 Gabriel Thieulin-Pardo
Gabriel Thieulin-Pardo Luisana Avilan
Luisana Avilan Mila Kojadinovic
Mila Kojadinovic Brigitte Gontero
Brigitte Gontero- UMR 7281, Centre National de la Recherche Scientifique, Aix-Marseille Université, Marseille, France
Intrinsically Disordered Proteins (IDPs), or protein fragments also called Intrinsically Disordered Regions (IDRs), display high flexibility as the result of their amino acid composition. They can adopt multiple roles. In globular proteins, IDRs are usually found as loops and linkers between secondary structure elements. However, not all disordered fragments are loops: some proteins bear an intrinsically disordered extension at their C- or N-terminus, and this flexibility can affect the protein as a whole. In this review, we focus on the disordered N- and C-terminal extensions of globular proteins from photosynthetic organisms. Using the examples of the A2B2-GAPDH and the α Rubisco activase isoform, we show that intrinsically disordered extensions can help regulate their “host” protein in response to changes in light, thereby participating in photosynthesis regulation. As IDPs are famous for their large number of protein partners, we used the examples of the NAC, bZIP, TCP, and GRAS transcription factor families to illustrate the fact that intrinsically disordered extremities can allow a protein to have an increased number of partners, which directly affects its regulation. Finally, for proteins from the cryptochrome light receptor family, we describe how a new role for the photolyase proteins may emerge by the addition of an intrinsically disordered extension, while still allowing the protein to absorb blue light. This review has highlighted the diverse repercussions of the disordered extension on the regulation and function of their host protein and outlined possible future research avenues.
Introduction
Proteins occupy a central position in the architecture and functioning of living matter. A major objective of protein biochemistry consists in explaining the physiological functions of these molecules by means of structural studies, also known as the “structure-function” relationship. Among others, X-ray crystallography is a powerful tool to solve macromolecular three-dimensional 3D structures. However, some proteins cannot be crystallized because they are fully disordered or possess disordered parts that are missing in the electron density map of the crystals. In the 1990s, Sedzik and Kirschner (1992) attempted to crystallize the myelin basic protein (MBP), the predominant extrinsic protein in both central and nervous system myelins. After several attempts, the authors concluded that MBP adopts a random coil conformation and that as long as its flexibility was not suppressed, it was not possible to obtain crystals (Sedzik and Kirschner, 1992). MBP was one of the first examples of many other “un-crystallizable” proteins. These proteins, originally named Intrinsically Unstructured Proteins (IUPs), are nowadays termed Intrinsically Disordered Proteins (IDPs) (Wright and Dyson, 1999; Dunker et al., 2001, 2008a,b). In 1998, Romero et al. showed that 15 000 proteins from the Swiss-Prot database contain one or more Intrinsically disordered regions (IDRs) comprising more than 40 amino acid residues (Romero et al., 1998). It was shown later that despite their lack of well-defined 3D structure, many partially or completely disordered proteins are functional (Wright and Dyson, 1999; Dunker et al., 2001, 2008a,b; Tompa, 2002). In the late 1990s, studies of disordered yet functional proteins emerged as a new research field, extending the traditional paradigm to include a more comprehensive view of protein structure-function (Wright and Dyson, 1999; Dunker et al., 2001; Tompa, 2002; Dunker et al., 2008a,b). In the past, different models have been proposed to explain protein functioning, and protein flexibility has appeared as a key point (Fersht, 1998). Among these models, the “induced-fit” model (Koshland et al., 1966) introduced the idea that protein conformational changes could be triggered upon ligand binding. These notions were applied to IDPs, and many of them were shown to undergo an “induced-folding” upon binding to their partners (Dunker et al., 2002). Short motifs called MoREs (Molecular Recognition Elements) are often involved in the interaction, involving disorder to order transitions (Fuxreiter et al., 2004, 2007; Oldfield et al., 2005; Mohan et al., 2006; Vacic et al., 2007; Hazy and Tompa, 2009). However, the idea that preformed binding elements exist before the binding, and even in the absence of a partner, led to the “conformational selection” model. In some cases, the IDP is not fully structured in the presence of its partner and the term “fuzziness” was coined by Fuxreiter and Tompa to describe such complexes (Tompa and Fuxreiter, 2008; Hazy and Tompa, 2009; Fuxreiter and Tompa, 2012). The flexibility of IDPs increases the chance of their polypeptide chains adopting the right conformations in the presence of their partners. Furthermore, the high ratio of hydrophilic residues in IDPs facilitates initial contacts with their partners. The interactions are also stronger with IDPs: their lack of structure or absence of rigidity increasing association constants (Dunker et al., 2001; Meszaros et al., 2007; Chouard, 2011). The ability of the IDPs to adopt multiple conformations allows the same region to adapt to different binding sites in many “induced-fits” and thus to have multiple partners (Uversky et al., 2000; Tompa, 2002; Uversky, 2002, 2011; Meszaros et al., 2007; Carmo-Silva and Salvucci, 2013). The discovery of IDPs and their singular lack of definite structure brought nuances to the “structure-function” dogma, showing that the same structure, or lack of one, could have multiple partners and thus multiple functions (Wright and Dyson, 1999; Dunker et al., 2001; Tompa, 2002; Uversky, 2002, 2011; Meszaros et al., 2007; Sun et al., 2013). In this regard, IDPs could be seen as the “master keys” of the protein-protein interaction network.
The ability of IDPs to bind to multiple partners makes them naturally good regulators, as they can modulate the activity of several proteins in a coordinated way (Dunker et al., 2001, 2002, 2005; Gavin et al., 2002, 2006; Haynes et al., 2006; Patil and Nakamura, 2006; Mittag et al., 2010; Uversky, 2010; Pancsa and Tompa, 2012). Therefore, multiple areas of the cellular response can be affected by a single signal allowing IDPs to play a major role in regulatory pathways (Tompa, 2002; Dunker et al., 2005; Haynes et al., 2006; Uversky, 2010; Pancsa and Tompa, 2012). The flexibility of IDPs can also be modified by the cellular environment or by post-translational modifications. IDPs and IDRs are often targets of different post-translational modifications (the most common being phosphorylation, methylation and ubiquitination) which can radically affect their affinity for their partners and their stability, thus multiplying the possibilities for a fine-tuned regulation (Tompa, 2002; Dunker et al., 2005; Haynes et al., 2006; Uversky, 2010; Pancsa and Tompa, 2012). These particularities make IDPs the hubs in a vast net of protein-protein interactions (Gavin et al., 2002, 2006). They carry out basic functions such as regulation of metabolic pathways, transcription, translation or cellular signal transduction; they can act as scavengers of toxic molecules and they play a key role in the assembly of multi-protein complexes (Uversky, 2011). Moreover, their roles in several diseases of major medical interest, such as cancer (Castillo et al., 2014; Saha et al., 2014; Xue et al., 2014), Alzheimer's disease (Uversky, 2009; Kovacech and Novak, 2010; Salminen et al., 2011; Karagoz and Rudiger, 2015) prion disease (Tompa, 2009; Uversky, 2009; Breydo and Uversky, 2011) or Parkinson's disease (Uversky, 2009; Hazy et al., 2011; Breydo et al., 2012; Alderson and Markley, 2013) have been extensively studied (Babu et al., 2011).
While the discovery and characterization of IDPs and IDRs is a rapidly growing, and an increasingly recognized, area of protein science, (Tompa, 2002; Uversky, 2010; Uversky and Dunker, 2010; Chouard, 2011), little information is available photosynthetic organisms, where IDPs have been described as central players in many responses such as biotic and abiotic stress, development, metabolism regulation, or adaptation to oxic atmosphere (Kragelund et al., 2012; Pancsa and Tompa, 2012; Yruela and Contreras-Moreira, 2012, 2013; Pietrosemoli et al., 2013; Sun et al., 2013; Panda and Ghosh, 2014). Published data mainly concern Arabidopsis thaliana, a higher plant model with one of the best-annotated sequenced genomes (Arabidopsis Genome Initiative, 2000). Yet, the recent analysis of 12 plant genomes revealed that the occurrence of disorder in plants is similar to the one found in other eukaryotes (Bracken et al., 1999; Yruela and Contreras-Moreira, 2012, 2013; Sun et al., 2013). An in silico analysis of plant nuclear proteomes suggested a higher disorder in the internal part of nuclear-encoded plant proteins rather than at their extremities, in contrast to the chloroplast- and mitochondrion-encoded proteomes (Yruela and Contreras-Moreira, 2012). This is also pointed by studies on prokaryotes showing that the IDRs may be more frequent at the extremities of the proteins that act as “molecular shields” such as chaperones (Krisko et al., 2010; Chakrabortee et al., 2012).
In this review, we describe several globular proteins with N- or C-terminal IDR extensions in photsynthetic organisms, as opposed to entirely disordered proteins or globular proteins containing one or more IDRs in the middle of their sequences. The aim of this work is not to give an exhaustive list of the roles undertaken by such disordered extensions, as this has recently been reviewed (Uversky, 2013). Instead, we focus on globular proteins or domains that acquired their disordered tails during evolution, using examples from photosynthetic organisms. The addition of a disordered extension to a globular protein created new regulation opportunities, making these proteins responsive to environmental factors through self-regulation, post-translational modifications or new protein-protein interactions. We illustrate the impact of disordered extensions by describing proteins involved in photosynthetic metabolism and regulation of gene expression (Table 1).
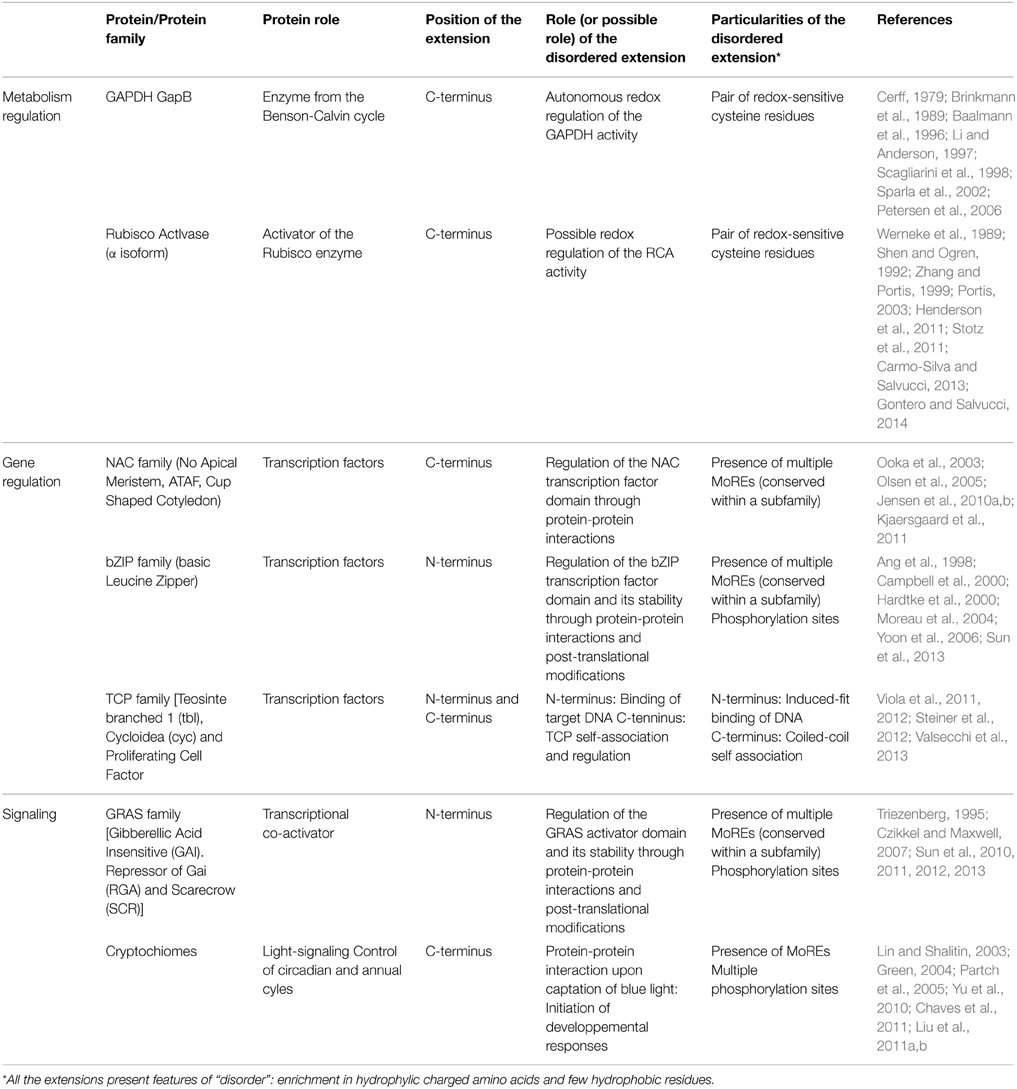
Table 1. Summary of the characteristics of the Intrinsically disordered extensions presented in this review.
GAPDH and CP12
As knowledge about proteins progressed, new results appeared showing that some enzymes were able to carry out more than one function in a single polypeptide chain and were classified as multifunctional proteins (Kirschner and Bisswanger, 1976). In many cases, the dual function resulted from the fusion of two genes that initially encoded different proteins. Later on, the term “moonlighting” (Jeffery, 1999) categorized proteins that have different functions. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a well-known moonlighting enzyme and has at least ten distinct, confirmed non-enzymatic activities apart from its enzymatic function (Sirover, 1999, 2011; Hildebrandt et al., 2015). The GAPDHs constitute a large and diverse family of dehydrogenases universally represented in living organisms. They catalyze the reductive dephosphorylation of 1, 3-bisphosphoglyceric acid (BPGA) producing glyceraldehyde-3-phosphate (GAP) and inorganic phosphate using NAD(P)H as a cofactor (Trost et al., 2006). Glycolytic GAPDHs(also named GapC) are NAD-specific and mainly found in the cytosol, but in land plants a second type of glycolytic GAPDH named GapCp is targeted to the chloroplast(Petersen et al., 2003; Marri et al., 2005; Munoz-Bertomeu et al., 2010). Both GapC and GapCp are NAD-specific and form homotetramers in vivo that are not subject to complex regulatory mechanisms. However, in photosynthetic organisms, another GAPDH catalyzes the unique reductive step of the Benson-Calvin cycle is present and uses both NADH and NADPH with a marked preference for NADPH (Falini et al., 2003).
Like all GAPDHs, the NADPH-dependent GAPDH is made up of two functional domains, one corresponding to the catalytic domain (residues 148–313 in spinach GAPDH) and the other one being the cofactor-binding domain, or Rossman fold (residues 1–147 and 313–334, respectively). The latter contains a flexible, arginine-rich region called the S-loop (Fermani et al., 2001). In higher plants, this GAPDH exists in different forms such as a heterotetramer made up of two GapA and two GapB subunits (A2B2), a homotetramer made up of four GapA subunits, and as a hexadecamer (A8B8) (Baalmann et al., 1994, 1995; Scheibe et al., 2002; Howard et al., 2011a,b). The GapB subunit is similar to the GapA subunit but bears a C-terminal extension which has a regulatory function (Cerff, 1979; Brinkmann et al., 1989; Baalmann et al., 1996; Li and Anderson, 1997; Scagliarini et al., 1998; Sparla et al., 2002) (Figure 1A). This subunit is thought to derive from a gene duplication event which occurred near the origin of Streptophyta (which include charophytes and land plants) (Petersen et al., 2006) and might be a construct of a GapA moiety fused at the C-terminus with the C-terminal half of the CP12, a 8.2–8.5 kDa Chloroplast Protein (Pohlmeyer et al., 1996). The portion of CP12 acquired by GapB subunits confers redox properties to GapB-containing GAPDH (Pupillo and Piccari, 1973; Pupillo and Giuliani Piccari, 1975; Wolosiuk and Buchanan, 1976, 1978; Trost and Pupillo, 1993; Baalmann et al., 1994, 1995; Sparla et al., 2002). Cyanobacteria, primitive photosynthetic eukaryotes (including the glaucophyte Cyanophora paradoxa), and red and green algae (except charophytes and sister lineages) seem to contain exclusively the GapA subunit (Petersen et al., 2006; Trost et al., 2006). However, the small prasinophyte green alga Ostreococcus tauri has been shown to possess GapA and GapB (Robbens et al., 2007).
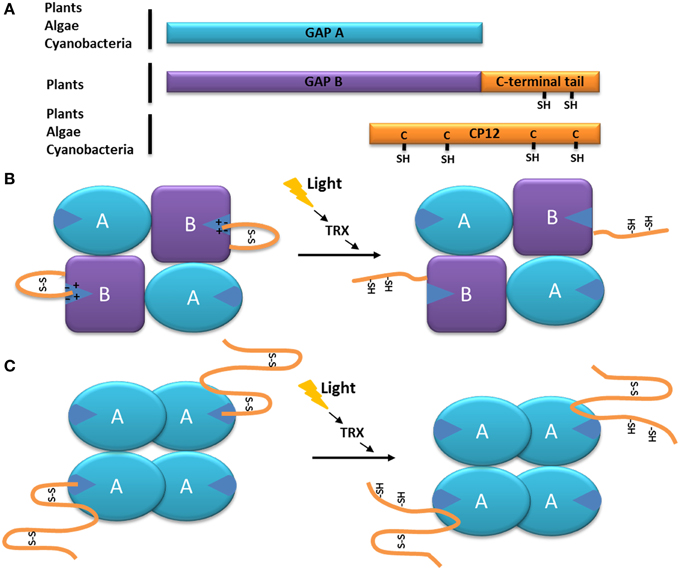
Figure 1. Model of the function of the C-terminal extension of the GAPDH subunit GapB. (A) Schematic organization of the GapB from higher plants and O. tauri (Robbens et al., 2007) subunit compared to the GapA and CP12 proteins. The C-terminal extension of GapB is homologous to the C-terminal of CP12 and present two regulatory cysteine residues. (B) Schematic representation of the autonomous redox regulation of the A2B2-GAPDH. When oxidized, the C-terminal extension of the GapB subunit presents a disulfide bridge, which places the C-terminal amino acids inside the active site of the enzyme, resulting in its inhibition. The disulfide bridge can be reduced by the thioredoxin f (TRX), and the enzyme becomes active. (C) Schematic representation of the autonomous redox regulation of the A4-GAPDH by CP12. When oxidized, the C-terminal part of the CP12 protein presents a disulfide bridge, which places its C-terminal amino acids inside the active site of GAPDH, resulting in its inhibition. The disulfide bridge can be reduced by the thoioredoxin f (TRX) or DTT and the enzyme becomes active.
CP12 is a protein of about 80 amino acid residues that was originally described by Pohlmeyer et al. (1996) and has been found in most photosynthetic organisms (Groben et al., 2010; Gontero and Maberly, 2012; Gontero and Salvucci, 2014; Lopez-Calcagno et al., 2014). The CP12 proteins show high primary sequence variability, in particular at the N-terminus. However, they share some remarkable common features.CP12 proteins have an amino acid composition poor in order-promoting residues although they contain cysteine residues (Groben et al., 2010), and behave abnormally under gel electrophoresis and size exclusion chromatographies (Gontero and Maberly, 2012) suggesting that they are IDPs. Moreover, recent data from fluorescence correlation spectroscopy (FCS) show that the hydrodynamic radius of CP12 from the green alga Chlamydomonas reinhardtii is large compared to that expected for globular proteins of this molecular mass (Moparthi et al., 2014). The cysteine residues are involved in the formation of disulfide bridges and peptide loops and are found as pairs at the C-terminus and/or at the N-terminus. When oxidized, CP12 proteins may present α helices maintained by the N-terminal disulfide bridge (Graciet et al., 2003a; Gardebien et al., 2006). The algal CP12 is a key component of a supra-molecular complex controlling the activity of the Benson-Calvin cycle by regrouping several key enzymes of the cycle, including GAPDH, phosphoribulokinase (PRK) and fructose 1,6-bisphosphate aldolase (FBP aldolase). Within the ternary GAPDH/CP12/PRK complex, both enzymes are strongly inhibited (Avilan et al., 1997; Lebreton et al., 1997; Graciet et al., 2003a; Erales et al., 2008; Marri et al., 2008). CP12 forms a fuzzy complex with the green alga C. reinhardtii A4-GAPDH, as revealed by EPR studies (Mileo et al., 2013) (see the Minireview by Lebreton et al. in this Topic Research). The ternary complex has also been found in the cyanobacterium Synechococcus elongatus and in the higher plant A. thaliana. The A4-GAPDH-CP12 sub-complex from these organisms have been crystallized but in both complexes, the first 50 amino acid residues were not visible in the density map, consistent with a high flexibility of this region in the crystal (Avilan et al., 2000; Matsumura et al., 2011; Fermani et al., 2012). Recently, it was also observed using FCS and FRET (Förster Resonance Energy Transfer) that the algal CP12 flexibility is not abolished by its interactions with either GAPDH or with PRK (Moparthi et al., 2014, 2015).
In the case of the GapB subunit of GAPDH from higher plants, the C-terminal end of the protein (ca 30 residues)is strongly homologous to the C-terminal part of CP12 (Pohlmeyer et al., 1996; Trost et al., 2006; Groben et al., 2010) (Figure 1A). The regulation of the A. thaliana A2B2-GAPDH activity by the C-terminal extension of the GapB subunit is now very well understood: upon oxidation (which happens during the day-night transition) the two cysteine residues of the CP12-like tail form a disulfide bridge that places the C-terminal ultimate glutamate residue (E362) inside the active site (stabilized by the electrostatic interactions with an arginine residue R77 involved in the NADP cofactor binding). Consequently, the NADPH cofactor is not able to enter the catalytic site and thus NAPDH-dependant A2B2-GAPDH activity is inhibited (Sparla et al., 2005; Fermani et al., 2007) (Figure 1B). In contrast, during the night-day transition, the disulfide bridge maintaining the C-terminal extension into the active site is reduced by thioredoxin f, thereby releasing the CP12-like tail and resulting in A2B2-GAPDH activity (Figure 1B) (Sparla et al., 2002; Trost et al., 2006; Fermani et al., 2007). This mechanism is very similar to the one observed in C. reinhardtii between the homotetrameric A4-GAPDH and free CP12, where the penultimate glutamate (E79) of the CP12 interacts with the arginine residue R82 of A4-GAPDH (Figure 1C) (Trost et al., 2006; Erales et al., 2011; Avilan et al., 2012). The reduction of the GAPDH-CP12 by dithiothreitol (DTT) in the alga results in a more active NADPH-GAPDH as a consequence of the rupture of disulfide bridges on CP12. Of interest, DTT, in vitro mimicks thioredoxins in vivo and it has been shown that CP12 can be reduced by thioredoxin f in the light (Marri et al., 2009).
In the higher plant, A. thaliana and in the green alga, C. reinhardtii, the stoichiometry of the oxidized A4-GAPDH-CP12 sub-complex is two CP12 molecules for one A4-GAPDH (Marri et al., 2008; Kaaki et al., 2013), while four CP12 molecules interact with each GAPDH tetramer in the cyanobacterium, S. elongatus (Matsumura et al., 2011). When interacting with CP12, A4-GAPDH activity decreased by two-fold (in the case of the C. reinhardtii proteins, the catalytic constant kcat of the free enzyme was 430 ± 17 s−1, and became 251 ± 9 s−1 in the presence of CP12), suggesting that only two of the four active sites were blocked (Graciet et al., 2003b) (Figure 1C). The same observation was made for the A. thaliana A2B2-GAPDH: upon oxidation, the A2B2-GAPDH activity decreased by 2-fold (its kcat changed from 59 ± 19 s−1 to 27 ± 10 s−1) although its catalytic constant in a reduced state (kcat = 59 ± 19 s−1) was comparable to the one of the free A4-GAPDH (kcat = 61 ± 4 s−1) (Sparla et al., 2004). The regulation of the plant A2B2-GAPDH and of the algal A4-GAPDH-CP12 complex is thus very similar. With the addition of the C-terminal extension within the GapB subunit, the A2B2-GAPDH has become autonomously redox-regulated, a property that was previously provided through interaction with CP12.
Although the appearance of the GapB subunit represents an important step in the evolution of the redox control of the Calvin-Benson cycle enzymes, this new autonomous regulation co-exists with the CP12-based one in higher plants (Scheibe et al., 2002), and a A2B2-GAPDH-PRK complex entirely devoid of CP12 has yet to be identified. The presence of CP12 is likely to be required for the assembly of larger supramolecular complex, and in C. reinhardtii, A4-GAPDH-CP12-PRK was shown to interact with the aldolase (Erales et al., 2008). In this regard, one may wonder how this system will continue to evolve, and if more enzymes of the Benson-Calvin cycle will also acquire similar CP12-like disordered extensions, possibly meaning that the CP12 protein will become redundant. However, CP12 seems to be a part of numerous other processes in photosynthetic organisms (Singh et al., 2008; Howard et al., 2011a,c; Stanley et al., 2013), so it is unlikely to disappear completely from higher plants genomes in the future.
Rubisco Activase
Ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) is the enzyme that catalyzes the formation of two molecules of phosphoglyceric acid using one molecule of ribulose 1,5-bisphosphate (RuBP) and one of carbon dioxide (CO2). As the primary CO2 acceptor of most photoautotrophic organisms, Rubisco can represent up to half of soluble proteins in higher plants, and is believed to be the most abundant protein on Earth (Ellis, 1979; Losh et al., 2013; Raven, 2013). Rubisco from most photosynthetic organisms, including plants and cyanobacteria, is a very large protein (550 kDa), composed of large (L, 52 kDa) and small (S, 12 kDa) subunits arranged as a L8S8 hexadecamer. For this enzyme to be active, a lysine residue inside the Rubisco active site (K201 in Nicotiana tobacum) must be carbamylated and bind a Mg2+ ion (Lorimer et al., 1976; Andersson and Backlund, 2008). The addition of the non-catalytic CO2 molecule to the active site is a spontaneous process, but the presence of RuBP or other sugar-phosphate at the active site decreases the carbamylation efficiency of Rubisco and thus its activity (Lorimer et al., 1976; Cleland et al., 1998). The Rubisco activases (RCAs) exhibit ATPase activity and were first characterized for their ability to promote the carbamylation of such RuBP-inhibited Rubisco (Portis et al., 1986). With time, it became clear that the RCAs allowed the CO2 to enter the active site of Rubisco by removing the hindering RuBP or its analog, carboxyarabinitol bisphosphate (CABP) (Figure 2A) (Portis et al., 2008). The presence of RCAs allows Rubisco to function at its maximal capacity in sub-optimal CO2 concentration that would normally not permit carbamylation in vivo (Portis et al., 1986). In higher plants, RCAs, as expected, are mostly present in the parts of plants involved in photosynthesis (Watillon et al., 1993; Liu et al., 1996), and their expression follows a daily cycle that is regulated by external factors like light and temperature (Martino-Catt and Ort, 1992; Watillon et al., 1993; Liu et al., 1996; To et al., 1999).
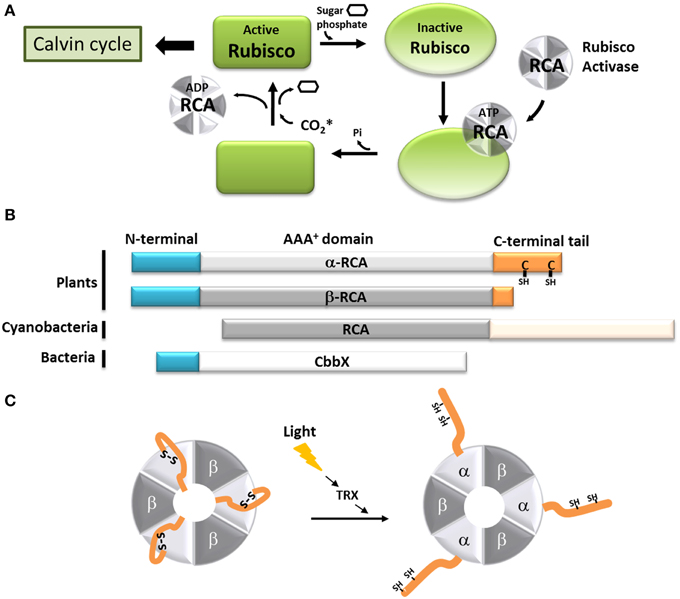
Figure 2. Model of the function of the C-terminal extension of the α-Rubisco activase. (A) Schematic representation of the Rubisco activase reaction with Rubisco. Only the carbamylated Rubisco is active and can participate to the Calvin-Benson cycle. However, the presence of sugar phosphate in the Rubisco active site prevents is carbamylation. Using ATP, the Rubisco activase facilitates the departure of the inhibiting sugar phosphate and promotes the Rubisco carbamylation. The CO2* represents the non-substrate molecule carbamylating Rubisco. (B) Schematic organization of the α and β Rubisco activases subunits from plants, β-cyanobacteria and CbbX. (C) Schematic representation of the light-dependent redox regulation of the α3β3 Rubisco activase. When oxidized, the C-terminal extension of the α-RCA subunit bears a disulfide bridge, which places the C-terminal amino acids inside the nucleotide-binding site of the protein, resulting in its inhibition. The disulfide bridge can be reduced by the thioredoxin f (TRX), and Rubisco becomes active.
In most organisms from the green lineage, two isoforms of RCA are found: an α isoform of 45–46 kDa and a β isoform of 41–43 kDa (Werneke et al., 1989; Rundle and Zielinski, 1991; To et al., 1999; Gontero and Salvucci, 2014). The only difference between the two RCA isoforms is the presence of a short C-terminal extension (ca 30 amino acid residues depending on the species) on the α isoform (Figure 2B). Both the α and β isoforms were found in Arabidopsis thaliana, spinach, and rice, although only one RCA gene is present (Werneke et al., 1988; To et al., 1999); in these species, the presence of the two RCA isoforms is the result of alternate splicing (Werneke et al., 1989; Rundle and Zielinski, 1991; To et al., 1999). On the other hand, other species like barley, cotton, maize and tobacco have multiple RCA genes (Rundle and Zielinski, 1991; Qian and Rodermel, 1993; Salvucci et al., 2003; Yin et al., 2010). In most cases, these organisms have separate genes coding for α and β RCAs without alternative splicing (Rundle and Zielinski, 1991), although all the genes identified in tobacco and cucumber appear to only encode the β isoform (Portis, 2003). To the best of our knowledge, the C-terminus of the α and β RCA isoforms were never tested for intrinsic disorder. Using several disorder predictors, including MeDor (Lieutaud et al., 2008) and MFDp2 (Mizianty et al., 2014), we were able to determine that the end of both the C-terminal part of the α and β RCAs (ca 50 residues for the α isoform and 20 residues for the β isoform) seem to be intrinsically disordered, including the entire C-terminal extension of the α RCA (Figure 3). The most remarkable features of this disordered tail are the two cysteine residues (C392 and C411 in the A. thaliana protein), that are highly conserved among the α RCA isoforms (Zhang and Portis, 1999).
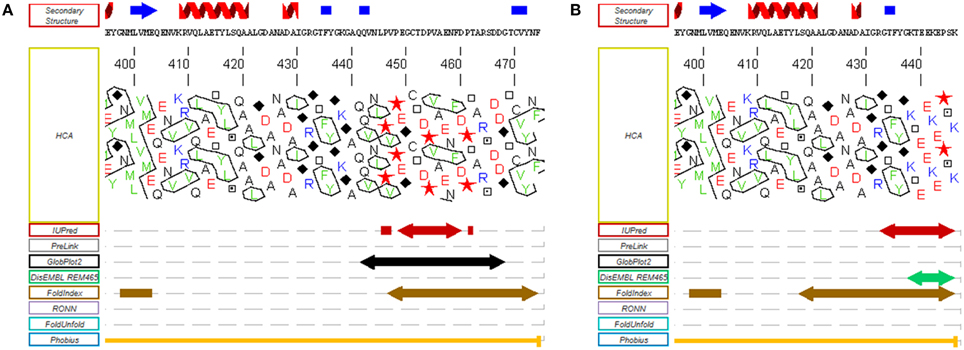
Figure 3. Disorder predictions of the C-terminal regions of the α and β RCA proteins from A. thaliana. MeDor (http://www.vazymolo.org/MeDor/) graphical output of the C-terminal part of the α (A) and β (B) Rubisco activase isoforms from A. thaliana. Predicted secondary structure elements and the HCA plot, are shown above and below the amino acid sequence, respectively. Arrows below the HCA plot correspond to regions of predicted disorder (Lieutaud et al., 2008).
The crystal structure of the tobacco β RCA was recently solved at 3 Å (Stotz et al., 2011), showing that RCA proteins are functional doughnut-shaped hexamers displaying an AAA+ fold, as was predicted using other AAA+ proteins (ATPases involved in a multitude of processes, Neuwald et al., 1999 as templates Portis, 2003). Interestingly, the last 23 residues of the protein were absent from the structure, indicating that this part of the molecule is very flexible and can adopt different conformations. Moreover, the substrate recognition site of the RCA from the creosote bush, Larrea tridentata, was solved at the atomic level (Henderson et al., 2011). Unfortunately, the structural studies were performed only on the β RCA isoform. As the α RCA core is identical, its structure should not be different from the β RCA, but the α RCA was shown to form functional αnβn heteromers rather than αn homomers (Crafts-Brandner et al., 1997; Zhang et al., 2001). In the light of these new structural data, we can suppose that α RCA can form heterohexamers α3β3 (Figure 2C). These structural data also show the presence of three loops containing hydrophilic amino acid residues lining the surface of a central pore. Site-directed mutations introduced in this part of the proteins severely diminished the Rubisco activation and the ATP hydrolysis by the RCA proteins (Stotz et al., 2011), confirming that this region is implicated in the binding of ATP (Salvucci et al., 1993; Li et al., 2006). Based on this information, the model that has been proposed for RCA interaction with Rubisco includes one face of the flat hexamer interacting with the surface of the Rubisco, while an exposed loop of the Rubisco protein could fit into the central hole. Minor conformational changes of this Rubisco loop, allowed by the ATP hydrolysis, would then be transmitted to Rubisco allowing the inhibiting RuBP to be released and Rubisco to be carbamylated (Stotz et al., 2011).
The activity of the α and β RCAs is classically described to be dependent on the ATP:ADP ratio (Zhang and Portis, 1999; Carmo-Silva and Salvucci, 2013), and is extremely sensitive to high temperatures (Portis, 2003; Salvucci, 2004). Moreover, the activity of the α RCA is regulated by light (Mächler and Nösberger, 1980; Perchorowicz et al., 1981). This observation is linked to the action of thioredoxin f on the two cysteine residues present on its C-terminal extension (Shen and Ogren, 1992; Zhang and Portis, 1999; Zhang et al., 2001, 2002; Portis, 2003; Wang and Portis, 2006). A site-directed mutagenesis study (Shen and Ogren, 1992) showed that the substitution of only one of the two cysteine residues was enough to abolish the light regulation of α RCA, implicating the involvement of a disulfide bridge. Several studies showed that the mechanism of inhibition involves the blocking of the ATP-binding region by the C-terminal extension upon oxidation. This self-inhibition would be stabilized by strong electrostatic forces between the negatively-charged tail and the positively charged nucleotide site (Shen and Ogren, 1992; Zhang and Portis, 1999; Zhang et al., 2001, 2002; Wang and Portis, 2006; Carmo-Silva and Salvucci, 2013) (Figure 2C). It was also observed that the β RCA, although devoid of regulatory cysteine residues, could be light-regulated in the presence of the α isoform (Zhang and Portis, 1999; Zhang et al., 2001). In the hypothesis that RCAs form α3β3 heterohexamers, we can assume that the combined bulk of C-terminal extensions efficiently inhibit the whole complex in dark conditions (Figure 2C). The RCA activity can be restored consequently by the reduction of the C-terminal disulfide bridge by the thioredoxin f, which occurs upon dark-light transitions (Carmo-Silva and Salvucci, 2013). In this case, the acquisition of a C-terminal tail, originally by alternate splicing, has allowed the RCA protein to fine-tune the activity of Rubisco in function of the light availability in addition to the energetic state of the cell.
In other photosynthetic organisms, the Rubisco activase system is different or works in a different way. In β-cyanobacteria, the RCA protein has the same main domains as plant RCAs, but lacks the N-terminal domain necessary for Rubisco activation found in plants and green algae (Van De Loo and Salvucci, 1996; Li et al., 1999; Stotz et al., 2011; Gontero and Salvucci, 2014; Mueller-Cajar et al., 2014). This could explain why no Rubisco activation has been observed using cyanobacterial RCAs (Li et al., 1999; Pearce, 2006). The latter also possess a very long (180 residues) intrinsically disordered C-terminal extension that seems to target the protein to the carboxysomes (Zarzycki et al., 2013) (Figure 2B). Organisms from the red lineage (α-proteobacteria, rhodophyta, heterokontophyta, etc.) do not have exactly the same Rubisco as the green lineage, and the so-called “Red Rubisco” has a slightly longer large subunit. These organisms do not have RCA genes, but the same Rubisco activase function is carried out by another protein, CbbX (Pearce, 2006; Gontero and Salvucci, 2014) (Figure 2B). The crystal structure of CbbX has recently been solved, showing that this protein is organized in hexamers arranged in a very comparable manner to green RCAs (Mueller-Cajar et al., 2011). It was also suggested that CbbX mechanisms are based on the same principles as the one of RCA, with the C-terminus of the large Rubisco subunit inserted into the central hole of CbbX (Mueller-Cajar et al., 2011). It should be noted that CbbX seems to have an IDR at its C-terminus, but its implications in CbbX activity is yet to be studied.
Rubisco activase is not the only “friendly” protein involved in the regulation of Rubisco, since other proteins are needed during its assembly and folding, including the cpn60 chaperone, which also has a disordered C-terminal tail (Goloubinoff et al., 1989; Cloney et al., 1992; Libich et al., 2013).
Three Transcription Factor Families (NAC, bZIP and TCP)
Disordered regions are ideal for proteins coordinating regulatory events and as such, transcription factors participating in regulation and signaling functions are enriched in IDRs.
The NAC family (named after No Apical Meristem, ATAF, Cup-Shaped Cotyledon) is one of the largest families of plant-specific transcription factors (Ooka et al., 2003; Olsen et al., 2005; Rushton et al., 2008; Sun et al., 2013). These family members are involved in a very large variety of processes, including plant development (Olsen et al., 2005), biotic and abiotic stress responses (Jensen et al., 2010b; Seo and Park, 2010; Seo et al., 2010) and leaf senescence (Kjaersgaard et al., 2011). The NAC transcription factors usually contain two domains: the N-terminal NAC domain and the C-terminal extremity domain (Figure 4A). The NAC domain is mainly conserved and well-ordered, displaying a typical structure comprising α helices flanking one β strand (Ernst et al., 2004). This domain binds the consensus DNA sequence CGT(GA) (Olsen et al., 2005). The C-terminal domain of the NAC proteins is highly variable within the family; however, some motifs in the C-terminus may display a sub-family-specific conservation (Jensen et al., 2010a). The C-terminal domains composition reveals a very high percentage of hydrophilic (Asp, Glu, Ser, Thr) and proline (Pro) residues, whereas the proportion of hydrophobic and aromatic residues is very low (Olsen et al., 2005; Jensen et al., 2010a). These specificities are typical of IDRs, and the C-terminal domain of some NAC proteins was experimentally characterized as an IDR (Jensen et al., 2010a,b). Despite this IDR feature, some hydrophobic and/or aromatic residues are present in this domain; interestingly, these amino acid residues are often conserved among a subfamily (Jensen et al., 2010a,b; Kjaersgaard et al., 2011). The IDR C-terminal domains of the NAC proteins are predicted to contain MoREs that are conserved in sub-families (Jensen et al., 2010a). It has been experimentally confirmed that these particular residues are very important to the specific function of each sub-group in the NAC family, and are essential to activation mechanisms often involving many different partners (Ooka et al., 2003; Ernst et al., 2004; Taoka et al., 2004; Olsen et al., 2005; Ko et al., 2007; Jensen et al., 2010a) (Figure 4B).
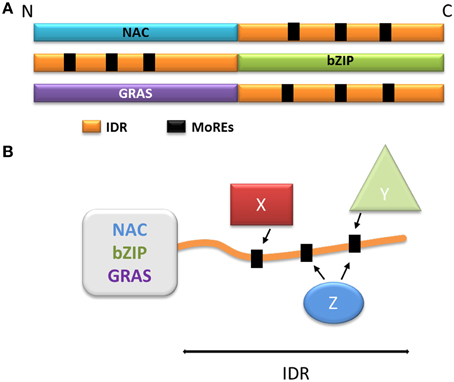
Figure 4. Model of the function of the disordered extremities of the NAC, bZIP, and GRAS proteins. (A) Schematic organization of the NAC, bZIP, and GRAS protein families. The disordered parts are schematized in orange, the MoREs are represented as black squares. (B) Schematization of the multiple protein-protein interactions involving the disordered extremity. X, Y, and Z represent different protein partners capable of interacting with one or more MoREs and regulate the behavior of the globular domain.
The bZIP (basic Leucine Zipper) transcription factors family is ubiquitous and is one of the largest families of transcription factors in eukaryotes. bZIP transcription factors take part in a multitude of regulatory pathways such as development, metabolism, circadian rhythm and response to stress (Sun et al., 2013). The bZIP proteins are composed of two domains: a C-terminal bZIP domain and a N-terminal activation domain (Figure 4A). The C-terminal bZIP domain gives its name to the family and displays large patches of basic residues and leucine zipper motifs (Ellenberger et al., 1992; Vinson et al., 1993). The leucine zipper regions are organized in α helices and are responsible for the dimerization of the proteins through the formation of a coiled-coil structure (Vinson et al., 1993; Yoon et al., 2006), while the basic regions bind to the DNA molecule (Ellenberger et al., 1992). Interestingly, the basic regions have been described either as fully ordered, very flexible or intrinsically disordered depending on the protein (Bracken et al., 1999; Podust et al., 2001; Moreau et al., 2004; Yoon et al., 2006). When bound to DNA, the basic regions have however been observed as α helices, suggesting that the interaction triggers folding in response to a specific DNA motif (Hollenbeck et al., 2002), illustrating once more the disorder to order transition (induced-fit). The N-terminal regions of bZIP proteins act as regulators (Ang et al., 1998; Sun et al., 2013), and are mostly intrinsically disordered (Campbell et al., 2000; Moreau et al., 2004; Yoon et al., 2006; Sun et al., 2013). These regions typically contain different MoREs, and their flexibility allows the interaction with multiple partners, again by adopting different secondary structures (Ang et al., 1998; Campbell et al., 2000; Oldfield et al., 2005; Yoon et al., 2006) (Figure 4B). Through these activating or inhibiting interactions, transcription of the genes targeted by bZIP proteins is effectively modulated in response to several signals. The N-terminal disordered domain also modulates the activity of bZIP transcription factors through post-translational modifications, and phosphorylation in particular. In plants, bZIP transcription factors can be phosphorylated in response to illumination, which disrupts the interactions between the bZIP proteins and their activating partners (Ciceri et al., 1997; Hardtke et al., 2000). The phosphorylated proteins also have lower affinity for their DNA targets, resulting in a decrease of gene activation (Ciceri et al., 1997; Hardtke et al., 2000). Interestingly, some bZIP proteins also display IDRs in their C-terminal domain. In the case of bZIP28 (initially a transmembrane protein), these IDRs are exposed to the lumen of the endoplasmic reticulum and allow the interaction, through MOREs with BIP, the majority reticulum chaperone. In response to stress, bZIP28 is relocated to the Golgi and the cytoplasmic domain is detached, allowing it to enter the nucleus and to control gene expression (Srivastava et al., 2013, 2014).
A recent study on TCP8, a transcription factor belonging to the TCP [Teosinte branched 1 (tb1), Cycloidea (cyc) and Proliferating Cell Factor (PCF)] family, showed the presence of three IDRs, two of them at the N- and C-terminal extremities (Valsecchi et al., 2013). While the N-terminus binds DNA in an induced-fit mechanism, the C-terminal region is involved in the TCP protein self-association in a coiled-coil structure (Valsecchi et al., 2013). Furthermore, it seems that different transcription factors from the TCP family can interact, modulating the response of different pathways to multiple stimuli (Baier and Latzko, 1975; Viola et al., 2011, 2012; Steiner et al., 2012; Valsecchi et al., 2013).
As illustrated in these examples, the disordered tails of transcription factors have an essential role in modulating their activities through protein-protein interactions with a wide range of activators and inhibitors. Moreover, these extensions are often prone to phosphorylation and constitute another level of regulation. Together, these IDRs form a complex signaling web, turning the transcription factors into hubs and allowing the genes involved in adaptive responses to be finely regulated.
GRAS Family
The GRAS family comprises proteins involved in numerous aspects of plant development and growth. This large family is named after its first members, Gibberellic Acid Insensitive (GAI), Repressor of Gai (RGA) and Scarecrow (SCR), and its members are mostly related to signaling in response to phytohormones [gibberellic acid (GA), auxin, brassinosteroids] and biotic and abiotic stress (Bolle, 2004; Sun et al., 2011). The GRAS family proteins are composed of one variable N-terminal region and a commonly conserved C-terminal GRAS domain (Figure 4A), and are divided into ten subfamilies based on phylogeny (Bolle, 2004; Tian et al., 2004; Lim et al., 2005; Sanchez et al., 2007; Sun et al., 2011). The conserved GRAS domain (ca 380 residues depending on the subfamilies) acts as a transcriptional co-activator (Heery et al., 1997) through leucine-rich motifs. GRAS domains typically contain two leucine-rich motifs, which are needed for specific protein-protein interactions(Cui et al., 2007; Vacic et al., 2007; Fode et al., 2008; Hirsch and Oldroyd, 2009; Hirsch et al., 2009; Hou et al., 2010). The GRAS proteins interact with a large number of nuclear proteins, most of which are transcription factors, thereby modulating their target activity (Hirsch and Oldroyd, 2009; Hirsch et al., 2009; Hou et al., 2010; Sun et al., 2012).
In contrast to the highly conserved GRAS domain, the N-terminal domains of the GRAS family proteins display a rich diversity at the sequence level, although the N-terminus is conserved within subfamilies (Sun et al., 2010, 2011, 2012, 2013). Moreover, these N-terminal domains have recently been identified as intrinsically disordered (Sun et al., 2010, 2011, 2012, 2013). Interestingly, patches of repeated hydrophobic and/or aromatic residues are found in the N-terminal region (Triezenberg, 1995; Sun et al., 2011). These patches are arranged in conserved motifs within subfamilies (Triezenberg, 1995; Sun et al., 2011), and are involved in specific multiple protein-protein interactions (Sun et al., 2010, 2012). In the case of the DELLA subfamily which has been intensively studied, the N-terminal domain can interact with the gibberellic acid receptor GIB1, but only when GIB1 has bound its ligand (Murase et al., 2008; Hirano et al., 2010; Sun et al., 2010, 2012). Moreover, each DELLA protein domain (N-terminal and C-terminal domains) can interact with several partners, making these proteins a hub at the center of the gibberellic acid response pathway. Other examples of GRAS proteins are important in other regulatory pathways, although subfamilies are always specialized in a precise type of stimulus (phytohormones, biotic and abiotic stress, etc…) (Sun et al., 2010, 2011, 2012, 2013).
A common feature of the GRAS proteins is their ability to acquire a structure when bound to a partner, unlike the fuzzy GAPDH/CP12 complex (Mileo et al., 2013). As mentioned above, MoREs are present in GRAS proteins; each one was predicted to occur within the N-terminal domains, and more specifically in the elements conserved within subfamilies, strengthening the idea that these motifs are the key to the specificity of GRAS proteins (Sun et al., 2011, 2012). In the case of the DELLA subfamily, the presence of the MoREs has been verified experimentally (Sun et al., 2010, 2011, 2012). Interestingly, the N-terminal domain of the GRAS proteins is also the target of phosphorylation, which again introduces another way to fine-tune the regulation of these proteins (Fu et al., 2002; Iakoucheva et al., 2004; Hussain et al., 2007; Mittag et al., 2010). Phosphorylation of the N-terminal domain is directly linked to the activity of the GRAS proteins, modulating the affinity of the N-terminus for its partners, and having a direct effect on the GRAS proteins stability through the control of their degradation (Day et al., 2004; Hussain et al., 2005; Itoh et al., 2005; Czikkel and Maxwell, 2007).
When considering the GRAS family as a whole, it is remarkable how conserved the GRAS domains and patterns are, while the N-terminal domains are highly variable. It seems that the addition of a disordered protein segment to the GRAS domain has increased its number of partners, and thus turned it into a signal-integration hub involved in many different pathways. On the other hand, one could consider that the addition of GRAS domains to pre-existing IDPs involved in the phytohormonal and/or stress responses has allowed these IDPs to control, even more directly, the cellular responses by acting on gene expression.
Cryptochrome
Cryptochromes are a group of proteins in which most members have an intrinsically disordered C-terminal tail that can have a profound impact on their overall function. Together with the photolyases, these proteins belong to the photolyase/cryptochrome family (Lin and Shalitin, 2003; Sancar, 2004; Chaves et al., 2006; Ozturk et al., 2007; Fortunato et al., 2015).
Photolyases are ancient enzymes that use blue light to catalyze the repair of DNA lesions caused by ultraviolet light. Lesions such as cyclobutane pyrimidine dimers (CPD) and pyrimidine-pyrimidone photoproducts are repaired by photolyases CPD and by photolyases 6–4, respectively. Photolyase capacity to use blue light is due to the presence of two chromophores: a photoantenna pterin (5,10-methenyltetrahydrofolateor a-hydroxy-5-deazaflavin) and flavin adenine dinucleotide (FAD). During the DNA repair, the two chromophores cofactors absorb blue photons and initiate splitting of the cyclobutane ring by a mechanism involving reactive radicals (Liu et al., 2011b).
Cryptochromes, the other group of proteins in the photolyase/cryptochrome family, have a photolyase homologous region (called PHR) and a C-terminal tail (Figure 5A) (Yu et al., 2010). Cryptochromes are able to absorb blue light in a very similar way to the photolyases. Another group within this family includes DASH-type cryptochromes named after the Drosophila, Arabidopsis, Synechocystis and Human. Members of this group are closer to photolyases than to cryptochromes, and are able to repair single-stranded DNA (Chaves et al., 2011) and may also have N-terminal and C-terminal disordered extensions.
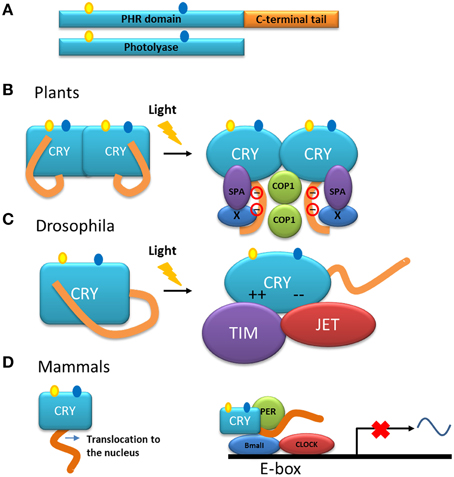
Figure 5. Models of the function of the C-terminal tail of cryptochromes. (A) Representation of cryptochrome (CRY) and photolyase. Cryptochromes have a photolyase-homologous region (PHR) and a C-terminal tail. The chromophore molecules of the PHR are shown. (B) Model of the action mechanism of cryptochromes from Arabidopsis. After absorption of light, the C-terminal tail is phosphorylated and a change in conformation is triggered in the entire molecule. The C-terminal tail is exposed at the surface of the protein and as a consequence interactions with partner proteins such as COP1 and SPA are induced (Liu et al., 2011a,b). (C) In darkness, the C-terminal tail of the cryptochrome from Drosophila inhibits the binding of the proteins involved in the circadian rhythm. After illumination, the inhibition by the tail is released and the PHR domain interacts through electrostatic interaction with the protein partners TIM and JET (Green, 2004; Czarna et al., 2013). (D) In mammals, cryptochrome is necessary for the translocation of the protein into the nucleus in which it is part of the core of the transcription/translation feedback that controls the circadian clock together with the proteins PER, BMAL, and CLOCK.
In contrast to photolyases, cryptochromes do not have the ability to repair DNA. However, in many organisms, the absorption of photons by the chromophores in the photolyase homologous region of these proteins, induces conformational change (through electron transfer and subsequent phosphorylation), which in turn trigger specialized signaling events through protein-protein interactions (Liu et al., 2011b). It has been shown that the function of cryptochromes resides mainly within their C-terminal tails (Yang et al., 2000; Green, 2004; Chaves et al., 2006, 2011; Yu et al., 2010). Interestingly, this tail is poorly conserved among groups of organisms. In Arabidopsis, two cryptochromes are present, CRY1 and CRY2, that have different C-terminal extensions although a DAS motif is found in both (Lin and Shalitin, 2003). The length of the C-terminal tail in cryptochromes of animals, plants and some unicellular organisms varies from 30 to 250 residues and, as mentioned above, is intrinsically disordered. This characteristic has been established by sequence analysis, biochemical methods such as analysis of the sensitivity to protease cleavage, and physical methods such as circular dichroism and nuclear magnetic resonance (NMR)on recombinant C-terminal extensions of both Arabidopsis and human cryptochromes (Partch et al., 2005). Comparison of the proteolysis susceptibility between full-length cryptochromes and their C-terminal tail showed that this tail interacts with the photolyase domain, causing it to adopt a tertiary structure. The susceptibility to proteolysis of the C-terminal tail of the CRY1 from A. thaliana increases after illumination, which is consistent with a conformational change (Partch et al., 2005). Indeed, the crystal structure of the complete cryptochrome from Drosophila confirmed that the C-terminal tail stays in a groove of the photolyase domain and mimics the recognition of photolyases with DNA (Zoltowski et al., 2011; Czarna et al., 2013).
In plants, cryptochromes play a role, together with other photoreceptors, in a variety of functions. In general, the cryptochromes of plants are involved in mechanisms that respond to blue light and their action has been explored in the inhibition of the elongation of hypocotyls, in the photoperiodic induction of flowering, in the circadian clock as in animals, and in other functions (Yu et al., 2010; Chaves et al., 2011; Liu et al., 2011b). These studies have been mainly performed in the model plant A. thaliana. Studies using transgenic plants overexpressing the C-terminal tail of CRY1 or CRY2, fused with β-glucuronidase (GUS) showed a constitutive morphogenic phenotype similar to that produced by blue light (Yang et al., 2000), indicating that, in the cryptochrome molecule, the C-terminal tail is responsible for the light-induced function. Moreover, NC80, an 80-residues segment present in the Arabidopsis protein, is responsible for the function of the C-terminal tail of CRY2 (Yu et al., 2007). The C-terminal tail of these proteins interacts with other proteins such as COP1 (constitutive photomorphogenic 1) (Wang et al., 2001; Yang et al., 2001), a multifunctional E3 ubiquitin ligase, and SPA1 (suppressor of phytochrome A 1) (Zuo et al., 2011; Liu et al., 2011a). This interaction is part of the initial steps for the light signaling and mechanisms to modulate the developmental process in the plant either by: (1) modulation of gene transcription or (2) suppression of proteolysis of regulators involved in development (i.e., flowering) (Liu et al., 2011a,b). Models have been proposed to explain the mode of action of plant cryptochromes (Lin and Shalitin, 2003; Partch et al., 2005; Yu et al., 2007; Liu et al., 2011a). In general, in these models, the photolyase domain and the C-terminal tail form a closed conformation in the dark. Upon illumination, an open and active conformation is adopted and, in this new conformation, the C-terminal tail is exposed allowing its interaction with other proteins to initiate signaling (Figure 5B). A model of action that includes dimerization and light dependent-phosphorylation that explains the exposure of the C-terminal tail as a result of charge repelling has also been proposed (Figure 5B) (Lin and Shalitin, 2003; Yu et al., 2007).
Although cryptochromes of plants are involved as photoreceptors in the circadian cycle, the molecular role of cryptochromes in relation to this cycle has been more elucidated in Drosophila. In this organism, the cryptochrome modulates the central oscillator, or clock, through the light-dependent interaction with the protein Timeless (TIM) (Busza et al., 2004), one of the components of the clock core. This interaction favors the degradation of both TIM and the cryptochrome itself, thus triggering the light/dark cycle each day by synchronization of the clock with the environment. The protein Jetlag (JET), an E3 ligase, also binds to the cryptochrome in a light-dependent manner and is responsible for the ubiquitination and subsequent proteolysis of both the cryptochrome and TIM (Peschel et al., 2009). In this case, and in contrast with the cryptochromes in Arabidopsis, the binding of the cryptochrome from Drosophila to its partners is performed by the photolyase domain of the protein (Figure 5C), whereas in the dark, the C-terminal tail inhibits this binding determining thus the photosensitivity of the circadian clock (Busza et al., 2004; Green, 2004).
In contrast to their homologs from plants and Drosophila, where the disordered C-terminal tail is used for light signaling, mammalian cryptochromes are light–independent transcriptional repressors in the core of the circadian clock (Figure 5D). Mammalian cryptochromes repress transcription processes that are dependent on the protein complex BMAL/CLOCK (Sancar, 2004; Chaves et al., 2011). In the case of these cryptochromes, the function of the C-terminal tail is more complex: (i) it is involved in the nuclear localization of the protein and (ii) with the photolyase domain, it also has a role in the interaction with other components of the clock such as BMAL (Chaves et al., 2006). Interestingly, the C-terminal tail also contributes to the circadian period length, since its phosphorylation affects the level of the protein, either promoting its own degradation in the case of CRY 2 (Harada et al., 2005) or stabilizing the protein as for CRY 1 (Gao et al., 2013).
It has been proposed that cryptochromes have evolved several times independently as an example of convergent evolution (Green, 2004). Only small changes have occurred in the photolyase domain, this part of the protein being conserved among cryptochromes and photolyases. One possible mechanism to explain the acquisition of C-terminal extensions in existing proteins would be through gene fusion (Marsh and Teichmann, 2010). If this mechanism had taken place at the origin of cryptochromes, it would suggest that proteins related to the C-terminal tail of cryptochromes already existed independently and had a function of their own. These independent domains became later associated to a photolyase domain providing them with the capacity to detect light. As mentioned above, the plant cryptochromes C-terminal domain is active and has the information needed to achieve signaling (Yang et al., 2000). During evolution, this protein could have fused with a duplicate of photolyase. In this hypothesis, the addition of the light-dependent photolyase module might be a way to adjust the physiology of the organisms to their environment through light perception. This could therefore be seen as an IDP having acquired a globular extension. Since in plants, a motif (DAS) within the C-terminal tail is conserved, it has been proposed that the ancestral plant cryptochrome emerged from a fusion of a photolyase with a protein containing the DAS motif (Lin and Shalitin, 2003). Another hypothesis that could explain the acquisition of the C-terminal tail in cryptochromes is by gene extension into a non-coding region (Marsh and Teichmann, 2010). The photolyase gene could thus have been extended through junk DNA. Analysis of phylogenetic relationships of gene families in animals showed that extension of an existing gene by “exonization” of a previous non-coding region seems to be an important evolutionary strategy to add a C-terminal disordered extension to proteins (Buljan et al., 2010). The high variability and different functions of the C-terminal tail of cryptochromes among plants and animals are in accordance with this hypothesis. Studies on the origin and evolution of the C-terminal tail of cryptochromes will give insights into the adaptation of organisms to light.
Conclusion
Within the present review, we tried to demonstrate the central and multiple roles of intrinsically disordered tails carried by certain globular proteins. Describing several examples of proteins displaying IDRs in photosynthetic organisms, we discussed how IDRs impact on both the functions and mechanisms of action of their “host” proteins. The examples of the A2B2-GAPDH and the α-Rubisco activase isoform show that their C-terminal disordered extensions participate in the light-dependent redox regulation of the photosynthetic metabolism. The cases of the multiple transcription factors with a disordered tail are very similar yet very different. In the few examples listed here, the disordered region plays a major role in the regulation of the DNA-binding domain through protein-protein interactions or post-translational modifications. Their sensitivity to a large number of signals allows the activity of the transcription factors to be modulated according to many factors (one to many), turning these proteins into hubs in a large signaling web. Lastly, the cryptochrome family is a prime example of a disordered extension changing the fundamental function of the initial photolyase into a light-dependent signaling protein, conserving the ability to absorb blue light and repurposing it.
The examples presented here are but a few of the multitude of proteins that have acquired a disordered extension (Uversky, 2013), although most examples do not usually come from the photosynthetic world. We can expect that in the years to come, an increasing number of these proteins will be identified. A great question that remains is how these proteins originated. While in some cases, the addition of an IDR seems to be quite recent like the GapB subunit. In other cases, this addition might be very ancient as in the NAC, bZIP, and GRAS families, in which there are multiple disordered extensions families that may derive from multiple fusion events, or a long succession of duplications followed by diverging evolution of the subfamilies. We hope that the expansion of the IDP field in general and specifically, the one involved in “green” biochemistry, will 1 day answer these questions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Studentship (GT) was given by Ecole Normale Supérieure, Cachan. This work (BG) was supported by the Centre National de la Recherche Scientifique (CNRS) and Aix-Marseille Université. LA got a fellowship from ANR-13-BIME-0001-02. We would like to thank Pr. Carla Green for her kind help. We also thank Pr. Stephen Maberly for editing the english.
References
Alderson, T. R., and Markley, J. L. (2013). Biophysical characterization of alpha-synuclein and its controversial structure. Intrinsically Disord. Proteins 1, 18–39. doi: 10.4161/idp.26255
Andersson, I., and Backlund, A. (2008). Structure and function of Rubisco. Plant Physiol. Biochem. 46, 275–291. doi: 10.1016/j.plaphy.2008.01.001
Ang, L. H., Chattopadhyay, S., Wei, N., Oyama, T., Okada, K., Batschauer, A., et al. (1998). Molecular interaction between COP1 and HY5 defines a regulatory switch for light control of Arabidopsis development. Mol. Cell 1, 213–222. doi: 10.1016/S1097-2765(00)80022-2
Arabidopsis Genome Initiative. (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815. doi: 10.1038/35048692
Avilan, L., Gontero, B., Lebreton, S., and Ricard, J. (1997). Memory and imprinting effects in multienzyme complexes–I. Isolation, dissociation, and reassociation of a phosphoribulokinase-glyceraldehyde-3-phosphate dehydrogenase complex from Chlamydomonas reinhardtii chloroplasts. Eur. J. Biochem. 246, 78–84. doi: 10.1111/j.1432-1033.1997.00078.x
Avilan, L., Lebreton, S., and Gontero, B. (2000). Thioredoxin activation of phosphoribulokinase in a bi-enzyme complex from Chlamydomonas reinhardtii chloroplasts. J. Biol. Chem. 275, 9447–9451. doi: 10.1074/jbc.275.13.9447
Avilan, L., Puppo, C., Erales, J., Woudstra, M., Lebrun, R., and Gontero, B. (2012). CP12 residues involved in the formation and regulation of the glyceraldehyde-3-phosphate dehydrogenase-CP12-phosphoribulokinase complex in Chlamydomonas reinhardtii. Mol. Biosyst. 8, 2994–3002. doi: 10.1039/c2mb25244a
Baalmann, E., Backhausen, J. E., Kitzmann, C., and Scheibe, R. (1994). Regulation of NADP-dependent glyceraldehyde 3-phosphate dehydrogenase activity in spinach chloroplast. Botanica Acta 107, 313–320. doi: 10.1111/j.1438-8677.1994.tb00801.x
Baalmann, E., Backhausen, J. E., Rak, C., Vetter, S., and Scheibe, R. (1995). Reductive modification and nonreductive activation of purified spinach chloroplast NADP-dependent glyceraldehyde-3-phosphate dehydrogenase. Arch. Biochem. Biophys. 324, 201–208. doi: 10.1006/abbi.1995.0031
Baalmann, E., Scheibe, R., Cerff, R., and Martin, W. (1996). Functional studies of chloroplast glyceraldehyde-3-phosphate dehydrogenase subunits A and B expressed in Escherichia coli: formation of highly active A4 and B4 homotetramers and evidence that aggregation of the B4 complex is mediated by the B subunit carboxy terminus. Plant Mol. Biol. 32, 505–513. doi: 10.1007/BF00019102
Babu, M. M., Van Der Lee, R., De Groot, N. S., and Gsponer, J. (2011). Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440. doi: 10.1016/j.sbi.2011.03.011
Baier, D., and Latzko, E. (1975). Properties and regulation of C-1-fructose-1,6-diphosphatase from spinach chloroplasts. Biochim. Biophys. Acta 396, 141–148. doi: 10.1016/0005-2728(75)90197-8
Bolle, C. (2004). The role of GRAS proteins in plant signal transduction and development. Planta 218, 683–692. doi: 10.1007/s00425-004-1203-z
Bracken, C., Carr, P. A., Cavanagh, J., and Palmer, A. G. III. (1999). Temperature dependence of intramolecular dynamics of the basic leucine zipper of GCN4: implications for the entropy of association with DNA. J. Mol. Biol. 285, 2133–2146. doi: 10.1006/jmbi.1998.2429
Breydo, L., and Uversky, V. N. (2011). Role of metal ions in aggregation of intrinsically disordered proteins in neurodegenerative diseases. Metallomics 3, 1163–1180. doi: 10.1039/c1mt00106j
Breydo, L., Wu, J. W., and Uversky, V. N. (2012). Alpha-synuclein misfolding and Parkinson's disease. Biochim. Biophys. Acta 1822, 261–285. doi: 10.1016/j.bbadis.2011.10.002
Brinkmann, H., Cerff, R., Salomon, M., and Soll, J. (1989). Cloning and sequence analysis of cDNAs encoding the cytosolic precursors of subunits GapA and GapB of chloroplast glyceraldehyde-3- phosphate dehydrogenase from pea and spinach. Plant Mol. Biol. 13, 81–94. doi: 10.1007/BF00027337
Buljan, M., Frankish, A., and Bateman, A. (2010). Quantifying the mechanisms of domain gain in animal proteins. Genome Biol. 11:R74. doi: 10.1186/gb-2010-11-7-r74
Busza, A., Emery-Le, M., Rosbash, M., and Emery, P. (2004). Roles of the two Drosophila CRYPTOCHROME structural domains in circadian photoreception. Science 304, 1503–1506. doi: 10.1126/science.1096973
Campbell, K. M., Terrell, A. R., Laybourn, P. J., and Lumb, K. J. (2000). Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry 39, 2708–2713. doi: 10.1021/bi9923555
Carmo-Silva, A. E., and Salvucci, M. E. (2013). The regulatory properties of Rubisco activase differ among species and affect photosynthetic induction during light transitions. Plant Physiol. 161, 1645–1655. doi: 10.1104/pp.112.213348
Castillo, P., Cetina, A. F., Mendez-Tenorio, A., Espinoza-Fonseca, L. M., and Barron, B. L. (2014). Papillomavirus binding factor (PBF) is an intrinsically disordered protein with potential participation in osteosarcoma genesis, in silico evidence. Theor. Biol. Med. Model. 11:51. doi: 10.1186/1742-4682-11-51
Cerff, R. (1979). Quaternary structure of higher plant glyceraldehyde-3-phosphate dehydrogenases. Eur. J. Biochem. 94, 243–247. doi: 10.1111/j.1432-1033.1979.tb12891.x
Chakrabortee, S., Tripathi, R., Watson, M., Schierle, G. S., Kurniawan, D. P., Kaminski, C. F., et al. (2012). Intrinsically disordered proteins as molecular shields. Mol. Biosyst. 8, 210–219. doi: 10.1039/C1MB05263B
Chaves, I., Pokorny, R., Byrdin, M., Hoang, N., Ritz, T., Brettel, K., et al. (2011). The cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364. doi: 10.1146/annurev-arplant-042110-103759
Chaves, I., Yagita, K., Barnhoorn, S., Okamura, H., Van Der Horst, G. T., and Tamanini, F. (2006). Functional evolution of the photolyase/cryptochrome protein family: importance of the C-terminus of mammalian CRY1 for circadian core oscillator performance. Mol. Cell. Biol. 26, 1743–1753. doi: 10.1128/MCB.26.5.1743-1753.2006
Chouard, T. (2011). Structural biology: breaking the protein rules. Nature 471, 151–153. doi: 10.1038/471151a
Ciceri, P., Gianazza, E., Lazzari, B., Lippoli, G., Genga, A., Hoscheck, G., et al. (1997). Phosphorylation of Opaque2 changes diurnally and impacts its DNA binding activity. Plant Cell 9, 97–108. doi: 10.1105/tpc.9.1.97
Cleland, W. W., Andrews, T. J., Gutteridge, S., Hartman, F. C., and Lorimer, G. H. (1998). Mechanism of Rubisco: the carbamate as general base. Chem. Rev. 98, 549–562. doi: 10.1021/cr970010r
Cloney, L. P., Bekkaoui, D. R., Wood, M. G., and Hemmingsen, S. M. (1992). Assessment of plant chaperonin-60 gene function in Escherichia coli. J. Biol. Chem. 267, 23333–23336.
Crafts-Brandner, S. J., Van De Loo, F. J., and Salvucci, M. E. (1997). The two forms of ribulose-1,5-bisphosphate carboxylase/oxygenase activase differ in sensitivity to elevated temperature. Plant Physiol. 114, 439–444.
Cui, H., Levesque, M. P., Vernoux, T., Jung, J. W., Paquette, A. J., Gallagher, K. L., et al. (2007). An evolutionarily conserved mechanism delimiting SHR movement defines a single layer of endodermis in plants. Science 316, 421–425. doi: 10.1126/science.1139531
Czarna, A., Berndt, A., Singh, H. R., Grudziecki, A., Ladurner, A. G., Timinszky, G., et al. (2013). Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell 153, 1394–1405. doi: 10.1016/j.cell.2013.05.011
Czikkel, B. E., and Maxwell, D. P. (2007). NtGRAS1, a novel stress-induced member of the GRAS family in tobacco, localizes to the nucleus. J. Plant Physiol. 164, 1220–1230. doi: 10.1016/j.jplph.2006.07.010
Day, R. B., Tanabe, S., Koshioka, M., Mitsui, T., Itoh, H., Ueguchi-Tanaka, M., et al. (2004). Two rice GRAS family genes responsive to N -acetylchitooligosaccharide elicitor are induced by phytoactive gibberellins: evidence for cross-talk between elicitor and gibberellin signaling in rice cells. Plant Mol. Biol. 54, 261–272. doi: 10.1023/B:PLAN.0000028792.72343.ee
Dunker, A. K., Brown, C. J., Lawson, J. D., Iakoucheva, L. M., and Obradovic, Z. (2002). Intrinsic disorder and protein function. Biochemistry 41, 6573–6582. doi: 10.1021/bi012159+
Dunker, A. K., Cortese, M. S., Romero, P., Iakoucheva, L. M., and Uversky, V. N. (2005). Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 272, 5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x
Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., et al. (2001). Intrinsically disordered protein. J. Mol. Graph. Model. 19, 26–59. doi: 10.1016/S1093-3263(00)00138-8
Dunker, A. K., Oldfield, C. J., Meng, J., Romero, P., Yang, J. Y., Chen, J. W., et al. (2008a). The unfoldomics decade: an update on intrinsically disordered proteins. BMC Genomics 9(Suppl. 2):S1. doi: 10.1186/1471-2164-9-S2-S1
Dunker, A. K., Silman, I., Uversky, V. N., and Sussman, J. L. (2008b). Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 18, 756–764. doi: 10.1016/j.sbi.2008.10.002
Ellenberger, T. E., Brandl, C. J., Struhl, K., and Harrison, S. C. (1992). The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted alpha helices: crystal structure of the protein-DNA complex. Cell 71, 1223–1237. doi: 10.1016/S0092-8674(05)80070-4
Ellis, R. J. (1979). The most abundant protein in the world. Trends Biochem. Sci. 4, 241–244. doi: 10.1016/0968-0004(79)90212-3
Erales, J., Avilan, L., Lebreton, S., and Gontero, B. (2008). Exploring CP12 binding proteins revealed aldolase as a new partner for the phosphoribulokinase/glyceraldehyde 3-phosphate dehydrogenase/CP12 complex - purification and kinetic characterization of this enzyme from Chlamydomonas reinhardtii. FEBS J. 275, 1248–1259. doi: 10.1111/j.1742-4658.2008.06284.x
Erales, J., Mekhalfi, M., Woudstra, M., and Gontero, B. (2011). Molecular mechanism of NADPH-glyceraldehyde-3-phosphate dehydrogenase regulation through the C-terminus of CP12 in Chlamydomonas reinhardtii. Biochemistry 50, 2881–2888. doi: 10.1021/bi1020259
Ernst, H. A., Olsen, A. N., Larsen, S., and Lo Leggio, L. (2004). Structure of the conserved domain of ANAC, a member of the NAC family of transcription factors. EMBO Rep. 5, 297–303. doi: 10.1038/sj.embor.7400093
Falini, G., Fermani, S., Ripamonti, A., Sabatino, P., Sparla, F., Pupillo, P., et al. (2003). Dual coenzyme specificity of photosynthetic glyceraldehyde 3-phosphate dehydrogenase interpreted by the crystal structure of A4 isoform complexed with NAD. Biochemistry 42, 4631–4639. doi: 10.1021/bi0272149
Fermani, S., Ripamonti, A., Sabatino, P., Zanotti, G., Scagliarini, S., Sparla, F., et al. (2001). Crystal structure of the non-regulatory A4 isoform of spinach chloroplast glyceraldehyde-3-phosphate dehydrogenase complexed with NADP. J. Mol. Biol. 314, 527–542. doi: 10.1006/jmbi.2001.5172
Fermani, S., Sparla, F., Falini, G., Martelli, P. L., Casadio, R., Pupillo, P., et al. (2007). Molecular mechanism of thioredoxin regulation in photosynthetic A2B2-glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 104, 11109–11114. doi: 10.1073/pnas.0611636104
Fermani, S., Trivelli, X., Sparla, F., Thumiger, A., Calvaresi, M., Marri, L., et al. (2012). Conformational selection and folding-upon-binding of the intrinsically disordered protein CP12 regulate photosynthetic enzymes assembly. J. Biol. Chem. 287, 21372–21383. doi: 10.1074/jbc.M112.350355
Fersht, A. (1998). Structure and Mechanism in Protein Science: a Guide to Enzyme Catalysis and Protein Folding. New York: W. H. Freeman and Company.
Fode, B., Siemsen, T., Thurow, C., Weigel, R., and Gatz, C. (2008). The Arabidopsis GRAS protein SCL14 interacts with class II TGA transcription factors and is essential for the activation of stress-inducible promoters. Plant Cell 20, 3122–3135. doi: 10.1105/tpc.108.058974
Fortunato, A. E., Annunziata, R., Jaubert, M., Bouly, J. P., and Falciatore, A. (2015). Dealing with light: the widespread and multitasking cryptochrome/photolyase family in photosynthetic organisms. J. Plant Physiol. 172, 42–54. doi: 10.1016/j.jplph.2014.06.011
Fu, X., Richards, D. E., Ait-Ali, T., Hynes, L. W., Ougham, H., Peng, J., et al. (2002). Gibberellin-mediated proteasome-dependent degradation of the barley DELLA protein SLN1 repressor. Plant Cell 14, 3191–3200. doi: 10.1105/tpc.006197
Fuxreiter, M., Simon, I., Friedrich, P., and Tompa, P. (2004). Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 338, 1015–1026. doi: 10.1016/j.jmb.2004.03.017
Fuxreiter, M., and Tompa, P. (2012). Fuzzy complexes: a more stochastic view of protein function. Adv. Exp. Med. Biol. 725, 1–14. doi: 10.1007/978-1-4614-0659-4_1
Fuxreiter, M., Tompa, P., and Simon, I. (2007). Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23, 950–956. doi: 10.1093/bioinformatics/btm035
Gao, P., Yoo, S. H., Lee, K. J., Rosensweig, C., Takahashi, J. S., Chen, B. P., et al. (2013). Phosphorylation of the cryptochrome 1 C-terminal tail regulates circadian period length. J. Biol. Chem. 288, 35277–35286. doi: 10.1074/jbc.M113.509604
Gardebien, F., Thangudu, R. R., Gontero, B., and Offmann, B. (2006). Construction of a 3D model of CP12, a protein linker. J. Mol. Graph. Model. 25, 186–195. doi: 10.1016/j.jmgm.2005.12.003
Gavin, A. C., Aloy, P., Grandi, P., Krause, R., Boesche, M., Marzioch, M., et al. (2006). Proteome survey reveals modularity of the yeast cell machinery. Nature 440, 631–636. doi: 10.1038/nature04532
Gavin, A. C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., et al. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147. doi: 10.1038/415141a
Goloubinoff, P., Christeller, J. T., Gatenby, A. A., and Lorimer, G. H. (1989). Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfoleded state depends on two chaperonin proteins and Mg-ATP. Nature 342, 884–889. doi: 10.1038/342884a0
Gontero, B., and Maberly, S. C. (2012). An intrinsically disordered protein, CP12: jack of all trades and master of the Calvin cycle. Biochem. Soc. Trans. 40, 995–999. doi: 10.1042/BST20120097
Gontero, B., and Salvucci, M. E. (2014). Regulation of photosynthetic carbon metabolism in aquatic and terrestrial organisms by Rubisco activase, redox-modulation and CP12. Aquat. Bot. 118, 14–23. doi: 10.1016/j.aquabot.2014.05.011
Graciet, E., Gans, P., Wedel, N., Lebreton, S., Camadro, J. M., and Gontero, B. (2003a). The small protein CP12: a protein linker for supramolecular assembly. Biochemistry 42, 8163–8170. doi: 10.1021/bi034474x
Graciet, E., Lebreton, S., Camadro, J. M., and Gontero, B. (2003b). Characterization of native and recombinant A4 glyceraldehyde 3- phosphate dehydrogenase. Eur. J. Biochem. 270, 129–136. doi: 10.1046/j.1432-1033.2003.03372.x
Green, C. B. (2004). Cryptochromes: tail-ored for distinct functions. Curr. Biol. 14, R847–R849. doi: 10.1016/j.cub.2004.09.040
Groben, R., Kaloudas, D., Raines, C. A., Offmann, B., Maberly, S. C., and Gontero, B. (2010). Comparative sequence analysis of CP12, a small protein involved in the formation of a Calvin cycle complex in photosynthetic organisms. Photosynth. Res. 103, 183–194. doi: 10.1007/s11120-010-9542-z
Harada, Y., Sakai, M., Kurabayashi, N., Hirota, T., and Fukada, Y. (2005). Ser-557-phosphorylated mCRY2 is degraded upon synergistic phosphorylation by glycogen synthase kinase-3 beta. J. Biol. Chem. 280, 31714–31721. doi: 10.1074/jbc.M506225200
Hardtke, C. S., Gohda, K., Osterlund, M. T., Oyama, T., Okada, K., and Deng, X. W. (2000). HY5 stability and activity in Arabidopsis is regulated by phosphorylation in its COP1 binding domain. EMBO J. 19, 4997–5006. doi: 10.1093/emboj/19.18.4997
Haynes, C., Oldfield, C. J., Ji, F., Klitgord, N., Cusick, M. E., Radivojac, P., et al. (2006). Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2:e100. doi: 10.1371/journal.pcbi.0020100
Hazy, E., Bokor, M., Kalmar, L., Gelencser, A., Kamasa, P., Han, K. H., et al. (2011). Distinct hydration properties of wild-type and familial point mutant A53T of alpha-synuclein associated with Parkinson's disease. Biophys. J. 101, 2260–2266. doi: 10.1016/j.bpj.2011.08.052
Hazy, E., and Tompa, P. (2009). Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem 10, 1415–1419. doi: 10.1002/cphc.200900205
Heery, D. M., Kalkhoven, E., Hoare, S., and Parker, M. G. (1997). A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387, 733–736. doi: 10.1038/42750
Henderson, J. N., Kuriata, A. M., Fromme, R., Salvucci, M. E., and Wachter, R. M. (2011). Atomic resolution x-ray structure of the substrate recognition domain of higher plant ribulose-bisphosphate carboxylase/oxygenase (Rubisco) activase. J. Biol. Chem. 286, 35683–35688. doi: 10.1074/jbc.C111.289595
Hildebrandt, T., Knuesting, J., Berndt, C., Morgan, B., and Scheibe, R. (2015). Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 396, 523–537. doi: 10.1515/hsz-2014-0295
Hirano, K., Asano, K., Tsuji, H., Kawamura, M., Mori, H., Kitano, H., et al. (2010). Characterization of the molecular mechanism underlying gibberellin perception complex formation in rice. Plant Cell 22, 2680–2696. doi: 10.1105/tpc.110.075549
Hirsch, S., Kim, J., Munoz, A., Heckmann, A. B., Downie, J. A., and Oldroyd, G. E. (2009). GRAS proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula. Plant Cell 21, 545–557. doi: 10.1105/tpc.108.064501
Hirsch, S., and Oldroyd, G. E. (2009). GRAS-domain transcription factors that regulate plant development. Plant Signal. Behav. 4, 698–700. doi: 10.4161/psb.4.8.9176
Hollenbeck, J. J., Mcclain, D. L., and Oakley, M. G. (2002). The role of helix stabilizing residues in GCN4 basic region folding and DNA binding. Protein Sci. 11, 2740–2747. doi: 10.1110/ps.0211102
Hou, X., Lee, L. Y., Xia, K., Yan, Y., and Yu, H. (2010). DELLAs modulate jasmonate signaling via competitive binding to JAZs. Dev. Cell 19, 884–894. doi: 10.1016/j.devcel.2010.10.024
Howard, T. P., Fryer, M. J., Singh, P., Metodiev, M., Lytovchenko, A., Obata, T., et al. (2011a). Antisense suppression of the small chloroplast protein CP12 in tobacco alters carbon partitioning and severely restricts growth. Plant Physiol. 157, 620–631. doi: 10.1104/pp.111.183806
Howard, T. P., Lloyd, J. C., and Raines, C. A. (2011b). Inter-species variation in the oligomeric states of the higher plant Calvin cycle enzymes glyceraldehyde-3-phosphate dehydrogenase and phosphoribulokinase. J. Exp. Bot. 62, 3799–3805. doi: 10.1093/jxb/err057
Howard, T. P., Upton, G. J., Lloyd, J. C., and Raines, C. A. (2011c). Antisense suppression of the small chloroplast protein CP12 in tobacco: a transcriptional viewpoint. Plant Signal. Behav. 6, 2026–2030. doi: 10.4161/psb.6.12.18055
Hussain, A., Cao, D., Cheng, H., Wen, Z., and Peng, J. (2005). Identification of the conserved serine/threonine residues important for gibberellin-sensitivity of Arabidopsis RGL2 protein. Plant J. 44, 88–99. doi: 10.1111/j.1365-313X.2005.02512.x
Hussain, A., Cao, D., and Peng, J. (2007). Identification of conserved tyrosine residues important for gibberellin sensitivity of Arabidopsis RGL2 protein. Planta 226, 475–483. doi: 10.1007/s00425-007-0497-z
Iakoucheva, L. M., Radivojac, P., Brown, C. J., O'connor, T. R., Sikes, J. G., Obradovic, Z., et al. (2004). The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 32, 1037–1049. doi: 10.1093/nar/gkh253
Itoh, H., Sasaki, A., Ueguchi-Tanaka, M., Ishiyama, K., Kobayashi, M., Hasegawa, Y., et al. (2005). Dissection of the phosphorylation of rice DELLA protein, SLENDER RICE1. Plant Cell Physiol. 46, 1392–1399. doi: 10.1093/pcp/pci152
Jeffery, C. J. (1999). Moonlighting proteins. Trends Biochem. Sci. 24, 8–11. doi: 10.1016/S0968-0004(98)01335-8
Jensen, M. K., Kjaersgaard, T., Nielsen, M. M., Galberg, P., Petersen, K., O'shea, C., et al. (2010a). The Arabidopsis thaliana NAC transcription factor family: structure-function relationships and determinants of ANAC019 stress signalling. Biochem. J. 426, 183–196. doi: 10.1042/BJ20091234
Jensen, M. K., Kjaersgaard, T., Petersen, K., and Skriver, K. (2010b). NAC genes: time-specific regulators of hormonal signaling in Arabidopsis. Plant Signal. Behav. 5, 907–910. doi: 10.4161/psb.5.7.12099
Kaaki, W., Woudstra, M., Gontero, B., and Halgand, F. (2013). Exploration of CP12 conformational changes and of quaternary structural properties using electrospray ionization traveling wave ion mobility mass spectrometry. Rapid. Commun. Mass Spectrom. 27, 179–186. doi: 10.1002/rcm.6442
Karagoz, G. E., and Rudiger, S. G. (2015). Hsp90 interaction with clients. Trends Biochem. Sci. 40, 117–125. doi: 10.1016/j.tibs.2014.12.002
Kirschner, K., and Bisswanger, H. (1976). Multifunctional proteins. Annu. Rev. Biochem. 45, 143–166. doi: 10.1146/annurev.bi.45.070176.001043
Kjaersgaard, T., Jensen, M. K., Christiansen, M. W., Gregersen, P., Kragelund, B. B., and Skriver, K. (2011). Senescence-associated barley NAC (NAM, ATAF1,2, CUC) transcription factor interacts with radical-induced cell death 1 through a disordered regulatory domain. J. Biol. Chem. 286, 35418–35429. doi: 10.1074/jbc.M111.247221
Ko, J. H., Yang, S. H., Park, A. H., Lerouxel, O., and Han, K. H. (2007). ANAC012, a member of the plant-specific NAC transcription factor family, negatively regulates xylary fiber development in Arabidopsis thaliana. Plant J. 50, 1035–1048. doi: 10.1111/j.1365-313X.2007.03109.x
Koshland, D. E. Jr., Nemethy, G., and Filmer, D. (1966). Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5, 365–385. doi: 10.1021/bi00865a047
Kovacech, B., and Novak, M. (2010). Tau truncation is a productive posttranslational modification of neurofibrillary degeneration in Alzheimer's disease. Curr. Alzheimer Res. 7, 708–716. doi: 10.2174/156720510793611556
Kragelund, B. B., Jensen, M. K., and Skriver, K. (2012). Order by disorder in plant signaling. Trends Plant Sci. 17, 625–632. doi: 10.1016/j.tplants.2012.06.010
Krisko, A., Smole, Z., Debret, G., Nikolic, N., and Radman, M. (2010). Unstructured hydrophilic sequences in prokaryotic proteomes correlate with dehydration tolerance and host association. J. Mol. Biol. 402, 775–782. doi: 10.1016/j.jmb.2010.08.012
Lebreton, S., Gontero, B., Avilan, L., and Ricard, J. (1997). Memory and imprinting effects in multienzyme complexes–II. Kinetics of the bienzyme complex from Chlamydomonas reinhardtii and hysteretic activation of chloroplast oxidized phosphoribulokinase. Eur. J. Biochem. 246, 85–91. doi: 10.1111/j.1432-1033.1997.t01-2-00085.x
Li, A. D., and Anderson, L. E. (1997). Expression and characterization of pea chloroplastic glyceraldehyde-3-phosphate dehydrogenase composed of only the B-subunit. Plant Physiol. 115, 1201–1209. doi: 10.1104/pp.115.3.1201
Li, C., Wang, D., and Portis, A. R. Jr. (2006). Identification of critical arginine residues in the functioning of Rubisco activase. Arch. Biochem. Biophys. 450, 176–182. doi: 10.1016/j.abb.2006.04.002
Li, L. A., Zianni, M. R., and Tabita, F. R. (1999). Inactivation of the monocistronic rca gene in Anabaena variabilis suggests a physiological ribulose bisphosphate carboxylase/oxygenase activase-like function in heterocystous cyanobacteria. Plant Mol. Biol. 40, 467–478. doi: 10.1023/A:1006251808625
Libich, D. S., Fawzi, N. L., Ying, J., and Clore, G. M. (2013). Probing the transient dark state of substrate binding to GroEL by relaxation-based solution NMR. Proc. Natl. Acad. Sci. U.S.A. 110, 11361–11366. doi: 10.1073/pnas.1305715110
Lieutaud, P., Canard, B., and Longhi, S. (2008). MeDor: a metaserver for predicting protein disorder. BMC Genomics 9(Suppl. 2):S25. doi: 10.1186/1471-2164-9-S2-S25
Lim, J., Jung, J. W., Lim, C. E., Lee, M. H., Kim, B. J., Kim, M., et al. (2005). Conservation and diversification of SCARECROW in maize. Plant Mol. Biol. 59, 619–630. doi: 10.1007/s11103-005-0578-y
Lin, C., and Shalitin, D. (2003). Cryptochrome structure and signal transduction. Annu. Rev. Plant Biol. 54, 469–496. doi: 10.1146/annurev.arplant.54.110901.160901
Liu, B., Zuo, Z., Liu, H., Liu, X., and Lin, C. (2011a). Arabidopsis cryptochrome 1 interacts with SPA1 to suppress COP1 activity in response to blue light. Genes Dev. 25, 1029–1034. doi: 10.1101/gad.2025011
Liu, H., Liu, B., Zhao, C., Pepper, M., and Lin, C. (2011b). The action mechanisms of plant cryptochromes. Trends Plant Sci. 16, 684–691. doi: 10.1016/j.tplants.2011.09.002
Liu, Z., Taub, C. C., and Mcclung, C. R. (1996). Identification of an Arabidopsis thaliana ribulose-1,5-bisphosphate carboxylase/oxygenase activase (RCA) minimal promoter regulated by light and the circadian clock. Plant Physiol. 112, 43–51. doi: 10.1104/pp.112.1.43
Lopez-Calcagno, P. E., Howard, T. P., and Raines, C. A. (2014). The CP12 protein family: a thioredoxin-mediated metabolic switch? Front. Plant. Sci. 5:9. doi: 10.3389/fpls.2014.00009
Lorimer, G. H., Badger, M. R., and Andrews, T. J. (1976). The activation of ribulose-1,5-bisphosphate carboxylase by carbon dioxide and magnesium ions. Equilibria, kinetics, a suggested mechanism, and physiological implications. Biochemistry 15, 529–536. doi: 10.1021/bi00648a012
Losh, J. L., Young, J. N., and Morel, F. M. (2013). Rubisco is a small fraction of total protein in marine phytoplankton. New Phytol. 198, 52–58. doi: 10.1111/nph.12143
Mächler, F., and Nösberger, J. (1980). Regulation of ribulose bisphosphate carboxylase activity in intact wheat leaves by light, CO2, and temperature. J. Exp. Bot. 31, 1485–1491. doi: 10.1093/jxb/31.6.1485
Marri, L., Sparla, F., Pupillo, P., and Trost, P. (2005). Co-ordinated gene expression of photosynthetic glyceraldehyde-3-phosphate dehydrogenase, phosphoribulokinase, and CP12 in Arabidopsis thaliana. J. Exp. Bot. 56, 73–80. doi: 10.1093/jxb/eri020
Marri, L., Trost, P., Trivelli, X., Gonnelli, L., Pupillo, P., and Sparla, F. (2008). Spontaneous assembly of photosynthetic supramolecular complexes as mediated by the intrinsically unstructured protein CP12. J. Biol. Chem. 283, 1831–1838. doi: 10.1074/jbc.M705650200
Marri, L., Zaffagnini, M., Collin, V., Issakidis-Bourguet, E., Lemaire, S. D., Pupillo, P., et al. (2009). Prompt and easy activation by specific thioredoxins of Calvin cycle enzymes of Arabidopsis thaliana associated in the GAPDH/CP12/PRK supramolecular complex. Mol. Plant 2, 259–269. doi: 10.1093/mp/ssn061
Marsh, J. A., and Teichmann, S. A. (2010). How do proteins gain new domains? Genome Biol. 11, 126. doi: 10.1186/gb-2010-11-7-126
Martino-Catt, S., and Ort, D. R. (1992). Low temperature interrupts circadian regulation of transcriptional activity in chilling-sensitive plants. Proc. Natl. Acad. Sci. U.S.A. 89, 3731–3735. doi: 10.1073/pnas.89.9.3731
Matsumura, H., Kai, A., Maeda, T., Tamoi, M., Satoh, A., Tamura, H., et al. (2011). Structure basis for the regulation of glyceraldehyde-3-phosphate dehydrogenase activity via the intrinsically disordered protein CP12. Structure 19, 1846–1854. doi: 10.1016/j.str.2011.08.016
Meszaros, B., Tompa, P., Simon, I., and Dosztanyi, Z. (2007). Molecular principles of the interactions of disordered proteins. J. Mol. Biol. 372, 549–561. doi: 10.1016/j.jmb.2007.07.004
Mileo, E., Lorenzi, M., Erales, J., Lignon, S., Puppo, C., Le Breton, N., et al. (2013). Dynamics of the intrinsically disordered protein CP12 in its association with GAPDH in the green alga Chlamydomonas reinhardtii: a fuzzy complex. Mol. Biosyst. 9, 2869–2876. doi: 10.1039/c3mb70190e
Mittag, T., Kay, L. E., and Forman-Kay, J. D. (2010). Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 23, 105–116. doi: 10.1002/jmr.961
Mizianty, M. J., Uversky, V., and Kurgan, L. (2014). Prediction of intrinsic disorder in proteins using MFDp2. Methods Mol. Biol. 1137, 147–162. doi: 10.1007/978-1-4939-0366-5_11
Mohan, A., Oldfield, C. J., Radivojac, P., Vacic, V., Cortese, M. S., Dunker, A. K., et al. (2006). Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 362, 1043–1059. doi: 10.1016/j.jmb.2006.07.087
Moparthi, S. B., Thieulin-Pardo, G., De Torres, J., Ghenuche, P., Gontero, B., and Wenger, J. (2015). FRET analysis of CP12 structural interplay by GAPDH and PRK. Biochem. Biophys. Res. Commun. doi: 10.1016/j.bbrc.2015.01.135
Moparthi, S. B., Thieulin-Pardo, G., Mansuelle, P., Rigneault, H., Gontero, B., and Wenger, J. (2014). Conformational modulation and hydrodynamic radii of CP12 protein and its complexes probed by fluorescence correlation spectroscopy. FEBS J. 281, 3206–3217. doi: 10.1111/febs.12854
Moreau, V. H., Da Silva, A. C., Siloto, R. M., Valente, A. P., Leite, A., and Almeida, F. C. (2004). The bZIP region of the plant transcription factor opaque-2 forms stable homodimers in solution and retains its helical structure upon subunit dissociation. Biochemistry 43, 4862–4868. doi: 10.1021/bi035905e
Mueller-Cajar, O., Stotz, M., and Bracher, A. (2014). Maintaining photosynthetic CO2 fixation via protein remodelling: the Rubisco activases. Photosyn. Res. 119, 191–201. doi: 10.1007/s11120-013-9819-0
Mueller-Cajar, O., Stotz, M., Wendler, P., Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Structure and function of the AAA+ protein CbbX, a red-type Rubisco activase. Nature 479, 194–199. doi: 10.1038/nature10568
Munoz-Bertomeu, J., Cascales-Minana, B., Alaiz, M., Segura, J., and Ros, R. (2010). A critical role of plastidial glycolytic glyceraldehyde-3-phosphate dehydrogenase in the control of plant metabolism and development. Plant Signal. Behav. 5, 67–69. doi: 10.4161/psb.5.1.10200
Murase, K., Hirano, Y., Sun, T. P., and Hakoshima, T. (2008). Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 456, 459–463. doi: 10.1038/nature07519
Neuwald, A. F., Aravind, L., Spouge, J. L., and Koonin, E. V. (1999). AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43.
Oldfield, C. J., Cheng, Y., Cortese, M. S., Romero, P., Uversky, V. N., and Dunker, A. K. (2005). Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 44, 12454–12470. doi: 10.1021/bi050736e
Olsen, A. N., Ernst, H. A., Leggio, L. L., and Skriver, K. (2005). NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 10, 79–87. doi: 10.1016/j.tplants.2004.12.010
Ooka, H., Satoh, K., Doi, K., Nagata, T., Otomo, Y., Murakami, K., et al. (2003). Comprehensive analysis of NAC family genes in Oryza sativa and Arabidopsis thaliana. DNA Res. 10, 239–247. doi: 10.1093/dnares/10.6.239
Ozturk, N., Song, S. H., Ozgur, S., Selby, C. P., Morrison, L., Partch, C., et al. (2007). Structure and function of animal cryptochromes. Cold Spring Harb. Symp. Quant. Biol. 72, 119–131. doi: 10.1101/sqb.2007.72.015
Pancsa, R., and Tompa, P. (2012). Structural disorder in eukaryotes. PLoS ONE 7:e34687. doi: 10.1371/journal.pone.0034687
Panda, A., and Ghosh, T. C. (2014). Prevalent structural disorder carries signature of prokaryotic adaptation to oxic atmosphere. Gene 548, 134–141. doi: 10.1016/j.gene.2014.07.002
Partch, C. L., Clarkson, M. W., Ozgur, S., Lee, A. L., and Sancar, A. (2005). Role of structural plasticity in signal transduction by the cryptochrome blue-light photoreceptor. Biochemistry 44, 3795–3805. doi: 10.1021/bi047545g
Patil, A., and Nakamura, H. (2006). Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 580, 2041–2045. doi: 10.1016/j.febslet.2006.03.003
Pearce, F. G. (2006). Catalytic by-product formation and ligand binding by ribulose bisphosphate carboxylases from different phylogenies. Biochem. J. 399, 525–534. doi: 10.1042/BJ20060430
Perchorowicz, J. T., Raynes, D. A., and Jensen, R. G. (1981). Light limitation of photosynthesis and activation of ribulose bisphosphate carboxylase in wheat seedlings. Proc. Natl. Acad. Sci. U.S.A. 78, 2985–2989. doi: 10.1073/pnas.78.5.2985
Peschel, N., Chen, K. F., Szabo, G., and Stanewsky, R. (2009). Light-dependent interactions between the Drosophila circadian clock factors cryptochrome, jetlag, and timeless. Curr. Biol. 19, 241–247. doi: 10.1016/j.cub.2008.12.042
Petersen, J., Brinkmann, H., and Cerff, R. (2003). Origin, evolution, and metabolic role of a novel glycolytic GAPDH enzyme recruited by land plant plastids. J. Mol. Evol. 57, 16–26. doi: 10.1007/s00239-002-2441-y
Petersen, J., Teich, R., Becker, B., Cerff, R., and Brinkmann, H. (2006). The GapA/B gene duplication marks the origin of Streptophyta (charophytes and land plants). Mol. Biol. Evol. 23, 1109–1118. doi: 10.1093/molbev/msj123
Pietrosemoli, N., Garcia-Martin, J. A., Solano, R., and Pazos, F. (2013). Genome-wide analysis of protein disorder in Arabidopsis thaliana: implications for plant environmental adaptation. PLoS ONE 8:e55524. doi: 10.1371/journal.pone.0055524
Podust, L. M., Krezel, A. M., and Kim, Y. (2001). Crystal structure of the CCAAT box/enhancer-binding protein beta activating transcription factor-4 basic leucine zipper heterodimer in the absence of DNA. J. Biol. Chem. 276, 505–513. doi: 10.1074/jbc.M005594200
Pohlmeyer, K., Paap, B. K., Soll, J., and Wedel, N. (1996). CP12: a small nuclear-encoded chloroplast protein provides novel insights into higher-plant GAPDH evolution. Plant Mol. Biol. 32, 969–978. doi: 10.1007/BF00020493
Portis, A. R., Jr. (2003). Rubisco activase - Rubisco's catalytic chaperone. Photosyn. Res. 75, 11–27. doi: 10.1023/A:1022458108678
Portis, A. R. Jr., Li, C., Wang, D., and Salvucci, M. E. (2008). Regulation of Rubisco activase and its interaction with Rubisco. J. Exp. Bot. 59, 1597–1604. doi: 10.1093/jxb/erm240
Portis, A. R., Salvucci, M. E., and Ogren, W. L. (1986). Activation of ribulosebisphosphate carboxylase/oxygenase at physiological CO2 and ribulosebisphosphate concentrations by Rubisco activase. Plant Physiol. 82, 967–971. doi: 10.1104/pp.82.4.967
Pupillo, P., and Giuliani Piccari, G. (1975). The reversible depolymerization of spinach chloroplast glyceraldehyde-phosphate dehydrogenase. Interaction with nucleotides and dithiothreitol. Eur. J. Biochem. 51, 475–482. doi: 10.1111/j.1432-1033.1975.tb03947.x
Pupillo, P., and Piccari, G. G. (1973). The effect of NADP on the subunit structure and activity of spinach chloroplast glyceraldehyde-3-phosphate dehydrogenase. Arch. Biochem. Biophys. 154, 324–331. doi: 10.1016/0003-9861(73)90064-7
Qian, J., and Rodermel, S. R. (1993). Ribulose-1,5-bisphosphate carboxylase/oxygenase activase cDNAs from Nicotiana tabacum. Plant Physiol. 102, 683–684. doi: 10.1104/pp.102.2.683
Raven, J. A. (2013). Rubisco: still the most abundant protein of Earth? New Phytol. 198, 1–3. doi: 10.1111/nph.12197
Robbens, S., Petersen, J., Brinkmann, H., Rouze, P., and Van De Peer, Y. (2007). Unique regulation of the Calvin cycle in the ultrasmall green alga Ostreococcus. J. Mol. Evol. 64, 601–604. doi: 10.1007/s00239-006-0159-y
Romero, P., Obradovic, Z., Kissinger, C. R., Villafranca, J. E., Garner, E., Guilliot, S., et al. (1998). Thousands of proteins likely to have long disordered regions. Pac. Symp. Biocomput. 3, 437–448. Available online at: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.132.4594
Rundle, S. J., and Zielinski, R. E. (1991). Organization and expression of two tandemly oriented genes encoding ribulosebisphosphate carboxylase/oxygenase activase in barley. J. Biol. Chem. 266, 4677–4685.
Rushton, P. J., Bokowiec, M. T., Han, S., Zhang, H., Brannock, J. F., Chen, X., et al. (2008). Tobacco transcription factors: novel insights into transcriptional regulation in the Solanaceae. Plant Physiol. 147, 280–295. doi: 10.1104/pp.107.114041
Saha, T., Kar, R. K., and Sa, G. (2014). Structural and sequential context of p53: a review of experimental and theoretical evidence. Prog. Biophys. Mol. Biol. 117, 250–263. doi: 10.1016/j.pbiomolbio.2014.12.002
Salminen, A., Ojala, J., Kaarniranta, K., Hiltunen, M., and Soininen, H. (2011). Hsp90 regulates tau pathology through co-chaperone complexes in Alzheimer's disease. Prog. Neurobiol. 93, 99–110. doi: 10.1016/j.pneurobio.2010.10.006
Salvucci, M. E. (2004). Potential for interactions between the carboxy- and amino-termini of Rubisco activase subunits. FEBS Lett. 560, 205–209. doi: 10.1016/S0014-5793(04)00111-5
Salvucci, M. E., Rajagopalan, K., Sievert, G., Haley, B. E., and Watt, D. S. (1993). Photoaffinity labeling of ribulose-1,5-bisphosphate carboxylase/oxygenase activase with ATP gamma-benzophenone. Identification of the ATP gamma-phosphate binding domain. J. Biol. Chem. 268, 14239–14244.
Salvucci, M. E., Van De Loo, F. J., and Stecher, D. (2003). Two isoforms of Rubisco activase in cotton, the products of separate genes not alternative splicing. Planta 216, 736–744. doi: 10.1007/s00425-002-0923-1
Sancar, A. (2004). Photolyase and cryptochrome blue-light photoreceptors. Adv. Protein Chem. 69, 73–100. doi: 10.1016/S0065-3233(04)69003-6
Sanchez, C., Vielba, J. M., Ferro, E., Covelo, G., Sole, A., Abarca, D., et al. (2007). Two SCARECROW-LIKE genes are induced in response to exogenous auxin in rooting-competent cuttings of distantly related forest species. Tree Physiol. 27, 1459–1470. doi: 10.1093/treephys/27.10.1459
Scagliarini, S., Trost, P., and Pupillo, P. (1998). The non-regulatory isoform of NADP(H)-glyceraldehyde 3-phosphate dehydrogenase from spinach chloroplasts. J. Exp. Bot. 49, 1307–1315. doi: 10.1093/jxb/49.325.1307
Scheibe, R., Wedel, N., Vetter, S., Emmerlich, V., and Sauermann, S. M. (2002). Co-existence of two regulatory NADP-glyceraldehyde 3-P dehydrogenase complexes in higher plant chloroplasts. Eur. J. Biochem. 269, 5617–5624. doi: 10.1046/j.1432-1033.2002.03269.x
Sedzik, J., and Kirschner, D. A. (1992). Is myelin basic protein crystallizable? Neurochem. Res. 17, 157–166. doi: 10.1007/BF00966794
Seo, P. J., Kim, M. J., Song, J. S., Kim, Y. S., Kim, H. J., and Park, C. M. (2010). Proteolytic processing of an Arabidopsis membrane-bound NAC transcription factor is triggered by cold-induced changes in membrane fluidity. Biochem. J. 427, 359–367. doi: 10.1042/BJ20091762
Seo, P. J., and Park, C. M. (2010). A membrane-bound NAC transcription factor as an integrator of biotic and abiotic stress signals. Plant Signal. Behav. 5, 481–483. doi: 10.4161/psb.11083
Shen, J. B., and Ogren, W. L. (1992). Alteration of spinach ribulose-1,5-bisphosphate carboxylase/oxygenase activase activities by site-directed mutagenesis. Plant Physiol. 99, 1201–1207. doi: 10.1104/pp.99.3.1201
Singh, P., Kaloudas, D., and Raines, C. A. (2008). Expression analysis of the Arabidopsis CP12 gene family suggests novel roles for these proteins in roots and floral tissues. J. Exp. Bot. 59, 3975–3985. doi: 10.1093/jxb/ern236
Sirover, M. A. (1999). New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim. Biophys. Acta 1432, 159–184. doi: 10.1016/S0167-4838(99)00119-3
Sirover, M. A. (2011). On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: biochemical mechanisms and regulatory control. Biochim. Biophys. Acta 1810, 741–751. doi: 10.1016/j.bbagen.2011.05.010
Sparla, F., Fermani, S., Falini, G., Zaffagnini, M., Ripamonti, A., Sabatino, P., et al. (2004). Coenzyme site-directed mutants of photosynthetic A4-GAPDH show selectively reduced NADPH-dependent catalysis, similar to regulatory A2B2-GAPDH inhibited by oxidized thioredoxin. J. Mol. Biol. 340, 1025–1037. doi: 10.1016/j.jmb.2004.06.005
Sparla, F., Pupillo, P., and Trost, P. (2002). The C-terminal extension of glyceraldehyde-3-phosphate dehydrogenase subunit B acts as an autoinhibitory domain regulated by thioredoxins and nicotinamide adenine dinucleotide. J. Biol. Chem. 277, 44946–44952. doi: 10.1074/jbc.M206873200
Sparla, F., Zaffagnini, M., Wedel, N., Scheibe, R., Pupillo, P., and Trost, P. (2005). Regulation of photosynthetic GAPDH dissected by mutants. Plant Physiol. 138, 2210–2219. doi: 10.1104/pp.105.062117
Srivastava, R., Deng, Y., and Howell, S. H. (2014). Stress sensing in plants by an ER stress sensor/transducer, bZIP28. Front. Plant Sci. 5:59. doi: 10.3389/fpls.2014.00059
Srivastava, R., Deng, Y., Shah, S., Rao, A. G., and Howell, S. H. (2013). BINDING PROTEIN is a master regulator of the endoplasmic reticulum stress sensor/transducer bZIP28 in Arabidopsis. Plant Cell 25, 1416–1429. doi: 10.1105/tpc.113.110684
Stanley, D. N., Raines, C. A., and Kerfeld, C. A. (2013). Comparative analysis of 126 cyanobacterial genomes reveals evidence of functional diversity among homologs of the redox-regulated CP12 protein. Plant Physiol. 161, 824–835. doi: 10.1104/pp.112.210542
Steiner, E., Efroni, I., Gopalraj, M., Saathoff, K., Tseng, T. S., Kieffer, M., et al. (2012). The Arabidopsis O-linked N-acetylglucosamine transferase SPINDLY interacts with class I TCPs to facilitate cytokinin responses in leaves and flowers. Plant Cell 24, 96–108. doi: 10.1105/tpc.111.093518
Stotz, M., Mueller-Cajar, O., Ciniawsky, S., Wendler, P., Hartl, F. U., Bracher, A., et al. (2011). Structure of green-type Rubisco activase from tobacco. Nat. Struct. Mol. Biol. 18, 1366–1370. doi: 10.1038/nsmb.2171
Sun, X., Jones, W. T., Harvey, D., Edwards, P. J., Pascal, S. M., Kirk, C., et al. (2010). N-terminal domains of DELLA proteins are intrinsically unstructured in the absence of interaction with GID1/gibberellic acid receptors. J. Biol. Chem. 285, 11557–11571. doi: 10.1074/jbc.M109.027011
Sun, X., Jones, W. T., and Rikkerink, E. H. (2012). GRAS proteins: the versatile roles of intrinsically disordered proteins in plant signalling. Biochem. J. 442, 1–12. doi: 10.1042/BJ20111766
Sun, X., Rikkerink, E. H., Jones, W. T., and Uversky, V. N. (2013). Multifarious roles of intrinsic disorder in proteins illustrate its broad impact on plant biology. Plant Cell 25, 38–55. doi: 10.1105/tpc.112.106062
Sun, X., Xue, B., Jones, W. T., Rikkerink, E., Dunker, A. K., and Uversky, V. N. (2011). A functionally required unfoldome from the plant kingdom: intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol. Biol. 77, 205–223. doi: 10.1007/s11103-011-9803-z
Taoka, K., Yanagimoto, Y., Daimon, Y., Hibara, K., Aida, M., and Tasaka, M. (2004). The NAC domain mediates functional specificity of CUP-SHAPED COTYLEDON proteins. Plant J. 40, 462–473. doi: 10.1111/j.1365-313X.2004.02238.x
Tian, C., Wan, P., Sun, S., Li, J., and Chen, M. (2004). Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol. Biol. 54, 519–532. doi: 10.1023/B:PLAN.0000038256.89809.57
To, K. Y., Suen, D. F., and Chen, S. C. (1999). Molecular characterization of ribulose-1,5-bisphosphate carboxylase/oxygenase activase in rice leaves. Planta 209, 66–76. doi: 10.1007/s004250050607
Tompa, P. (2002). Intrinsically unstructured proteins. Trends Biochem. Sci. 27, 527–533. doi: 10.1016/S0968-0004(02)02169-2
Tompa, P. (2009). Structural disorder in amyloid fibrils: its implication in dynamic interactions of proteins. FEBS J. 276, 5406–5415. doi: 10.1111/j.1742-4658.2009.07250.x
Tompa, P., and Fuxreiter, M. (2008). Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 33, 2–8. doi: 10.1016/j.tibs.2007.10.003
Triezenberg, S. J. (1995). Structure and function of transcriptional activation domains. Curr. Opin. Genet. Dev. 5, 190–196. doi: 10.1016/0959-437X(95)80007-7
Trost, P., Fermani, S., Marri, L., Zaffagnini, M., Falini, G., Scagliarini, S., et al. (2006). Thioredoxin-dependent regulation of photosynthetic glyceraldehyde-3-phosphate dehydrogenase: autonomous vs. CP12-dependent mechanisms. Photosyn. Res. 89, 1–13. doi: 10.1007/s11120-006-9099-z
Trost, P., and Pupillo, P. (1993). Inhibition of spinach D-glyceraldehyde 3-phosphate: NADP+ oxidoreductase (nonphosphorylating) by adenylate compounds: the effect of dead-end inhibitors on a steady state random reaction mechanism. Arch. Biochem. Biophys. 306, 76–82. doi: 10.1006/abbi.1993.1483
Uversky, V. N. (2002). What does it mean to be natively unfolded? Eur. J. Biochem. 269, 2–12. doi: 10.1046/j.0014-2956.2001.02649.x
Uversky, V. N. (2009). Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 14, 5188–5238. doi: 10.2741/3594
Uversky, V. N. (2010). The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010:568068. doi: 10.1155/2010/568068
Uversky, V. N. (2011). Multitude of binding modes attainable by intrinsically disordered proteins: a portrait gallery of disorder-based complexes. Chem. Soc. Rev. 40, 1623–1634. doi: 10.1039/C0CS00057D
Uversky, V. N. (2013). The most important thing is the tail: multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 587, 1891–1901. doi: 10.1016/j.febslet.2013.04.042
Uversky, V. N., and Dunker, A. K. (2010). Understanding protein non-folding. Biochim. Biophys. Acta 1804, 1231–1264. doi: 10.1016/j.bbapap.2010.01.017
Uversky, V. N., Gillespie, J. R., and Fink, A. L. (2000). Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41, 415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7
Vacic, V., Oldfield, C. J., Mohan, A., Radivojac, P., Cortese, M. S., Uversky, V. N., et al. (2007). Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 6, 2351–2366. doi: 10.1021/pr0701411
Valsecchi, I., Guittard-Crilat, E., Maldiney, R., Habricot, Y., Lignon, S., Lebrun, R., et al. (2013). The intrinsically disordered C-terminal region of Arabidopsis thaliana TCP8 transcription factor acts both as a transactivation and self-assembly domain. Mol. Biosyst. 9, 2282–2295. doi: 10.1039/c3mb70128j
Van De Loo, F. J., and Salvucci, M. E. (1996). Activation of ribulose-1,5-biphosphate carboxylase/oxygenase (Rubisco) involves Rubisco activase Trp16. Biochemistry 35, 8143–8148. doi: 10.1021/bi9604901
Vinson, C. R., Hai, T., and Boyd, S. M. (1993). Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: prediction and rational design. Genes Dev. 7, 1047–1058. doi: 10.1101/gad.7.6.1047
Viola, I. L., Reinheimer, R., Ripoll, R., Manassero, N. G., and Gonzalez, D. H. (2012). Determinants of the DNA binding specificity of class I and class II TCP transcription factors. J. Biol. Chem. 287, 347–356. doi: 10.1074/jbc.M111.256271
Viola, I. L., Uberti Manassero, N. G., Ripoll, R., and Gonzalez, D. H. (2011). The Arabidopsis class I TCP transcription factor AtTCP11 is a developmental regulator with distinct DNA-binding properties due to the presence of a threonine residue at position 15 of the TCP domain. Biochem. J. 435, 143–155. doi: 10.1042/BJ20101019
Wang, D., and Portis, A. R. Jr. (2006). Increased sensitivity of oxidized large isoform of ribulose-1,5-bisphosphate carboxylase/oxygenase (rubisco) activase to ADP inhibition is due to an interaction between its carboxyl extension and nucleotide-binding pocket. J. Biol. Chem. 281, 25241–25249. doi: 10.1074/jbc.M604756200
Wang, H., Ma, L. G., Li, J. M., Zhao, H. Y., and Deng, X. W. (2001). Direct interaction of Arabidopsis cryptochromes with COP1 in light control development. Science 294, 154–158. doi: 10.1126/science.1063630
Watillon, B., Kettmann, R., Boxus, P., and Burny, A. (1993). Developmental and circadian pattern of rubisco activase mRNA accumulation in apple plants. Plant Mol. Biol. 23, 501–509. doi: 10.1007/BF00019298
Werneke, J. M., Chatfield, J. M., and Ogren, W. L. (1988). Catalysis of ribulosebisphosphate carboxylase/oxygenase activation by the product of a Rubisco activase cDNA clone expressed in Escherichia coli. Plant Physiol. 87, 917–920. doi: 10.1104/pp.87.4.917
Werneke, J. M., Chatfield, J. M., and Ogren, W. L. (1989). Alternative mRNA splicing generates the two ribulosebisphosphate carboxylase/oxygenase activase polypeptides in spinach and Arabidopsis. Plant Cell 1, 815–825. doi: 10.1105/tpc.1.8.815
Wolosiuk, R. A., and Buchanan, B. B. (1976). Studies on the regulation of chloroplast NADP-linked glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 251, 6456–6461.
Wolosiuk, R. A., and Buchanan, B. B. (1978). Regulation of chloroplast phosphoribulokinase by the ferredoxin/thioredoxin system. Arch. Biochem. Biophys. 189, 97–101. doi: 10.1016/0003-9861(78)90119-4
Wright, P. E., and Dyson, H. J. (1999). Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331. doi: 10.1006/jmbi.1999.3110
Xue, B., Ganti, K., Rabionet, A., Banks, L., and Uversky, V. N. (2014). Disordered interactome of human papillomavirus. Curr. Pharm. Des. 20, 1274–1292. doi: 10.2174/13816128113199990072
Yang, H. Q., Tang, R. H., and Cashmore, A. R. (2001). The signaling mechanism of Arabidopsis CRY1 involves direct interaction with COP1. Plant Cell 13, 2573–2587. doi: 10.1105/tpc.13.12.2573
Yang, H. Q., Wu, Y. J., Tang, R. H., Liu, D., Liu, Y., and Cashmore, A. R. (2000). The C-termini of Arabidopsis cryptochromes mediate a constitutive light response. Cell 103, 815–827. doi: 10.1016/S0092-8674(00)00184-7
Yin, Z., Meng, F., Song, H., Wang, X., Xu, X., and Yu, D. (2010). Expression quantitative trait loci analysis of two genes encoding rubisco activase in soybean. Plant Physiol. 152, 1625–1637. doi: 10.1104/pp.109.148312
Yoon, M. K., Shin, J., Choi, G., and Choi, B. S. (2006). Intrinsically unstructured N-terminal domain of bZIP transcription factor HY5. Proteins 65, 856–866. doi: 10.1002/prot.21089
Yruela, I., and Contreras-Moreira, B. (2012). Protein disorder in plants: a view from the chloroplast. BMC Plant Biol. 12:165. doi: 10.1186/1471-2229-12-165
Yruela, I., and Contreras-Moreira, B. (2013). Genetic recombination is associated with intrinsic disorder in plant proteomes. BMC Genomics 14:772. doi: 10.1186/1471-2164-14-772
Yu, X., Liu, H., Klejnot, J., and Lin, C. (2010). The cryptochrome blue light receptors. Arabidopsis Book 8:e0135. doi: 10.1199/tab.0135
Yu, X., Shalitin, D., Liu, X., Maymon, M., Klejnot, J., Yang, H., et al. (2007). Derepression of the NC80 motif is critical for the photoactivation of Arabidopsis CRY2. Proc. Natl. Acad. Sci. U.S.A. 104, 7289–7294. doi: 10.1073/pnas.0701912104
Zarzycki, J., Axen, S. D., Kinney, J. N., and Kerfeld, C. A. (2013). Cyanobacterial-based approaches to improving photosynthesis in plants. J. Exp. Bot. 64, 787–798. doi: 10.1093/jxb/ers294
Zhang, N., Kallis, R. P., Ewy, R. G., and Portis, A. R. Jr. (2002). Light modulation of Rubisco in Arabidopsis requires a capacity for redox regulation of the larger Rubisco activase isoform. Proc. Natl. Acad. Sci. U.S.A. 99, 3330–3334. doi: 10.1073/pnas.042529999
Zhang, N., and Portis, A. R. Jr. (1999). Mechanism of light regulation of Rubisco: a specific role for the larger Rubisco activase isoform involving reductive activation by thioredoxin-f. Proc. Natl. Acad. Sci. U.S.A. 96, 9438–9443. doi: 10.1073/pnas.96.16.9438
Zhang, N., Schürmann, P., and Portis, A. Jr. (2001). Characterization of the regulatory function of the 46-kDa isoform of Rubisco activase from Arabidopsis. Photosyn. Res. 68, 29–37. doi: 10.1023/A:1011845506196
Zoltowski, B. D., Vaidya, A. T., Top, D., Widom, J., Young, M. W., and Crane, B. R. (2011). Structure of full-length Drosophila cryptochrome. Nature 480, 396–399. doi: 10.1038/nature10618
Keywords: intrinsically disordered proteins, GAPDH, CP12, Rubisco activase, cryptochromes, transcription factors
Citation: Thieulin-Pardo G, Avilan L, Kojadinovic M and Gontero B (2015) Fairy “tails”: flexibility and function of intrinsically disordered extensions in the photosynthetic world. Front. Mol. Biosci. 2:23. doi: 10.3389/fmolb.2015.00023
Received: 25 February 2015; Accepted: 04 May 2015;
Published: 19 May 2015.
Edited by:
Peter Tompa, Flanders Institute of Biotechnology (VIB), BelgiumReviewed by:
Anastassios Papageorgiou, University of Turku, FinlandEsko Oksanen, European Spallation Source, Sweden
Copyright © 2015 Thieulin-Pardo, Avilan, Kojadinovic and Gontero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brigitte Gontero, Centre National de la Recherche Scientifique-BIP, 31 Chemin Joseph Aiguier, 13 402 Marseille, France,Ym1ldW5pZXJAaW1tLmNucnMuZnI=