 Douglas B. Young
Douglas B. Young Iñaki Comas
Iñaki Comas Luiz P. S. de Carvalho
Luiz P. S. de Carvalho- 1Division of Mycobacterial Research, MRC National Institute for Medical Research, London, UK
- 2FISABIO Public Health, Valencia, Spain
Comparison of genome sequences from clinical isolates of Mycobacterium tuberculosis with phylogenetically-related pathogens Mycobacterium marinum, Mycobacterium kansasii, and Mycobacterium leprae reveals diversity amongst genes associated with vitamin B12-related metabolism. Diversity is generated by gene deletion events, differential acquisition of genes by horizontal transfer, and single nucleotide polymorphisms (SNPs) with predicted impact on protein function and transcriptional regulation. Differences in the B12 synthesis pathway, methionine biosynthesis, fatty acid catabolism, and DNA repair and replication are consistent with adaptations to different environmental niches and pathogenic lifestyles. While there is no evidence of further gene acquisition during expansion of the M. tuberculosis complex, the emergence of other forms of genetic diversity provides insights into continuing host-pathogen co-evolution and has the potential to identify novel targets for disease intervention.
Strains belonging to the Mycobacterium tuberculosis complex evolved by clonal expansion from a common ancestral population shared with Mycobacterium canettii, an occasional human pathogen closely resembling M. tuberculosis in terms of disease and evolution. M. tuberculosis and M. canettii share close phylogenetic links to other pathogens including Mycobacterium leprae, Mycobacterium marinum [and the derived Mycobacterium ulcerans (Doig et al., 2012)], and Mycobacterium kansasii (Figure 1) (Cole et al., 1998; Stinear et al., 2008; Supply et al., 2013; Blouin et al., 2014). There was an early division of the M. tuberculosis complex into two major branches (Brosch et al., 2002). The branch characterized by a 2 kb RD9 deletion and a single nucleotide polymorphism (SNP) that inactivates pyruvate kinase (Keating et al., 2005) gave rise to two lineages associated with human tuberculosis in West Africa (referred to as M. africanum) and a series of animal-adapted variants including M. bovis, M. orygis, M. microti, and M. pinnipedii (Comas et al., 2013). The branch with an intact pyruvate kinase diversified into five human lineages with patterns of geographic distribution and phylogenetic coalescence consistent with their having co-evolved with populations of modern humans migrating out of Africa around the Indian Ocean and across Eurasia (Comas et al., 2013). While the animal strains often retain the ability to cause sporadic cases of human disease (Bos et al., 2014), they do not generally establish an effective transmission cycle. Similarly, while human strains of M. tuberculosis can be isolated from cattle lesions in low prevalence herds (Berg et al., 2009), a high prevalence of bovine tuberculosis is always associated with cattle-adapted M. bovis (Firdessa et al., 2012). Epidemiological evidence suggests that individual human lineages are optimized for transmission within particular ethnic groups, providing a further indication of co-evolution between host and pathogen (Gagneux et al., 2006; Fenner et al., 2013).
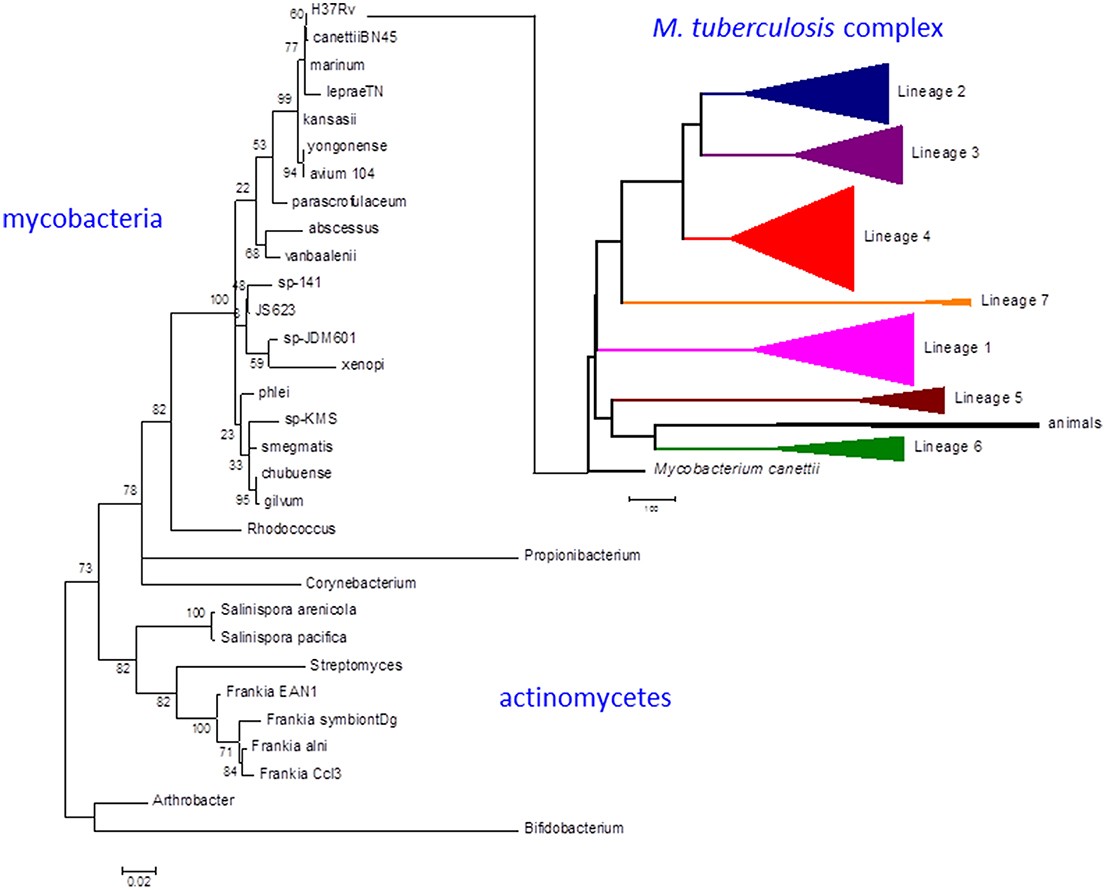
Figure 1. Phylogeny of M. tuberculosis and related actinomycetes. The M. tuberculosis complex emerged by clonal expansion of a strain closely related to the M. canettii cluster of slow-growing mycobacteria. The actinomycete phylogenetic tree is based on 16S rRNA sequences. Briefly, the corresponding sequences were aligned using the 16sRNA alignment tool available in the RDP database (Cole et al., 2014). The resulting alignment was analyzed with MEGA6 to infer a maximum likelihood phylogeny (Tamura et al., 2013). The best-fit model of nucleotide evolution after Akaike information criterion was Tamura-Nei with Gamma and invariants to model rate variation. Thousand bootstrap pseudo-replicates were used to give statistical support to the clades of the maximum likelihood topology. The M. tuberculosis complex tree is based on whole genome SNP analysis as obtained in Comas et al. (2013). Scale bar for the 16sRNA phylogeny reflects number of substitutions per site. In the case of the whole genome MTBC phylogeny reflects number of single nucleotide polymorphism. Numbers in the nodes of the 16sRNA topology reflects the percentage of bootstrap replicates supporting each node. No bootstrap support is shown for the MTBC phylogeny as all the case were higher than 95% as shown in Comas et al. (2013).
Despite sustained efforts to deliver optimal therapy over the last two decades, tuberculosis remains a major global health problem. Treatment of clinical disease undoubtedly saves lives and reduces suffering, but there are limitations to its effectiveness as a strategy to block transmission and there is a need for vaccines and preventive therapies that will arrest the disease process prior to development of an infectious state (Dye et al., 2013). Identification of molecular determinants that influence the transmission efficiency of the different host-adapted genotypes may point the way to novel interventions targeted toward disease control at a population level. It is anticipated that genetic changes that have a functional impact on pathogenesis will influence the repertoire of molecules that interact with the host immune system and the metabolic networks that support growth and survival in particular environmental niches. In this review we investigate the potential of combining phylogenetic and metabolic approaches to dissect events in the evolution of M. tuberculosis.
Biosynthesis of Vitamin B12
Vitamin B12 (also known as cobalamin, Figure 2) refers to a family of cobalt-containing, water soluble vitamins which are required in several, unrelated metabolic pathways. Due to the reactivity of its carbon-cobalt bond, B12-dependent enzymes are able to catalyze isomerizations, methyltransfers, and dehalogenations. Vitamin B12 presents an interesting focus for exploration of the metabolic phylogeny of host-pathogen interactions. It is likely that the progenitor eukaryotic cell included B12-dependent enzymes but lacked the complex set of genes required for B12 biosynthesis. Only bacteria and archaea have the machinery to synthesize vitamin B12 (Zhang et al., 2009; Doxey et al., 2015). While animals continued to use B12-dependent enzymes for methionine synthesis and methylmalonate metabolism, land plants and fungi rely on B12-independent enzymes for these pathways. Divergent evolution of algae generated both B12-dependent and B12-independent species (Helliwell et al., 2011). A fundamental feature inherent in the choice of a B12-dependent lifestyle is that it involves commitment to symbiosis with B12-producing bacteria, establishing a platform for the subsequent evolution of pathogens. Many prokaryotes have also elected to rely on community-acquired B12; bioinformatic analysis of whole genome sequences identifies an intact B12 biosynthesis pathway in only half of the bacteria that have B12-dependent enzymes (Zhang et al., 2009). Supply and utilization of vitamin B12 by bacteria can have an important impact on the metabolism of eukaryotic partners in co-culture (Wang et al., 2014; Watson et al., 2014). While it is unlikely that the relatively small numbers of tuberculosis bacteria will influence overall B12 levels in infected individuals, vitamin B12 produced or consumed by mycobacterial pathogens may have a local effect on the metabolic environment of human granulomatous lesions.
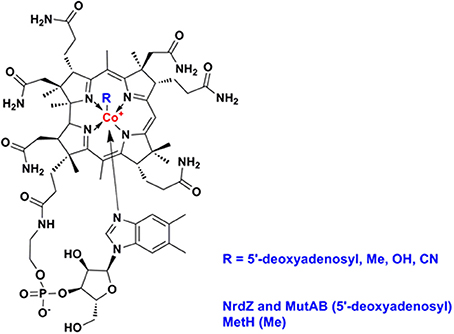
Figure 2. Structure of vitamin B12 (cobalamin).
Mycobacteria and related actinomycetes generally retain the ability to synthesize B12, predominantly by the aerobic pathway, and Propionibacterium shermanii is used for production of the vitamin (Martens et al., 2002). More distantly related actinomycetes such as Arthrobacter and Bifidobacteria lack a B12 pathway, though some Arthrobacter have a B12-dependent methionine synthase. As highlighted by Boritsch et al. (2014) distinctive feature of M. tuberculosis is that the cobF gene—encoding a precorrin-6a synthase required for B12 synthesis—was deleted during evolution from the M. canettii-like ancestor, along with two further genes and N- and C-terminal portions of flanking genes Rv0943c and Rv0944 (Supply et al., 2013). While this may not entirely ablate B12 biosynthesis [it has been suggested that alternative methyltransferases might partially compensate for the loss of CobF (Rodionov et al., 2003; Gopinath et al., 2013a)], it suggests that M. tuberculosis may have come to rely on the host environment as a source of vitamin. Although classically, NrdZ and MutAB utilize adenosylcobalamin and MetH employs methylcobalamin, cofactor specificity for mycobacterial enzymes has not been investigated. This is important in all organisms that might synthesize one form of the cofactors and uptake another. Importantly, species specific variation in these cofactors, such as differences in the exact structure of the lower ligand, could indicate that mycobacterial enzymes must have greater ability to accommodate more than one form of the cofactor. Similarly, while non-typhoidal Salmonellae retain a B12 pathway, this is inactivated in human-adapted typhoidal serovars (Gal-Mor et al., 2014; Nuccio and Baumler, 2014), and Yersinia pestis and Yersinia pseudotuberculosis lack most of the B12 biosynthesis genes present in Yersinia enterocolitica. The RD9 deletion causes further attrition of the B12 pathway, removing the amino terminus of CobL, with likely polar effects on expression of CobM and CobK, and a predictive algorithm based on primary sequence conservation (SIFT; Ng and Henikoff, 2003) identifies three SNPs that are likely to impair cobalamin biosynthesis enzymes in Lineage 5 isolates (CobM D53G, CobO Q202R, CobU H50R). In the case of CobN/Rv2026c, screening a panel of more than 200 clinical isolates representative of the global diversity of M. tuberculosis (Comas et al., 2013) identifies 17 SNPs predicted to have an impact on protein function, two strains with >1 kb deletion, and truncated proteins generated by frameshift events. Historical experience suggests that the incidence of active tuberculosis is markedly reduced in the context of pernicious anemia (B12-deficiency) (Barron, 1933), and analysis of recent data shows a slight increase in the risk of tuberculosis during the first year after treatment for pernicious anemia (Ramagopalan et al., 2013). These clinical findings are consistent with a model in which cobalamin availability is a limiting factor for the growth of M. tuberculosis in vivo. Several studies have highlighted a complex effect of nutritional factors on susceptibility to tuberculosis, including a potential links to low B12 availability in vegetarian diets (Chanarin and Stephenson, 1988; Cegielski and McMurray, 2004). The obligate pathogen M. leprae has a severely reduced genome (Cole et al., 2001) lacking all of the B12 pathway with the exception of a set of genes required for scavenging exogenous B12 precursors (Rodionov et al., 2003; Zhang et al., 2009; Gopinath et al., 2013a): cobT (ML0868), ML1149/Rv1314c (ATP:cobalamin adenosyltransferase), and the BacA transporter ML2084/Rv1819c (Gopinath et al., 2013b). The B12 pathway is intact in M. marinum and M. kansasii.
These mutations suggest that M. leprae has fully adapted to reliance on host-derived B12, that M. tuberculosis is at least partially reliant on the host, and that M. marinum and M. kansasii synthesize their own B12. We next screened for diversity in the three pathways that utilize B12-dependent enzymes. In each case the bacteria have the choice of favoring the B12-dependent pathway or a parallel B12-independent mechanism.
Biosynthesis of Methionine
B12-Dependent Methionine Synthesis
M. tuberculosis has a B12-dependent methionine synthase (Rv2124c) with sequence characteristics matching the predominant form of MetH found in most actinomycetes (MetHa). MetH catalyzes the B12-dependent synthesis of L-methionine (EC 2.1.1.13), utilizing L-homocysteine and 5-methyltetrahydrofolate (N5-MeTHF) as substrates and generating tetrahydrofolate (THF) as a co-product (Figure 3A). While MetHa shares key catalytic residues and structural features with the common form of MetH found in most bacteria and animals, sequence alignment—30% identity with E. coli and 29% identity with human MetH—shows it to have a distinct phylogenetic origin (Figure 4A). MetHa is always located on the chromosome adjacent to a gene with homology to ML1306 (Rv2125 in M. tuberculosis), one of 24 conserved “signature proteins” that define the Actinobacteria phylum (Gao and Gupta, 2012). The function of the ML1306 proteins is unclear. Primary sequence analysis and structural modeling suggest some similarity to chaperones involved in assembly of the archaeal proteasome, but functional studies in Streptomyces demonstrate a role in chromosome segregation (Ditkowski et al., 2010), and structural analysis of the paralogous protein Rv2714 (another actinobacteria signature protein) revealed features reminiscent of purine nucleoside phosphorylases, carboxypeptidases, and bacterial peptidyl-tRNA hydrolases (Grana et al., 2009).
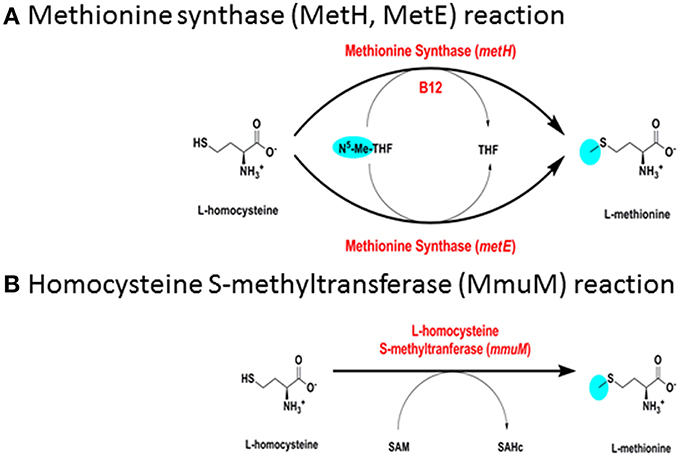
Figure 3. Biosynthesis of methionine. (A) Methionine synthase (MetH, MetE) reaction. (B) Homocysteine S-methyltransferase (MmuM) reaction.
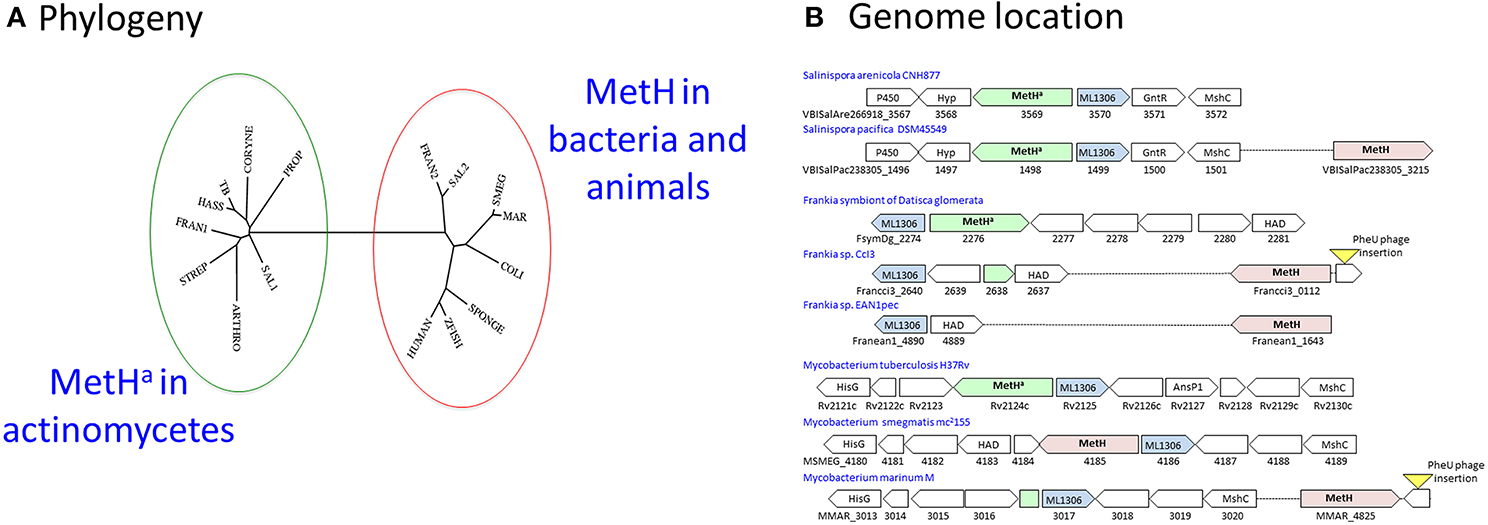
Figure 4. MetH switching in actinomycetes. (A) Phylogeny: The MetHa protein of M. tuberculosis (TB) is similar to that commonly found in actinomycetes including Streptomyces (STREP), Corynebacteria (CORYNE), Propionibacteria (PROP), and Arthrobacter (ARTHRO). In rapid-growing mycobacteria such as M. smegmatis (SMEG) MetH is related to the conventional variant found in other bacteria and animals. M. hassiacum (HASS) is an example of a rapid-growing mycobacterium with the MetHa variant, while slow-growing M. marinum (MAR) has MetH. One or both variants are found in Frankia (FRAN1/2) and Salinispora (SAL1/2) isolates. The phylogenetic tree was generated using the phylogeny.fr webserver (Dereeper et al., 2008). (B) Genome location: MetHa is always located adjacent to the actinomycete signature protein ML1306. Isolates of Salinispora pacifica include a second MetH variant in a separate location. Acquisition of a MetH variant in Frankia species is associated with deletion of MetHa. In M. marinum and Frankia sp. CcI3, the ML1306 site contains a fragment of MetHa; in both species an intact conventional MetH is present adjacent to a prophage insertion into tRNA-Phe-GAA, suggesting its possible introduction by horizontal transfer. Most rapid-growing mycobacteria have a MetH variant at the ML1306 site.
Some actinomycetes have acquired an additional metH gene resembling the common bacterial variant. Members of the genus Salinispora are obligate marine actinomycetes associated with sea sponges (Ziemert et al., 2014). While all of the sequenced Salinispora genomes have an ML1306-linked MetHa, the majority of S. pacifica isolates have an additional second MetH located elsewhere on the genome (Figure 4B). Frankia are actinomycete symbionts of plants (Tisa et al., 2013). While the obligate Frankia symbiont of Datisca glomerata has a typical actinomycete MetHa, other Frankia genomes have an alternative variant. Genome comparisons indicate that the original metHa gene was lost in most Frankia species as part of a 2.5 kb deletion between the ML1306 homolog and a conserved gene encoding an enzyme belonging to the haloalkanoic dehalogenase (HAD) superfamily, leaving only the acquired MetH variant (Figure 4B).
Mycobacterium marinum (Stinear et al., 2008) resembles Frankia sp. CcI3 in having an alternative MetH (MMAR_4825) with only a residual fragment of the actinomycete metHa sequence found adjacent to ML1306 homolog (MMAR_3107) (Figure 4B). In most rapid-growing mycobacteria, as well as in members of the Mycobacterium avium-intracellulare complex, the ML1306 site is occupied by a non-actinomycete MetH (Figure 4B). Exceptions include rapid-growing M. hassiacum and M. thermoresistibile which have a MetHa similar to M. tuberculosis, M. canettii, M. kansasii, M. xenopi, and M. leprae.
Comparison of genome sequences from clinical isolates reveals extensive MetHa polymorphism within the M. tuberculosis complex. Isolates from the sub-branch of EuroAmerican Lineage 4 that includes the well-characterized strain CDC1551 have a deletion that generates a non-functional MetHa (Warner et al., 2007). A further 20 non-synonymous SNPs spread throughout the protein are predicted to have an effect on enzyme function (Table 1). With the exception of E220D in Lineage 5, these SNPs occur close to the tips of the phylogenetic tree affecting only one or a few isolates and suggesting that they are of relatively recent origin. The frequent occurrence of functional SNPs—in comparison to MetE, for example—is consistent with the proposal of Warner et al. (2007) that loss of MetHa might have some selective benefit.
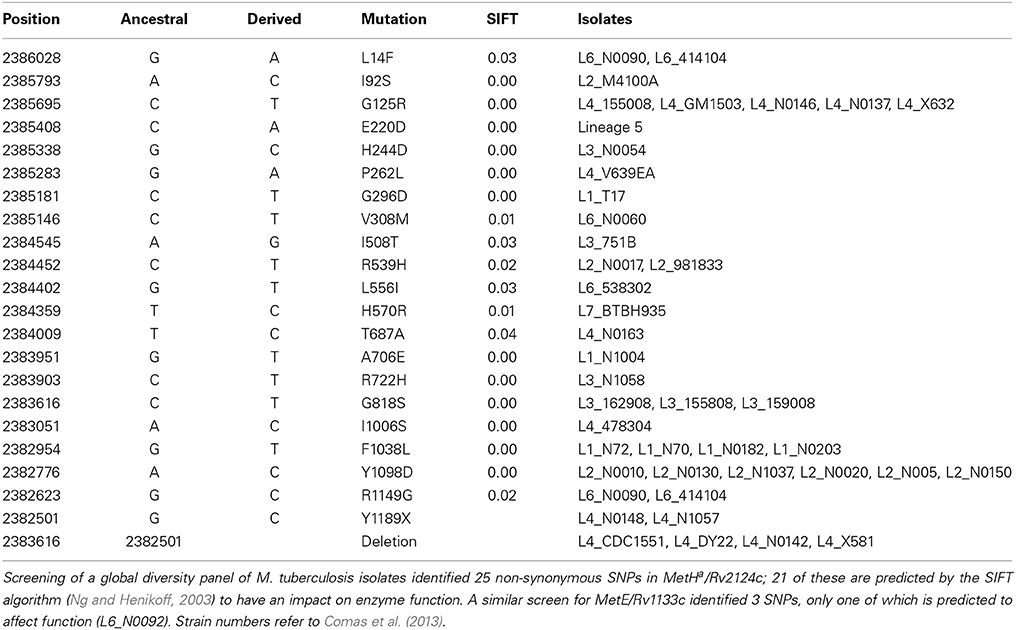
Table 1. M. tuberculosis MetHa SNPs with predicted functional impact.
B12-Independent Methionine Synthesis
M. tuberculosis has a putative B12-independent methionine synthase (EC 2.1.1.14), Rv1133/MetE, similar to that found in a wide range of bacteria. MetE catalyzes the B12-independent synthesis of L-methionine from L-homocysteine (Figure 3A), with N5-MeTHF as the methyl donor, similarly to MetH. A B12-independent reaction (EC 2.1.1.10) can also be achieved by transfer of a methyl group from S-adenosylmethionine by homocysteine S-methyltransferase (Rv2458/MmuM), as shown in Figure 3B. Rv2458 encodes an MmuM homolog in M. tuberculosis but does not substitute for MetE activity in a MetH knockout background (Warner et al., 2007). This is likely a reflection of the co-substrate needed in the reaction catalyzed by MmuM; S-adenosylmethionine versus N5-MeTHF utilized by MetH and MetE. As S-adenosylmethionine is synthesized by condensation of methionine and ATP it is impossible for this pathway to supply enough methionine for physiologic demands, as it will always consume one equivalent of S-adenosylmethione per methionine made, which in turn needs methionine to be regenerated. MmuM is conserved in the reduced genome of M. leprae but is absent in rapid-growing mycobacteria and has been deleted from M. marinum. In common with other actinomycetes, M. tuberculosis has a predicted protein with sequence match to the C-terminal catalytic domain of MetE (Rv3015c) that is often annotated as a MetE homolog. However, the canonical MetE enzyme has a duplicated domain structure (Pejchal and Ludwig, 2005) and the functional activity of the single domain proteins is unclear.
The catalytic turnover rate of E. coli MetH is 100-fold higher than MetE (Gonzalez et al., 1992), conferring a clear metabolic and ecological advantage in use of the B12-dependent pathway when B12 is available. Equivalent kinetic and thermodynamic constants for mycobacterial MetH, MetHa, and MetE are unknown; their determination might provide a functional rationale for MetH switching events. Expression of MetE in both E. coli and M. tuberculosis is repressed in the presence of B12 by binding of the cofactor to a regulatory riboswitch upstream of the metE coding sequence (Vitreschak et al., 2003; Warner et al., 2007). Warner et al. showed that in vitro culture of MetHa-negative M. tuberculosis in the presence of exogenous B12 resulted in selection of mutations in the MetE riboswitch (Warner et al., 2007), but there is no evidence of equivalent SNPs in clinical isolates with MetHa deletions or mutations (Figure 5).
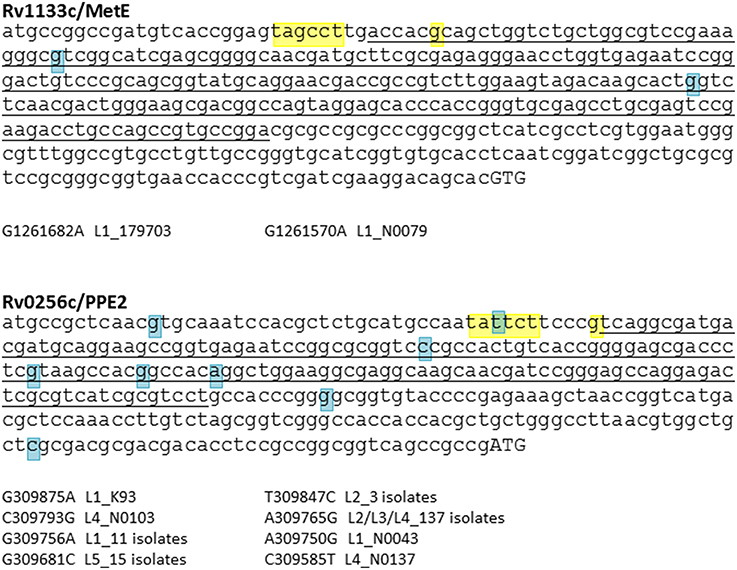
Figure 5. SNP analysis of B12 riboswitches. M. tuberculosis has two B12-dependent riboswitches, located upstream of Rv1133c (MetE) and Rv0256c (PPE2). Riboswitch sequences identified by Vitreschak et al. (2003) are underlined, −10 motifs and transcription start sites (Cortes et al., 2013) are highlighted in yellow, and start codons in uppercase. Blue highlights show SNPs identified in a panel of clinical isolates representative of the global diversity of M. tuberculosis (see Comas et al., 2013, for details of isolates). While there is evidence of diversification of the Rv0256c riboswitch, the MetE riboswitch has been conserved during expansion of the M. tuberculosis complex.
In contrast to MetE, accumulation of SNPs is observed in the case of a second M. tuberculosis B12 riboswitch located upstream of PPE2/Rv0256c (Vitreschak et al., 2003; Warner et al., 2007) (Figure 5). The function of Rv0256c is unclear. The presence of the riboswitch, together with a predicted membrane-spanning structure and potential operon link to B12 biosynthesis genes, led to a suggested role in cobalt transport (“CbtG”) (Rodionov et al., 2003). A recent elegant genetic study identified the BacA transporter as the sole determinant of cobalamin uptake by M. tuberculosis (Gopinath et al., 2013b). Riboswitch-regulated Rv0256c is retained in other mycobacteria including M. leprae, though the operon link to cob genes is variable.
During exponential growth of M. tuberculosis H37Rv, metE and metHa transcripts are present at a similar abundance around 5-fold higher than the median for all genes (Cortes et al., 2013). In terms of protein abundance, however, MetE is in 100-fold excess compared to MetHa (Cortes et al., 2013), suggesting that the MetHa protein is either translated less efficiently, or is more readily degraded. Proclivity to degradation might be a consequence of poor stability of MetH in the absence of B12, and it can be anticipated that addition of vitamin B12 to the growth medium might alter the protein ratio. In M. marinum, the metH transcript has an abundance matching the median level for all genes, with metE transcript 10-fold lower (Wang et al., 2013). This is consistent with the notion that contrary to M. tuberculosis, M. marinum constitutively produces B12.
Methionine and Folate Metabolism
The methyl group for the MetH/MetE reaction is provided by N5-MeTHF, and imbalance of vitamin B12 metabolism in humans is often associated with imbalance in folate metabolism. Similarly, disruption of methionine levels is observed in para-aminosalicylic acid treated M. tuberculosis (Chakraborty et al., 2013), indicating a tight coupling between folate biosynthesis and methionine levels. N5-MeTHF is generated from 5,10-methylenetetrahydrofolate by the MetF reductase, and mycobacteria are unusual amongst the actinomycetes in lacking an annotated copy of MetF. An exception is Mycobacterium abscessus, in which a MetF homolog is present between genes encoding lipoprotein LppM and geranylgeranyl pyrophosphate synthase IdsA. Other mycobacteria have a conserved hypothetical in this location; Rv2172c in M. tuberculosis. While Rv2172c shows no significant primary sequence homology with MetF, fold-recognition and structural modeling (Kelley and Sternberg, 2009) reveals a significant match with the crystal structure of MetF from Thermus thermophilus, identifying Rv2172c as an atypical MetF homolog.
Propionyl-CoA Catabolism
Methylmalonate Pathway (B12-Dependent)
Toxicity linked to the accumulation of propionyl-CoA derivatives during catabolism of odd-chain fatty acids is countered by their metabolism through either the B12-dependent methylmalonate pathway or the B12-independent methylcitrate cycle. One or other of the two pathways is essential for growth of M. tuberculosis on propionate (Munoz-Elias et al., 2006; Savvi et al., 2008; Eoh and Rhee, 2014). Methylmalonyl-CoA is also an important substrate for biosynthesis of cell wall lipids (Rainwater and Kolattukudy, 1983, 1985; Azad et al., 1997; Fernandes and Kolattukudy, 1998).
The methylmalonate pathway (Figure 6A) including B12-dependent MutAB methylmalonyl-CoA mutase (EC 5.4.99.2) is conserved across all mycobacteria including M. leprae. Propionyl-CoA enters the methylmalonate pathway through an essential acyl-CoA carboxylase comprising alpha (AccA3), beta (AccD5), and epsilon (AccE5) subunits (Gago et al., 2006; Lyonnet et al., 2014). Rv3281, encoding the epsilon subunit, is highly polymorphic between mycobacterial species and across multiple M. tuberculosis isolates, with a predicted N-terminal domain consisting of a variable number of repeat elements generating protein products that range from 85 to 203 amino acids. Functional activity of the epsilon subunit is retained in a 76 amino acid fragment from the conserved C-terminus [using valine-122 as start codon (Gago et al., 2006)] and the role—if any—of the variable N-terminus is unclear. Shotgun proteome analysis of M. tuberculosis H37Rv detected five peptides spanning the sequence from valine-63 to the C-terminus (Cortes et al., 2013) showing that at least part of the variable N-terminus is translated into protein.
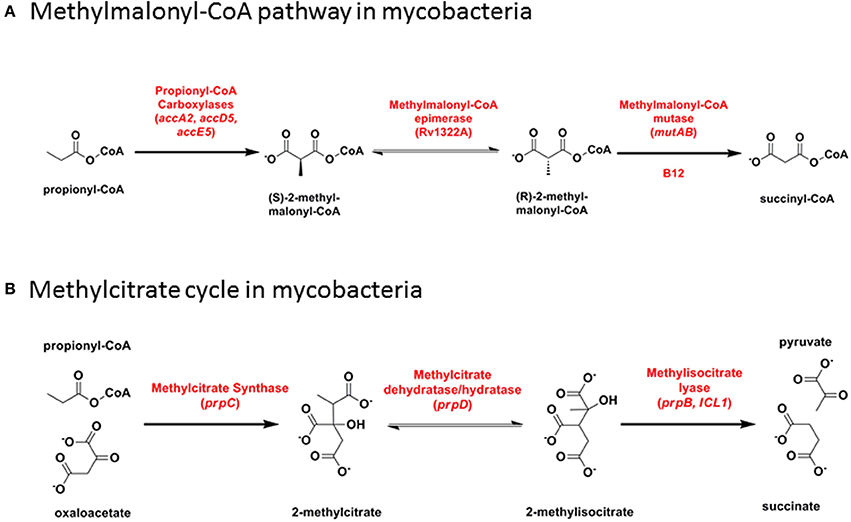
Figure 6. Propionyl-CoA catabolism. (A) Methylmalonyl-CoA pathway in mycobacteria. (B) Methylcitrate cycle in mycobacteria.
Mycobacterial Rv1492/MutA and Rv1493/MutB are presumed to form an alpha-beta heterodimer structure analogous to the well-characterized B12-dependent methylmalonyl-CoA mutase from Propionibacterium shermanii (Cracan and Banerjee, 2012); there is no evidence of diversification during the evolution of M. tuberculosis. M. canettii, and hence M. tuberculosis, lacks a locus containing a second mycobacterial methylmalonyl-CoA mutase (McmA2, MMAR_4797/4798 in M. marinum). This is a split-domain enzyme with substrate and B12 cofactor binding sites located on separate polypeptides. McmA2 is located in a ~150-gene region between Rv0838/LpqR and Rv0852/FadD16 that includes six conserved cytochrome P450 genes and an MCE locus. M. tuberculosis has only 15 genes in this region, M. leprae has eight genes and seven pseudogenes, and a 50-gene deletion removes the MCE locus and three of the P450s in M. ulcerans. The presence of a series of lipid-related enzymes in the MmcA2 region suggests that the role of the mutase in this case may be to generate methylmalonyl-CoA units for biosynthesis of a secondary metabolite or complex lipid that is missing from M. tuberculosis.
Methylcitrate Cycle (B12-Independent)
In the methylcitrate cycle (Figure 6B), 2-methylcitrate synthase (PrpC) combines propionyl-CoA with oxaloacetate to form 2-methylcitrate (EC 2.3.3.5), which is converted to methylisocitrate by PrpD dehydratase/hydratase (EC 4.2.1.79 and 4.2.1.99). These two steps resemble the first two reactions in the Krebs cycle, catalyzed by citrate synthase and aconitase, respectively. Methylisocitrate lyase (MCL, PrpB) converts methylisocitrate to pyruvate and succinate (EC 4.1.3.30) in a reaction analogous to that catalyzed by isocitrate lyase (ICL) in the glyoxylate shunt. In M. canettii/M. tuberculosis and in M. kansasii, a deletion between PrpD/Rv1130 and PrpC/Rv1131 results in loss of MCL. Differences in the length of residual intergenic sequence in the two species—54 bp in M. tuberculosis and 130 bp in M. kansasii—suggests that this may have occurred through two independent events. The methylcitrate cycle remains fully functional in M. tuberculosis, however, with ICL catalyzing the final step (Munoz-Elias et al., 2006; Eoh and Rhee, 2014). MCL is also disrupted by a frameshift mutation in M. ulcerans (Doig et al., 2012).
Mycobacteria have two ICLs. Icl1 (Rv0467 in M. tuberculosis H37Rv) is a typical bacterial ICL, clustering with related enzymes from other actinomycetes, Gram-positive and Gram-negative bacteria (Figure 7). The second ICL—generally annotated as AceA (Rv1915/Rv1916 in H37Rv)—is phylogenetically distinct from other bacterial and fungal ICLs. A single nucleotide deletion causes a frameshift in the aceA gene of H37Rv, resulting in its expression as two separate polypeptides which are unable to complete the methylcitrate cycle in an Icl1 mutant (Munoz-Elias et al., 2006).
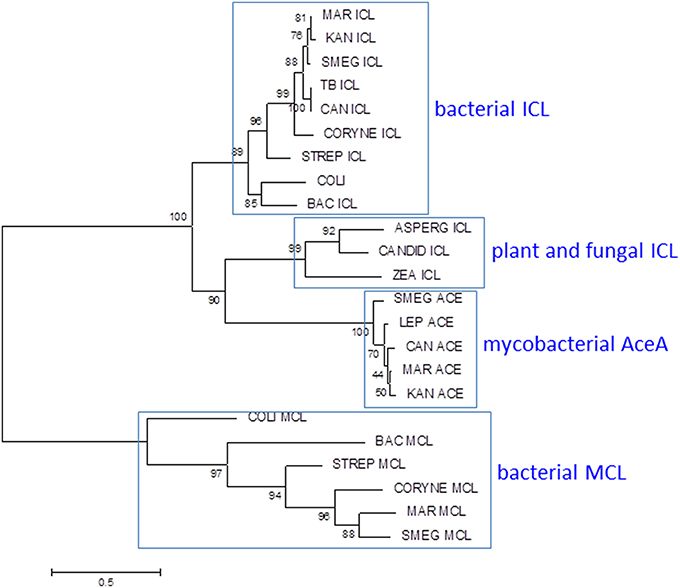
Figure 7. Phylogenetic analysis of mycobacterial isocitrate lyase (ICL) and 2-methylisocitrate lyase (MCL). Most mycobacteria have ICL and MCL enzymes similar to those found in other bacteria. MCL has been deleted from M. tuberculosis and M. kansasii and M. leprae lacks both MCL and ICL1. Mycobacteria also have a second isocitrate lyase (AceA) with a distinct phylogenetic origin. The ICL/MCL phylogeny was obtained after homolog searching of the ICL and MCL proteins present in M. tuberculosis and M. marinum. Muscle was used for amino acid alignment and a maximum likelihood phylogeny inferred as implemented in MEGA6 (Tamura et al., 2013). The JTT model of amino acid substitution incorporating gamma and invariant categories for rate variation was used. Thousand bootstrap pseudoreplicates were analyzed to obtain statistical support for the clades observed in the maximum likelihood topology.
Evolution of MCL activity from an ICL template has been studied in fungi and relevant amino acid changes have been mapped (Muller et al., 2011). Comparison of the ICL sequence from MCL-positive M. marinum with those from MCL-negative M. tuberculosis and M. kansasii reveals no obvious adaptive mutations, however, suggesting that the mycobacterial ICL has an intrinsic ability to utilize either substrate (Gould et al., 2006). The primary role of ICL is to catalyze conversion of isocitrate to succinate and glyoxylate to maintain energy generation and gluconeogenesis during growth on fatty acids. It can be anticipated that assigning a dual role to ICL in the methylcitrate cycle in addition to the glyoxylate shunt may require alterations in regulatory networks. Activation of the glyoxylate shunt in response to low glucose is controlled by the transcriptional regulator RamB (Rv0465c) (Micklinghoff et al., 2009), while expression of genes involved in the methylcitrate cycle is regulated by PrpR (Rv1129c) (Masiewicz et al., 2012). Both regulators bind to overlapping sites 80 bp upstream of the icl1 transcription start site. The binding site is conserved between MCL-positive and MCL-negative mycobacteria, however, suggesting that while there is an interplay between the two regulators, this predates the loss of MCL. Sequence comparisons within the M. tuberculosis complex highlight an intriguing diversity in the amino acid sequence of the RamB regulator. Four lineage-associated SNPs are predicted to have an impact on RamB function: N36D in Lineage 6, R106C in Lineage 4, Q121R in Lineage 1, and K229T in Lineage 5. Impaired function of RamB would reduce repression of ICL in the presence of glucose, potentially enhancing availability of ICL for the methylcitrate cycle. Therefore, species-specific alterations in regulatory networks further diversified by strain-specific SNPs in transcriptional regulators allow expression of an originally fatty acid catabolic pathway during growth on carbohydrates. Changes such as these are believed to confer on M. tuberculosis the unique ability to grow optimally with multiple types of carbon sources (de Carvalho et al., 2010; Rhee et al., 2011), which is believed to be the case during infection. Further rewiring of metabolic circuits linking glucose and fatty acid metabolism is likely to accompany the inactivation of pyruvate kinase in Lineage 5, Lineage 6, and the associated animal-adapted strains (Keating et al., 2005).
The origin and role of the second ICL in mycobacteria is unclear. While the methylcitrate cycle genes have been deleted and icl1 and ramB are reduced to pseudogenes in M. leprae, aceA and malate synthase remain intact, providing the potential for a functional glyoxylate shunt. Pyruvate kinase is intact in M. leprae, but pyruvate carboxylase is a pseudogene.
Biosynthesis of Deoxyribonucleotides
Ribonucleotide reductases (RNRs) are essential enzymes in all living cells, responsible for reduction of ribonucleotides to deoxyribonucleotides (EC 1.17.4.1) (Figure 8); the building blocks required for the synthesis of DNA.
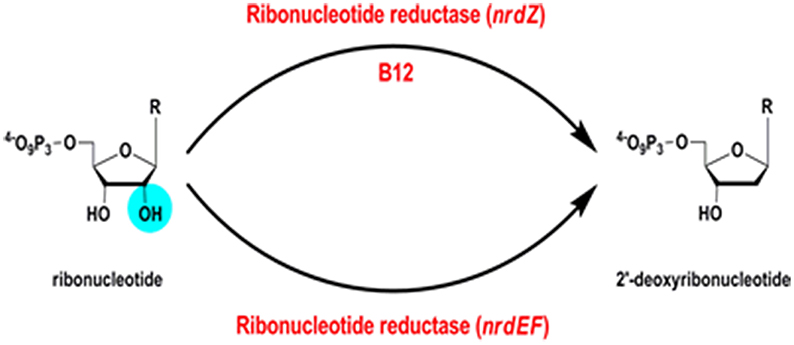
Figure 8. Ribonucleotide reductase reaction.
B12-Dependent Ribonucleotide Reductase
A B12-independent class Ib ribonucleotide reductase encoded by Rv3051c/nrdE and Rv3048c/nrdF2 is both necessary and sufficient to support growth of M. tuberculosis in a mouse model of infection (Dawes et al., 2003; Mowa et al., 2009). In addition, M. tuberculosis has a B12-dependent class II RNR, Rv0570 annotated as NrdZ (generally referred to as NrdJ in other bacteria). While the class I RNR requires oxygen, the class II enzyme has the potential to contribute an additional function under anaerobic conditions. In Pseudomonas aeruginosa, for example, a class II RNR is required to support biofilm growth under anaerobic conditions (Lee et al., 2012). It is important to note that B12 biosynthesis in mycobacteria requires oxygen, indicating that an anaerobic B12-dependent RNR would have to obtain B12 from exogenous sources under hypoxia.
Class II RNRs resemble class I and class III structurally (Sintchak et al., 2002) and the enzyme from Lactobacillus leichmannii is a monomer, contrasting with the oligomeric class I RNRs. In class II RNRs, the B12 cofactor is in its adenosylcobalamin form and is responsible for generation of the radical needed for the deoxygenation of the ribonucleotide. Reducing equivalents for the reaction are provided by oxidation of a pair of cysteine residues in class II RNRs (Booker et al., 1994), similarly to the mechanism described in class I RNRs.
NrdZ is differentially distributed amongst the mycobacteria in a pattern that is inconsistent with conventional phylogeny (Figure 9A); while it is present in slow-growing M. tuberculosis and M. kansasii and rapid-growing M. phlei, for example, it is absent from M. marinum and M. smegmatis. The mycobacterial class II RNR clusters separately from analogous enzymes in other actinomycetes, with a stronger resemblance to archaeal NrdJ (Figure 9B), suggesting its possible acquisition by horizontal transfer. Consistent with this, NrdJ homologs are found in the genomes of multiple mycobacteriophages (Dwivedi et al., 2013). In a phylogenetic analysis mycobacteriophage NrdZs cluster separately from chromosomal NrdZs; copies of the phage variant are integrated in the genomes of M. hassiacum (which also has an NdrZ) and M. rhodesiae JS60.
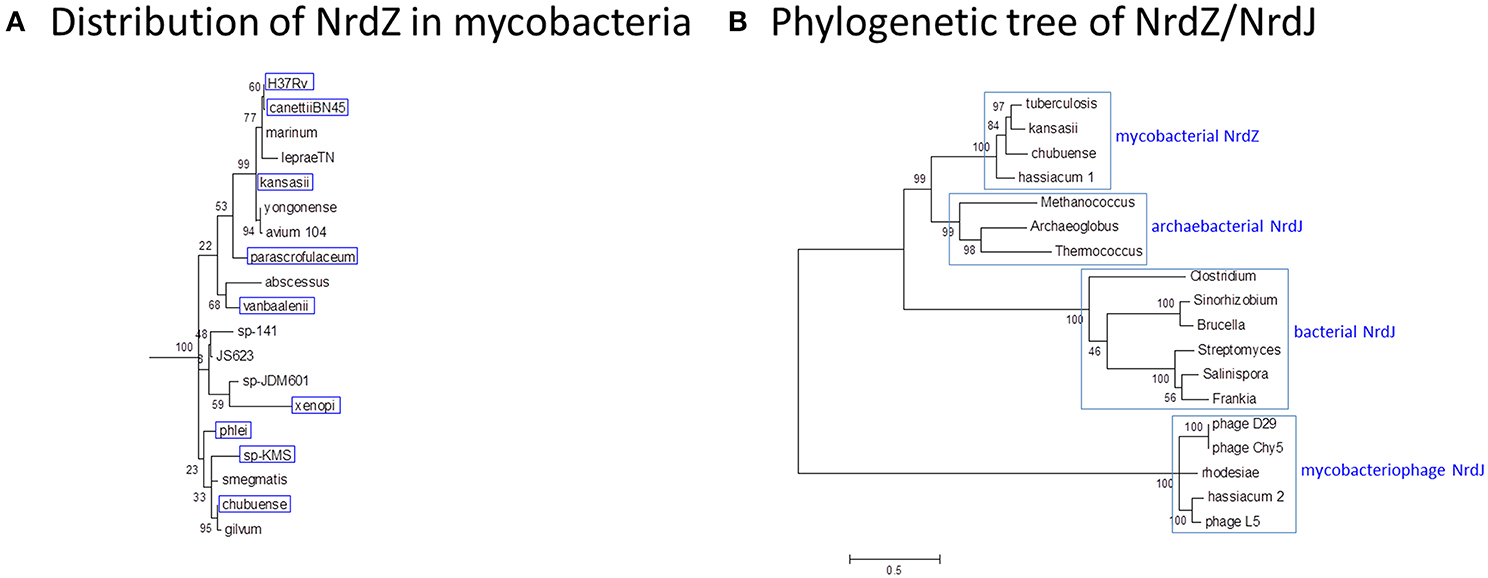
Figure 9. Phylogeny of B12-dependent ribonucleotide reductase (NrdZ). (A) Distribution of NrdZ in mycobacteria. NrdZ-positive mycobacteria are boxed in blue. The distribution of NrdZ in mycobacteria does not align with a conventional phylogeny. The mycobacteria phylogenetic tree is based on 16S rRNA sequences as described for Figure 1. (B) Phylogenetic tree of NrdZ/NrdJ. Mycobacterial NrdZ has a phylogenetic origin distinct from that of class II RNRs in other eubacteria, clustering more closely with archaebacterial NrdJ. Copies of NrdJ carried by mycobacteriophages and occasionally inserted into mycobacterial genomes also have a separate phylogenetic origin. The NrdZ phylogeny was obtained after homolog searching of the NrdZ protein present in M. tuberculosis. Muscle was used for amino acid alignment and a maximum likelihood phylogeny inferred as implemented in MEGA6 (Tamura et al., 2013). The JTT model of amino acid substitution incorporating gamma and invariant categories for rate variation was used. Thousand bootstrap pseudoreplicates were analyzed to obtain statistical support for the clades observed in the maximum likelihood topology.
Phylogenetic analysis reveals an extensive network of horizontal exchange amongst the mycobacteria (Becq et al., 2007; Stinear et al., 2008; Veyrier et al., 2009; Supply et al., 2013; Boritsch et al., 2014; Wang and Behr, 2014). Features associated with virulence attributes in M. tuberculosis—including Type VII secretion and MCE loci—are commonly found in the sequence of plasmids isolated from rapid-growing mycobacteria, and genes present in multiple copies on the M. tuberculosis genome—cytochrome P450s, adenylate cyclases, toxin-antitoxin modules, and WhiB transcription factors—are present on plasmids, mycobacteriophages, and integrated conjugative elements (Bordeleau et al., 2012). These may provide a pool of initially unassigned functional units that can be customized for specific purpose in a particular niche, or else discarded. While horizontal acquisition made an important contribution in evolution of the common ancestor with M. canettii, there is little or no evidence of horizontal transfer during diversification of the M. tuberculosis complex. Most strikingly, in spite of strong selective pressure there is no evidence of exchange of drug-resistance determinants. Presumably the pathogenic lifestyle of M. tuberculosis has resulted in its isolation from opportunities for horizontal acquisition.
NrdZ is part of the DosR regulon in M. tuberculosis, which comprises a set of genes that are induced in response to stress stimuli sensed by a two-component regulator (Park et al., 2003). Characterization of the DosR regulon in mycobacteria and related actinomycetes has identified a set of common “core” genes encoding universal stress proteins, α-crystallin chaperones, nitroreductase-domain proteins, triacylglyceride synthases, and YfiA-like hibernation factors that are presumed to facilitate transition to a survival phenotype, together with a set of “peripheral” genes with restricted distribution (Gerasimova et al., 2011). The DosR regulon in M. tuberculosis is triggered by exposure to nitric oxide, carbon monoxide, or hypoxia. NrdZ is a peripheral component of the DosR regulon and may have specific relevance for hypoxic survival of M. tuberculosis. In M. tuberculosis, NrdZ is located in a 63-gene region between Rv0568/Cyp136B and Rv0629/RecD, that includes seven toxin-antitoxin modules and an mce locus. This region has only 17 genes in M. marinum, while the corresponding 61-gene region in M. kansasii includes NrdZ (MKAN_19005) and other genes linked to anaerobic metabolism. These anaerobic functions are missing from M. marinum and M. leprae and may have been acquired by horizontal transfer.
The NrdZ locus in M. tuberculosis and M. kansasii also includes the peripheral DosR regulon gene pncB2 (Rv0573c, MKAN_19060) encoding a phosphoribosyltransferase involved in nicotinamide salvage. Again, PncB2 is absent from M. marinum. Nicotinamide provides another example of a vitamin that can be shared by host and pathogen. While M. tuberculosis has the potential to synthesize nicotinamide adenine dinucleotide (NAD+) de novo, growth of an nadABC deletion mutant in a mouse model (Vilcheze et al., 2010) shows that—in common with other human pathogens including Shigella flexneri, Streptococci and Staphylococcus aureus (Prunier et al., 2007; Sorci et al., 2013)—M. tuberculosis is able to scavenge nicotinamide from the host during infection. However, while M. leprae has preserved the capacity for de novo synthesis, it has lost the pncA and pncB salvage pathway genes. PncA is inactivated by an H57D SNP in cattle-adapted M. bovis, an L177R SNP is predicted to impact PncB2 function in Lineage 5, and PncA mutations are selected in pyrazinamide-resistant strains of M. tuberculosis without incurring any loss in pathogenesis, suggesting that retention of de novo synthesis is preferred over host scavenging as a source of the vitamin. Retention of different pathways by different pathogens may reflect selective pressures linked to nicotinamide availability in particular colonization niches.
B12-Independent Ribonucleotide Reductase
Genes encoding components of the major class Ib RNR—NrdE (Rv3051c) and NrdF2 (Rv3048c), with electrons supplied by NrdH (Rv3053c) and NrdI (Rv3052c)—are conserved in all mycobacteria, and additional genes with sequence homology to the thioredoxin/glutaredoxin-like NrdH are found on plasmids and mycobacteriophages (Dwivedi et al., 2013). In addition, in common with some other mycobacteria, M. tuberculosis has a second beta subunit gene (NrdF1/Rv1981c) and a gene encoding a third beta subunit that resembles a class Ic RNR (NrdB/Rv0233). While the alternative beta subunits are not essential for normal growth (Mowa et al., 2009), NrdF1 is active in a biochemical assay (Hammerstad et al., 2014) and it is possible that they make some contribution to survival in particular environments. The differential distribution of NrdB amongst mycobacterial species and the presence of a homolog on a plasmid from M. yongonense (pMyong1, OEM_p100240) suggest that it is also subject to exchange by horizontal transfer.
Expression of the class Ib RNR is regulated by transcription factor NrdR binding to characteristic tandem sites upstream of nrdF2 and nrdH (Rodionov and Gelfand, 2005). Consistent with the demonstration of repressor function for NrdR (Mowa et al., 2009), the binding sites are located at positions +15.5/+47.5 (nrdF2) and +15.5/+60.5 (nrdH) with respect to transcription start sites at 3409418 and 3415168, respectively (Cortes et al., 2013). The 32 bp spacing between tandem NrdR sites upstream of nrdH (Rodionov and Gelfand, 2005) is extended in M. tuberculosis H37Rv by duplication of a 13 bp sequence. In isolates belonging to “modern” Eurasian Lineages 2, 3, and 4 and Ethiopian Lineage 7, a C/T SNP at position 3415332 generates a new TANNNT −10 consensus motif (Rose et al., 2013) and an additional transcription start site upstream of NrdH providing the potential for NrdR-independent expression.
Utilization of Ethanolamine
The ability to utilize ethanolamine is an important virulence determinant for enteric pathogens (Garsin, 2010) and M. marinum has eutB and eutC genes encoding a predicted B12-dependent ethanolamine ammonia-lyase. These genes are inserted along with an ethanolamine transporter and an adenylate cyclase between the end of a carbon monoxide dehydrogenase locus (MMAR_0662/Rv0368c) (Cook et al., 2014) and a conserved glycosyl hydrolase (MMAR_0667/Rv0365c). In M. tuberculosis this location is occupied by genes encoding a zeta-epsilon toxin-antitoxin pair (Rv0367c-Rv0366c). Amongst the slow-growing mycobacteria, the three ethanolamine utilization genes are also present in M. gastri, and M. parascrofulaceum has copies of eutB and eutC. In host-adapted M. ulcerans, eutB, the ethanolamine transporter and the adenylate cyclase are present in the form of pseudogenes. A subset of rapid-growing mycobacteria, including M. smegmatis mc2155, have the potential for ethanolamine utilization, with the relevant enzymes likely sequestered within specialized microcompartments (Axen et al., 2014).
Concluding Comments
An important barrier to understanding the biology of infection—and design of effective antibacterial agents—is that the physiology of bacteria growing in culture differs from that of the same bacteria in the host. Direct characterization of the in vivo metabolome faces the technical limitation that the vast majority of metabolites are common to both host and pathogen and, at least in the case of tuberculosis, that the heterogeneity of lesions is likely to be mirrored by a corresponding heterogeneity in relevant metabolic phenotypes (Barry et al., 2009). Evolutionary biology offers an alternative approach of using phylogenetic evidence of adaptation to identify areas of metabolism that may play a crucial role in successful pathogenesis. We have used vitamin B12-dependent metabolism in M. tuberculosis to explore this strategy, allowing us to sample adaptations to metabolic pathways involved in methionine synthesis, methylmalonate processing, and DNA metabolism. Figure 10 summarizes differences in B12-related metabolism between M. tuberculosis and related mycobacterial pathogens M. marinum and M. leprae.
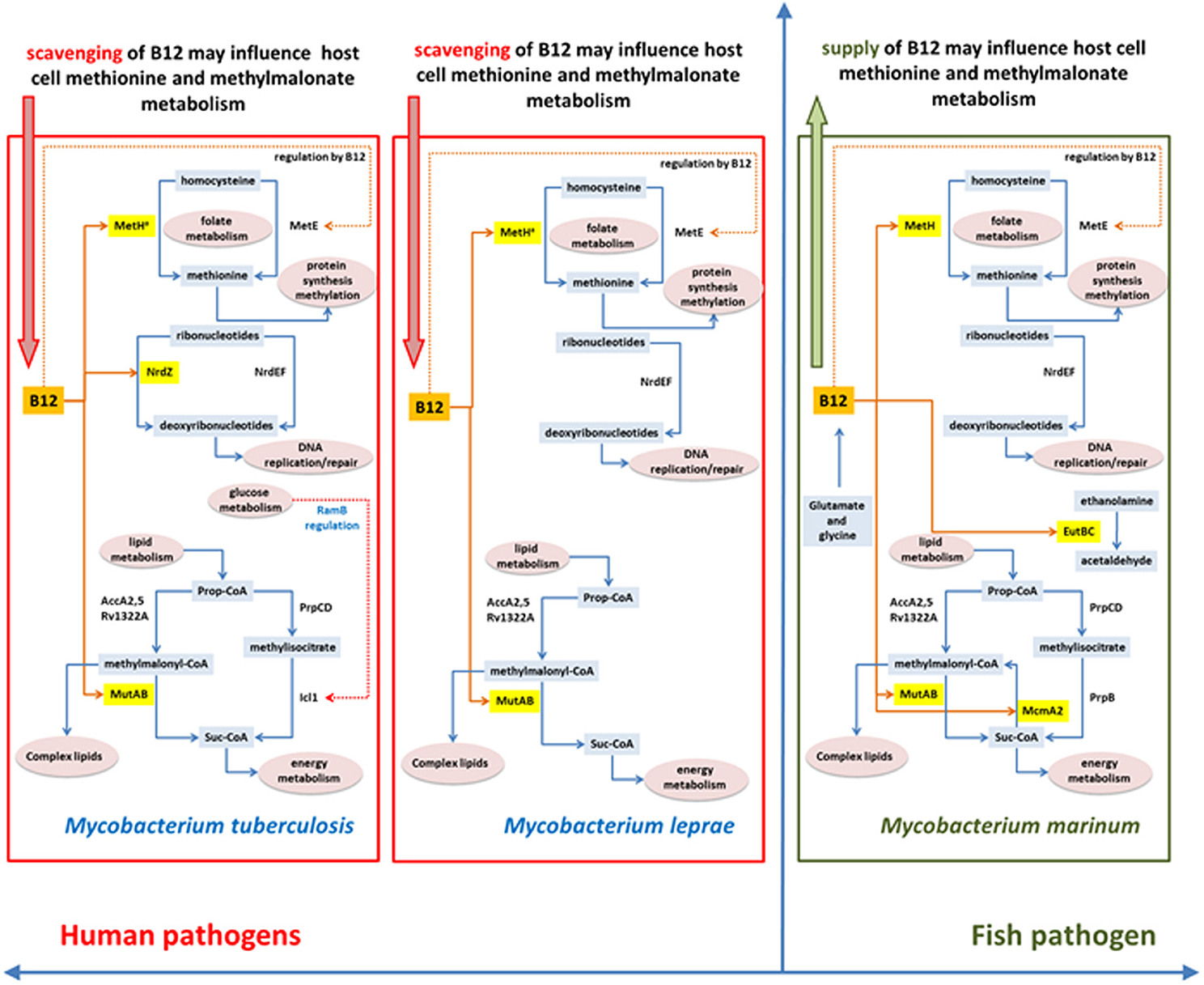
Figure 10. Differences and similarities between B12-related metabolism between M. tuberculosis, M. leprae and M. marinum.
Phylogenetic analysis highlights a marked difference in the evolution of anaerobic metabolism between closely-related pathogens. While M. marinum lacks a class II ribonucleotide reductase and constitutive as well as DosR-regulated enzymes for nitrate respiration, these genes are all present in M. tuberculosis together with a fumarate reductase that contributes to reductive operation of the TCA cycle (Rv1552-Rv1555, likely to have been acquired by horizontal transfer) (Watanabe et al., 2011). Further mutations that enhance constitutive expression of the NarG operon and the DosR regulon have been acquired by modern lineages of M. tuberculosis (Rose et al., 2013) suggesting an increasing reliance on anaerobic metabolism. This is consistent with inferences from experimental models (Via et al., 2008), but the phylogenetic evidence provides an additional important link to the role of hypoxia in the long-term context of host-pathogen co-evolution in human populations. While M. marinum has attractive properties as a surrogate model for tuberculosis drug discovery (Takaki et al., 2012), it would not be an appropriate choice in the case of drugs targeting hypoxic phenotypes. Similar to M. marinum, the M. leprae genome shows no evidence of a role for anaerobic metabolism.
Phylogenetic analysis highlights a dynamic exchange of genes amongst mycobacteria by horizontal transfer mediated by phages, plasmids, and conjugative elements that is likely to have played a crucial role in pre-adaptation of M. tuberculosis to its pathogenic role. While several M. tuberculosis virulence determinants can be traced back to horizontal acquisition by the M. canettii-like ancestor (Veyrier et al., 2009; Supply et al., 2013; Wang and Behr, 2014), it cannot be assumed that all such acquisitions reflect positive selection—many unique loci may simply be “passing through,” unlinked to functional biology of the host organism. The absence of further gene acquisition during diversification of the M. tuberculosis complex is a fundamentally important aspect of the biology of tuberculosis. M. tuberculosis is forced to rely on alterations to its existing genetic complement to optimize survival in an evolving human host, and is denied access to mobile drug-resistance elements. It has been proposed that M. tuberculosis has adapted to a more virulent phenotype over the course of its co-evolution with human populations (Comas et al., 2013); changes in vitamin B12 availability during the Neolithic transition from predominantly meat-eating hunter gatherers to vegetarian farmers represents one potential stimulus for mycobacterial adaptation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Axen, S. D., Erbilgin, O., and Kerfeld, C. A. (2014). A taxonomy of bacterial microcompartment loci constructed by a novel scoring method. PLoS Comput. Biol. 10:e1003898. doi: 10.1371/journal.pcbi.1003898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Azad, A. K., Sirakova, T. D., Fernandes, N. D., and Kolattukudy, P. E. (1997). Gene knockout reveals a novel gene cluster for the synthesis of a class of cell wall lipids unique to pathogenic mycobacteria. J. Biol. Chem. 272, 16741–16745. doi: 10.1074/jbc.272.27.16741
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barron, M. (1933). Pernicious anemia and tuberculosis: is there an antagonism? JAMA 100, 1590–1592. doi: 10.1001/jama.1933.02740200024007
Barry, C. E. III, Boshoff, H. I., Dartois, V., Dick, T., Ehrt, S., Flynn, J., et al. (2009). The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 7, 845–855. doi: 10.1038/nrmicro2236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becq, J., Gutierrez, M. C., Rosas-Magallanes, V., Rauzier, J., Gicquel, B., Neyrolles, O., et al. (2007). Contribution of horizontally acquired genomic islands to the evolution of the tubercle bacilli. Mol. Biol. Evol. 24, 1861–1871. doi: 10.1093/molbev/msm111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berg, S., Firdessa, R., Habtamu, M., Gadisa, E., Mengistu, A., Yamuah, L., et al. (2009). The burden of mycobacterial disease in ethiopian cattle: implications for public health. PLoS ONE 4:e5068. doi: 10.1371/annotation/f7240b30-f202-45c5-aacb-4fda2efb8b3d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blouin, Y., Cazajous, G., Dehan, C., Soler, C., Vong, R., Hassan, M. O., et al. (2014). Progenitor “Mycobacterium canettii” clone responsible for lymph node tuberculosis epidemic, Djibouti. Emerg. Infect. Dis. 20, 21–28. doi: 10.3201/eid2001.130652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Booker, S., Licht, S., Broderick, J., and Stubbe, J. (1994). Coenzyme B12-dependent ribonucleotide reductase: evidence for the participation of five cysteine residues in ribonucleotide reduction. Biochemistry 33, 12676–12685. doi: 10.1021/bi00208a019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bordeleau, E., Ghinet, M. G., and Burrus, V. (2012). Diversity of integrating conjugative elements in actinobacteria: coexistence of two mechanistically different DNA-translocation systems. Mob. Genet. Elem. 2, 119–124. doi: 10.4161/mge.20498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boritsch, E. C., Supply, P., Honore, N., Seeman, T., Stinear, T. P., and Brosch, R. (2014). A glimpse into the past and predictions for the future: the molecular evolution of the tuberculosis agent. Mol. Microbiol. 93, 835–852. doi: 10.1111/mmi.12720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bos, K. I., Harkins, K. M., Herbig, A., Coscolla, M., Weber, N., Comas, I., et al. (2014). Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature 514, 494–497. doi: 10.1038/nature13591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brosch, R., Gordon, S. V., Marmiesse, M., Brodin, P., Buchrieser, C., Eiglmeier, K., et al. (2002). A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. U.S.A. 99, 3684–3689. doi: 10.1073/pnas.052548299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cegielski, J. P., and McMurray, D. N. (2004). The relationship between malnutrition and tuberculosis: evidence from studies in humans and experimental animals. Int. J. Tuberc. Lung Dis. 8, 286–298.
Chakraborty, S., Gruber, T., Barry, C. E. III, Boshoff, H. I., and Rhee, K. Y. (2013). Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91. doi: 10.1126/science.1228980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chanarin, I., and Stephenson, E. (1988). Vegetarian diet and cobalamin deficiency: their association with tuberculosis. J. Clin. Pathol. 41, 759–762. doi: 10.1136/jcp.41.7.759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., McGarrell, D. M., Sun, Y., et al. (2014). Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., et al. (2001). Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011. doi: 10.1038/35059006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Comas, I., Coscolla, M., Luo, T., Borrell, S., Holt, K. E., Kato-Maeda, M., et al. (2013). Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182. doi: 10.1038/ng.2744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cook, G. M., Hards, K., Vilcheze, C., Hartman, T., and Berney, M. (2014). Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol. Spectr. 2, 1–30. doi: 10.1128/microbiolspec.MGM2-0015-2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cortes, T., Schubert, O. T., Rose, G., Arnvig, K. B., Comas, I., Aebersold, R., et al. (2013). Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 5, 1121–1131. doi: 10.1016/j.celrep.2013.10.031
Cracan, V., and Banerjee, R. (2012). Novel coenzyme B12-dependent interconversion of isovaleryl-CoA and pivalyl-CoA. J. Biol. Chem. 287, 3723–3732. doi: 10.1074/jbc.M111.320051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dawes, S. S., Warner, D. F., Tsenova, L., Timm, J., McKinney, J. D., Kaplan, G., et al. (2003). Ribonucleotide reduction in Mycobacterium tuberculosis: function and expression of genes encoding class Ib and class II ribonucleotide reductases. Infect. Immun. 71, 6124–6131. doi: 10.1128/IAI.71.11.6124-6131.2003
de Carvalho, L. P., Fischer, S. M., Marrero, J., Nathan, C., Ehrt, S., and Rhee, K. Y. (2010). Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17, 1122–1131. doi: 10.1016/j.chembiol.2010.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dereeper, A., Guignon, V., Blanc, G., Audic, S., Buffet, S., Chevenet, F., et al. (2008). Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469. doi: 10.1093/nar/gkn180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ditkowski, B., Troc, P., Ginda, K., Donczew, M., Chater, K. F., Zakrzewska-Czerwinska, J., et al. (2010). The actinobacterial signature protein ParJ (SCO1662) regulates ParA polymerization and affects chromosome segregation and cell division during Streptomyces sporulation. Mol. Microbiol. 78, 1403–1415. doi: 10.1111/j.1365-2958.2010.07409.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doig, K. D., Holt, K. E., Fyfe, J. A., Lavender, C. J., Eddyani, M., Portaels, F., et al. (2012). On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genomics 13:258. doi: 10.1186/1471-2164-13-258
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doxey, A. C., Kurtz, D. A., Lynch, M., Sauder, L. A., and Neufeld, J. D. (2015). Aquatic metagenomes implicate Thaumarchaeota in global cobalamin production. ISME J. 9, 461–471. doi: 10.1038/ismej.2014.142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dwivedi, B., Xue, B., Lundin, D., Edwards, R. A., and Breitbart, M. (2013). A bioinformatic analysis of ribonucleotide reductase genes in phage genomes and metagenomes. BMC Evol. Biol. 13:33. doi: 10.1186/1471-2148-13-33
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dye, C., Glaziou, P., Floyd, K., and Raviglione, M. (2013). Prospects for tuberculosis elimination. Annu. Rev. Public Health 34, 271–286. doi: 10.1146/annurev-publhealth-031912-114431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eoh, H., and Rhee, K. Y. (2014). Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids. Proc. Natl. Acad. Sci. U.S.A. 111, 4976–4981. doi: 10.1073/pnas.1400390111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fenner, L., Egger, M., Bodmer, T., Furrer, H., Ballif, M., Battegay, M., et al. (2013). HIV infection disrupts the sympatric host-pathogen relationship in human tuberculosis. PLoS Genet. 9:e1003318. doi: 10.1371/journal.pgen.1003318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fernandes, N. D., and Kolattukudy, P. E. (1998). A newly identified methyl-branched chain fatty acid synthesizing enzyme from Mycobacterium tuberculosis var. bovis BCG. J. Biol. Chem. 273, 2823–2828. doi: 10.1074/jbc.273.5.2823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Firdessa, R., Tschopp, R., Wubete, A., Sombo, M., Hailu, E., Erenso, G., et al. (2012). High prevalence of bovine tuberculosis in dairy cattle in central ethiopia: implications for the dairy industry and public health. PLoS ONE 7:e52851. doi: 10.1371/journal.pone.0052851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gagneux, S., Deriemer, K., Van, T., Kato-Maeda, M., De Jong, B. C., Narayanan, S., et al. (2006). Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2869–2873. doi: 10.1073/pnas.0511240103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gago, G., Kurth, D., Diacovich, L., Tsai, S. C., and Gramajo, H. (2006). Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J. Bacteriol. 188, 477–486. doi: 10.1128/JB.188.2.477-486.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gal-Mor, O., Boyle, E. C., and Grassl, G. A. (2014). Same species, different diseases: how and why typhoidal and non-typhoidal Salmonella enterica serovars differ. Front. Microbiol. 5:391. doi: 10.3389/fmicb.2014.00391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, B., and Gupta, R. S. (2012). Phylogenetic framework and molecular signatures for the main clades of the phylum Actinobacteria. Microbiol. Mol. Biol. Rev. 76, 66–112. doi: 10.1128/MMBR.05011-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garsin, D. A. (2010). Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat. Rev. Microbiol. 8, 290–295. doi: 10.1038/nrmicro2334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gerasimova, A., Kazakov, A. E., Arkin, A. P., Dubchak, I., and Gelfand, M. S. (2011). Comparative genomics of the dormancy regulons in mycobacteria. J. Bacteriol. 193, 3446–3452. doi: 10.1128/JB.00179-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez, J. C., Banerjee, R. V., Huang, S., Sumner, J. S., and Matthews, R. G. (1992). Comparison of cobalamin-independent and cobalamin-dependent methionine synthases from Escherichia coli: two solutions to the same chemical problem. Biochemistry 31, 6045–6056. doi: 10.1021/bi00141a013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gopinath, K., Moosa, A., Mizrahi, V., and Warner, D. F. (2013a). Vitamin B(12) metabolism in Mycobacterium tuberculosis. Future Microbiol. 8, 1405–1418. doi: 10.2217/fmb.13.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gopinath, K., Venclovas, C., Ioerger, T. R., Sacchettini, J. C., McKinney, J. D., Mizrahi, V., et al. (2013b). A vitamin B(1)(2) transporter in Mycobacterium tuberculosis. Open Biol. 3:120175. doi: 10.1098/rsob.120175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gould, T. A., Van De Langemheen, H., Munoz-Elias, E. J., McKinney, J. D., and Sacchettini, J. C. (2006). Dual role of isocitrate lyase 1 in the glyoxylate and methylcitrate cycles in Mycobacterium tuberculosis. Mol. Microbiol. 61, 940–947. doi: 10.1111/j.1365-2958.2006.05297.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grana, M., Bellinzoni, M., Miras, I., Fiez-Vandal, C., Haouz, A., Shepard, W., et al. (2009). Structure of Mycobacterium tuberculosis Rv2714, a representative of a duplicated gene family in Actinobacteria. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 65, 972–977. doi: 10.1107/S1744309109035027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hammerstad, M., Rohr, A. K., Andersen, N. H., Graslund, A., Hogbom, M., and Andersson, K. K. (2014). The class Ib ribonucleotide reductase from Mycobacterium tuberculosis has two active R2F subunits. J. Biol. Inorg. Chem. 19, 893–902. doi: 10.1007/s00775-014-1121-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Helliwell, K. E., Wheeler, G. L., Leptos, K. C., Goldstein, R. E., and Smith, A. G. (2011). Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Mol. Biol. Evol. 28, 2921–2933. doi: 10.1093/molbev/msr124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keating, L. A., Wheeler, P. R., Mansoor, H., Inwald, J. K., Dale, J., Hewinson, R. G., et al. (2005). The pyruvate requirement of some members of the Mycobacterium tuberculosis complex is due to an inactive pyruvate kinase: implications for in vivo growth. Mol. Microbiol. 56, 163–174. doi: 10.1111/j.1365-2958.2005.04524.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kelley, L. A., and Sternberg, M. J. (2009). Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371. doi: 10.1038/nprot.2009.2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, K. M., Go, J., Yoon, M. Y., Park, Y., Kim, S. C., Yong, D. E., et al. (2012). Vitamin B12-mediated restoration of defective anaerobic growth leads to reduced biofilm formation in Pseudomonas aeruginosa. Infect. Immun. 80, 1639–1649. doi: 10.1128/IAI.06161-11
Lyonnet, B. B., Diacovich, L., Cabruja, M., Bardou, F., Quemard, A., Gago, G., et al. (2014). Pleiotropic effect of AccD5 and AccE5 depletion in acyl-coenzyme A carboxylase activity and in lipid biosynthesis in mycobacteria. PLoS ONE 9:e99853. doi: 10.1371/journal.pone.0099853
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martens, J. H., Barg, H., Warren, M. J., and Jahn, D. (2002). Microbial production of vitamin B12. Appl. Microbiol. Biotechnol. 58, 275–285. doi: 10.1007/s00253-001-0902-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Masiewicz, P., Brzostek, A., Wolanski, M., Dziadek, J., and Zakrzewska-Czerwinska, J. (2012). A novel role of the PrpR as a transcription factor involved in the regulation of methylcitrate pathway in Mycobacterium tuberculosis. PLoS ONE 7:e43651. doi: 10.1371/journal.pone.0043651
Micklinghoff, J. C., Breitinger, K. J., Schmidt, M., Geffers, R., Eikmanns, B. J., and Bange, F. C. (2009). Role of the transcriptional regulator RamB (Rv0465c) in the control of the glyoxylate cycle in Mycobacterium tuberculosis. J. Bacteriol. 191, 7260–7269. doi: 10.1128/JB.01009-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mowa, M. B., Warner, D. F., Kaplan, G., Kana, B. D., and Mizrahi, V. (2009). Function and regulation of class I ribonucleotide reductase-encoding genes in mycobacteria. J. Bacteriol. 191, 985–995. doi: 10.1128/JB.01409-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, S., Fleck, C. B., Wilson, D., Hummert, C., Hube, B., and Brock, M. (2011). Gene acquisition, duplication and metabolic specification: the evolution of fungal methylisocitrate lyases. Environ. Microbiol. 13, 1534–1548. doi: 10.1111/j.1462-2920.2011.02458.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munoz-Elias, E. J., Upton, A. M., Cherian, J., and McKinney, J. D. (2006). Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol. Microbiol. 60, 1109–1122. doi: 10.1111/j.1365-2958.2006.05155.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ng, P. C., and Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nuccio, S. P., and Baumler, A. J. (2014). Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. MBio 5, e00929–e00914. doi: 10.1128/mBio.00929-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Park, H. D., Guinn, K. M., Harrell, M. I., Liao, R., Voskuil, M. I., Tompa, M., et al. (2003). Rv3133c/dosR is a transcription factor that mediates the hypoxic response of Mycobacterium tuberculosis. Mol. Microbiol. 48, 833–843. doi: 10.1046/j.1365-2958.2003.03474.x
Pejchal, R., and Ludwig, M. L. (2005). Cobalamin-independent methionine synthase (MetE): a face-to-face double barrel that evolved by gene duplication. PLoS Biol. 3:e31. doi: 10.1371/journal.pbio.0030031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prunier, A. L., Schuch, R., Fernandez, R. E., Mumy, K. L., Kohler, H., McCormick, B. A., et al. (2007). nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology 153, 2363–2372. doi: 10.1099/mic.0.2007/006916-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rainwater, D. L., and Kolattukudy, P. E. (1983). Synthesis of mycocerosic acids from methylmalonyl coenzyme A by cell-free extracts of Mycobacterium tuberculosis var. bovis BCG. J. Biol. Chem. 258, 2979–2985.
Rainwater, D. L., and Kolattukudy, P. E. (1985). Fatty acid biosynthesis in Mycobacterium tuberculosis var. bovis Bacillus Calmette-Guerin. Purification and characterization of a novel fatty acid synthase, mycocerosic acid synthase, which elongates n-fatty acyl-CoA with methylmalonyl-CoA. J. Biol. Chem. 260, 616–623.
Ramagopalan, S. V., Goldacre, R., Skingsley, A., Conlon, C., and Goldacre, M. J. (2013). Associations between selected immune-mediated diseases and tuberculosis: record-linkage studies. BMC Med. 11:97. doi: 10.1186/1741-7015-11-97
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rhee, K. Y., De Carvalho, L. P., Bryk, R., Ehrt, S., Marrero, J., Park, S. W., et al. (2011). Central carbon metabolism in Mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol. 19, 307–314. doi: 10.1016/j.tim.2011.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodionov, D. A., and Gelfand, M. S. (2005). Identification of a bacterial regulatory system for ribonucleotide reductases by phylogenetic profiling. Trends Genet. 21, 385–389. doi: 10.1016/j.tig.2005.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodionov, D. A., Vitreschak, A. G., Mironov, A. A., and Gelfand, M. S. (2003). Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J. Biol. Chem. 278, 41148–41159. doi: 10.1074/jbc.M305837200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rose, G., Cortes, T., Comas, I., Coscolla, M., Gagneux, S., and Young, D. B. (2013). Mapping of genotype-phenotype diversity among clinical isolates of Mycobacterium tuberculosis by sequence-based transcriptional profiling. Genome Biol. Evol. 5, 1849–1862. doi: 10.1093/gbe/evt138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Savvi, S., Warner, D. F., Kana, B. D., McKinney, J. D., Mizrahi, V., and Dawes, S. S. (2008). Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J. Bacteriol. 190, 3886–3895. doi: 10.1128/JB.01767-07
Sintchak, M. D., Arjara, G., Kellogg, B. A., Stubbe, J., and Drennan, C. L. (2002). The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat. Struct. Biol. 9, 293–300. doi: 10.1038/nsb774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sorci, L., Blaby, I. K., Rodionova, I. A., De Ingeniis, J., Tkachenko, S., De Crecy-Lagard, V., et al. (2013). Quinolinate salvage and insights for targeting NAD biosynthesis in group A streptococci. J. Bacteriol. 195, 726–732. doi: 10.1128/JB.02002-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stinear, T. P., Seemann, T., Harrison, P. F., Jenkin, G. A., Davies, J. K., Johnson, P. D., et al. (2008). Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18, 729–741. doi: 10.1101/gr.075069.107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Supply, P., Marceau, M., Mangenot, S., Roche, D., Rouanet, C., Khanna, V., et al. (2013). Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat. Genet. 45, 172–179. doi: 10.1038/ng.2517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takaki, K., Cosma, C. L., Troll, M. A., and Ramakrishnan, L. (2012). An in vivo platform for rapid high-throughput antitubercular drug discovery. Cell Rep. 2, 175–184. doi: 10.1016/j.celrep.2012.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tisa, L. S., Beauchemin, N., Gtari, M., Sen, A., and Wall, L. G. (2013). What stories can the Frankia genomes start to tell us? J. Biosci. 38, 719–726. doi: 10.1007/s12038-013-9364-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veyrier, F., Pletzer, D., Turenne, C., and Behr, M. A. (2009). Phylogenetic detection of horizontal gene transfer during the step-wise genesis of Mycobacterium tuberculosis. BMC Evol. Biol. 9:196. doi: 10.1186/1471-2148-9-196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Via, L. E., Lin, P. L., Ray, S. M., Carrillo, J., Allen, S. S., Eum, S. Y., et al. (2008). Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect. Immun. 76, 2333–2340. doi: 10.1128/IAI.01515-07
Vilcheze, C., Weinrick, B., Wong, K. W., Chen, B., and Jacobs, W. R. Jr. (2010). NAD+ auxotrophy is bacteriocidal for the tubercle bacilli. Mol. Microbiol. 76, 365–377. doi: 10.1111/j.1365-2958.2010.07099.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vitreschak, A. G., Rodionov, D. A., Mironov, A. A., and Gelfand, M. S. (2003). Regulation of the vitamin B12 metabolism and transport in bacteria by a conserved RNA structural element. RNA 9, 1084–1097. doi: 10.1261/rna.5710303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H., Tomasch, J., Jarek, M., and Wagner-Dobler, I. (2014). A dual-species co-cultivation system to study the interactions between Roseobacters and dinoflagellates. Front. Microbiol. 5:311. doi: 10.3389/fmicb.2014.00311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, J., and Behr, M. A. (2014). Building a better bacillus: the emergence of Mycobacterium tuberculosis. Front. Microbiol. 5:139. doi: 10.3389/fmicb.2014.00139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, S., Dong, X., Zhu, Y., Wang, C., Sun, G., Luo, T., et al. (2013). Revealing of Mycobacterium marinum transcriptome by RNA-seq. PLoS ONE 8:e75828. doi: 10.1371/journal.pone.0075828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warner, D. F., Savvi, S., Mizrahi, V., and Dawes, S. S. (2007). A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in Mycobacterium tuberculosis: implications for differential methionine synthase function in strains H37Rv and CDC1551. J. Bacteriol. 189, 3655–3659. doi: 10.1128/JB.00040-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Watanabe, S., Zimmermann, M., Goodwin, M. B., Sauer, U., Barry, C. E. III, and Boshoff, H. I. (2011). Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 7:e1002287. doi: 10.1371/journal.ppat.1002287
Watson, E., Macneil, L. T., Ritter, A. D., Yilmaz, L. S., Rosebrock, A. P., Caudy, A. A., et al. (2014). Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell 156, 759–770. doi: 10.1016/j.cell.2014.01.047
Zhang, Y., Rodionov, D. A., Gelfand, M. S., and Gladyshev, V. N. (2009). Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC Genomics 10:78. doi: 10.1186/1471-2164-10-78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ziemert, N., Lechner, A., Wietz, M., Millan-Aguinaga, N., Chavarria, K. L., and Jensen, P. R. (2014). Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. U.S.A. 111, E1130–E1139. doi: 10.1073/pnas.1324161111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Mycobacterium tuberculosis, phylogenetic analysis, vitamin B12, metabolism, mycobacteria
Citation: Young DB, Comas I and de Carvalho LPS (2015) Phylogenetic analysis of vitamin B12-related metabolism in Mycobacterium tuberculosis. Front. Mol. Biosci. 2:6. doi: 10.3389/fmolb.2015.00006
Received: 24 November 2014; Paper pending published: 16 December 2014;
Accepted: 09 February 2015; Published online: 04 March 2015.
Edited by:
Sandra Macedo-Ribeiro, Instituto de Biologia Molecular e Celular, PortugalReviewed by:
Digby Warner, University of Cape Town, South AfricaNuno Empadinhas, Center for Neuroscience and Cell Biology, Portugal
Copyright © 2015 Young, Comas and de Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luiz P. S. de Carvalho, Division of Mycobacterial Research, MRC National Institute for Medical Research, The Ridgeway, Mill Hill, London, NW7 1AA, UK e-mail:bHVpei5wZWRyb0BuaW1yLm1yYy5hYy51aw==