 Dimce Micevski
Dimce Micevski Jessica E. Zammit
Jessica E. Zammit Kaye N. Truscott
Kaye N. Truscott David A. Dougan
David A. Dougan- Department of Biochemistry, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, VIC, Australia
In Escherichia coli, σS is the master regulator of the general stress response. The level of σS changes in response to multiple stress conditions and it is regulated at many levels including protein turnover. In the absence of stress, σS is rapidly degraded by the AAA+ protease, ClpXP in a regulated manner that depends on the adaptor protein RssB. This two-component response regulator mediates the recognition of σS and its delivery to ClpXP. The turnover of σS however, can be inhibited in a stress specific manner, by one of three anti-adaptor proteins. Each anti-adaptor binds to RssB and inhibits its activity, but how this is achieved is not fully understood at a molecular level. Here, we describe details of the interaction between each anti-adaptor and RssB that leads to the stabilization of σS. By defining the domains of RssB using partial proteolysis we demonstrate that each anti-adaptor uses a distinct mode of binding to inhibit RssB activity. IraD docks specifically to the N-terminal domain of RssB, IraP interacts primarily with the C-terminal domain, while IraM interacts with both domains. Despite these differences in binding, we propose that docking of each anti-adaptor induces a conformational change in RssB, which resembles the inactive dimer of RssB. This dimer-like state of RssB not only prevents substrate binding but also triggers substrate release from a pre-bound complex.
Introduction
In their natural environment, bacteria are constantly exposed to changing and sometimes harsh environmental conditions. To survive these changes they have developed sophisticated stress response pathways to regulate the expression of specific genes that either restore cellular homeostasis or enable the bacteria to adapt to their new conditions. A key transcription factor or master regulator generally orchestrates these programmed changes. In Escherichia coli and related enteric bacteria, the cells response to a wide variety of different stress conditions (often referred to as the general stress response) is coordinated by a single transcription factor, σS (also known as σ38 or RpoS) (Hengge-Aronis, 2002). As such, the cellular levels of σS are highly regulated, not only at the transcriptional and translational levels, but also at the post-translational level through regulated proteolysis (Hengge, 2009; Battesti et al., 2011; Micevski and Dougan, 2013). In the absence of stress, σS levels remain low, largely as a result of its rapid turnover by the AAA+ (ATPases associated with a variety of cellular activities) protease ClpXP. Importantly, the turnover of σS is mediated by a specialized adaptor protein, RssB (also known as SprE) (Muffler et al., 1996; Pratt and Silhavy, 1996). RssB is a member of the response regulator (RR) family of proteins and like most RRs it is composed of two domains; a receiver domain and an effector (or output) domain (Galperin, 2010). However, in the case of RssB these domains have yet to be biochemically defined. As with most RRs the receiver domain of RssB is proposed to act as a phosphorylation-mediated switch, which regulates the activity of the effector domain (Gao and Stock, 2010). Consistent with this idea, RssB can be phosphorylated on a highly conserved Asp residue (Asp58). To date however, the physiological role of RssB-phosphorylation as a means to regulate σS levels remains controversial, as σS is only partially stabilized in cells carrying a non-phosphorylatable mutant of RssB (Peterson et al., 2004; Zhou and Gottesman, 2006). It has recently been proposed that cellular ATP levels directly control σS stability (Peterson et al., 2012). Independent of this control, both the recognition of σS (by RssB) and its degradation (by ClpXP) in vitro can be enhanced by phosphorylation of RssB. However, the mechanistic details by which RssB binds to σS and delivers it to ClpXP remains poorly understood (Hengge, 2009; Micevski and Dougan, 2013).
The stability of σS is also regulated by a group of unrelated proteins, which collectively have been termed anti-adaptors as they inhibit the activity of the adaptor protein RssB. Currently, three anti-adaptors have been identified in E. coli, all of which inhibit the turnover of σS in response to a specific stress (Bougdour et al., 2006, 2008). Related anti-adaptor proteins have also been identified in Salmonella, however despite their sequence similarity with E. coli anti-adaptors, these proteins are regulated by different stress conditions (Bougdour et al., 2008; Merrikh et al., 2009a,b). In E. coli the different anti-adaptors have been named, IraP (Inhibitor of RssB activity during phosphate starvation) which as the name suggests is specifically induced in response to phosphate starvation, IraD which is specifically induced in response to DNA damage and IraM which is specifically induced in response to magnesium starvation.
Although the transcriptional regulation of all three anti-adaptors has been extensively studied and is currently well understood (Bougdour et al., 2006, 2008; Bougdour and Gottesman, 2007; Merrikh et al., 2009a,b; Battesti et al., 2012), the mechanism of action of these proteins remains poorly defined. Here we show via a detailed biochemical analysis, that all three anti-adaptors use distinct modes of binding to inhibit RssB activity.
Results
RssB is Composed of Two Domains
To dissect the mode of action of the three known anti-adaptor proteins, we first experimentally determined the domain boundaries of RssB. To do so, we performed limited proteolysis on untagged RssB (Figure 1). In the presence of thermolysin, untagged RssB (~36 kDa) was rapidly and specifically cleaved into two fragments (Figure 1A). N-terminal sequencing of the larger (~24 kDa) fragment (RVEEEE, corresponding to residues 131–136 of RssB) revealed this fragment to be the C-terminal effector domain. Therefore, in this study we defined the C-terminal domain of RssB (RssBC) as residues 131–337 and the N-terminal domain (RssBN) as residues 1–129 (Figure 1B). Next, untagged versions of each domain (RssBN and RssBC) were purified (Figure S1A) using the ubiquitin (Ub) fusion system (Catanzariti et al., 2004). To assess the activity of these domains, a series of competition-based degradation experiments were performed, in which the RssB-mediated turnover of σS by ClpXP was monitored either in the absence or presence of RssBN or RssBC (Figure S1B). Importantly, neither RssBN (Figure S1B, lower panel) nor RssBC (Figure S1B, middle panel) altered the RssB-mediated turnover of σS (Figure S1B, upper panel). These findings validated the use of these domains to assess their ability to bind to and hence inhibit the activity of each anti-adaptor in competition assays.
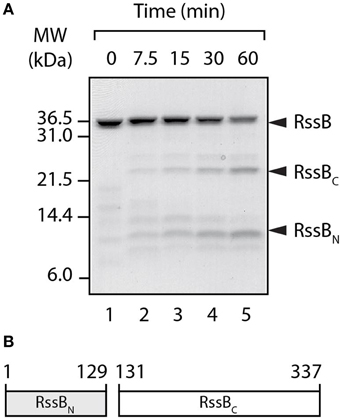
Figure 1. RssB is composed of two domains. (A) In the presence of thermolysin, RssB was digested into two stable fragments (RssBN and RssBC). Proteins were separated by SDS-PAGE and stained with CBB. (B) Schematic diagram of recombinant RssBN (residues 1–129) and RssBC, (residues 131–337) used in this study.
IraD Binds to the N-Terminal Response Regulator Domain of RssB
First we asked the question, does IraD interact with an individual domain of RssB? To address this question we monitored the ability of IraD to inhibit the RssB-mediated turnover of σS, either in the absence of added domains [Figure 2A, (ii) and B, open circles] or in the presence of RssBN [Figure 2A, (vi) and B, open triangles] or RssBC [Figure 2A, (iv) and B, open diamonds]. As expected IraD was able to inhibit the RssB-mediated turnover of σS, however in the presence of RssBN, the IraD-mediated inhibition of σS turnover was completely reversed. Importantly, this effect was specific for RssBN, as addition of RssBC was unable to relieve IraD-mediated inhibition. These data indicate that only RssBN can compete with full length RssB for binding to IraD and suggest that IraD binds specifically to the N-domain of RssB. To confirm if this relief of IraD inhibition on σS degradation was due to a specific interaction between IraD and the N-domain of RssB, a series of pull-down experiments were performed. In these experiments purified His10-tagged IraD was immobilized to Ni-NTA agarose beads and assessed for its ability to bind purified recombinant untagged RssB domains. As expected full-length RssB was specifically eluted from Ni-NTA beads containing immobilized IraD (Figure 2C, lane 11) and not from beads lacking immobilized protein (Figure 2C, lane 9). Importantly, only RssBN was specifically eluted from beads containing immobilized IraD (Figure 2C, lane 13) confirming that the N-terminal domain of RssB is sufficient for interaction with IraD.
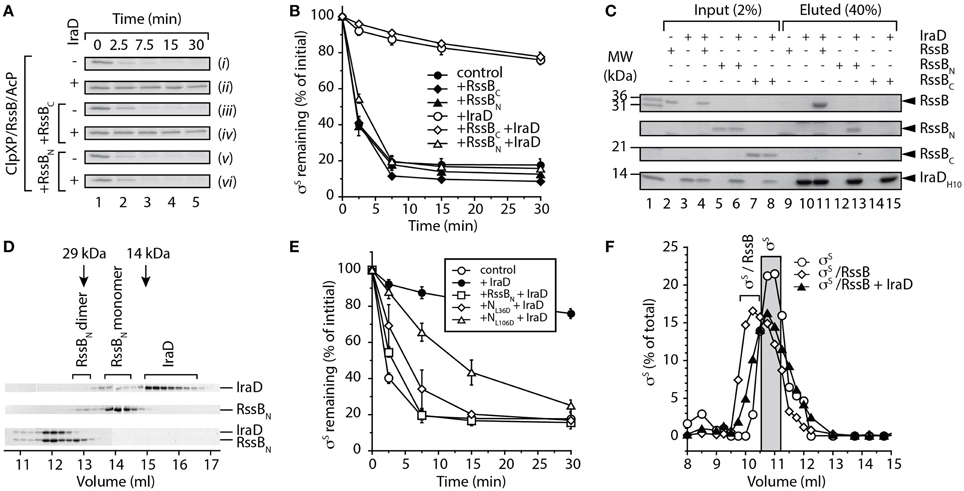
Figure 2. IraD specifically interacts with the N-terminal domain of RssB. (A) The RssB-mediated turnover of σS by ClpXP, was monitored in the absence of added domains (i and ii), or in the presence of either RssBC (iii and iv) or RssBN (v and vi), with (ii, iv, and vi) or without (i, iii, and v) IraD. Strips show σS following separation by SDS-PAGE and staining with CBB. (B) The RssB-mediated turnover of σS by ClpXP (control) was monitored either in the presence of RssBC (diamonds) or RssBN (triangles), with (open symbols) or without (filled symbols) IraD as shown in (A). Quantification (using GelEval) of the relative amount of σS remaining, from three independent experiments (n = 3). Error bars represent the standard error of the mean (s.e.m.). (C) Ni-NTA agarose beads either lacking or containing immobilized IraD were incubated with either RssB, RssBN or RssBC (lanes 2–8) and following washing the bound proteins were eluted with imidazole (lanes 9–15). Proteins were separated by SDS-PAGE and visualized with CBB. (D) CBB-stained strips, from multiple SDS-PAGE gels showing fractions following separation of 5 nmol of either IraD (top panel), RssBN (middle panel) or the IraD-RssBN complex (lower panel), by SEC using Superdex 75. (E) The RssB-mediated turnover of σS by ClpXP (control) was monitored either in the absence (open circles) or presence (filled circles) of IraD. The IraD-inhibited turnover of σS was monitored in the presence of wild type (open squares), L36D (open diamonds) or L106D (open triangles) RssBN (labeled RssBN, NL36D and NL106D, respectively). Quantification (using GelEval) of the relative amount of σS remaining, from three independent experiments (n = 3). Error bars represent the s.e.m. (F) Following preincubation of σS with RssB (+AcP), IraD was added to the reaction prior to separation of the proteins using Superdex 200. Following separation of the proteins by SEC, the fractions were analyzed by SDS-PAGE and the amount of σS quantified using GelEval. The elution profile of σS (open circles) is shifted in the presence of RssB (open diamonds). The addition of IraD to a pre-formed RssB/σS complex prevents this shift of σS (filled triangles).
To validate this mode of binding and estimate the stoichiometry of the complex we performed size exclusion chromatography (SEC). In the absence of RssBN, IraD eluted in two peaks (Figure 2D, upper panel); the predominant peak (~15 ml) representing a monomer of IraD, and a minor peak (~14 ml) representing the IraD dimer. Similarly, in the absence of IraD, RssBN also eluted in two peaks (Figure 2D, middle panel) a monomer at ~14 ml, and a minor dimeric peak (at ~13 ml). Importantly, when both proteins were incubated together and analyzed by SEC, both IraD and RssBN (Figure 2D, lower panel) co-eluted, in a single peak at ~12 ml. Based on the molecular weight of this peak (estimated using protein standards) and the intensity of the two protein bands we propose that IraD and RssBN form a heterodimer. Interestingly, a shift in the homodimer of RssBN (albeit a minor component) was not observed, suggesting that this form of RssBN is unable to interact with IraD (Figure 2D). To investigate a possible role of the RssBN dimerization interface in IraD binding, we generated a mutant of RssBN lacking the β5-α5 segment referred to here as RssBN1−104. Based on the structure of Pseudomonas aeruginosa RssB (PDB: 3EQ2; Levchenko et al., unpublished) this segment of RssB forms the dimerization interface. Analysis by SEC (data not shown) revealed that the elution profile of RssBN1−104 was not altered in the presence of IraD consistent with a loss of interaction. Collectively these data suggest that IraD binds to RssB through its dimerization interface. Importantly, the dimer of RssB is unable to recognize or deliver σS to ClpXP for degradation (Figure S2). Hence, we propose that IraD-binding, to the dimerization interface, triggers a switch in the conformation of RssB, which mimics the domain arrangement present in the dimeric form. We refer to this conformation as the “off” state.
To further validate this interaction site, we generated two single point mutations in RssBN, which based on its similarity to other RRs are located on opposite faces of the protein. In each case, a central hydrophobic residue (L36 within the N-terminal 1-2-2 interface or L106 within the C-terminal 4-5-5 interface) was replaced with aspartate (Figure S3). To monitor the effect of these single point mutations on the interaction with IraD we compared the ability of wild type and mutant N-domains (referred to here as NL36D and NL106D, respectively) to relieve the IraD-mediated inhibition of σS degradation (Figure 2E). This analysis revealed that NL106D exhibited a reduced ability to relieve the IraD-mediated inhibition of σS degradation in comparison to wild type RssBN (Figure 2E, compare open triangles with open squares). In contrast, NL36D retained the ability to inhibit IraD-activity (Figure 2E, open diamonds). Hence, these data confirm that IraD binds to N-terminal domain of RssB through the dimerization interface, which is directly linked to the C-terminal domain. Consequently, we propose that IraD-binding to the N-terminal domain may modulate communication to the C-terminal effector domain of RssB, where σS docking occurs (manuscript in preparation).
Next to determine if IraD was able to trigger release of σS from RssB we monitored the elution profile of the RssB/σS complex by SEC, before and after the addition of IraD (Figure 2F). In the absence of IraD, σS co-eluted with RssB in a single peak at ~10 ml (Figure 2F, open diamonds). However, following the addition of IraD to a preformed RssB/σS complex, the elution profile of σS (Figure 2F, filled triangles) resembled that of σS alone (Figure 2F, open circles). These data suggest that binding of IraD to the dimerization interface of RssB stabilizes the adaptor protein in a dimer-like conformation that triggers release of σS.
IraP Binds to the C-Terminal Effector Domain of RssB
Next, we asked the question, does IraP use a similar or distinct mechanism to inhibit the RssB-mediated degradation of σS? To address this question we monitored the ability of IraP to inhibit the RssB-mediated turnover of σS, in the absence or presence of a 2.5-fold excess of either RssBN or RssBC (Figures 3A, B). Consistent with published data (Bougdour et al., 2006, 2008), recombinant IraP was able to inhibit the RssB-mediated degradation of σS by ClpXP [Figure 3A, compare (ii) with (i)], however in comparison to IraD, IraP was a less potent inhibitor of σS degradation (compare Figures 2B, 3B). Interestingly, in contrast to the analysis of IraD (Figure 2B, compare open diamonds with open circles) the addition of RssBC was sufficient to partially inhibit IraP-activity as seen by the loss of inhibition of σS turnover (Figure 3B, compare open diamonds with open circles), while RssBN had no effect on the turnover of σS (Figure 3B, compare open triangles with open circles). To examine more directly the mode of interaction between IraP and RssB, we monitored the binding of IraP with each RssB domain, using a series of pull-down experiments, in which purified recombinant IraP was immobilized to Ni-NTA agarose beads, via a C-terminal His10-tag (Figure 3C). Consistent with the competition degradation assays (Figures 3A, B), an interaction between RssBC and immobilized IraP was observed (Figure 3C, lane 14). Although a specific interaction between RssBN and IraP was not detected in most pull-down experiments as shown in the representative example (Figure 3C, lane 12) “weak” binding was observed in a single experiment. Collectively, these data clearly indicate that IraP docks onto the C-terminal effector domain of RssB however a potential role, for the N-terminal domain could not be completely excluded. To further examine any possible contribution of the N-terminal domain of RssB in binding to IraP, we again employed the competition-based degradation assay to monitor σS turnover. In this case, the turnover of σS was monitored in the presence of increasing concentrations of RssBN (up to a 20-fold excess), either in the absence or presence of IraP (Figure 3D). Consistent with a weak interaction between IraP and RssBN the rate of σS turnover (in the presence of IraP) increased in the presence of higher concentrations of RssBN (Figure 3D, columns 7–10), while the same concentrations of RssBN did not affect the rate of σS turnover in the absence of IraP (Figure 3D, columns 3–6). Collectively these data suggest that in contrast to IraD, IraP interacts with both domains of RssB. However, based on the relative ability of each domain to inhibit IraP activity, IraP appears to bind with higher affinity to the C-terminal domain of RssB.
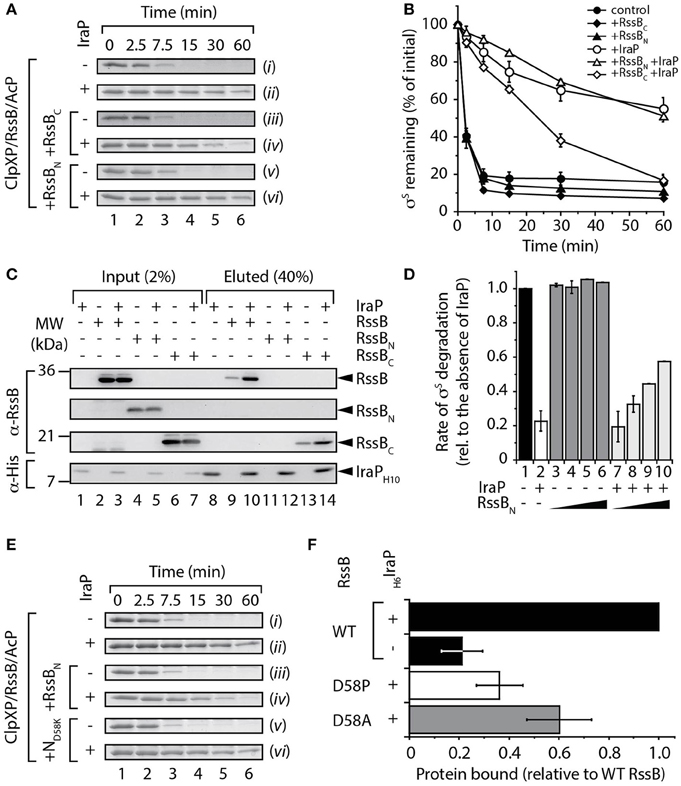
Figure 3. Interaction of IraP with RssB. (A,B) The RssB-mediated turnover of σS by ClpXP (control), was monitored in the absence of added domains (i and ii), or in the presence of either RssBC (iii and iv) or RssBN (v and vi), with (ii, iv, and vi) or without (i, iii, and v) IraP. Proteins were separated by SDS-PAGE and stained with CBB. Only the σS-strip of the gel is shown. (B) The amount of σS remaining was quantified from three independent experiments. Error bars represent s.e.m. (C) Ni-NTA agarose beads either lacking or containing immobilized IraP were incubated with RssB, RssBN, or RssBC (lanes 1–7) and following washing the bound proteins were eluted with imidazole (lanes 8–14). Proteins were detected by immunoblotting (as indicated) following separation by SDS-PAGE. (D) The initial rate of σS degradation in the absence of IraP (lane 1) was compared to the rate of degradation in the presence of IraP (lane 2) or with increasing concentrations (5, 10, 15, and 20 μM) of RssBN either in the absence (lanes 3–6) or presence (lanes 7–10) of IraP. n = 3, and error bars represent the s.e.m. (E) The RssB-mediated turnover of σS by ClpXP, was monitored either in the absence of added domains (i and ii), or in the presence of wild type RssBN (iii and iv) or ND58K (v and vi), with (ii, iv, and vi) or without (i, iii, and v) IraP and separated by 15% SDS-PAGE. (F) The interaction of His6-IraP with wild type or mutant RssB was monitored by pull-down. Recovery of wild type RssB (black bar), D58P (white bar) or D58A (gray bar) was determined from the quantitation of three independent experiments. Error bars represent the s.e.m.
Given the apparently weak interaction of IraP with the N-terminal domain, we chose to further clarify this interaction by examining the role of phosphorylation (of Asp58) on IraP-binding. In this case, we initially generated a non-phosphorylatable mutant of RssBN in which Asp58 was replaced with Lys (here referred to as ND58K) and compared the ability of this mutant (relative to wild type RssBN) to inhibit IraP binding to full-length RssB in the presence of the phospho-donor, AcP (Figure 3E, S4). Importantly, given that this mutant cannot be phosphorylated, this permitted a direct comparison of RssBN phosphorylation and IraP binding in the presence of AcP. Consistent with our previous findings (Figure 3D), a 5-fold excess of RssBN resulted in a ~2-fold increase in the rate of σS turnover in the presence of IraP (Figure S4). In contrast, a 5-fold excess of ND58K did not alter the IraP-mediated inhibition (Figure 3E, S4). Next we performed a series of pull-down experiments to directly monitor the interaction between IraP and RssB (Figure 3F). In this case, we replaced Asp58 with either Pro or Ala to generate two well-characterized non-phosphorylatable mutants of RssB, RssB(D58P) or RssB(D58A) (Bouche et al., 1998; Peterson et al., 2004), here referred to as D58P and D58A, respectively. Specifically, wild type or mutant RssB was expressed in presence or absence of His6-tagged IraP, the cell lysate was then applied to Ni-NTA beads and after extensive washing to remove non-specifically bound proteins, IraP was eluted with imidazole and the relative amount of RssB (wild type or mutant) specifically co-eluting with IraP determined. Consistent with the in vitro pull-down (shown in Figure 3C) a small amount of RssB was found to interact non-specifically with the beads however the levels of RssB recovered were significantly increased in the presence of IraP (Figure 3F, compare black bars). Importantly, mutation of Asp58 (to either Pro or Ala) resulted in a dramatic loss in the amount of RssB bound to IraP (Figure 3F, white or gray bars, respectively). Collectively, these data suggest that IraP docks to the C-terminal domain of RssB, however its binding appears be stabilized by the phosphorylation state of the N-terminal domain. Next, to confirm if the stabilized binding of IraP to RssBC was driven by a conformational change in the C-terminal domain of RssB induced by phosphorylation of the N-domain we examined if binding of RssBC (or indeed RssBN) could be enhanced in trans (Figure S5). To do so, we monitored the binding of IraP to each RssB domain, either alone or together (Figure S5). Consistent with the idea that phosphorylation of the receiver domain drives a conformational change in the effector domain, which triggers improved interaction with IraP, the addition of RssBN in trans did not enhance the recovery of RssBC. In contrast, the recovery of RssBN was reduced in the presence of RssBC, confirming that the N-domain alone interacts only weakly with IraP (Figure S5, lane 5).
IraM Binds to Both Domains of RssB
To determine if the third anti-adaptor, IraM, mimicked the action of IraD or IraP, its ability to bind to the domains of RssB was examined. In this case, due to poor recovery of recombinant IraM, we chose to monitor the binding of IraM to the different domains of RssB via pull-down experiments using cell lysates in which the proteins of interest were co-expressed. Specifically, we generated a series of clones, which enabled the overexpression of untagged RssB (full-length or individual domains) in the absence or presence of overexpressed His6-IraM. Following preliminary evaluation of the levels of soluble IraM and RssB (within the cell lysate) via SDS-PAGE (Figure S6) the appropriate amount of each lysate was applied to Ni-NTA agarose beads and the pull-down performed. The eluted proteins were evaluated by immunoblotting (Figure 4A) using the appropriate antibody. As expected, full-length RssB was specifically recovered, from the column containing immobilized His6-IraM (Figure 4A, lane 3). Interestingly, in contrast to both IraD and IraP, IraM appeared to form a stable interaction with both domains of RssB (Figure 4A). Indeed, based on the recovery of each domain in comparison to the input, both domains appear to contribute equally to IraM binding (Figure 4B). These data indicate that IraM imposes its inhibitory effect on RssB activity through docking to both domains of RssB. Although this mode of binding is similar to that of IraP, the relative contribution of each domain appears to be quite different. Next, we examined if both domains can function in trans. To do so we compared the binding of each domain to IraM, either alone or in the presence of the other domain (Figure S7). Unexpectedly, and in contrast to IraP (Figure S5), we observed an improved recovery of RssBN (when incubated in the presence of RssBC). These data suggest that the C-terminal domain of RssB can promote binding of RssBN to IraM, by stabilizing RssBN (or IraM) in a conformation that is permissive for interaction with the other component. Collectively, these data suggest that all three anti-adaptors use distinct modes of binding to inhibit RssB-activity.
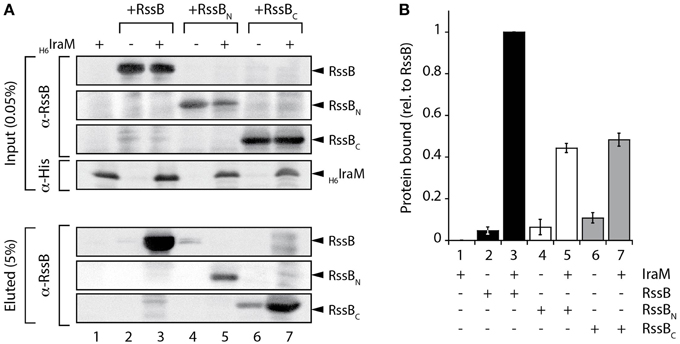
Figure 4. Interaction of IraM and RssB. (A) Following incubation of Ni-NTA agarose beads with lysates either lacking (lanes 2, 4, and 6) or containing (lanes 1, 3, 5, and 7) overexpressed His6-IraM, in the presence of recombinant RssB (lanes 2 and 3), RssBN (lanes 4 and 5) or RssBC (lanes 6 and 7) the beads were washed extensively then IraM, together with interacting proteins were eluted with imidazole. Proteins were detected by immunoblotting (as indicated) following separation by 16.5% Tris-Tricine SDS-PAGE. (B) The recovery of RssBN (white bar) or RssBC (gray bar) relative to RssB (black bar) was determined from the quantitation of three independent experiments. Error bars represent the s.e.m.
Anti-Adaptor Binding Triggers a Conformational Change in RssB
While each anti-adaptor exhibited a different mode of binding to RssB it still remained to be determined how they inhibit the RssB-mediated recognition of σS. One possibility is that each anti-adaptor simply inhibits substrate engagement by RssB via steric hindrance due to the presence of the bound anti-adaptor. This however, at least in the case of IraD, appears unlikely as the anti-adaptor binds specifically to RssBN and this binding is sufficient to trigger release of σS from the C-domain of RssB, in a preformed RssB/σS complex. An alternative possibility is that anti-adaptor binding triggers a conformational change in RssB that promotes release of σS from a second site. To gain further insight into the mode of action of the RssB anti-adaptors we repeated the partial proteolysis experiments, in the absence or presence of either IraD (Figure 5A) or IraP (Figure 5C). Consistent with our previous data (Figure 1A), in the absence of anti-adaptor, RssB was rapidly cleaved into two stable domains upon addition of thermolysin (Figure 5A, top panel). In contrast to RssB, IraD was rapidly and completely degraded following the addition of thermolysin (Figure 5A, middle panel). However, when IraD was incubated with RssB, both proteins were clearly protected from cleavage by thermolysin (Figure 5A, lower panel). In the case of RssB, the entire protein was completely stable while in the case of IraD only a fragment of the protein (here termed IraDcore) remained stable throughout the experiment. To determine if stabilization of the IraDcore fragment was solely due to its interaction with the N-terminal domain of RssB, we repeated the IraD partial proteolysis experiment in the presence of RssBN (Figure 5B). These data clearly show, that RssBN is not only sufficient to stabilize the core fragment of IraD, but also verifies that IraDcore migrates with a different mobility to RssBN. Collectively these data suggest that binding of IraD to the N-terminal domain of RssB is sufficient to trigger a conformational change in RssB, which protects it from cleavage by thermolysin. Next we examined whether IraP also protected RssB in the limited proteolysis assay, in the same manner as IraD. Consistent with the effect of IraD, IraP also stabilized full-length RssB, however in contrast to IraD, a concomitant stabilization of the anti-adaptor was not observed (Figure 5C). These data suggest that only a short fragment of IraP is required for binding to RssB, and that this fragment is sufficient to stabilize the protease-protected or inactive conformation of RssB. Importantly, in contrast to both IraD and IraP, σS did not protect RssB from cleavage by thermolysin (Figure 5D). These data suggest that in comparison to anti-adaptor docking, substrate binding to RssB either occurs through a different site on RssB or alternatively does not trigger the same conformational change.
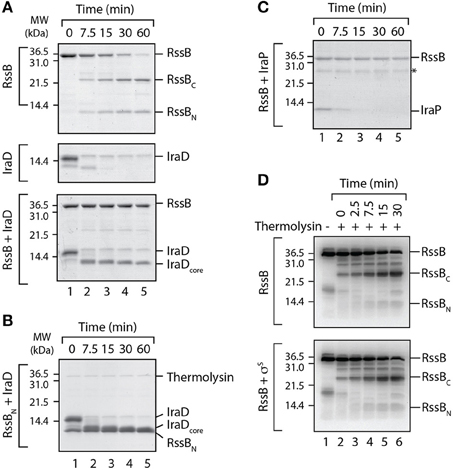
Figure 5. IraD and IraP trigger a conformational change in RssB. (A) The domain structure of RssB was analyzed by partial proteolysis using thermolysin in the absence (top panel) or presence (lower panel) of IraD. As a control IraD (alone) was also analyzed in the presence of thermolysin (middle panel). (B) The core domain of IraD (IraDcore) is stabilized by RssBN (from partial proteolysis with thermolysin). (C) The domain structure of RssB was monitored in the presence of IraP, using thermolysin. (A–C) Proteins were separated by SDS-PAGE and stained with CBB. (D) SigmaS (σS) is unable to protect the RssB linker region from cleavage by thermolysin. The domain structure of RssB was analyzed by partial proteolysis with thermolysin in the absence (top panel) or presence (bottom panel) of σS. Following partial proteolysis in the presence of thermolysin (lanes 2–6), the protein fragments were separated by SDS-PAGE, transferred to PVDF membrane and immunodecorated with α-RssB antisera, to specifically detect the RssB fragments.
Discussion
RpoS is the central regulator of the general stress response in E. coli. In the absence of stress, the cellular levels of σS are undetectable (< 1 molecule/cell), however in the presence of stress the level of σS increases rapidly, up to ~ 230 molecules/cell during stationary phase (Jishage and Ishihama, 1995; Maeda et al., 2000). This rapid change in cellular concentration of σS is largely achieved by regulating its turnover. Under normal conditions σS is rapidly degraded by the AAA+ protease ClpXP, which is mediated by a specialized adaptor protein RssB. However, upon exposure to stress the RssB-mediated turnover of σS is inhibited by one of three stress-specific anti-adaptor proteins IraD, IraP or IraM. Although the molecular components of this regulated protein degradation pathway have been reconstituted in vitro, currently little progress has been made toward determining the molecular basis of inhibition by these anti-adaptors.
Here, we have experimentally determined the domain structure of RssB (Figure 1) and defined the first mechanistic details of each anti-adaptor. Interestingly, each anti-adaptor interacts with RssB in a specific manner. Consistent with recent data from Gottesman and colleagues (Battesti et al., 2013) we found that IraD docks exclusively to the N-terminal domain (Figure 2) and IraM interacts with both domains (Figure 4). However, in contrast to Gottesman and colleagues (Battesti et al., 2013), we find that IraP interacts primarily with the C-terminal domain (Figure 3) although the interaction appears to be modulated by phosphorylation of the N-domain of RssB. Importantly, our data suggests that anti-adaptor docking, to either domain, triggers a conformation change in RssB (Figure 5), which results in substrate release. Specifically, we propose that IraD binding, to the dimerization interface of RssB, stabilizes an inactive conformation of RssB, which triggers release of σS from the C-terminal effector domain. Consistent with this model, binding of IraP to the C-terminal domain of RssB appears to induce a similar conformational change and hence we speculate that all three anti-adaptors, independent of their mode of docking to RssB, induce a conformational change in RssB, that mimics the inactive dimer-like state of RssB, which is unable to bind (or deliver) σS (this model is summarized in Figure 6).
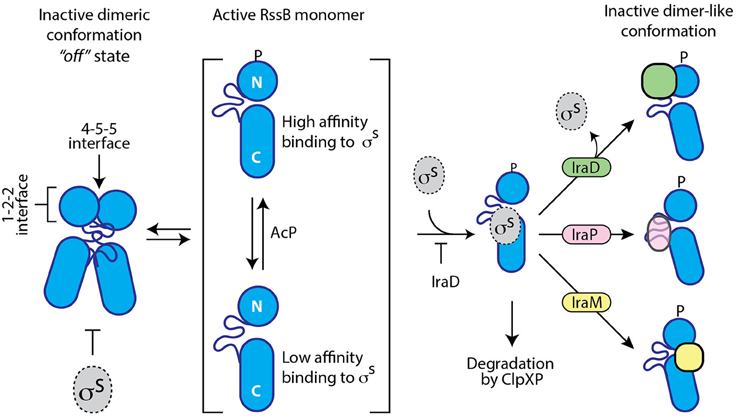
Figure 6. Cartoon illustrating a model of the different modes of action of the three anti-adaptors, IraD (green), IraP (pink), and IraM (yellow). In the absence of anti-adaptors, RssB (blue) is able to recognize the transcription factor σS (gray). This recognition is inhibited by dimerization of RssB and enhanced by phosphorylation of the N-terminal domain of RssB. Anti-adaptor binding not only inhibits the recognition of σS, but in the case of IraD, also triggers the release of σS. Binding of IraD (to RssBN) and IraP (to RssBC) triggers a conformational change in RssB, which we propose resembles the dimer-like state or RssB. Binding of IraM to both domains inhibits σS interaction via an unknown mechanism.
In the case of IraP, a short region of this anti-adaptor appears to be sufficient to stabilize the inactive conformation of RssB. Consistent with this idea, a short region (hexapeptide) of the Bacillus subtilis anti-adaptor–ComS, is sufficient to displace the substrate ComK from the adaptor protein MecA, and hence inhibit its degradation by the ClpCP protease (Turgay et al., 1998; Prepiak and Dubnau, 2007). In this case however, the anti-adaptor appears to compete directly with the substrate for binding to the same site on MecA and as a result ComS is also degraded by the ClpCP/MecA protease. In contrast to MecA, we propose that RssB interacts with the anti-adaptors (IraD and IraP) and the substrate (σS) through distinct binding sites, as degradation of the RssB-anti-adaptors by their cognate protease has not been observed.
Based on bioinformatic analysis of the IraP amino acid sequence, the short region of IraP responsible for interaction with RssB could be located within the N-terminus of the anti-adaptor, as this region is predicted to form a coiled-coil (residues 1–35). Similarly a short segment (residues 127–155) in the C-terminal domain of RssB is also predicted to form a coiled-coil. Given that coiled-coils often play important roles in biological interactions, one possibility is that both coiled-coils drive the formation of the heterodimeric complex. This interaction could result in a conformational change to RssB, or alternatively it could stabilize a conformation of RssB, which prevents substrate binding. Consistent with this idea, a single point mutant (L9S) within the coiled-coil region of IraP reduces its inhibitory activity (Bougdour et al., 2006). Interestingly, the structure of full-length RssB from P. aeruginosa (3EQ2) also contains a coiled-coil region, located between the N- and C-terminal domains, which appears to mediate homodimer formation. Based on our biochemical data, the dimeric conformation of E. coli RssB is unable to deliver σS to ClpXP for degradation. Therefore, we propose that IraP, similar to IraD, stabilizes RssB in a dimer-like conformation, which renders RssB unable to recognize or deliver σS to ClpXP for degradation. This mode of inhibition shares some striking similarities to the regulation of a quorum sensing transcriptional activator (TraR) in bacteria such as Agrobacterium tumefaciens, which is controlled through its interaction with a quorum sensing anti-activator, known as TraM. Similar to IraP, TraM also contains a coiled-coil region, which is involved not only in homodimer formation (Chen et al., 2004), but also in binding to TraR resulting in the formation of the TraR-TraM anti-activation complex (Chen et al., 2007). In this case, TraM binding to one site in TraR, has been shown to cause a conformational change in TraR that prevents substrate binding at a second site.
Collectively, our data shows that each anti-adaptor forms a stable complex with RssB, albeit through a unique mode of binding. Atomic resolution structures of each anti-adaptor, both alone and in complex with the adaptor protein RssB, are eagerly awaited to better understand the nature of these interactions.
Experimental Procedures
Protein Purification and Size Exclusion Chromatography
His6-tagged ClpX and ClpP were overexpressed in E. coli and purified as described previously (Dougan et al., 2003). His10-tagged IraP and IraD were overexpressed in E. coli and purified essentially as described by Bougdour et al. (2008). Untagged σS, RssB, RssBC, and RssBN (wild type and specific point mutants) were generated using the Ub-fusion system (Catanzariti et al., 2004) and purified essentially as described previously (Ninnis et al., 2009), using a combination of IMAC and preparative grade SEC to separate monomeric and dimeric RssB. To examine the different protein complexes, analytical SEC was performed. All columns were pre-equilibrated in chilled GF buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 140 mM NaCl, 5% (v/v) glycerol, 0.005% (v/v) Triton X-100).
In Vitro Degradation Assays
The in vitro σS degradation assays were performed essentially as described (Zhou et al., 2001) with minor modifications. All reactions were performed in degradation buffer (20 mM Tris-HCl pH 7.5, 140 mM NaCl, 10 mM MgCl2, 0.1 mM EDTA, 5% (v/v) glycerol, 0.005% (v/v) Triton X-100, 1 mM DTT) and contained 1 μM σS. Unless otherwise stated reactions containing σS were pre-incubated at 30°C, with 1 μM ClpX, 1 μM ClpP and 0.08 μM monomeric RssB and 20 mM of the phospho donor, acetyl phosphate (AcP) (Bouche et al., 1998). Where indicated IraD (1 μM), IraP (1 μM), RssBN (2.5–20 μM), and RssBC (2.5 μM) were included. All reactions were initiated with the addition of 2 mM ATP and samples collected at the specified time-points. Samples were separated using SDS-PAGE and visualized using Coomassie Brilliant Blue (CBB) staining.
Limited Proteolysis
Limited proteolysis experiments were performed using thermolysin as described previously (Lowth et al., 2012). Following a short pre-incubation (2 min at 25°C) in the absence of the protease, the substrate (i.e., RssB with or without anti-adaptor) was incubated with the protease. Reactions were stopped with the addition of PMSF (6 mM) and 1 × SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE and visualized by staining with CBB.
In Vitro “Pull-Down” Experiments
To examine the interaction of RssB (either full-length protein or individual domains) with purified IraD or IraP, in vitro “pull-down” experiments were performed as previously described (Geissler et al., 2002). To examine the interaction of IraM or IraP with RssB (full-length protein (wild type or mutant) and individual domains), “pull-down” experiments were performed using soluble protein extracts isolated from BL21(DE3) codon+ cells in which untagged RssB (full-length (wild type or mutant) or individual domains) was overexpressed in the absence or presence of either His6-IraM or His6-IraP. Following overexpression of the proteins, a series of soluble lysates were prepared in extraction buffer (20 mM Tris-HCl pH 7.9, 500 mM NaCl, 10% (v/v) glycerol, 10 mM imidazole, 1% (v/v) Triton X-100). Based on the amount of IraM present within each soluble lysate, between 40 and 160 mg of total protein was applied to 100 μl of pre-equilibrated Ni-NTA agarose beads. To monitor the in trans binding of each RssB domain to the immobilized anti-adaptor proteins (IraP or IraM) the appropriate lysate was supplemented with 250 μg of RssBN. Soluble protein lysates (in the presence or absence of additional RssBN) and Ni-NTA agarose beads were then incubated for 30 min at 4°C. The beads were then transferred to individual MoBiTec columns. Further washes and recovery of bound proteins was performed as described (Geissler et al., 2002).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Susan Gottesman for comments on the manuscript. DM and JEZ were supported by La Trobe University Postgraduate Scholarships. KNT is an Australian Research Council (ARC) Future Fellow. DAD is an ARC Australian Research Fellow. This work was funded by ARC Discovery Project DP110103936. The authors have no conflict of interest to declare.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmolb.2015.00015/abstract
References
Battesti, A., Hoskins, J. R., Tong, S., Milanesio, P., Mann, J. M., Kravats, A., et al. (2013). Anti-adaptors provide multiple modes for regulation of the RssB adaptor protein. Genes Dev. 27, 2722–2735. doi: 10.1101/gad.229617.113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Battesti, A., Majdalani, N., and Gottesman, S. (2011). The RpoS-mediated general stress response in Escherichia coli. Annu. Rev. Microbiol. 65, 189–213. doi: 10.1146/annurev-micro-090110-102946
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Battesti, A., Tsegaye, Y. M., Packer, D. G., Majdalani, N., and Gottesman, S. (2012). H-NS regulation of IraD and IraM antiadaptors for control of RpoS degradation. J. Bacteriol. 194, 2470–2478. doi: 10.1128/JB.00132-12
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bouche, S., Klauck, E., Fischer, D., Lucassen, M., Jung, K., and Hengge-Aronis, R. (1998). Regulation of RssB-dependent proteolysis in Escherichia coli: a role for acetyl phosphate in a response regulator-controlled process. Mol. Microbiol. 27, 787–795. doi: 10.1046/j.1365-2958.1998.00725.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bougdour, A., Cunning, C., Baptiste, P. J., Elliott, T., and Gottesman, S. (2008). Multiple pathways for regulation of sigmaS (RpoS) stability in Escherichia coli via the action of multiple anti-adaptors. Mol. Microbiol. 68, 298–313. doi: 10.1111/j.1365-2958.2008.06146.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bougdour, A., and Gottesman, S. (2007). ppGpp regulation of RpoS degradation via anti-adaptor protein IraP. Proc. Natl. Acad. Sci. U.S.A. 104, 12896–12901. doi: 10.1073/pnas.0705561104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bougdour, A., Wickner, S., and Gottesman, S. (2006). Modulating RssB activity: IraP, a novel regulator of sigma(S) stability in Escherichia coli. Genes Dev. 20, 884–897. doi: 10.1101/gad.1400306
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Catanzariti, A. M., Soboleva, T. A., Jans, D. A., Board, P. G., and Baker, R. T. (2004). An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 13, 1331–1339. doi: 10.1110/ps.04618904
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, G., Jeffrey, P. D., Fuqua, C., Shi, Y., and Chen, L. (2007). Structural basis for antiactivation in bacterial quorum sensing. Proc. Natl. Acad. Sci. U.S.A. 104, 16474–16479. doi: 10.1073/pnas.0704843104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, G., Malenkos, J. W., Cha, M. R., Fuqua, C., and Chen, L. (2004). Quorum-sensing antiactivator TraM forms a dimer that dissociates to inhibit TraR. Mol. Microbiol. 52, 1641–1651. doi: 10.1111/j.1365-2958.2004.04110.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dougan, D. A., Weber-Ban, E., and Bukau, B. (2003). Targeted delivery of an ssrA-tagged substrate by the adaptor protein SspB to its cognate AAA+ protein ClpX. Mol. Cell 12, 373–380. doi: 10.1016/j.molcel.2003.08.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Galperin, M. Y. (2010). Diversity of structure and function of response regulator output domains. Curr. Opin. Microbiol. 13, 150–159. doi: 10.1016/j.mib.2010.01.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gao, R., and Stock, A. M. (2010). Molecular strategies for phosphorylation-mediated regulation of response regulator activity. Curr. Opin. Microbiol. 13, 160–167. doi: 10.1016/j.mib.2009.12.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Geissler, A., Chacinska, A., Truscott, K. N., Wiedemann, N., Brandner, K., Sickmann, A., et al. (2002). The mitochondrial presequence translocase: an essential role of Tim50 in directing preproteins to the import channel. Cell 111, 507–518. doi: 10.1016/S0092-8674(02)01073-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hengge, R. (2009). Proteolysis of sigmaS (RpoS) and the general stress response in Escherichia coli. Res. Microbiol. 160, 667–676. doi: 10.1016/j.resmic.2009.08.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hengge-Aronis, R. (2002). Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev. 66, 373–395. doi: 10.1128/MMBR.66.3.373-395.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jishage, M., and Ishihama, A. (1995). Regulation of RNA polymerase sigma subunit synthesis in Escherichia coli: intracellular levels of sigma 70 and sigma 38. J. Bacteriol. 177, 6832–6835.
Lowth, B. R., Kirstein-Miles, J., Saiyed, T., Brotz-Oesterhelt, H., Morimoto, R. I., Truscott, K. N., et al. (2012). Substrate recognition and processing by a Walker B mutant of the human mitochondrial AAA+ protein CLPX. J. Struct. Biol. 179, 193–201. doi: 10.1016/j.jsb.2012.06.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maeda, H., Fujita, N., and Ishihama, A. (2000). Competition among seven Escherichia coli sigma subunits: relative binding affinities to the core RNA polymerase. Nucliec Acids Res. 28, 3497–3503. doi: 10.1093/nar/28.18.3497
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merrikh, H., Ferrazzoli, A. E., Bougdour, A., Olivier-Mason, A., and Lovett, S. T. (2009a). A DNA damage response in Escherichia coli involving the alternative sigma factor, RpoS. Proc. Natl. Acad. Sci. U.S.A. 106, 611–616. doi: 10.1073/pnas.0803665106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merrikh, H., Ferrazzoli, A. E., and Lovett, S. T. (2009b). Growth phase and (p)ppGpp control of IraD, a regulator of RpoS stability, in Escherichia coli. J. Bacteriol. 191, 7436–7446. doi: 10.1128/JB.00412-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Micevski, D., and Dougan, D. A. (2013). Proteolytic regulation of stress response pathways in Escherichia coli. Subcell Biochem. 66, 105–128. doi: 10.1007/978-94-007-5940-4_5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Muffler, A., Fischer, D., Altuvia, S., Storz, G., and Hengge-Aronis, R. (1996). The response regulator RssB controls stability of the sigma(S) subunit of RNA polymerase in Escherichia coli. EMBO J. 15, 1333–1339.
Ninnis, R. L., Spall, S. K., Talbo, G. H., Truscott, K. N., and Dougan, D. A. (2009). Modification of PATase by L/F-transferase generates a ClpS-dependent N-end rule substrate in Escherichia coli. EMBO J. 28, 1732–1744. doi: 10.1038/emboj.2009.134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peterson, C. N., Levchenko, I., Rabinowitz, J. D., Baker, T. A., and Silhavy, T. J. (2012). RpoS proteolysis is controlled directly by ATP levels in Escherichia coli. Genes Dev. 26, 548–553. doi: 10.1101/gad.183517.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peterson, C. N., Ruiz, N., and Silhavy, T. J. (2004). RpoS proteolysis is regulated by a mechanism that does not require the SprE (RssB) response regulator phosphorylation site. J. Bacteriol. 186, 7403–7410. doi: 10.1128/JB.186.21.7403-7410.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pratt, L. A., and Silhavy, T. J. (1996). The response regulator SprE controls the stability of RpoS. Proc. Natl. Acad. Sci. U.S.A. 93, 2488–2492. doi: 10.1073/pnas.93.6.2488
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Prepiak, P., and Dubnau, D. (2007). A peptide signal for adapter protein-mediated degradation by the AAA+ protease ClpCP. Mol. Cell 26, 639–647. doi: 10.1016/j.molcel.2007.05.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turgay, K., Hahn, J., Burghoorn, J., and Dubnau, D. (1998). Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 17, 6730–6738. doi: 10.1093/emboj/17.22.6730
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, Y., and Gottesman, S. (2006). Modes of regulation of RpoS by H-NS. J. Bacteriol. 188, 7022–7025. doi: 10.1128/JB.00687-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, Y., Gottesman, S., Hoskins, J. R., Maurizi, M. R., and Wickner, S. (2001). The RssB response regulator directly targets sigma(S) for degradation by ClpXP. Genes Dev. 15, 627–637. doi: 10.1101/gad.864401
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: anti-adaptor, regulation, general stress response, AAA+ protease, degradation
Citation: Micevski D, Zammit JE, Truscott KN and Dougan DA (2015) Anti-adaptors use distinct modes of binding to inhibit the RssB-dependent turnover of RpoS (σS) by ClpXP Front. Mol. Biosci. 2:15. doi: 10.3389/fmolb.2015.00015
Received: 23 January 2015; Accepted: 08 April 2015;
Published: 23 April 2015.
Edited by:
Walid A. Houry, University of Toronto, CanadaReviewed by:
Carolyn K. Suzuki, Rutgers University, USAPierre Genevaux, Centre National de la Recherche Scientifique, France
Copyright © 2015 Micevski, Zammit, Truscott and Dougan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Dougan, La Trobe University, Melbourne, 3086, VIC, AustraliaZC5kb3VnYW5AbGF0cm9iZS5lZHUuYXU=