 Shima Shahjouei1
Shima Shahjouei1 Saeed Ansari
Saeed Ansari- 1Department of Neurosurgery, Tehran University of Medical Sciences, Tehran, Iran
- 2Department of Neurology, University of Tennessee Health Science Center, Memphis, TN, USA
- 3Department of Microbiology, Immunology, and Biochemistry, University of Tennessee Health Science Center, Memphis, TN, USA
- 4Biocomplexity Institute, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA
Adropin is a 4.9 kDa peptide that is important for maintenance of metabolic and non-metabolic homeostasis. It regulates glucose and fatty acid metabolism and is involved in endothelial cell function and endothelial nitric oxide (NO) synthase bioactivity as well as physical activity and motor coordination. Adropin is expressed in many tissues and organs including central nervous system (CNS). This peptide plays a crucial role in the development of various CNS disorders such as stroke, schizophrenia, bipolar disorder as well as Alzheimer's, Parkinson's, and Huntington's diseases. In this comprehensive review, the potential roles of adropin in cellular signaling pathways that lead to pathogenesis and/or treatment of CNS disorders will be discussed.
Introduction
Adropin is a 4.9 kDa peptide encoded by Energy Homeostasis Associated gene (Enho) located on chromosome 9 (Kumar et al., 2008; Aydin, 2014). A variety of organs including central nervous system (neurons, neuroglial cells, pia mater, vascular area, Purkinje cells, and granular layer), heart, kidney, liver, pancreas, and human umbilical vein synthesize adropin (Lovren et al., 2010; Aydin et al., 2013, 2014).
Constantly new functions for adropin are identified. Adropin's function as a regulator of glucose and lipid homeostasis and insulin sensitivity was initially described in 2008 by Kumar et al. (2008) and later by Aydin (2014). Lovren et al. (2010) demonstrated the endothelial protective potentials of adropin in 2010. Adropin activates vascular endothelial growth factor receptor 2 (VEGFR2) and its two downstream signaling pathways—phosphatidylinositol-3 kinase/ serine, threonine kinase (PI3K/Akt) and extracellular signal-regulated kinases 1/2 (ERK 1/2) (Figure 1). Therefore, adropin modulates expression of endothelial nitric oxide synthase (eNOS) (Lovren et al., 2010). Also, adropin increases the endothelial cells proliferation, migration and potential to form capillary-like structures (Lovren et al., 2010). Recently, it is found that adropin reduces the endothelial permeability (Lovren et al., 2010; Yang et al., 2016).
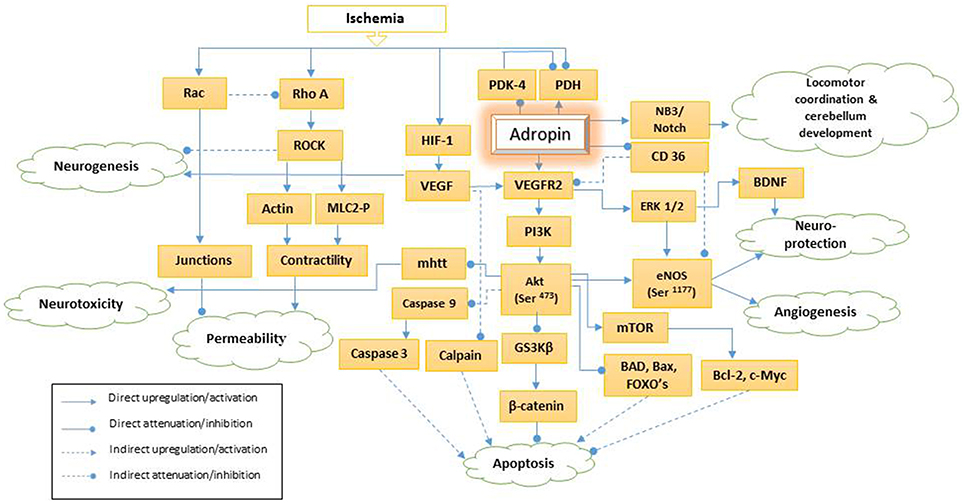
Figure 1. Schematic presentation of adropin signaling pathways. PDH, pyruvate dehydrogenase; PDK-4, pyruvate dehydrogenase kinase-4; HIF-1, hypoxia-inducible factor-1a; NB3, neural recognition molecule 3; VEGF, vascular endothelial growth factor; mhtt, mutant huntingtin; VEGFR2, vascular endothelial growth factor receptor 2; PI3K, phosphatidylinositol-3 kinase; Akt (Ser-473), phosphorylation of serine 473 of serine, threonine kinase; GS3Kβ, glycogen synthase kinase 3β; CD36, Cluster of Differentiation36; ERK 1/2, extracellular signal-regulated kinases 1/2; Mtor, mammalian target of rapamycin; BAD, Bcl-2 associated death protein; eNOS (Ser-1177), phosphorylation of endothelial nitric synthase on serine 1177; BNDF, brain-derived neurotrophic factor; FOXO, Forkhead box O; Bcl-2: B-cell lymphoma 2; Bax, Bcl-2 associated X protein; ROCK, Rho-associated protein kinase; MLC2-P, Phosphorylated myosin light chain 2.
Adropin enhances mitochondrial function and activates pyruvate dehydrogenase (PDH)—a rate-limiting enzyme in glucose oxidation. Further, adropin suppresses two key enzymes in fatty acid utilization: carnitine palmitoyltransferase-1B (CPT-1B) and Cluster of Differentiation 36 (CD36) (Gao et al., 2015); thus, it plays a role in fatty acid oxidation.
Adropin may act as a potential protective regulator of atherogenesis and cardiovascular diseases (Wu et al., 2014; Zhao et al., 2015b; Li et al., 2016). Serum adropin level is inversely associated with severity of coronary atherosclerosis and serum level of homocysteine—a potential risk factor for atherosclerosis and cardiovascular diseases (Zhao et al., 2015a). The serum adropin level is diminished in patients with cardiac syndrome X and stable coronary artery disease (Celik et al., 2013; Zhao et al., 2015b). At the onset of acute myocardial infarction, serum adropin level is usually lower than controls (Yu et al., 2014); however, it raises between 1 and 24 h following myocardial infarction (Aydin et al., 2014). Plasma adropin level has a positive association with severity of heart failure and negative correlation with left ventricular ejection fraction (Lian et al., 2011). Low level of plasma adropin is predictive of pseudoexfoliation (Oğurel et al., 2016), coronary slow flow phenomenon (Demircelik and Kurtul, 2015), saphenous vein graft occlusion following coronary artery bypass grafting (Demircelik, 2014), as well as pediatric obstructive sleep apnea in the presence of endothelial dysfunction (Gozal et al., 2013). While Gu et al. (2015) described plasma adropin level as an independent indicator of hypertension, other studies failed to show this association (Altincik and Sayin, 2015).
Adropin, as a membrane-anchored protein modulates the Notch1 signaling pathway via neural recognition molecule 3 (NB3) (Figure 1). NB-3 belongs to the contactin family and acts as a membrane-tethered Notch1 ligand that mediates cell surface interaction during nervous system development. An animal study demonstrated that adropin regulates locomotor activity and motor coordination via the NB3/Notch signaling pathway and plays an important role in cerebellum development (Wong et al., 2014). In this review, we discuss various roles of adropin in central nervous system pathogenesis via different intra and extra cellular signaling pathways as well as its therapeutic potentials.
Adropin and Vascular Endothelial Growth Factor Receptor 2 (VEGFR2)
VEGFR2—a tyrosine kinase receptor—is especially expressed in endothelial cells and regulates endothelial function and angiogenesis. Adropin strongly upregulates this receptor, activates PI3K/Akt and ERK1/2 pathways, and enhances eNOS thus, modulating NO bioavailability (Lovren et al., 2010; Figure 1). Hypoxic insults enhance hypoxia-inducible factor-1a (HIF-1a), and VEGF gene expression as its downstream signaling pathway (Mu et al., 2003). VEGF is involved in neurogenesis and has a neuroprotection function. This has been discussed under “Adropin and Neurogenesis” section in more details.
Role of Adropin in Activation of PI3K/Akt Signaling Pathway
PI3K induces the phosphorylation of Akt (also known as protein kinase B) under the effect of growth factors such as VEGF, cytokines, insulin, and other cellular stimuli (Figure 1). Activation of Akt requires consequent phosphorylation on Thr-308 and Ser-473. Once Ser-473 is phosphorylated, Akt is fully activated regardless of Thr-308 phosphorylation status (Wang et al., 2009). Adropin can activate Akt by stimulating Ser-473 phosphorylation (Lovren et al., 2010).
Phosphorylated-Akt provokes cell cycle progression, proliferation, differentiation, and survival (Blanco-aparicio et al., 2007; Manning and Cantley, 2007). Moreover, this pathway triggers intracellular ligands such as mammalian target of rapamycin (mTOR)—which plays an important role in angiogenesis, neuronal regeneration, synaptic plasticity, inflammatory responses, and apoptosis (Annovazzi et al., 2009; Chen et al., 2012; Li et al., 2015). Thereby, PI3K/Akt/mTOR pathway may be a target of stroke therapeutic agents (Li et al., 2015).
Neurodegenerative conditions such as Alzheimer's, Parkinson's and Huntington's diseases are associated with defective Akt signaling (Colin et al., 2005; Griffin et al., 2005; Timmons et al., 2009; Giralt et al., 2010). Similarly, damaged Akt/GSK3β (the serine/threonine kinase glycogen synthase kinase 3β) signaling pathway plays a role in the pathophysiology of neuropsychiatric disorders such as schizophrenia and bipolar disorders (Emamian et al., 2004; Jope, 2011). Since, variation in AKT1—one of the three genes encoding Akt—has been associated with schizophrenia and bipolar disorders (Ikeda et al., 2004; Karege et al., 2012), PI3K/Akt activation by adropin might also have a therapeutic potential in disorders such as Parkinson's (Burke, 2007; Timmons et al., 2009) and schizophrenia (Schwab et al., 2005) as discussed below:
Ischemic Insult
Cerebral ischemic injuries cause neural loss secondary to apoptosis or necrosis—which can be triggered by oxidative stress, metabolic compromise and disruption of calcium homeostasis at the cellular level (Mattson et al., 2001). Altintas et al. demonstrated that infarct size is positively correlated with blood adropin level in animal models of cerebral ischemia (Altintas et al., 2016). Activation of Akt by adropin can prevent neuronal and cellular death, (Chong et al., 2005) and might contribute to neuro-protective effect of ischemic postconditioning (Gao et al., 2008; Wang et al., 2009). PI3K/Akt pathway induces mTOR and also attenuates apoptotic proteins such as GSK3β and forkhead family of transcription factor. Thereby, inactivation of Akt might contribute to neuronal apoptosis and pathogenesis of ischemic stroke (Noshita et al., 2001; Franke et al., 2003; Hanumanthappa et al., 2014; Li et al., 2015).
Huntington Disease (HD)
Abnormal expansion of a polyglutamine stretch in the N terminus of protein huntingtin is responsible for neuropathology of HD (Humbert et al., 2002). Induction of Akt Ser-473 phosphorylation attenuates mutant huntingtin toxicity and makes the cell more resistance to apoptotic signals by modulating proteins such as GSK3β and FOXO1 (Humbert et al., 2002; Manning and Cantley, 2007). In addition, activated Akt decreases intranuclear inclusions of mutant huntingtin (Humbert et al., 2002). It was demonstrated that maintaining high levels of activated Akt may delay cell death and allow the recovery of neuronal viability after mutant huntingtin silencing (Canals, 2004).
Parkinson's Disease (PD)
Timmons and colleagues showed that Akt is expressed at high levels in tyrosine hydroxylase dopaminergic neurons. Selective loss of these neurons and diminished phosphorylated Akt at Ser-473 is obvious in the brain of patients with Parkinson's disease (Timmons et al., 2009). The glial cell line-derived neurotrophic factor (GDNF) as the downstream of phosphorylated Akt has neuroprotective effect against dopaminergic neurodegeneration (Ries et al., 2006). Thus, medications like adropin that target the dopaminergic system via Akt activation or those with the potential to increase the phosphorylated Akt have neuroprotective characteristics in PD (Ries et al., 2006; Burke, 2007; Levy et al., 2009; Timmons et al., 2009).
Schizophrenia
AKT1 gene single nucleotide polymorphisms (SNPs) and haplotype studies indicated the involvement of Akt in Schizophrenia (Ikeda et al., 2004; Schwab et al., 2005; Thiselton et al., 2008). Expression or activity of AKT1 and phosphorylation of its substrate—GSK3β—is reduced in Schizophrenic patients (Emamian et al., 2004; Kalkman, 2006). As summarized by Beaulieu and colleagues, many of the antipsychotics and psychoactive substances modulate dopamine-dependent behaviors through Akt/GSK3β signaling pathway (Beaulieu et al., 2007). In addition, Schizophrenia is associated with insulin receptor deficit, disruptive insulin dependent Akt signaling and insulin resistance (Zhao et al., 2006). Adropin might be a potent therapeutic agent in Schizophrenia while it enhances Akt phosphorylation (Lovren et al., 2010) and prevents insulin resistance (Ganesh Kumar et al., 2012).
Alzheimer's Disease (AD)
Activation of PI3K/Akt/Wnt/β-catenin signaling induces neurogenesis and reverse cognitive deficit in AD animal models (Tiwari et al., 2015). In addition, reduced phospho-Akt and increased FOXO3a levels in the nuclei of neurons where proapototic genes were activated can cause adipokine dyshomeostasis, oxidative stress, mitochondrial dysfunction, and eventually neurodegeneration (Nuzzo et al., 2015). These data suggest Akt might be the link between insulin resistance, obesity, and AD.
Bipolar Disorder
Regulation of Akt/mTOR pathway is critical in synaptic neurotransmission and plasticity, as well as modulating cell proliferation and migration. There is evidence of excitotoxicity, neuroinflammation, and brain atrophy in BD due to apoptosis and disturbed synaptic function. A cadaver study on BD postmortem prefrontal cortex demonstrated an elevation in protein and mRNA levels of the pro-apoptotic factors (Bax, BAD, caspase-9 and caspase-3) and reduction in anti-apoptotic factors (BDNF and Bcl-2) and the synaptic markers (synaptophysin and drebrin) (Kim et al., 2010). The Bax/Bcl-2 ratio appeared to be crucial in deciding the life or death of a cell and was increased in the above study. In another study, blood AKT1and mTOR mRNA expression decreased in BD during depressive episodes comparing to healthy controls, supporting an integrated Akt/mTOR signaling pathway activity in the pathogenesis of BD (Machado-Vieira et al., 2015). In accordance, activation of mTOR by N-methyl-D-aspartate (NMDA) antagonists results in rapid antidepressant effect in animal models (Li et al., 2010).
Study on animals under high-fat diet showed that obesity may desensitize serotonin-dependent Akt/GSK3β signaling and impair cell proliferation in the dentate gyrus of the hippocampus, and cause depression (Papazoglou et al., 2014). Available evidence support the notion that enhancing the inhibitory control of Akt/GSK3β is a key component of the therapeutic actions of drugs used to treat mood disorders (Li and Jope, 2010).
Adropin and Extracellular Signal-Regulated Kinases 1/2 (ERK1/2)
ERK 1/2 is a member of the mitogen-activated protein kinase family. Adropin via VEGFR2 can activate ERK 1/2 and its downstream cascades of substances such as brain-derived neurotrophic factor (BDNF) (Figure 1; Lovren et al., 2010). BNDF promotes neuronal development, differentiation, survival and neurological function improvement following brain injury and ischemia (Zhu et al., 2013; Zhao et al., 2014; Wu et al., 2015). Ischemic postconditioning, both early and delayed, may further reduce reperfusion injury via ERK 1/2 and BDNF activation (Wu et al., 2015). In contrast, post-ischemic inhibition of ERK 1/2 in diabetic rats may mitigate DNA repairing ability, accelerated apoptosis and aggravate neuronal loss (Zhao et al., 2014). In addition, ERK 1/2 activation induces nuclear factor erythroid 2-related factor2 (Nrf2) and protects neurons against beta-amyloid-induced cell death and oxidative stress.
Adropin and Nitric Oxide Synthase (NOS)
One of the endothelial protective functions of adropin is regulation of nitric oxide (NO) bioavailability (Lovren et al., 2010). NO promotes angiogenesis, reparative vasculogenesis and acts as an anti-atherosclerotic, anti-inflammatory and anti-thrombotic factor.
NO is generated by nitric oxide synthase (NOS) that is upregulated by PI3K/Akt and ERK 1/2 signaling pathways (Figure 1) (Lovren et al., 2010; Peng et al., 2012). NOS polymorphisms and diminished endothelial NOS expression are associated with spontaneous cerebral thrombosis and infarction, progressive cerebral amyloid angiopathy, blood brain barrier breakdown, and cognitive impairment—characteristics of cerebral small vessel disease, stroke and neurodegenerative diseases such as Alzheimer's disease (Hassan, 2004; Jeynes and Provias, 2009; Tan et al., 2015). Additionally, Tan et al. evidenced that this vascular occlusion occurs exclusively at the same hypoperfused areas identified in preclinical Alzheimer's disease (temporoparietal and retrosplenial granular cortexes, and hippocampus; Tan et al., 2015).
Adropin directly upregulates NOS expression in both in-vivo and in-vitro endothelial cells resulting in proliferation, migration, and capillary-like tube formation and diminished permeability and apoptosis of these cells (Lovren et al., 2010). Moreover, upregulation of NOS increases cerebral blood flow and prevents stress-induced hypotension, inflammation, apoptosis and cerebral ischemia (Lin et al., 2010). Thus, early administration of nitric oxide or its precursor to patients with acute stroke has been shown to affect lesion size, cerebral blood flow, mood, cognition and quality of life (Willmot et al., 2005; Woodhouse et al., 2015).
Adropin and Cluster of Differentiation 36 (CD36)
CD36 is a member of the class B scavenger receptor family and is activated by various ligands with diverse cellular responses—such as the production of free radicals, induction of inflammatory responses, and endothelial dysfunction (Cho, 2005, 2012). CD36 has anti-angiogenic nature and downregulates VEGFR2 phosphorylation, (Primo et al., 2005) and through its ligands such as oxLDL (a major factor in the development of atherosclerosis) causes endothelial cell stiffness and atherosclerosis (Shentu et al., 2010). Adropin downregulates CD36 gene expression and cell surface CD36 protein levels which indicate a potential reduction of muscle fatty acid uptake (Gao et al., 2015). Alongside, adropin treatment has been shown to downregulate peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) that regulates expression of CD36 (Gao et al., 2015).
CD36 is known to be one of the underlying causes of cerebrovascular and neurodegenerative diseases. Accumulation of β-Amyloid (a CD36 ligand) in the vicinity of plaques of Alzheimer's disease, and in the cerebrovascular wall of hemorrhagic stroke had been described (Winkler et al., 2001; Hernandez-Guillamon et al., 2012). Increased CD36 gene expression following blood-brain barrier damage and circulating amyloid β protein following ischemic insult might contribute to the pathogenesis of vascular dementia and bridge the gap between vascular dementia and Alzheimer's disease (Lee et al., 2005; Ueno et al., 2016).
Adropin and Glucose Oxidation
Adropin upregulates glucose oxidation via decreasing acetylation of pyruvate dehydrogenase complex (PDHC, a rate-limiting enzyme in glucose oxidation) and down-regulating pyruvate dehydrogenase kinase-4 (PDK-4)- a PDHC inhibitor. PDHC is a mitochondrial matrix enzyme complex that catalyzes oxidative decarboxylation of pyruvate to produce acetyl-CoA, which plays a critical role in cerebral aerobic energy metabolism (Cardell et al., 1989; Martin et al., 2005). Impaired cerebral energy metabolism and PDHC activity are seen in acute brain injury and chronic neurodegenerative conditions such as Alzheimer's disease and Wernicke-Korsakoff syndrome (Martin et al., 2005). PDHC activity is attenuated after brain ischemia (Cardell et al., 1989; Martin et al., 2005). This reperfusion dependent suppression might be due to the depressed activity of pyruvate dehydrogenase phosphatase or oxidative stress (because of hyperoxic resuscitation) (Martin et al., 2005). Inactivation of PDHC can be a possible cause of post-ischemic metabolic depression, prolonged intracellular lactic acidosis, and secondary tissue energy depletion, which contribute to neuronal injury and neurological impairment (Cardell et al., 1989; Martin et al., 2005). In addition, compensating the enzyme activity by administration of acetyl-L-carnitine which is converted to acetyl-Co or dichloroacetate (DCA) improves neurologic outcome (Rosenthal et al., 1992; Martin et al., 2005). Adropin treatment in animal studies increases the ratio of CoA/acetyl-CoA which directly promote PDHC activity and pyruvate oxidation (Gao et al., 2015).
Adropin and Endothelial Permeability
The involvement of Adropin in endothelial permeability was originally described by Lovren and coworkers in 2010 (Lovren et al., 2010). Adropin attenuates the hypoxic/low glycemic induced paracellular permeability by inhibiting ROCK/MLC2 signaling pathway (Figure 1; Yang et al., 2016). As described by Wojciak-Stothard and Ridley, the endothelial permeability is determined by intercellular junctions integrity and basal intracellular actinomyosin contractility (Wojciak-Stothard and Ridley, 2002). Rho GTPases such as Rac 1 and Rho A act antagonistically to regulate endothelial permeability (Wojciak-Stothard and Ridley, 2002; Wojciak-Stothard et al., 2006). Rac 1 enhances the cellular junction and adherence, (Wojciak-Stothard et al., 2006) and inhibits Rho under chronic ischemia (Wojciak-Stothard et al., 2005). In contrast, Rho A and its downstream Rho-associated protein kinase (ROCK) enhance the marginal cell isometric tension and actinomysin contractility (Wojciak-Stothard et al., 2006). Hypoxic/hypoglycemic condition induces activation of Rho/ROCK signaling pathway by stimulating K-ras effector pathways independent of HIF-1 (Mizukami et al., 2006; Wojciak-Stothard et al., 2006; Yang et al., 2016) (Figure 1). Activated ROCK promotes direct phosphorylation of myosin light chain 2 (MLC2) at Ser19 site and inhibition of myosin light chain phosphatase (MLCP). Phosphorylated MLC2 enhances actinomyosin contractility, intracellular tension and increases cellular permeability (Yang et al., 2016). In addition, down regulation of Rac 1 induces actin formation via Rho activation and intensifies contractility (Wojciak-Stothard et al., 2006; Weidemann et al., 2013).
Adropin and Neurogenesis
Induction of mesenchymal cells with inhibitors of prolyl hydroxylase—a key enzyme in HIF-1α degradation—promotes mesenchymal cells differentiation to morphologically neuron-like cells (Pacary et al., 2006). HIF-1α production under ischemic conditions induces potentially neurogenic factors—EPO (erythropoietin), p21 and VEGF (Jin et al., 2002; Yu et al., 2002; Pacary et al., 2006). Animal models of ischemic stroke demonstrated functions for VEGF in neuroprotection (better neurological outcomes and smaller infarct volume), neurogenesis (in both early and delayed phases in neuronal precursors) and in angiogenesis (endothelial cell proliferation, migration, survival and vascular permeability) (Jin et al., 2001; Sun et al., 2003; Shimotake et al., 2010). Although neurogenesis and angiogenesis are known to be coupled, the neurotrophic potential of VEGF might be independent of angiogenesis: VEGF induces axonal outgrowth—by acting on growing axons and nerve cell bodies—and suppresses the cell-death pathways mediated by calpain-dependent and caspase-3-dependent mechanisms (Sondell et al., 2000; Jin et al., 2001; Shimotake et al., 2010).
Recent studies demonstrated that inhibition of Rho/ROCK signaling pathway enhances HIF-1 activity and upregulates EPO, VEGF and p21, and consequently potentiates neurogenesis (Pacary et al., 2007, 2008). Adropin might be a novel candidate to promote neurogenesis as it can inhibit the Rho/ ROCK pathway without affecting VEGF level (Yang et al., 2016).
Adropin and Orphan G Protein-Coupled Receptor (GPR19)
Stein et al. discovered GPR19 as a potential adropin receptor (Stein et al., 2016). GPR19 is a transmembrane receptor similar to the neuropeptide Y receptors and the dopamine D2 receptor family (O'Dowd et al., 1996). GPR19 is more likely expressed in cerebellum, caudate, putamen, thalamus, hypothalamus, hippocampus, frontal cortex and olfactory bulb (O'Dowd et al., 1996; Hoffmeister-Ullerich et al., 2004). Transcripts of GPR19 can be detected in neuroectodermal origin tissues in early embryogenesis, and they are gradually restricted to the regions of the developing brain (Hoffmeister-Ullerich et al., 2004). Signal transduction through GPR19 enhances ERK and Akt phosphorylation in cerebral neurons (Hossain et al., 2016). Recently, Stein et al. described the adropin function in water intake inhibition through GPR19 (Stein et al., 2016). However, the distribution of GPR19 and potency of its downstream signaling pathways suggest more critical actions for adropin in neuronal development and protection.
Conclusion
Studies regarding the effects of adropin in different organs are still in infancy stage, but increasing evidence suggest that this peptide has unique effects on endothelial cell function via upregulating eNOS expression through the VEGFR2-PI3K-Akt, VEGFR2-ERK 1/2 pathways and inhibition of Rho/ROCK pathway. However, our current knowledge mainly comes from animal studies or treatment with the putative secreted domain of adropin. Whether these findings are transferable to clinical studies needs to be determined. Moreover, adropin may be utilized as a promising biomarker for CNS disease risk stratification or diagnosis, and/or a potential therapeutic candidate in CNS injuries. Although adropin seems to be a novel target to limit vascular diseases, in parallel with the documented effects on metabolic modulation, further investigations are needed to elucidate the specific mechanism underlying the association between adropin and CNS diseases.
Author Contributions
Study concept and design: SA, RZ. Acquisition of data: SS, SA, TP, RZ. Drafting and critical revision of manuscript: SS, SA, TP, RZ. Study supervision: TP, RZ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Altincik, A., and Sayin, O. (2015). Evaluation of the relationship between serum adropin levels and blood pressure in obese children. J. Pediatr. Endocrinol. Metab. 28, 1095–1100. doi: 10.1515/jpem-2015-0051
Altintas, O., Kumas, M., and Altintas, M. (2016). Neuroprotective effect of ischemic preconditioning via modulating the expression of adropin and oxidative markers against transient cerebral ischemia in diabetic rats. Peptides 79, 31–38. doi: 10.1016/j.peptides.2016.03.011
Annovazzi, L., Mellai, M., Caldera, V., Valente, G., Tessitore, L., and Schiffer, D. (2009). mTOR, S6 and AKT expression in relation to proliferation and apoptosis/autophagy in glioma. Anticancer Res. 29, 3087–3094.
Aydin, S. (2014). Three new players in energy regulation: preptin, adropin and irisin. Peptides 56, 94–110. doi: 10.1016/j.peptides.2014.03.021
Aydin, S., Kuloglu, T., Aydin, S., Eren, M. N., Yilmaz, M., Kalayci, M., et al. (2013). Expression of adropin in rat brain, cerebellum, kidneys, heart, liver, and pancreas in streptozotocin-induced diabetes. Mol. Cell. Biochem. 380, 73–81. doi: 10.1007/s11010-013-1660-4
Aydin, S., Kuloglu, T., Aydin, S., Kalayci, M., and Yilmaz, M., Çakmak, T., et al. (2014). Elevated adropin: a candidate diagnostic marker for myocardial infarction in conjunction with troponin-I. Peptides 58, 91–97. doi: 10.1016/j.peptides.2014.06.001
Beaulieu, J.-M., Gainetdinov, R. R., and Caron, M. G. (2007). The Akt–GSK-3 signaling cascade in the actions of dopamine. Trends Pharmacol. Sci. 28, 166–172. doi: 10.1016/j.tips.2007.02.006
Blanco-aparicio, C., Renner, O., Leal, J. F. M., and Carnero, A. (2007). PTEN, more than the AKT Pathway. Carcinogenesis 28, 1379–1386. doi: 10.1093/carcin/bgm052
Burke, R. E. (2007). Inhibition of mapk and stimulation of akt kinase signaling pathways: two approaches with therapeutic potential in the treatment of neurodegenerative disease. Pharmacol. Ther. 114, 261–277. doi: 10.1016/j.pharmthera.2007.02.002
Canals, J. M. (2004). Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in huntington's disease. J. Neurosci. 24, 7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004
Cardell, M., Koide, T., and Wieloch, T. (1989). Pyruvate dehydrogenase activity in the rat cerebral cortex following cerebral ischemia. J. Cereb. Blood Flow Metab. 9, 350–357. doi: 10.1038/jcbfm.1989.53
Celik, A., Balin, M., Kobat, M. A., Erdem, K., Baydas, A., Bulut, M., et al. (2013). Deficiency of a new protein associated with cardiac syndrome X; called adropin. Cardiovasc. Ther. 31, 174–178. doi: 10.1111/1755-5922.12025
Chen, H., Qu, Y., Tang, B., Xiong, T., and Mu, D. (2012). Role of mammalian target of rapamycin in hypoxic or ischemic brain injury: potential neuroprotection and limitations. Rev. Neurosci. 23, 279–287. doi: 10.1515/revneuro-2012-0001
Cho, S. (2005). The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J. Neurosci. 25, 2504–2512. doi: 10.1523/JNEUROSCI.0035-05.2005
Cho, S. (2012). CD36 as a therapeutic target for endothelial dysfunction in stroke. Curr. Pharm. Des. 18, 3721–3730. doi: 10.2174/138161212802002760
Chong, Z. Z., Li, F., and Maiese, and, K. (2005). Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 20, 299–315.
Colin, E., Régulier, E., Perrin, V., Dürr, A., Brice, A., Aebischer, P., et al. (2005). Akt is altered in an animal model of Huntington's disease and in patients. Eur. J. Neurosci. 21, 1478–1488. doi: 10.1111/j.1460-9568.2005.03985.x
Demircelik, B. (2014). Adropin: a new marker for predicting late saphenous vein graft disease after coronary artery bypass. Grafting 37, 338–344.
Demircelik, B., and Kurtul, A. (2015). The relationship between adropin levels and the slow coronary flow phenomenon. Ind. J. Clin. Biochem. 30, 412–417. doi: 10.1007/s12291-014-0470-0
Emamian, E. S., Hall, D., Birnbaum, M. J., Karayiorgou, M., and Gogos, J., a (2004). Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat. Genet. 36, 131–137. doi: 10.1038/ng1296
Franke, T. F., Hornik, C. P., Segev, L., Shostak, G. A., and Sugimoto, C. (2003). PI3K/Akt and apoptosis: size matters. Oncogene 22, 8983–8998. doi: 10.1038/sj.onc.1207115
Ganesh Kumar, K., Zhang, J., Gao, S., Rossi, J., McGuinness, O. P., Halem, H. H., et al. (2012). Adropin deficiency is associated with increased adiposity and insulin resistance. Obesity 20, 1394–1402. doi: 10.1038/oby.2012.31
Gao, S., Mcmillan, R. P., Zhu, Q., Lopaschuk, G. D., Hulver, M. W., and Butler, A. A. (2015). Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet- induced obese mice with insulin resistance. Mol. Metab. 4, 1–15. doi: 10.1016/j.molmet.2015.01.005
Gao, X., Zhang, H., Takahashi, T., Hsieh, J., Liao, J., Steinberg, G., et al. (2008). The Akt signaling pathway contributes to postconditioning's protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem. 105, 943–955. doi: 10.1111/j.1471-4159.2008.05218.x
Giralt, A., Torres-peraza, J. F., Canals, J. M., and D.I. M. (2010). PH domain leucine-rich repeat protein phosphatase 1 contributes to maintain the activation of the PI3K / Akt pro-survival pathway in Huntington's disease striatum. Cell Death Differ. 17, 324–335. doi: 10.1038/cdd.2009.127
Gozal, D., Kheirandish-Gozal, L., Bhattacharjee, R., Molero-Ramirez, H., Tan, H.-L., and Bandla, H. P. R. (2013). Circulating adropin concentrations in pediatric obstructive sleep apnea: potential relevance to endothelial function. J. Pediatr. 163, 1122–1126. doi: 10.1016/j.jpeds.2013.05.040
Griffin, R. J., Moloney, A., Kelliher, M., Johnston, J. A., Ravid, R., Dockery, P., et al. (2005). Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J. Neurochem. 93, 105–117. doi: 10.1111/j.1471-4159.2004.02949.x
Gu, X., Li, H., Zhu, X., Gu, H., and Chen, J. (2015). Inverse correlation between plasma adropin and ET-1 levels in essential hypertension a cross-sectional study. Medicine (Baltimore). 94, 1–5. doi: 10.1097/MD.0000000000001712
Hanumanthappa, P., Densi, A., and Krishnamurthy, R. G. (2014). Glycogen Synthase Kinase-β3 in Ischemic Neuronal Death. Curr. Neurovasc. Res. 11, 271–278. doi: 10.2174/1567202611666140520151002
Hassan, A. (2004). Endothelial nitric oxide gene haplotypes and risk of cerebral small-vessel disease. Stroke 35, 654–659. doi: 10.1161/01.STR.0000117238.75736.53
Hernandez-Guillamon, M., Martinez-Saez, E., Delgado, P., Domingues-Montanari, S., Boada, C., Penalba, A., et al. (2012). MMP-2/MMP-9 plasma level and brain expression in cerebral amyloid angiopathy-associated hemorrhagic stroke. Brain Pathol. 22, 133–141. doi: 10.1111/j.1750-3639.2011.00512.x
Hoffmeister-Ullerich, S. A. H., Süsens, U., and Schaller, H. C. (2004). The orphan G-protein-coupled receptor GPR19 is expressed predominantly in neuronal cells during mouse embryogenesis. Cell Tissue Res. 318, 459–463. doi: 10.1007/s00441-004-0948-9
Hossain, M. S., Mineno, K., and Katafuchi, T. (2016). Neuronal orphan g-protein coupled receptor proteins mediate plasmalogens-induced activation of ERK and Akt signaling. PLoS ONE 11:e0150846. doi: 10.1371/journal.pone.0150846
Humbert, S., Bryson, E., A., Cordelières, F. P., Connors, N. C., Datta, S. R., Finkbeiner, S., et al. (2002). The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves huntingtin phosphorylation by Akt. Dev. Cell 2, 831–837. doi: 10.1016/S1534-5807(02)00188-0
Ikeda, M., Iwata, N., Suzuki, T., Kitajima, T., Yamanouchi, Y., Kinoshita, Y., et al. (2004). Association of AKT1 with schizophrenia confirmed in a Japanese population. Biol. Psychiatry 56, 698–700. doi: 10.1016/j.biopsych.2004.07.023
Jeynes, B., and Provias, J. (2009). Significant negative correlations between capillary expressed eNOS and Alzheimer lesion burden. Neurosci. Lett. 463, 244–248. doi: 10.1016/j.neulet.2009.07.091
Jin, K., Mao, X. O., Batteur, S. P., McEachron, E., Leahy, A., and Greenberg, D. A. (2001). Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience 108, 351–358. doi: 10.1016/S0306-4522(01)00154-3
Jin, K., Zhu, Y., Sun, Y., Mao, X. O., Xie, L., and Greenberg, D. A. (2002). Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 99, 11946–11950. doi: 10.1073/pnas.182296499
Jope, R. S. (2011). Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front. Mol. Neurosci. 4:16. doi: 10.3389/fnmol.2011.00016
Kalkman, H. O. (2006). The role of the phosphatidylinositide 3-kinase-protein kinase B pathway in schizophrenia. Pharmacol. Ther. 110, 117–134. doi: 10.1016/j.pharmthera.2005.10.014
Karege, F., Méary, A., Perroud, N., Jamain, S., Leboyer, M., Ballmann, E., et al. (2012). Genetic overlap between schizophrenia and bipolar disorder: a study with AKT1 gene variants and clinical phenotypes. Schizophr. Res. 135, 8–14. doi: 10.1016/j.schres.2011.12.015
Kim, H.-W., Rapoport, S. I., and Rao, J. S. (2010). Altered expression of apoptotic factors and synaptic markers in postmortem brain from bipolar disorder patients. Neurobiol. Dis. 37, 596–603. doi: 10.1016/j.nbd.2009.11.010
Kumar, K. G., Trevaskis, J. L., Lam, D. D., Sutton, G. M., Koza, R. A., Chouljenko, V. N., et al. (2008). Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 8, 468–481. doi: 10.1016/j.cmet.2008.10.011
Lee, P. H., Bang, O. Y., Hwang, E. M., Lee, J. S., Joo, U. S., Mook-Jung, I., et al. (2005). Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural Transm. 112, 1371–1379. doi: 10.1007/s00702-004-0274-0
Levy, O., a., Malagelada, C., and Greene, L., A. (2009). Cell death pathways in Parkinson's disease: proximal triggers, distal effectors, and final steps. Apoptosis 14, 478–500. doi: 10.1007/s10495-008-0309-3
Li, L., Xie, W., Zheng, X. L., Yin, W. D., and Tang, C. K. (2016). A novel peptide adropin in cardiovascular diseases. Clin. Chim. Acta 453, 107–113. doi: 10.1016/j.cca.2015.12.010
Li, N., Lee, B., Liu, R.-J., Banasr, M., Dwyer, J. M., Iwata, M., et al. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959. doi: 10.1126/science.1190287
Li, W., Yang, Y., Hu, Z., Ling, S., and Fang, M. (2015). Neuroprotective effects of DAHP and Triptolide in focal cerebral ischemia via apoptosis inhibition and PI3K/Akt/mTOR pathway activation. Front. Neuroanat. 9:48. doi: 10.3389/fnana.2015.00048
Li, X., and Jope, R. S. (2010). Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology 35, 2143–2154. doi: 10.1038/npp.2010.105
Lian, W., Gu, X., Qin, Y., and Zheng, X. (2011). Elevated plasma levels of adropin in heart failure patients. Intern. Med. 50, 1523–1527. doi: 10.2169/internalmedicine.50.5163
Lin, H., Wu, C., and Huang, C. (2010). The Akt-endothelial nitric oxide synthase pathway in hypoxic – ischemic tolerance in the neonatal rat brain. Stroke 1543–1551. doi: 10.1161/STROKEAHA.109.574004
Lovren, F., Pan, Y., Quan, A., Singh, K. K., Shukla, P. C., Gupta, M., et al. (2010). Adropin is a novel regulator of endothelial function. Circulation 122, S185–S192. doi: 10.1161/CIRCULATIONAHA.109.931782
Machado-Vieira, R., Zanetti, M. V., Teixeira, A. L., Uno, M., Valiengo, L. L., Soeiro-de-Souza, M. G., et al. (2015). Decreased AKT1/mTOR pathway mRNA expression in short-term bipolar disorder. Eur. Neuropsychopharmacol. 25, 468–473. doi: 10.1016/j.euroneuro.2015.02.002
Manning, B. D., and Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129, 1261–1274. doi: 10.1016/j.cell.2007.06.009
Martin, E., Rosenthal, R. E., and Fiskum, G. (2005). Pyruvate dehydrogenase complex: Metabolic link to ischemic brain injury and target of oxidative stress. J. Neurosci. Res. 79, 240–247. doi: 10.1002/jnr.20293
Mattson, M. P., Duan, W., Pedersen, W. A., and Culmsee, C. (2001). Neurodegenerative disorders and ischemic brain diseases. Apoptosis 6, 69–81. doi: 10.1023/A:1009676112184
Mizukami, Y., Fujiki, K., Duerr, E.-M., Gala, M., Jo, W.-S., Zhang, X., et al. (2006). Hypoxic regulation of vascular endothelial growth factor through the induction of phosphatidylinositol 3-kinase/Rho/ROCK and c-Myc. J. Biol. Chem. 281, 13957–13963. doi: 10.1074/jbc.M511763200
Mu, D., Jiang, X., Sheldon, R. A., Fox, C. K., Hamrick, S. E. G., Vexler, Z. S., et al. (2003). Regulation of hypoxia-inducible factor 1α and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol. Dis. 14, 524–534. doi: 10.1016/j.nbd.2003.08.020
Noshita, N., Lewén, A., Sugawara, T., and Chan, P. H. (2001). Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 21, 1442–1450. doi: 10.1097/00004647-200112000-00009
Nuzzo, D., Picone, P., Baldassano, S., Caruana, L., Messina, E., Gammazza, A. M., et al. (2015). Insulin resistance as common molecular denominator linking obesity to Alzheimer's disease. Curr. Alzheimer Res. 12, 723–35. doi: 10.2174/1567205012666150710115506
O'Dowd, B. F., Nguyen, T., Lynch, K. R., Kolakowski, L. F., Thompson, M., Cheng, R., et al. (1996). A novel gene codes for a putative G protein-coupled receptor with an abundant expression in brain. FEBS Lett. 394, 325–329. doi: 10.1016/0014-5793(96)00901-5
Oğurel, T., Oğurel, R., and Topuz, M., Örnek, N., and Örnek, K. (2016). Plasma adropin level in patients with pseudoexfoliation. Int. Ophthalmol. doi: 10.1007/s10792-016-0185-8. [Epub ahead of print].
Pacary, E., Legros, H., Valable, S., Duchatelle, P., Lecocq, M., Petit, E., et al. (2006). Synergistic effects of CoCl(2) and ROCK inhibition on mesenchymal stem cell differentiation into neuron-like cells. J. Cell Sci. 119, 2667–2678. doi: 10.1242/jcs.03004
Pacary, E., Petit, E., and Bernaudin, M. (2008). Concomitant inhibition of prolyl hydroxylases and ROCK initiates differentiation of mesenchymal stem cells and PC12 towards the neuronal lineage. Biochem. Biophys. Res. Commun. 377, 400–406. doi: 10.1016/j.bbrc.2008.09.145
Pacary, E., Tixier, E., Coulet, F., Roussel, S., Petit, E., and Bernaudin, M. (2007). Crosstalk between HIF-1 and ROCK pathways in neuronal differentiation of mesenchymal stem cells, neurospheres and in PC12 neurite outgrowth. Mol. Cell. Neurosci. 35, 409–423. doi: 10.1016/j.mcn.2007.04.002
Papazoglou, I. K., Jean, A., Gertler, A., Taouis, M., and Vacher, C.-M. (2014). Hippocampal GSK3β as a molecular link between obesity and depression. Mol. Neurobiol. 363–374. doi: 10.1007/s12035-014-8863-x
Peng, B., Guo, Q., He, Z., Ye, Z., Yuan, Y., Wang, N., et al. (2012). Remote ischemic postconditioning protects the brain from global cerebral ischemia / reperfusion injury by up-regulating endothelial nitric oxide synthase through the PI3K / Akt pathway. Brain Res. 1445, 92–102. doi: 10.1016/j.brainres.2012.01.033
Primo, L., Ferrandi, C., Roca, C., Marchiò, S., di Blasio, L., Alessio, M., et al. (2005). Identification of CD36 molecular features required for its in vitro angiostatic activity. FASEB J. 19, 1713–1715. doi: 10.1096/fj.05-3697fje
Ries, V., Henchcliffe, C., Kareva, T., Rzhetskaya, M., Bland, R., During, M. J., et al. (2006). Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 18757–18762. doi: 10.1073/pnas.0606401103
Rosenthal, R. E., Williams, R., Bogaert, Y. E., Getson, P. R., and Fiskum, G. (1992). Prevention of postischemic canine neurological injury through potentiation of brain energy metabolism by acetyl-L-carnitine. Stroke 23, 1312–1318. doi: 10.1161/01.STR.23.9.1312
Schwab, S. G., Hoefgen, B., Hanses, C., Hassenbach, M. B., Albus, M., Lerer, B., et al. (2005). Further evidence for association of variants in the AKT1 gene with schizophrenia in a sample of European sib-pair families. Biol. Psychiatry 58, 446–450. doi: 10.1016/j.biopsych.2005.05.005
Shentu, T. P., Titushkin, I., Singh, D. K., Gooch, K. J., Subbaiah, P. V., Cho, M., et al. (2010). oxLDL-induced decrease in lipid order of membrane domains is inversely correlated with endothelial stiffness and network formation. Am. J. Physiol. Cell Physiol. 299, C218–C229. doi: 10.1152/ajpcell.00383.2009
Shimotake, J., Derugin, N., Wendland, M., Vexler, Z. S., and Donna, M., Ferriero (2010). Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke 41, 343–349. doi: 10.1161/STROKEAHA.109.564229
Sondell, M., Sundler, F., and Kanje, M. (2000). Vascular endothelial growth factor is a neurotrophic factor which stimulates axonal outgrowth through the flk-1 receptor. Eur. J. Neurosci. 12, 4243–4254. doi: 10.1046/j.0953-816X.2000.01326.x
Stein, L. M., Yosten, G. L. C., and Samson, W. K. (2016). Adropin acts in brain to inhibit water drinking: potential interaction with the orphan G protein-coupled receptor, GPR19. Am. J. Physiol. Regul. Integr. Comp. Physiol. 310, R476–R480. doi: 10.1152/ajpregu.00511.2015
Sun, Y., Jin, K., Xie, L., Childs, J., Mao, X. O., Logvinova, A., et al. (2003). VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Invest. 111, 1843–1851. doi: 10.1172/JCI200317977
Tan, X.-L., Xue, Y.-Q., Ma, T., Wang, X., Li, J. J., Lan, L., et al. (2015). Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol. Neurodegener. 10, 24. doi: 10.1186/s13024-015-0020-0
Thiselton, D. L., Vladimirov, V. I., Kuo, P.-H., McClay, J., Wormley, B., Fanous, A., et al. (2008). AKT1 Is associated with schizophrenia across multiple symptom dimensions in the irish study of high density schizophrenia families. Biol. Psychiatry 63, 449–457. doi: 10.1016/j.biopsych.2007.06.005
Timmons, S., Coakley, M. F., Moloney, A. M., and Neill, C. O. (2009). Akt signal transduction dysfunction in Parkinson' s disease. Neurosci. Lett. 467, 30–35. doi: 10.1016/j.neulet.2009.09.055
Tiwari, S. K., Seth, B., Agarwal, S., Yadav, A., Karmakar, M., Gupta, S. K., et al. (2015). Ethosuximide induces hippocampal neurogenesis and reverses cognitive deficits in an Amyloid-β toxin-induced alzheimer rat model via the phosphatidylinositol 3-kinase (PI3K)/Akt/Wnt/β-Catenin Pathway. J Biol Chem 290, 28540–28558. doi: 10.1074/jbc.M115.652586
Ueno, M., Chiba, Y., Matsumoto, K., Murakami, R., Fujihara, R., Kawauchi, M., et al. (2016). Blood-brain barrier damage in vascular dementia. Neuropathology 36, 115–124. doi: 10.1111/neup.12262
Wang, H., Wang, G., Yu, Y., and Wang, Y. (2009). The role of phosphoinositide-3-kinase/Akt pathway in propofol-induced postconditioning against focal cerebral ischemia-reperfusion injury in rats. Brain Res. 1297, 177–184. doi: 10.1016/j.brainres.2009.08.054
Weidemann, A., Breyer, J., Rehm, M., Eckardt, K.-U., Daniel, C., Cicha, I., et al. (2013). HIF-1α activation results in actin cytoskeleton reorganization and modulation of Rac-1 signaling in endothelial cells. Cell Commun. Signal. 11, 80. doi: 10.1186/1478-811X-11-80
Willmot, M., Gray, L., Gibson, C., Murphy, S., and Bath, P. M. W. (2005). A systematic review of nitric oxide donors and l-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide 12, 141–149. doi: 10.1016/j.niox.2005.01.003
Winkler, D. T., Bondolfi, L., Herzig, M. C., Jann, L., Calhoun, M. E., Wiederhold, K. H., et al. (2001). Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 21, 1619–1627.
Wojciak-Stothard, B., and Ridley, A. J. (2002). Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 39, 187–199. doi: 10.1016/S1537-1891(03)00008-9
Wojciak-Stothard, B., Tsang, L. Y. F., and Hawrth, S. G. (2005). Rac and Rho play opposing roles in the regulation of hypoxia/reoxygenation-induced permeability changes in pulmonary artery endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 288, L749–L760. doi: 10.1152/ajplung.00361.2004
Wojciak-Stothard, B., Tsang, L. Y. F., Paleolog, E., Hall, S. M., and Haworth, S. G. (2006). Rac1 and RhoA as regulators of endothelial phenotype and barrier function in hypoxia-induced neonatal pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L1173–L1182. doi: 10.1152/ajplung.00309.2005
Wong, C.-M., Wang, Y., Lee, J. T. H., Huang, Z., Wu, D., Xu, A., et al. (2014). Adropin is a brain membrane-bound protein regulating physical activity via the NB-3/notch signaling pathway in mice. J. Biol. Chem. 289, 25976–25986. doi: 10.1074/jbc.M114.576058
Woodhouse, L., Scutt, P., Krishnan, K., Berge, E., Gommans, J., Ntaios, G., et al. (2015). Effect of hyperacute administration (within 6 hours) of transdermal glyceryl trinitrate, a nitric oxide donor, on outcome after stroke. Stroke 46, 00–00. doi: 10.1161/STROKEAHA.115.009647
Wu, H., Yang, S., Dai, J., Qiu, Y., Miao, Y., and Zhang, X. (2015). Combination of early and delayed ischemic postconditioning enhances brain-derived neurotrophic factor production by upregulating the ERK-CREB pathway in rats with focal ischemia. Mol. Med. Rep. 6427–6434. doi: 10.3892/mmr.2015.4327
Wu, L., Fang, J., Chen, L., Zhao, Z., Luo, Y., Lin, C., et al. (2014). Low serum adropin is associated with coronary atherosclerosis in type 2 diabetic and non-diabetic patients. Clin. Chem. Lab. Med. 52, 751–758. doi: 10.1515/cclm-2013-0844
Yang, C., DeMars, K. M., Hawkins, K. E., and Candelario-Jalil, E. (2016). Adropin reduces paracellular permeability of rat brain endothelial cells exposed to ischemia-like conditions. Peptides 81, 29–37. doi: 10.1016/j.peptides.2016.03.009
Yu, H., Zhao, P., Wu, M., Liu, J., and Yin, W. (2014). Serum adropin levels are decreased in patients with acute myocardial infarction. Regul. Pept. 190, 46–49. doi: 10.1016/j.regpep.2014.04.001
Yu, X. B., Shacka, J. J., Eells, J. B., Suarez-Quian, C., Przygodzki, R. M., Beleslin-Cokic, B., et al. (2002). Erythropoietin receptor signalling is required for normal brain development. Development 129, 505–516.
Zhao, L. P., Xu, W. T., Wang, L., You, T., Chan, S. P., Zhao, X., et al. (2015b). Serum adropin level in patients with stable coronary artery disease. Hear. Lung Circ. 24, 975–979. doi: 10.1016/j.hlc.2015.03.008
Zhao, L. P., You, T., Chan, S. P., Chen, J. C., and Xu, W. T. (2015a). Adropin is associated with hyperhomocysteine and coronary atherosclerosis. Exp. Ther. Med. 1065–1070.
Zhao, Y., Li, J., Tang, Q., Zhang, P., Jing, L., Chen, C., et al. (2014). Regulation of extracellular signal?regulated kinase 1/2 influences hippocampal neuronal survival in a rat model of diabetic cerebral ischemia. Neural Regen. Res. 9, 749–756. doi: 10.4103/1673-5374.131581
Zhao, Z., Ksiezak-Reding, H., Riggio, S., Haroutunian, V., and Pasinetti, G. M. (2006). Insulin receptor deficits in schizophrenia and in cellular and animal models of insulin receptor dysfunction. Schizophr. Res. 84, 1–14. doi: 10.1016/j.schres.2006.02.009
Keywords: adropin, neurodegenerative disease, neuroprotection, biomarker, predictor, therapeutic, cellular signaling pathways
Citation: Shahjouei S, Ansari S, Pourmotabbed T and Zand R (2016) Potential Roles of Adropin in Central Nervous System: Review of Current Literature. Front. Mol. Biosci. 3:25. doi: 10.3389/fmolb.2016.00025
Received: 21 March 2016; Accepted: 27 May 2016;
Published: 27 June 2016.
Edited by:
Megha Agrawal, University of Illinois at Chicago, USAReviewed by:
Alireza Noorian, Kaiser Permanente Orange County Stroke Program, USALeili Shahgholi, University of Florida, USA
Bardia Nourbakhsh, University of California, San Francisco, USA
Copyright © 2016 Shahjouei, Ansari, Pourmotabbed and Zand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramin Zand, cnphbmRAdXRoc2MuZWR1