 Shuai Liu
Shuai Liu Xiaosu Zhou1†
Xiaosu Zhou1† Qijun Chen
Qijun Chen- 1MOH Key Laboratory of Systems Biology of Pathogens, Institute of Pathogen Biology, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2College of Life Science and Technology, Southwest University for Nationalities, Chengdu, China
- 3Key Laboratory of Zoonosis, Shenyang Agriculture University, Shenyang, China
Alternative splicing (AS), as one of the most important topics in the post-genomic era, has been extensively studied in numerous organisms. However, little is known about the prevalence and characteristics of AS in Echinococcus species, which can cause significant health problems to humans and domestic animals. Based on high-throughput RNA-sequencing data, we performed a genome-wide survey of AS in two major pathogens of echinococcosis-Echinococcus granulosus and Echinococcus multilocularis. Our study revealed that the prevalence and characteristics of AS in protoscoleces of the two parasites were generally consistent with each other. A total of 6,826 AS events from 3,774 E. granulosus genes and 6,644 AS events from 3,611 E. multilocularis genes were identified in protoscolex transcriptomes, indicating that 33–36% of genes were subject to AS in the two parasites. Strikingly, intron retention instead of exon skipping was the predominant type of AS in Echinococcus species. Moreover, analysis of the Kyoto Encyclopedia of Genes and Genomes pathway indicated that genes that underwent AS events were significantly enriched in multiple pathways mainly related to metabolism (e.g., purine, fatty acid, galactose, and glycerolipid metabolism), signal transduction (e.g., Jak-STAT, VEGF, Notch, and GnRH signaling pathways), and genetic information processing (e.g., RNA transport and mRNA surveillance pathways). The landscape of AS obtained in this study will not only facilitate future investigations on transcriptome complexity and AS regulation during the life cycle of Echinococcus species, but also provide an invaluable resource for future functional and evolutionary studies of AS in platyhelminth parasites.
Introduction
Cystic echinococcosis and alveolar echinococcosis are potentially fatal chronic and progressive zoonoses affecting humans and domestic animals (Eckert and Deplazes, 2004; Craig and Larrieu, 2006). These diseases are caused by the metacestode larval stage of two related cestodes, Echinococcus granulosus and Echinococcus multilocularis. The life cycles of both of the parasitic cestodes require intermediate hosts (wild or domesticated ungulates, rodents, and humans are the intermediate hosts for larval parasites), and definitive hosts (domestic dogs, foxes, and other wild carnivores are the definitive hosts for adult parasites). The intermediate hosts are infected by the oral uptake of eggs, which contain oncospheres. Once the eggs are activated within the host’s digestive system, the oncospheres hatch from the eggs, penetrate the intestinal wall, and are carried by the bloodstream to the liver, lungs, or other organs, turning into hydatid cysts filled with fluid and protoscoleces. Protoscoleces, as the key infectious component of the larval stages, can interact with both definitive and intermediate hosts. They can asexually proliferate in intermediate hosts and mature into adult parasites when the hydatid cysts are swallowed by the definitive host (McManus et al., 2003). The parasites have exploited daedal strategies to adapt to the distinct environments of different hosts and to evade host immune responses, yet the concrete mechanisms of these strategies remain poorly understood.
Alternative splicing, as an essential characteristic of eukaryotic gene expression, is a post-transcriptional process that enables one gene to encode two or more mature mRNAs (Nilsen and Graveley, 2010). It plays a key role in the transmission of genetic information from DNA to proteins by expanding the coding capacity of genomes (Naftelberg et al., 2015). Alternative splice products may generate proteins with different structures, sub-cellular localizations, and molecular functions, some of which are putative targets for vaccines or drugs (Keren et al., 2010; Yeoh et al., 2015). Regulation of AS is as important as regulation of transcription to determine cell- and tissue-specific features, normal cell functioning, and responses of eukaryotic cells to external cues (Kornblihtt et al., 2013; Naftelberg et al., 2015). For the parasite, AS may play important roles in the host–parasite interplay through generating alternative isoforms with different functions (Hull and Dlamini, 2014). The discovery of AS giving rise to different isoforms of antigenic proteins seems to be indicative of a further means of immune evasion by the parasite (DeMarco et al., 2010; Hull and Dlamini, 2014).
As one of the next-generation sequencing approaches, RNA-Seq has been applied in numerous studies that have altered our view of the extent and complexity of eukaryotic transcriptomes (Wang et al., 2009; Marguerat and Bahler, 2010). This technique enables researchers to survey the entire transcriptome in a very high-throughput and quantitative manner, which provides inviting information about transcriptional and post-transcriptional gene regulation, and gives unique insight into the variety of transcript structures and processing on a global scale (Mortazavi et al., 2008; Sultan et al., 2008). In particular, RNA-Seq data can be directly used to identify AS events in transcriptomes by searching for sequence reads that span splice junctions, which has dramatically enriched our knowledge of AS in the eukaryotic kingdom (Martin and Wang, 2011; Kornblihtt et al., 2013; Staiger and Brown, 2013; Hull and Dlamini, 2014; Filichkin et al., 2015). Recently, the decoding and availability of the genome sequences of two Echinococcus species has proved pivotal for the systematic dissection of the biology of the parasites (Tsai et al., 2013; Zheng et al., 2013). The main obstacle lies in that a precise estimate of AS events cannot be obtained merely based on genomic sequence analysis. As far as we know, deep-sequencing research explicitly related to AS in Echinococcus species has not been conducted, even though a few studies focusing on transcriptome profile analyses of E. granulosus (Pan et al., 2014) or E. multilocularis (Huang et al., 2016) have been reported using next-generation sequencing approaches.
In the present study, we have investigated AS events in protoscolex transcriptomes of both E. granulosus and E. multilocularis using a high-throughput RNA-Seq approach. We identified 1000s of AS events in the two parasites, which can be classified into seven splicing patterns. Interestingly, intron retention was found to be the predominant splicing type in protoscoleces. This provides a valuable basis for a more precise, dynamic understanding of the transcriptome in Echinococcus species.
Materials and Methods
Ethics Statement
All procedures performed on animals in this study were conducted following the animal husbandry guidelines of the Chinese Academy of Medical Sciences and with permission from the Experimental Animal Committee of the Chinese Academy of Medical Sciences with the Ethical Clearance Number IPB-2011-8.
Parasites
All parasite materials were collected from the Xinjiang Uyghur Autonomous Region, China. For E. granulosus transcriptome sequencing, a unilocular cyst was isolated from a sheep liver infected with E. granulosus. After rinsing 10 times with normal saline, protoscoleces were aspirated from the hydatid cysts, washed three times with normal saline and then soaked in RNAlater solution (Ambion, CA, United States); samples were stored at -80°C until total RNA was isolated. Hydatid cysts of E. multilocularis were freshly isolated from male gerbils at 5–6 months after infection with E. multilocularis. After rinsing with normal saline, the cysts were opened and homogenized to a smooth consistency in 200 ml normal saline. The suspension was successively passed through 40, 80, 140, and 325 mesh metal sieves. The protoscoleces on the 325 mesh metal sieve were collected into a beaker and were washed five times with ice-cold normal saline. The storage method was as above.
RNA Extraction and Deep Sequencing of mRNA
For mRNA library construction and deep sequencing, total RNA was isolated using an RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Potential contaminating genomic DNA was removed from RNA samples using a Turbo DNA-free kit (Ambion, CA, United States). The quantity and integrity of the total RNA was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, United States). Normalized starting quantities of total RNA were used to prepare two biological replicates of Illumina sequencing libraries for each species using a TruSeqTM RNA sample preparation kit (Illumina, San Diego, CA, United States). Following the manufacturer’s instructions, libraries were constructed and sequenced using a paired-end read protocol with 100 bp of data collected per run on an Illumina HiSeqTM 2000 instrument housed at MininGene Biotechnology Co. Ltd, Beijing, China. Data analyses and base calling were performed using the Illumina instrument software.
mRNA Transcriptome Data Processing and Bioinformatic Analyses
A Perl program was written to remove low-quality reads (more than half of the bases had a quality value less than 5). The retained high-quality reads were mapped to the published genomes of E. granulosus (Tsai et al., 2013; Zheng et al., 2013) and E. multilocularis (Tsai et al., 2013) by HISAT (Kim et al., 2015), and then assembled with StringTie (Pertea et al., 2015) to construct unique transcript sequences, using the parameters: -g -b -u -o. AS events in a gene were detected by SUPPA (Alamancos et al., 2015). Cuffquant (Trapnell et al., 2010) was used to quantify the expression of genes and transcripts, and the raw expression values were normalized by Cuffnorm. The expression values were calculated for each sample based on the number of fragments per kilobase of exon per million reads mapped. InterPro domain information (Mulder et al., 2003) was annotated by InterProScan (release 53.0) (Zdobnov and Apweiler, 2001) and functional assignments mapped with GO (Harris et al., 2004). WEGO (v3.3) (Ye et al., 2006) was used for GO classification. To obtain an overview of the gene pathway networks, KEGG analysis was performed by comparing the transcripts with the KEGG database (release 58) (Kanehisa et al., 2006) using BLASTX (Altschul et al., 1997; Ouyang et al., 2007) at E-values ≤ 1E-10. A Perl script was used to retrieve KO information from the BLAST results, and then establish pathway associations between the transcripts and database. A Perl script was written to count the number of total genes and the number of AS genes in a certain functional pathway, then a significant score P-value was calculated using the R scripts under hypergeometric distribution. A function pathway with P-value < 0.05 was deemed as enrichment in AS genes.
Verification of AS Events by RT-PCR
To validate AS events experimentally, RT-PCR was performed for eight pairs of homologous genes that underwent different AS types in the two parasites. For each sample, 1 μg total RNA was reverse transcribed into first-strand cDNA using a SuperScript III Reverse Transcriptase kit (Invitrogen, Carlsbad, CA, United States) with oligo(dT)12-18 primer. Primers were designed based on the consensus gene sequences of the two parasites, and the primer sequences are listed in Supplementary Table S1. PCR was performed in a 25 μl reaction system using High-Fidelity PCR Master Mix (NEB) and the procedure was as follows: initial denaturation at 98°C for 1 min; 98°C for 10 s, 60°C for 30 s, and 72°C for 30 s, for 35 cycles; and a final extension at 72°C for 5 min. The PCR products were visualized by 1.5% agarose gel electrophoresis analysis.
Identification and Phylogenetic Analysis of the Echinococcus Venom-Allergen-Like Protein (VAL) Gene Family
Using protein sequences of Schistosoma mansoni VALs (Chalmers et al., 2008) as query sequences, the initial protein sequences of Echinococcus VALs were retrieved from the non-redundant protein sequence database of the National Center for Biotechnology Information by BLASTP search (E-value cut-off: 10-5). CD-HIT v4.5.4 software1 was then used to eliminate redundant protein sequences. All remaining protein sequences were further examined for the presence of SCP/TAPS-representative protein domains by searching the Conserved Domain Database (Marchler-Bauer et al., 2011) of the National Center for Biotechnology Information. A phylogenetic relationship tree was built using the full-length amino acid sequences of Echinococcus VALs in the following steps. First, sequences were aligned using ClustalX (Larkin et al., 2007), then they were refined manually, and a phylogenetic tree was finally generated using MEGA 5.0 software (Tamura et al., 2011) by the neighbor-joining method (the bootstrap test was performed with 1000 replicates).
Results
Transcriptome Sequencing and Assembly
In this study, a total of 43,509,848 and 44,379,588 high-quality reads (100 bp/read) were obtained from worms of E. granulosus and E. multilocularis, respectively. Low-quality sequences were removed by a Perl program, and the retained high-quality reads were mapped to the published genomes of E. granulosus and E. multilocularis to construct unique transcript sequences. A total of 34,717,856 reads (79.79%) mapped to the E. granulosus genome reported by Tsai et al. (2013), 31,156,736 reads (71.61%) mapped to the E. granulosus genome reported by Zheng et al. (2013), and 38,882,179 reads (87.61%) mapped to the E. multilocularis genome (Tsai et al., 2013) (Table 1). Therefore, all the subsequent assemblies of reads and bioinformatics analyses, such as gene annotation and AS events, were based on the first mapping results for E. granulosus. Using the software StringTie, 24,550 (7,925 known and 16,625 novel transcripts) and 23,771 transcripts (8,432 known and 15,339 novel transcripts) were assembled for E. granulosus and E. multilocularis, respectively, and the assembly yielded 11,330 genes (6,815 known and 4,515 novel genes) for E. granulosus and 10,101 genes (7,051 known and 3,050 novel genes) for E. multilocularis (Table 1 and Supplementary Tables S2, S3), compared with the reference genome data. The mean length of transcripts for the two parasites was 2016 and 2086 bp, respectively (Table 1). All sequence data has been deposited in the GEO database2 with an accession number of GSE59173.
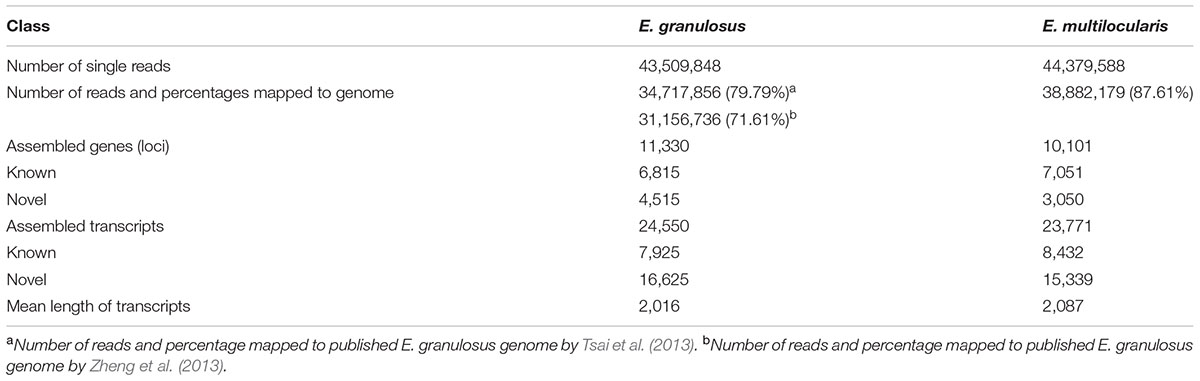
TABLE 1. Summary data of the transcriptome analysis.
The sequence reads could be classified into four types: exon, intron, intergenic, and spliced. The proportions of the four sequence types in the E. granulosus transcriptome were 56% (exons), 4% (introns), 15% (intergenic), and 25% (spliced) (Figure 1). The percentages of exons and introns in the E. multilocularis transcriptome were 55 and 3%, which is similar to the E. granulosus transcriptome; while the proportions of intergenic and spliced transcripts in the E. multilocularis transcriptome were 24 and 18%, which is significantly different from the proportions in the E. granulosus transcriptome (Figure 1). In our transcriptome database, the total exon lengths for E. granulosus and E. multilocularis were 22,249,494 bp (19.80% of the genome) and 21,805,251 bp (19.17% of the genome), which were longer than the total exon lengths in the reference genome databases for E. granulosus (15,213,358 bp, 13.54%) and E. multilocularis (16,112,293 bp, 14.16%) (Tsai et al., 2013).
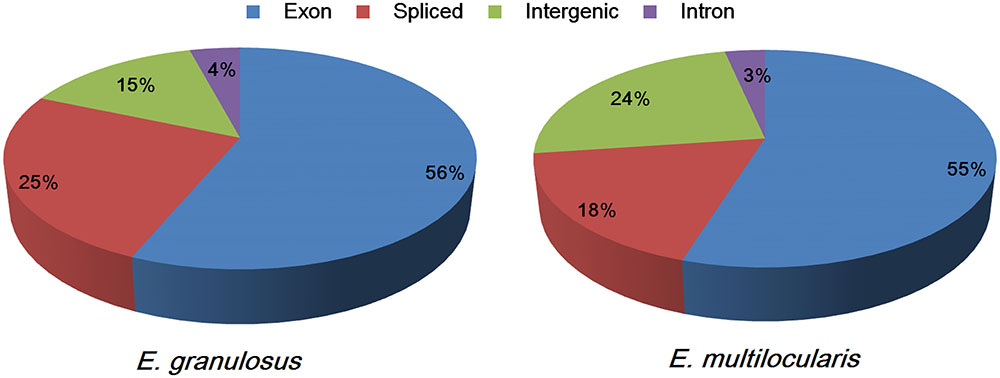
FIGURE 1. Proportions of the four types of sequence reads mapping to the genomes of E. granulosus and E. multilocularis. The percentages of exon (the highest) and intron (the lowest) sequences were similar between the two parasites, whereas the percentages of spliced and intergenic sequences were significantly different between the two parasites.
A Large Number of AS Events Were Identified in E. granulosus and E. multilocularis
A total of 6,826 AS events from 3,774 E. granulosus genes (33.31%) and 6,644 AS events in 3,611 E. multilocularis genes (35.75%) were bioinformatically predicted (Figure 2A and Supplementary Tables S4, S5). These AS events could be classified into seven types: intron retention, exon skipping, alternative donor site, alternative acceptor site, alternative first exon, alternative last exon, and mutually exclusive exon. The results showed that the proportions of the seven AS types were similar in E. granulosus and E. multilocularis. Intron retention (39 and 42%) was the most common AS type, followed by alternative donor site (21 and 17%), alternative acceptor site (17 and 18%), exon skipping (both 16%), and the least common splicing forms were alternative first exon (both 4%), alternative last exon (both 2%), and mutually exclusive exon (both 1%) in the two parasites, E. granulosus and E. multilocularis, respectively (Figure 2B and Supplementary Tables S4, S5).
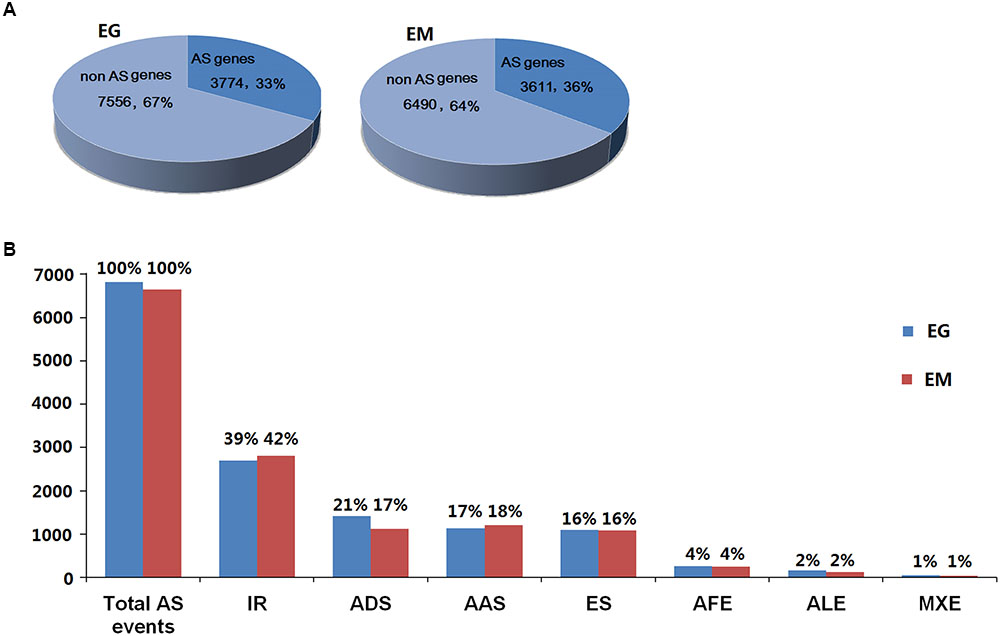
FIGURE 2. General information about AS events in E. granulosus and E. multilocularis. (A) Prevalence of AS in protoscolex transcriptomes of the two parasites. (B) Proportions of the seven types of AS in protoscolex transcriptomes of the two parasites. EG, E. granulosus; EM, E. multilocularis; IR, intron retention; ADS, alternative donor site; AAS, alternative acceptor site; ES, exon skipping; AFE, alternative first exon; ALE, alternative last exon; MXE, mutually exclusive exon.
A Panel of AS Events Were Validated by RT-PCR
To validate the AS events predicted by bioinformatic analysis, RT-PCR was carried out on eight pairs of homologous genes producing different types of AS events in the two parasites. Four pairs of homologous genes with intron-retention events were serine/arginine-rich splicing factor (EG.4660 and EM.3827), SNARE protein Sec22 (EG.5720 and EM.2557), hypothetical protein (EG.3709 and EM.5622), and hypothetical protein (EG.5720 and EM.2557). The other four pairs of homologous genes with exon skipping events were peroxidasin (EG.9768 and EM.6188), filamin (EG.8983 and EM.6048), adducin-related protein (EG.7075 and EM.1228), and heat shock protein 70 (EG.583 and EM.6781). Agarose gel electrophoresis showed that bands corresponding with the predicted AS events could be detected in the RT-PCR products (Figures 3, 4). Notably, some extra bands detected in the RT-PCR products were proved to be additional AS events from the corresponding genes by Sanger sequencing method (Figures 3, 4). The results were indicative that the bioinformatic prediction based on the primary sequencing data was reliable, and additional AS events for some genes, which could not be sequenced nor predicted in the present study, also occurred in the two parasites.
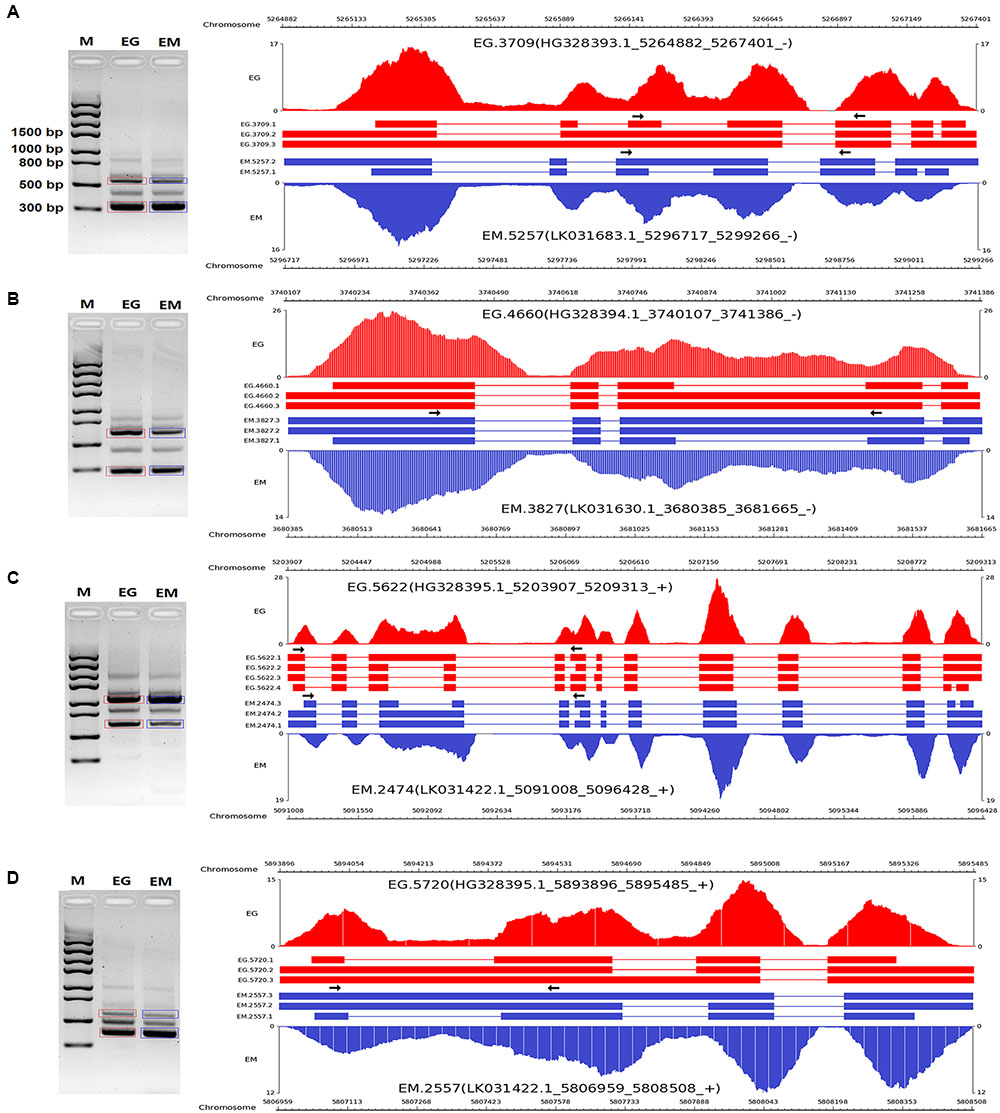
FIGURE 3. RT-PCR validation of intron-retention events in E. granulosus and E. multilocularis. A subset of intron-retention events (A–D) in protoscoleces were confirmed by RT-PCR. Paired arrows in the gene frames indicate where primers were designed for each AS event. RT-PCR products were visualized with 1.5% agarose gels. Electrophoretic bands corresponding with predicted AS events in E. granulosus and E. multilocularis are marked by red and blue boxes, respectively. M, marker; EG, E. granulosus; EM, E. multilocularis.
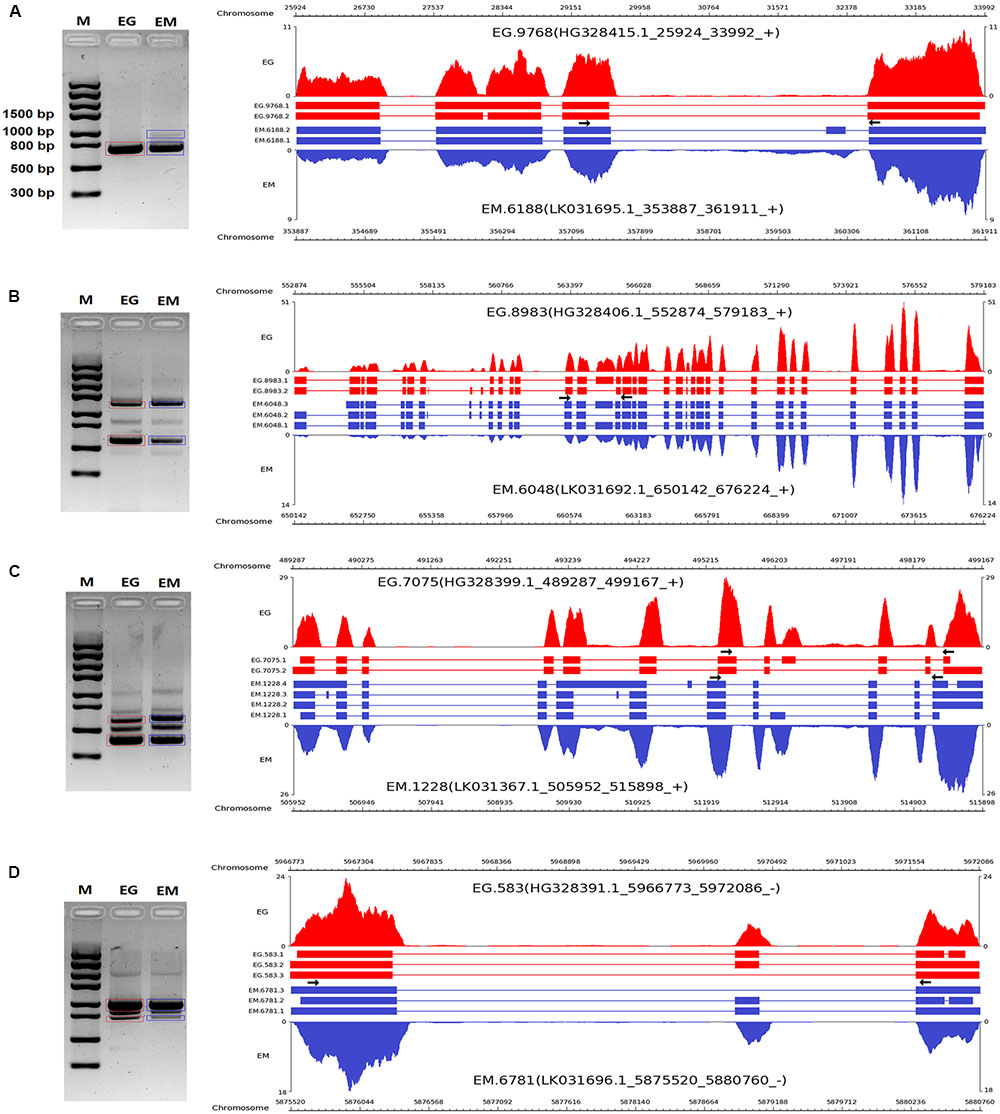
FIGURE 4. RT-PCR validation of exon skipping events in E. granulosus and E. multilocularis. A subset of four exon skipping events (A–D) in protoscoleces were confirmed by RT-PCR. Paired arrows in the gene frames indicate where primers were designed for each AS event. RT-PCR products were visualized with 1.5% agarose gels. Electrophoretic bands corresponding with predicted AS events in E. granulosus and E. multilocularis are marked by red and blue boxes, respectively. M, marker; EG, E. granulosus; EM, E. multilocularis.
GO and KEGG Pathway Analyses of AS Genes
Gene Ontology analysis was performed to summarize and explore the functional categories of the genes that underwent AS events in the two parasites. A total of 2,364 AS genes of E. granulosus and 2,376 AS genes of E. multilocularis were annotated for different GO terms (Supplementary Tables S3, S4). The results showed that the proportions of enriched GO terms for AS genes were globally similar between E. granulosus and E. multilocularis based on biological process (Figure 5A), molecular function (Figure 5B), and cellular component (Figure 5C). For biological processes, genes with AS events were predominantly enriched in GO terms that were relevant to cellular process and metabolic process. For the molecular function category, the majority of AS genes were annotated with the term binding, followed by the term catalytic activity.
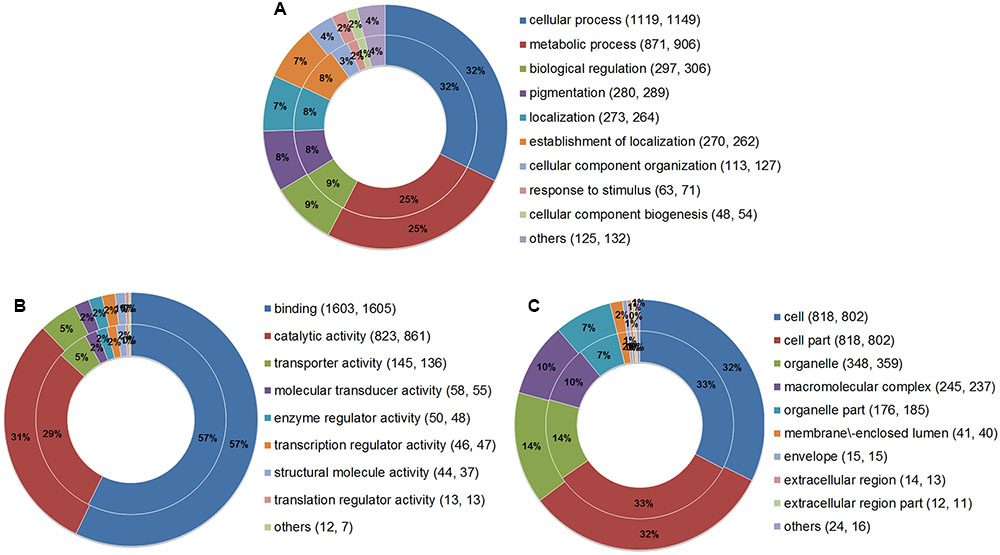
FIGURE 5. Comparative GO analysis of genes with AS events between E. granulosus and E. multilocularis. GO terms were summarized according to three categories: biological process (A), molecular function (B), and cellular component (C). The outer ring represents AS genes of E. multilocularis, and the inner ring represents AS genes of E. granulosus. Numbers in brackets represent the AS genes of E. granulosus and E. multilocularis falling into the corresponding GO terms.
Moreover, 1,099 and 1,115 AS genes were annotated for KEGG pathways. The enrichment pathway analysis showed that these genes were significantly enriched in 26 pathways relating to cellular processes, environmental information processing, genetic information processing, metabolism, and organismal systems (P < 0.05, Table 2). For environmental information processing, the AS genes were mainly enriched in the phosphatidylinositol signaling system, cellular antigens, and Jak-STAT, VEGF, and Notch signaling pathways, suggesting that AS may play important roles in the response to host environmental stress. For metabolism, the most significantly enriched pathways were involved in purine, fatty acid, galactose, glycerolipid, or N-glycan metabolism or biosynthesis. For genetic information processing, the enriched AS genes were grouped mainly in basal transcription factors, RNA transport, and the mRNA surveillance pathway. For organismal systems, the significant enrichment of AS genes included those relating to development (osteoclast differentiation), the immune system (Fc epsilon RI, T cell receptor, and Toll-like receptor signaling pathway), and the endocrine system (GnRH signaling pathway) (Table 2). Further experimental verification is necessary to clarify the biological regulation mechanisms of these AS events in Echinococcus species.
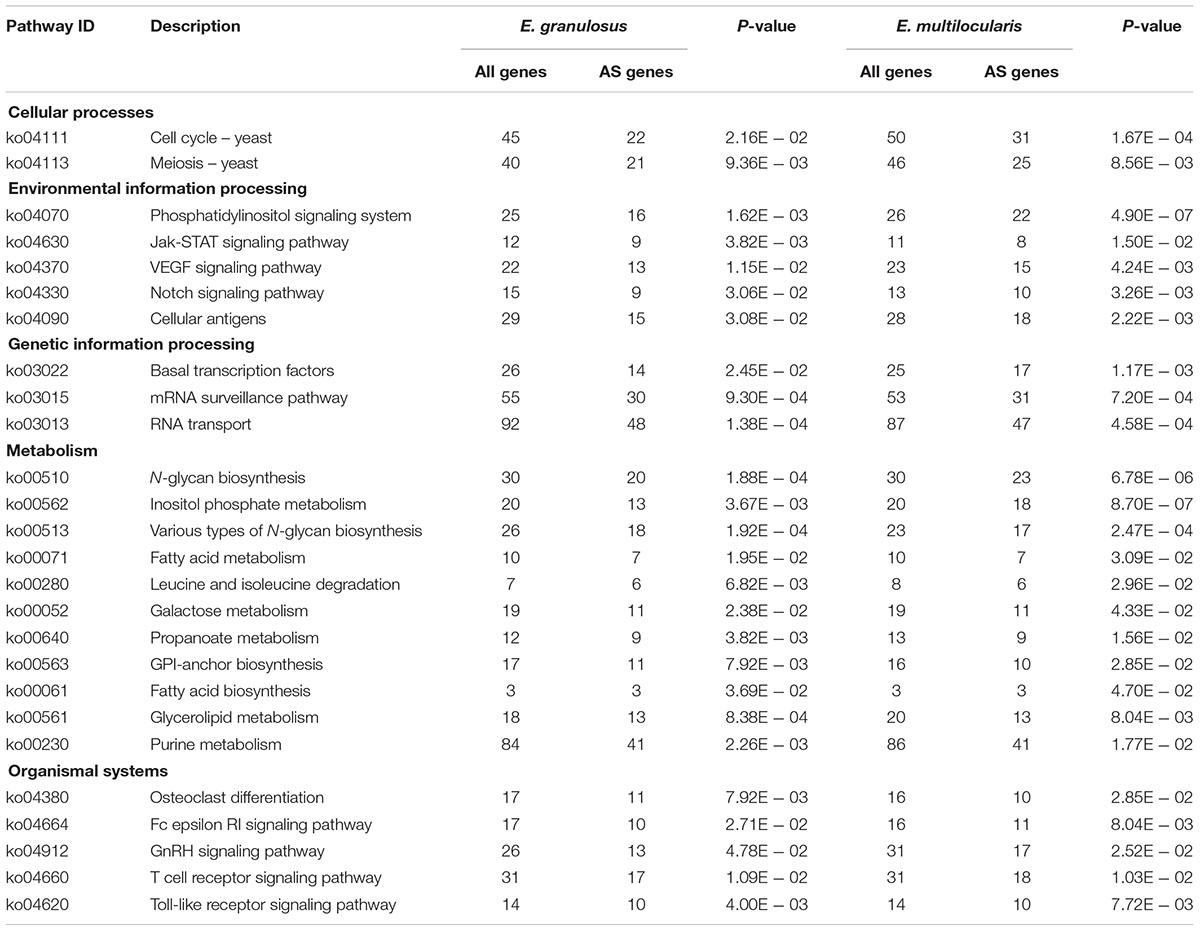
TABLE 2. Pathway enrichment analysis of AS genes in E. granulosus and E. multilocularis.
Sequence Conservation Analysis of Echinococcus AS Genes
Using AS gene sequences of E. multilocularis as the query database, we performed a BLASTX search to retrieve homologous genes in the following five organisms: E. granulosus, S. japonicum, S. mansoni, Caenorhabditis elegan, and Homo sapiens. A total of 3389 of E. multilocularis AS genes were identified as having homologous genes in the five organisms. Venn diagram analysis showed that there were three major groups of E. multilocularis AS genes: group I (conserved genes, 158 genes) (Figure 6A and Supplementary Table S6), for which homologous genes could be retrieved in all five organisms; group II (Platyhelminthes genes, 300 genes) (Figure 6A and Supplementary Table S7), for which homologous genes could only be retrieved in E. granulosus, S. japonicum, and S. mansoni; and group III (Cestoidea genes, 2360 genes) (Figure 6A and Supplementary Table S8), for which homologous genes could only be retrieved in E. granulosus. Moreover, the percentages of E. granulosus genes with AS events from the three groups were compared to investigate whether AS events were related to their gene sequence conservation. In group I, 63.3% of E. granulosus homologs with AS events were identified, which was lower than the percentages for group II (66.7%), and group III (67.8%) (Figure 6B). Meanwhile, we compared the proportions of S. japonicum genes with AS events from group I and group II using our published S. japonicum transcriptome data (Piao et al., 2014). The percentage of S. japonicum homologs with AS events in group I (67.1%) was lower than the percentage in group II (73.0%) (Figure 6C). The representative homologous gene pairs with AS events from the three groups are listed in Table 3.
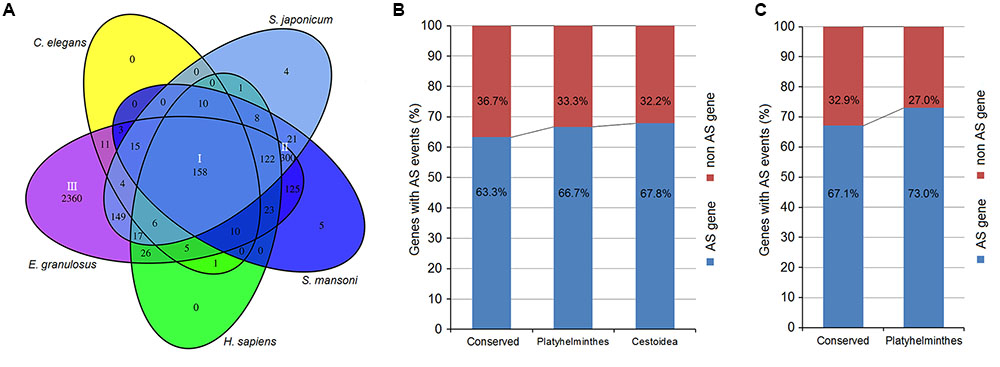
FIGURE 6. Sequence conservation analysis of Echinococcus AS genes. (A) Venn diagram of Echinococcus AS genes with homologous genes in the five organisms. (B) The percentages of E. granulosus genes with AS events from the three groups. (C) The percentages of S. japonicum genes with AS events from groups I and II.
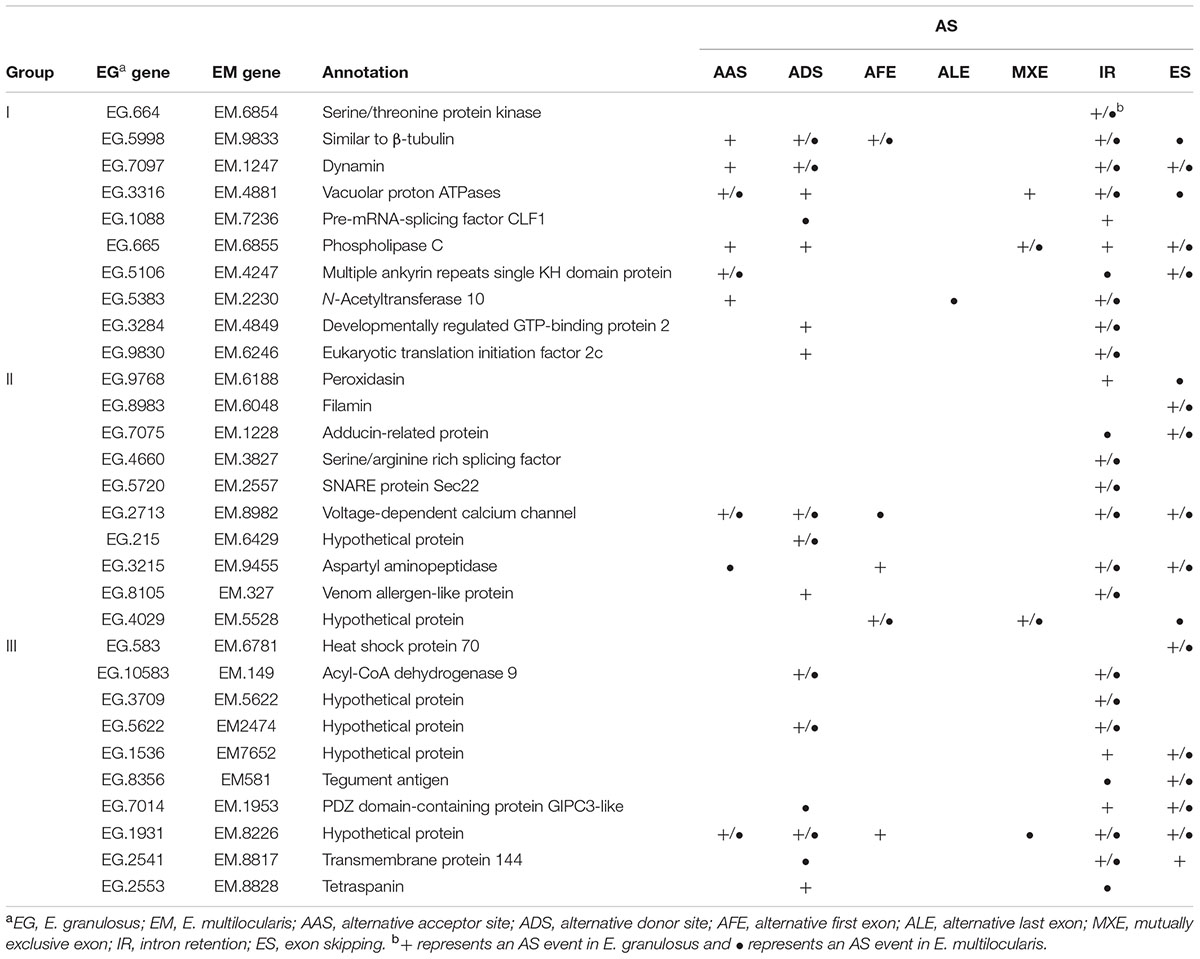
TABLE 3. Representative homologous gene pairs with AS events from groups I, II, and III.
Phylogenetic Relationship and AS Analyses of the Echinococcus VAL Gene Family
The VAL genes are members of the sperm-coating protein/Tpx-1/Ag5/PR-1/Sc7 (SCP/TAPS) superfamily, which may be important for chronic host/parasite interactions (Chalmers et al., 2008; Farias et al., 2012). It has been found that members of the VAL family exhibit multiple expression patterns and AS in the life cycle of S. mansoni (Chalmers et al., 2008) and S. japonicum (Liu et al., 2015). Based on the genome databases of the two species, we identified 22 E. granulosus VALs and 19 E. multilocularis VALs with complete SCP/TAPS domains. Next, a phylogenetic tree was reconstructed to examine the phylogenetic relationships among VALs in the two Echinococcus species. The result showed that the Echinococcus VAL family was divided into three major groups, and the majority of genes had homologous counterparts between the two Echinococcus species (Figure 7). Using these nucleotide sequences of Echinococcus VALs as query sequences, local BLASTN searches were performed to identify the corresponding mRNA sequences in our RNA-Seq data. Nine E. granulosus VALs and nine E. multilocularis VALs were proved to be transcribed in protoscoleces (Figure 7), and the others may be expressed in other developmental stages or the amount of their transcription was too low to be detected by our study. Interestingly, AS transcripts of E. granulosus VALs (CDS24051.1 and CDS22593.1) and the E. multilocularis VAL (CDS35801.1) from group II were discovered in our RNA-Seq data (Figure 7). Therefore, AS events may be ubiquitous in the VAL family of parasitic platyhelminths, and is well worth further study.
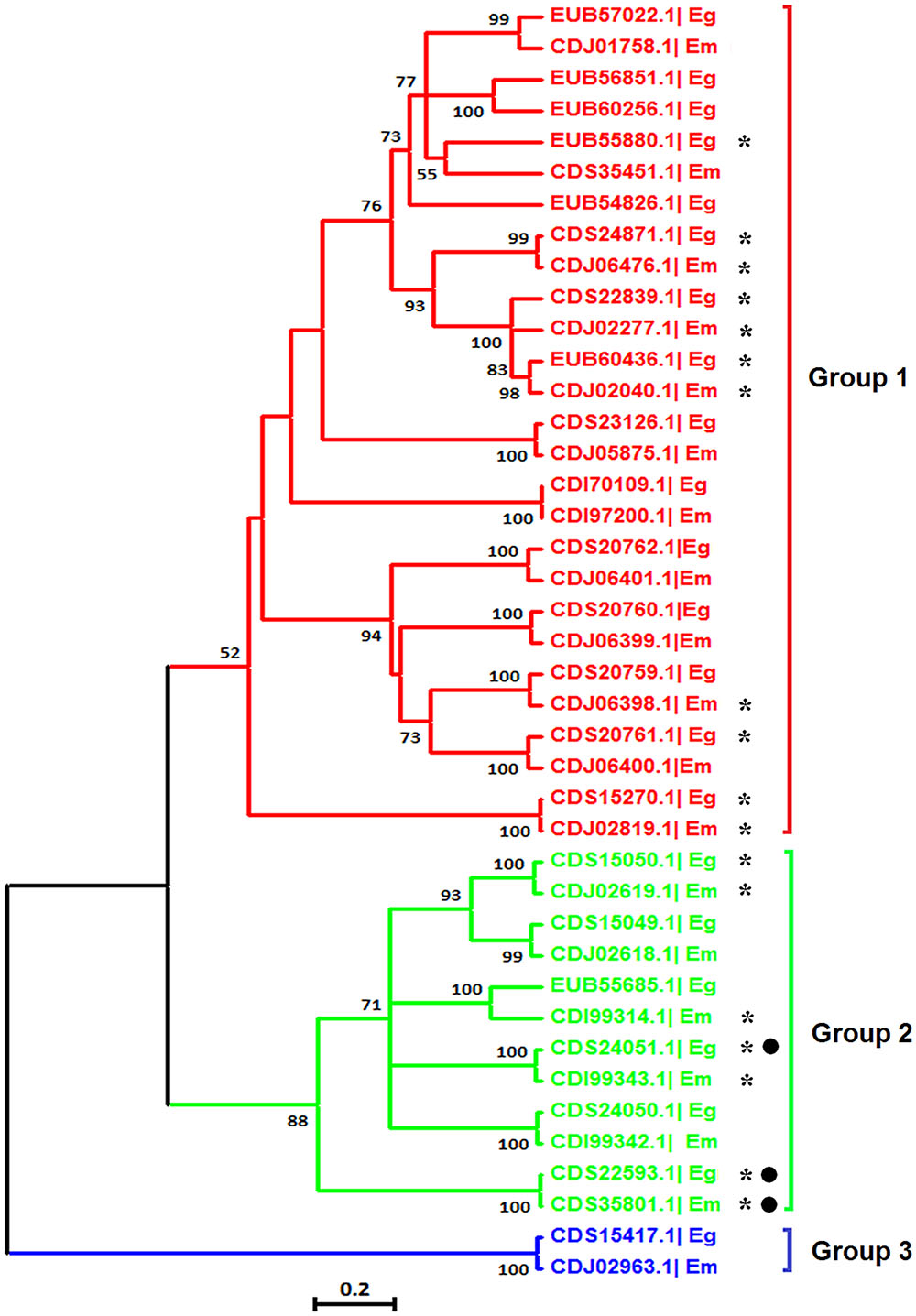
FIGURE 7. Molecular phylogenetic relationship of the VAL gene family of E. granulosus and E. multilocularis. The tree was divided into three phylogenetic groups and the bootstrap values (more than 50) are shown at the nodes. Asterisks represent genes transcribed in protoscoleces and black dots represent genes with AS. Eg, E. granulosus; Em, E. multilocularis.
Discussion
As one of major fields of enquiry in the post-genomic era, AS has been investigated in an increasing number of species over the past decade (Kalsotra and Cooper, 2011; Filichkin et al., 2015; Lunghi et al., 2016). It has been shown that AS plays a crucial role in enhancing transcriptomic and proteomic diversity, and its prevalence and characteristics vary considerably at many different levels, such as in species, tissues, and developmental stage (Kornblihtt et al., 2013). In this study, to the best of our knowledge, we have presented for the first time, a genome-wide analysis of AS events in protoscolex transcriptomes of E. granulosus and E. multilocularis. We found that approximately 33–36% of genes in the two Echinococcus species underwent AS events. This result is close to the AS percentage in S. japonicum (35%) (Piao et al., 2014) and Drosophila melanogaster (31%) (Graveley et al., 2011), and higher than the percentages in Danio rerio (17%) (Lu et al., 2010) and C. elegans (25%) (Ramani et al., 2011), but far below the percentage in H. sapiens (over 90%) (Pan et al., 2008; Wang et al., 2008). It is known that AS within the same species can be affected by developmental stage, tissue type, and growth conditions. Indeed, a higher AS percentage was obtained in schistosomulum and adult stages of S. japonicum (Wang et al., 2015), compared with the AS percentage in the adult stage only (Piao et al., 2014). Our estimate of AS events in Echinococcus species could be underestimated, since the parasite experiences several distinct environments during its complex life cycle, yet we only sequenced samples for one stage. We would expect a higher AS percentage to be observed in Echinococcus species when more developmental stages (such as adult, egg, and oncosphere) are analyzed.
Alternative splicing events can be commonly classified into seven types: intron retention, exon skipping, alternative donor site, alternative acceptor site, alternative first exon, alternative last exon, and mutually exclusive exon (Black, 2003). Previous studies have revealed that exon skipping, alternative donor site, alternative acceptor site, and intron retention are the four basic AS types (Nilsen and Graveley, 2010), and the overall AS patterns vary across species, tissue types, and developmental stages (Wang et al., 2008; Gibilisco et al., 2016). Exon skipping is thought to be the most prevalent AS type in animals, whereas intron retention represents the most common AS form in plants and unicellular eukaryotes (Barbazuk et al., 2008). Unexceptionally, the four major types of AS covered the vast majority of AS events in this study. Intron retention is a process by which specific introns, or fragments thereof, remain unspliced in polyadenylated transcripts. It is noteworthy that intron retention, but not exon skipping, was the predominant AS type in protoscoleces based on our high-coverage poly(A)+ RNA-Seq data. The advent of new sequencing technologies and algorithms is providing growing evidence that intron retention is a universal mechanism common to many eukaryotic organisms (Lunghi et al., 2016). By applying a new pipeline for intron-retention detection to high-coverage poly(A)+ RNA-Seq data, Braunschweig et al. (2014) found that intron retention is far more frequent in mammals than previously believed, affecting transcripts from as many as three-quarters of intron-containing genes. Meanwhile, previous studies have shown that intron retention is developmentally regulated and specific to the developmental phase or tissue (Boothby et al., 2013; Wang et al., 2016). In a study by Dvinge and Bradley (2015), it was found that an increase in retained introns is common in most cancer transcriptomes affecting genes involved in RNA processing and nuclear export. Moreover, the identification of increasing numbers of regulated intron-retention events in multi-stage unicellular parasites and plants suggests that these mechanisms can facilitate adaptation to acute host environmental changes, or duly supply key molecules to invade and exploit different ecological niches (Filichkin et al., 2015; Lunghi et al., 2016). The amazingly high frequency of intron retention in protoscoleces of Echinococcus species revealed that intron retention may play crucial roles in the development of the parasite and in host–parasite interaction. It will be fascinating to investigate the diversity of AS occurring in different developmental stages of Echinococcus species in the future. Deeper mechanistic research of intron retention will provide novel insight into its role in regulating gene expression during the life cycle of the parasites. Intron-retention mechanisms deserve to be elevated from paltry biological events to key elements that facilitate the functional tuning of cells, tissues or even developmental stages of numerous eukaryotes, especially for animals.
Gene Ontology analysis was performed to summarize and explore the functional categories of AS genes identified in this study. The results indicated that the proportions of enriched GO categories between the two parasites were generally consistent with each other in this study. Meanwhile, the KEGG pathway enrichment analysis showed that these AS genes tended to be enriched in multiple pathways, such as numerous metabolic pathways (e.g., purine, fatty acid, galactose, and glycerolipid metabolism) and key signaling pathways (e.g., Jak-STAT, VEGF, Notch, and GnRH signaling pathways). Interestingly, we found that 30 genes that underwent AS events were significantly enriched in mRNA surveillance pathways (P < 0.001), which are utilized by organisms to ensure the quality and fidelity of mRNA. As one of the well-known mRNA surveillance pathways (Lykke-Andersen and Jensen, 2015), the NMD pathway not only targets mRNAs containing premature termination codons for degradation, but also plays a key role in the regulation of physiological gene expression (Chang et al., 2007; Fatscher et al., 2015; Hug et al., 2016). Studies by Filichkin et al. (2010) and Sorber et al. (2011) are suggestive that a proportion of premature termination codon-containing transcripts generated by AS events may either produce truncated protein isoforms or tune gene expression post-transcriptionally by the NMD pathway. Although this highly conserved pathway has been reported in numerous eukaryotes, the NMD pathway has yet to be shown in Echinococcus species. Using human core members (UPF1, UPF2, and UPF3) of the NMD pathway as query sequences, we identified homologs to all three in the genomes of E. granulosus (CDS17643.1, CDS16560.1, and CDS18511.1) (Tsai et al., 2013; Zheng et al., 2013) and E. multilocularis (CDS42690.1, CDS41342.1, and CUT99229.1) (Tsai et al., 2013), suggesting the NMD pathway exists in Echinococcus species.
Conclusion
We have performed what is, to the best of our knowledge, the first genome-wide survey of AS events in Echinococcus species using a next-generation sequencing technology. We found that the AS prevalence was about 33–36%, and intron retention was the predominant AS type in the protoscolex transcriptomes of E. granulosus and E. multilocularis. Genes that underwent AS events were significantly enriched in dozens of pathways mainly related to metabolism, signal transduction, and genetic information processing. These results will not only significantly improve the re-annotation of Echinococcus genomes, but also provide an invaluable resource for future functional and evolutionary studies of AS in the platyhelminth parasites.
Author Contributions
Experiments were conceived and designed by SL and QC. Experiments were performed by XZ, SL, LH, XP, and NH. Data was analyzed by SL and XZ. The paper was written by SL, XZ, and QC.
Funding
This work was supported by the Special Fund for Health Research in the Public Interest (grant no. 201202019, and 201402001), the National S & T Major Program (grant no. 2012ZX10004-220), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13007).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors thank the Genome Sequencing and Functional Analysis Consortium for making the genomes of E. granulosus and E. multilocularis available publicly online.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00929/full#supplementary-material
Abbreviation
AS, alternative splicing; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; NMD, nonsense-mediated mRNA decay; RNA-Seq, RNA-sequencing.
Footnotes
References
Alamancos, G. P., Pages, A., Trincado, J. L., Bellora, N., and Eyras, E. (2015). Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA 21, 1521–1531. doi: 10.1261/rna.051557.115
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Barbazuk, W. B., Fu, Y., and Mcginnis, K. M. (2008). Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res. 18, 1381–1392. doi: 10.1101/gr.053678.106
Black, D. L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336. doi: 10.1146/annurev.biochem.72.121801.161720
Boothby, T. C., Zipper, R. S., Van Der Weele, C. M., and Wolniak, S. M. (2013). Removal of retained introns regulates translation in the rapidly developing gametophyte of Marsilea vestita. Dev. Cell 24, 517–529. doi: 10.1016/j.devcel.2013.01.015
Braunschweig, U., Barbosa-Morais, N. L., Pan, Q., Nachman, E. N., Alipanahi, B., Gonatopoulos-Pournatzis, T., et al. (2014). Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 24, 1774–1786. doi: 10.1101/gr.177790.114
Chalmers, I. W., Mcardle, A. J., Coulson, R. M., Wagner, M. A., Schmid, R., Hirai, H., et al. (2008). Developmentally regulated expression, alternative splicing and distinct sub-groupings in members of the Schistosoma mansoni venom allergen-like (SmVAL) gene family. BMC Genomics 9:89. doi: 10.1186/1471-2164-9-89
Chang, Y. F., Imam, J. S., and Wilkinson, M. F. (2007). The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51–74. doi: 10.1146/annurev.biochem.76.050106.093909
Craig, P. S., and Larrieu, E. (2006). Control of cystic echinococcosis/hydatidosis: 1863-2002. Adv. Parasitol. 61, 443–508. doi: 10.1016/S0065-308X(05)61011-1
DeMarco, R., Mathieson, W., Manuel, S. J., Dillon, G. P., Curwen, R. S., Ashton, P. D., et al. (2010). Protein variation in blood-dwelling schistosome worms generated by differential splicing of micro-exon gene transcripts. Genome Res. 20, 1112–1121. doi: 10.1101/gr.100099.109
Dvinge, H., and Bradley, R. K. (2015). Widespread intron retention diversifies most cancer transcriptomes. Genome Med 7:45. doi: 10.1186/s13073-015-0168-9
Eckert, J., and Deplazes, P. (2004). Biological, epidemiological, and clinical aspects of echinococcosis, a zoonosis of increasing concern. Clin. Microbiol. Rev. 17, 107–135. doi: 10.1128/CMR.17.1.107-135.2004
Farias, L. P., Rodrigues, D., Cunna, V., Rofatto, H. K., Faquim-Mauro, E. L., and Leite, L. C. (2012). Schistosoma mansoni venom allergen like proteins present differential allergic responses in a murine model of airway inflammation. PLoS Negl. Trop. Dis. 6:e1510. doi: 10.1371/journal.pntd.0001510
Fatscher, T., Boehm, V., and Gehring, N. H. (2015). Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell Mol. Life Sci. 72, 4523–4544. doi: 10.1007/s00018-015-2017-9
Filichkin, S., Priest, H. D., Megraw, M., and Mockler, T. C. (2015). Alternative splicing in plants: directing traffic at the crossroads of adaptation and environmental stress. Curr. Opin. Plant Biol. 24, 125–135. doi: 10.1016/j.pbi.2015.02.008
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R., Bryant, D. W., Fox, S. E., et al. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20, 45–58. doi: 10.1101/gr.093302.109
Gibilisco, L., Zhou, Q., Mahajan, S., and Bachtrog, D. (2016). Alternative splicing within and between Drosophila species, sexes, tissues, and developmental stages. PLoS Genet. 12:e1006464. doi: 10.1371/journal.pgen.1006464
Graveley, B. R., Brooks, A. N., Carlson, J. W., Duff, M. O., Landolin, J. M., Yang, L., et al. (2011). The developmental transcriptome of Drosophila melanogaster. Nature 471, 473–479. doi: 10.1038/nature09715
Harris, M. A., Clark, J., Ireland, A., Lomax, J., Ashburner, M., Foulger, R., et al. (2004). The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261.
Huang, F., Dang, Z., Suzuki, Y., Horiuchi, T., Yagi, K., Kouguchi, H., et al. (2016). Analysis on gene expression profile in oncospheres and early stage metacestodes from Echinococcus multilocularis. PLoS Negl. Trop. Dis. 10:e0004634. doi: 10.1371/journal.pntd.0004634
Hug, N., Longman, D., and Caceres, J. F. (2016). Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483–1495. doi: 10.1093/nar/gkw010
Hull, R., and Dlamini, Z. (2014). The role played by alternative splicing in antigenic variability in human endo-parasites. Parasit. Vectors 7:53. doi: 10.1186/1756-3305-7-53
Kalsotra, A., and Cooper, T. A. (2011). Functional consequences of developmentally regulated alternative splicing. Nat. Rev. Genet. 12, 715–729. doi: 10.1038/nrg3052
Kanehisa, M., Goto, S., Hattori, M., Aoki-Kinoshita, K. F., Itoh, M., Kawashima, S., et al. (2006). From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357. doi: 10.1093/nar/gkj102
Keren, H., Lev-Maor, G., and Ast, G. (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 11, 345–355. doi: 10.1038/nrg2776
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kornblihtt, A. R., Schor, I. E., Allo, M., Dujardin, G., Petrillo, E., and Munoz, M. J. (2013). Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 14, 153–165. doi: 10.1038/nrm3525
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Liu, S., Zhou, X., Piao, X., Wu, C., Hou, N., and Chen, Q. (2015). Comparative analysis of transcriptional profiles of adult Schistosoma japonicum from different laboratory animals and the natural host, water buffalo. PLoS Negl. Trop. Dis. 9:e0003993. doi: 10.1371/journal.pntd.0003993
Lu, J., Peatman, E., Wang, W., Yang, Q., Abernathy, J., Wang, S., et al. (2010). Alternative splicing in teleost fish genomes: same-species and cross-species analysis and comparisons. Mol. Genet. Genomics 283, 531–539. doi: 10.1007/s00438-010-0538-3
Lunghi, M., Spano, F., Magini, A., Emiliani, C., Carruthers, V. B., and Di Cristina, M. (2016). Alternative splicing mechanisms orchestrating post-transcriptional gene expression: intron retention and the intron-rich genome of apicomplexan parasites. Curr. Genet. 62, 31–38. doi: 10.1007/s00294-015-0506-x
Lykke-Andersen, S., and Jensen, T. H. (2015). Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 16, 665–677. doi: 10.1038/nrm4063
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., Deweese-Scott, C., et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229. doi: 10.1093/nar/gkq1189
Marguerat, S., and Bahler, J. (2010). RNA-seq: from technology to biology. Cell Mol. Life Sci. 67, 569–579. doi: 10.1007/s00018-009-0180-6
Martin, J. A., and Wang, Z. (2011). Next-generation transcriptome assembly. Nat. Rev. Genet. 12, 671–682. doi: 10.1038/nrg3068
McManus, D. P., Zhang, W., Li, J., and Bartley, P. B. (2003). Echinococcosis. Lancet 362, 1295–1304. doi: 10.1093/nar/gkg046
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Mulder, N. J., Apweiler, R., Attwood, T. K., Bairoch, A., Barrell, D., Bateman, A., et al. (2003). The InterPro Database, 2003 brings increased coverage and new features. Nucleic Acids Res. 31, 315–318.
Naftelberg, S., Schor, I. E., Ast, G., and Kornblihtt, A. R. (2015). Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 84, 165–198. doi: 10.1146/annurev-biochem-060614-034242
Nilsen, T. W., and Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463. doi: 10.1038/nature08909
Ouyang, S., Zhu, W., Hamilton, J., Lin, H., Campbell, M., Childs, K., et al. (2007). The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res. 35, D883–D887. doi: 10.1093/nar/gkl976
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. doi: 10.1038/ng.259
Pan, W., Shen, Y., Han, X., Wang, Y., Liu, H., Jiang, Y., et al. (2014). Transcriptome profiles of the protoscoleces of Echinococcus granulosus reveal that excretory-secretory products are essential to metabolic adaptation. PLoS Negl. Trop. Dis. 8:e3392. doi: 10.1371/journal.pntd.0003392
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Piao, X., Hou, N., Cai, P., Liu, S., Wu, C., and Chen, Q. (2014). Genome-wide transcriptome analysis shows extensive alternative RNA splicing in the zoonotic parasite Schistosoma japonicum. BMC Genomics 15:715. doi: 10.1186/1471-2164-15-715
Ramani, A. K., Calarco, J. A., Pan, Q., Mavandadi, S., Wang, Y., Nelson, A. C., et al. (2011). Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res. 21, 342–348. doi: 10.1101/gr.114645.110
Sorber, K., Dimon, M. T., and Derisi, J. L. (2011). RNA-Seq analysis of splicing in Plasmodium falciparum uncovers new splice junctions, alternative splicing and splicing of antisense transcripts. Nucleic Acids Res. 39, 3820–3835. doi: 10.1093/nar/gkq1223
Staiger, D., and Brown, J. W. (2013). Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 25, 3640–3656. doi: 10.1105/tpc.113.113803
Sultan, M., Schulz, M. H., Richard, H., Magen, A., Klingenhoff, A., Scherf, M., et al. (2008). A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science 321, 956–960. doi: 10.1126/science.1160342
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Tsai, I. J., Zarowiecki, M., Holroyd, N., Garciarrubio, A., Sanchez-Flores, A., Brooks, K. L., et al. (2013). The genomes of four tapeworm species reveal adaptations to parasitism. Nature 496, 57–63. doi: 10.1038/nature12031
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Wang, H., Chen, Y., Li, X., Chen, G., Zhong, L., Chen, G., et al. (2016). Genome-wide analysis of alternative splicing during human heart development. Sci. Rep. 6:35520. doi: 10.1038/srep35520
Wang, X., Xu, X., Lu, X., Zhang, Y., and Pan, W. (2015). Transcriptome bioinformatical analysis of vertebrate stages of Schistosoma japonicum reveals alternative splicing events. PLoS ONE 10:e0138470. doi: 10.1371/journal.pone.0138470
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Ye, J., Fang, L., Zheng, H., Zhang, Y., Chen, J., Zhang, Z., et al. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, W293–W297. doi: 10.1093/nar/gkl031
Yeoh, L. M., Goodman, C. D., Hall, N. E., Van Dooren, G. G., Mcfadden, G. I., and Ralph, S. A. (2015). A serine-arginine-rich (SR) splicing factor modulates alternative splicing of over a thousand genes in Toxoplasma gondii. Nucleic Acids Res. 43, 4661–4675. doi: 10.1093/nar/gkv311
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Keywords: Echinococcus granulosus, Echinococcus multilocularis, transcriptome, alternative splicing, nextgeneration sequencing
Citation: Liu S, Zhou X, Hao L, Piao X, Hou N and Chen Q (2017) Genome-Wide Transcriptome Analysis Reveals Extensive Alternative Splicing Events in the Protoscoleces of Echinococcus granulosus and Echinococcus multilocularis. Front. Microbiol. 8:929. doi: 10.3389/fmicb.2017.00929
Received: 03 February 2017; Accepted: 08 May 2017;
Published: 23 May 2017.
Edited by:
Bang Shen, Huazhong Agricultural University, ChinaReviewed by:
Paras Jain, Albert Einstein College of Medicine, United StatesSi-Yang Huang, Chinese Academy of Agricultural Sciences, China
Carlos Robello, Institut Pasteur de Montevideo, Uruguay
Copyright © 2017 Liu, Zhou, Hao, Piao, Hou and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuai Liu, bGl1c2h1YWlAaXBiY2Ftcy5hYy5jbg== Qijun Chen, cWlqdW4uY2hlbkBpcGJjYW1zLmFjLmNu
†These authors have contributed equally to this work.