 Patrícia Shigunov*
Patrícia Shigunov* Bruno Dallagiovanna*
Bruno Dallagiovanna*- Stem Cells Basic Biology Laboratory, Carlos Chagas Institute, Oswaldo Cruz Foundation, Curitiba, Brazil
Stem cells are undifferentiated cells with the ability to self-renew and the potential to differentiate into all body cell types. Stem cells follow a developmental genetic program and are able to respond to alterations in the environment through various signaling pathways. The mechanisms that control these processes involve the activation of transcription followed by a series of post-transcriptional events. These post-transcriptional steps are mediated by the interaction of RNA-binding proteins (RBPs) with defined subpopulations of RNAs creating a regulatory gene network. Characterizing these RNA-protein networks is essential to understanding the regulatory mechanisms underlying the control of stem cell fate. Ribonomics is the combination of classical biochemical purification protocols with the high-throughput identification of transcripts applied to the functional characterization of RNA-protein complexes. Here, we describe the different approaches that can be used in a ribonomic approach and how they have contributed to understanding the function of several RBPs with central roles in stem cell biology.
Introduction
Stem cells are undifferentiated cells that are found in multicellular organisms both throughout embryonic development and in adult tissues. These cells have the ability to self-renew and the potential to differentiate into all body cell types. This plasticity makes stem cells especially attractive for use in cell therapies.
Embryonic stem cells respond to and follow a developmental genetic program that is triggered by a complex cascade of regulatory molecules. Adult stem cells remain in small amounts in adult tissues, where they are responsible for tissue repair and homeostasis (Zummo et al., 2007). Adult stem cells are able to perceive the environment through various signaling pathways that are activated by extracellular molecules and respond to these stimuli by changing their quiescent state via the activation of proliferation or differentiation (Dalton, 2013; Watt and Huck, 2013). The mechanisms that control these traits in embryonic and adult stem cells involve several steps of regulation, starting with the activation of transcription, followed by a series of post-transcriptional events (Cassar and Stanford, 2012; Cheung and Rando, 2013; Christie et al., 2013). RNA-binding proteins (RBPs) are essential mediators in post-transcriptional regulation. Interaction of RBPs with mRNAs result in complex genetic networks, and their characterization is essential to understand stem cell commitment. Here, we describe the current scenario of RNA-protein networks in stem cells and the different ribonomic approaches used in their identification.
Stem Cells and the Post-transcriptional Regulation of Gene Expression
The importance of post-transcriptional regulation has been gaining prominence since it was demonstrated that, in most cases, the transcriptome does not correlate with the proteome. This comparison contributed to the significance of post-transcriptional and translational regulation in the control of protein expression (Futcher et al., 1999; Gygi et al., 1999; Tenenbaum et al., 2000; Jayaseelan et al., 2014). In eukaryotes, transcription occurs in the nucleus, and mRNAs are translated in the cytoplasm. This spatial localization allows several sequential steps of regulation in order to achieve a fine-tuning regulation of the fate of cellular mRNAs (Glisovic et al., 2008). The desired transcript expression is mediated by different trans-acting regulators, such as RBPs and regulatory non-coding RNAs, which are organized in ribonucleoprotein complexes (RNPs). RBPs influence the structure and interactions of mRNAs and play critical roles in their biogenesis, stability, function, transport, and cellular localization (Lunde et al., 2007).
The diversity of RBPs allows cells to use them in an enormous array of combinations, giving rise to a unique RNP for each mRNA (MacKereth and Sattler, 2012). The orchestration of different RNPs in response to various stimuli prompted the concept of the RNA Regulon (Keene, 2007).
Technological advances have enabled the development of several strategies to identify and characterize RBPs and the RNAs with which they interact. In recent years, the Ribonomic approach has been applied to the functional characterization of RNPs in a wide range of eukaryotic model organisms. Ribonomics is defined by the combination of classical biochemical purification protocols with the high-throughput identification of transcripts (Tenenbaum et al., 2002).
Different strategies have been used to isolate the population of mRNAs bound by an RBP, which differ in complexity and in the ability to identify true interactions. Classical RNA Pull-Down approaches (Einarson et al., 2007) involve the use of recombinant-tagged proteins that are immobilized onto different types of supports and purify mRNA in an in vitro affinity chromatography assay (Figure 1A).
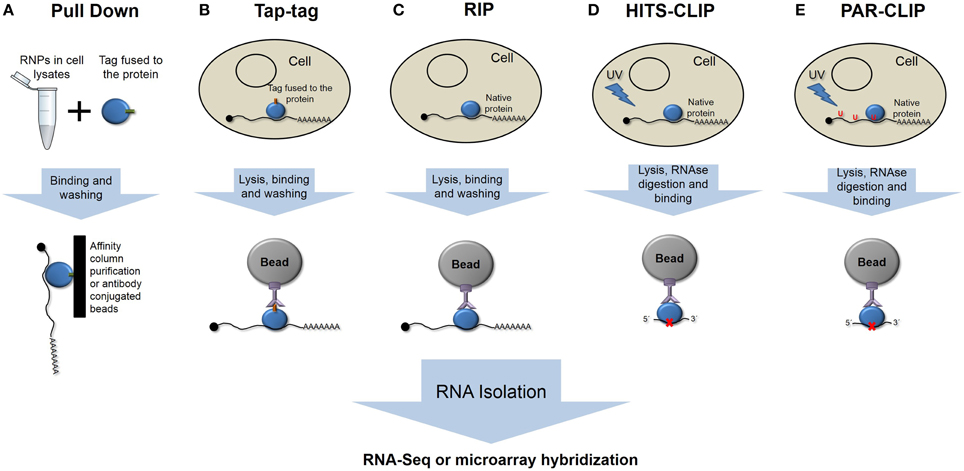
Figure 1. Schematic representation of the different main ribonomic strategies. (A) RNA pulldown in vitro purification. (B) Tandem affinity purification of tagged proteins. (C) RNA-protein immunoprecipitation. (D) High-throughput sequencing of RNA isolated by crosslinking and immunoprecipitation. (E) Photoactivatable-ribonucleoside- enhanced crosslinking and immunoprecipitation.
The tandem affinity purification (TAP-tag) method involves the fusion of a double-tag either at the amino or carboxy terminus of the protein followed by transfection of the studied cell type (Figure 1B). In vivo-formed RNA-protein complexes are purified by two-step affinity chromatography with tag-specific binding proteins (Puig et al., 2001; Gerber et al., 2006). However, the presence of the tag could interfere with native interactions, yielding false, or at least incomplete patterns of binding.
RNA targets of RBPs have been identified by immunoprecipitation assays, followed by genomic analysis using microarrays, known as RIP-Chip (RNA ImmunoPrecipitation and microchip hybridization), or more recently using next-generation sequencing methods, RIP-Seq (Figure 1C) (Tenenbaum et al., 2002; Jayaseelan et al., 2011, 2014). These techniques could be considered gold-standard techniques, as we are isolating the native complexes under normal conditions. The effectiveness of this approach relies in the purity and affinity of the antibodies that are used.
One major unwanted output of these techniques is the co-purification of non-specific and usually abundant transcripts. To isolate only specific transcripts that are bound by RBPs, different strategies have been developed to improve the RIP analysis. The refinement of this technique involves the cross-linking of RNA-protein complexes prior to purification (CLIP) (Figure 1D). Ultraviolet light causes the formation of covalent bonds between RNAs and proteins in direct or close contact. CLIP assays reduce the rate of false positives, and when combined with nuclease treatments, they can give precise information about the RNA element that is recognized and bound by RBPs (Ule et al., 2003; Jensen and Darnell, 2008). HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation) or CLIP-Seq represent high-throughput methods that were developed to generate genome-wide RNA–protein interaction maps (Licatalosi et al., 2008).
To map the binding site, RNA is digested up to a length of ~50 nt, reverse transcribed after RNA adapter ligation, and amplified to prior sequencing. One limitation is represented by the low efficiency of crosslinking using UV light. PAR-CLIP (PhotoActivatable Ribonucleoside-enhanced CrossLinking and ImmunoPrecipitation) (Hafner et al., 2010a) has been developed to more precisely map the exact binding sites at the nucleotide resolution and to increase the efficiency of the crosslinking (Figure 1E). PAR-CLIP is based on the incorporation of photoreactive ribonucleoside analogs (4-thiouridine or 6-thioguanosine) into newly synthesized RNAs. The use of ribonucleoside analogs has two advantages: they allow crosslinking with UV light at 365 nm, and they lead to a base transition during reverse transcription (thymidine to cysteine or guanosine to adenosine when using 4-thiouridine or 6-thioguanosine, respectively), which can be used to define the crosslink site at a nucleotide resolution (Hafner et al., 2010b).
These techniques allow the identification of subsets of RNAs that have related functions and are potentially co-regulated (Jayaseelan et al., 2014). In this review, we focus on recent insights from the RNA target of RBPs studies carried out in stem cells and progenitor cells that have contributed to understanding the post-transcriptional regulation of self-renewal and cell differentiation processes.
Stem Cell Ribonomics and Gene Regulatory Networks in Self-renewal and Differentiation
The ribonomic approach has been used to identify the gene regulatory networks that are formed by the RBPs and mRNAs that are involved in the commitment of stem cell differentiation. Nevertheless, other strategies have been used that identify bona fide targets of RBPs. Mex3 is a maternal totipotency factor that controls mRNA metabolism in oocytes and germline stem cells (Draper et al., 1996). This protein has a conserved dual KH domain that is involved in the direct binding of the 3′UTR of transcripts (Jadhav et al., 2008). In C. elegans, the Mex3 binding site was determined using the SELEX approach (Pagano et al., 2009). The defined Mex3 recognition element was used to search a C. elegans transcript database for putative target mRNAs. Hits were filtered to include only transcripts that are expressed in embryonic stem cells identifying 214 candidate Mex3 targets. A detailed analysis of these targets showed the presence of multiple regulatory RBPs, such as members of the PUF family, GLD1 and Nanos2, and essential pluripotency factors, such as SOX2. These results allowed the authors to understand the dual role of Mex3 in regulating germline stem cell totipotency and embryonic cell fate specification.
The RIP assay has also been combined with other techniques, not as powerful as high-throughput sequencing that allowed the identification of mRNA targets. The DEAD END (DND1) protein is essential for the maintenance of viable stem cells. This protein is expressed in human embryonic stem cells, where the population of associated mRNAs was identified (Zhu et al., 2011). The strategy was to overexpress a hemagglutinin (HA)-tagged protein in human embryonic stem cells. Bound transcripts were identified by RIP, followed by RT-PCR. Transcripts encoding pluripotency factors, such as Oct4, Sox2, and Nanog, were identified as associated with the protein. This approach showed that DND1 is a post-transcriptional regulator of the expression of pluripotency factors that are essential for stem cell maintenance.
The identification of target mRNAs showed new functions of RBPs that were not evident or were shaded by a dominant phenotype in reverse genetic assays. There are several examples where the identification of RBP-associated transcripts led to the discovery of essential pathways in stem cell fate regulation.
A deficiency in the fragile X mental retardation protein (FMRP) is responsible for fragile X syndrome. FMRP contains multiple domains that directly interact with RNA (Li and Zhao, 2014). FMRP regulates mRNA expression by repressing translation (Napoli et al., 2008) and by interacting with the miRNA machinery (Caudy et al., 2002; Ishizuka et al., 2002). FMRP is essential for the maintenance of germ stem cells and adult and embryonic stem cells (Li and Zhao, 2014). In Drosophila germ stem cells, RIP assays were used to purify small RNAs that were bound by FMRP. TaqMan assays were then used to identify 72 different miRNAs (Yang et al., 2009). The authors found that the bantam miRNA was responsible, along with FMRP, for the maintenance of the germ stem cell population. In mouse adult neural stem cells, FMRP regulates genes that are critical for stem cell function (Luo et al., 2010). RIP-Chip assays showed the presence of genes that are classified as enhancers of cell cycle progression and Wnt signaling. Genetic analysis confirmed the role of FMRP in the control of cell specification and Wnt signaling. Moreover, an analysis of the regulatory pathways that are controlled by FMRP suggests that this protein plays an important role in learning and memory formation.
Identifying associated mRNAs can also suggest the mechanisms of gene regulation of an RBP. In neural progenitor stem cells, Boris association with mRNA was analyzed by native RIP and hybridization to Affymetrix gene expression arrays (Ogunkolade et al., 2013). Transcripts corresponding to the ribosomal RNAs (rRNAs) 18S and 28S were overrepresented, suggesting the association of Boris with translating ribosomes, which was confirmed by the western blotting of polysomal fractions. Genes that are involved in the Wnt and cadherin signaling pathways were overrepresented, as well as many genes that are classified as encoding RBPs. These results were confirmed, as the overexpression of Boris leads to the activation of the Wnt canonical pathway. Hence, not only were the putative targets of Boris identified, but the presence of rRNAs suggests that Boris is a translational regulator that exerts its function through association with translating polysomes.
Combining ribonomic results with cell transcriptomics could also define a protein's function. ZFP36l2 is an RBP that is involved in the self-renewal of mouse hematopoietic stem cells. Associated mRNAs that were identified by RIP-Chip showed the presence of AU-rich motifs in their 3′UTR. These mRNAs are induced or preferentially expressed during erythroid differentiation. ZFP36l2 expression is negatively correlated with the expression pattern of its target transcripts, strongly suggesting that it is a negative regulator at the post-transcriptional level (Zhang et al., 2013).
The emergence of large-scale sequencing rendered a new powerful tool to identify not only already known transcripts but also unannotated and non-coding RNAs. RbFox2 is involved in linking non-sense-mediated decay (NMD) mechanisms with alternative splicing regulation. In mouse embryonic stem cells, a stringent purification method was used when iCLIP libraries were generated by sequential Flag and HA tag immunoprecipitation (Jangi et al., 2014). Through RNA-Seq analysis, the authors were able to identify hundreds of splicing events that were associated with RbFox2 RNA binding to intron regions. Moreover, an unexpected enrichment in genes that are regulated by AS-NMD, particularly RBPs, was observed. This observation, that hundreds of silent splicing events bound by Rbfox2 are putative AS-NMD cassette exons suggests that functional splicing regulatory activity can be attributed to the majority of Rbfox2-binding events.
Specific functions of large multifunctional protein complexes can also be dissected using the ribonomic approach. Polycomb proteins play essential roles in stem cell renewal. Antibodies against the EZH2 subunit, which interacts directly with RNA, were used for native RIP-Seq in mouse eukaryotic stem cells (Zhao et al., 2010). A total of 9788 transcripts were found as putative targets, including poly-adenylated transcripts, non-coding RNAs and unannotated sequences (Lee, 2010). The characterization of a polycomb “transcriptome” suggests that the existence of RNA cofactors may be a general feature of polycomb regulation. The ribonomic approach could be useful to identify RNA cofactors for other chromatin modifiers.
Another example of how characterizing RNA-protein interactions could help to understand the biological role of an RBP in stem cells is the Musashi family of proteins (MSI). The Musashi proteins are found in stem and progenitor cells and are overexpressed in several cancer cells, with a well-defined role in the regulation of the undifferentiated state. A pulldown assay (RNA bind and seq) using RNA from mouse neural stem cells identified the population of MSI1-bound transcripts (Katz et al., 2014). The MSI1 protein was fused to a streptavidin-binding peptide, and RNA was pulled down and sequenced under high throughput. This strategy defined a binding element that was present in the 3′UTR of mRNAs. Regarding MSI2, ribonomic assays were performed in hematopoietic stem cells. Park et al. (2014) overexpressed a flag-tagged MSI2 protein and used HITS-CLIP to identify 1097 putative targets of the protein. Among the subpopulation of mRNAs were RNA fate regulators and genes that are involved in the regulation of cell signaling and developmental pathways (Park et al., 2014). The gene network that was defined by the target genes showed that MSI2 is involved in stem cell self-renewal and TGFβ signaling.
The PUF (Pumilio/FBF1) Family of RNA-binding Proteins and the Control of Stem Cell Proliferation
PUF (Pumilio and FBF) proteins are mRNA regulators with a conserved role in the maintenance of mitotic division, resulting in the self-renewal of stem cells (Wickens et al., 2002). Putative PUF target mRNAs have been identified on a genomic scale in budding yeast, human HeLa cells, human adipose-derived stem cells and fly ovaries and embryos (Gerber et al., 2004, 2006; Galgano et al., 2008; Morris et al., 2008; Shigunov et al., 2012). The composition of the associated population of mRNAs depends on the cell transcriptome (Shigunov et al., 2012; Abil et al., 2014). Nevertheless, a comparison of putative FBF and PUF targets in metazoans revealed 40 common transcripts, including well-established stem cell fate regulators (Kershner and Kimble, 2010).
PUF proteins were immunoprecipitated with bound mRNAs, and those RNAs were then used to probe microarrays (RIP-Chip) (Galgano et al., 2008; Kershner and Kimble, 2010; Shigunov et al., 2012). The FBF target mRNAs represent ~7% of the C. elegans protein-coding genes, and PUF proteins in humans and Drosophila likely control a similar proportion (7–11%) of their respective transcriptomes (Gerber et al., 2006; Galgano et al., 2008; Morris et al., 2008; Kershner and Kimble, 2010). Putative PUM1 and PUM2 targets have been analyzed in HeLa and HEK293 cells, showing that these proteins bind a large set of transcripts with a large overlap of putative targets (Galgano et al., 2008; Morris et al., 2008; Hafner et al., 2010b). One of the common biological processes of PUF target mRNAs is cell proliferation control, which has been demonstrated by several groups (Kennedy et al., 1997; Crittenden et al., 2002; Lee et al., 2007; Ariz et al., 2009; Kalchhauser et al., 2011; Chen et al., 2012; Racher and Hansen, 2012; Van Etten et al., 2012).
Our group was able to identify approximately 300 putative PUM2 targets in adipose-derived stem cells using RIP-Chip (Figures 2A,B) (Shigunov et al., 2012). Cellular growth and proliferation were associated as network functions of putative PUM2 targets by a highest score analysis. Among the putative PUM2 targets, we also found several transcripts encoding proteins that are involved in the negative regulation of proliferation. Several transcripts encoding cell cycle-related proteins were found to be associated with PUF proteins in different organisms, suggesting that PUF proteins could be directly involved in the control of progression through the cell cycle (Galgano et al., 2008; Morris et al., 2008; Hafner et al., 2010b; Kershner and Kimble, 2010).
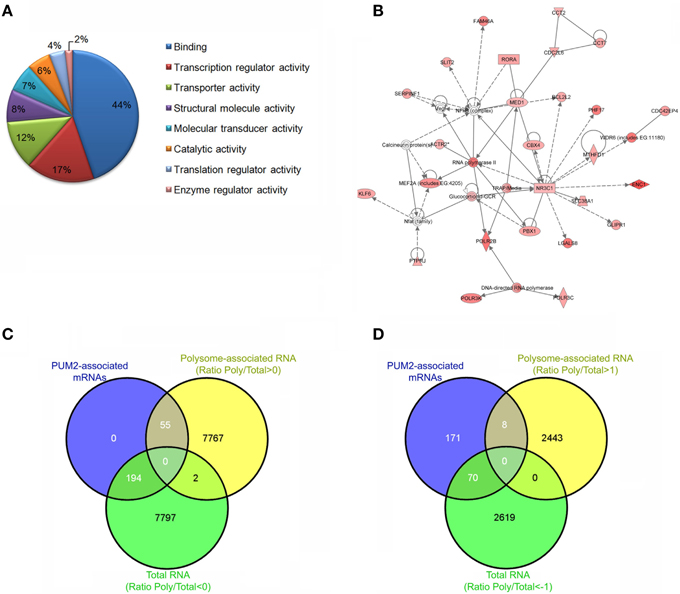
Figure 2. Steps to Ribonomic analysis of PUM2-associated mRNAs. Ensure that the protein of interest was specifically immunoprecipitated using western blotting. Identify and cluster the mRNAs according to their enrichment and the biological and technical replicates. (A) Cluster the mRNAs according to biological process, cellular component, and molecular function. (B) Identify networks of mRNAs that are associated with PUM2. (C) Comparison of the PUM2-associated mRNAs with Total RNA (ratio < 0) and polysome-associated mRNAs (ratio > 0). Ratio polysome-associated RNAs/Total RNA. (D) Comparison of the PUM2-associated mRNAs with Total RNA and polysome-associated mRNAs enriched < 1 and >1, respectively.
In another study from our group, we evaluated the total RNAs and polysome-associated mRNAs in adipose-derived stem cells using polysome profiling (Spangenberg et al., 2013). We reanalyzed the data (ArrayExpress E-MTAB-1366) to identify RNAs that were enriched in the polysomal fraction by calculating the ratio between polysome-associated and total RNA. When we compared the previously described PUM2-associated mRNAs with total RNAs and polysome-associated mRNAs (Oliveros, 2007), we found only 22% of PUM2-associated mRNAs in the polysomal fraction (Figure 2C). If we look at the elements that are enriched by fold change (1 > log < −1), this number decreases to 10% (Figure 2D). Pumilio represses the translation of specific mRNAs by recruiting factors that control RNA stability (Goldstrohm et al., 2006), and Pum2 represses translation by competing with eIF4E to bind the cap (Cao et al., 2010). Our results suggest that PUM2 could prevent the mRNAs from associating with ribosomes by competing with eIF4E. PUMILIO-1 and 2 RBPs have been previously identified as regulators that are involved in stem cell proliferation in invertebrates (Catelain et al., 2014). PUF proteins in concert with other proteins coordinate the temporal or spatial pattern of translation of a large set of mRNAs. Experiments to identify tissue-specific mRNA targets of PUF will also allow the determination of the landscape of mRNAs and partners that are unique to each cell type, contributing to the understanding of protein function.
Conclusion
The identification of which RNAs are associated with post-transcriptional regulatory proteins and the genetic network that they form is important to understand the essential steps in stem cell differentiation and the mechanisms that are responsible for the maintenance of the undifferentiated state. The combination of affinity purification methods and large-scale identification of transcripts, plus the development of powerful bioinformatics tools will lead to a systematic characterization of these networks. Though much effort has been devoted to this challenge, there is still much work in the future that is needed to understand the complex mechanisms underlying post-transcriptional regulation.
Author Contributions
The authors contributed equally to write the mini review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from Fundação Araucária and FIOCRUZ. B. D. received fellowship from Conselho Nacional de Desenvolvimento Cientifico e Tecnológico—CNPq.
References
Abil, Z., Denard, C. A., and Zhao, H. (2014). Modular assembly of designer PUF proteins for specific post-transcriptional regulation of endogenous RNA. J. Biol. Eng. 8:7. doi: 10.1186/1754-1611-8-7
Ariz, M., Mainpal, R., and Subramaniam, K. (2009). C. elegans RNA-binding proteins PUF-8 and MEX-3 function redundantly to promote germline stem cell mitosis. Dev. Biol. 326, 295–304. doi: 10.1016/j.ydbio.2008.11.024
Cao, Q., Padmanabhan, K., and Richter, J. D. (2010). Pumilio 2 controls translation by competing with eIF4E for 7-methyl guanosine cap recognition. RNA 16, 221–227. doi: 10.1261/rna.1884610
Cassar, P. A., and Stanford, W. L. (2012). Integrating post-transcriptional regulation into the embryonic stem cell gene regulatory network. J. Cell. Physiol. 227, 439–449. doi: 10.1002/jcp.22787
Catelain, C., Michelet, F., Hattabi, A., Poirault-Chassac, S., Kortulewski, T., Tronik-Le Roux, D., et al. (2014). The Notch Delta-4 ligand helps to maintain the quiescence and the short-term reconstitutive potential of haematopoietic progenitor cells through activation of a key gene network. Stem Cell Res. 13, 431–441. doi: 10.1016/j.scr.2014.10.002
Caudy, A. A., Myers, M., Hannon, G. J., and Hammond, S. M. (2002). Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 16, 2491–2496. doi: 10.1101/gad.1025202
Chen, D., Zheng, W., Lin, A., Uyhazi, K., Zhao, H., and Lin, H. (2012). Pumilio 1 suppresses multiple activators of p53 to safeguard spermatogenesis. Curr. Biol. 22, 420–425. doi: 10.1016/j.cub.2012.01.039
Cheung, T. H., and Rando, T. A. (2013). Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 14, 329–340. doi: 10.1038/nrm3591
Christie, K. J., Emery, B., Denham, M., Bujalka, H., Cate, H. S., and Turnley, A. M. (2013). Transcriptional regulation and specification of neural stem cells. Adv. Exp. Med. Biol. 786, 129–155. doi: 10.1007/978-94-007-6621-1_8
Crittenden, S. L., Bernstein, D. S., Bachorik, J. L., Thompson, B. E., Gallegos, M., Petcherski, A. G., et al. (2002). A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature 417, 660–663. doi: 10.1038/nature754
Dalton, S. (2013). Signaling networks in human pluripotent stem cells. Curr. Opin. Cell Biol. 25, 241–246. doi: 10.1016/j.ceb.2012.09.005
Draper, B. W., Mello, C. C., Bowerman, B., Hardin, J., and Priess, J. R. (1996). MEX-3 is a KH domain protein that regulates blastomere identity in early C. elegans embryos. Cell 87, 205–216. doi: 10.1016/S0092-8674(00)81339-2
Einarson, M. B., Pugacheva, E. N., and Orlinick, J. R. (2007). GST Pull-down. CSH Protoc. 2007:pdb.prot4757. doi: 10.1101/pdb.prot4757
Futcher, B., Latter, G. I., Monardo, P., McLaughlin, C. S., and Garrels, J. I. (1999). A sampling of the yeast proteome. Mol. Cell. Biol. 19, 7357–7368. doi: 10.1128/MCB.19.11.7357
Galgano, A., Forrer, M., Jaskiewicz, L., Kanitz, A., Zavolan, M., and Gerber, A. P. (2008). Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS ONE 3:e3164. doi: 10.1371/journal.pone.0003164
Gerber, A. P., Herschlag, D., and Brown, P. O. (2004). Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2:E79. doi: 10.1371/journal.pbio.0020079
Gerber, A. P., Luschnig, S., Krasnow, M. A., Brown, P. O., and Herschlag, D. (2006). Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 103, 4487–4492. doi: 10.1073/pnas.0509260103
Glisovic, T., Bachorik, J. L., Yong, J., and Dreyfuss, G. (2008). RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 582, 1977–1986. doi: 10.1016/j.febslet.2008.03.004
Goldstrohm, A. C., Hook, B. A., Seay, D. J., and Wickens, M. (2006). PUF proteins bind Pop2p to regulate messenger RNAs. Nat. Struct. Mol. Biol. 13, 533–539. doi: 10.1038/nsmb1100
Gygi, S. P., Rochon, Y., Franza, B. R., and Aebersold, R. (1999). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730. doi: 10.1128/MCB.19.3.1720
Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., et al. (2010a). PAR-CliP–a method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 41:e2034. doi: 10.3791/2034
Hafner, M., Landthaler, M., Burger, L., Khorshid, M., Hausser, J., Berninger, P., et al. (2010b). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. doi: 10.1016/j.cell.2010.03.009
Ishizuka, A., Siomi, M. C., and Siomi, H. (2002). A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 16, 2497–2508. doi: 10.1101/gad.1022002
Jadhav, S., Rana, M., and Subramaniam, K. (2008). Multiple maternal proteins coordinate to restrict the translation of C. elegans nanos-2 to primordial germ cells. Development 135, 1803–1812. doi: 10.1242/dev.013656
Jangi, M., Boutz, P. L., Paul, P., and Sharp, P. A. (2014). Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 28, 637–651. doi: 10.1101/gad.235770.113
Jayaseelan, S., Doyle, F., Currenti, S., and Tenenbaum, S. A. (2011). RIP: an mRNA localization technique. Methods Mol. Biol. 714, 407–422. doi: 10.1007/978-1-61779-005-8_25
Jayaseelan, S., Doyle, F., and Tenenbaum, S. A. (2014). Profiling post-transcriptionally networked mRNA subsets using RIP-Chip and RIP-Seq. Methods 67, 13–19. doi: 10.1016/j.ymeth.2013.11.001
Jensen, K. B., and Darnell, R. B. (2008). CLIP: crosslinking and immunoprecipitation of in vivo RNA targets of RNA-binding proteins. Methods Mol. Biol. 488, 85–98. doi: 10.1007/978-1-60327-475-3_6
Kalchhauser, I., Farley, B. M., Pauli, S., Ryder, S. P., and Ciosk, R. (2011). FBF represses the Cip/Kip cell-cycle inhibitor CKI-2 to promote self-renewal of germline stem cells in C. elegans. EMBO J. 30, 3823–3829. doi: 10.1038/emboj.2011.263
Katz, Y., Li, F., Lambert, N. J., Sokol, E. S., Tam, W. L., Cheng, A. W., et al. (2014). Musashi proteins are post-transcriptional regulators of the epithelial-luminal cell state. Elife 3:e03915. doi: 10.7554/eLife.03915
Keene, J. D. (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543. doi: 10.1038/nrg2111
Kennedy, B. K., Gotta, M., Sinclair, D. A., Mills, K., McNabb, D. S., Murthy, M., et al. (1997). Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89, 381–391. doi: 10.1016/S0092-8674(00)80219-6
Kershner, A. M., and Kimble, J. (2010). Genome-wide analysis of mRNA targets for Caenorhabditis elegans FBF, a conserved stem cell regulator. Proc. Natl. Acad. Sci. U.S.A. 107, 3936–3941. doi: 10.1073/pnas.1000495107
Lee, J. T. (2010). The X as model for RNA's niche in epigenomic regulation. Cold Spring Harb. Perspect. Biol. 2:a003749. doi: 10.1101/cshperspect.a003749
Lee, M. H., Hook, B., Pan, G., Kershner, A. M., Merritt, C., Seydoux, G., et al. (2007). Conserved regulation of MAP kinase expression by PUF RNA-binding proteins. PLoS Genet. 3:e233. doi: 10.1371/journal.pgen.0030233
Li, Y., and Zhao, X. (2014). Concise review: fragile X proteins in stem cell maintenance and differentiation. Stem Cells 32, 1724–1733. doi: 10.1002/stem.1698
Licatalosi, D. D., Mele, A., Fak, J. J., Ule, J., Kayikci, M., Chi, S. W., et al. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469. doi: 10.1038/nature07488
Lunde, B. M., Moore, C., and Varani, G. (2007). RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 8, 479–490. doi: 10.1038/nrm2178
Luo, Y., Shan, G., Guo, W., Smrt, R. D., Johnson, E. B., Li, X., et al. (2010). Fragile x mental retardation protein regulates proliferation and differentiation of adult neural stem/progenitor cells. PLoS Genet. 6:e1000898. doi: 10.1371/journal.pgen.1000898
MacKereth, C. D., and Sattler, M. (2012). Dynamics in multi-domain protein recognition of RNA. Curr. Opin. Struct. Biol. 22, 287–296. doi: 10.1016/j.sbi.2012.03.013
Morris, A. R., Mukherjee, N., and Keene, J. D. (2008). Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol. Cell. Biol. 28, 4093–4103. doi: 10.1128/MCB.00155-08
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., et al. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134, 1042–1054. doi: 10.1016/j.cell.2008.07.031
Ogunkolade, B. W., Jones, T. A., Aarum, J., Szary, J., Owen, N., Ottaviani, D., et al. (2013). BORIS/CTCFL is an RNA-binding protein that associates with polysomes. BMC Cell Biol. 14:52. doi: 10.1186/1471-2121-14-52
Oliveros, J. C. (2007). VENNY. An Interactive Tool for Comparing Lists with Venn Diagrams. Available online at: http://bioinfogp.cnb.csic.es/tools/venny/index.html
Pagano, J. M., Farley, B. M., Essien, K. I., and Ryder, S. P. (2009). RNA recognition by the embryonic cell fate determinant and germline totipotency factor MEX-3. Proc. Natl. Acad. Sci. U.S.A. 106, 20252–20257. doi: 10.1073/pnas.0907916106
Park, S. M., Deering, R. P., Lu, Y., Tivnan, P., Lianoglou, S., Al-Shahrour, F., et al. (2014). Musashi-2 controls cell fate, lineage bias, and TGF-β signaling in HSCs. J. Exp. Med. 211, 71–87. doi: 10.1084/jem.20130736
Puig, O., Caspary, F., Rigaut, G., Rutz, B., Bouveret, E., Bragado-Nilsson, E., et al. (2001). The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24, 218–229. doi: 10.1006/meth.2001.1183
Racher, H., and Hansen, D. (2012). PUF-8, a Pumilio homolog, inhibits the proliferative fate in the Caenorhabditis elegans germline. G3 (Bethesda) 2, 1197–1205. doi: 10.1534/g3.112.003350
Shigunov, P., Sotelo-Silveira, J., Kuligovski, C., De Aguiar, A. M., Rebelatto, C. K., Moutinho, J. A., et al. (2012). PUMILIO-2 is involved in the positive regulation of cellular proliferation in human adipose-derived stem cells. Stem Cells Dev. 21, 217–227. doi: 10.1089/scd.2011.0143
Spangenberg, L., Shigunov, P., Abud, A. P., Cofré, A. R., Stimamiglio, M. A., Kuligovski, C., et al. (2013). Polysome profiling shows extensive posttranscriptional regulation during human adipocyte stem cell differentiation into adipocytes. Stem Cell Res. 11, 902–912. doi: 10.1016/j.scr.2013.06.002
Tenenbaum, S. A., Carson, C. C., Lager, P. J., and Keene, J. D. (2000). Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc. Natl. Acad. Sci. U.S.A. 97, 14085–14090. doi: 10.1073/pnas.97.26.14085
Tenenbaum, S. A., Lager, P. J., Carson, C. C., and Keene, J. D. (2002). Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods 26, 191–198. doi: 10.1016/S1046-2023(02)00022-1
Ule, J., Jensen, K. B., Ruggiu, M., Mele, A., Ule, A., and Darnell, R. B. (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215. doi: 10.1126/science.1090095
Van Etten, J., Schagat, T. L., Hrit, J., Weidmann, C. A., Brumbaugh, J., Coon, J. J., et al. (2012). Human Pumilio proteins recruit multiple deadenylases to efficiently repress messenger RNAs. J. Biol. Chem. 287, 36370–36383. doi: 10.1074/jbc.M112.373522
Watt, F. M., and Huck, W. T. (2013). Role of the extracellular matrix in regulating stem cell fate. Nat. Rev. Mol. Cell Biol. 14, 467–473. doi: 10.1038/nrm3620
Wickens, M., Bernstein, D. S., Kimble, J., and Parker, R. (2002). A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet. 18, 150–157. doi: 10.1016/S0168-9525(01)02616-6
Yang, Y., Xu, S., Xia, L., Wang, J., Wen, S., Jin, P., et al. (2009). The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet. 5:e1000444. doi: 10.1371/journal.pgen.1000444
Zhang, L., Prak, L., Rayon-Estrada, V., Thiru, P., Flygare, J., Lim, B., et al. (2013). ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature 499, 92–96. doi: 10.1038/nature12215
Zhao, J., Ohsumi, T. K., Kung, J. T., Ogawa, Y., Grau, D. J., Sarma, K., et al. (2010). Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 40, 939–953. doi: 10.1016/j.molcel.2010.12.011
Zhu, R., Iacovino, M., Mahen, E., Kyba, M., and Matin, A. (2011). Transcripts that associate with the RNA binding protein, DEAD-END (DND1), in embryonic stem (ES) cells. BMC Mol. Biol. 12:37. doi: 10.1186/1471-2199-12-37
Keywords: ribonomics, RNA-binding proteins, stem cells, gene network, differentiation
Citation: Shigunov P and Dallagiovanna B (2015) Stem Cell Ribonomics: RNA-Binding Proteins and Gene Networks in Stem Cell Differentiation. Front. Mol. Biosci. 2:74. doi: 10.3389/fmolb.2015.00074
Received: 24 September 2015; Accepted: 07 December 2015;
Published: 22 December 2015.
Edited by:
Ghanshyam Upadhyay, City College of New York, USAReviewed by:
Sandeep Kumar, State University of New York College of Optometry, USAMahendra Pratap Kashyap, University of Pittsburgh, USA
Deepak Gurbani, University of Texas Southwestern Medical Center, USA
Israr Ahmad, University of Alabama at Birmingham, USA
Copyright © 2015 Shigunov and Dallagiovanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrícia Shigunov, cGF0cmljaWEuc2hpZ3Vub3ZAZmlvY3J1ei5icg==;
Bruno Dallagiovanna, YnJ1bm9kQGZpb2NydXouYnI=