 Ildefonso M. De la Fuente
Ildefonso M. De la Fuente- 1Department of Cell Biology and Immunology, Institute of Parasitology and Biomedicine “López-Neyra,” Consejo Superior de Investigaciones Científicas, Granada, Spain
- 2Department of Mathematics, University of the Basque Country, UPV/Euskal Herriko Unibertsitatea, Leioa, Spain
A large number of studies have demonstrated the existence of metabolic covalent modifications in different molecular structures, which are able to store biochemical information that is not encoded by DNA. Some of these covalent mark patterns can be transmitted across generations (epigenetic changes). Recently, the emergence of Hopfield-like attractor dynamics has been observed in self-organized enzymatic networks, which have the capacity to store functional catalytic patterns that can be correctly recovered by specific input stimuli. Hopfield-like metabolic dynamics are stable and can be maintained as a long-term biochemical memory. In addition, specific molecular information can be transferred from the functional dynamics of the metabolic networks to the enzymatic activity involved in covalent post-translational modulation, so that determined functional memory can be embedded in multiple stable molecular marks. The metabolic dynamics governed by Hopfield-type attractors (functional processes), as well as the enzymatic covalent modifications of specific molecules (structural dynamic processes) seem to represent the two stages of the dynamical memory of cellular metabolism (metabolic memory). Epigenetic processes appear to be the structural manifestation of this cellular metabolic memory. Here, a new framework for molecular information storage in the cell is presented, which is characterized by two functionally and molecularly interrelated systems: a dynamic, flexible and adaptive system (metabolic memory) and an essentially conservative system (genetic memory). The molecular information of both systems seems to coordinate the physiological development of the whole cell.
Enzymatic Self-Organization
Multienzymatic Complexes
Living cells are essentially highly evolved dynamic metabolic reactors, in which most of the biomolecules are synthesized and destroyed by means of interrelated enzymatic processes, which are densely integrated in modular networks that shape one of the most complex dynamic systems in nature (De la Fuente, 2014).
From a functional point of view, enzymes are the main molecules of this amazing biochemical reactor. They are responsible for almost all biomolecular transformations, which, when globally considered, are called cellular metabolism. Most enzymes are proteins, but a few RNA molecules called ribozymes, also manifest catalytic activity. How the enzymes are organized under the complex conditions prevailing inside the cell is a crucial issue for understanding the fundamental functional architecture of cellular life.
For many years, it has been assumed that enzymes work in an isolated way. However, intensive studies of protein-protein interactions have shown that the internal cellular medium is an assembly of supra-molecular protein complexes (Gavin and Superti-Furga, 2003; Segura et al., 2010; Völkel et al., 2010; Ramisetty and Washburn, 2011), where homologous or heterologous protein-protein interactions are the rule (Pang et al., 2008). Thus, analyses of the proteome of Saccharomyces cerevisiae have shown that at least 83% of proteins form complexes comprised of 2–83 proteins (Gavin et al., 2002). This kind of associative organization occurs in all sorts of cells, both eukaryotes and prokaryotes (Uetz et al., 2000; Ito et al., 2001; Ho et al., 2002; Bobik, 2006; Sutter et al., 2008; Yeates et al., 2008).
Complexes formed by multienzymatic associations may allow for substrate channeling, i.e., direct transfer of intermediate metabolites from the active site of one enzyme to the next without prior diffusion into the bulk medium. Metabolic channeling decreases the transit time of reaction substrates, thus increasing the speed and efficiency of catalysis by avoiding delays due to the diffusion of reaction intermediates and their eventual loss (Clegg and Jackson, 1990; Ovadi and Srere, 2000; Jorgensen et al., 2005; Jovanović et al., 2007). Substrate channeling may also occur within channels or on the electrostatic surface of enzyme complexes (Milani et al., 2003; Ishikawa et al., 2004; Beg et al., 2007).
Additionally, reversible interactions between multienzyme complexes and structural proteins or membranes occur frequently in eukaryotic cells leading to the emergence of metabolic microcompartments (Verkman, 2002; Lunn, 2007; Monge et al., 2008, 2009; Saks et al., 2009). For instance, the nuclear processes such as DNA replication, DNA repair and transcription of RNAs are organized in dynamic microenvironments that are functionally connected to the ubiquitin-proteasome system (Zhao et al., 2009). Microcompartmentation also occurs in prokaryotes, but in this case it consists of protein “shells” composed of thousands of protein subunits (Yeates et al., 2007; Fan et al., 2010). The dynamics of molecular processes that intervene in microcompartmentation and its maintenance remain unclear.
The concept of multienzymatic associations was first conceived in 1970 by Kuzin (1970) and adopted in 1972 by Srere (1972, 1985) who suggested the use of the term metabolon to refer to an organized multienzyme complex belonging to a metabolic sequence. A wide variety of studies have revealed the importance of multienzymatic associations in almost all cellular processes (Veliky et al., 2007; Pflieger et al., 2011). Various examples of such associations are detailed in Table 1.
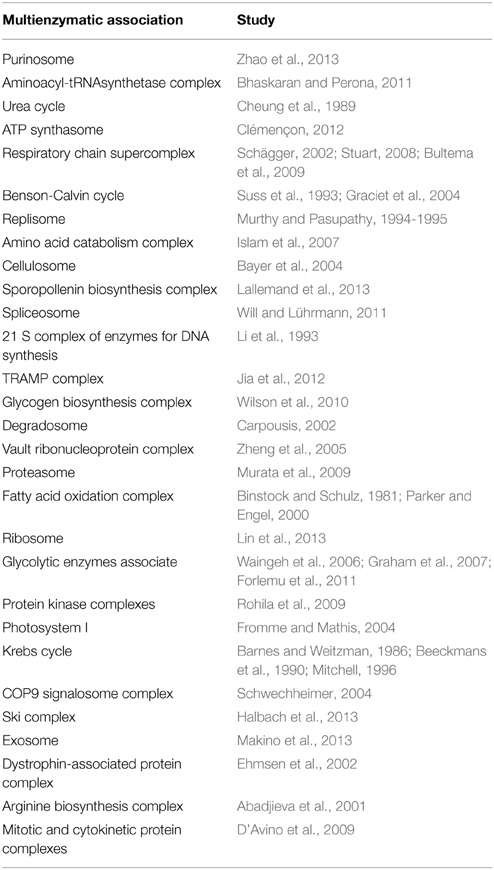
Table 1. Examples of multienzymatic associations in published studies.
Recently, analysis of conserved sequences in biochemical reactions has shown that cellular metabolism contains basic functional units which tend to correspond to traditional metabolic pathways. These functional multi-enzymatic units seem to be the catalytic building blocks of cellular metabolism (Kanehisa, 2013; Muto et al., 2013). Another kind of functional catalytic association is the interactome which refers to protein–protein interaction networks (unlike metabolic pathways, that represent a sequence of molecular interactions leading to a final product) (Van Leene et al., 2007; Bonetta, 2010; Gautam et al., 2012; Sharma et al., 2013).
Temporal Self-Organization of Multienzymatic Processes
Enzymatic organization at the molecular level presents another relevant characteristic: the emergence of dissipative catalytic structures. Experimental observations have shown that enzymes shape functional catalytic associations in which molecular rhythms and quasi-steady states may spontaneously emerge far from thermodynamic equilibrium. In fact, under cellular conditions, the temporal evolution of practically all metabolite concentrations exhibits marked variations, presenting transitions between quasi-steady states and oscillatory behaviors (De la Fuente, 2014). In the quasi-stationary states, the concentration of any given metabolite drifts over time in a non-oscillatory way. Not long ago, the use of nanobiosensors to provide real-time measurements of specific intracellular metabolites in intact cells, revealed complex oscillations and quasi-steady states, but importantly, these patterns were never found to be constant (Ozalp et al., 2010).
Metabolic rhythms constitute a genuine manifestation of dissipative self-organization in the enzymatic activities of the cell. In general terms, self-organization can be defined as the spontaneous emergence of macroscopic non-equilibrium dynamic structures, as a result of the collective behavior of elements interacting nonlinearly with each other, to generate a system which increases its structural and functional complexity, driven by energy dissipation (Halley and Winkler, 2008; Misteli, 2009; De la Fuente, 2014).
An important ingredient of metabolic self-organization is the nonlinear interaction between metabolites and enzymes, involving autocatalysis, feed-back processes, and allosteric regulation, among others. It is well established that non-equilibrium states can be a source of order in the sense that the irreversible processes may lead to a new type of dynamic state in which the system becomes ordered in space and time. In fact, the non-linearity associated with irreversible enzymatic reactions seems to be an essential mechanism which may allow dissipative metabolic organization far from thermodynamic equilibrium (Goldbeter, 2002, 2007).
Self-organization is based on the concept of dissipative structures, and its theoretical roots can be traced back to the Nobel Prize Laureate in Chemistry Ilya Prigogine (Nicolis and Prigogine, 1977). There exist different kinds of metabolic dissipative structures under cellular conditions, including oscillations in metabolite concentrations, molecular circadian rhythms and spatial biochemical waves. During the last four decades, extensive studies of biochemical dynamics both in prokaryotic and eukaryotic cells have revealed that most of the fundamental metabolic processes exhibit oscillations (temporal self-organizations). Various examples of molecular oscillations are detailed in Table 2.
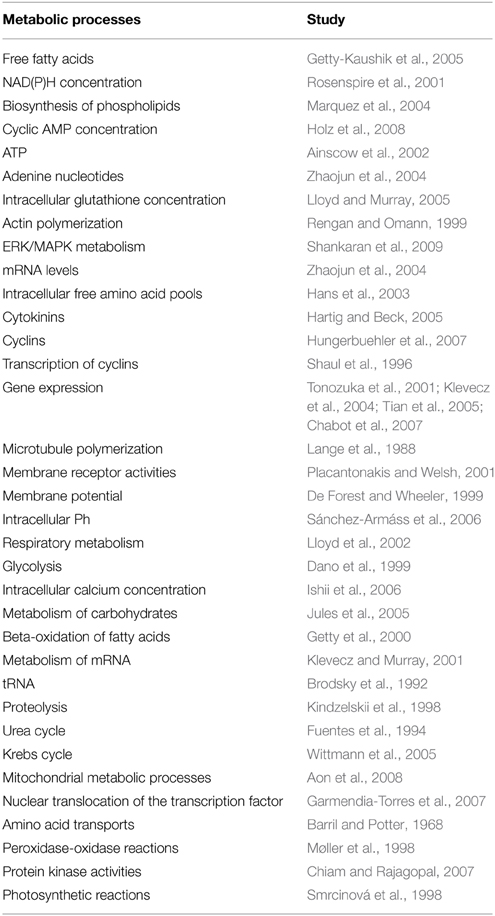
Table 2. Examples of temporal self-organization in metabolic processes.
Experimental observations of S. cerevisiae during continuous culture have also revealed the presence of oscillatory dynamics in most of the transcriptome and metabolome (Klevecz et al., 2004; Murray et al., 2007). In some studies, it has been observed that at least 60% of transcripts exhibit oscillations with an approximate period of 300 min (Tu et al., 2005). Moreover, other studies have shown that the entire transcriptome presents low-amplitude oscillatory behavior (Lloyd and Murray, 2006) and this phenomenon has been described as a genome-wide oscillation (Klevecz et al., 2004; Lloyd and Murray, 2005, 2006; Oliva et al., 2005; Tu et al., 2005; Murray et al., 2007).
Evidence that cells exhibit multi-oscillatory metabolic processes with fractal properties has also been reported. This dynamic behavior appears to be consistent with scale-free dynamics spanning a wide range of frequencies of at least three orders of magnitude (Aon et al., 2008).
The temporal organization of metabolic processes in terms of rhythmic phenomena spans periods ranging from milliseconds (Aon et al., 2006), to seconds (Roussel et al., 2006), minutes (Chance et al., 1973; Berridge and Galione, 1988) and hours (Brodsky, 2006). Oscillatory behavior from simple patterns to complex temporal structures (MacDonald et al., 2003), including bursting oscillations, with one large spike and a series of secondary oscillations (Dekhuijzen and Bagust, 1996), have often been observed. Multiple autonomous oscillatory behaviors emerge simultaneously in the cell, so that global metabolism can be considered to be a complex multi-oscillator super-system (Lloyd and Murray, 2005, 2006; Murray et al., 2007; Aon et al., 2008; Sasidharan et al., 2012; Amariei et al., 2014; De la Fuente et al., 2014).
Numerous mathematical studies of temporal metabolic rhythms have contributed to a better understanding of the self-organized enzymatic functionality which is apparent under cellular conditions (Boiteux et al., 1980; Tyson, 1991; De la Fuente et al., 1996, 1999; Heinrich and Schuster, 1996; Novak et al., 2007; Zhou et al., 2009; Goldbeter, 2013). Dissipative self-organization at the enzymatic level presents other kinds of emergent dynamic structures, such as circadian rhythms (Schibler and Sassone-Corsi, 2002; Cardone et al., 2005; De la Fuente, 2010) and spatial biochemical waves (Petty and Kindzelskii, 2001; Bernardinelli et al., 2004; Asano et al., 2008; De la Fuente, 2010). Metabolic rhythms constitute one of the hallmark properties of multienzymatic dynamics. The conditions required for the emergence and sustainability of these rhythms, and how they are regulated, represent a biological problem of the highest significance. However, in spite of its physiological importance, many aspects of cellular dissipative structures are still poorly understood and thus deserve further attention.
Dissipative Multienzymatic Complexes: Metabolic Subsystems
Self-organized multienzymatic complexes can be viewed as dissipative structures in which molecular rhythms and functional integrative processes can emerge, increasing the efficiency and control of the biochemical reactions involved (De la Fuente, 2010; De la Fuente and Cortés, 2012). This concept was conceived in 1999 and was termed metabolic subsystem to refer to a dissipatively structured enzymatic set in which molecular oscillations and steady state patterns may emerge spontaneously (De la Fuente et al., 1999).
Thermodynamically, metabolic subsystems represent advantageous biochemical organizations, forming unique, well-defined dynamic systems, in which catalytic activity is autonomous with respect to other enzymatic associations. Therefore, each self-organized enzymatic set shapes a catalytic entity as a whole and carries out its activities relatively independently of others (De la Fuente, 2010, 2014). The presence of some regulatory processes (enzymes of both allosteric and covalent modulation) in each metabolic subsystem makes sophisticated interconnections among them possible. As a result of variable combinations in the different input conditions (substrate fluxes, allosteric signals, specific post-translational modulations, etc.), a rich variety of dynamic patterns, each corresponding to distinct catalytic activity regimes, can emerge in these metabolic subsystems (De la Fuente et al., 2013, 2014).
This huge variety of catalytic patterns is a sine qua non condition for complex information properties (with a capacity to store functional catalytic patterns) that can emerge in the cellular metabolic networks (see Section Metabolic Networks and Information Properties for more details).
Metabolic subsystems represent highly efficient nanomachines which can execute autonomous and discrete biological functions, underlying the structural and functional dynamic organization of the cell. Associations between metabolic subsystems can form higher level complex molecular organizations, which appear to occur for example, in intracellular energetic units (ICEU) (Saks et al., 2006) and synaptosomes (Monge et al., 2008).
In essence, metabolic subsystems are self-organized biochemical machines which seem to constitute a fundamental and elemental catalytic unit of the cellular biochemical reactor (De la Fuente, 2014).
Functional Organization of Enzymes in Metabolic Networks
Metabolic Functionality
All physiological processes rely on metabolic functionality, which is a key element for understanding the dynamic principles of cellular life at all levels of its biochemical organization (Noble et al., 2014).
Enzymatic activity depends on different factors, mainly substrate concentration, temperature, pH, enzyme concentration and the presence of any inhibitors or activators. When an enzymatic system operates sufficiently far from equilibrium, oscillatory catalytic patterns can emerge. Such dynamic behaviors find their roots in non-linear regulatory processes, e.g., feedback loops, cooperativity and stoichiometric autocatalysis (Goldbeter, 2002). In addition, due to collective interactions under cellular conditions, substrate fluxes and regulatory signals shape complex topological structures in which metabolite concentration is continuously changing over time because of the dynamics that emerge in the catalytic networks (De la Fuente, 2014). This collective enzymatic connectivity makes the rate at which enzymatic reactions proceed highly complex.
As a consequence, the functional architecture of the cell is a result of all these different factors, and in particular, of the complex dynamic interactions between enzymatic processes integrated in biochemical networks. Thus, metabolic functionality at a systemic level prominently depends on the collective architecture (structural and functional) of the catalytic reactions, which is defined by the two essential principles that link the different modes of enzymatic connectivity, i.e., metabolic segregation and metabolic integration.
Metabolic Segregation
The separation of metabolic tasks and catalytic roles requires that the enzymes with correlated biochemical activities are grouped together. As a consequence, the entire metabolism is functionally segregated at multiple levels of specialization (e.g., signal transduction, oxidative phosphorylation, lysosomal metabolic activity, etc.), originating different kinds of specific modular structures. A metabolic module can be considered to be a discrete functional entity, which performs relatively autonomous processes with specific and coherent dynamic activities (Hartwell et al., 1999).
A wide variety of studies have shown that cellular metabolic networks are organized in a specialized modular fashion; this constitutes a defining characteristic of metabolic organization in the cell (Ravasz et al., 2002; Nurse, 2008; Ravasz, 2009; Hao et al., 2012; Kaltenbach and Stelling, 2012; Geryk and Slanina, 2013). Modular segregations have been observed in all the central physiological processes, e.g., the enzymatic organization of metabolic pathways (Muto et al., 2013), chaperone activities (Korcsmáros et al., 2007), chemotaxis (Postma et al., 2004; Shimizu et al., 2010), apoptosis (Harrington et al., 2008), the cell cycle (Boruc et al., 2010; Hsu et al., 2011), the Golgi apparatus (Nakamura et al., 2012), and kinetochore organization (Petrovic et al., 2014). Specific modular metabolic networks also participate in the transcriptional system, such as miRNA regulatory networks (Gennarino et al., 2012), transcription factor networks (Neph et al., 2012), RNA polymerase complexes (Schubert, 2014), and mRNA dynamics (Vlasova-St Louis and Bohjanen, 2011). Metabolic subsystems, i.e., self-organized multienzymatic associations, can be considered to be elemental catalytic modules in the cell (De la Fuente et al., 2008, 2010, 2014).
Metabolic networks may also contain canonical motifs, which represent recurrent and statistically significant sub-graphs or patterns (Milo et al., 2002; Kashtan and Alon, 2005; Alon, 2007; Masoudi-Nejad et al., 2012). These sub-graphs that repeat themselves in a specific network are defined by a specific pattern of interactions, which may reflect particular functional properties but, in contrast to modules, motifs do not function in isolation (Lacroix et al., 2006).
The functional role played by specialized metabolic networks is defined largely by its structural connections. However, at least three main kinds of metabolic connectivity have to be considered: structural connectivity, which is formed by structural molecular links such as substrate fluxes and regulatory signals, functional connectivity, which describes statistical dependencies (non-causal) between the activity patterns, and effective connectivity, which rests on the causal effects between different biochemical activities (De la Fuente et al., 2011, 2013). A number of information-based dynamic tools for the functionality and correlations between biochemical processes have been proposed. However, functional correlations (functional connectivity) do not imply effective connectivity because most synchronization measures do not distinguish between causal and non-causal interactions. In recent studies, transfer entropy (TE) has been proposed to be a rigorous, robust and self-consistent method for the causal quantification of functional information flow between nonlinear processes (effective connectivity).
The quantitative study of enzymatic effective connectivity can be considered at different biochemical organization levels, e.g., in a biochemical pathway (De la Fuente and Cortés, 2012) and in a metabolic network (De la Fuente et al., 2011). In particular, in order to investigate the functional structure (both functional and effective connectivity) of a large number of self-organized enzymes, the effective connectivity has been analyzed by means of transfer entropy in dissipative metabolic networks (DMNs) under different external conditions (De la Fuente et al., 2011).
Essentially, a DMN is a set of interconnected nodes, each one corresponding to a specific metabolic subsystem, which can be considered to be an elemental self-organized catalytic module (see Section Enzymatic Self-Organization for more details). The DMN nodes are interconnected through a complex topological structure (structural connectivity) of substrate fluxes and different regulatory mechanisms: activatory and inhibitory allosteric modulations, and post-translational modulations. In keeping with experimental observations, the emergent output activity of each enzymatic subsystem in the DMN's can be either oscillatory or steady state and comprises an infinite number of distinct activity regimes (De la Fuente et al., 2008, 2010; De la Fuente and Cortés, 2012).
Transfer entropy analysis of the DMN showed that in addition to the network's topological structure, characterized by the specific location of enzymatic subsystems, molecular substrate fluxes and regulatory signals, there is another functional structure of biomolecular information flow which is modular, dynamic and able to modify the catalytic activities of all the enzymatic sets (De la Fuente et al., 2011). In particular, certain self-organized enzymatic sets are spontaneously clustered forming functional metabolic sub-networks. Some modules are preserved under different external conditions, whereas others are preserved but with the fluxes inverted and yet other modules emerge only under specific external perturbations. These modules work efficiently: very concrete enzymatic subsystems are connected with others by means of effective information flow allowing high coordination and precise catalytic regulation. Dynamic changes between the functional modules correspond to metabolic switches, which allow for critical transitions in enzymatic activities. The modules coordinated by effective connectivity and the functional switches seem to be important regulatory mechanisms which are able to modify the catalytic activity of all the enzymatic sets (De la Fuente et al., 2011).
Metabolic Integration
In addition to functional segregation, cellular enzymatic processes exhibit another fundamental principle of metabolic connectivity, i.e., functional integration.
Catalytic systems organized into distinct modular networks seem to be integrated; as a consequence, systemic responses emerge from the concerted actions of specialized metabolic processes (Almaas et al., 2004, 2005; Yoon et al., 2007; Buescher et al., 2012; San Román et al., 2014). Whereas functional segregation is an expression of the relative autonomy of specialized biochemical networks, metabolic integration is a complementary principle that refers to significant deviations from modular network autonomy, so that the collective catalytic behavior generates coherent global patterns and molecular information that is simultaneously highly diversified and highly integrated (De la Fuente et al., 2011).
Different mechanisms are responsible for establishing functional metabolic integration. Studies of transfer entropy in DMNs have shown that the integration of specialized biochemical modules is mediated by effective connectivity processes which lead to the emergence of a systemic functional structure that encompasses the whole metabolic system. This global structure is characterized by a small set of different enzymatic processes which are always locked into active states (metabolic core), whereas the remaining catalytic reactions present on-off dynamics (see Figure 1A) (De la Fuente et al., 2011).
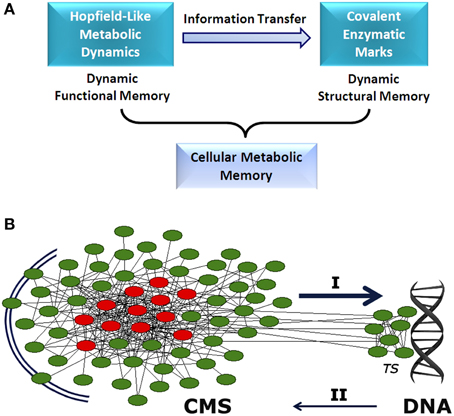
Figure 1. Cellular metabolic memory. (A) Hopfield-like attractor dynamics can emerge in metabolic networks. They have the capacity to store functional catalytic patterns which can be correctly recovered by specific input stimuli. These metabolic dynamics are stable and can be maintained as long-term memory. Specific information associated with these memories can be transferred to the enzymatic processes involved in covalent post-translational modulation. Thus, specific functional stored information can be reversibly embedded in the form of structural molecular marks. These two dynamical processes, storing of catalytic patterns in the form of metabolic attractors (functional processes) and stabilizing metabolic information as molecular marks (structural processes), represent the two stages of the dynamic memory of cellular metabolism. (B) Between the extracellular environment and DNA, there appears to exist a cellular metabolic structure (CMS) which behaves as a very complex decentralized information processing system with the capacity to store metabolic memory (A). The CMS generates sets of biochemical instructions that make each enzymatic activity evolve with a particular and precise dynamic of change, allowing self-regulation and adaptation to the external medium. In addition, the CMS permanently sends a flow of molecular signals to DNA-associated metabolism, which shapes the complex transcriptional system. These molecular flows allow accurate regulation of gene expression, so that only specific polypeptides that are required for adaptive maintenance of the CMS are synthesized. Together, both informative systems (CMS and DNA) coordinate the physiological development of the cell. I: determinative molecular flux for the regulation of the transcriptional system. II: polypeptide reposition. TS: transcriptional system. The CMS is represented by a network. Red circles, metabolic core; green circles, on-off metabolic processes.
As early as in 1999, in an exhaustive analysis of several million different DMNs, the emergence of this functional structure at a global level was observed for the first time (De la Fuente et al., 1999). Afterwards, different studies implementing flux balance analysis of experimental data added new evidence of this systemic metabolic structure in the cell in which a small set of enzymatic reactions belonging to different metabolic processes remain active under all investigated growth conditions (metabolic core), and the remaining enzymatic reactions stay only intermittently active. The emergence of this global catalytic structure was verified for E. coli, H. pylori, and S. cerevisiae (Almaas et al., 2004, 2005). In these studies, the reactions present in the metabolic core were also characterized, and were found to be essential for biomass formation and for ensuring optimal metabolic performance. Likewise, the percentage of enzymatic reactions belonging to the core turned out to be small: 36.2% of all different reactions in H. pylori, 11.9% in E. coli and 2.8% in S. cerevisiae.
Similar results regarding the size of metabolic cores have been reported following extensive numerical analysis of DMNs, in which the percentages of metabolic subsystems pertaining to the core ranged from 44.6 to 0.7%. Nevertheless, their distribution was found to be skewed toward the lower percentages, since 71% of the studied networks showed metabolic core sizes ranging between 7.6 and 0.7% of all metabolic subsystems, with a mean size of 2% (De la Fuente et al., 2009).
In addition to the emergence of a global metabolic structure (De la Fuente et al., 2008), metabolic analysis by means of flux balance analysis and other studies of effective connectivity in DMNs have shown that two main dynamic mechanisms are involved in systemic metabolic self-regulation: flux plasticity and structural plasticity (Almaas et al., 2004, 2005; De la Fuente et al., 2011). Flux plasticity represents changes in the intensity of the substrate fluxes, whereas structural plasticity results in persistent changes in the dynamic structure conformed by the catalytic processes that present on-off and on changing states.
Another key player involved in functional metabolic integration is energy. Living cells require a permanent generation of energy flow to sustain the functionality of their complex metabolic structures, which integrate a large ensemble of modular networks. Practically all bioenergetic processes are coupled with each other via adenosine nucleotides, which are consumed or regenerated by different enzymatic reactions. Moreover, adenosine nucleotides also act as an allosteric control of numerous regulatory enzymes allowing changes in ATP, ADP, and AMP levels to regulate the functional activity of those metabolic subsystems that exhibit this allosteric mechanism. At a systemic level, the temporal dynamics of the concentrations of adenosine nucleotides seem to be determined by the adenylate energy system which represents the synthesis of ATP coupled to energy-consumption processes through a complex network of enzymatic reactions which interconvert ATP, ADP, and AMP (De la Fuente et al., 2014). Experimental studies have shown that adenosine nucleotide dynamics present complex transitions over time. Thus, the use of nanobiosensors has permitted real-time-resolved measurements of intracellular ATP concentrations in intact cells. These experiments revealed the rhythmic behavior of the concentration of ATP and complex quasi steady-state dynamics with large variations over time but, importantly, the concentration of ATP was never found to be constant (Ozalp et al., 2010; Ytting et al., 2012).
The concentration levels of ATP, ADP, and AMP roughly reflect the energetic status of the cell, and a specific systemic ratio between them was proposed by Atkinson as the adenylate energy charge (AEC) (Atkinson and Walton, 1967) which is a scalar index ranging between 0 and 1. When the total adenine nucleotide pool is in form of AMP, the energy charge is zero, and the system is completely discharged (zero concentrations of ATP and ADP). With only ADP, the energy charge is 0.5. If the whole adenine nucleotide pool is in form of ATP, the AEC is 1. Under physiological growth conditions, organisms seem to maintain their AEC within narrow values (between 0.7 and 0.95), despite extremely large fluctuations in adenine nucleotide concentrations. This systemic physiological phenomenon constitutes a key property of the functional integration of metabolic processes in the cell (De la Fuente et al., 2014). The principal biological significance of maintained AEC levels appears to be the necessity of having the adenylate pool highly phosphorylated, keeping the rate of adenylate energy production similar to that of adenylate energy expenditure (De la Fuente et al., 2014). In-depth experimental studies have shown that the physiological AEC ratio is practically preserved in all unicellular organisms, both eukaryotes and prokaryotes. Besides, this ratio is also strictly maintained during all metabolic transformations that occur during the cell cycle, thereby appearing to be a manifestation of the integrative processes of systemic metabolism (De la Fuente et al., 2014).
Functional specialization and integration are not exclusive; rather, they are two dynamic complementary principles, and the interplay between them seems to be an essential element for the functional collective architecture of systemic metabolism in the cell.
Metabolic Networks and Information Properties
Experimental Findings of Biomolecular Information Processing in Unicellular Organisms
A large number of experimental studies have demonstrated that the processing and storing of some kind of biomolecular information is a fundamental requirement for cells to be able to exhibit complex systemic behavior.
One of the most studied examples of complex systemic behavior in a single cell is Physarum polycephalum. This unicellular species, belonging to the Amoebozoa phylum, is an important branch of eukaryote evolution, characterized by a large cytoplasm with many nuclei that remain suspended in a single contiguous protoplasmic volume called plasmodium. The body of the plasmodium contains a complex dynamic network of tubular structures by means of which nutrients and biomolecular elements circulate in an effective manner (Shirakawa and Gunji, 2007).
The movement of the plasmodium is termed shuttle streaming, characterized by the rhythmic back-and-forth flow of biomolecular substances. The dynamically reconfiguring network of tubular structures can redirect the flow of protoplasm toward the plasmatic membrane, causing the movement of a mass of flowing pseudopods, and as a result, the organism can crawl over the ground at a speed of approximately 1–5 cm/h (Kessler, 1982). The complex protoplasm exhibits rich spatio-temporal oscillatory behavior, and rhythmically synchronized streams with intracellular oscillatory patterns such as oscillations in ATP concentration, the plasmagel/plasmasol exchange rhythm and oscillations in the cytoplasmic free calcium level (Ueda et al., 1986).
Over the last several years, a number of studies of Physarum polycephalum have shown that this cell is able to store information, learn and recall past events. For example:
- This unicellular organism has the ability to find the minimum-length solution between two points in a maze; this feature is considered to require cellular information processing (Nakagaki et al., 2000, 2001); it has also been verified that the shortest path problem in the maze requires a mathematically rigorous solution (Miyaji and Ohnishi, 2008).
- Physarum constructs appropriate networks for maximizing nutrient uptake, achieving better network configurations than those based on the shortest connections of Steiner's minimum tree (Nakagaki et al., 2004a,b).
- In particular, high quality solutions for the 8-city traveling salesman problem (TSP), which is known to be NP-hard and hence computationally difficult, have been found by using Physarum Polycephalum (Aono et al., 2011a,b; Zhu et al., 2011).
- Other direct experimental studies found evidence that this kind of amoeba can memorize sequences of periodic environmental changes and recall past events during adaptation to different stimuli (it can even anticipate a previously applied 1 h cold-dry pattern) (Saigusa et al., 2008).
- Despite being a single multinucleate cell, the plasmodium can be used to control autonomous robots (Tsuda et al., 2007; Gough et al., 2009).
- This cell is able to form an optimized network closely approaching the purposely designed Tokyo railway system (Tero et al., 2010).
- Furthermore, it has also been verified that the plasmodium can solve complex multiobjective foraging problems (Dussutour et al., 2010; Bonner, 2010; Latty and Beekman, 2011).
- Self-organization, self-optimization and resolution of complex optimization problems by means of adaptive network development have also been reported in Physarum Polycephalum (Marwan, 2010).
All of these experimental results point to the need for an intrinsic memory storage mechanism (Marwan, 2010) and a “primitive intelligence” (Nakagaki et al., 2000; Nakagaki, 2001; Nakagaki and Guy, 2008; Saigusa et al., 2008; Pershin et al., 2009).
Many other experimental studies have shown that other unicellular organisms also exhibit sophisticated behavior including information storage. A pioneer scientist in revealing such behavior was Jennings, who showed many years ago that a single cell such as Paramecium can exhibit a primitive kind of learning and memory (Jennings, 1905). Neutrophils also exhibit a rudimentary memory system by which they are able to “recall” past directions (Albrecht and Petty, 1998). Dictyostelium and Polysphondylium amoebae seem to have a rudimentary memory and they can show long directional persistence (~10 min), being able to remember the last direction in which they turned (Li et al., 2008). Even individual neurons seem to display short-term memory, and permanent information can be stored when nerve cells in the brain reorganize and strengthen their connections with one another (Sidiropoulou et al., 2009).
At a molecular level, information processing (Ausländer et al., 2012; Daniel et al., 2013) has been observed in numerous processes, such as the reversible phosphorylation of proteins (Thomson and Gunawardena, 2009); microtubule dynamics (Faber et al., 2006); enzymatic processes (Baron et al., 2006; Katz and Privman, 2010); redox regulation (Dwivedi and Kemp, 2012); transcription (Mooney et al., 1998); genetic regulatory networks (Qi et al., 2013); input signal transduction (Roper, 2007); biochemical networks (Bowsher, 2011); NF-kappaB dynamics (Tay et al., 2010); intracellular signaling reactions (Kamimura and Kobayashi, 2012; Purvis and Lahav, 2013); metabolic switches (Ramakrishnan and Bhalla, 2008); the chemotaxis pathway (Shimizu et al., 2010); network motifs (Alon, 2007), and other cellular processes (Ben-Jacob, 2009).
Quantitative Studies for Molecular Information Processing in Self-Organized Enzymatic Sets and in Dissipative Metabolic Networks
Metabolic patterns may have information which can be captured by transfer entropy which allows the quantification of biomolecular information flows in bits (Schreiber, 2000). In 2012, the molecular informative properties of a single self-organized multienzymatic set (a metabolic subsystem) were analyzed in silico by using transfer entropy for the first time (De la Fuente and Cortés, 2012). According to this analysis, the information patterns contained in the different substrate fluxes are continually integrated and transformed by the set of enzymes that form part of the metabolic subsystem. Transfer entropy analysis also showed that in the integrative catalytic processes, a dynamic functional structure for molecular information processing emerges, characterized by changes in informative flows that reflect the modulation of the enzymatic kinetic activities. The level of functional influence in terms of causal interaction between the enzymes is not always the same, but varies depending on information fluxes and the particular dynamic regime of the catalytic system. The multienzymatic set shapes a functional dynamic structure, which is able to permanently send biomolecular information between its different enzymatic parts, in such a way that the activity of each enzyme could be considered an information event.
As a result of the overall catalytic process, enzymatic activities are functionally coordinated, and the metabolic subsystem operates as an information processing system, which at each moment redefines sets of biochemical instructions that make each enzyme evolve in a particular and precise catalytic pattern. In terms of information theory, the metabolic subsystem performs three simultaneous functions: signal reception, signal integration, and the generation of new molecular information. In light of this research, the self-organized enzymatic sets (metabolic subsystems) seem to represent the most basic units for information processing in cells (De la Fuente and Cortés, 2012).
On the other hand, when considering the activity of an interrelated set of different metabolic subsystems, a highly parallel, super-complex structure of molecular information processing emerges in the network. This was evidenced in a numerical study of dissipative metabolic networks in which the biomolecular information flows were quantified in bits between the metabolic subsystems by using transfer entropy (De la Fuente et al., 2011). Specifically, the results showed that the network as a whole integrates and transforms the molecular information coming from both the external environment and the individually assembled metabolic subsystems. The basic units of information processing (each self-organized multienzymatic set) work in parallel and coupled with each other, and as a consequence of these collective processes, functional modules, metabolic switches, highly dynamical metabolic synapses and an information processing super-system emerge in the network.
In addition to the topological network structure, characterized by the specific location of each multienzymatic set, molecular substrate fluxes and regulatory signals (mainly allosteric and covalent modulations), a functional network structure of effective connectivity emerges which is dynamic and characterized by having significant variations of biomolecular information flows between the metabolic subsystems. The dissipative network behaves as a very complex decentralized information processing super-system which generates molecular information flows between the self-organized enzymatic sets, and forces them to be functionally interlocked, i.e., each catalytic set is conditioned to cooperate with others at a precise and specific activity regime in concordance with the activity system as a whole. As a result of the overall functional process, the network defines sets of biochemical instructions at each moment that make every metabolic subsystem evolve with a particular and precise dynamic change of catalytic patterns. Quantitative analysis also revealed that the functional network is able to self-regulate against external perturbations by means of this information processing super-system (De la Fuente et al., 2011).
Dynamic Metabolic Memories
Memory is a fundamental requirement for efficient information processing. Recently, catalytic network dynamics have been numerically analyzed using statistical mechanics tools. In particular, an equivalent Hopfield network has been obtained using a Boltzmann machine, showing that enzymatic activities are governed by systemic attractors with the capacity to store functional metabolic patterns (De la Fuente et al., 2013). This stored molecular information can be correctly recovered from specific input stimuli (Table 3).

Table 3. Metabolic dynamics and functional memory.
In short, the main conclusions of the statistical mechanics analysis were the following:
I. The catalytic dynamics of the dissipative metabolic network are dependent on the Lyapunov function i.e., the energy function of the biochemical system, which is a fundamental element in the regulation of enzymatic activities. Starting in any initial state following an external stimulus, and due to collective enzymatic dynamics, the metabolic network evolves in order to reduce the energy function, which in the absence of noise will decrease in a monotone way until achieving a final system state that is a local minimum of the Lyapunov function. When the biochemical system state reaches a local minimum, it becomes a (locally) stable state for the enzymatic network. The dissipative metabolic network has multiple stable states, each of which corresponds to a specific global enzymatic pattern of the biochemical system.
II. Enzymatic activities of the metabolic network are governed by systemic attractors, which are locally stable states in which all enzymatic dynamics are stabilized. The minima of the Lyapunov function corresponds to these emergent systemic attractors, which regulate the metabolic patterns of the subsystems and determine the enzymatic network to operate as an individual, completely integrated system. The energy function, on which the dynamics of the metabolic network depend, has a complex landscape with multiple attractors (local minima).
III. The emerging systemic attractors correspond to Hopfield-like attractors in which metabolic information patterns can be stored (Hopfield, 1982). The biochemical information contained in the attractors behaves as a functional metabolic memory i.e., enzymatic activity patterns stored as stable states, which regulate both the permanent catalytic changes in the internal medium, as well as the metabolic responses originated to properly integrate the perturbations coming from the external environment. When an external stimulus pattern is presented to the system, the network states are driven by the intrinsic enzymatic dynamics toward a determined systemic attractor which corresponds to a set of memorized biochemical patterns. The formation of the attractor landscape is achieved by metabolic synaptic modifications, i.e., the functional and molecular connections between enzymatic sets, essentially through substrate fluxes, covalent modulations (post-translational modulations) and allosteric regulations. These connectivity patterns can be oscillatory or steady states and comprises an infinite number of distinct activity behaviors. Each biochemical synapse is involved in the storage of the metabolic memory, and metabolic synaptic plasticity seems to be the basis of the memories stored in the metabolic network.
IV. A key attribute of the analyzed systemic attractors is the presence of “associative memory.” When the metabolic network is stimulated by an input pattern, enzymatic activities converge to the stored pattern, which most closely resembles the input, i.e., the attractors have associative memory. Metabolic network attractors seem to store functional enzymatic patterns which can be correctly recovered from specific input patterns. The results explicitly show that there are indeed memory attractors and a capacity to retrieve information patterns (from the weights) which are local minima of the catalytic activity in the network. Thus, if the initial condition of the network dynamics starts within the basin of attraction of such a pattern, the dynamics will converge to the originally stored pattern. As it is well known, this class of “pattern-completion” dynamics has been addressed in Hopfield network studies for years, and it is generally named “associative memory” (Amit, 1989).
The Mean Field Approximation
In metabolic memory analysis, a mean field assumption was computed to describe the equivalent-Hopfield network depicting metabolic dynamics (De la Fuente et al., 2013). The results indicated that information can be stored and eventually retrieved by the local minima of the metabolic network energy; this is what researchers in neural network studies refer to as “having associative memory” (Amit, 1989). Notice that the original dissipative metabolic network can have much more complicated energy landscapes than the ones approached using the mean field solution. For more details about information encoding and retrieving (associative memory) and the capacity for pattern storage of a metabolic network see Supplementary Material S1.
Post-Translational Modifications and Metabolic Memories
The dynamics of metabolic synaptic plasticity governed by Hopfield-type attractors are the fundamental element essential for storing functional metabolic memories in enzymatic networks. However, from a bioenergetic point of view, Hopfield-like metabolic memory is expensive since it requires permanent activity to maintain network functionality, spending substantial amounts of energy mainly in the form of ATP.
Recent investigations have shown that certain enzymes responsible for post-translational modulation can act on multimeric proteins which undergo different modification states that may have slow relaxation dynamics under specific conditions. The activity of such post-translational enzymes depends crucially on the temporal history of the molecular information they receive, and this information can be stored as a slow relaxation process in which slowness enables long-term maintenance of an embedded state (Hatakeyama and Kaneko, 2014). This property permits an efficient information transfer from the Hopfield-like metabolic dynamics to the enzymes involved in covalent post-translational modulation, so that functional memory can be embedded in multiple stable molecular marks.
Molecular information transfer from the functional metabolic memories to the covalent marks requires much less energy than the maintenance of emergent attractor dynamics in metabolic networks. Thus, the post-translational modification of proteins seems to be an essential mechanism for the maintenance of Hopfield-like memories in a structurally and energetically efficient way.
Post-translational modification (PTM) is a highly dynamic, essential enzymatic process which modifies protein functionality, reversibly transforming its structure by adding biochemical marks. Consequently, a single protein subjected to PTM may show a broad range of molecular functions and provoke profound physiological effects in cells (Cohen, 2000; Impens et al., 2014). In fact, PTM is a metabolic mechanism present in all cell types, which can affect the protein structures involved in most cellular processes, e.g., the regulation of the microtubule cytoskeleton (Janke and Bulinski, 2011); signal integration (Deribe et al., 2010); chromatin regulation (Bannister and Kouzarides, 2011); modifications of RNA silencing factors (Heo and Kim, 2009); modulation of mitochondrial membrane activities (Distler et al., 2009); regulation of different metabolic processes in the nucleus (Gloire and Piette, 2009); changes of cytochrome activity (Aguiar et al., 2005); regulation of the mitochondrial translation machinery (Koc and Koc, 2012) as well as other modulations of metabolic processes (Running, 2014). There are different types of PTMs, e.g., phosphorylation, methylation, glycosylation, acetylation, ubiquitination, SUMOylation, etc., and the number and abundance of these modifications is constantly being updated (Khoury et al., 2011).
Protein phosphorylation is one of the most extensively studied PTMs, and the phosphorylation-dephosphorylation mechanism is assumed to take place on a fast time scale in comparison to protein half-lives. Different sites on the same protein can be consecutively phosphorylated-dephosphorylated. These events are not distributed along the whole protein structure but are instead constrained to sites of high accessibility and structural flexibility (Gnad et al., 2007), which allows molecular information encoding, by adding or removing molecular marks (Thomson and Gunawardena, 2009).
While bare DNA encodes 2 bits of information per nucleotide, even in a single protein reversible chemical modifications occur at multiple sites, allowing the encoding of a potentially large amount of information. A protein with n phosphorylated sites has an exponential number (2∧ n) of phospho-forms; the number of phosphorylated sites on a protein shows a significant increase from prokaryotes (with n = 7 sites) to eukaryotes, with examples having n = 150 sites, implying that a protein can encode a large amount of molecular information as a function of a varying number of covalent phospho-marks (Thomson and Gunawardena, 2009). The reversible structural changes in regulatory networks of proteins through PTM allow molecular information encoding and complex information processing, which can originate flexibility and adaptive metabolic responses in the cell (Sims and Reinberg, 2008; Sunyer et al., 2008; Thomson and Gunawardena, 2009).
Attractor network dynamics followed by structural modifications of proteins would represent the two stages of the dynamic memory of cellular metabolism. In accordance with both processes, metabolic memories are derived from continuous changes in the functionality of the network dynamic (due to metabolic synaptic plasticity) linked to structural molecular modifications (covalent marks).
Examples of Post-Translational Modifications and Molecular Information Storage in the Cell
The Escherichia coli chemotaxis network represents one of the best studied examples of a biochemical dynamic system with the capacity to store functional information using molecular information processing linked to post-translational modification dynamics (Tu et al., 2008; Greenfield et al., 2009; Li and Stock, 2009; Shimizu et al., 2010; Stock and Zhang, 2013). Bacterial cell swimmers integrate environmental information accurately in order to make proper decisions for their own survival, in such a way that they can move toward favorable sites and away from unfavorable environments by changing their swimming patterns. As a result of the chemotaxis system, the signal transduction process translates information into appropriate motor responses and the bacterial cells traverse gradients of chemical attractants, displaying an efficient directional sensing and movement by performing temporal comparisons of ligand concentrations. Thus, the cell detects extracellular chemical gradients and senses very small fractional changes, even at nanomolar concentrations (Sourjik and Berg, 2002, 2004).
Since the pioneering studies by Julius Adler in the 1960's (Adler, 1966; Adler and Dahl, 1967; Adler and Templeton, 1967), the biochemical mechanisms that underlie sensory-motor regulation in E. coli have been extensively investigated and their principal characteristics have been detailed (Baker et al., 2006; Hazelbauer et al., 2008). Membrane receptors responsible for signal transduction assemble into large clusters of interacting proteins, shaping a complex modular network (Greenfield et al., 2009; Shimizu et al., 2010; Hamadeh et al., 2011) with one main feedback loop (Wadhams and Armitage, 2004; Emonet and Cluzel, 2008). The network consists of several thousand alpha-helical transmembrane protein fibers that interact with one another forming a cortical structure below the cytoplasmic membrane. The fiber ends that pass through the cytoplasmic membrane interact with a complex layer of sensory receptor domains.
E. coli sensory–motor functionality is regulated by two reversible protein modification processes: phosphorylation and carboxyl methylation. The phosphorylation processes provide a direct connection between sensory receptor complexes and motor responses. The transmembrane protein fibers shape a four-helix bundle with at least eight potentially anionic glutamate side chains that can be either exposed as a negative charge or neutralized by methylation. Each fiber can potentially be in any one of 2∧ 8 different states of modification, and the increments or decrements in attractant concentration produce behavioral responses that lead to changes in methylation dynamics (Stock and Zhang, 2013).
The dynamic changes in chemotaxis network functionality provoke specific molecular information processing and, as a consequence, structural molecular modifications in the form of methylation. They are carried out in such a way that the cell can record the recent chemical past by using this reversible methylation process in determined glutamic acid residues (Li and Stock, 2009). The dynamic methylation-demethylation patterns linked to information processing serve as a structural and functional memory allowing the detection of attractant gradients, by comparing current concentrations to those encountered in the past. The carboxyl methylation mechanism stores information concerning environmental conditions that the bacterium has experienced, and these dynamic patterns of glutamyl modifications act as a structural dynamic memory which allows cells to respond efficiently to continual changes in attractant concentrations (Greenfield et al., 2009; Li and Stock, 2009; Stock and Zhang, 2013).
The E. coli chemotaxis network exhibits a rich variety of behavior as well as dynamic properties such as robustness (Alon et al., 1999; von Dassow et al., 2000; Kollmann et al., 2005), signal amplification (Tu, 2013), molecular information processing (Shimizu et al., 2010), fast response, and ultra-sensitive adaptation (Bray et al., 1998; Wadhams and Armitage, 2004; Sourjik and Berg, 2004; Kollmann et al., 2005). The essential components of the E. coli chemotaxis system are highly conserved among all motile prokaryotes (Wuichet et al., 2007). However, many species have chemotaxis networks that are much more complex than that of E. coli (Hamadeh et al., 2011).
Although the chemistry of signal transduction in prokaryotic and eukaryotic cells differs substantially, there are numerous fundamental similarities (Stock et al., 2002). As in prokaryotes, signal transduction in eukaryotic cells generally involves phosphotransfer mechanisms and other post-translational modifications that couple metabolic energy to information processing (Li and Stock, 2009).
In particular, the biochemical systems of molecular information processing linked to post-translational modification dynamics are highly evolved in neuronal cells. Neurons are connected through synapses that undergo long-lasting activity dependent alterations in their transmission efficacy, permitting learning and response to experience. One of the principal means of controlling synaptic transmission is regulation of neurotransmitter receptors, especially the AMPA-type glutamate receptors which represent the principal excitatory neurotransmitter receptors in neurons. The number of AMPA receptors at the synapse is extremely dynamic and strictly regulated through the addition or removal of receptors resulting in long-term synaptic potentiation or depression (LTP or LTD, respectively) (Anggono and Huganir, 2012). These synaptic plasticity events are thought to underlie information storage. In particular, reversible post-translational modifications of AMPA receptors, including phosphorylation (Wang et al., 2005), palmitoylation (Hayashi et al., 2005), ubiquitination (Schwarz et al., 2010), SUMOylation (Luo et al., 2013), N-glycosylation (Tucholski et al., 2014) and O-GlcNAcylation (Taylor et al., 2014) are important regulatory mechanisms of synaptic AMPA receptor expression and function.
By controlling the expression and function of AMPA receptors, these multiple forms of post-translational modification can vigorously regulate plasticity events and many important functions linked to molecular information processing, including learning and memory (Takeuchi et al., 2014). While prevailing models of long-lasting information storage identify mRNA translation as essential for learning and memory in neural cells, there is evidence that memory storage at the PTM level can occur in the absence of protein synthesis (Routtenberg and Rekart, 2005; Routtenberg, 2008).
Long-Term and Short-Term Memory
Since the beginning of the neuronal network modeling of associative memory, the connectivity matrix in the Hopfield network was assumed to result from a long-term memory learning process, occurring over a much slower time scale than neuronal dynamics (Willshaw et al., 1969, 1970; Amari, 1975; Hopfield, 1982; Amit, 1989; Hertz et al., 1991). Therefore, it is well accepted that the attractors emerging in neuronal dynamics described by Hopfield networks are the result of a long-term memory process.
Besides, extensive physiological recordings of neuronal processes have revealed some type of memory structure. Among others, there are at least two commonly used indicators for this kind of memory: the Hurst exponent (Das et al., 2003; De la Fuente et al., 2006) and the presence of long range correlations in plasticity dynamics for measured synaptic weight, for instance, long tails in the synaptic distribution of weights (Barbour et al., 2007). Several phenomenological models have shown that this persistence in the dynamics of weights underlying short-term memory might eventually be translated into a stable long-term memory after memory consolidation (Fusi et al., 2005; Mongillo et al., 2008; Itskov et al., 2011; Romani et al., 2013), but the precise mechanisms making this transition possible are not yet well understood.
Long-term correlations (mimicking short-term memory in neuronal systems) have also been analyzed in different metabolic processes not belonging to neuronal cell-type. One of the most studied is the calcium-activated potassium channels which has been observed in Leydig cells (Varanda et al., 2000; Bandeira et al., 2008), kidney Vero cells (Kazachenko et al., 2007), and human bronchial epithelial cells (Wawrzkiewicz et al., 2012). Other biochemical processes also present long-term correlation as for example, the intracellular transport pathway of Chlamydomonas reinhardtii (Ludington et al., 2013), the NADPH series of mouse liver cells (Ramanujan et al., 2006), and the mitochondrial membrane potential of cardiomyocytes (Aon et al., 2008). Moreover, different biochemical numerical studies on enzymatic pathways and dissipative metabolic networks also show long-term correlation (De la Fuente et al., 1998a,b, 1999). These studies support the thesis that, at the molecular level, metabolic memories exhibit both long-term and short-term memory, but this issue requires further research.
The Cellular Metabolic Structure—DNA System
Elements of the Cellular Metabolic Structure
Metabolism can be considered to be the largest known source of complexity in nature. Cellular enzymatic processes shape a highly structured, dynamic super-system with different levels of organization, which have a large number of adaptive molecular mechanisms acquired throughout biological evolution. On the way to structural and functional complexity, metabolic evolution has given rise to a large diversity of biochemical organizational forms, ranging from sophisticated bacterial metabolism, through the complex enzymatic networks belonging to protists, to the extraordinary structures and processes derived from multicellular eukaryotic organization, such as embryogenesis or the neural networks present in higher mammals. The millions of living metabolic species represent the inexhaustible source of complexity developed by the dynamic forces that structure the metabolic systems.
Enzymes are fundamental molecules for metabolic life. At a basic level, they can shape dissipatively structured functional associations (metabolic subsystems), which seem to constitute elemental catalytic units of the cell reactor. A rich variety of dynamic catalytic patterns emerges in each metabolic subsystem (including oscillatory and quasi-steady states), which correspond to distinct activity regimes (see Section Enzymatic Self-Organization). Catalytic changes allow metabolic synapses, i.e., the structural and functional dynamic connections between enzymatic sets. As a result of flexible activity regimes, flux plasticity and structural plasticity also appear in the cell (see Section Functional Organization of Enzymes in Metabolic Networks). At a superior level, enzymatic processes are organized into distinct modular metabolic networks, which work with relative independence of the global system. In turn, the whole metabolism seems to be functionally integrated, generating systemic responses from the concerted actions of modular networks, shaping a complex dynamic super-system, the Cellular Metabolic Structure (CMS). Therefore, the two principles that link the main modes of metabolic connectivity in the cell are functional segregation and functional systemic integration (see Section Functional Organization of Enzymes in Metabolic Networks).
The CMS is an integrated catalytic entity that acts as an information processing system capable of storing functional biochemical patterns. Metabolic synaptic dynamics governed by Hopfield-type attractors and structural covalent modifications represent the two stages of the dynamic memory of cellular metabolism. Plasticity seems to be the key essential property for the emergence of metabolic memory (see Section Metabolic Networks and Information Properties). All these properties constitute some of the main elemental principles of the CMS, which allow the self-organization, self-regulation and adaptation of the cell to the external medium.
Self-Regulation of the Cellular Metabolic Structure
Cells are open metabolic systems. In order to maintain self-organized biochemical functionality, the CMS needs a permanent exchange of energy and matter with the external medium. As a consequence, metabolic life would be impossible in the absence of a capacity to adequately respond to the large number of combinations of external variables to which it is exposed.
The CMS is a sophisticated, sensitive system, whose enzymatic processes change immediately in response to variations in environmental conditions (Almaas et al., 2004, 2005; Papp et al., 2004; Wang and Zhang, 2009). When external perturbations are small, the CMS changes accordingly, and slowly reverts back to the pre-stimulus dynamic by reorganizing and adjusting catalytic patterns and metabolic fluxes (Inoue and Kaneko, 2013). If external perturbations are more intense, e.g., the replacement of a major source of nutrients with another, the CMS computes the various possible ways to efficiently process the molecular substrates, turning off certain metabolic activities (even completely eliminating specific enzymatic sets), and adding new reactive processes and alternative routes. As a result, metabolism shows extensive reorganization in its functionality, resulting in a rewiring of global network fluxes (Storlien et al., 2004; Buescher et al., 2012; San Román et al., 2014; Zhang et al., 2014).
When the metabolic system experiences extreme external conditions, e.g. nutrient deprivation, the CMS generates very complex enzymatic reorganization involving the entire system. In general, the autophagy interaction network is activated in response to starvation (Behrends et al., 2010), and as a consequence, proteins, organelles and other cellular substructures are sequestered in autophagosomal vesicles to be later degraded. This is one of the major mechanisms by means of which a starving cell reallocates nutrients from unnecessary metabolic pathways to more-essential processes (Shintani and Klionsky, 2004). Under starvation conditions, the CMS computes the efficient rates of molecular turnover, modulates systemic enzymatic processes with extreme precision, and adjusts the number of different organelles to match available resources. For instance, the CMS profoundly regulates different mechanisms such as ribosome biosynthesis and degradation (Pestov and Shcherbik, 2012), nucleolar proteome dynamics (Andersen et al., 2005), protein turnover (Neher et al., 2003), lipid degradation (Holdsworth and Ratledge, 1988), peroxisomal number (Mijaljica et al., 2006), and global cellular energy (Boender et al., 2011), among others.
There are endless possible combinations of environmental variables (physical, chemical, nutritional and biological) to which the cell must give an adequate response. The CMS has the properties to self-adapt in response to this plethora of external variables, displaying an impressive capacity to regulate thousands of metabolic processes simultaneously, computing the appropriate regulatory responses at each moment through complex and specific catalytic dynamic patterns that must represent an appropriate response to each set of external perturbations. Furthermore, these self-regulations occur very quickly. For instance, the metabolic dynamics of Escherichia coli were studied in cultures in which the glucose concentration was constantly changing, following an up-and-down pattern. The authors found that the systemic energetic state of the cell (measured by the adenylate energy charge) self-adjusted as a whole within only 2 min of the external perturbations (Weber et al., 2005).
In addition, metabolism also exhibits complex and continuous cycles of molecular turnover, including that of the complete proteome, glycome, lipidome, metabolome, and transcriptome of the cell. More specifically, enzymes, the essential molecular actors for cell functionality, are continually synthesized and destroyed in a dynamic turnover process in which all proteins are broken down partially into peptides, or completely, into amino acids for later de novo regeneration (Doherty and Beynon, 2006; Doherty et al., 2009; Li, 2010). As a result of the global metabolic dynamic that encompasses reactive molecular transformations, including protein turnover, specific polypeptides must be suitably synthetized when required by physiological conditions. Therefore adequate molecular information flow from the CMS to DNA is essential for ensuring adequate polypeptide replacement.
Nexus between the Cellular Metabolic Structure and DNA
Just over 40 years ago, the Nobel Prize Laureate John Gurdon demonstrated the existence of a molecular flow from the cytoplasm into the nucleus involved in the differential expression of certain genes during early embryonic development (Gurdon, 1971). His experiments involved transplantation of the nucleus of a cell, which was carrying out a specific dynamic activity, into an enucleated cytoplasm whose metabolic activity was quite another. These studies were an extension of Briggs and King's work on transplanting nuclei from embryonic blastula cells (Briggs and King, 1959).
Gurdon reached a fundamental conclusion on the basis of his experiments: that some cytoplasmic molecules control the expression of certain genes (or their repression) in an independent way. In all hybrid cells, the transplanted nucleus did not continue its previous activity, but rather changed its function to conform to that of the host cytoplasmic metabolism to which it had been exposed. For instance: the mid-blastula nucleus synthesizes DNA but not RNA, and when it is implanted into the cytoplasm of a growing oocyte whose nucleus does not synthesize DNA, but rather RNA, the new hybrid cell stops synthesizing DNA and starts synthesizing RNA. If this same mid-blastula nucleus is injected into the cytoplasm of a mature oocyte which is not synthesizing any nucleic acid and whose chromosomes are placed in spindle phase, the implanted nucleus ends up synthesizing DNA and the chromosomes arrange in the spindle of cell division. When an adult neuron nucleus, which synthesizes RNA but rarely synthesizes DNA, is injected into the cytoplasm of an unfertilized but activated egg whose nucleus would normally synthesize DNA but not RNA, the transplanted nucleus stops RNA synthesis and begins DNA synthesis (Gurdon, 1971).
Another one of Gurdon's experimental series proved that certain cytoplasmic components select which genes will be activated and/or repressed at any given time and consequently, it was deduced that major changes in different kinds of gene activity are a response to molecular signals coming from cytoplasmic metabolism (Gurdon, 1971). Analogous experiments have since been conducted in protozoans, amphibians, plants and in different mammals such as cats, mice, monkeys, rabbits, horses, donkeys, pigs, goats, and cows; even a mouse was cloned using the nucleus of an olfactory neuron (Eggan et al., 2004). Of all these, the most popular example is Dolly, who was the result of an experiment in which a single nucleus from a differentiated mammary cell was successfully fused with an enucleated unfertilized egg from another adult sheep (Wilmut et al., 1997).
According to contemporary paradigms, DNA contains all the essential information for the maintenance and development of the cell, and therefore the mammary nucleus should be able to modify the metabolic activity of the cytoplasm into which it has been injected, so that the new hybrid organism should act as a mammary cell. But what happened in the Dolly experiment was precisely the opposite: the transferred nucleus underwent a radical transformation via the molecular information flux which originated from the host cytoplasmic metabolism. This information reprogrammed genetic expression in the implanted nucleus which then began to express transcription patterns which were completely different to those associated with the previous, non-transplanted conditions. Overall, these experiments demonstrated that this new program of molecular instructions comes, not from the nucleus, but rather from the metabolic structure of the host cytoplasm.
In the Dolly experiment, gene expression patterns were regulated by the cytoplasmic metabolic structure of the cell into which the mammary nucleus had been injected. The hybrid cell no longer behaved like a mammary cell, but rather, it started to divide according to a new molecular program, that is, the sequence of instructions to perform a specified biochemical task, executed by cytoplasmic metabolism. During the first three cell divisions, the new cell replicates its DNA without expressing any genes, and this is followed by successive cell divisions and progressive differentiation, first into the early embryo and subsequently into all of the cell types that characterize the animal. Cellular Metabolic Structure (involving all the metabolism in both the cytoplasm and nucleoplasm) is able to reprogram the gene expression of mammary DNA, and both the CMS and the DNA coordinate together the development of an entire organism.
Extensive experimental observations of molecular flow from cytoplasmic metabolism to the nucleus of eukaryotic cells, date back to as early as the 1960s (Zetterberg, 1966; Arms, 1968; Gurdon and Weir, 1969; Merriam, 1969) and continue to be reported (Cook et al., 2007; Wimmer et al., 2013; Tamura and Hara-Nishimura, 2014). Many different experimental approaches have shown that molecular flow from cytoplasmic metabolism to the nucleoplasm consists of ordinary components of cellular metabolism. Thousands of proteins, metabolites, cofactors and ions are transferred to the nucleoplasm with remarkable efficiency and specificity (Kuersten et al., 2001; Cook et al., 2007; Gerhäuser, 2012), and the different molecular combinations, each with precise concentrations, lead to the modulation of DNA-associated metabolism, which regulates gene activity and other processes.
Extensive experimental studies both in prokaryotes and eukaryotes have uncovered the same fundamental process: genes exhibiting transcriptional induction in response to metabolic changes, having their origins in self-adaptation to a wide range of external factors such as nutrient deprivation, chemical agents (hormones, pH, ionic strength, among others) and physical fluctuations, including temperature shock, drought, high-intensity light etc. (Causton et al., 2001; Gasch and Werner-Washburne, 2002; Kreps et al., 2002; Hazen et al., 2003; Liu et al., 2005; Deutscher et al., 2006; Stern et al., 2007; Swindell et al., 2007; Wilson and Nierhaus, 2007; Angers et al., 2010; Mirouze and Paszkowski, 2011). For instance, under glucose starvation, the stringent reduction of ATP turnover at the metabolic level was accompanied by a strong down-regulation of genes involved in protein synthesis (Boender et al., 2011).
The traffic from the CMS to DNA-associated metabolism ensures the intimate subordination of the transcriptional system to the needs of metabolic activity, at each moment. The transcriptional system seems to be one of the most complex dynamic structures of the cell. It is made up of different, highly coordinated metabolic networks which integrate a large variety of signals and process molecular information from both the DNA and the CMS. Unfortunately, most of the dynamic regulatory aspects of the metabolic transcriptional system are still poorly understood.
A huge variety of biochemical mechanisms participates in the control of gene expression, including those which involve topoisomerases, transcription factors, RNA polymerases, small RNAs, the translation system, the turnover rate of proteins, the dynamic levels of adenylate energy charge, etc. These metabolic processes are in turn modulated by complex molecular networks (Balázsi et al., 2005; Missiuro et al., 2009) such as, for instance, those associated with miRNAs (Dong et al., 2010; Gennarino et al., 2012; Schulz et al., 2013), transcription factor processes (Walhout, 2006; Neph et al., 2012), RNA polymerase complexes (Schubert, 2014), and mRNA dynamics (Maquat and Gong, 2009; Schott and Stoecklin, 2010; Vlasova-St Louis and Bohjanen, 2011) among others.
Chromosome packaging is another essential biochemical control process for gene expression, and is common to all living organisms. In eukaryotes, nucleosome-based DNA organization allows for the reversible access of different metabolic regulatory mechanisms in order to modulate specific DNA sequences. In fact, the tails of the four core histones are subject to a variety of enzyme-catalyzed, post-translational modifications (Turner, 2005). Networks of biochemical factors that functionally interact with the nucleosome, reversibly remodeling it, have also been reported (Burgio et al., 2008). Prokaryotic cells, despite exhibiting a very different DNA organization to that of eukaryotes, also present a protein collectivity referred to as nucleoid-associated proteins, which contributes to the compaction and regulation of the genome (Rimsky and Travers, 2011; Takeda et al., 2011).
In addition, gene expression can also be modulated through methylation-demethylation dynamics on DNA nucleotides, mostly at CpG sites to convert cytosine to 5-methylcytosine. The enzymatic systems for these covalent marks are also integrated in DNA-associated metabolism, and seem to be directly involved in maintaining dynamic metabolic memories in the transcriptional system.
In brief, it appears that both eukaryotes and prokaryotes have a common mechanism to coordinate the two information storage systems in the cell: the CMS and DNA (Figure 1B). The CMS integrates and processes stimuli coming from the environment, generating self-regulatory metabolic responses and the traffic of molecular information to DNA-associated metabolism. This information flux ensures the intimate integration and subordination of the transcriptional system to the rest of the metabolic activity of the cell. The molecular flow allows the appropriate orchestration of the whole transcriptional system and, as a consequence, specific parts of the genome are activated and deactivated at strategic times so that, in accordance with the requirements of adaptive maintenance of cellular metabolism at each moment, gene expression is accurately regulated and only specific polypeptides are synthesized. Other important implications of the CMS in the regulation of DNA-associated metabolism, such as the generation of new polypeptidic information by means of alternative splicing or directed mutations, are beyond the scope of the present work.
Duality of Cellular Hereditary Information
Many different experimental approaches have provided ample evidence that specific functional metabolic processes codified via covalent mark patterns (epigenetic changes), can be preserved when cells divide and thus can be transferred to the next generation, in a manner which is different of DNA-based inheritance, i.e., this intergenerational preservation is not encoded in the DNA sequence.
For instance, epigenetic changes in response to environmental impacts on cellular metabolism have been observed to be transmitted to the offspring in different kinds of cells (Pembrey et al., 2006; Gluckman et al., 2007; Nadeau, 2009; Jiminez-Chirallon et al., 2009; Carone et al., 2010; Grossniklaus, 2011). Alterations in available nutrients may also induce specific metabolic processes that seem to be transmitted to the next generation by means of covalent marks that do not involve changes in the DNA sequence (Kaati et al., 2007; Waterland et al., 2007). Moreover, experimental findings regarding cells under stress shed new light on the transgenerational inheritance of metabolic memory processes (Seong et al., 2011; Crews et al., 2012; Pecinka and Mittelsten Scheid, 2012). Likewise, persistent and stable transmission of epigenetic information across generations has been reported. For instance, in Arabidopsis thaliana, environmental stress induces genomic flexibility (increasing the hyper-recombination state), which persists in a stable way for up to four untreated generations (Molinier et al., 2006).
A stably inherited DNA methylation pattern has also been reported in a variant of Linaria vulgaris originally described more than 260 years ago by Linnaeus (1749). In this example, the fundamental symmetry of the flower was found to change from bilateral to radial as a result of extensive methylation of the Lcyc gene that controls the formation of dorsal petals (Cubas et al., 1999).
The complex reprogramming of metabolic information marks in primordial germ cells according to their parental origin is another fine example of heritable biochemical patterns which are not caused by changes in the DNA sequence (Seisenberger et al., 2012; Hanna and Kelsey, 2014). The transmission of epigenetic metabolic information across generations has also been observed in bacteria (Casadesús and Low, 2013; Gonzalez et al., 2014).
Overall, epigenetic processes seem to be the manifestation of cellular metabolic memory. The covalent modification of histones and the dynamics of methylation-demethylation of specific DNA nucleotides appear to represent the storage of specific functional biochemical memories in a structural way, and these marks can emerge as a consequence of Hopfield-type metabolic dynamics.
There is thus ample evidence that cells have two types of information storage system which are capable of transmitting specific information to the following generations, i.e., the CMS and DNA systems. The molecular information stored in both systems may well be sufficient to coordinate the physiological development of the entire cell.
Concluding Remarks
The observations and consequent reflections I present here concerning numerous cellular processes and different quantitative biomolecular approaches, both presumably interrelated, have led me to consider, without underestimating the enormous importance of DNA, that the elements responsible for the organization and regulation of fundamental biological processes are to be found in determined properties and characteristics of cellular metabolism.
As far as I have been able to ascertain, I find it probable that the essential attributes upon which cellular functionality lies have their basis in the as yet insufficiently understood Cellular Metabolic Structure. In this paper, I have tried, in a conceptual manner, to present the basic elements of a dualist framework concerning the information stored in the cell, according to which the dynamic metabolic memory and the genetic memory seem to be the manifestation of two systems of information storage in the cell. From this perspective, rather than a genoteque1 in evolution, the cell could be considered to be a singular metabolic entity, able to permanently self-organize and self-regulate, as well as to adapt to the environment.
The work presented herein is necessarily and predominantly an exercise of multidisciplinary integration, where concepts from different branches of biological knowledge—mainly Biochemistry, Physiology, Cell Biology and Molecular Biology—are addressed within the approach of Systems Biology. Moreover, during the development and completion of quantitative studies, it has been necessary to analyze many experimental and numerical data with different analytic methods coming from several disciplines such as differential calculus, statistical mechanics, discrete mathematics, computing techniques and artificial intelligence, as well as other physico-mathematical tools. However, in this manuscript, mathematical formulation has been omitted in favor of a more didactic character of the text. That being said, it is necessary to stress that, in my opinion, without mathematics—the language of nature—it would not be possible to precisely understand metabolic dynamics and the catalytic functionality that sustains them. Fortunately, today the necessary application of mathematics to all fields of human knowledge, biology in particular, is widely accepted. In the autobiographical recollections that Charles Darwin wrote in 1876 for his children, he expressed the importance of understanding “something of the great leading principles of mathematics, for men thus endowed seem to have an extra sense” (Darwin, 1905).
The novel dual framework of functional and structural information stored in the cell which I present here could open new perspectives on the comprehension of subjects of notable interest, as yet not well understood, such as cellular differentiation, to name but one. Thus, cells with a different Cellular Metabolic Structure and, therefore, different catalytic activity programs, but with the same genetic heritage, can express different genes and manifest different physiological behavior. Something along these lines could be said about the Theory of Evolution, in terms of how it was presented by the endearing Charles Darwin, and the possibility that cells themselves are able to self-adapt to the environment permanently through the dynamic properties of cellular metabolism and the dual character of cellular hereditary information.
Frankly, I have to admit that dealing with these overwhelmingly complex issues of systemic cell biology with the desired clarity and precision is an extraordinarily difficult undertaking. In addition, our current comprehension of dynamic biological phenomena in its most elemental aspects is still very incomplete and insufficient. It is truly extraordinary that in such a reduced space of merely a few microns, millions of biochemical processes form a metabolic entity that is sensitive, self-organized, self-regulated, able to process and store information, and perpetuate itself in time.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmolb.2015.00016/abstract
Footnotes
1. ^Literally, a box of genes.
References
Abadjieva, A., Pauwels, K., Hilven, P., and Crabeel, M. (2001). A new yeast metabolon involving at least the two first enzymes of arginine biosynthesis: acetylglutamate synthase activity requires complex formation with acetylglutamate kinase. J. Biol. Chem. 276, 42869–42880. doi: 10.1074/jbc.M103732200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adler, J. (1966). Chemotaxis in bacteria. Science 153, 708–716. doi: 10.1126/science.153.3737.708
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adler, J., and Dahl, M. M. (1967). A method for measuring the motility of bacteria and for comparing random and non-random motility. J. Gen. Microbiol. 46, 161–173. doi: 10.1099/00221287-46-2-161
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Adler, J., and Templeton, B. (1967). The effect of environmental conditions on the motility of Escherichia coli. J. Gen. Microbiol. 46, 175–184. doi: 10.1099/00221287-46-2-175
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aguiar, M., Masse, R., and Gibbs, B. F. (2005). Regulation of cytochrome P450 by post-translational modification. Drug. Metab. Rev. 37, 379–404. doi: 10.1081/DMR-200046136
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ainscow, E. K., Mirsham, S., Tang, T., Ashford, M. L. J., and Rutter, G. A. (2002). Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: evidence for ATP independent control of ATP-sensitive K+ channels. J. Physiol. 544(Pt 2), 429–445. doi: 10.1113/jphysiol.2002.022434
Albrecht, E., and Petty, R. H. (1998). Cellular memory: neutrophil orientation reverses during temporally decreasing chemoattractant concentrations. Proc. Natl. Acad. Sci. U.S.A. 95, 5039–5044. doi: 10.1073/pnas.95.9.5039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Almaas, E., Kovacs, B., Vicsek, T., Oltvai, Z. N., and Barabási, A. L. (2004). Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 427, 839–843. doi: 10.1038/nature02289
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Almaas, E., Oltvai, Z. N., and Barabási, A. L. (2005). The activity reaction core and plasticity of metabolic networks. PLoS Comput. Biol. 1:e68. doi: 10.1371/journal.pcbi.0010068
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alon, U. (2007). Network motifs: theory and experimental approaches. Nat. Rev. Genet. 8, 450–461. doi: 10.1038/nrg2102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Alon, U., Surette, M. G., Barkai, N., and Leibler, S. (1999). Robustness in bacterial chemotaxis. Nature 397, 168–117. doi: 10.1038/16483
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amari, S. (1975). Homogeneous nets of neuron-like elements. Biol. Cybern. 17, 211–220. doi: 10.1007/BF00339367
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Amariei, C., Tomita, M., and Murray, D. B. (2014). Quantifying periodicity in omics data. Front. Cell Dev. Biol. 2:40. doi: 10.3389/fcell.2014.00040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Andersen, J. S., Lam, Y. W., Leung, A. K., Ong, S. E., Lyon, C. E., Lamond, A. I., et al. (2005). Nucleolar proteome dynamics. Nature 433, 77–83. doi: 10.1038/nature03207
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Angers, B., Castonguay, E., and Massicotte, R. (2010). Environmentally induced phenotypes and DNA methylation: how to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 19, 1283–1295. doi: 10.1111/j.1365-294X.2010.04580.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Anggono, V., and Huganir, R. L. (2012). Regulation of AMPA receptor trafficking and synaptic plasticity. Curr. Opin. Neurobiol. 22, 461–469. doi: 10.1016/j.conb.2011.12.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aon, M. A., Cortassa, S., and O'Rourke, B. (2006). The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys. J. 91, 4317–4327. doi: 10.1529/biophysj.106.087817
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aon, M. A., Roussel, M. R., Cortassa, S., O'Rourke, B., Murray, D. B., Beckmann, M., et al. (2008). The scale-free dynamics of eukaryotic cells. PLoS ONE 3:e3624. doi: 10.1371/journal.pone.0003624
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aono, M., Zhu, L., and Hara, M. (2011a). Amoeba-based neurocomputing for 8-city traveling salesman problem. Int. J. Unconv. Comput. 7, 463–480. doi: 10.2420/AF03.2008.36
Aono, M., Zhu, L., Kim, S. J., and Hara, M. (2011b). Performance enhancement of amoeba-based neurocomputer for 8-city traveling salesman problem. Proc. NOLTA. 104–107.
Arms, K. (1968). Cytonucleoproteins in cleaving eggs of Xenopus laevis. J. Embryol. Exp. Morphol. 20, 367–374.
Asano, Y., Nagasaki, A., and Uyeda, T. Q. P. (2008). Correlated waves of actin filaments and PIP3 in Dictyostelium cells. Cell Mot. Cytosk. 65, 923–934. doi: 10.1002/cm.20314
Atkinson, D. E., and Walton, G. M. (1967). Adenosine triphosphate conservation in metabolic regulation. Rat liver citrate cleavage enzyme. J. Biol. Chem. 242, 3239–3241.
Ausländer, S., Ausländer, D., Müller, M., Wieland, M., and Fussenegger, M. (2012). Programmable single-cell mammalian biocomputers. Nature 487, 123–127. doi: 10.1038/nature11149
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baker, M. D., Wolanin, P. M., and Stock, J. B. (2006). Signal transduction in bacterial chemotaxis. Bioessays 28, 9–22. doi: 10.1002/bies.20343
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Balázsi, G., Barabási, A. L., and Oltvai, Z. N. (2005). Topological units of environmental signal processing in the transcriptional regulatory network of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 7841–7846. doi: 10.1073/pnas.0500365102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bandeira, H. T., Barbosa, C. T., De Oliveira, R. A., Aguiar, J. F., and Nogueira, R. A. (2008). Chaotic model and memory in single calcium-activated potassium channel kinetics. Chaos 18, 033136. doi: 10.1063/1.2944980
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell. Res. 21, 381–395. doi: 10.1038/cr.2011.22
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barbour, B., Brunel, N., Hakim, V., and Nadal, J. P. (2007). What can we learn from synaptic weight distributions? Trends Neurosci. 30, 622–629. doi: 10.1016/j.tins.2007.09.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barnes, S. J., and Weitzman, P. D. (1986). Organization of citric acid cycle enzymes into a multienzyme cluster. FEBS Lett. 201, 267–270. doi: 10.1016/0014-5793(86)80621-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Baron, R., Lioubashevski, O., Katz, E., Niazov, T., and Willner, I. (2006). Logic gates and elementary computing by enzymes. J. Phys. Chem. A. 110, 8548–8553. doi: 10.1021/jp0568327
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barril, E. F., and Potter, A. R. (1968). Systematic oscillations of amino acid transport in liver from rats adapted to controlled feeding schedules. J. Nutr. 95, 228–237.
Bayer, E. A., Belaich, J. P., Shoham, Y., and Lamed, R. (2004). The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu. Rev. Microbiol. 58, 521–554. doi: 10.1146/annurev.micro.57.030502.091022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beeckmans, S., Van Driessche, E., and Kanarek, L. (1990). Clustering of sequential enzymes in the glycolytic pathway and the citric acid cycle. J. Cell. Biochem. 43, 297–306. doi: 10.1002/jcb.240430402
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beg, Q. K., Vazquez, A., Ernst, J., de Menezes, M. A., Bar-Joseph, Z., Barabási, A. L., et al. (2007). Intracellular crowding defines the mode and sequence of substrate uptake by Escherichia coli and constrains its metabolic activity. Proc. Natl. Acad. Sci. U.S.A. 104, 12663–12668. doi: 10.1073/pnas.0609845104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Behrends, C., Sowa, M. E., Gygi, S. P., and Harper, J. W. (2010). Network organization of the human autophagy system. Nature 466, 68–76. doi: 10.1038/nature09204
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ben-Jacob, E. (2009). Learning from bacteria about natural information processing. Ann. N.Y. Acad. Sci. 1178, 78–90. doi: 10.1111/j.1749-6632.2009.05022.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bernardinelli, Y., Magistretti, P. J., and Chatton, J. Y. (2004). Astrocytes generate Na+-mediated metabolic waves. Proc. Natl. Acad. Sci. U.S.A. 101, 14937–14942. doi: 10.1073/pnas.0405315101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bhaskaran, H., and Perona, J. J. (2011). Two-step aminoacylation of tRNA without channeling in Archaea. J. Mol. Biol. 411, 854–869. doi: 10.1016/j.jmb.2011.06.039
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Binstock, J. F., and Schulz, H. (1981). Fatty acid oxidation complex from Escherichia coli. Methods Enzymol. 71(Pt C), 403–411. doi: 10.1016/0076-6879(81)71051-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bobik, T. A. (2006). Polyhedral organelles compartmenting bacterial metabolic processes. Appl. Microbiol. Biotechnol. 70, 517–525. doi: 10.1007/s00253-005-0295-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boender, L. G., Almering, M. J., Dijk, M., van Maris, A. J., de Winde, J. H., Pronk, J. T., et al. (2011). Extreme calorie restriction and energy source starvation in Saccharomyces cerevisiae represent distinct physiological states. Biochim. Biophys. Acta 1813, 2133–2144. doi: 10.1016/j.bbamcr.2011.07.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boiteux, A., Hess, B., and Sel'kov, E. E. (1980). Creative functions of instability and oscillations in metabolic systems. Curr. Top. Cell. Reg. 17, 171–203. doi: 10.1016/B978-0-12-152817-1.50010-9
Bonetta, L. (2010). Protein-protein interactions: interactome under construction. Nature 468, 851–854. doi: 10.1038/468851a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bonner, J. T. (2010). Brainless behavior: a myxomycete chooses a balanced diet. Proc. Natl. Acad. Sci. U.S.A. 107, 5267–5268. doi: 10.1073/pnas.1000861107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Boruc, J., Van den Daele, H., Hollunder, J., Rombauts, S., Mylle, E., Hilson, P., et al. (2010). Functional modules in the Arabidopsis core cell cycle binary protein-protein interaction network. Plant Cell. 22, 1264–1280. doi: 10.1105/tpc.109.073635
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bowsher, C. G. (2011). Information processing by biochemical networks: a dynamic approach. J. R. Soc. Interface 8, 186–200. doi: 10.1098/rsif.2010.0287
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bray, D., Levin, M. D., and Morton-Firth, C. J. (1998). Receptor clustering as a cellular mechanism to control sensitivity. Nature 393, 85–88. doi: 10.1038/30018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Briggs, B., and King, T. J. (1959). “Nucleocytoplasmic interactions in egg and embryos,” in The Cell, Vol. 1, eds J. Brachet and A. E. Mirsky (New York, NY: Acad. Press), 537–617.
Brodsky, V., Boikov, P. Y., Nechaeva, N. V., Yurovitsky, Y. G., Novikova, T. E., Fateeva, V. I., et al. (1992). The rhythm of protein synthesis does not depend on oscillations of ATP level. J. Cell Sci. 103(Pt 2), 363–370.
Brodsky, V. Y. (2006). Direct cell-cell communication: a new approach derived from recent data on the nature and self-organisation of ultradian (circahoralian) intracellular rhythms. Biol. Rev. Camb. Philos. Soc. 81, 143–162. doi: 10.1017/S1464793105006937
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buescher, J. M., Liebermeister, W., Jules, M., Uhr, M., Muntel, J., Botella, E., et al. (2012). Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science 335, 1099–1103. doi: 10.1126/science.1206871
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bultema, J. B., Braun, H. P., Boekema, E. J., and Kouril, R. (2009). Megacomplex organization of the oxidative phosphorylation system by structural analysis of respiratory supercomplexes from potato. Biochim. Biophys. Acta 1787, 60–67. doi: 10.1016/j.bbabio.2008.10.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Burgio, G., La Rocca, G., Sala, A., Arancio, W., Di Gesù, D., Collesano, M., et al. (2008). Genetic identification of a network of factors that functionally interact with the nucleosome remodeling ATPase ISWI. PLoS Genet. 4:e1000089. doi: 10.1371/journal.pgen.1000089
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cardone, L., Hirayama, J., Giordano, F., Tamaru, T., Palvimo, J. J., and Sassone-Corsi, P. (2005). Circadian clock control by SUMOylation of BMAL1. Science 309, 1390–1394. doi: 10.1126/science.1110689
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carone, B. R., Fauquier, L., Habib, N., Shea, J. M., Hart, C. E., Li, R., et al. (2010). Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143, 1084–1096. doi: 10.1016/j.cell.2010.12.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Carpousis, A. J. (2002). The Escherichia coli RNA degradosome: structure, function and relationship in other ribonucleolytic multienzyme complexes. Biochem. Soc. Trans. 30, 150–155. doi: 10.1042/BST0300150
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Casadesús, J., and Low, D. A. (2013). Programmed heterogeneity: epigenetic mechanisms in bacteria. J. Biol. Chem. 288, 13929–13935. doi: 10.1074/jbc.R113.472274
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Causton, H. C., Ren, B., Koh, S. S., Harbison, C. T., Kanin, E., Jennings, E. G., et al. (2001). Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell. 12, 323–337. doi: 10.1091/mbc.12.2.323
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chabot, J. R., Pedraza, J. M., Luitel, P., and van Oudenaarden, A. (2007). Stochastic gene expression out-of-steady-state in the cyanobacterial circadian clock. Nature 450, 1249–1252. doi: 10.1038/nature06395
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chance, B., Pye, E. K., Ghosh, A. D., and Hess, B. (eds.). (1973). Biological and Biochemical Oscillations. New York, NY: Academic Press.
Cheung, C. W., Cohen, N. S., and Raijman, L. (1989). Channeling of urea cycle intermediates in situ in permeabilized hepatocytes. J. Biol. Chem. 264, 4038–4044.
Chiam, H. K., and Rajagopal, G. (2007). Oscillations in intracellular signaling cascades. Phys. Rev. E. 75(Pt 1):061901. doi: 10.1103/PhysRevE.75.061901
Clegg, J. S., and Jackson, S. A. (1990). Glucose metabolism and the channelling of glycolytic intermediates in permeabilized L-929 cells. Arch. Biochem. Biophys. 278, 452–460. doi: 10.1016/0003-9861(90)90284-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Clémençon, B. (2012). Yeast mitochondrial interactome model: metabolon membrane proteins complex involved in the channeling of ADP/ATP. Int. J. Mol. Sci. 13, 1858–1885. doi: 10.3390/ijms13021858
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cohen, P. (2000). The regulation of protein function by multisite phosphorylation - a 25 year update. Trends Biochem. Sci. 25, 596–601. doi: 10.1016/S0968-0004(00)01712-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cook, A., Bono, F., Jinek, M., and Conti, E. (2007). Structural biology of nucleocytoplasmic transport. Annu. Rev. Biochem. 76, 647–671. doi: 10.1146/annurev.biochem.76.052705.161529
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crews, D., Gillette, R., Scarpino, S. V., Manikkam, M., Savenkova, M. I., and Skinner, M. K. (2012). Epigenetic transgenerational inheritance of altered stress responses. Proc. Natl. Acad. Sci. U.S.A. 109, 9143–9148. doi: 10.1073/pnas.1118514109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi: 10.1038/43657
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Daniel, R., Rubens, J. R., Sarpeshkar, R., and Lu, T. K. (2013). Synthetic analog computation in living cells. Nature 497, 619–623. doi: 10.1038/nature12148
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dano, S., Sorensen, P., and Hynne, F. (1999). Sustained oscillations in living cells. Nature 402, 320–322. doi: 10.1038/46329
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Das, M., Gebber, G. L., Barman, S. M., and Lewis, C. D. (2003). Fractal properties of sympathetic nerve discharge. J. Neurophysiol. 89, 833–840. doi: 10.1152/jn.00757.2002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
D'Avino, P. P., Archambault, V., Przewloka, M. R., Zhang, W., Laue, E. D., and Glover, D. M. (2009). Isolation of protein complexes involved in mitosis and cytokinesis from Drosophila cultured cells. Methods Mol. Biol. 545, 99–112. doi: 10.1007/978-1-60327-993-2_6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De Forest, M., and Wheeler, C. J. (1999). Coherent oscillations in membrane potential synchronize impulse bursts in central olfactory neurons of the crayfish. J. Neurophysiol. 81, 1231–1241.
De la Fuente, I. M. (2010). Quantitative analysis of cellular metabolic dissipative, self-organized structures. Int. J. Mol. Sci. 11, 3540–3599. doi: 10.3390/ijms11093540
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M. (2014). “Metabolic Dissipative Structures,” in Systems Biology of Metabolic and Signaling Networks: Energy, Mass and Information Transfer, eds M. A. Aon, V. Saks, and U. Schlattner (New York, NY: Springer Books), 179–212.
De la Fuente, I. M., Benítez, N., Santamaría, A., Veguillas, J., and Aguirregabiria, J. M. (1999). Persistence in metabolic nets. Bull. Mathemat. Biol. 61, 573–595. doi: 10.1006/bulm.1999.0103
De la Fuente, I. M., and Cortés, J. M. (2012). Quantitative analysis of the effective functional structure in yeast glycolysis. PLoS ONE 7:e30162. doi: 10.1371/journal.pone.0030162
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Cortés, J., Pérez-Pinilla, M., Ruiz-Rodríguez, V., and Veguillas, J. (2011). The metabolic core and catalytic switches are fundamental elements in the self-regulation of the systemic metabolic structure of cells. PLoS ONE 6:e2722. doi: 10.1371/journal.pone.0027224
De la Fuente, I. M., Cortes, J. M., Pelta, D. A., and Veguillas, J. (2013). Attractor metabolic networks. PLoS ONE 8:e58284. doi: 10.1371/journal.pone.0058284
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Cortés, J. M., Valero, E., Desroches, M., Rodrigues, S., Malaina, I., et al. (2014). On the dynamics of the adenylate energy system: homeorhesis vs homeostasis. PLoS ONE 9:e108676. doi: 10.1371/journal.pone.0108676
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Martínez, L., Aguirregabiria, J. M., and Veguillas, J. (1998a). R/S analysis in strange attractors. Fractals 6, 95–100. doi: 10.1142/S0218348X98000110
De la Fuente, I. M., Martínez, L., Benitez, N., Veguillas, J., and Aguirregabiria, J. M. (1998b). Persistent behavior in a phase-shift sequence of periodical biochemical oscillations. Bull. Mathemat. Biol. 60, 689–702. doi: 10.1006/bulm.1997.0036
De la Fuente, I. M., Martínez, L., Pérez-Samartín, A. L., Ormaetxea, L., Amezaga, C., and Vera-López, A. (2008). Global self-organization of the cellular metabolic structure. PLoS ONE 3:e3100. doi: 10.1371/journal.pone.0003100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Martinez, L., Veguillas, J., and Aguirregabiria, J. M. (1996). Quasiperiodicity route to chaos in a biochemical system. Biophys J. 71, 2375–2379. doi: 10.1016/S0006-3495(96)79431-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Pérez-Samartín, A., Martínez, L., García, M. A., and Vera-López, A. (2006). Long-range correlations in rabbit brain neural activity. Ann. Biomed. Eng. 34, 295–299. doi: 10.1007/s10439-005-9026-z
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Vadillo, F., Pérez-Pinilla, M. B., Vera-López, A., and Veguillas, J. (2009). The number of catalytic elements is crucial for the emergence of metabolic cores. PLoS ONE 4:e7510. doi: 10.1371/journal.pone.0007510
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
De la Fuente, I. M., Vadillo, F., Pérez-Samartín, A. L., Pérez-Pinilla, M.-B., and Bidaurrazaga, J. (2010). Global self-regulations of the cellular metabolic structure. PLoS ONE 5, e9484:1–e9484:15. doi: 10.1371/journal.pone.0009484
Dekhuijzen, A., and Bagust, J. (1996). Analysis of neural bursting: nonrhythmic and rhythmic activity in isolated spinal cord. J. Neurosci. Methods 67, 141–147. doi: 10.1016/0165-0270(96)00033-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deribe, Y. L., Pawson, T., and Dikic, I. (2010). Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 17, 666–672. doi: 10.1038/nsmb.1842
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Deutscher, J., Francke, C., and Postma, P. W. (2006). How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70, 939–1031. doi: 10.1128/MMBR.00024-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Distler, A. M., Kerner, J., Lee, K., and Hoppel, C. L. (2009). Post-translational modifications of mitochondrial outer membrane proteins. Methods Enzymol. 457, 97–115. doi: 10.1016/S0076-6879(09)05006-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Doherty, M. K., and Beynon, R. J. (2006). Protein turnover on the scale of the proteome. Expert Rev. Proteomics. 3, 97–110. doi: 10.1586/14789450.3.1.97
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Doherty, M. K., Hammond, D. E., Clague, M. J., Gaskell, S. J., and Beynon, R. J. (2009). Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J. Proteome Res. 8, 104–112. doi: 10.1021/pr800641v
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dong, J., Jiang, G., Asmann, Y. W., Tomaszek, S., Jen, J., Kislinger, T., et al. (2010). MicroRNA networks in mouse lung organogenesis. PLoS ONE 5:e10854. doi: 10.1371/journal.pone.0010854
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dussutour, A., Latty, T., Beekman, M., and Simpson, S. J. (2010). Amoeboid organism solves complex nutritional challenges. Proc. Natl. Acad. Sci. U.S.A. 107, 4607–4611. doi: 10.1073/pnas.0912198107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Dwivedi, G., and Kemp, M. L. (2012). Systemic redox regulation of cellular information processing. Antioxidant. Redox Signal. 16, 374. doi: 10.1089/ars.2011.4034
Eggan, K., Baldwin, K., Tackett, M., Osborne, J., Gogos, J., Chess, A., et al. (2004). Mice cloned from olfactory sensory neurons. Nature 428, 44–49. doi: 10.1038/nature02375
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ehmsen, J., Poon, E., and Davies, K. (2002). The dystrophin-associated protein complex. J. Cell. Sci. 115(Pt 14), 2801–2803.
Emonet, T., and Cluzel, P. (2008). Relationship between cellular response and behavioral variability in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 105, 3304–3309. doi: 10.1073/pnas.0705463105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Faber, J., Portugal, R., and Rosa, L. P. (2006). Information processing in brain microtubules. Biosystems 83, 1–9. doi: 10.1016/j.biosystems.2005.06.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fan, C., Cheng, S., Liu, Y., Escobar, C. M., and Crowley, C. S. (2010). Short N-terminal sequences package proteins into bacterial microcompartments. Proc. Natl. Acad. Sci. U.S.A. 107, 7509–7514. doi: 10.1073/pnas.0913199107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Forlemu, N. Y., Njabon, E. N., Carlson, K. L., Schmidt, E. S., Waingeh, V. F., and Thomasson, K. A. (2011). Ionic strength dependence of F-actin and glycolytic enzyme associations: a Brownian dynamics simulations approach. Proteins 79, 2813–2827. doi: 10.1002/prot.23107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fromme, P., and Mathis, P. (2004). Unraveling the photosystem I reaction center: a history, or the sum of many efforts. Photosyn. Res. 80, 109–124. doi: 10.1023/B:PRES.0000030657.88242.e1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fuentes, J. M., Pascual, M. R., Salido, G., and Soler, J. A. (1994). Oscillations in rat liver cytosolic enzyme activities of the urea cycle. Arch. Int. Physiol. Biochim. Biophys. 102, 237–241. doi: 10.3109/13813459409003936
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fusi, S., Drew, P. J., and Abbott, L. F. (2005). Cascade models of synaptically stored memories. Neuron 45, 599–611. doi: 10.1016/j.neuron.2005.02.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Garmendia-Torres, C., Goldbeter, A., and Jacquet, M. (2007). Nucleocytoplasmic oscillations of the yeast transcription factor Msn2: evidence for periodic PKA activation. Curr. Biol. 17, 1044–1049. doi: 10.1016/j.cub.2007.05.032
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gasch, A. P., and Werner-Washburne, M. (2002). The genomics of yeast responses to environmental stress and starvation. Funct. Integr. Genomics 2, 181–192. doi: 10.1007/s10142-002-0058-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gautam, V. K., Costantini, S., Paladino, A., and Colonna, G. (2012). Interactomic and pharmacological insights on human Sirt-1. Front. Pharmacol. 3:40. doi: 10.3389/fphar.2012.00040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gavin, A. C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., et al. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147. doi: 10.1038/415141a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gavin, A. C., and Superti-Furga, G. (2003). Protein complexes and proteome organization from yeast to man. Curr. Opin. Chem. Biol. 7, 21–27. doi: 10.1016/S1367-5931(02)00007-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gennarino, V. A., D'Angelo, G., Dharmalingam, G., Fernandez, S., Russolillo, G., Sanges, R., et al. (2012). Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res. 22, 1163–1172. doi: 10.1101/gr.130435.111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gerhäuser, C. (2012). Cancer cell metabolism, epigenetics and the potential influence of dietary components - A perspective. Biomed. Res. 23, 1–21.
Geryk, J., and Slanina, F. (2013). Modules in the metabolic network of E. coli with regulatory interactions. Int. J. Data Min. Bioinform. 8, 188–202. doi: 10.1504/IJDMB.2013.055500
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Getty, L., Panteleon, A. E., Mittelman, S. D., Dea, M. K., and Bergman, R. N. (2000). Rapid oscillations in omental lipolysis are independent of changing insulin levels in vivo. J. Clin. Invest. 106, 421–430. doi: 10.1172/JCI7815
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Getty-Kaushik, L., Richard, A. M., and Corkey, B. E. (2005). Free fatty acid regulation of glucose-dependent intrinsic oscillatory lipolysis in perfused isolated rat adipocytes. Diabetes 54, 629–637. doi: 10.2337/diabetes.54.3.629
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gloire, G., and Piette, J. (2009). Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid. Redox. Signal. 11, 2209–2222. doi: 10.1089/ars.2009.2463
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gluckman, P. D., Hanson, M. A., and Beedle, A. S. (2007). Non-genomic transgenerational inheritance of disease risk. Bioessays 29, 145–154. doi: 10.1002/bies.20522
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gnad, F., Ren, S., Cox, J., Olsen, J. V., Macek, B., Oroshi, M., et al. (2007). PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 8:R250. doi: 10.1186/gb-2007-8-11-r250
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goldbeter, A. (2002). Computational approaches to cellular rhythms. Nature 420, 238–245. doi: 10.1038/nature01259
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Goldbeter, A. (2007). Biological rhythms as temporal dissipative structures. Adv. Chem. Phys. 135, 253–295. doi: 10.1002/9780470121917.ch8
Goldbeter, A. (2013). Oscillatory enzyme reactions and Michaelis-Menten kinetics. FEBS Lett. 587, 2778–2784. doi: 10.1016/j.febslet.2013.07.031
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gonzalez, D., Kozdon, J. B., McAdams, H. H., Shapiro, L., and Collier, J. (2014). The functions of DNA methylation by CcrM in Caulobacter crescentus: a global approach. Nucleic Acids Res. 42, 3720–3735. doi: 10.1093/nar/gkt1352
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gough, J., Jones, G., Lovell, C., Macey, P., Morgan, H., Revilla, F. D., et al. (2009). Integration of cellular biological structures into robotic systems. Acta Futura 3, 43–49.
Graciet, E., Lebreton, S., and Gontero, B. (2004). Emergence of new regulatory mechanisms in the Benson-Calvin pathway via protein-protein interactions: a glyceraldehyde-3-phosphate dehydrogenase/CP12/phosphoribulokinase complex. J. Exp. Bot. 55, 1245–1254. doi: 10.1093/jxb/erh107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Graham, J. W., Williams, T. C., Morgan, M., Fernie, A. R., Ratcliffe, R. G., and Sweetlove, L. J. (2007). Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. Plant Cell. 19, 3723–3738. doi: 10.1105/tpc.107.053371
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Greenfield, D., McEvoy, A. L., Shroff, H., Crooks, G. E., Wingreen, N. S., Betzig, E., et al. (2009). Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 7:e1000137. doi: 10.1371/journal.pbio.1000137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Grossniklaus, U. (2011). Plant germline development: a tale of cross-talk, signaling, and cellular interactions. Sex. Plant Reprod. 24, 91–95. doi: 10.1007/s00497-011-0170-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gurdon, J. B. (1971). Transplant of Nuclei and Cell Differentiation. Moscow: Znanie, Series 10 (Biology).
Gurdon, J. B., and Weir, R. S. (1969). Cytoplasmic proteins and the control of nuclear activity in early amphibian development. Biochem. J. 114, 52–53.
Halbach, F., Reichelt, P., Rode, M., and Conti, E. (2013). The yeast ski complex: crystal structure and RNA channeling to the exosome complex. Cell 154, 814–826. doi: 10.1016/j.cell.2013.07.017
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Halley, J. D., and Winkler, D. A. (2008). Consistent concepts of self-organization and self-assembly. Complexity 14, 10–17. doi: 10.1002/cplx.20235
Hamadeh, A., Roberts, M. A. J., August, E., McSharry, P. E., Maini, P. K., Armitage, J. P., et al. (2011). Feedback control architecture and the bacterial chemotaxis network. PLoS Comput. Biol. 7:e1001130. doi: 10.1371/journal.pcbi.1001130
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hanna, C. W., and Kelsey, G. (2014). The specification of imprints in mammals. Heredity (Edinb.) 113, 176–183. doi: 10.1038/hdy.2014.54
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hans, M. A., Heinzle, E., and Wittmann, C. (2003). Free intracellular amino acid pools during autonomous oscillations in Saccharomyces cerevisiae. Biotechnol. Bioeng. 82, 143–151. doi: 10.1002/bit.10553
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hao, D., Ren, C., and Li, C. (2012). Revisiting the variation of clustering coefficient of biological networks suggests new modular structure. BMC Syst. Biol. 6:34. doi: 10.1186/1752-0509-6-34
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Harrington, H. A., Ho, K. L., Ghosh, S., and Tung, K. C. (2008). Construction and analysis of a modular model of caspase activation in apoptosis. Theor. Biol. Med. Model. 5:26. doi: 10.1186/1742-4682-5-26
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hartig, K., and Beck, E. (2005). Endogenous cytokinin oscillations control cell cycle progression of tobacco BY-2 cells. Plant Biol. 7, 33–40. doi: 10.1055/s-2004-830474
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hartwell, L. H., Hopfield, J. J., Leibler, S., and Murray, A. W. (1999). From molecular to modular cell biology. Nature 402, C47–C52. doi: 10.1038/35011540
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hatakeyama, T. S., and Kaneko, K. (2014). Kinetic memory based on the enzyme-limited competition. PLoS Comput. Biol. 10:e1003784. doi: 10.1371/journal.pcbi.1003784
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hayashi, T., Rumbaugh, G., and Huganir, R. L. (2005). Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron 47, 709–723. doi: 10.1016/j.neuron.2005.06.035
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hazelbauer, G. L., Falke, J. J., and Parkinson, J. S. (2008). Bacterial chemoreceptors: high-performance signaling in networked arrays. Trends Biochem. Sci. 33, 9–19. doi: 10.1016/j.tibs.2007.09.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hazen, S. P., Wu, Y., and Kreps, J. A. (2003). Gene expression profiling of plant responses to abiotic stress. Funct. Integr. Genom. 3, 105–111. doi: 10.1007/s10142-003-0088-4
Heinrich, R., and Schuster, S. (1996). The Regulation of Cellular Systems. New York, NY: Chapman and Hall.
Heo, I., and Kim, V. N. (2009). Regulating the regulators: post-translational modifications of RNA silencing factors. Cell 139, 28–31. doi: 10.1016/j.cell.2009.09.013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hertz, J., Krogh, A., and Palmer, R. G. (1991). Introduction to the Theory of Neural Computation. Boston, MA: Addison-Wesley Longman Publishing Co.
Ho, Y., Gruhler, A., Heilbut, A., Bader, G. D., Moore, L., Adams, S. L., et al. (2002). Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415, 180–183. doi: 10.1038/415180a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Holdsworth, J. E., and Ratledge, C. (1988). Lipid turnover in oleaginous yeasts. J. Gen. Microbiol. 134, 339–346. doi: 10.1099/00221287-134-2-339
Holz, G. G., Heart, E., and Leech, C. A. (2008). Synchronizing Ca2+ and cAMP oscillations in pancreatic beta cells: a role for glucose metabolism and GLP-1 receptors? Focus on “Regulation of cAMP dynamics by Ca2+ and G protein-coupled receptors in the pancreatic β-cell: a computational approach.” Am. J. Physiol. Cell Physiol. 294, C4–C6. doi: 10.1152/ajpcell.00522.2007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hopfield, J. J. (1982). Neural networks and physical systems with emergent collective computational abilities. Proc. Nat. Acad. Sci. U.S.A. 79, 2554–2558. doi: 10.1073/pnas.79.8.2554
Hsu, J. T., Peng, C. H., Hsieh, W. P., Lan, C. Y., and Tang, C. Y. (2011). A novel method to identify cooperative functional modules: study of module coordination in the Saccharomyces cerevisiae cell cycle. BMC Bioinformatics 12:281. doi: 10.1186/1471-2105-12-281
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hungerbuehler, A. K., Philippsen, P., and Gladfelter, A. S. (2007). Limited functional redundancy and oscillation of cyclins in multinucleated Ashbya gossypii fungal cells. Eukaryot. Cell. 6, 473–486. doi: 10.1128/EC.00273-06
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Impens, F., Radoshevich, L., Cossart, P., and Ribet, D. (2014). Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. U.S.A. 111, 12432–12437. doi: 10.1073/pnas.1413825111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Inoue, M., and Kaneko, K. (2013). Cooperative adaptive responses in gene regulatory networks with many degrees of freedom. PLoS Comput. Biol. 9:e1003001. doi: 10.1371/journal.pcbi.1003001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishii, K., Hirose, K., and Iino, M. (2006). Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep. 7, 390–396. doi: 10.1038/sj.embor.7400620
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ishikawa, M., Tsuchiya, D., Oyama, T., Tsunaka, Y., and Morikawa, K. (2004). Structural basis for channelling mechanism of a fatty acid bold beta-oxidation multienzyme complex. EMBO J. 23, 2745–2754. doi: 10.1038/sj.emboj.7600298
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Islam, M. M., Wallin, R., Wynn, R. M., Conway, M., Fujii, H., Mobley, J. A., et al. (2007). A novel branched-chain amino acid metabolon. Protein-protein interactions in a supramolecular complex. J. Biol. Chem. 282, 11893–11903. doi: 10.1074/jbc.M700198200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ito, T., Chiba, T., Ozawa, R., Yoshida, M., Hattori, M., and Sakaki, Y. (2001). A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U.S.A. 98, 4569–4574. doi: 10.1073/pnas.061034498
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Itskov, V., Hansel, D., and Tsodyks, M. (2011). Short-term facilitation may stabilize parametric working memory trace. Front. Comp. Neurosci. 5:40. doi: 10.3389/fncom.2011.00040
Janke, C., and Bulinski, J. C. (2011). Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell. Biol. 12, 773–786. doi: 10.1038/nrm3227
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jennings, H. S. (1905). Behavior of Lower Organisms. Bloomington, IN: Indiana University Press, Reprint Edition.
Jia, H., Wang, X., Anderson, J. T., and Jankowsky, E. (2012). RNA unwinding by the Trf4/Air2/Mtr4 polyadenylation (TRAMP) complex. Proc. Natl. Acad. Sci. U.S.A. 109, 7292–7297. doi: 10.1073/pnas.1201085109
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jiminez-Chirallon, J. C., Isganaitis, E., Charalambous, M., Gesta, S., Pentinat-Pelegrin, T., Faucette, R. R., et al. (2009). Intergenerational transmission of glucose intolerance and obesity by in utero under-nutrition in mice. Diabetes 58, 460–468. doi: 10.2337/db08-0490
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jorgensen, K., Rasmussen, A. V., Morant, M., Nielsen, A. H., Bjarnholt, N., and Zagrobelny, M. (2005). Metabolon formation and metabolic channeling in the biosynthesis of plant natural products. Curr. Opin. Plant Biol. 8, 280–291. doi: 10.1016/j.pbi.2005.03.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jovanović, S., Jovanović, A., and Crawford, M. R. (2007). M-ldh serves as a regulatory subunit of the cytosolic substrate-channelling complex in vivo. J. Mol. Biol. 371, 349–361. doi: 10.1016/j.jmb.2007.05.081
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jules, M., Francois, J., and Parrou, J. L. (2005). Autonomous oscillations in Saccharomyces cerevisiae during batch cultures on trehalose. FEBS J. 272, 1490–1500. doi: 10.1111/j.1742-4658.2005.04588.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaati, G., Bygren, L. O., Pembrey, M., and Sjöström, M. (2007). Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. 15, 784–790. doi: 10.1038/sj.ejhg.5201832
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kaltenbach, H. M., and Stelling, J. (2012). Modular analysis of biological networks. Adv. Exp. Med. Biol. 736, 3–17. doi: 10.1007/978-1-4419-7210-1_1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kamimura, A., and Kobayashi, T. J. (2012). Information processing and integration with intracellular dynamics near critical point. Front. Physiol. 3:203. doi: 10.3389/fphys.2012.00203
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kanehisa, M. (2013). Chemical and genomic evolution of enzyme-catalyzed reaction networks. FEBS Lett. 587, 2731–2737. doi: 10.1016/j.febslet.2013.06.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kashtan, N., and Alon, U. (2005). Spontaneous evolution of modularity and network motifs. Proc. Natl. Acad. Sci. U.S.A. 102, 13773–13778. doi: 10.1073/pnas.0503610102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Katz, E., and Privman, V. (2010). Enzyme-based logic systems for information processing. Chem. Soc. Rev. 39, 1835–1857. doi: 10.1039/b806038j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kazachenko, V. N., Astashev, M. E., and Grinevic, A. A. (2007). Multifractal analysis of K+ channel activity. Biochemistry 2, 169–175. doi: 10.1134/S1990747807020109
Kessler, D. (1982). “Plasmodial structure and motility,” in Cell Biology of Physarum and Didymium, Organisms, Nucleus, and Cell Cycle, Vol. 1., eds H. C. Aldrich and J. W. Daniel (New York, NY: Academic Press), 145–208.
Khoury, G. A., Baliban, R. C., and Floudas, C. A. (2011). Proteome-wide post-translational modification statistics: frequency analysis and curation of the Swiss-prot database. Sci. Rep. 1:90. doi: 10.1038/srep00090
Kindzelskii, A. L., Zhou, M. J., Haugland, P. R., Boxer, A. L., and Petty, R. H. (1998). Oscillatory pericellular proteolysis and oxidant deposition during neutrophil locomotion. Biophys. J. 74, 90–97. doi: 10.1016/S0006-3495(98)77770-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klevecz, R. R., Bolen, J., Forrest, G., and Murray, D. B. (2004). A genomewide oscillation in transcription gates DNA replication and cell cycle. Proc. Natl. Acad. Sci. U.S.A. 101, 1200–1205. doi: 10.1073/pnas.0306490101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Klevecz, R. R., and Murray, D. B. (2001). Genome wide oscillations in expression –Wavelet analysis of time series data from yeast expression arrays uncovers the dynamic architecture of phenotype. Mol. Biol. Rep. 28, 73–82. doi: 10.1023/A:1017909012215
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Koc, E. C., and Koc, H. (2012). Regulation of mammalian mitochondrial translation by post-translational modifications. Biochim. Biophys. Acta 1819, 1055–1066. doi: 10.1016/j.bbagrm.2012.03.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kollmann, M., Løvdok, L., Bartholomé, K., Timmer, J., and Sourjik, V. (2005). Design principles of a bacterial signalling network. Nature 438, 504–507. doi: 10.1038/nature04228
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Korcsmáros, T., Kovács, I. A., Szalay, M. S., and Csermely, P. (2007). Molecular chaperones: the modular evolution of cellular networks. J. Biosci. 32, 441–446. doi: 10.1007/s12038-007-0043-y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kreps, J. A., Wu, Y., Hur-Song, C., Zhu, T., Wang, X., and Harper, J. F. (2002). Transcriptomic changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Phys. 130, 2129–2141. doi: 10.1104/pp.008532
Kuersten, S., Ohno, M., and Mattaj, I. W. (2001). Nucleocytoplasmic transport: ran, beta and beyond. Trends Cell Biol. 11, 497–503. doi: 10.1016/S0962-8924(01)02144-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lacroix, V., Fernandes, C. G., and Sagot, M. F. (2006). Motif search in graphs: application to metabolic networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 3, 360–368. doi: 10.1109/TCBB.2006.55
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lallemand, B., Erhardt, M., Heitz, T., and Legrand, M. (2013). Sporopollenin biosynthetic enzymes interact and constitute a metabolon localized to the endoplasmic reticulum of tapetum cells. Plant Physiol. 162, 616–625. doi: 10.1104/pp.112.213124
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lange, G., Mandelkow, E. M., Jagla, A., and Mandelklow, E. (1988). Tubulin oligomers and microtubule oscillations Antagonistic role of microtubule stabilizers and destabilizers. Eur. J. Biochem. 178, 61–69. doi: 10.1111/j.1432-1033.1988.tb14429.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Latty, T., and Beekman, M. (2011). Speed-accuracy trade-offs during foraging decisions in the acellular slime mould Physarum polycephalum. Proc. Biol. Sci. 278, 539–545. doi: 10.1098/rspb.2010.1624
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, C., Cao, L. G., Wang, Y. L., and Baril, E. F. (1993). Further purification and characterization of a multienzyme complex for DNA synthesis in human cells. J. Cell. Biochem. 53, 405–419. doi: 10.1002/jcb.240530418
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, L., Nørrelykke, S. F., and Cox, E. C. (2008). Persistent cell motion in the absence of external signals: a search strategy for eukaryotic cells. PLoS ONE 3:e2093. doi: 10.1371/journal.pone.0002093
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Q. (2010). Advances in protein turnover analysis at the global level and biological insights. Mass Spectrom. Rev. 29, 717–736. doi: 10.1002/mas.20261
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Li, Z., and Stock, J. B. (2009). Protein carboxyl methylation and the biochemistry of memory. Biol. Chem. 390, 1087–1096. doi: 10.1515/BC.2009.133
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lin, J., Lu, J., Feng, Y., Sun, M., and Ye, K. (2013). An RNA-binding complex involved in ribosome biogenesis contains a protein with homology to tRNA CCA-adding enzyme. PLoS Biol. 11:e1001669. doi: 10.1371/journal.pbio.1001669
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Liu, F., VanToai, T., Moy, L. P., Bock, G., Linford, L. D., and Quackenbush, J. (2005). Global transcription profiling reveals comprehensive insight into hypoxic response in Arabidopsis. Plant Phys. 137, 1115–1129. doi: 10.1104/pp.104.055475
Lloyd, D., Eshantha, L., Salgado, J., Turner, M. P., and Murray, D. B. (2002). Respiratory oscillations in yeast: clock-driven mitochondrial cycles of energization. FEBS Lett. 519, 41–44. doi: 10.1016/S0014-5793(02)02704-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lloyd, D., and Murray, D. B. (2005). Ultradian metronome: timekeeper for orchestration of cellular coherence. Trends Biochem. Sci. 30, 373–377. doi: 10.1016/j.tibs.2005.05.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lloyd, D., and Murray, D. B. (2006). The temporal architecture of eukaryotic growth. FEBS Lett. 580, 2830–2835. doi: 10.1016/j.febslet.2006.02.066
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ludington, W. B., Wemmer, K. A., Lechtreck, K. F., Witman, G. B., and Marshall, W. F. (2013). Avalanche-like behavior in ciliary import. Proc. Natl. Acad. Sci. U.S.A. 110, 3925–3930. doi: 10.1073/pnas.1217354110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lunn, E. J. (2007). Compartmentation in plant metabolism. J. Exp. Bot. 58, 35–47. doi: 10.1093/jxb/erl134
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, J., Ashikaga, E., Rubin, P. P., Heimann, M. J., Hildick, K. L., Bishop, P., et al. (2013). Receptor trafficking and the regulation of synaptic plasticity by SUMO. Neuromol. Med. 15, 692–706. doi: 10.1007/s12017-013-8253-y
MacDonald, M. J., Fahien, L. A., Buss, J. D., Hasan, N. M., Fallon, M. J., Kendrick, M. A., et al. (2003). Citrate oscillates in liver and pancreatic beta cell mitochondria and in INS-1 insulinoma cells. J. Biol. Chem. 278, 51894–51900. doi: 10.1074/jbc.M309038200
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Makino, D. L., Baumgärtner, M., and Conti, E. (2013). Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature 495, 70–75. doi: 10.1038/nature11870
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Maquat, L. E., and Gong, C. (2009). Gene expression networks: competing mRNA decay pathways in mammalian cells. Biochem. Soc. Trans. 37(Pt 6), 1287–1292. doi: 10.1042/BST0371287
Marquez, S., Crespo, P., Carlini, V., Garbarino-Pico, E., Baler, R., Caputto, B. L., et al. (2004). The metabolism of phospholipids oscillates rhythmically in cultures of fibroblasts and is regulated by the clock protein PERIOD 1. FASEB J. 18, 519–521. doi: 10.1096/fj.03-0417fje
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Marwan, W. (2010). Systems biology. Amoeba-inspired network design. Science 327, 419–420. doi: 10.1126/science.1185570
Masoudi-Nejad, A., Schreiber, F., and Kashani, Z. R. (2012). Building blocks of biological networks: a review on major network motif discovery algorithms. IET Syst. Biol. 6, 164–174. doi: 10.1049/iet-syb.2011.0011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merriam, R. W. (1969). Movement of cytoplasmic proteins into nuclei induced to enlarge and initiate DNA or RNA synthesis. J. Cell Sci. 5, 333–349.
Mijaljica, D., Prescott, M., and Devenish, R. J. (2006). Endoplasmic reticulum and Golgi complex: contributions to, and turnover by, autophagy. Traffic 7, 1590–1595. doi: 10.1111/j.1600-0854.2006.00495.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Milani, M., Pesce, A., Bolognesi, M., Bocedi, A., and Ascenzi, P. (2003). Substrate channeling: molecular bases. Biochem. Mol. Biol. Educ. 31, 228–233. doi: 10.1002/bmb.2003.494031040239
Milo, R., Shen-Orr, S., Itzkovitz, S., Kashtan, N., Chklovskii, D., and Alon, U. (2002). Network motifs: simple building blocks of complex networks. Science 298, 824–827. doi: 10.1126/science.298.5594.824
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mirouze, M., and Paszkowski, J. (2011). Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 14, 267–274. doi: 10.1016/j.pbi.2011.03.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Missiuro, P. V., Liu, K., Zou, L., Ross, B. C., Zhao, G., Liu, J. S., et al. (2009). Information flow analysis of interactome networks. PLoS Comput. Biol. 5:e1000350. doi: 10.1371/journal.pcbi.1000350
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Misteli, T. (2009). Self-organization in the genome. Proc. Natl. Acad. Sci. U.S.A. 106, 6885–6886. doi: 10.1073/pnas.0902010106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mitchell, C. G. (1996). Identification of a multienzyme complex of the tricarboxylic acid cycle enzymes containing citrate synthase isoenzymes from Pseudomonas aeruginosa. Biochem. J. 313(Pt 3), 769–774.
Miyaji, T., and Ohnishi, I. (2008). Physarum can solve the shortest path problem on riemannian surface mathematically rigourously. Int. J. Pure App. Math. 47, 353–369.
Molinier, J., Ries, G., Zipfel, C., and Hohn, B. (2006). Transgeneration memory of stress in plants. Nature 442, 1046–1049. doi: 10.1038/nature05022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Møller, A. C., Hauser, M. J. B., and Olsen, L. F. (1998). Oscillations in peroxidase-catalyzed reactions and their potential function in vivo. Biophys. Chem. 72, 63–72. doi: 10.1016/S0301-4622(98)00123-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Monge, C., Beraud, N., Kuznetsov, A. V., Rostovtseva, T., and Sackett, D. (2008). Regulation of respiration in brain mitochondria and synaptosomes: restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol. Cell. Biochem. 318, 147–165. doi: 10.1007/s11010-008-9865-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Monge, C., Grichine, A., Rostovtseva, T., Sackett, P., and Saks, V. A. (2009). Compartmentation of ATP in cardiomyocytes and mitochondria. Kinetic studies and direct measurements. Biophys. J. 96, 241a. doi: 10.1016/j.bpj.2008.12.1188
Mongillo, G., Barak, O., and Tsodyks, M. (2008). Synaptic theory of working memory. Science 319, 1543–1546. doi: 10.1126/science.1150769
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mooney, R. A., Artsimovitch, I., and Landick, R. (1998). Information processing by RNA polymerase: recognition of regulatory signals during RNA chain elongation. J. Bacteriol. 180, 3265–3275.
Murata, S., Yashiroda, H., and Tanaka, K. (2009). Molecular mechanisms of proteasome assembly. Nat. Rev. Mol. Cell. Biol. 10, 104–115. doi: 10.1038/nrm2630
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murray, D., Beckmann, M., and Kitano, H. (2007). Regulation of yeast oscillatory dynamics. Proc. Natl. Acad. Sci. U.S.A. 104, 2241–2246. doi: 10.1073/pnas.0606677104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murthy, V., and Pasupathy, K. (1994-1995). Isolation and characterization of a multienzyme complex containing DNA replicative enzymes from mitochondria of S. cerevisiae. Multienzyme complex from yeast mitochondria. Mol. Biol. Rep. 20, 135–141. doi: 10.1007/BF00990545
Muto, A., Kotera, M., Tokimatsu, T., Nakagawa, Z., Goto, S., and Kanehisa, M. (2013). Modular architecture of metabolic pathways revealed by conserved sequences of reactions. J. Chem. Inf. Model. 53, 613–622. doi: 10.1021/ci3005379
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nadeau, J. H. (2009). Transgenerational genetic effects on phenotypic variation and disease risk. Hum. Mol. Genet. 18, R202–R210. doi: 10.1093/hmg/ddp366
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagaki, T. (2001). Smart behavior of true slime mold in a labyrinth. Res. Microbiol. 152, 767–770. doi: 10.1016/S0923-2508(01)01259-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagaki, T., and Guy, R. D. (2008). Intelligent behaviors of amoeboid movement based on complex dynamics of soft matter. Soft Matter. 4, 57–67. doi: 10.1039/B706317M
Nakagaki, T., Kobayashi, R., Ueda, T., and Nishiura, Y. (2004a). Obtaining multiple separate food sources: behavioural intelligence in the Physarum plasmodium. Proc. Biol. Sci. 271, 2305–2310. doi: 10.1098/rspb.2004.2856
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagaki, T., Yamada, H., and Toth, A. (2000). Maze-solving by an amoeboid organism. Nature 407, 470–470. doi: 10.1038/35035159
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagaki, T., Yamada, H., and Hara, M. (2004b). Smart network solutions in an amoeboid organism. Biophys. Chem. 107, 1–5. doi: 10.1016/S0301-4622(03)00189-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakagaki, T., Yamada, H., and Toth, A. (2001). Path-finding by tube morphogenesis in an amoeboid organism. Biophys. Chem. 92, 47–52. doi: 10.1016/S0301-4622(01)00179-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakamura, N., Wei, J. H., and Seemann, J. (2012). Modular organization of the mammalian Golgi apparatus. Curr. Opin. Cell. Biol. 24, 467–474. doi: 10.1016/j.ceb.2012.05.009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neher, S. B., Sauer, R. T., and Baker, T. A. (2003). Distinct peptide signals in the UmuD and UmuD' subunits of UmuD/D' mediate tethering and substrate processing by the ClpXP protease. Proc. Natl. Acad. Sci. U.S.A. 100, 13219–13224. doi: 10.1073/pnas.2235804100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Neph, S., Stergachis, A. B., Reynolds, A., Sandstrom, R., Borenstein, E., and Stamatoyannopoulos, J. A. (2012). Circuitry and dynamics of human transcription factor regulatory networks. Cell 150, 1274–1286. doi: 10.1016/j.cell.2012.04.040
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nicolis, G., and Prigogine, I. (1977). Self-Organization in Nonequilibrium Systems. From Dissipative Structures to Order Through Fluctuations. New York, NY: Wiley.
Noble, D., Jablonka, E., Joyner, M. J., Müller, G. B., and Omholt, S. W. (2014). Evolution evolves: physiology returns to centre stage. J. Physiol. 592(Pt 11), 2237–2244. doi: 10.1113/jphysiol.2014.273151
Novak, B., Tyson, J. J., Gyorffy, B., and Csikasz-Nagy, A. (2007). Irreversible cell-cycle transitions are due to systems-level feedback. Nat. Cell Biol. 9, 724–728. doi: 10.1038/ncb0707-724
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nurse, P. (2008). Life, logic and information. Nature 454, 424–426. doi: 10.1038/454424a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Oliva, A., Rosebrock, A., Ferrezuelo, F., Pyne, S., Chen, H., Skiena, S., et al. (2005). The cell cycle-regulated genes of Schizosaccharomyces pombe. PLoS Biol. 3:e225. doi: 10.1371/journal.pbio.0030225
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ovadi, J., and Srere, P. A. (2000). Macromolecular compartmentation and channeling. Int. Rev. Cytol. 192, 255–280. doi: 10.1016/S0074-7696(08)60529-X
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ozalp, V. C., Pedersen, T. R., Nielsen, L. J., and Olsen, L. F. (2010). Time-resolved measurements of intracellular ATP in the yeast Saccharomyces cerevisiae using a new type of nanobiosensor. J. Biol. Chem. 285, 37579–37588. doi: 10.1074/jbc.M110.155119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pang, C. N., Krycer, J. R., Lek, A., and Wilkins, M. R. (2008). Are protein complexes made of cores, modules and attachments? Proteomics 8, 425–434. doi: 10.1002/pmic.200700801
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Papp, B., Pál, C., and Hurst, L. D. (2004). Metabolic network analysis of the causes and evolution of enzyme dispensability in yeast. Nature 429, 661–664. doi: 10.1038/nature02636
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parker, A., and Engel, P. C. (2000). Preliminary evidence for the existence of specific functional assemblies between enzymes of the beta-oxidation pathway and the respiratory chain. Biochem. J. 345(Pt 3), 429–435. doi: 10.1042/0264-6021:3450429
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petrovic, A., Mosalaganti, S., Keller, J., Mattiuzzo, M., Overlack, K., and Krenn, V. (2014). Modular assembly of RWD domains on the Mis12 complex underlies outer kinetochore organization. Mol. Cell. 53, 591–605. doi: 10.1016/j.molcel.2014.01.019
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pecinka, A., and Mittelsten Scheid, O. (2012). Stress-induced chromatin changes: a critical view on their heritability. Plant Cell Physiol. 53, 801–808. doi: 10.1093/pcp/pcs044
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pembrey, M. E., Bygren, L. O., Kaati, G., Edvinsson, S., Northstone, K., Sjöström, M., et al. (2006). Sex-specific, male-line transgenerational responses in humans. Eur. J. Hum. Genet. 14, 159–166. doi: 10.1038/sj.ejhg.5201538
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pershin, Y. V., Fontaine, S., and Di Ventra, M. (2009). Memristive model of amoeba learning. Phy. Rev. E. Stat. Nonlin. Soft Matter Phys. 80:021926. doi: 10.1103/PhysRevE.80.021926
Pestov, D. G., and Shcherbik, N. (2012). Rapid cytoplasmic turnover of yeast ribosomes in response to rapamycin inhibition of TOR. Mol. Cell. Biol. 32, 2135–2144. doi: 10.1128/MCB.06763-11
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Petty, H. R., and Kindzelskii, A. L. (2001). Dissipative metabolic patterns respond during neutrophil transmembrane signaling. Proc. Natl. Acad. Sci. U.S.A. 98, 3145–3149. doi: 10.1073/pnas.061014298
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pflieger, D., Bigeard, J., and Hirt, H. (2011). Isolation and characterization of plant protein complexes by mass spectrometry. Proteomics 11, 1824–1833. doi: 10.1002/pmic.201000635
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Placantonakis, D. G., and Welsh, J. P. (2001). Two distinct oscillatory states determined by the NMDA receptor in rat inferior olive. J. Physiol. 534(Pt 1), 123–140. doi: 10.1111/j.1469-7793.2001.t01-1-00123.x
Postma, M., Bosgraaf, L., Loovers, H. M., and Van Haastert, P. J. (2004). Chemotaxis: signalling modules join hands at front and tail. EMBO Rep. 5, 35–40. doi: 10.1038/sj.embor.7400051
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Purvis, J. E., and Lahav, G. (2013). Encoding and decoding cellular information through signaling dynamics. Cell 152, 945–956. doi: 10.1016/j.cell.2013.02.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Qi, H., Blanchard, A., and Lu, T. (2013). Engineered genetic information processing circuits. Wiley Interdiscip. Rev. Syst. Biol. Med. 5, 273–287. doi: 10.1002/wsbm.1216
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramakrishnan, N., and Bhalla, U. S. (2008). Memory switches in chemical reaction space. PLoS Comput. Biol. 4:e1000122. doi: 10.1371/journal.pcbi.1000122
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramanujan, V. K., Biener, G., and Herman, B. (2006). Scaling behavior in mitochondrial redox fluctuations. Biophys. J. 90, L70–L72. doi: 10.1529/biophysj.106.083501
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ramisetty, S. R., and Washburn, M. P. (2011). Unraveling the dynamics of protein interactions with quantitative mass spectrometry. Crit. Rev. Biochem. Mol. Biol. 46, 216–228. doi: 10.3109/10409238.2011.567244
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ravasz, E. (2009). Detecting hierarchical modularity in biological networks. Methods Mol. Biol. 541, 145–160. doi: 10.1007/978-1-59745-243-4_7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ravasz, E., Somera, A. L., Mongru, D. A., Oltvai, Z. N., and Barabási, A. L. (2002). Hierarchical organization of modularity in metabolic networks. Science 297, 1551–1555. doi: 10.1126/science.1073374
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rengan, R., and Omann, G. M. (1999). Regulation of oscillations in filamentous actin content in polymorphonuclear leukocytes stimulated with leukotriene B4 and platelet-activating factor. Biochem. Biophys. Res. Commun. 262, 479–486. doi: 10.1006/bbrc.1999.1222
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rimsky, S., and Travers, A. (2011). Pervasive regulation of nucleoid structure and function by nucleoid-associated proteins. Curr. Opin. Microbiol. 14, 136–141. doi: 10.1016/j.mib.2011.01.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rohila, J. S., Chen, M., Chen, S., Chen, J., Cerny, R. L., Dardick, C., et al. (2009). Protein-protein interactions of tandem affinity purified protein kinases from rice. PLoS ONE 4:e6685. doi: 10.1371/journal.pone.0006685
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Romani, S., Pinkoviezky, I., Rubin, A., and Tsodyks, M. (2013). Scaling laws of associative memory retrieval. Neural Comput. 25, 2523–2544. doi: 10.1162/NECO_a_00499
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Roper, S. D. (2007). Signal transduction and information processing in mammalian taste buds. Pflugers Arch. 454, 759–776. doi: 10.1007/s00424-007-0247-x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rosenspire, A. J., Kindzelskii, A. L., and Petty, H. (2001). Pulsed DC electric fields couple to natural NAD(P)H oscillations in HT-1080 fibrosarcoma cells. J. Cell. Sci. 114(Pt 8), 1515–1520.
Roussel, M. R., Ivlev, A. A., and Igamberdiev, A. U. (2006). Oscillations of the internal CO2 concentration in tobacco leaves transferred to low CO2. J. Plant Physiol. 34, 1188–1196. doi: 10.1016/j.jplph.2006.08.004
Routtenberg, A. (2008). The substrate for long-lasting memory: if not protein synthesis, then what?. Neurobiol. Learn. Mem. 89, 225–233. doi: 10.1016/j.nlm.2007.10.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Routtenberg, A., and Rekart, J. L. (2005). Post-translational protein modification as the substrate for long-lasting memory. Trends Neurosci. 28, 12–19. doi: 10.1016/j.tins.2004.11.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Running, M. P. (2014). The role of lipid post-translational modification in plant developmental processes. Front. Plant Sci. 5:50. doi: 10.3389/fpls.2014.00050
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saigusa, T., Tero, A., Nakagaki, T., and Kuramoto, Y. (2008). Amoebae anticipate periodic events. Phys. Rev. Lett. 100:018101. doi: 10.1103/PhysRevLett.100.018101
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saks, V., Dzeja, P., Schlattner, U., Vendelin, M., Terzic, A., and Wallimann, T. (2006). Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J. Physiol. 571(Pt 2), 253–273. doi: 10.1113/jphysiol.2005.101444
Saks, V., Monge, C., and Guzun, R. (2009). Philosophical basis and some historical aspects of systems biology: from Hegel to Noble - applications for bioenergetic research. Int. J. Mol. Sci. 10, 1161–1192. doi: 10.3390/ijms10031161
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
San Román, M., Cancela, H., and Acerenza, L. (2014). Source and regulation of flux variability in Escherichia coli. BMC Syst. Biol. 8:67. doi: 10.1186/1752-0509-8-67
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sánchez-Armáss, S., Sennoune, S. R., Maiti, D., Ortega, F., and Martínez-Zaguila, R. (2006). Spectral imaging microscopy demonstrates cytoplasmic pH oscillations in glial cells. Am. J. Physiol. Cell Physiol. 290, C524–C538. doi: 10.1152/ajpcell.00290.2005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sasidharan, K., Tomita, M., Aon, M., Lloyd, D., and Murray, D. B. (2012). Time-structure of the yeast metabolism in vivo. Adv. Exp. Med. Biol. 736, 359–379. doi: 10.1007/978-1-4419-7210-1_21
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schägger, H. (2002). Respiratory chain supercomplexes of mitochondria and bacteria. Biochim. Biophys. Acta 1555, 154–159. doi: 10.1016/S0005-2728(02)00271-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schibler, U., and Sassone-Corsi, P. (2002). A web of circadian pacemakers. Cell 111, 919–922. doi: 10.1016/S0092-8674(02)01225-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schott, J., and Stoecklin, G. (2010). Networks controlling mRNA decay in the immune system. Wiley Interdiscip. Rev. RNA 1, 432–456. doi: 10.1002/wrna.13
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schreiber, T. (2000). Measuring information transfer. Phys. Rev. Lett. 85, 461–464. doi: 10.1103/PhysRevLett.85.461
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schubert, V. (2014). RNA polymerase II forms transcription networks in rye and Arabidopsis nuclei and its amount increases with endopolyploidy. Cytogenet. Genome Res. 143, 69–77. doi: 10.1159/000365233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schulz, M. H., Pandit, K. V., Lino Cardenas, C. L., Ambalavanan, N., Kaminski, N., and Bar-Joseph, Z. (2013). Reconstructing dynamic microRNA-regulated interaction networks. Proc. Natl. Acad. Sci. U.S.A. 110, 15686–15691. doi: 10.1073/pnas.1303236110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwarz, L. A., Hall, B. J., and Patrick, G. N. (2010). Activity-dependent ubiquitination of GluA1 mediates a distinct AMPA receptor endocytosis and sorting pathway. J. Neurosci. 30, 16718–16729. doi: 10.1523/JNEUROSCI.3686-10.2010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwechheimer, C. (2004). The COP9 signalosome (CSN): an evolutionary conserved proteolysis regulator in eukaryotic development. Biochim. Biophys. Acta 1695, 45–54. doi: 10.1016/j.bbamcr.2004.09.023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Segura, M. P., Lilley, K. S., and Dupree, P. (2010). Proteomic complex detection using sedimentation (ProCoDeS): screening for proteins in stable complexes and their candidate interaction partners. Biochem. Soc. Trans. 38, 923–927. doi: 10.1042/BST0380923
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seisenberger, S., Andrews, S., Krueger, F., Arand, J., Walter, J., Santos, F., et al. (2012). The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 48, 849–862. doi: 10.1016/j.molcel.2012.11.001
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Seong, K. H., Li, D., Shimizu, H., Nakamura, R., and Ishii, S. (2011). Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145, 1049–1061. doi: 10.1016/j.cell.2011.05.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shankaran, H., Ippolito, D. L., Chrisler, W. B., Resat, H., Bollinger, N., Opresko, L. K., et al. (2009). Rapid and sustained nuclear–cytoplasmic ERK oscillations induced by epidermal growth factor. Mol. Syst. Biol. 5, 332. doi: 10.1038/msb.2009.90
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sharma, A., Costantini, S., and Colonna, G. (2013). The protein-protein interaction network of the human Sirtuin family. Biochim. Biophys. Acta. 1834, 1998–2009. doi: 10.1016/j.bbapap.2013.06.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shaul, O., Mironov, V., Burssens, S., Van Montagu, M., and Inze, D. (1996). Two Arabidopsis cyclin promoters mediate distinctive transcriptional oscillation in synchronized tobacco BY-2 cells. Proc. Natl. Acad. Sci. U.S.A. 93, 4868–4872. doi: 10.1073/pnas.93.10.4868
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shimizu, T. S., Tu, Y., and Berg, H. C. (2010). A modular gradient-sensing network for chemotaxis in Escherichia coli revealed by responses to time-varying stimuli. Mol. Syst. Biol. 6, 382. doi: 10.1038/msb.2010.37
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shintani, T., and Klionsky, D. J. (2004). Autophagy in health and disease: a double-edged sword. Science 306, 990–995. doi: 10.1126/science.1099993
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Shirakawa, T., and Gunji, Y. P. (2007). Emergence of morphological order in the network formation of Physarum polycephalum. Biophys. Chem. 128, 253–260. doi: 10.1016/j.bpc.2007.04.010
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sidiropoulou, K., Lu, F. M., Fowler, M. A., Xiao, R., Phillips, C., Ozkan, E. D., et al. (2009). Dopamine modulates an mGluR5-mediated depolarization underlying prefrontal persistent activity. Nat. Neurosci. 12, 190–199. doi: 10.1038/nn.2245
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sims, R. J., and Reinberg, D. (2008). Is there a code embedded in proteins that is based on post-translational modification?. Nat. Rev. Mol. Cell. Biol. 9, 815–820. doi: 10.1038/nrm2502
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Smrcinová, M., Sørensen, P. G., Krempasky, J., and Ballo, P. (1998). Chaotic oscillations in a chloroplast system under constant illumination. Inter. J. Bifurcation Chaos. 8, 2467–2470. doi: 10.1142/S0218127498001984
Sourjik, V., and Berg, H. (2002). Receptor sensitivity in bacterial chemotaxis. Proc. Natl. Acad. Sci. U.S.A. 99, 123–127. doi: 10.1073/pnas.011589998
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sourjik, V., and Berg, H. (2004). Functional interactions between receptors in bacterial chemotaxis. Nature 428, 437–441. doi: 10.1038/nature02406
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Srere, P. A. (1972). “Is there an organization of Krebs cycle enzymes in the mitochondrial matrix?,” in Energy Metabolism and the Regulation of Metabolic Processes in Mitochondria, eds R. W. Hanson and W. A. Mehlman (New York, NY: Academic Press), 79–91.
Srere, P. A. (1985). The metabolon. Trends. Biochem. Sci. 10, 109–110. doi: 10.1016/0968-0004(85)90266-X
Stern, S., Dror, T., Stolovicki, E., Brenner, N., and Braun, E. (2007). Genome-wide transcriptional plasticity underlies cellular adaptation to novel challenge. Mol. Syst. Biol. 3, 106. doi: 10.1038/msb4100147
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stock, J. B., Levit, M. N., and Wolanin, P. M. (2002). Information processing in bacterial chemotaxis. Sci. STKE. 2002:pe25. doi: 10.1126/stke.2002.132.pe25
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stock, J. B., and Zhang, S. (2013). The biochemistry of memory. Curr. Biol. 23, R741–R745. doi: 10.1016/j.cub.2013.08.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Storlien, L., Oakes, N. D., and Kelley, D. E. (2004). Metabolic flexibility. Proc. Nutr. Soc. 63, 363–368. doi: 10.1079/PNS2004349
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stuart, R. A. (2008). Supercomplex organization of the oxidative phosphorylation enzymes in yeast mitochondria. J. Bioenerg. Biomembr. 40, 411–417. doi: 10.1007/s10863-008-9168-4
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sunyer, B., Diao, W., and Lubec, G. (2008). The role of post-translational modifications for learning and memory formation. Electrophoresis 29, 2593–2602. doi: 10.1002/elps.200700791
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Suss, K. H., Arkona, C., Manteuffel, R., and Adler, K. (1993). Calvin cycle multienzyme complexes are bound to chloroplast thylakoid membranes of higher plants in situ. Proc. Natl. Acad. Sci. U.S.A. 90, 5514–5518. doi: 10.1073/pnas.90.12.5514
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Sutter, M., Boehringer, D., Gutmann, S., Günther, S., Prangishvili, D., Loessner, M. J., et al. (2008). Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 15, 939–947. doi: 10.1038/nsmb.1473
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Swindell, W. R., Huebner, M., and Weber, A. P. (2007). Plastic and adaptive gene expression patterns associated with temperature stress in Arabidopsis thaliana. Heredity (Edinb.) 99, 143–150. doi: 10.1038/sj.hdy.6800975
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takeda, T., Yun, C.-S., Shintani, M., Yamane, H., and Nojiri, H. (2011). Distribution of genes encoding nucleoid-associated protein homologs in plasmids. Int. J. Evol. Biol. 2011:685015. doi: 10.4061/2011/685015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takeuchi, T., Duszkiewicz, A. J., and Morris, R. G. (2014). The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369:20130288. doi: 10.1098/rstb.2013.0288
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tamura, K., and Hara-Nishimura, I. (2014). Functional insights of nucleocytoplasmic transport in plants. Front. Plant Sci. 5:118. doi: 10.3389/fpls.2014.00118
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tay, S., Hughey, J. J., Lee, T. K., Lipniacki, T., Quake, S. R., and Covert, M. W. (2010). Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 466, 267–271. doi: 10.1038/nature09145
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Taylor, E. W., Wang, K., Nelson, A. R., Bredemann, T. M., Fraser, K. B., Clinton, S. M., et al. (2014). O-GlcNAcylation of AMPA receptor GluA2 is associated with a novel form of long-term depression at hippocampal synapses. J. Neurosci. 34, 10–21. doi: 10.1523/JNEUROSCI.4761-12.2014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tero, A., Takagi, S., Saigusa, T., Ito, K., Bebber, D. P., Fricker, M. D., et al. (2010). Rules for biologically inspired adaptive network design. Science 327, 439–442. doi: 10.1126/science.1177894
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Thomson, M., and Gunawardena, J. (2009). Unlimited multistability in multisite phosphorylation systems. Nature 460, 274–277. doi: 10.1038/nature08102
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tian, B., Nowak, D. E., and Brasier, A. R. (2005). A TNF-induced gene expression program under oscillatory NF-κB control. BMC Genomics 6:137. doi: 10.1186/1471-2164-6-137
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tonozuka, H., Wang, J., Mitsui, K., Saito, T., and Hamada, Tsurugi, K. (2001). Analysis of the upstream regulatory region of the GTS1 gene required for its oscillatory expression. J. Biochem. 130, 589–595. doi: 10.1093/oxfordjournals.jbchem.a003023
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tsuda, S., Zauner, K. P., and Gunji, Y. P. (2007). Robot control with biological cells. Biosystems 87, 215–223. doi: 10.1016/j.biosystems.2006.09.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tu, B. P., Kudlicki, A., Rowicka, M., and McKnight, S. L. (2005). Logic of the yeast metabolic cycle: temporal compartmentalization of cellular processes. Science 310, 1152–1158. doi: 10.1126/science.1120499
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tu, Y. (2013). Quantitative modeling of bacterial chemotaxis: signal amplification and accurate adaptation. Annu. Rev. Biophys. 42, 337–359. doi: 10.1146/annurev-biophys-083012-130358
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tu, Y., Shimizu, T. S., and Berg, H. C. (2008). Modeling the chemotactic response of Escherichia coli to time-varying stimuli. Proc. Natl. Acad. Sci. U.S.A. 105, 14855–14860. doi: 10.1073/pnas.0807569105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tucholski, J., Pinner, A. L., Simmons, M. S., and Meador-Woodruff, J. H. (2014). Evolutionarily conserved pattern of AMPA receptor subunit glycosylation in mammalian frontal cortex. PLoS ONE 9:e94255. doi: 10.1371/journal.pone.0094255
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Turner, B. M. (2005). Reading signals on the nucleosome with a new nomenclature for modified histones. Nat. Struct. Mol. Biol. 12, 110–112. doi: 10.1038/nsmb0205-110
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Tyson, J. J. (1991). Modeling the cell division cycle: cdc2 and cyclin interactions. Proc. Natl. Acad. Sci. U.S.A. 88, 7328–7332. doi: 10.1073/pnas.88.16.7328
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ueda, T., Matsumoto, K., Akitaya, T., and Kobatake, Y. (1986). Spatial and temporal organization of intracellular adenine nucleotides and cyclic nucleotides in relation to rhythmic motility in Physarum plasmodium. Exp. Cell. Res. 162, 486–494. doi: 10.1016/0014-4827(86)90352-6
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Uetz, P., Giot, L., Cagney, G., Mansfield, T. A., Judson, R. S., Knight, J. R., et al. (2000). A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627. doi: 10.1038/35001009
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Van Leene, J., Stals, H., Eeckhout, D., Persiau, G., Van De Slijke, E., Van Isterdael, G., et al. (2007). A tandem affinity purification-based technology platform to study the cell cycle interactome in Arabidopsis thaliana. Mol. Cell. Proteom. 6, 1226–1238. doi: 10.1074/mcp.M700078-MCP200
Varanda, W. A., Liebovitch, L. S., Figueiroa, J. N., and Nogueira, R. A. (2000). Hurst analysis applied to the study of single calcium-activated potassium channel kinetics. J. Theor. Biol. 206, 343–353. doi: 10.1006/jtbi.2000.2131
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Veliky, M. M., Starikovich, L. S., Klimishin, N. I., and Chayka Ya, P. (2007). Molecular Mechanisms in the Integration of Metabolism. Lviv: Lviv National University).
Verkman, A. S. (2002). Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem. Sci. 27, 27–33. doi: 10.1016/S0968-0004(01)02003-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vlasova-St Louis, I., and Bohjanen, P. R. (2011). Coordinate regulation of mRNA decay networks by GU-rich elements and CELF1. Curr. Opin. Genet. Dev. 21, 444–451. doi: 10.1016/j.gde.2011.03.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Völkel, P., Le Faou, P., and Angrand, P. O. (2010). Interaction proteomics: characterization of protein complexes using tandem affinity purification-mass spectrometry. Biochem. Soc. Trans. 38, 883–887. doi: 10.1042/BST0380883
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
von Dassow, G., Meir, E., Munro, E. M., and Ordell, G. M. (2000). The segment polarity network is a robust developmental module. Nature 406, 188–191. doi: 10.1038/35018085
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wadhams, G. H., and Armitage, J. P. (2004). Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell. Biol. 5, 1024–1037. doi: 10.1038/nrm1524
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Waingeh, V. F., Gustafson, C. D., Kozliak, E. I., Lowe, S. L., Knull, H. R., and Thomasson, K. A. (2006). Glycolytic enzyme interactions with yeast and skeletal muscle F-actin. Biophys. J. 90, 1371–1384. doi: 10.1529/biophysj.105.070052
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Walhout, A. J. (2006). Unraveling transcription regulatory networks by protein-DNA and protein-protein interaction mapping. Genome Res. 16, 1445–1454. doi: 10.1101/gr.5321506
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, J. Q., Arora, A., Yang, L., Parelkar, N. K., Zhang, G., Liu, X., et al. (2005). Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol. Neurobiol. 32, 237–249. doi: 10.1385/MN:32:3:237
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wang, Z., and Zhang, J. (2009). Abundant indispensable redundancies in cellular metabolic networks. Genome Biol. Evol. 1, 23–33. doi: 10.1093/gbe/evp002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Waterland, R. A., Travisano, M., and Tahiliani, K. G. (2007). Diet-induced hypermethylation at agouti viable yellow is not inherited transgenerationally through the female. FASEB J. 21, 3380–3385. doi: 10.1096/fj.07-8229com
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wawrzkiewicz, A., Pawelek, K., Borys, P., Dworakowska, B., and Grzywna, Z. J. (2012). On the simple random-walk models of ion-channel gate dynamics reflecting long-term memory. Eur. Biophys. J. 41, 505–526. doi: 10.1007/s00249-012-0806-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Weber, J., Kayser, A., and Rinas, U. (2005). Metabolic flux analysis of Escherichia coli in glucose-limited continuous culture. II. Dynamic response to famine and feast, activation of the methylglyoxal pathway and oscillatory behaviour. Microbiology 151(Pt 3), 707–716. doi: 10.1099/mic.0.27482-0
Will, C. L., and Lührmann, R. (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3:a003707. doi: 10.1101/cshperspect.a003707
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Willshaw, D. J., Buneman, O. P., and Longuet-Higgins, H. C. (1969). Non-holographic associative memory. Nature 222, 960–962. doi: 10.1038/222960a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Willshaw, D. J., Longuet-Higgins, H. C., and Buneman, O. P. (1970). Models for the brain. Nature 225, 177–178. doi: 10.1038/225178a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilmut, I., Schnieke, A. E., McWhir, J., Kind, A. J., and Campbell, K. H. (1997). Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810–813. doi: 10.1038/385810a0
Wilson, D. N., and Nierhaus, K. H. (2007). The weird and wonderful world of bacterial ribosome regulation. Crit. Rev. Biochem. Mol. Biol. 42, 187–219. doi: 10.1080/10409230701360843
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wilson, W. A., Roach, P. J., Montero, M., Baroja-Fernández, E., Muñoz, F. J., and Eydallin, G. (2010). Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol. Rev. 34, 952–985. doi: 10.1111/j.1574-6976.2010.00220.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wimmer, C., Platzer, S., Hillen, W., and Klotzsche, M. (2013). A novel method to analyze nucleocytoplasmic transport in vivo by using short peptide tags. J. Mol. Biol. 425, 1839–1845. doi: 10.1016/j.jmb.2013.02.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wittmann, C., Hans, M., Van Winden, A. W., Ras, C., and Heijnen, J. J. (2005). Dynamics of intracellular metabolites of glycolysis and TCA cycle during cell-cycle-related oscillation in Saccharomyces cerevisiae. Biotechnol. Bioeng. 89, 839–847. doi: 10.1002/bit.20408
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Wuichet, K., Alexander, R. P., and Zhulin, I. B. (2007). Comparative genomic and protein sequence analyses of a complex system controlling bacterial chemotaxis. Methods Enzymol. 422, 1–31. doi: 10.1016/S0076-6879(06)22001-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yeates, T. O., Tsai, Y., Tanaka, S., Sawaya, M. R., and Kerfeld, C. A. (2007). Self-assembly in the carboxysome: a viral capsid-like protein shell in bacterial cells. Biochem. Soc. Trans. 35(Pt 3), 508–511. doi: 10.1042/BST0350508
Yeates, T. O., Kerfeld, C. A., Heinhorst, S., Cannon, G. C., and Shively, J. M. (2008). Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat. Rev. Microbiol. 6, 681–691. doi: 10.1038/nrmicro1913
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Yoon, J., Si, Y., Nolan, R., and Lee, K. (2007). Modular decomposition of metabolic reaction networks based on flux analysis and pathway projection. Bioinformatics 23, 2433–2440. doi: 10.1093/bioinformatics/btm374
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Ytting, C. K., Fuglsang, A. T., Hiltunen, J. K., Kastaniotis, A. J., Özalp, V. C., Nielsen, L. J., et al. (2012). Measurements of intracellular ATP provide new insight into the regulation of glycolysis in the yeast Saccharomyces cerevisiae. Integr. Biol. (Camb.) 4, 99–107. doi: 10.1039/C1IB00108F
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zetterberg, A. (1966). Protein migration between cytoplasm and cell nucleus during interphase in mouse fibroblasts in vitro. Expl. Cell Res. 43, 526–536. doi: 10.1016/0014-4827(66)90023-1
Zhang, S., Hulver, M. W., McMillan, R. P., Cline, M. A., and Gilbert, E. R. (2014). The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. (Lond.) 11:10. doi: 10.1186/1743-7075-11-10
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, H., French, J. B., Fang, Y., and Benkovic, S. J. (2013). The purinosome, a multi-protein complex involved in the de novo biosynthesis of purines in humans. Chem. Commun. (Camb.) 49, 4444–4452. doi: 10.1039/c3cc41437j
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhao, R., Bodnar, M. S., and Spector, D. L. (2009). Nuclear neighborhoods and gene expression. Curr. Opin. Genet. Dev. 19, 172–919. doi: 10.1016/j.gde.2009.02.007
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhaojun, X. U., So-ichi, Y., and Kunio, T. (2004). Gts1p stabilizes oscillations in energy metabolism by activating the transcription of TPS1 encoding trehalose-6-phosphate synthase 1 in the yeast Saccharomyces cerevisiae. Biochem. J. 383(Pt 1), 171–178. doi: 10.1039/c1ib00108f
Zheng, C. L., Sumizawa, T., Che, X. F., Tsuyama, S., Furukawa, T., and Haraguchi, M. (2005). Characterization of MVP and VPARP assembly into vault ribonucleoprotein complexes. Biochem. Biophys. Res. Commun. 326, 100–107. doi: 10.1016/j.bbrc.2004.11.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zhou, L., Cortassa, S., Wei, A. C., Aon, M. A., Winslow, R. L., and O'Rourke, B. (2009). Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys J. 97, 1843–1852. doi: 10.1016/j.bpj.2009.07.029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: systems biology, metabolic networks, systemic metabolism, Hopfield dynamics, dissipative processes, self-organization
Citation: De la Fuente IM (2015) Elements of the cellular metabolic structure. Front. Mol. Biosci. 2:16. doi: 10.3389/fmolb.2015.00016
Received: 29 December 2014; Accepted: 12 April 2015;
Published: 28 April 2015.
Edited by:
Elena Papaleo, University of Copenhagen, DenmarkReviewed by:
Miyako Kusano, University of Tsukuba/RIKEN Center for Sustainable Resource Science, JapanThomas Nägele, University of Vienna, Austria
Copyright © 2015 De la Fuente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ildefonso M. De la Fuente, Instituto de Parasitología y Biomedicina “López-Neyra,” Consejo Superior de Investigaciones Científicas, Parque Tecnológico de Ciencias de la Salud, Avda. del Conocimiento s/n, E-18016 Granada, Spain,bXRwbWFkZWlAZWh1LmVz