 Parik Kakani
Parik Kakani Sneha Suman
Sneha Suman Lalita Gupta
Lalita Gupta Sanjeev Kumar
Sanjeev Kumar- Molecular Parasitology and Vector Biology Lab, Department of Biological Sciences, Birla Institute of Technology and Science, Pilani, India
The life cycle of Plasmodium in two evolutionary distant hosts, mosquito, and human, is a complex process. It is regulated at various stages of developments by a number of diverged mechanisms that ultimately determine the outcome of the disease. During the development processes, Plasmodium invades a variety of cells in two hosts. The invaded cells tend to undergo apoptosis and are subsequently removed from the system. This process also eliminates numerous parasites along with these apoptotic cells as a part of innate defense against the invaders. Plasmodium should escape the invaded cell before it undergoes apoptosis or it should manipulate host cell apoptosis for its survival. Interestingly, both these phenomena are evident in Plasmodium at different stages of development. In addition, the parasite also exhibits altruistic behavior and triggers its own killing for the selection of the best ‘fit’ progeny, removal of the ‘unfit’ parasites to conserve the nutrients and to support the host survival. Thus, the outcomes of cell apoptosis are ambivalent, favorable as well as unfavorable during malaria progression. Here we discuss that the manipulation of host cell apoptosis might be helpful in the regulation of Plasmodium development and will open new frontiers in the field of malaria research.
Introduction
Arthropods are infamous vectors for numerous human diseases that are major public health hazards throughout the world. The causative agents of these diseases include helminths, protozoa, bacteria, and viruses. Among the large number of vector-borne infections, malaria is included in the list of top five infectious diseases. It is caused by an apicomplexan parasite Plasmodium and transmitted by the Anopheles mosquito among humans. This disease is reported in over 90 countries primarily in tropical and subtropical regions in sub-Saharan Africa, Central and South America, the Caribbean island of Hispaniola, the Middle East, the Indian subcontinent, South-East Asia, and Oceania. On an average more than 198 million people are infected and 584,000 people die due to malaria every year worldwide (World Health Organization [WHO], 2014).
The malaria parasite Plasmodium completes its life cycle in two evolutionary distant hosts; mosquito and a vertebrate such as human. The sexual life cycle is completed in female Anopheles mosquito (a definitive primary host) and asexual life cycle predominates in human (a secondary host). The mosquito ingests Plasmodium gametocytes stages that undergo fertilization and form zygote inside the midgut lumen. Further, in the same compartment, the formation of ookinetes from zygote takes place near about 15h after ingestion. Ookinete traverses the midgut epithelium around 24h of ingestion and develops into an oocyst in the space between midgut epithelium and basal lamina (Figure 1). Oocyst matures approximately after 10 days and then releases 1000s of sporozoites into the mosquito haemocoel. Sporozoites circulate throughout the hemolymph and some of them end up in salivary glands. Sporozoites entered the salivary gland further undergo maturation and are ready to be injected into the new human host during the subsequent feeding (Zheng, 1997; Beier, 1998; Sinden, 2002).
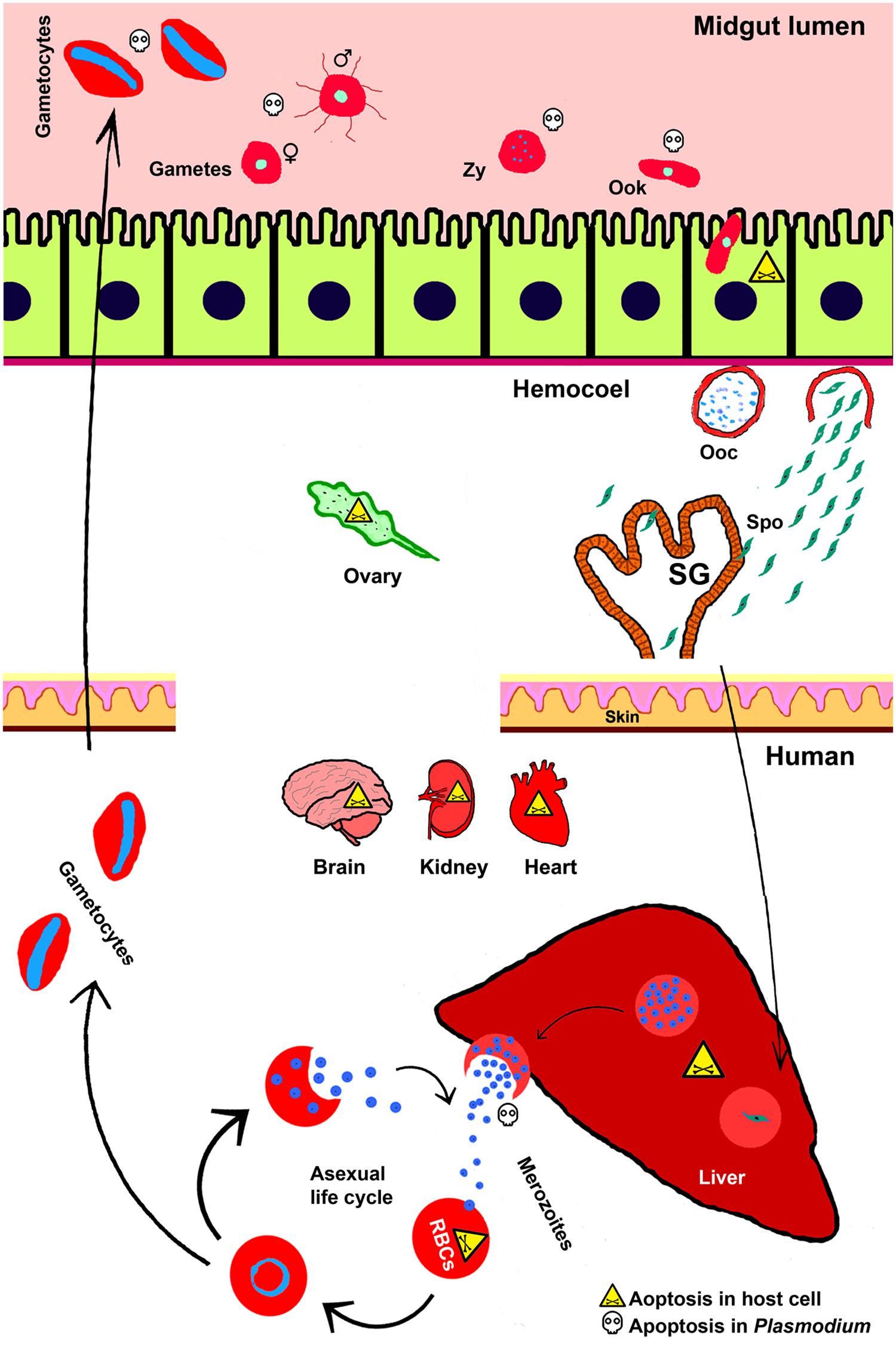
FIGURE 1. Apoptosis during Plasmodium life cycle in invertebrate and vertebrate hosts. Life cycle of Plasmodium is completed in two hosts. The sexual life cycle occurs in mosquito and asexual life cycle takes place in the vertebrate host such as, human. Various stages of Plasmodium development and the phenomena of cell death in both the hosts and parasites are depicted here. Zy, Zygote; Ook, ookinetes; Ooc, oocysts; Spo, sporozoites; SG, salivary gland.
Plasmodium cycle in human host begins with the entry of mosquito-injected sporozoites into the circulating blood. These sporozoites migrate toward the liver and initiate pre-erythrocytic cycle in hepatocytes that continue approximately for 6–15 days. During this phase, Plasmodium undergoes extensive growth and division and at the end, 1000s of merozoites are released into the blood. These merozoites further invade red blood cells (RBCs) and initiate erythrocytic cycle (Frevert, 2004). This asexual erythrocytic cycle further produces more merozoites and at 48 or 72 h, depending on the Plasmodium species, they are released from the RBC and immediately invade new erythrocytes. In continuation of the erythrocytic cycle some merozoites differentiate into gametocytes and after ingestion by another female mosquito they continue the sexual cycle as discussed above (Cowman et al., 2012).
It is noteworthy to mention that in both the hosts, Plasmodium development takes place in compartments-specific manner and during this process parasite interacts with diverse cell types. During these interactions, apoptosis takes place in the parasites as well as the host cells. Apoptosis is a genetically orchestrated type of cell death that involves numerous distinguishable morphological and cellular alterations (Kerr et al., 1972; Cowman et al., 2012). Generally, the advanced stages of apoptosis are associated with the removal of dying cell from the body system and its replacement by a new cell. Studies revealed that pathogens are eliminated along with the dying cell in case of several diseases, including malaria (Williams, 1994; Sinai et al., 2004).
Cell apoptosis plays an important role in establishment of host–pathogen relationships. The present review identifies the sites of apoptosis in malaria parasite and host cells during Plasmodium life cycle. It also highlights the effect of apoptosis on the parasite or host as a strategy by which the parasites proliferate in a healthy environment or a self-defense/repair mechanism by the host. Here, apoptosis of host cells, Plasmodium and the outcome of this phenomenon in disease progression has been discussed. Apoptosis of immune cells in both the hosts is not considered as part of this review.
Apoptosis in Plasmodium During Sexual Stages of Development
Plasmodium encounters a harsh environment inside the mosquito gut that creates somewhat a bottleneck situation to the parasite numbers at different stages of development such as, gamete, zygote, and ookinete stages (Chose et al., 2003). Majority of gametocytes that arrive in the midgut lumen fail to develop further and on an average approximately 80% of them undergo apoptosis (Sinden, 1999; Sinden and Billingsley, 2001). Studies analyzed the mode of cell death in Plasmodium berghei (mouse malaria) sexual stages found that zygotes and ookinetes exhibit the characteristic apoptotic cell morphology revealed by DNA fragmentation, chromation condensation and phosphatidylserine (PS) translocation (Al-Olayan et al., 2002).
The apoptosis in Plasmodium may be a natural way to select the most potent or best ‘fit’ parasite that can carry forward the malaria cycle in mosquito where it may also reduce the parasite burden for better survival of the insect host. This indicates that an increased load of Plasmodium infection might have deleterious effects in the mosquito host. Similar parasitic behavior is also reported in Leishmania where promostigote stage reveals apoptotic features during their development inside the sand fly gut. Apoptotic parasites die and show philanthropic behavior toward the viable parasites. This phenomenon maintains a vital association for the survival of both parasites and sand fly (Shaha, 2006; Van Zandbergen et al., 2006).
It is also possible that mosquito internal environment factors such as, immune components of the ingested blood, the natural gut symbionts or mosquito innate immunity might be regulating the process of apoptosis in Plasmodium. The immune components in mosquito ingested blood include white blood cells (WBCs), complement system proteins, cytokines and reactive oxygen or nitrogen species (Lensen et al., 1998; Ramiro et al., 2011; Simon et al., 2013). Previous reports found that WBCs have phagocytic activity against P. falciparum and P. berghei gametocytes/gametes in Anopheles gambiae midgut. The removal of WBCs form the ingested blood significantly reduced the number of apoptotic ookinetes. In addition, the nitric oxides (NO) generated by activated WBCs also induce parasite death (Lensen et al., 1997; Muniz-Junqueira et al., 2001). Cytokines such as, TNF-α and TGF-β1 present in the ingested blood meal also have anti-plasmodial effects. TNF-α inhibits the male gamete formation (called exflagellation) in P. berghei that is mediated through leucocytes generated reactive nitrogen species (RNS; Naotunne et al., 1993; Ali et al., 2010; Ramiro et al., 2011). TGF-β1 in fact induces A. stephensi nitric oxide synthase (NOS) at low concentrations and suppresses the parasite numbers (Luckhart et al., 2003). The human complement system remains active up to 6 h in the mosquito midgut. A direct effect of the complement system in preventing gametogenesis has been observed in case of P. falciparum development. However, oocysts and zygotes have learned to nullify the complement-mediated lysis through surface binding of factor H that inactivates complement effector protein C3b. This seems to be a co-evolved mechanism of protection in these complement-sensitive stages (Simon et al., 2013). The role of mosquito gut flora in the regulation of Plasmodium development is reported by several laboratories. In a simple way, the reduction of microbial communities in antibiotics fed Anopheles mosquitoes increases their susceptibility to malaria parasite infection (Boissière et al., 2012; Minard et al., 2013; Kajla et al., 2015a). These observations are important in terms of manipulating the endogenous microbial flora of the mosquito to control their vectorial capacity and this area is under investigation by many researchers.
Gene silencing studies revealed that a number of mosquito immune genes regulate parasite load. For example, clip-domain serine protease (CLIPC2) plays an anti-plasmodial role (Blumberg et al., 2013). Similarly, thioester containing protein 1 (TEP1), an opsonin, induces parasite melanization and inhibits Plasmodium development (Blandin et al., 2004; Blumberg et al., 2013). In addition, mosquito generated NO and reactive oxygen species (ROS) have been reported to trigger apoptosis in Plasmodium ookinetes. Studies demonstrated that in vitro exposure of Plasmodium ookinetes with sodium nitroprusside, the NO producer, induced caspase-like activity (Ali et al., 2010). On the other hand, feeding L-NAME (N-nitro-L-arginine methyl ester), the inhibitor of NO producing key enzyme NOS, along with P. falciparum infected blood reduced the levels of apoptotic ookinetes and ultimately increased the number of developing oocysts (Luckhart et al., 1998). In addition, the reduction of NOS gene expression after silencing the mosquito STAT, produced similar effect on Plasmodium as observed above in case of L-NAME treatment (Gupta et al., 2009). Thus, we can conclude that the regulation of Plasmodium development inside the mosquito midgut is a multifaceted phenomenon and has been exploited by a number of transmission blocking strategies. For example, the mosquito midgut specific molecules such as alanyl amino peptidase 1 (APN1), carboxypeptidase B (CPB) and a heme peroxidase HPX15 are proposed to design transmission blocking vaccines because inhibition of these molecules suppressed Plasmodium development (Lavazec et al., 2007; Atkinson et al., 2015; Kajla et al., 2015b).
Apoptosis in Ookinete-Invaded Midgut Epithelial Cells is Beneficial for the Mosquito Host
Plasmodium ookinetes are motile and cross the midgut epithelium around 20 h after ingestion and targeted by the mosquito immune system. Numerous mosquito immune pathways such as, peptidoglycan recognition proteins (PGRP), scavenger receptors (SCRs), C-type lectins (CTLs), and the genes regulating melanization cascade are reported to induce apoptosis in ookinetes during their traversal to the mosquito midgut epithelium (Michel and Kafatos, 2005). Ookinete invasion-induced expression of NOS and peroxidases mediate the nitration of invaded midgut cells. This nitration process activates mosquito complement TEP1 that mediate ookinete death (Kumar et al., 2004; De Almeida Oliveira et al., 2012; Garver et al., 2013). Ookinete invasion also induces apoptosis in midgut epithelial cells that is revealed by characteristic apoptotic features and commonly observed in A. gambiae- P. berghei, A. gambiae- P. falciparum, and Aedes aegypti- P. gallinaceum combinations. At the last stage of apoptosis, the invaded cells extrude from the epithelial layer into the gut lumen and are replaced by the new cells (Han et al., 2000; Zieler and Dvorak, 2000; Kumar et al., 2004; Gupta et al., 2005). It is important that for a successful invasion the traversing ookinetes must escape unharmed before the invaded cells extrude into the lumen.
The apoptosis of midgut cells is beneficial for the mosquito host because it tends to remove the ‘slow moving ookinetes’ that are entrapped inside the damaged cells. Studies found that in case of A. stephensi, the bulged out epithelial cells are found in midgut lumen and P. falciparum ookinete are entrapped inside them (Baton and Ranford-Cartwright, 2004, 2005). Our observations also revealed that in the same mosquito midgut cells undergo apoptosis soon after they are invaded by the P. falciparum ookinetes (Kumar and Barillas-Mury, 2005). Although the P. berghei ookinete invasion induces cell death in mosquito midgut epithelium, however, in this case the ookinetes entrapped inside the dead cells are rarely observed. It may be simply due to the reason that P. falciparum infected mosquitoes are maintained at 28°C and P. berghei infected mosquitoes at 20°C. This lower temperature maintenance might slow down the midgut cell death processes as reported in case of some mammalian cells (Sakurai et al., 2005).
The invasion of Anopheles mosquito midgut by P. falciparum and P. berghei ookinetes is also different in many ways. In the former case, generally less number of ookinetes successfully completes the invasion and develops into the oocysts. However, in the case of P. berghei the rate of ookinete invasion is mostly higher in comparison to the P. falciparum infection (Smith et al., 2014). The rapid turnover of mosquito midgut epithelial cells in case of P. falciparum, due to their maintenance at 28°C, may be associated with less successful invasion events. In this case, the damaged cells undergo a rapid removal from the epithelial surface and are replaced by the new cells. These dead cells also take away the pathogens that invade them as the part of an intrinsic defense mechanism. The phenomenon of rapid epithelial turnover in reducing the infection by microbial intruders has been observed in other enteric infections. For example, in case of gastrointestinal habitants Helicobacter pylori and Trichuris muris nematode infections, the gastric epithelial cells undergo apoptosis and expulsion. The increased rate of epithelial turnover makes the mouse resistant against the worm infection (Moss et al., 1996; Kim et al., 1998; Cliffe et al., 2005).
It is of note that the mosquito midgut has few hundred cells; thus, the death in large extend of these cells during ookinete invasion may also be deleterious to the insect host. In other words, the higher density of Plasmodium infectious stages in the ingested blood might have deleterious effects on mosquito host. Thus, the balanced regulation of midgut epithelium apoptosis in these mosquitoes is important for their survival after Plasmodium infection.
Ookinete Tends to Suppress Midgut Cell Apoptosis for Its Own Survival
It is clear from the above discussion that to establish infection in the mosquito midgut, ookinetes should escape before they are destroyed or extruded along with the damaged epithelial cell. We believe that in this situation Plasmodium must be manipulating either the midgut epithelial immunity or apoptosis in the target cell. Studies carried in A. gambiae mosquito revealed that the ookinete protein Pfs47 prevents the activation of caspases and inhibits Jun-N-terminal kinase-mediated activation of apoptosis in invaded mosquito midgut cells (Ramphul et al., 2015). In this study, the wild type parasites regulated broad changes in gene expression profile of the mosquito midgut, however, the Pfs47 mutants failed to do so. Evidences obtained from the above study revealed that the Plasmodium death reduced the parasite load in mosquito midgut to support the survival of the vector, completion of sexual life cycle and disease transmission to the vertebrate host. On the other hand, Plasmodium mediated interference to the apoptosis of mosquito midgut epithelium also supports its own survival. It manipulates the expression of peroxidase/oxidase system responsible for the nitration of ookinetes, which causes parasite lysis by TEP1 (Ramphul et al., 2015).
The Death of Oocysts is Mediated by the Mosquito Late Phase Immunity
The ookinete that successfully traverses the midgut are further transformed into the oocyst. The phenomenon of cell apoptosis is not observed in oocysts. However, recent studies reveal that in A. gambiae the oocysts encounter a late phase immunity that is mediated through STAT pathway. Silencing of STAT gene in these mosquitoes increased the number of oocysts survival against controls, which was mediated through the reduced expression of NOS, an effector gene of STAT pathway (Gupta et al., 2009). In the same mosquito, an LPS-induced TNFα transcription factor (LITAF)-like 3 (LL3) is also reported to mediate the late phase immunity. LL3 binds to the promoter region of an anti-plasmodial gene termed as serine protease inhibitor 6 (SRPN6) and modulates its expression. Silencing of LL3 gene restricts the differentiation of hemocytes and their responses to parasite infection that results in the increased number of oocysts in the silenced mosquitoes. Thus, LL3 implicates late-phase immunity against Plasmodium oocysts through hemocytes (Smith et al., 2012, 2015). These observations might call upon to understand the correlation between the STAT and SRPN 6 pathways in terms of regulating the late phase immunity against Plasmodium oocysts.
Death Process in the Sporozoites
Sporozoites released from the oocyst after several rounds of mitotic divisions although undergo a death process; however, this is a type of non-apoptotic death. They are released in 1000s and 10–20% of them finally invade the salivary glands. Rests of the sporozoites are cleared from the mosquito circulation by hemocytes-mediated phagocytosis (Foley, 1978; Hernández-Martínez et al., 2002; Hillyer et al., 2003, 2007). In salivary glands, Plasmodium uptake occurs via a receptor-mediated endocytosis or through a specific ligand interaction. No cytoskeleton rearrangement or apoptosis has been observed in the gland epithelium (Ghosh and Jacobs-Lorena, 2009; Mueller et al., 2010).
Plasmodium entry in salivary gland epithelial layer is dissimilar to the midgut epithelium invasion. Salivary glands mount an acute immune response against invading sporozoites. Studies found that some common genes exhibit similar regulation in both epithelia. For example, Plasmodium invasion downregulates a fatty acid synthase (AGAP009176) however, GTP-binding nuclear protein, lysosomal thioreductase precursor, and SRPN6 were up-regulated in both the tissues (Rosinski-Chupin et al., 2007). Interestingly, the silencing of SRPN6 gene in mosquito midgut and salivary gland increased the number of parasites in the respective organ (Abraham et al., 2005; Pinto et al., 2008). This indicates that mosquito innate immunity regulates Plasmodium number at different stages of development. This provides an opportunity to manipulate mosquito immunity to control the vectorial competence.
Plasmodium Infection Mediated Apoptosis in Non-target Mosquito Cells
Plasmodium does not directly interact with some mosquito cells; however, the infection exhibits an indirect effect on the apoptosis of some other host cells. For example, P. yoelii nigeriensis infection in A. stephensi resulted in characteristic apoptosis of cells in ovaries or the follicular epithelium and reduced egg production by the gravid females (Hopwood et al., 2001; Ahmed and Hurd, 2006). In addition, the follicular epithelial cell apoptosis is also the major trigger of follicular reabsorption. The follicles showing reabsorption are detected at 12 h that gradually increases to maximum at 24 h post infected blood feeding. Interestingly, at 12 h post feeding the Plasmodium development taking place in midgut bolus and at 24 h the ookinete invades the midgut (Han et al., 2000; Kumar et al., 2010). These findings might indicate the advanced detection of Plasmodium-induced factors by the mosquito system. In parallel, the anti-plasmodial immunity of mosquito may be responsible for follicular apoptosis through the generation of ROS or RNS. These assumptions are supported by experimental findings where high levels of NO, nitrites/nitrates or ROS are found in Plasmodium infected mosquito midgut as well as hemolymph (Luckhart et al., 1998; Crampton and Luckhart, 2001; Kumar et al., 2003). This may be also possible that during the ookinete invasion some bolus bacteria are exposed to the mosquito immune system and the reactive products of innate immunity (ROS and RNS) mediate the degenerative effects in ovaries. The induction of mosquito immunity after lipopolysaccharides (LPS) inoculation into the hemocoel also caused follicular resorption and reduction of fecundity (Ahmed et al., 2002; Hurd, 2003). On the other hand, Plasmodium might have developed some mechanisms to induce follicular resorption and channelizing the eggs stored energy for its own development. The understanding of these mechanisms requires further investigations and it can provide a ground to control mosquito fecundity and their population.
Plasmodium Asexual Life Cycle and Cell Apoptosis in Vertebrate Host
Plasmodium sporozoites initiate a silent infectious phase called pre-erythrocytic stage inside the liver cells (hepatocytes) and ultimately develop into exo-erythrocytic merozoites (Prudêncio et al., 2011). The sporozoites delivered after mosquito bite in the vertebrate host travel through blood stream and reach liver sinusoids. In the liver sinusoids, sporozoites come to arrest through the interactions of their surface circumsporozoite protein (CSP) with the glycosaminoglycans (GAGs) of stellate cells. According to the gateway hypothesis, the sporozoite glides along the sinusoid wall to locate a Kupffer cell, traverse it and subsequently invade the underlying hepatocyte (Frevert et al., 2005). Recent findings revealed that a heavily glycosylated protein CD68, present exclusively on the surface of the Kupffer cells, acts like a candidate receptor for sporozoite entry. The sporozoites are also reported to remain enclosed within the CD68 endosome that protects them against lysosomal attack within the Kupffer cell. Although the ligand for CD68 receptor is unknown; however, the Kupffer cells of CD68 knockout mouse imposed a barrier for sporozoite invasion (Cha et al., 2015). These findings are important for developing vaccines to block liver stages (LSs) of sporozoites development.
Interestingly, the CD68+ Kupffer cells are known to exhibit both phagocytic activity and ROS production capacity (Kinoshita et al., 2010). Thus, the survival of traversing sporozoites must be dependent on the modulation of immune pathways in these macrophages. Studies found that the sporozoite CSP interaction with heparan sulfate proteoglycans (HSPGs) and the low-density lipoprotein receptor-related protein LRP-1 on the surface of these Kupffer cells activates adenylyl cyclase (AC) enzyme. This enzyme, in turn, upregulates cAMP activity and inhibits NADPH oxidase-mediated generation of ROS by the Kupffer cells (Usynin et al., 2007; Kinoshita et al., 2010; Tavares et al., 2013; Cha et al., 2015). These mechanisms create an anti-inflammatory environment that protects sporozoites from the immune attacks in the liver. It is also observed that Kupffer cells exhibit the sign of apoptosis during transformation of sporozoites into early exo-erythrocytic forms (Usynin et al., 2007; Klotz and Frevert, 2008). In conclusion, the sporozoite not only induces desensitization of Kupffer cells to pro-inflammatory stimuli, it also forces their programmed cell death for its own survival.
Plasmodium Manipulates Host Immunity and Apoptosis of Hepatocytes
The intracellular sporozoites are protected from the host immune responses; however, the infected hepatocytes have other mechanisms to eliminate them. Studies have shown that infected hepatocytes undergo apoptosis without external triggers. When sporozoites traverse the hepatocytes, they deform the hepatocyte morphology that results in wounding. This wounding is the trigger for induction of apoptosis in hepatocytes. The apoptosis in infected hepatocytes has been confirmed by previous studies that observed the uptake of fluorescein isothiocyanate (FITC) labeled dextran and propidium iodide by these cells (Mota et al., 2001; Kaushansky et al., 2013). Blocking the apoptosis process in hepatocytes increases LS parasite burden in mice. This suggests that apoptosis of hepatocytes has negative effect on Plasmodium progression through hepatocytic stages (Kaushansky et al., 2013). In other words, apoptosis of infected hepatocytes is beneficial for vertebrate host but detrimental for the sporozoites. It is of note that the apoptosis of Plasmodium infected hepatocytes exposes the parasitic antigens to initiate protective immune responses in the host (Leiriao et al., 2005b). Therefore, for a successful completion of the LSs, sporozoites must be able to modulate the host immunity and the process of cell death in hepatocytes.
Sporozoites induce the release of hepatocyte growth factor (HGF) from the traversed cells that inhibits tumour necrosis factor (TNF)-mediated apoptosis of hepatocytes. This process helps in successful establishment of the liver infection (Carrolo et al., 2003; Huh et al., 2004; Leiriao et al., 2005a). In addition, sporozoites also inhibit the translocation of PS into the outer leaflet of the cell membrane to block the ‘eat me’ signal displayed by infected hepatocytes to the Kupffer cells. In fact, the swapping of PS into the outer leaflet is driven by the increased cytosolic Ca2+. The hepatic stages of parasite sequester this intracellular Ca2+ and ultimately inhibit the cellular autophage in hepatocytes (Sturm et al., 2006). A similar mechanism is also displayed by intra-erythrocytic parasites as discussed later.
Plasmodium sporozoites are also equipped with other specific mechanisms to manipulate the apoptosis of infected hepatocytes and completing the LSs. Sporozoites mostly invade those hepatocytes that express high levels of EphA2, a transmembrane receptor tyrosine kinase (RTK). EphA2 is generally expressed in the majority of epithelial cells and the interaction of this receptor with its ligand ephrin leads to the contact-dependent cell–cell communication (Park et al., 2013). Interestingly, in hepatocytes the sporozoite remains enclosed inside a parasitophorous vacuole (PV) that is the part of hepatocyte plasma membrane. Recent studies revealed that the interaction of sporozoite surface protein P36 with hepatocyte EphA2 determines the formation of PV. The formation of PV is a viable process to establish a tolerant environment for the replication of sporozoite in the intracellular compartment. In the absence of PV formation, the sporozoites infected hepatocytes suffer extensive cell death and reduce the burden of LSs (Mueller et al., 2005; Kaushansky et al., 2015). Experimental evidences also revealed that the LS infection of Plasmodium was largely decreased in EphA2-/- mice in comparison to wild-type mice. This indicated that EphA2 receptor mediated invasion is helpful in the survival of sporozoites and manipulation of apoptosis in the hepatocytes (Kaushansky et al., 2015). Furthermore, studies carried with P. berghei-hepatocyte infection stages revealed that the parasite surface protein P36p, the member of P48/45 family of proteins, is responsible for delaying the apoptosis in hepatocytes. It is of note that P36p-deficient P. berghei sporozoites are capable of infecting HepG2 cells in culture as well as mouse liver cells in vivo. However, the mutant parasites are eliminated faster due to an increased apoptosis in the infected hepatocytes in comparison to the wild type parasite infection. In addition, the mutant parasites also developed the protective immunity in vivo (Van Dijk et al., 2005). On the other hand, sporozoite secreted cysteine protease inhibitor also blocks the cysteine proteases of the invaded hepatocytes and suppress their apoptosis (Rennenberg et al., 2010). These findings might conclude that it is a must for the sporozoites to either delay or block the apoptosis of the infected hepatocytes. If the infected liver cell undergoes apoptosis, it will help the antigen presenting cells to display parasite antigens to the acquired immune system. These findings provide newer ways to develop protective immunity against the LSs of Plasmodium through manipulation of apoptosis in hepatocytes and demands further exploring this area in details.
Apoptosis in Erythrocytes Has Dual Effects on Malaria Progression
The enucleated RBCs exhibit programmed cell death known as eryptosis. This phenomenon is similar to apoptosis and characterized by the cell shrinkage, membrane bebbling, and exposure of PS at the cell surface. A major signal of eryptosis is an increased cytosolic Ca2+ that involves the cell membrane scrambling in a way similar to the liver cells as discussed before (Föller et al., 2008; Lang et al., 2008). In fact, Plasmodium induces oxidative burst that, in turn, activates ion channels in the infected RBC for the uptake of nutrients, Na+ and Ca2+ ions and excretion of waste product. This Ca2+ entry is the major signal for inducing eryptosis (Brand et al., 2003; Tanneur et al., 2006). The phenomenon of eryptosis has been observed in P. yoelii 17XL and P. berghei ANKA infected RBCs in vivo as well as in P. falciparum culture. Studies carried in case of P. yoelii 17XL revealed that in addition to the parasitized RBCs (pRBCs), non-parasitized RBCs (nRBC) also undergo eryptosis in response to a high parasite load (Eda and Sherman, 2002; Koka et al., 2008; Totino et al., 2010, 2013).
Apoptosis of RBCs has dual role in the advancement of malaria disease. In one way, infected cells undergoing apoptosis are removed by splenic phagocytosis and this process controls the parasite load as well as contributes to the anaemic conditions (Lang et al., 2009; Totino et al., 2010). The death of infected RBCs is the sign of risk for Plasmodium development. Thus, to avoid splenic phagocytosis, Plasmodium-infected RBCs adhere to the microvascular endothelium via PS and chondroitin sulfate A (CSA) and ends up in more complications such as cerebral malaria, blood–brain barrier (BBB) dysfunction and multiple organ failure (Eda and Sherman, 2002; Setty et al., 2002; Hunt and Grau, 2003; Tripathi et al., 2007; Craig et al., 2012). To overcome the eryptosis-associated side effects, Plasmodium tends to delay this process by reducing both the intra-erythrocytic Ca2+ levels and the activity of erythrocyte Ca2+ pump (Duranton et al., 2003; Huber et al., 2005). Thus, Plasmodium should manage the apoptosis of RBCs to balance between the progression of disease and death of the vertebrate host. These mechanisms are important to control erythrocytic stages of Plasmodium development. Target specific blockers may be synthesized to manipulate the Plasmodium-induced channels in RBC membrane. This will open new opportunities in this field. Several research labs are actively engaged in this area to develop these channel blockers to inhibit the progression of erythrocytic cycle in human malaria (Kang et al., 2005).
Cell Apoptosis in General is Associated with Severe Complications
Severe complications in malaria are associated with the sequestration of Plasmodium-infected red blood cells (pRBCs) in the brain and other organs. Sequestration involves the cytoadherence of pRBCs, which causes over-expression of inflammatory cytokines and target cell apoptosis (Hunt and Grau, 2003; Pino et al., 2003; Wilson et al., 2008). In the severe malaria, vascular integrity is altered as a result of endothelial cell (EC) activation and death, which is caused by the activation of Plasmodium apoptosis-linked pathogenicity factors (PALPF), PALPF-2, PALPF-5, and PF11_0521 (N’Dilimabaka et al., 2014). Some findings also suggested that glycosylphosphatidylinositol (GPI) of P. falciparum is responsible for the cardiomyocyte apoptosis that reveals malaria associated severity (Wennicke et al., 2008). Acute organ failure (for example, kidney or liver) during complicated malaria is associated with the sequestration and adhesion of pRBCs to the ECs in target organs. In P. berghei ANKA infected BALB/c mice high expression of ICAM-1 was found on renal tissues, which is responsible for the interaction of pRBCs with them. This cytoadherence causes change in vascular permeability and recruits inflammatory cells, which contributes to the increased rate of cell apoptosis (Elias et al., 2012). Hence, switching from uncomplicated to severe malaria is possibly caused by the activation of cell apoptosis in different organs. Thus, the target-specific strategies against these major factors that are associated with the disease ‘severity’ might be helpful to control complications during malaria infections.
Apoptosis in Plasmodium is Crucial for Host Survival
Plasmodium demonstrates stress responses in vertebrate host and this may result in inducing its own apoptosis. The progression of Plasmodium merozoites through intraerythrocytic development includes high fever where temperature of the host body raises up to 41°C. The rise in body temperature is associated with erythrocyte rupture and release of new merozoites. This high temperature can be a natural stimulus for Plasmodium to undergo programmed cell death process. In vitro studies demonstrated that the DNA fragmentation takes place in P. falciparum strain 3D7 after 2 h at 41°C (Oakley et al., 2007). We believe that Plasmodium must be equipped by the mechanism that can provide temperature tolerance for its survival.
If the parasite numbers increase beyond an optimal level, it may cause stress and death of the host. In this situation, parasite should adopt a strategy to regulate its own number. The induction of self-apoptosis by the Plasmodium may be one of the ways to achieve this goal. In vitro Plasmodium culture studies revealed that a growth arrested phenomenon through apoptosis occurs when the parasites undergo a certain percentage of parasitaemia. For example, highly synchronous culture of P. falciparum strains Dd2 and 3D7 exhibit cell apoptosis at >11% parasitaemia. Interestingly, these parasites continued their life cycle in the ring stages and failed to progress to the trophozoites and schizonts stages (Mutai and Waitumbi, 2010). This phenomenon of arresting self-development may also reveal a spontaneous judgment by the parasite to protect the host. Host environment also plays an important role in the regulation of Plasmodium development. The heme degraded product bilirubin is reported to induce apoptosis in Plasmodium. In vitro studies demonstrated that the addition of bilirubin to P. falciparum culture increased mRNA expression of apoptosis-related genes in the parasites (Kumar et al., 2008). This further suggests that regulating the phenomenon of self-apoptosis in Plasmodium might be a good way to control the disease progression in human host. This proposal demands further research in this area.
Conclusion
The interaction of Plasmodium with vertebrate and invertebrate systems is very complex. At different stages of malaria infection, the Plasmodium number is regulated by the host immune system or sometime it is a self-directed decision by the parasite. In addition, the program cell death in both the hosts also contributes to this phenomenon; however, Plasmodium is capable of suppressing this event to confer its successful development. Thus, the manipulation of apoptosis in Plasmodium or host cell might be a promising strategy to control the disease.
Author Contributions
PK and SS carried the literature survey and contributed to the initial draft. LG and SK wrote the manuscript with input from others. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
Authors would like to acknowledge the Department of Science and Technology (DST), Government of India for providing the fellowship to PK. This work was supported by the Department of Science and Technology (DST), Government of India (Grant number SR/SO/HS/131/2010).
References
Abraham, E. G., Pinto, S. B., Ghosh, A., Vanlandingham, D. L., Budd, A., Higgs, S., et al. (2005). An immune-responsive serpin, SRPN6, mediates mosquito defense against malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 102, 16327–16332. doi: 10.1073/pnas.0508335102
Ahmed, A. M., Baggott, S. L., Maingon, R., and Hurd, H. (2002). The costs of mounting an immune response are reflected in the reproductive fitness of the mosquito Anopheles gambiae. Oikos 97, 371–377. doi: 10.1034/j.1600-0706.2002.970307
Ahmed, A. M., and Hurd, H. (2006). Immune stimulation and malaria infection impose reproductive costs in Anopheles gambiae via follicular apoptosis. Microbes Infect. 8, 308–315. doi: 10.1016/j.micinf.2005.06.026
Ali, M., Al-Olayan, E. M., Lewis, S., Matthews, H., and Hurd, H. (2010). Naturally occurring triggers that induce apoptosis-like programmed cell death in Plasmodium berghei ookinetes. PLoS ONE 5:e12634. doi: 10.1371/journal.pone.0012634
Al-Olayan, E. M., Williams, G. T., and Hurd, H. (2002). Apoptosis in the malaria protozoan, Plasmodium berghei: a possible mechanism for limiting intensity of infection in the mosquito. Int. J. Parasitol. 32, 1133–1143. doi: 10.1016/S0020-7519(02)00087-5
Atkinson, S. C., Armistead, J. S., Mathias, D. K., Sandeu, M. M., Tao, D., Borhani-Dizaji, N., et al. (2015). The Anopheles-midgut APN1 structure reveals a new malaria transmission-blocking vaccine epitope. Nat. Struct. Mol. Biol. 22, 532–539. doi: 10.1038/nsmb.3048
Baton, L. A., and Ranford-Cartwright, L. C. (2004). Plasmodium falciparum ookinete invasion of the midgut epithelium of Anopheles stephensi is consistent with the Time Bomb model. Parasitology 129, 663–676. doi: 10.1017/S0031182004005979
Baton, L. A., and Ranford-Cartwright, L. C. (2005). How do malaria ookinetes cross the mosquito midgut wall? Trends Parasitol. 21, 22–28. doi: 10.1016/j.pt.2004.11.001
Beier, J. C. (1998). Malaria parasite development in mosquitoes. Annu. Rev. Entomol. 43, 519–543. doi: 10.1146/annurev.ento.43.1.519
Blandin, S., Shiao, S. H., Moita, L. F., Janse, C. J., Waters, A. P., Kafatos, F. C., et al. (2004). Complement-like protein TEP1 is a determinant of vectorial capacity in the malaria vector Anopheles gambiae. Cell 116, 661–670. doi: 10.1016/S0092-8674(04)00173-4
Blumberg, B. J., Trop, S., Das, S., and Dimopoulos, G. (2013). Bacteria- and IMD pathway-independent immune defenses against Plasmodium falciparum in Anopheles gambiae. PLoS ONE 8:e72130. doi: 10.1371/journal.pone.0072130
Boissière, A., Tchioffo, M. T., Bachar, D., Abate, L., Marie, A., Nsango, S. E., et al. (2012). Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog. 8:e1002742. doi: 10.1371/journal.ppat.1002742
Brand, V. B., Sandu, C. D., Duranton, C., Tanneur, V., Lang, K. S., Huber, S. M., et al. (2003). Dependence of Plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell. Physiol. Biochem. 13, 347–356. doi: 10.1159/000075122
Carrolo, M., Giordano, S., Cabrita-Santos, L., Corso, S., Vigario, A. M., Silva, S., et al. (2003). Hepatocyte growth factor and its receptor are required for malaria infection. Nat. Med. 9, 1363–1369. doi: 10.1038/nm947
Cha, S. J., Park, K., Srinivasan, P., Schindler, C. W., van Rooijen, N., Stins, M., et al. (2015). CD68 acts as a major gateway for malaria sporozoite liver infection. J. Exp. Med. 212, 1391–1403. doi: 10.1084/jem.20110575
Chose, O., Sarde, C. O., Gerbod, D., Viscogliosi, E., and Roseto, A. (2003). Programmed cell death in parasitic protozoans that lack mitochondria. Trends Parasitol. 19, 559–564. doi: 10.1016/j.pt.2003.09.016
Cliffe, L. J., Humphreys, N. E., Lane, T. E., Potten, C. S., Booth, C., and Grencis, R. K. (2005). Accelerated intestinal epithelial cell turnover: a new mechanism of parasite expulsion. Science 308, 1463–1465. doi: 10.1126/science.1108661
Cowman, A. F., Berry, D., and Baum, J. (2012). The cellular and molecular basis for malaria parasite invasion of the human red blood cell. J. Cell Biol. 198, 961–971. doi: 10.1083/jcb.201206112
Craig, A. G., Khairul, M. F., and Patil, P. R. (2012). Cytoadherence and severe malaria. Malays. J. Med. Sci. 19, 5–18.
Crampton, A., and Luckhart, S. (2001). The role of As60A, a TGF-beta homolog, in Anopheles stephensi innate immunity and defense against Plasmodium infection. Infect. Genet. Evol. 1, 131–141. doi: 10.1016/S1567-1348(01)00017
De Almeida Oliveira, G., Lieberman, J., and Barillas-Mury, C. (2012). Epithelial nitration by a Peroxidase/NOX5 system mediates mosquito antiplasmodial immunity. Science 335, 856–859. doi: 10.1126/science.1209678
Duranton, C., Huber, S., Tanneur, V., Lang, K., Brand, V., Sandu, C., et al. (2003). Electrophysiological properties of the Plasmodium falciparum-induced cation conductance of human erythrocytes. Cell. Physiol. Biochem. 13, 189–198. doi: 10.1159/000072421
Eda, S., and Sherman, I. W. (2002). Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell. Physiol. Biochem. 12, 373–384. doi: 10.1159/000067908
Elias, R. M., Correa-Costa, M., Barreto, C. R., Silva, R. C., Hayashida, C. Y., Castoldi, Â, et al. (2012). Oxidative stress and modification of renal vascular permeability are associated with acute kidney injury during P. berghei ANKA infection. PLoS ONE 7:e44004. doi: 10.1371/journal.pone.0044004
Foley, D. A. (1978). “Innate cellular defense by mosquito hemocytes,” in Comparative Pathobiology: Invertebrate Models for Biomedical Research, ed. L. A. Bulla Jr. (New York, NY: Plenum Press), 113–144.
Föller, M., Huber, S. M., and Lang, F. (2008). Erythrocyte programmed cell death. IUBMB Life 60, 661–668. doi: 10.1002/iub.106
Frevert, U. (2004). Sneaking in through the back entrance: the biology of malaria liver stages. Trends Parasitol. 20, 417–424. doi: 10.1016/j.pt.2004.07.007
Frevert, U., Engelmann, S., Zougbédé, S., Stange, J., Ng, B., Matuschewski, K., et al. (2005). Intravital observation of Plasmodium berghei sporozoite infection of the liver. PLoS Biol. 3:e192. doi: 10.1371/journal.pbio.0030192
Garver, L. S., Oliveira, G. A., and Barillas-Mury, C. (2013). The JNK pathway is a key mediator of Anopheles gambiae antiplasmodial immunity. PLoS Pathog. 9:e1003622. doi: 10.1371/journal.ppat.1003622
Ghosh, A. K., and Jacobs-Lorena, M. (2009). Plasmodium sporozoite invasion of the mosquito salivary gland. Curr. Opin. Microbiol. 12, 394–400. doi: 10.1016/j.mib.2009.06.010
Gupta, L., Kumar, S., Han, Y. S., Pimenta, P. F. P., and Barillas-Mury, C. (2005). Midgut epithelial responses of different mosquito–Plasmodium combinations: the actin cone zipper repair mechanism in Aedes aegypti. Proc. Natl. Acad. Sci. U.S.A. 102, 4010–4015. doi: 10.1073/pnas.0409642102
Gupta, L., Molina-Cruz, A., Kumar, S., Rodrigues, J., Dixit, R., Zamora, R. E., et al. (2009). The STAT pathway mediates late phase immunity against Plasmodium in the mosquito Anopheles gambiae. Cell Host Microbe 5, 498–507. doi: 10.1016/j.chom.2009.04.003
Han, Y. S., Thompson, J., Kafatos, F. C., and Barillas-Mury, C. (2000). Molecular interactions between Anopheles stephensi midgut cells and Plasmodium berghei: the time bomb theory of ookinete invasion of mosquitoes. EMBO J. 19, 6030–6040. doi: 10.1093/emboj/19.22.6030
Hernández-Martínez, S., Lanz, H., Rodríguez, M. H., González-Ceron, L., and Tsutsumi, V. (2002). Cellular-mediated reactions to foreign organisms inoculated into the hemocoel of Anopheles albimanus (Diptera: Culicidae). J. Med. Entomol. 39, 61–69. doi: 10.1603/0022-2585-39.1.61
Hillyer, J. F., Barreau, C., and Vernick, K. D. (2007). Efficiency of salivary gland invasion by malaria sporozoites is controlled by rapid sporozoite destruction in the mosquito hemocoel. Int. J. Parasitol. 37, 673–681. doi: 10.1016/j.ijpara.2006.12.00
Hillyer, J. F., Schmidt, S. L., and Christensen, B. M. (2003). Rapid phagocytosis and melanization of bacteria and Plasmodium sporozoites by hemocytes of the mosquito Aedes aegypti. J. Parasitol. 89, 62–69. doi: 10.1645/0022-3395(2003)089[0062:RPAMOB]2.0.CO;2
Hopwood, J. A., Ahmed, A. M., Polwart, A., Williams, G. T., and Hurd, H. (2001). Malaria-induced apoptosis in mosquito ovaries: a mechanism to control vector egg production. J. Exp. Biol. 204, 2773–2780.
Huber, S. M., Duranton, C., and Lang, F. (2005). Patch-clamp analysis of the “new permeability pathways” in malaria-infected erythrocytes. Int. Rev. Cytol. 246, 59–134. doi: 10.1016/S0074-7696(05)46003-9
Huh, C. G., Factor, V. M., Sanchez, A., Uchida, K., Conner, E. A., and Thorgeirsson, S. S. (2004). Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc. Natl. Acad. Sci. U.S.A. 101, 4477–4482. doi: 10.1073/pnas.0306068101
Hunt, N. H., and Grau, G. (2003). Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 24, 491–499. doi: 10.1016/S1471-4906(03)00229-1
Hurd, H. (2003). Manipulation of medically important insect vectors by their parasites. Annu. Rev. Entomol. 48, 141–161. doi: 10.1146/annurev.ento.48.091801.112722
Kajla, M., Gupta, K., Gupta, L., and Kumar, S. (2015a). A fine-tuned management between physiology and immunity maintains the gut microbiota in insects. Biochem. Physiol. 4, 182–190. doi: 10.4172/2168-9652.1000182
Kajla, M., Gupta, K., Kakani, P., Dhawan, R., Choudhury, T. P., Gupta, L., et al. (2015b). Identification of an Anopheles lineage-specific unique Heme Peroxidase HPX15: a plausible candidate for arresting malaria parasite development. J. Phylogenetics Evol. Biol. 3, 160. doi: 10.4172/2329-9002.1000160
Kang, M., Lisk, G., Hollingworth, S., Baylor, S. M., and Desai, S. A. (2005). Malaria parasites are rapidly killed by dantrolene derivatives specific for the plasmodial surface anion channel. Mol. Pharmacol. 68, 34–40.
Kaushansky, A., Douglass, A. N., Arang, N., Vigdorovich, V., Dambrauskas, N., Kain, H. S., et al. (2015). Malaria parasites target the hepatocyte receptor EphA2 for successful host infection. Science 350, 1089–1092. doi: 10.1126/science.aad3318
Kaushansky, A., Metzger, P. G., Douglass, A. N., Mikolajczak, S. A., Lakshmanan, V., Kain, H. S., et al. (2013). Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis. 4, e762. doi: 10.1038/cddis.2013.286
Kerr, J. F. R., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. doi: 10.1038/bjc.1972.33
Kim, J. M., Eckmann, L., Savidge, T. C., Lowe, D. C., Witthöft, T., and Kagnoff, M. F. (1998). Apoptosis of human intestinal epithelial cells after bacterial invasion. J. Clin. Invest. 102, 1815–1823. doi: 10.1172/JCI2466
Kinoshita, M., Uchida, T., Sato, A., Nakashima, M., Nakashima, H., Shono, S., et al. (2010). Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J. Hepatol. 53, 903–910. doi: 10.1016/j.jhep.2010.04.037
Klotz, C., and Frevert, U. (2008). Plasmodium yoelii sporozoites modulate cytokine profile and induce apoptosis in murine Kupffer cells. Int. J. Parasitol. 38, 1639–1650. doi: 10.1016/j.ijpara.2008.05.018
Koka, S., Lang, C., Boini, K. M., Bobbala, D., Huber, S. M., and Lang, F. (2008). Influence of chlorpromazine on eryptosis, parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 22, 261–268. doi: 10.1159/000149804
Kumar, S., and Barillas-Mury, C. (2005). Cell biology of Plasmodium falciparum midgut invasion of Anopheline mosquitoes. Am. J. Trop. Med. Hyg. 73, 153.
Kumar, S., Christophides, G. K., Cantera, R., Charles, B., Han, Y. S., Meister, S., et al. (2003). The role of reactive oxygen species on Plasmodium melanotic encapsulation in Anopheles gambiae. Proc. Natl. Acad. Sci. U.S.A. 100, 14139–14144. doi: 10.1073/pnas.2036262100
Kumar, S., Guha, M., Choubey, V., Maity, P., Srivastava, K., Puri, S. K., et al. (2008). Bilirubin inhibits Plasmodium falciparum growth through the generation of reactive oxygen species. Free Radic. Biol. Med. 44, 602–613. doi: 10.1016/j.freeradbiomed.2007.10.057
Kumar, S., Gupta, L., Han, Y. S., and Barillas-Mury, C. (2004). Inducible peroxidases mediate nitration of Anopheles midgut cells undergoing apoptosis in response to Plasmodium invasion. J. Biol. Chem. 279, 53475–53482. doi: 10.1074/jbc.M409905200
Kumar, S., Molina-Cruz, A., Gupta, L., Rodrigues, J., and Barillas-Mury, C. (2010). A peroxidase/dual oxidase system modulates midgut epithelial immunity in Anopheles gambiae. Science 327, 1644–1648. doi: 10.1126/science.1184008
Lang, F., Gulbins, E., Lerche, H., Huber, S. M., Kempe, D. S., and Foller, M. (2008). Eryptosis, a window to systemic disease. Cell. Physiol. Biochem. 22, 373–380. doi: 10.1159/000185448
Lang, P. A., Kasinathan, R. S., Brand, V. B., Duranton, C., Lang, C., Koka, S., et al. (2009). Accelerated clearance of Plasmodium-infected erythrocytes in sickle cell trait and annexin-A7 deficiency. Cell. Physiol. Biochem. 24, 415–428. doi: 10.1159/000257529
Lavazec, C., Boudin, C., Lacroix, R., Bonnet, S., Diop, A., Thiberge, S., et al. (2007). Carboxypeptidases B of Anopheles gambiae as targets for a Plasmodium falciparum transmission-blocking vaccine. Infect. Immun. 75, 1635–1642. doi: 10.1128/IAI.00864-06
Leiriao, P., Albuquerque, S. S., Corso, S., van Gemert, G. J., Sauerwein, R. W., Rodriguez, A., et al. (2005a). HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell. Microbiol. 7, 603–609. doi: 10.1111/j.1462-5822.2004.00490.x
Leiriao, P., Mota, M. M., and Rodriguez, A. (2005b). Apoptotic Plasmodium-infected hepatocytes provide antigens to liver dendritic cells. J. Infect. Dis. 191, 1576–1581. doi: 10.1086/429635
Lensen, A., Mulder, L., Tchuinkam, T., Willemsen, L., Eling, W., and Sauerwein, R. (1998). Mechanisms that reduce transmission of Plasmodium falciparum malaria in semi immune and non immune persons. J. Infect. Dis. 177, 1358–1363. doi: 10.1086/515263
Lensen, A. H., Bolmer-Van de Vegte, M., van Gemert, G. J., Eling, W. M., and Sauerwein, R. W. (1997). Leukocytes in a Plasmodium falciparum-infected blood meal reduce transmission of malaria to Anopheles mosquitoes. Infect. Immun. 65, 3834–3837.
Luckhart, S., Crampton, A. L., Zamora, R., Lieber, M. J., Dos Santos, P. C., Peterson, T. M. L., et al. (2003). Mammalian transforming growth factor β1 activated after ingestion by Anopheles stephensi modulates mosquito immunity. Infect. Immun. 71, 3000–3009. doi: 10.1128/IAI.71.6.3000-3009.2003
Luckhart, S., Vodovotz, Y., Cui, L., and Rosenberg, R. (1998). The mosquito Anopheles stephensi limits malaria parasite development with inducible synthesis of nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 95, 5700–5705. doi: 10.1073/pnas.95.10.5700
Michel, K., and Kafatos, F. C. (2005). Mosquito immunity against Plasmodium. Insect Biochem. Mol. Biol. 35, 677–689. doi: 10.1016/j.ibmb.2005.02.009
Minard, G., Mavingui, P., and Moro, C. V. (2013). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors 6, 146. doi: 10.1186/1756-3305-6-146
Moss, S. F., Calam, J., Agarwal, B., Wang, S., and Holt, P. R. (1996). Induction of gastric epithelial apoptosis by Helicobacter pylori. Gut 38, 498–501. doi: 10.1136/gut.38.4.498
Mota, M. M., Pradel, G., Vanderberg, J. P., Hafalla, J. C., Frevert, U., Nussenzweig, R. S., et al. (2001). Migration of Plasmodium sporozoites through cells before infection. Science 291, 141–144. doi: 10.1126/science.291.5501.141
Mueller, A.-K., Camargo, N., Kaiser, K., Andorfer, C., Frevert, U., Matuschewski, K., et al. (2005). Plasmodium liver stage developmental arrest by depletion of a protein at the parasite–host interface. Proc. Natl. Acad. Sci. U.S.A. 102, 3022–3027. doi: 10.1073/pnas.0408442102
Mueller, A.-K., Kohlhepp, F., Hammerschmidt, C., and Michel, K. (2010). Invasion of mosquito salivary glands by malaria parasites: prerequisites and defense strategies. Int. J. Parasitol. 40, 1229–1235. doi: 10.1016/j.ijpara.2010.05.005
Muniz-Junqueira, M. I., dos Santos-Neto, L. L., and Tosta, C. E. (2001). Influence of tumor necrosis factor-alpha on the ability of monocytes and lymphocytes to destroy intraerythrocytic Plasmodium falciparum in vitro. Cell. Immunol. 208, 73–79. doi: 10.1006/cimm.2001.1770
Mutai, B. K., and Waitumbi, J. N. (2010). Apoptosis stalks Plasmodium falciparum maintained in continuous culture condition. Malar. J. 9, S6. doi: 10.1186/1475-2875-9-S3-S6
Naotunne, T. S., Karunaweera, N. D., Mendis, K. N., and Carter, R. (1993). Cytokine-mediated inactivation of malarial gametocytes is dependent on the presence of white blood cells and involves reactive nitrogen intermediates. Immunology 78, 555–562.
N’Dilimabaka, N., Taoufiq, Z., Zougbédé, S., Bonnefoy, S., Lorthiois, A., Couraud, P. O., et al. (2014). P. falciparum isolate-specific distinct patterns of induced apoptosis in pulmonary and brain endothelial Cells. PLoS ONE. 9:e90692. doi: 10.1371/journal.pone.0090692
Oakley, M. S. M., Kumar, S., Anantharaman, V., Zheng, H., Mahajan, B., Haynes, J. D., et al. (2007). Molecular factors and biochemical pathways induced by febrile temperature in intraerythrocytic Plasmodium falciparum parasites. Infect. Immun. 75, 2012–2025. doi: 10.1128/IAI.01236-06
Park, J. E., Son, A. I., and Zhou, R. (2013). Roles of EphA2 in development and disease. Genes 4, 334–357. doi: 10.3390/genes4030334
Pino, P., Vouldoukis, I., Kolb, J. P., Mahmoudi, N., Desportes-Livage, I., Bricaire, F., et al. (2003). Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J. Infect. Dis. 187, 1283–1290. doi: 10.1086/373992
Pinto, S. B., Kafatos, F. C., and Michel, K. (2008). The parasite invasion marker SRPN6 reduces sporozoite numbers in salivary glands of Anopheles gambiae. Cell. Microbiol. 10, 891–898. doi: 10.1111/j.1462-5822.2007.01091.x
Prudêncio, M., Mota, M. M., and Mendes, A. M. (2011). A toolbox to study liver stage malaria. Trends Parasitol. 27, 565–574. doi: 10.1016/j.pt.2011.09.004
Ramiro, R. S., Alpedrinha, J., Carter, L., Gardner, A., and Reece, S. E. (2011). Sex and death: the effects of innate immune factors on the sexual reproduction of malaria parasites. PLoS Pathog. 7:e1001309. doi: 10.1371/journal.ppat.1001309
Ramphul, U. N., Garver, L. S., Molina-Cruz, A., Canepa, G. E., and Barillas-Mury, C. (2015). Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. U.S.A. 112, 1273–1280. doi: 10.1073/pnas.1423586112
Rennenberg, A., Lehmann, C., Heitmann, A., Witt, T., Hansen, G., Nagarajan, K., et al. (2010). Exoerythrocytic Plasmodium parasites secrete a cysteine protease inhibitor involved in sporozoite invasion and capable of blocking cell death of host hepatocytes. PLoS Pathog. 6:e1000825. doi: 10.1371/journal.ppat.1000825
Rosinski-Chupin, I., Briolay, J., Brouilly, P., Perrot, S., Gomez, S. M., Chertemps, T., et al. (2007). SAGE analysis of mosquito salivary gland transcriptomes during Plasmodium invasion. Cell. Microbiol. 9, 708–724. doi: 10.1111/j.1462-5822.2006.00822.x
Sakurai, T., Itoh, K., Liu, Y., Higashitsuji, H., Sumitomo, Y., Sakamaki, K., et al. (2005). Low temperature protects mammalian cells from apoptosis initiated by various stimuli in vitro. Exp. Cell Res. 309, 264–272. doi: 10.1016/j.yexcr.2005.06.002
Setty, B. N. Y., Kulkarni, S., and Stuart, M. J. (2002). Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 99, 1564–1571. doi: 10.1182/blood.V99.5.1564
Shaha, C. (2006). Apoptosis in Leishmania species and its relevance to disease pathogenesis. Indian J. Med. Res. 123, 233–244.
Simon, N., Lasonder, E., Scheuermayer, M., Kuehn, A., Tews, S., Fischer, R., et al. (2013). Malaria parasites co-opt human factor H to prevent complement-mediated lysis in the mosquito midgut. Cell Host Microbe 13, 29–41. doi: 10.1016/j.chom.2012.11.013
Sinai, A. P., Payne, T. M., Carmen, J. C., Hardi, L., Watson, S. J., and Molestina, R. E. (2004). Mechanisms underlying the manipulation of host apoptotic pathways by Toxoplasma gondii. Int. J. Parasitol. 34, 381–391. doi: 10.1016/j.ijpara.2003.11.009
Sinden, R. E. (2002). Molecular interactions between Plasmodium and its insect vectors. Cell. Microbiol. 4, 713–724. doi: 10.1046/j.1462-5822.2002.00229.x
Sinden, R. E., and Billingsley, P. F. (2001). Plasmodium invasion of mosquito cells: hawk or dove? Trends Parasitol. 17, 209–211. doi: 10.1016/S1471-4922(01)01928-6
Smith, R. C., Barillas-Mury, C., and Jacobs-Lorena, M. (2015). Hemocyte differentiation mediates the mosquito late-phase immune response against Plasmodium in Anopheles gambiae. Proc. Natl. Acad. Sci. U.S.A. 112, 3412–3420. doi: 10.1073/pnas.1420078112
Smith, R. C., Eappen, A. G., Radtke, A. J., and Jacobs-Lorena, M. (2012). Regulation of anti-Plasmodium immunity by a LITAF-like transcription factor in the malaria vector Anopheles gambiae. PLoS Pathog. 8:e1002965. doi: 10.1371/journal.ppat.1002965
Smith, R. C., Vega-Rodríguez, J., and Jacobs-Lorena, M. (2014). The Plasmodium bottleneck: malaria parasite losses in the mosquito vector. Mem. Inst. Oswaldo Cruz 109, 644–661. doi: 10.1590/0074-0276130597
Sturm, A., Amino, R., van de Sand, C., Regen, T., Retzlaff, S., Rennenberg, A., et al. (2006). Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 313, 1287–1290. doi: 10.1126/science.1129720
Tanneur, V., Duranton, C., Brand, V. B., Sandu, C. D., Akkaya, C., Kasinathan, R. S., et al. (2006). Purinoceptors are involved in the induction of an osmolyte permeability in malaria-infected and oxidized human erythrocytes. FASEB J. 20, 133–135. doi: 10.1096/fj.04-3371fje
Tavares, J., Formaglio, P., Thiberge, S., Mordelet, E., Van Rooijen, N., Medvinsky, A., et al. (2013). Role of host cell traversal by the malaria sporozoite during liver infection. J. Exp. Med. 210, 905–915. doi: 10.1084/jem.20121130
Totino, P. R., Magalhães, A. D., Silva, L. A., Banic, D. M., Daniel-Ribeiro, C. T., Ferreira-da-Cruz, M., et al. (2010). Apoptosis of non-parasitized red blood cells in malaria: a putative mechanism involved in the pathogenesis of anaemia. Malar. J. 9, 350. doi: 10.1186/1475-2875-9-350
Totino, P. R. R., Pinna, R. A., De-Oliveira, A. C. A. X., Banic, D. M., Daniel-Ribeiro, C. T., Ferreira-da-Cruz, M., et al. (2013). Apoptosis of non-parasitised red blood cells in Plasmodium yoelii malaria. Mem. Inst. Oswaldo Cruz 108, 686–690. doi: 10.1590/0074-0276108062013003
Tripathi, A. K., Sullivan, D. J., and Stins, M. F. (2007). Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J. Infect. Dis. 195, 942–950. doi: 10.1086/512083
Usynin, I., Klotz, C., and Frevert, U. (2007). Malaria circumsporozoite protein inhibits the respiratory burst in Kupffer cells. Cell. Microbiol. 9, 2610–2628. doi: 10.1111/j.1462-5822.2007.00982
Van Dijk, M. R., Douradinha, B., Franke-Fayard, B., Heussler, V., van Dooren, M. W., van Schaijk, B., et al. (2005). Genetically attenuated, P36p-deficient malarial sporozoites induce protective immunity and apoptosis of infected liver cells. Proc. Natl. Acad. Sci. U.S.A. 102, 12194–12199. doi: 10.1073/pnas.0500925102
Van Zandbergen, G., Bollinger, A., Wenzel, A., Kamhawi, S., Voll, R., Klinger, M., et al. (2006). Leishmania disease development depends on the presence of apoptotic promastigotes in the virulent inoculum. Proc. Natl. Acad. Sci. U.S.A. 103, 13837–13842. doi: 10.1073/pnas.0600843103
Wennicke, K., Debierre-Grockiego, F., Wichmann, D., Brattig, N. W., Pankuweit, S., Maisch, B., et al. (2008). Glycosylphosphatidylinositol-induced cardiac myocyte death might contribute to the fatal outcome of Plasmodium falciparum malaria. Apoptosis 13, 857–866. doi: 10.1007/s10495-008-0217-6
Williams, G. T. (1994). Programmed cell death: a fundamental protective response to pathogens. Trends Microbiol. 2, 463–464. doi: 10.1016/0966-842X(94)90648-3
Wilson, N. O., Huang, M.-B., Anderson, W., Bond, V., Powell, M., Thompson, W. E., et al. (2008). Soluble factors from Plasmodium falciparum-infected erythrocytes induce apoptosis in human brain vascular endothelial and neuroglia cells. Mol. Biochem. Parasitol. 162, 172–176. doi: 10.1016/j.molbiopara.2008.09.003
World Health Organization [WHO] (2014). “World Malaria Report: 2014,” in WHO Global Malaria Programme. Geneva: World Health Organization.
Zheng, L. (1997). Molecular approaches to mosquito parasite interactions. Arch. Insect Biochem. Physiol. 34, 1–18. doi: 10.1002/(SICI)1520-6327(1997)34:1<1::AID-ARCH1>3.3.CO;2-R
Keywords: cell apoptosis, Plasmodium, mosquito, midgut epithelium, hepatocytes, erythrocytes
Citation: Kakani P, Suman S, Gupta L and Kumar S (2016) Ambivalent Outcomes of Cell Apoptosis: A Barrier or Blessing in Malaria Progression. Front. Microbiol. 7:302. doi: 10.3389/fmicb.2016.00302
Received: 19 December 2015; Accepted: 24 February 2016;
Published: 15 March 2016.
Edited by:
Alexandre Morrot, Federal University of Rio de Janeiro, BrazilReviewed by:
Giampietro Corradin, University of Lausanne, SwitzerlandWai-Hong Tham, The Walter and Eliza Hall Institute, Australia
Copyright © 2016 Kakani, Suman, Gupta and Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjeev Kumar, c2FuamVldkBwaWxhbmkuYml0cy1waWxhbmkuYWMuaW4=