakamura et al., 2012).
The retrieved aph(3′)-IIa sequence homologs comprised a wide range of variant sequences originating from a broad variety of environmental sources including soil, water, marine sediments, manure, sewage sludge, and diverse human (gut, skin, urinary tract, lung, brain) and animal habitats (birds, pigs, cows).
For recombination analysis, the aph(3′)-IIa homologs were grouped into 3 datasets originating from duck pathogens (dataset 1) and river water (dataset 2) as representatives for sequences from bacteria living in a common habitat with the obvious physical property to exchange gene fragments. The remaining unique full length aph(3′)-IIa homologs were from bacteria of diverse animal, human or genuine environmental origins which could not be allocated to a common biotope (dataset 3). A combined analysis of sequences from dataset 3 comprising such different ecosystems is valid since lateral transfer of fragments in the evolution of a gene of interest can be assessed by sequence comparison without the prerequisite that the source organisms are of the same species or have been isolated from a common habitat. For example Oggioni et al. discovered mosaic patterns in tetracycline resistance genes by comparing previously published sequences of tetracycline resistant Enterococcus faecalis, S. pneumoniae, Staphylococcus aureus, Ureaplasma urealyticum, and Neisseria spp. isolates (Oggioni et al., 1996). Similarly Boc et al. detected numerous recombination events in the evolution of the rubisco gene rbcL by comparison of amino acid sequences from various photosynthetic bacteria and algae (Boc and Makarenkov, 2011).
The theoretical lower limit for most of the RDP4 algorithms applied for the identification of a mosaic gene (i.e., a gene affected by intragenic recombination) is three (Martin, 2010). Many publications refer to approx. 8–12 sequences to be sufficient for a reliable identification of mosaic genes (Oggioni et al., 1996; Dowson et al., 1997; Filipe et al., 2000; King et al., 2005): Oggioni et al. used a total of eight sequences to identify tet(M) as mosaic gene in silico with high significance (Oggioni et al., 1996). Filipe et al. tested 12 murM alleles (Filipe et al., 2000), Dowson et al. 8 pbp2b alleles (Dowson et al., 1997), and King et al. used 12 novel 5′ and 10 novel 3′ nanA alleles to establish gene mosaicism (King et al., 2005). Our efforts are far exceeding any data collections used so far for the detection of mosaic genes in a single approach.
In the analysis of our third dataset, seven sequence comparison algorithms of the RDP4 suite provided evidence for a recombination event. The risk of identifying false positives, i.e., of mistaking mutation for recombination events, is inherent to any in silico recombination detection strategy (Martin, 2010; Boc and Makarenkov, 2011). Therefore, it is current practice to confirm calculated recombination events with several methods, including phylogeny-based and substitution distribution-based algorithms (Bay and Bielawski, 2011; Boc and Makarenkov, 2011). The described recombination event in the aph(3′)-IIa gene dataset is supported by three phylogeny-based methods (RDP, BootScan, SiScan), 4 substitution distribution-based methods (MaxChi, GeneConv, Chimera, 3Seq) and to some extent also by the phylogeny-based genetic algorithm (GARD). In bacterial multi-locus sequence typing (MLST), a major application area of the RDP4 software, many authors have convened to accept a software reported recombination event, if it is detected by at least three methods with a Bonferroni-corrected p-value < 0.05 (Keymer and Boehm, 2011; Smith et al., 2012; Alvarez-Perez et al., 2013). This criterion is met by the recombination event described here. The different methods agreed on the exchanged gene region and on the recombination partners involved in this event, but proposed different positions as recombination breakpoints. This reflects the different aspects of information each algorithm is targeting in a sequence alignment (Martin, 2010).
The mosaic aph(3′)-IIa gene identified in our third dataset is located in a Tn5 similar cassette on pOZ176, an incP-2 plasmid from the multidrug-resistant isolate P. aeruginosa PA96 (Xiong et al., 2013). Plasmid pOZ176 is of environmental origin showing homologies with a vector from the plant pathogen P. fluorescens and to genomic islands present in the environmental bacteria Ralstonia solanacearum and Azotobacter vinelandii. Codon usage analysis indicated that most of the resistance genes of pOZ176 were not originally from P. aeruginosa but acquired by horizontal gene transfer from other species indicating a long history of DNA rearrangements most probably driven by antibiotic selection (Xiong et al., 2013).
PA96 is reported to be phenotypically resistant to at least 13 antibiotics (including amikacin and gentamicin) from three different substance classes [ß-lactams (pencillins, cephalosporins, carbapenems), fluoroquinolones, and aminoglycosides] (Xiong et al., 2013). Whole genome sequencing revealed that PA96 is carrier of aph(3′)-IIb (Deraspe et al., 2014), which mediates resistance to kanamycin, neomycin, and butirosin (Zeng and Jin, 2003) potentially masking an antibiotic activity of the newly discovered aph(3′)-IIa mosaic gene. At present there is no information available whether this novel mosaic gene on pOZ176 is functionally active and expressing any antibiotic resistance phenotype.
Antibiotic resistance marker genes used in transgenic plants are in several cases plant-codon optimized versions of their bacterial counterparts (Roa-Rodriguez and Nottenburg, 2003). The plant-derived aph(3′)-IIa variant of the transgenic potato line EH92-527-1 (Amflora) contains a characteristic mutation. Alignment of the recombinant aph(3′)-IIa gene of pOZ176 with the plant-derived transgenic variant of aph(3′)-IIa from EH92-527-1 revealed an absence of the plant allele-specific mutation in pOZ176 and vice versa an absence of the mutations distinctive for the recombinant aph(3′)-IIa allele in the transgenic counterpart (data not shown due to confidential business information restrictions). These observations indicate that an involvement of this transgenic allele in the evolution of aph(3′)-IIa of pOZ176 is unlikely.
Compared to the complex recombination history of known mosaic genes such as pbp2b (Dowson et al., 1997), murM (Filipe et al., 2000), or tet(M) (Oggioni et al., 1996), the observed recombination frequency among aph(3′)-IIa homologs was low. Although intragenic recombination is thought to be a frequent process during bacterial evolution (Didelot and Maiden, 2010) our report is presenting the first evidence for only a single mosaic formation event among aph(3′)-IIa homologs. To verify the sensitivity of our approach, we analyzed sequence collections of pbp2b, murM, and tet(M) with RDP4 using the same settings, and detected a multitude of recombination breakpoints and corresponding p-values several orders of magnitude lower than those obtained with the aph(3′)-IIa datasets (data not shown). According to the currently available sequence information in GenBank and compared to typical mosaic genes aph(3′)-IIa appears to be less prone for intragenic recombination. However, it is important to realize that novel aph(3′)-IIa sequence variants becoming prospectively available may change the outcome of the in silico recombination analysis.
We conclude that a recombination event has occurred during the evolution of an aph(3′)-IIa homolog present on a plasmid of environmental origin in a pathogenic multi-resistant strain of P. aeruginosa. The observed number of variant aph(3′)-IIa sequences is low and their diversity appears to be not primarily driven by intragenic recombinations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by the Austrian Ministry of Health (project grant No. GZ 70420/0118-II/B/15/2013).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.00442/abstract
References
Altamia, M. A., Wood, N., Fung, J. M., Dedrick, S., Linton, E. W., Concepcion, G. P., et al. (2014). Genetic differentiation among isolates of Teredinibacter turnerae, a widely occurring intracellular endosymbiont of shipworms. Mol. Ecol. 23, 1418–1432. doi: 10.1111/mec.12667
Alvarez-Perez, S., de Vega, C., and Herrera, C. M. (2013). Multilocus sequence analysis of nectar pseudomonads reveals high genetic diversity and contrasting recombination patterns. PLoS ONE 8:e75797. doi: 10.1371/journal.pone.0075797
Bay, R. A., and Bielawski, J. P. (2011). Recombination detection under evolutionary scenarios relevant to functional divergence. J. Mol. Evol. 73, 273–286. doi: 10.1007/s00239-011-9473-0
Boc, A., and Makarenkov, V. (2011). Towards an accurate identification of mosaic genes and partial horizontal gene transfers. Nucleic Acids Res. 39, e144. doi: 10.1093/nar/gkr735
Boc, A., Philippe, H., and Makarenkov, V. (2010). Inferring and validating horizontal gene transfer events using bipartition dissimilarity. Syst. Biol. 59, 195–211. doi: 10.1093/sysbio/syp103
Boni, M. F., Posada, D., and Feldman, M. W. (2007). An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176, 1035–1047. doi: 10.1534/genetics.106.068874
Bush, K., Courvalin, P., Dantas, G., Davies, J., Eisenstein, B., Huovinen, P., et al. (2011). Tackling antibiotic resistance. Nat. Rev. Microbiol. 9, 894–896. doi: 10.1038/nrmicro2693
Chee-Sanford, J. C., Mackie, R. I., Koike, S., Krapac, I. G., Lin, Y. F., Yannarell, A. C., et al. (2009). Fate and transport of antibiotic residues and antibiotic resistance genes following land application of manure waste. J. Environ. Qual. 38, 1086–1108. doi: 10.2134/jeq2008.0128
Chen, J., Jin, M., Qiu, Z. G., Guo, C., Chen, Z. L., Shen, Z. Q., et al. (2012). A survey of drug resistance bla genes originating from synthetic plasmid vectors in six chinese rivers. Environ. Sci. Technol. 46, 13448–13454. doi: 10.1021/es302760s
Deraspe, M., Alexander, D. C., Xiong, J., Ma, J. H., Low, D. E., Jamieson, F. B., et al. (2014). Genomic analysis of Pseudomonas aeruginosa PA96, the host of carbapenem resistance plasmid pOZ176. FEMS Microbiol. Lett. 356, 212–216. doi: 10.1111/1574-6968.12435
de Vries, J., and Wackernagel, W. (2002). Integration of foreign DNA during natural transformation of Acinetobacter sp. by homology-facilitated illegitimate recombination. Proc. Natl. Acad. Sci. U.S.A. 99, 2094–2099. doi: 10.1073/pnas.042263399
Didelot, X., and Maiden, M. C. (2010). Impact of recombination on bacterial evolution. Trends Microbiol. 18, 315–322. doi: 10.1016/j.tim.2010.04.002
Doern, G. V., Heilmann, K. P., Huynh, H. K., Rhomberg, P. R., Coffman, S. L., and Brueggemann, A. B. (2001). Antimicrobial resistance among clinical isolates of Streptococcus pneumoniae in the United States during 1999–2000, including a comparison of resistance rates since 1994–1995. Antimicrob. Agents Chemother. 45, 1721–1729. doi: 10.1128/AAC.45.6.1721-1729.2001
Dowson, C. G., Barcus, V., King, S., Pickerill, P., Whatmore, A., and Yeo, M. (1997). Horizontal gene transfer and the evolution of resistance and virulence determinants in Streptococcus. Soc. Appl. Bacteriol. Symp. Ser. 26, 42S–51S. doi: 10.1046/j.1365-2672.83.s1.5.x
Dowson, C. G., Coffey, T. J., and Spratt, B. G. (1994). Origin and molecular epidemiology of penicillin-binding-protein-mediated resistance to beta-lactam antibiotics. Trends Microbiol. 2, 361–366. doi: 10.1016/0966-842X(94)90612-2
Durante-Mangoni, E., Grammatikos, A., Utili, R., and Falagas, M. E. (2009). Do we still need the aminoglycosides? Int. J. Antimicrob. Agents 33, 201–205. doi: 10.1016/j.ijantimicag.2008.09.001
Duron, O. (2014). Arsenophonus insect symbionts are commonly infected with APSE, a bacteriophage involved in protective symbiosis. FEMS Microbiol. Ecol. 90, 184–194. doi: 10.1111/1574-6941.12381
Filipe, S. R., Severina, E., and Tomasz, A. (2000). Distribution of the mosaic structured murM genes among natural populations of Streptococcus pneumoniae. J. Bacteriol. 182, 6798–6805. doi: 10.1128/JB.182.23.6798-6805.2000
Fraser, C., Hanage, W. P., and Spratt, B. G. (2007). Recombination and the nature of bacterial speciation. Science 315, 476–480. doi: 10.1126/science.1127573
Freel, K. C., Millan-Aguinaga, N., and Jensen, P. R. (2013). Multilocus sequence typing reveals evidence of homologous recombination linked to antibiotic resistance in the genus Salinispora. Appl. Environ. Microbiol. 79, 5997–6005. doi: 10.1128/AEM.00880-13
Gibbs, M. J., Armstrong, J. S., and Gibbs, A. J. (2000). Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16, 573–582. doi: 10.1093/bioinformatics/16.7.573
Hakenbeck, R. (2000). Transformation in Streptococcus pneumoniae: mosaic genes and the regulation of competence. Res. Microbiol. 151, 453–456. doi: 10.1016/S0923-2508(00)00170-4
Hall, T. (2007). BioEdit Version 7.0.0. Available online at: http://www.mbio.ncsu.edu/bioedit/biodoc.pdf (Accessed August 14, 2014).
Hanage, W. P., Fraser, C., and Spratt, B. G. (2006). The impact of homologous recombination on the generation of diversity in bacteria. J. Theor. Biol. 239, 210–219. doi: 10.1016/j.jtbi.2005.08.035
Heinemann, J. A., and Traavik, T. (2004). Problems in monitoring horizontal gene transfer in field trials of transgenic plants. Nat. Biotechnol. 22, 1105–1109. doi: 10.1038/nbt1009
Hester, S. E., Park, J., Goodfield, L. L., Feaga, H. A., Preston, A., and Harvill, E. T. (2013). Horizontally acquired divergent O-antigen contributes to escape from cross-immunity in the classical bordetellae. BMC Evol. Biol. 13:209. doi: 10.1186/1471-2148-13-209
Hollingshead, S. K., Becker, R., and Briles, D. E. (2000). Diversity of PspA: Mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 68, 5889–5900. doi: 10.1128/IAI.68.10.5889-5900.2000
Holmes, E. C., Worobey, M., and Rambaut, A. (1999). Phylogenetic evidence for recombination in dengue virus. Mol. Biol. Evol. 16, 405–409. doi: 10.1093/oxfordjournals.molbev.a026121
Hulter, N., and Wackernagel, W. (2008). Double illegitimate recombination events integrate DNA segments through two different mechanisms during natural transformation of Acinetobacter baylyi. Mol. Microbiol. 67, 984–995. doi: 10.1111/j.1365-2958.2007.06096.x
Johansen, B. K., Wasteson, Y., Granum, P. E., and Brynestad, S. (2001). Mosaic structure of Shiga-toxin-2-encoding phages isolated from Escherichia coli O157: H7 indicates frequent gene exchange between lambdoid phage genomes. Microbiology 147, 1929–1936. Available online at: http://mic.sgmjournals.org/content/147/7/1929.full.pdf
Keymer, D. P., and Boehm, A. B. (2011). Recombination shapes the structure of an environmental Vibrio cholerae population. Appl. Environ. Microbiol. 77, 537–544. doi: 10.1128/AEM.02062-10
King, S. J., Whatmore, A. M., and Dowson, C. G. (2005). NanA, a neuraminidase from Streptococcus pneumoniae, shows high levels of sequence diversity, at least in part through recombination with Streptococcus oralis. J. Bacteriol. 187, 5376–5386. doi: 10.1128/JB.187.15.5376-5386.2005
Kocabiyik, S., and Perlin, M. (1992). Altered substrate specificity by substitutions at Tyr218 in bacterial aminoglycoside 3′-phosphotransferase-II. FEMS Microbiol. Lett. 72, 199–202. doi: 10.1016/0378-1097(92)90529-W
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. (2006a). GARD: a genetic algorithm for recombination detection. Bioinformatics 22, 3096–3098. doi: 10.1093/bioinformatics/btl474
Kosakovsky Pond, S. L., Posada, D., Gravenor, M. B., Woelk, C. H., and Frost, S. D. W. (2006b). Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901. doi: 10.1093/molbev/msl051
Le, P. T., Pontarotti, P., and Raoult, D. (2014). Alphaproteobacteria species as a source and target of lateral sequence transfers. Trends Microbiol. 22, 147–156. doi: 10.1016/j.tim.2013.12.006
Leff, L. G., Dana, J. R., McArthur, J. V., and Shimkets, L. J. (1993). Detection of Tn5-like sequences in kanamycin-resistant stream bacteria and environmental DNA. Appl. Environ. Microbiol. 59, 417–421.
Ma, B. L., Blackshaw, R. E., Roy, J., and He, T. (2011). Investigation on gene transfer from genetically modified corn (Zea mays L.) plants to soil bacteria. J. Environ. Sci. Health B 46, 590–599. doi: 10.1080/03601234.2011.586598
Martin, D. P. (2010). RDP3 Instruction Manual. Available online at: http://web.cbio.uct.ac.za/~darren/RDPManual.pdf (Accessed September 22, 2014).
Martin, D. P., Lemey, P., Lott, M., Moulton, V., Posada, D., and Lefeuvre, P. (2010). RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26, 2462–2463. doi: 10.1093/bioinformatics/btq467
Martin, D. P., Posada, D., Crandall, K. A., and Williamson, C. (2005). A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 21, 98–102. doi: 10.1089/aid.2005.21.98
Miki, B., and McHugh, S. (2004). Selectable marker genes in transgenic plants: applications, alternatives and biosafety. J. Biotechnol. 107, 193–232. doi: 10.1016/j.jbiotec.2003.10.011
Murray, P. R., Baron, E. J., Pfaller, M. A., Tenover, F. C., and Yolken, R. H. (1999). Manual of Clinical Microbiology, 7th Edn. Washington, DC: ASM Press.
Nakamura, K., Kohda, T., Shibata, Y., Tsukamoto, K., Arimitsu, H., Hayashi, M., et al. (2012). Unique biological activity of botulinum D/C mosaic neurotoxin in murine species. Infect. Immun. 80, 2886–2893. doi: 10.1128/IAI.00302-12
Oggioni, M. R., Dowson, C. G., Smith, J. M., Provvedi, R., and Pozzi, G. (1996). The tetracycline resistance gene tet(M) exhibits mosaic structure. Plasmid 35, 156–163. doi: 10.1006/plas.1996.0018
Padidam, M., Sawyer, S., and Fauquet, C. M. (1999). Possible emergence of new geminiviruses by frequent recombination. Virology 265, 218–225. doi: 10.1006/viro.1999.0056
Patterson, A. J., Rincon, M. T., Flint, H. J., and Scott, K. P. (2007). Mosaic tetracycline resistance genes are widespread in human and animal fecal samples. Antimicrob. Agents Chemother. 51, 1115–1118. doi: 10.1128/AAC.00725-06
Peirano, G., Agerso, Y., Aarestrup, F. M., Dos Reis, E. M., and Dos Prazeres Rodrigues, D. (2006). Occurrence of integrons and antimicrobial resistance genes among Salmonella enterica from Brazil. J. Antimicrob. Chemother. 58, 305–309. doi: 10.1093/jac/dkl248
Perlin, M. H., and Lerner, S. A. (1986). High-level amikacin resistance in Escherichia coli due to phosphorylation and impaired aminoglycoside uptake. Antimicrob. Agents Chemother. 29, 216–224. doi: 10.1128/AAC.29.2.216
Pontiroli, A., Simonet, P., Frostegard, A., Vogel, T. M., and Monier, J. M. (2007). Fate of transgenic plant DNA in the environment. Environ. Biosafety Res. 6, 15–35. doi: 10.1051/ebr:2007037
Posada, D., and Crandall, K. A. (2001). Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc. Natl. Acad. Sci. U.S.A. 98, 13757–13762. doi: 10.1073/pnas.241370698
Prudhomme, M., Libante, V., and Claverys, J. P. (2002). Homologous recombination at the border: insertion-deletions and the trapping of foreign DNA in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. U.S.A. 99, 2100–2105. doi: 10.1073/pnas.032262999
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Roa-Rodriguez, C., and Nottenburg, C. (2003). Antibiotic Resistance Genes and their Uses in Genetic Transformation, Especially in Plants. CAMBIA. Available online at: http://www.bios.net/daisy/Antibiotic/752.html
Shakya, T., Stogios, P. J., Waglechner, N., Evdokimova, E., Ejim, L., Blanchard, J. E., et al. (2011). A small molecule discrimination map of the antibiotic resistance kinome. Chem. Biol. 18, 1591–1601. doi: 10.1016/j.chembiol.2011.10.018
Shaw, K. J., Rather, P. N., Hare, R. S., and Miller, G. H. (1993). Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 57, 138–163.
Smalla, K., van Overbeek, L. S., Pukall, R., and van Elsas, J. D. (1993). The prevalence of nptII and Tn5 in kanamycin-resistant bacteria from different environments. FEMS Microbiol. Lett. 13, 47–58. doi: 10.1111/j.1574-6941.1993.tb00050.x
Smith, J. M. (1992). Analyzing the mosaic structure of genes. J. Mol. Evol. 34, 126–129. doi: 10.1007/BF00182389
Smith, J. M., Dowson, C. G., and Spratt, B. G. (1991). Localized sex in bacteria. Nature 349, 29–31. doi: 10.1038/349029a0
Smith, S. E., Showers-Corneli, P., Dardenne, C. N., Harpending, H. H., Martin, D. P., and Beiko, R. G. (2012). Comparative genomic and phylogenetic approaches to characterize the role of genetic recombination in mycobacterial evolution. PLoS ONE 7:e50070. doi: 10.1371/journal.pone.0050070
Spratt, B. (1994). Resistance to antibiotics mediated by target alterations. Science 264, 388–393. doi: 10.1126/science.8153626
Spratt, B. G., Zhang, Q. Y., Jones, D. M., Hutchison, A., Brannigan, J. A., and Dowson, C. G. (1989). Recruitment of a penicillin-binding protein gene from Neisseria flavescens during the emergence of penicillin resistance in Neisseria meningitidis. Proc. Natl. Acad. Sci. U.S.A. 86, 8988–8992. doi: 10.1073/pnas.86.22.8988
Thomas, J. C., Godfrey, P. A., Feldgarden, M., and Robinson, D. A. (2012). Draft genome sequences of Staphylococcus aureus sequence type 34 (ST34) and ST42 hybrids. J. Bacteriol. 194, 2740–2741. doi: 10.1128/JB.00248-12
Weiller, G. F. (1998). Phylogenetic profiles: a graphical method for detecting genetic recombinations in homologous sequences. Mol. Biol. Evol. 15, 326–335. doi: 10.1093/oxfordjournals.molbev.a025929
WHO (2011). Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis - 2011 Update. Available online at: http://whqlibdoc.who.int/publications/2011/9789241501583_eng.pdf (Accessed Mar 4, 2013).
WHO (2012). Critically Important Antimicrobials for Human Medicine. 3rd Revision 2011. Geneva: WHO Press; World Health Organization. Available online at: http://www.who.int/foodborne_disease/resistance/cia/en/ (Accessed Oct 7, 2013).
Woegerbauer, M., Zeinzinger, J., Springer, B., Hufnagl, P., Indra, A., Korschineck, I., et al. (2014). Prevalence of the aminoglycoside phosphotransferase genes aph(3′)-IIIa and aph(3′)-IIa in Escherichia coli, Enterococcus faecalis, Enterococcus faecium, Pseudomonas aeruginosa, Salmonella enterica subsp. enterica and Staphylococcus aureus isolates in Austria. J. Med. Microbiol. 63, 210–217. doi: 10.1099/jmm.0.065789-0
Xiong, J., Alexander, D. C., Ma, J. H., Deraspe, M., Low, D. E., Jamieson, F. B., et al. (2013). Complete sequence of pOZ176, a 500-kilobase IncP-2 plasmid encoding IMP-9-mediated carbapenem resistance, from outbreak isolate Pseudomonas aeruginosa 96. Antimicrob. Agents Chemother. 57, 3775–3782. doi: 10.1128/AAC.00423-13
Yang, F. F., Sun, Y. N., Li, J. X., Wang, H., Zhao, M. J., Su, J., et al. (2012). Detection of aminoglycoside resistance genes in Riemerella anatipestifer isolated from ducks. Vet. Microbiol. 158, 451–452. doi: 10.1016/j.vetmic.2012.02.027
Zeng, L., and Jin, S. (2003). aph(3′)-IIb, a gene encoding an aminoglycoside-modifying enzyme, is under the positive control of surrogate regulator HpaA. Antimicrob. Agents Chemother. 47, 3867–3876. doi: 10.1128/AAC.47.12.3867-3876.2003
Keywords: antibiotic resistance, horizontal gene transfer, mosaic genes, homologous recombination, nptII
Citation: Woegerbauer M, Kuffner M, Domingues S and Nielsen KM (2015) Involvement of aph(3′)-IIa in the formation of mosaic aminoglycoside resistance genes in natural environments. Front. Microbiol. 6:442. doi: 10.3389/fmicb.2015.00442
Received: 13 March 2015; Accepted: 24 April 2015;
Published: 19 May 2015.
Edited by:
Fiona Walsh, National University of Ireland Maynooth, IrelandReviewed by:
Carlos F. Amábile-Cuevas, Fundación Lusara, MexicoCarmen Losasso, Istituto Zooprofilattico Sperimentale delle Venezie, Italy
Copyright © 2015 Woegerbauer, Kuffner, Domingues and Nielsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Woegerbauer, Integrative Risk Assessment – Data – Statistics, Austrian Agency for Health and Food Safety, Spargelfeldstrasse 191, 1220 Vienna, Austria,bWFya3VzLndvZWdlcmJhdWVyQGFnZXMuYXQ=
†Present Address: Kaare M. Nielsen, Department for Life Sciences and Health, Oslo and Akershus University College of Applied Sciences, Oslo, Norway