 Elisabeth Grohmann
Elisabeth Grohmann Nikolaus Goessweiner-Mohr3,4,5,6*
Nikolaus Goessweiner-Mohr3,4,5,6* Sabine Brantl
Sabine Brantl- 1Division of Infectious Diseases, University Medical Center Freiburg, Freiburg im Breisgau, Germany
- 2Life Sciences and Technology, Beuth University of Applied Sciences Berlin, Berlin, Germany
- 3Center for Structural System Biology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 4Deutsches Elektronen-Synchrotron, Hamburg, Germany
- 5Institute of Molecular Biotechnology, Austrian Academy of Sciences, Vienna, Austria
- 6Research Institute of Molecular Pathology, Vienna, Austria
- 7Lehrstuhl für Genetik, Biologisch-Pharmazeutische Fakultät, AG Bakteriengenetik, Friedrich-Schiller-Universität Jena, Jena, Germany
pIP501 is a Gram-positive broad-host-range model plasmid intensively used for studying plasmid replication and conjugative transfer. It is a multiple antibiotic resistance plasmid frequently detected in clinical Enterococcus faecalis and Enterococcus faecium strains. Replication of pIP501 proceeds unidirectionally by a theta mechanism. The minimal replicon of pIP501 is composed of the repR gene encoding the essential rate-limiting replication initiator protein RepR and the origin of replication, oriR, located downstream of repR. RepR is similar to RepE of related streptococcal plasmid pAMβ1, which has been shown to possess RNase activity cleaving free RNA molecules in close proximity of the initiation site of DNA synthesis. Replication of pIP501 is controlled by the concerted action of a small protein, CopR, and an antisense RNA, RNAIII. CopR has a dual function: It acts as transcriptional repressor at the repR promoter and, in addition, prevents convergent transcription of RNAIII and repR mRNA (RNAII), which indirectly increases RNAIII synthesis. CopR binds asymmetrically as a dimer at two consecutive binding sites upstream of and overlapping with the repR promoter. RNAIII induces transcriptional attenuation within the leader region of the repR mRNA (RNAII). Deletion of either control component causes a 10- to 20-fold increase of plasmid copy number, while simultaneous deletions have no additional effect. Conjugative transfer of pIP501 depends on a type IV secretion system (T4SS) encoded in a single operon. Its transfer host-range is considerably broad, as it has been transferred to virtually all Gram-positive bacteria including Streptomyces and even the Gram-negative Escherichia coli. Expression of the 15 genes encoding the T4SS is tightly controlled by binding of the relaxase TraA, the transfer initiator protein, to the operon promoter overlapping with the origin of transfer (oriT). The T4SS operon encodes the DNA-binding proteins TraJ (VirD4-like coupling protein) and the VirB4-like ATPase, TraE. Both proteins are actively involved in conjugative DNA transport. Moreover, the operon encodes TraN, a small cytoplasmic protein, whose specific binding to a sequence upstream of the oriT nic-site was demonstrated. TraN seems to be an effective repressor of pIP501 transfer, as conjugative transfer rates were significantly increased in an E. faecalis pIP501ΔtraN mutant.
Introduction
Plasmids are extrachromosomal elements, by definition not encoding any essential functions for the bacterial host but rather contributing additional traits, which can be advantageous or even essential for survival under particular conditions, e.g., in the presence of antibiotic pressure. pIP501 is a considerably small, 30.6-kb broad-host-range self-transmissible plasmid, which was isolated from a clinical Streptococcus agalactiae strain (Evans and Macrina, 1983). It belongs to incompatibility group Inc18 and encodes resistance to antibiotics of the macrolide/lincosamide/streptogramin (MLS) group and to chloramphenicol.
Inc18 plasmids encode an efficient plasmid stabilization system, the ε-θ ζ locus functioning as a toxin-antitoxin system (Ceglowski et al., 1993). The ϖ–ε-ζ operon of pSM19035 and of other Inc18 plasmids is a novel proteic plasmid addiction system in which the ε and ζ genes code for an antitoxin and a toxin, respectively, while ϖ- plays an autoregulatory role. Broad-host-range efficiency of the ϖ–ε-ζ cassette has been demonstrated in eight different Gram-positive bacteria, including among others the human pathogens, E. faecalis, S. agalactiae, Streptococcus pyogenes, and Staphylococcus aureus (Brzozowska et al., 2012). Expression of toxin Zeta was shown to be bactericidal for Gram-positive bacteria and bacteriostatic for the Gram-negative Escherichia coli, thus stabilizing plasmids in E. coli less efficiently than in Gram-positive bacteria (Zielenkiewicz and Ceglowski, 2005).
pIP501 replicons stabilized by this toxin-antitoxin system have been frequently encountered in E. faecium isolates from geographically diverse clinical, human community and poultry fecal origin (Rosvoll et al., 2010). In addition, pIP501-like replicons are often linked with the vancomycin resistance phenotype encoded by vanA (Rosvoll et al., 2010).
pIP501 is characterized by a replicon, which is tightly controlled on several levels by protein and RNA key players, and a conjugative transfer (tra) region comprising almost half of the plasmid genome encoding 15 putative Tra factors making up a Gram-positive T4SS. The review summarizes (i) key findings on replication and copy number control processes that involve DNA-binding proteins and (ii) current knowledge on key factors of the pIP501 T4SS whose activity involves interaction with DNA. The review ends with a Conclusion and Perspective section on urgent future research needs in the field of plasmid biology.
pIP501 Replication and Copy Number Control
Plasmid pIP501 from S. agalactiae belongs, together with pAMβ1 from E. faecalis and pSM19035 from S. pyogenes to the Inc18 family of plasmids that replicate unidirectionally by the theta mechanism (Brantl et al., 1990; Bruand et al., 1993) in a multitude of Gram-positive bacteria, including Bacillus subtilis. All three plasmids show a high degree of sequence identity in their replication regions (Brantl et al., 1989, 1990; Swinfield et al., 1990).
The RepR Protein
The minimal pIP501 replicon comprises the repR gene encoding the essential replication initiator protein RepR (57.4 kDa) and the replication origin, oriR (Brantl et al., 1990) located downstream of the repR gene (see Figure 1). The minimal origin oriR has been narrowed down to 52 bp and includes an inverted repeat, both branches of which are essential (Brantl and Behnke, 1992a). The RepR protein is rate-limiting for pIP501 replication (Brantl and Behnke, 1992c) and can both act in cis and in trans at oriR. The repR promoter pII is located 300 bp upstream of the Shine-Dalgarno (SD) sequence of the repR gene, and this leader region proved to be essential for replication control (see below). RepR of pIP501 has not been analyzed in detail. However, the highly similar RepE protein from pAMβ1 has been biochemically characterized (Le Chatelier et al., 2001): RepE is a monomer and binds specifically, rapidly and durably to the origin oriEpAMβ1 at a unique binding site immediately upstream of the replication initiation site. RepE binding induces only a weak bend. In addition, it also binds non-specifically to single stranded (ss) DNA with a 2- to 4-fold greater affinity than for double stranded (ds) oriE. RepE binding to oriEpAMβ1 causes denaturation of the AT-rich sequence downstream of its binding site yielding an open complex that is atypical: Its formation does not require multiple RepE binding sites or a strong oriE bending or any co-factors, and its melted region acts as substrate for RepE binding. These properties and the requirement of transcription through the origin for DNA polymerase I to initiate replication as well as a primosome to load the replisome indicate that RepE might assist primer generation at the origin. It has been hypothesized that it might cleave its own repE mRNA downstream of the ORF to generate the replication primer. As RepRpIP501 and RepEpAMβ1 display 97% sequence identity, it can be assumed that these characteristics also apply for RepRpIP501.
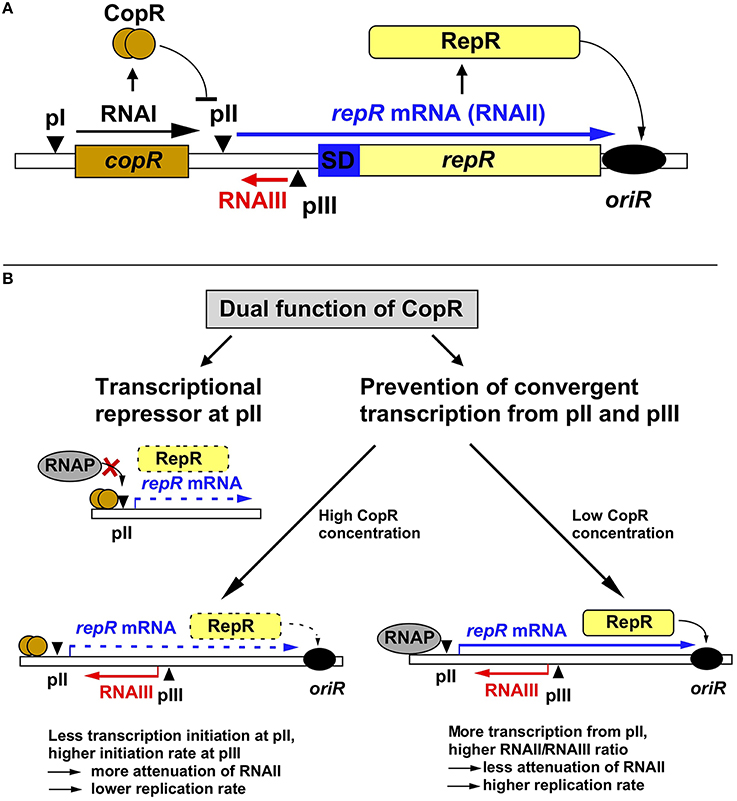
Figure 1. Regulation of pIP501 replication. (A) Working model on regulation of pIP501 replication. The minimal replicon with the copR and repR genes is shown, separated by the 329-bp long leader region. (B) Dual function of CopR. On the one hand, CopR represses transcription from the repR promoter pII by binding to its operator region upstream of the −35 box, thereby inhibiting RNAP binding. On the other hand, CopR prevents convergent transcription from pII and pIII that, in its absence reduces initiation at the supercoiling-sensitive antisense promoter pIII which results in lower RNAIII compared to RNAII levels, yielding a lower attenuation rate, and, hence, higher replication rate. Additionally, the higher amount of RNAII (repR mRNA) titrates the remaining long-lived RNAIII, which further reduces the amount of the inhibitor. In the presence of CopR, RNAP transcribes less frequently through pIII, which allows higher initiation rates at pIII resulting in increased premature termination of RNAII transcription and, consequently, lower replication rates. Left, the plasmid-copy number is 10-fold lower than right, but relatively more RNAIII is present, the amount of RNAIII is (as determined in Northern blots) approximately the same in both cases, reflected by the same thickness of the red arrows symbolizing RNAIII.
Regulation of pIP501 Replication by Two Components
Replication of pIP501 is regulated by the products of two non-essential genes, copR and rnaIII (Brantl, 2014, 2015), which control the synthesis of the rate-limiting replication initiator protein RepR (Brantl and Behnke, 1992c). RNAIII is a 136-nt long antisense RNA and CopR is a small protein composed of 92 amino acids (aa, see below). RNAIII induces premature termination (attenuation) of transcription of the essential repR mRNA (Brantl et al., 1993; Brantl and Wagner, 1994, 1996; Heidrich and Brantl, 2003, 2007). CopR acts as transcriptional repressor at the essential repR promoter pII (Brantl, 1994). Point mutations and deletions in either rnaIII or copR result in the same 10- to 20-fold increase in the copy number of pIP501 derivatives (Brantl and Behnke, 1992b). However, simultaneous deletions do not display additive effects suggesting the involvement of a limiting host factor. Surprisingly, the half-life of RNAIII is with 30 min unusually long (Brantl and Wagner, 1996). Such a long-lived antisense RNA should presumably be a poor regulator since fortuitous decreases in plasmid copy number in individual cells could only be slowly corrected, resulting in unstable maintenance. However, unstable maintenance of pIP501 derivatives was never observed, likely because the second regulator CopR in fact has a dual function and thus provides pIP501 with a strategy to cope with the risk of unstable inheritance (Figure 1). CopR exerts its effect, by the same molecular event, on two levels: transcriptional repression of repR mRNA synthesis (see below), and accumulation of RNAIII by prevention of convergent transcription, thereby indirectly increasing transcription initiation from the antisense promoter pIII (Brantl and Wagner, 1997). The discovery of a second function for CopR was initiated by the surprising finding that high copy number pIP501 derivatives lacking copR and low copy number derivatives containing copR produce the same intracellular amounts of RNAIII. Transcriptional pI-lacZ fusions revealed that CopR does not activate its own promoter pI (Brantl, 1994) and half-life measurements indicated that CopR does not affect the half-life of RNAIII. Instead, in the presence of both sense promoter pII and antisense promoter pIII in cis, CopR provided in cis or in trans causes an increase in the intracellular concentration of RNAIII. This effect can be attributed to the CopR protein and not the copR mRNA (Brantl and Wagner, 1997). Apparently, in the absence of CopR, the increased (de-repressed) RNAII transcription interferes, in cis, with initiation of RNAIII transcription (“convergent transcription”), yielding a lower RNAIII/plasmid ratio. The crucial factor in convergent transcription is the movement of the RNA polymerase toward or through the pIII promoter region, whereas it does not proceed through pII. Promoter pII as well as promoter pIII are supercoiling sensitive indicated by the effect of the gyrase inhibitor novobiocin on the accumulation of both RNAII and RNAIII (Brantl and Wagner, 1997). Therefore, in the absence of CopR, transcription from pII reduces initiation at pIII by inducing positive supercoils. By contrast, in the presence of CopR, promoter pII is 10-fold repressed, so that convergent transcription is mostly abolished. Consequently, more transcription from promoter pIII can be initiated resulting in increased RNAIII/plasmid ratios. Therefore, we propose the following model for pIP501 copy number control: RNAIII alone is able to adjust increases in copy number. At higher plasmid concentrations, more RNAIII is synthesized which in turn increases transcriptional attenuation of RNAII thus decreasing the replication frequency. In contrast, fortuitous copy number decreases cannot rapidly be corrected by RNAIII, since its long half-life (Brantl and Wagner, 1996) will result in high concentrations of the inhibitor, which threatens to yield a replication frequency inappropriately low for the current copy number. CopR, due to its dual function, can correct downward fluctuations of the plasmid copy number: Decreased synthesis of CopR de-represses promoter pII. This has two consequences (Brantl, 1994): (1) enhanced transcription of RNAII and (2) convergent transcription, which reduces RNAIII transcription. Both effects enhance RepR synthesis resulting in a higher replication frequency. The molecular event of pII de-repression works as an amplifier. In summary, the concerted action of two regulatory components, RNAIII and CopR, efficiently regulates pIP501 replication and ensures stable plasmid maintenance.
Biochemical Characterization of the CopR Protein
Three Inc18 Family Plasmids Encode Almost Identical Cop Proteins
Two almost identical Cop proteins with the same functions are encoded by the related streptococcal plasmids pAMβ1 (CopF) and pSM19035 (CopS) that share a high degree of sequence similarity with CopR at the aa level (Swinfield et al., 1990; Ceglowski et al., 1993): only two conservative aa exchanges at positions 51 and 80 are present (Brantl et al., 1994) and 2 additional aa (CopF) or two lacking aa (CopS) are found at the C-terminal end. To date, CopR is the best characterized Cop protein of this family. The repressor activity of CopF has been demonstrated and its operator sequence was narrowed down to a region of 31 bp (Le Chatelier et al., 1994). CopS has not been characterized in detail.
Identification of Bases and Phosphate Residues Contacted by CopR
The gene product of the copR gene is a small protein composed of 92 aa (predicted MW 10.4 kDa) that acts as transcriptional repressor at the essential repR promoter pII. CopR binds to a 44-bp region containing inverted repeat IR1 upstream of pII (Brantl, 1994). It does not autoregulate its own promoter pI nor does it activate the antisense RNA promoter pIII (Brantl, 1994). To identify bases and phosphates at the backbone directly contacted by CopR, chemical footprinting studies were performed (Steinmetzer and Brantl, 1997; Figure 2A). Methylation interference identified three guanines (G240, G242, and G251) and one cytosine (C239) in the top strand and two guanines (G252 and G254) and one cytosine (C255) in the bottom strand that are contacted by CopR in the major groove of DNA (Brantl et al., 1994). Furthermore, missing base interference uncovered the contribution of the bases adjacent to these guanines to the specific DNA-protein contacts. To determine phosphate residues in the DNA backbone essential for CopR binding ethylation interference experiments were employed. In the top strand, ethylation of C239, G240, and T241 interfered strongly with CopR binding while in the bottom strand, ethylation of T253, G254, and C255 affected binding. The recognition sequence of CopR is situated at the center of inverted repeat IR1. The protein contacts two consecutive major grooves (site I and II) on the same face of the DNA. Both binding sites share the common sequence motif 5′CGTG3′, and the outermost G is most important for CopR binding. A243 and G/C251 located within the loop region of the inverted repeat IR1 evoke an imperfect symmetry within the binding sequence (Figure 2). The sequence of the CopR operator was narrowed down to 17 bp. Gel filtration and native gel electrophoresis revealed that CopR is mainly dimeric under the conditions assayed (Steinmetzer and Brantl, 1997). An initially obtained sigmoidal binding curve proved to be the result of two coupled equilibria, on the one hand dimerization of CopR monomers and on the other hand CopR dimer-DNA binding. Using analytical ultracentrifugation, a KDimer-value of 1.44 ± 0.49 × 10−6 M for CopR dimers was determined (Steinmetzer et al., 1998) indicating relatively weak interactions between the two monomers. Using the KDimer− value and the binding curve, the equilibrium dissociation constant K2 for the CopR-DNA complex was calculated to be 4 ± 1.3 × 10−10 M, i.e., ≈ 0.4 nM. In this concentration range, CopR is mostly monomeric. By quantitative Western blotting, the intracellular concentration of CopR in B. subtilis carrying low copy number (copR+ rnaIII+) pIP501 derivatives was determined to be 20–30 μM. As this value is 10- to 20-fold higher than the KDimer, CopR is preferentially present as a dimer in the cell. Using gel-shift assays with wild-type and a C-terminally truncated CopR species (CopΔ20), it was demonstrated that CopR also binds to the DNA as a preformed dimer (Steinmetzer et al., 1998).
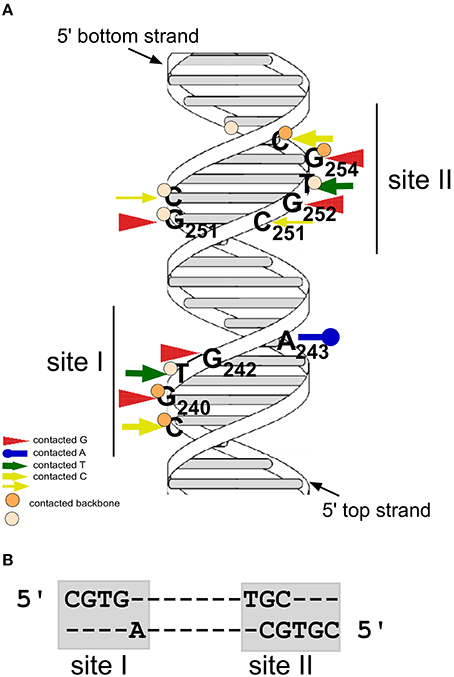
Figure 2. CopR contacts two consecutive sites at the major groove of DNA. (A) Model of the CopR DNA target with the two binding sites. Arrows denote bases contacted by CopR, circles represent phosphate groups of the DNA backbone contacted by CopR. Positions of bound Gs and As are indicated. Dark orange circles and thick yellow arrows represent strong contacts, light orange circles and thin yellow arrows weak contacts. Positions of the two binding sites are indicated. (B) DNA sequences of CopR binding sites I and II.
3D Model of CopR and Identification of Residues Involved in DNA Recognition and Dimerization
A structural model encompassing the N-terminal 63 aa of CopR was constructed (Figure 3). This model was based on the rather low (14%) sequence similarity to the P22 c2 repressor (Steinmetzer et al., 2000b). The model lacks the C-terminal 29 aa that had been found previously to be not important for DNA binding. In analogy to the P22 c2 repressor this model proposes that CopR is a HTH (helix-turn-helix) protein and describes the property of the protein to bind to DNA as a dimer at two consecutive major grooves (Steinmetzer et al., 1998). The protein backbone is built up by five α-helices, two of which are involved in DNA binding. Helix I is situated between aa 5 and 13. Helix II containing aa 18–25 is proposed to be the stabilization helix, while helix III comprising aa 29–37 is suggested to be the recognition helix. Moreover, aa 44–54 and 58–62 are predicted to form helices IV and V. The model proposes that aa R29, S30, S33, and R34 in the recognition-helix contact defined bases in the DNA sequence-specifically. In addition, residues K10, K18, K20, S28, N31, and S40 are supposed to contact the DNA-phosphate backbone sequence-unspecifically. E36 is near K10, R13, and K18 that, in the model, are close together in space and in contact with the phosphate backbone of the DNA. Residues F5, L9, F21, L25, Y32, I35, P42, L47, I50, and L53 build the hydrophobic core of CopR. Residues E2, F5, I44, K45, L47, L58, V59, and L62 are located on the protein surface and are suggested to be part of the dimeric interface. The real conformation of the fifth helix involving residues L58–I63 may—due to the uncertainties in the sequence alignment—differ from the model.
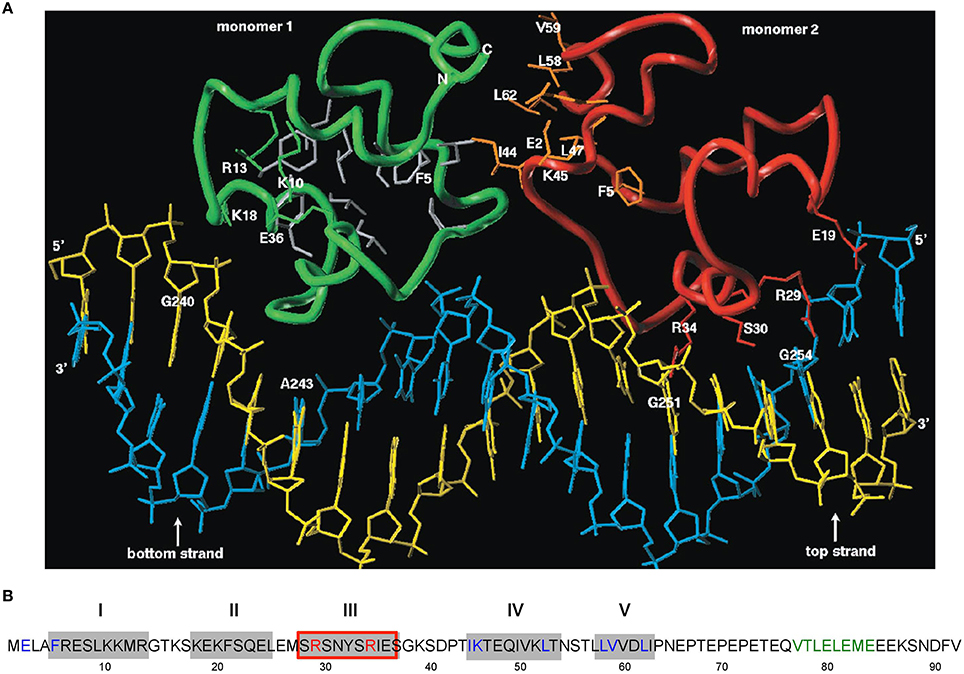
Figure 3. Amino acid sequence of CopR and 3D model of the CopR-DNA complex. (A) Model of the CopR-DNA complex. Green and red, CopR monomer I and II, respectively. Blue, DNA bottom strand. Yellow, DNA top strand. Gray, aa forming the hydrophobic core (for reasons of clarity shown only for monomer I). Orange, aa at the dimeric interface, shown only for monomer II. The presented DNA conformation is speculative. (B) Amino acid sequence of CopR (Brantl et al., 1994). Predicted α-helices are shown as gray boxes and numbered above the boxes. Red-framed box, DNA recognition helix. Red letters, aa involved in specific DNA recognition. Blue letters, aa predicted and shown to be involved in dimerization. Green letters, alternating hydrophobic and hydrophilic aa at the C-terminus forming a β-strand that stabilizes CopR.
Based on experimental footprinting data (Steinmetzer and Brantl, 1997), the CopR homology model and the crystal structure of the 434 c1 repressor-DNA complex, a model for the complex of CopR with the DNA target was generated (Figure 3A). To test the function of aa involved in sequence and non-sequence specific DNA recognition as well as aa important for correct protein folding, site-directed mutagenesis was employed. CD measurements of CopR variants were carried out to detect structural changes resulting from the mutations. In addition, dimerization was monitored by glutardialdehyde cross-linking and analytical ultracentrifugation. This approach allowed to localize the predicted HTH motif between aa 18 and 37 and to determine two aa within the recognition helix that make specific contacts with the DNA, R29 and R34 (Steinmetzer et al., 2000b; Figure 3A). Variants R29Q and R34Q showed only non-specific DNA binding at very high (micromolar) concentrations, while the protein structure was not affected. Furthermore, mutations of aa predicted to be involved in non-specific binding of the DNA backbone (S28T and K10Q) led to decreased binding affinity while maintaining selectivity. Additionally, substitution of aa necessary for proper folding—E36 and F5—caused significant structural changes. Taken together, these data support the model of CopR as a HTH protein that belongs to the λ repressor superfamily and uses α-helix III as recognition helix.
To verify the model predictions on aa involved in dimerization of CopR monomers, a combination of site-directed mutagenesis, EMSA, dimerization studies using sedimentation equilibrium centrifugation and CD measurements was used (Steinmetzer et al., 2000a). This allowed locating the dimeric interface between aa I44 and L62 (Figure 3A). As aa F5 situated at the N-terminus is needed for proper folding, it could not be unambigously assigned to the dimeric interface. CD measurements at protein concentrations below the KDimer value demonstrated that the monomer of CopR is folded. As in the first analysis, double and triple variants were constructed that all exhibited drastically increased dimerization constants, the analysis was complemented with single variants in the dimeric interface (Steinmetzer et al., 2002b). DNA binding and dimerization constants were calculated, urea-induced denaturation experiments were applied to evaluate the in vitro stability, and CD spectra of all mutated CopR proteins were measured. Variants I44D, L58D, V59S, and L62D had 4- to 50-fold increased KDimer-values and bound the CopR operator only non-specifically. Thereby, the substitution of aa L58 or L62 that were predicted to form several close interface contacts severely diminished dimerization, while mutation of the weakly interacting aa V59 did not significantly affect dimerization. Whereas the CD spectra did not show drastic structural alterations, the denaturation data revealed that the four variants unfold differently compared with the wild-type. Our results reveal that the four analyzed aa are engaged in dimerization as well as in folding of the monomer, i.e., they stabilize the monomer and, in addition, the dimeric interface (Steinmetzer et al., 2002b). Possibly, for economic reasons, some aa have dual functions in a small protein like CopR. Our data obtained with the four variants carrying single amino acid exchanges indicate that conformational changes are indeed necessary for dimerization. Furthermore, we observed that a single aa can on the one hand contribute to intra-monomeric contacts, when the protein is present as a monomer, and on the other hand contribute to inter-monomeric contacts when the protein dimerizes.
Structure of the DNA and Shape of the Protein in the CopR-DNA Complex
To determine the DNA conformation in the CopR-DNA complex, a combination of hydroxyl radical footprinting and fluorescence resonance energy transfer (FRET) measurements (Steinmetzer et al., 2002a) was employed. The footprints of CopR covered in total 29 bp and showed three defined areas of protection for each strand. This is comparable with the results obtained for the λ-repressor, 434-repressor, and for phage Φ105 repressor from B. subtilis (Tullius and Dombroski, 1986; Van Kaer et al., 1989; Ramesh and Nagaraja, 1996). The area of protection was significantly larger than that calculated earlier by chemical interference experiments, where the distance between the outermost contacts made up 17 bp (Steinmetzer and Brantl, 1997). Protected sites I and II were consistent with the previously identified contacted sites I and II. By contrast, the outer site III had not been identified before. For site III of the bottom strand, protection was weaker. This confirms our former observation that the interaction between CopR and the DNA is slightly asymmetric and also reflects the imperfect symmetry of the operator sequence (Steinmetzer and Brantl, 1997). FRET measurements revealed a bending angle of 20–25° for the DNA around the CopR protein, which is similar to that observed in the 434 c1 repressor-DNA complex and the λ c1 repressor-DNA complex. Furthermore, sedimentation velocity experiments demonstrated an extended shape of CopR dimers which accounts for the relatively large protection area detected with hydroxyl radical footprinting. To determine the global shape of the DNA in complex with CopR, FRET experiments with two DNA fragments were performed: A 19-bp DNA-fragment comprises only the minimal operator sequence (+2 bp for stabilization) and a 34-bp-fragment includes also the outer contact sites. For both fragments, bending angles of 20–25° were measured. This demonstrates that the center of the DNA bending is within the 17-bp sequence constituting the minimal operator and that the additional outer base contacts did not increase the DNA bending beyond 40–50°. Both outer binding sites do not add more than 10–15° to the overall bent. A slight bent is also in agreement with the fact that no hypersensitive sites were observed in hydroxyl radical footprinting indicating that CopR binding does not cause a drastic distortion of the DNA backbone. Analytical ultracentrifugation revealed that CopR dimers have an extended shape with a size of 8.4 nm for the fully hydrated protein. Due to this extended shape, only a gentle bending of the DNA is needed to enable CopR to make additional contacts outside of its 17-bp operator that reinforce the protein-DNA interaction. The CopR operator contains—similar to the operator of the 434 repressor—two TG-steps 11 bp apart that may constitute bending points by providing the flexibility required for the conformational changes of the DNA (Tzou and Hwang, 1999). As the CopR model does not include the 29 C-terminal aa, it can be assumed that these residues contact the outer binding sites III. Interestingly, variant CopRΔ27 lacking the 27 C-terminal aa has a 10-fold increased KD value (3.8 nM instead of 0.4 nM) for the CopR-DNA complex (Kuhn et al., 2000). This corroborates the formation of additional contacts between aa of the full-length CopR-C-terminus and the DNA backbone.
The C-terminus of CopR is Structured and Important for Protein Stability
Previous results showed that the C-terminal 27 aa of CopR were neither necessary for DNA binding nor for dimerization (Steinmetzer et al., 1998, 2000b). However, CopRΔ27 was 5-fold impaired in copy number control in vivo compared to both the wild-type and CopRΔ20. Interestingly, the C-terminus of CopR is very acidic comprising 10 Glu and one Asp residues. Therefore, a series of CopR variants truncated at the C-terminus were investigated for their half-life in vivo as well as for dimerization, DNA binding, structure and stability in vitro (Kuhn et al., 2000). The last 28 aa were apparently not required for DNA binding and dimerization, although the KD was 10-fold increased for CopRΔ27. Progressive deletions from the C-terminus significantly shortened the half-life of CopR: The half-life decreased from 42 min (wild-type CopR) over 24 min (CopRΔ7), ≈4.75 min (CopRΔ20), to ≈0.3 min (CopRΔ27). Guanidine-HCl denaturation assays corroborated that variants with shortened half-lives were also less stable in vitro. These results indicate that the C-terminus of CopR is required for protein stability. Amino acid substitutions within the C-terminus indicated that neither length nor charge is important for stabilization. CD measurements revealed that the C-terminus of CopR that contains alternating hydrophilic and hydrophobic aa residues is structured and forms a β-strand (Kuhn et al., 2000). Further analysis of the stabilizing motifs within the C-terminus (Kuhn et al., 2001) showed that both the wild-type (QVTLELEME, Figure 3A) and an artificial (QVTVTVTVT) β-strand structure (variant CopRVT) between aa 76 and 84 stabilized the corresponding protein derivatives. By contrast, replacement of the β-strand by an α-helix or an unstructured sequence significantly or moderately destabilized the protein. A second stabilization motif was identified in the 7 C-terminal aa, as their deletion from CopR or CopRVT reduced the half-life of the corresponding pIP501 derivatives to ≈50% (Kuhn et al., 2001). Our hypothesis is that the structured C-terminus of CopR interacts with other aa sequences in the core protein, thereby preventing its proteolytic degradation.
Surprisingly, variant CopRΔ20 with a 10-fold reduced half-life was fully functional in vivo in copy number control. The intracellular concentration of this variant was with 1 μM 15-fold lower than that of wild-type CopR (Kuhn et al., 2000). Why does wild-type CopR have such a long half-life, if a half-life of 4.75 min is completely sufficient for proper control? de la Hoz and colleagues investigating CopS from related plasmid pSM19035 found that the copS promoter is 8-fold down-regulated by the plasmid encoded ϖ- protein (de la Hoz et al., 2000). They suggested that ϖ- might represent a global regulator linking copy-number control with better than random segregation of pSM19035. As pIP501 derivatives lacking ϖ did not display defects in replication control (Brantl and Behnke, 1992b), an ϖ- homolog is apparently not required for replication control of pIP501. An 8-fold down-regulation of copR would still result in an intracellular CopR concentration of >2 μM, i.e., twice the amount determined for CopRΔ20. In case ϖ were included in pIP501 replication control and repressed copR 8-fold, the long CopR half-life would still ensure that sufficient CopR molecules are present to warrant proper control.
Evolution of CopR Resulted in Maximal DNA Binding Affinity
When pIP501 evolved in its original host, S. agalactiae, selection was, apparently, for a low, but not the lowest possible, copy number, that was optimal under the conditions experienced by this bacterial host. This assumption is based on the independent in vivo selection of three almost identical (in their core sequences) operators of the related streptococcal plasmids pIP501, pSM19035 and pAMβ1. CopR, CopS, and CopF have similar Cop operators with identical binding sites I and II. Only the spacer regions of the copR and copS operator differ (G244A and T247A), and the flanking sequences of the copR and copF binding sites display two nt exchanges (T236G and A260G).
One instrument to adjust the copy number of pIP501 is the KD value of the CopR-operator DNA complex. Based on the data summarized above we wondered if the copR operator found in nature (in pIP501) was optimized for strong DNA binding or if it would be possible to select an operator sequence that is bound more efficiently by CopR and, if yes, how such an operator would behave in vivo. To this end, we employed a SELEX experiment with copR operator sequences of different lengths combined with subsequent EMSAs with mutated operator fragments, copy-number determinations, and in vitro transcription (Freede and Brantl, 2004). Four experiments were performed: SELEX 1 with a randomized 7-bp spacer region, SELEX 2 with a randomized 17-bp fragment spanning the minimal operator, SELEX 3 with a longer operator (30 bp), and SELEX 4 with randomized 5-bp operator flanking regions. Our results demonstrate that the optimal spacer sequence between the two CopR binding sites comprises 7 bp, is AT rich and requires an A/T and T at the 3′ positions. By contrast, broad variations in the sequences flanking the minimal 17-bp operator did not affect CopR binding. These results show that the sequence differences between the copR, copS, and copF operator can be neglected. SELEX 2 for the minimal 17-bp copR operator yielded the same sequences as in vivo selection except that the completely symmetrical operator was found, too. Three simultaneous nucleotide exchanges outside the bases directly contacted by CopR selected in SELEX 3 did only slightly affect CopR binding in vitro or copy numbers in vivo. Therefore, we can conclude that in vivo evolution of the copR operator sequence was for maximal binding affinity.
Transcriptional Repressor CopR Acts by Inhibiting RNA Polymerase Binding
To investigate the complexes formed by the B. subtilis RNA polymerase (RNAP) at the repR promoter pII and to elucidate the mechanism exerted by CopR to repress transcription, a combination of DNase I footprinting, EMSA and KMnO4 footprinting was used (Licht et al., 2011). As shown by DNase I footprinting, the binding sites for CopR and RNAP overlap. EMSA confirmed that CopR and B. subtilis RNAP can not bind simultaneously. Instead, they compete for binding at promoter pII. Apparently, CopR prevents the access of RNAP to the promoter region by steric exclusion. We assume that CopR competes with the αCTD of the RNAP. Additionally, KMnO4 footprinting experiments revealed that prevention of open complex formation at pII does not further increase the repression effect of CopR. Furthermore, CopR-operator complexes were 18-fold less stable than RNAP-pII complexes in competition assays. However, due to its higher intracellular concentration CopR can effectively compete with RNAP for binding to the same region, where promoter and operator overlap. What are the consequences for copy number control? The half-lives of both CopR-pII and RNAP-pII complexes provide the time window for regulation. As CopR is produced constitutively and has a much higher intracellular concentration than the RNAP, repression can occur quickly inspite of the long half-life of the RNAP-pII complex. However, upon cell division the CopR concentration decreases, the repressor can be displaced by the RNAP—due to the much shorter half-life of the CopR-DNA complexes—and transcription of repR mRNA will be resumed immediately.
pIP501 Conjugative Transfer
pIP501 encodes a Gram-positive T4SS, whose key characteristics include the lack of a putative inner membrane transport channel owed to the different membrane composition of Gram-positive organisms and the lack of a third putative conjugative ATPase, a VirB11-like protein (Bhatty et al., 2013). The whole T4SS is encoded by the tra operon coding for 15 putative Tra proteins, seven of these show sequence or structural homology with Vir proteins of the Gram-negative prototype T4SS from Agrobacterium tumefaciens (Figure 4). Expression of the tra operon is controlled by the transfer initiator protein, TraA.
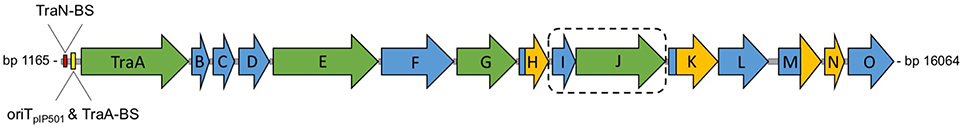
Figure 4. Genetic organization of the pIP501 tra operon. Genetic organization of the pIP501 tra operon. Proteins with known function are colored in green; the potential two-protein fusion coupling protein (consisting of TraIpIP501 and TraJpIP501) is indicated by a dashed box. Domains or proteins which have been structurally characterized are colored in yellow; TraA binding site, TraN binding site and oriTpIP501 are indicated upstream of traA. The genes of the pIP501 tra operon are drawn to scale. BS, binding site.
Biochemical Characterization of the TraA Relaxase
The TraA protein belongs to the family of IncQ-type relaxases, which includes the relaxases of the Gram-positive plasmids pGO1, pSK41, and pMRC01 as well as those of plasmids RSF1010, pSC101, and pTF1 of Gram-negative bacterial origin. The prototype of this relaxase family is the MobA protein encoded by the mobilizable plasmid RSF1010. MobA is a multifunctional protein consisting of an N-terminal relaxase domain and a C-terminal DNA primase domain (Scherzinger et al., 1991; Henderson and Meyer, 1996).
To confirm the postulated relaxase activity of TraA, supercoiled plasmid pVA2241 which contains a 309-bp fragment encompassing oriTpIP501 (Wang and Macrina, 1995) was used as a substrate in an in vitro cleavage assay with purified TraA protein. TraA sequence-specifically cleaved the oriTpIP501 containing supercoiled DNA (Kurenbach et al., 2002). TraA relaxase activity was optimal between 42 and 45°C, with the reactions being less efficient at temperatures below 37°C. TraA-mediated cleavage of supercoiled DNA was strictly dependent on Mg2+ or Mn2+. Mg+2 optimum was 5 mM, and optimal Mn2+ concentration was 10 mM. As was the case with MobM of pMV158 (Guzman and Espinosa, 1997), the TraI-TraJ oriT complexes from plasmid RP4 (Pansegrau et al., 1990), TrwC from R388 (Llosa et al., 1995), and TraI of F (Matson and Morton, 1991), the maximum amount of form FII (relaxed plasmid form) produced by TraA was about 55%. Total DNA relaxation was never obtained.
Interestingly, the N-terminal part of TraA comprising the first 293 aa also cleaved supercoiled oriTpIP501 containing DNA, albeit less efficiently (Approximately 25% conversion) than the full-length protein. These data coincide with those of MobA from plasmid RSF1010. Experiments with a C-terminally truncated MobA protein demonstrated that MobA-dependent oriT nicking activity resides within the first 34% (243 aa) of the 78-kDa MobA protein (Scherzinger et al., 1992).
pIP501 tra Operon Expression is Not Growth-Phase Dependent
The compact organization of the pIP501 oriT region is similar to that of rolling-circle-replicating plasmid pMV158, which was shown to be efficiently mobilized by pIP501 (van der Lelie et al., 1990; Kurenbach et al., 2003). The two regions are similar, meaning that the oriT nic-region, where the relaxase binds to its cognate DNA (Grohmann et al., 1999), lies within the respective promoter region (Farías et al., 1999). This configuration suggests autoregulation of the putative pIP501 tra operon consisting of the genes traA to traO (Figure 4) by the DNA relaxase TraA.
To study co-transcription of traA to traO, we conducted Reverse Transcription PCR (RT-PCR) with RNA isolated from E. faecalis (pIP501) cells harvested during mid exponential growth phase. Primer pairs were selected to amplify two successive genes of the tra region. RT-PCR resulted in products of the expected size (Kurenbach et al., 2002, 2006). We also tested for the existence of transcription products beyond traO using primers which would generate a traO/copR product of 480 bp. Using RNA as template, the respective product was never observed. Transcription of the pIP501 tra operon appears to be terminated by a strong rho-independent transcriptional terminator (Kurenbach et al., 2006).
To test the potential impact of the growth phase on the transcription of the tra genes, total RNA from E. faecalis (pIP501) was isolated at three different time-points: in the early exponential, the mid exponential, and the stationary growth phase (OD600 = 1.0). First, we looked if the tra genes are transcribed in all three growth phases. The selected RT-PCR amplicons from traC to traD, traF to traG, and traM to traN were generated with RNA from all three time-points (Kurenbach et al., 2006). Semi-quantitative RT-PCRs were carried out for traA to traB, and for traM to traN to study the transcription levels of different tra genes under differing physiological conditions. As a control, the constitutively expressed GAP-DH gene was amplified by RT-PCR, with RNA from E. faecalis cells harvested at the respective time-points as template. Densitometric analysis of the amplification products did not show any significant differences with respect to the growth phase, the same picture was obtained, as expected, for the constitutively expressed GAP-DH (Kurenbach et al., 2006).
However, we cannot exclude that tra gene transcription declines at a later stage in stationary phase, as we have seen slightly lower transfer frequencies (2- to 3-fold decrease) for donors and/or recipients at high cell densities (OD600 > 1) (Kurenbach et al., 2006). However, a phenomenon like “F2 phenocopies,” meaning that F+ cells get transfer-deficient in stationary phase (Hayes, 1964), was not observed. Transcription of several F-encoded tra genes decreases in mid-exponential or stationary phase, which is in agreement with a rapid decrease in transfer frequency in mid-exponential phase (Frost and Manchak, 1998). We conclude that the pIP501 tra genes are transcribed during the whole growth cycle of E. faecalis and that their level of expression does not depend on the growth phase.
TraA Relaxase Binds to the Ptra Promoter
The compact structure of the pIP501 oriT region (Figure 5), in the sense that the Ptra −10 and −35 boxes overlap with the left half repeat of inverted repeat structures (IR-1 and IR-2), likely representing the TraA recognition and binding site (Kopec et al., 2005), suggests autoregulation of the tra operon by TraA relaxase. To study TraA binding to the Ptra promoter, three DNA fragments were selected, the first comprising the −35 and −10 region, the second only the −35 region, and the third the −10 region alone. The shortest N-terminal TraA portion exhibiting relaxase activity, TraAN246 (Kopec et al., 2005), was used in band shift assays with ds oligonucleotides comprising the different parts of the Ptra promoter. Applying increasing TraAN246 concentrations to the −10 fragment, we detected one retarded DNA–protein complex. Binding affinity for the −35 region and for the whole promoter region was weaker than for the −10 region fragment. This could be due to presence of the complete left half repeat of IR-2 in the −10 region fragment. This complete left half repeat was present in all ss oligonucleotides that bound TraAN246 and TraA. An oligonucleotide similar to the −10 region fragment, but additionally comprising the right half repeat, resulted in similar binding affinity (Kopec et al., 2005). For all tested promoter fragments, TraA exhibited similar binding affinities as its N-terminal domain TraAN246. We conclude that TraA relaxase binds to the Ptra promoter region and that only the N-terminal TraA relaxase portion, TraAN246, is required for efficient binding (Kopec et al., 2005).
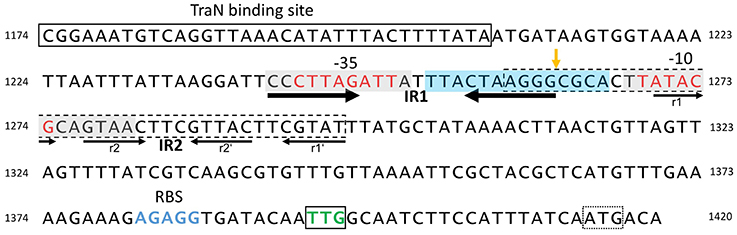
Figure 5. Structure of the pIP501 oriT region on DNA level. oriT region of pIP501 (nucleotides 1174–1420, GenBank L39769.1). Putative −10 and −35 promoter elements are colored in red; the TraN binding site is boxed. The perfect IR (IR1) is indicated with thick arrows; the second, imperfect IR (IR2) is marked with thin arrows—its repeating segments are individually named. Both repeats have the potential to generate a secondary structure. The areas that showed to be either protected from (gray box, mainly on the cleaved strand) or hypersensitive to (blue box, mainly on the non-cleaved strand) DNaseI cleavage in TraA footprinting assays (Kurenbach et al., 2006) are marked. oriTpIP501 core sequence (GenBank L39769.1, bp 1259–1296) is indicated with a dashed box; oriT nic-site is shown by an orange vertical arrow. tra operon ribosomal binding site (RBS) is colored in blue; the start codon of the first tra gene (traA, TTG) is boxed and colored in green; a second, potential start codon (ATG) is marked with a dotted box.
DNase I footprinting with a 250-bp DNA fragment encompassing Ptra and the complete IR-1 and IR-2 sequences demonstrated protection of the −35 and −10 region, with hypersensitive sites on the non-cleaved strand in vicinity of the nic site, at the nic site and two nucleotides 3′ of the −10 region (Kurenbach et al., 2006). DNase I protection on the cleaved strand extended eight nucleotides to the nic site, the nic site itself appeared as hypersensitive site. The DNase I hypersensitive sites are likely due to a conformational change of the oriT region induced by TraA binding, making the DNA better accessible for DNase I attack.
We have demonstrated that the left half repeats of IR-1 and IR-2 are the preferential binding sites for TraA. We postulate that binding of TraA to its target DNA is required for recognition and cleavage of DNA at the 5′-GpC-3′ dinucleotide in the nic site, which would remain accessible to the enzymatic activity of TraA.
Expression of the tra Genes is Controlled by TraA Relaxase
To confirm that TraA binding to the Ptra promoter region affects promoter activity, we cloned the promoterless lacZ gene in plasmid pQF120 under control of Ptra. E. coli cells with the construct, pQF120-Ptra::lacZ, gave blue colonies on LB X-Gal plates and generated β-galactosidase activity of 401 Miller units (Kurenbach et al., 2006). The impact of traA expression in trans on Ptra activity was studied by co-transformation of E. coli with pQF120-Ptra::lacZ and pACYC184-Ptac::GST-traA which expresses traA under control of the tac promoter. Upon induction of traA expression β-galactosidase activity dropped to 6 Miller units. As a control, the effect of co-resident pACYC184-Ptac::GST on Ptra activity of pQF120-Ptra::lacZ was analyzed. No significant change in β-galactosidase activity (407 Miller units) was observed. The data clearly demonstrated that the tra operon is regulated at the transcriptional level by TraA relaxase (Kurenbach et al., 2006).
For Mob, the mobilization protein encoded by the mobilizable broad-host-range plasmid pBBR1, whose nic site is identical with that of pMV158, autoregulation by binding of Mob to its promoter region overlapping with oriT has also been demonstrated (Szpirer et al., 2001).
Autoregulation of tra gene expression mediated by the transfer initiator protein, the DNA relaxase, seems to be an efficient mechanism to shut down plasmid transfer at a very early stage of conjugation, and is likely destined to obtain an optimum balance between the maximum transfer potential and the lowest burden for the host.
Two Conjugative ATPases Show Non-specific DNA Binding Activity
The pIP501 tra region comprises a Gram-positive conjugative T4SS, encoding, like most of the related systems in Gram-positive bacteria, two ATPases, VirB4-like ATPase, TraE, and VirD4-like coupling protein, TraJ (for a recent review on pIP501 T4SS see Goessweiner-Mohr et al., 2013, 2014a). However, in contrast to coupling proteins from other T4SSs from Gram-negative and Gram-positive bacteria alike, pIP501 appears to encode the first coupling protein consisting of two proteins, the TraJ protein and the TraI protein. TraJ has ATP-binding and low ATPase activity in vitro, and the membrane-associated TraI protein, encoded immediately upstream of traJ in the tra operon (Figure 4), presumably recruits TraJ via protein-protein interaction to its putative site of action, the cytoplasmic membrane.
Already in 2002, Llosa and coworkers postulated that during conjugative plasmid transfer, ss-plasmid DNA is “actively pushed into the recipient cell by action of the coupling protein” (Llosa et al., 2002). In the case of pIP501, the energy for this process could be generated by ATP hydrolysis, mediated by TraJ. EMSAs with purified TraJ on ssDNA and dsDNA containing the minimal oriTpIP501 region (GenBank L39769.1, bp 1259–1296) or random DNA of the same size with no similarity to oriTpIP501 were performed. The DNA substrates were 42 bases or 42 bp long; the random 42-mer lacked the ability to form a hairpin-like secondary structure (Kopec et al., 2005), one of the prototypical characteristics of oriT regions. TraJ bound non-sequence specifically to both ssDNA substrates, whereas binding to dsDNA substrates was not observed, not even at very high TraJ concentrations (Arends, 2010). These observations are in agreement with the postulated function of TraJ as a conjugative coupling protein, connecting the relaxosome consisting of the TraA relaxase covalently bound to the 5′-end of the processed ss pIP501 DNA with the mating pair formation complex.
The VirB4-like ATPase, TraE, which showed higher in vitro ATPase activity than the coupling protein (Çelik, 2011) also bound non-sequence specifically to ss oriT 42-mer DNA and random 42-mer DNA in EMSAs performed similarly to those described above for TraJ. As demonstrated for TraJ, dsDNA was no substrate for TraE (Çelik, 2011). TraE and TraJ could be both actively involved in generating energy for the T4SS process, presumably each ATPase producing energy for different step(s) in the T4S process. Details on these processes have not been unraveled so far.
TraN is a Putative Transfer Repressor
TraN Binds Sequence-Specifically to oriTpIP501 DNA
TraN is a small (14.4 kDa) soluble cytoplasmic protein encoded by traN, the penultimate gene of the pIP501 tra operon. The structure of TraN was solved to 1.35 Å resolution. It contains an internal dimer fold with antiparallel β-sheets in the center and a HTH motif at both ends (Goessweiner-Mohr et al., 2012, 2014b).
Because TraN co-purified with DNA, we investigated if it can interact with radiolabelled ssDNA and dsDNA oligonucleotides. By applying the identical oligonucleotides as described for the EMSAs with TraE and TraJ, TraN showed only a slight shift for the ssDNA oligonucleotides, whereas the dsDNA fragments were significantly shifted. The random and the oriTpIP501 containing oligonucleotide showed the same binding affinity (Goessweiner-Mohr et al., 2014b).
To search for a potential sequence-specific TraN binding site, we conducted EMSAs with dsDNA fragments encompassing the oriTpIP501 and sequences upstream and downstream of this region. At high TraN concentrations, all DNA fragments were cooperatively shifted. A small but significant stepwise shift using an equimolar protein:DNA ratio was visible only for fragments comprising a common 149-bp sequence 5′ of the oriT sequence, for which we postulate a preferred TraN binding site (Goessweiner-Mohr et al., 2014b).
To delimit the specific TraN binding site within the 149-bp sequence, we designed a new footprinting assay which is based upon 5′-to-3′ exonuclease digestion. The TraN binding site was localized to a 34-bp sequence located 55 bp 5′ of oriTpIP501 nic-site. Interestingly, the TraN binding site has no direct or inverted repeats but is A/T rich (Goessweiner-Mohr et al., 2014b).
Thermal stability of TraN was studied in presence and absence of DNA with a Thermofluor-based assay. The melting temperature (Tm) of TraN alone amounted to 54.3°C; the binding of a non-specific (random) 34-mer dsDNA oligonucleotide raised the Tm to 65.2°C, whereas addition of DNA containing the specific binding site increased Tm to 70.4°C. The stabilizing effect indicates an enhanced binding affinity for the specific site compared with the random DNA (Goessweiner-Mohr et al., 2014b).
To determine whether there is a difference in the molar ratio of the TraN–DNA interaction between the random and the specific oligonucleotides, as well as to obtain information on the respective binding constants, isothermal titration calorimetry analyses with the oligonucleotide encompassing the binding site and the non-specific (random) oligonucleotides used in the Thermofluor experiments were carried out. When titrating with non-specific DNA, two TraN molecules bound to one dsDNA fragment (in a 2:1 ratio), whereas, as expected, equimolar stoichiometry (1:1 ratio) was observed for the specific interaction. TraN was found to bind to the specific binding site exothermically with a binding constant of 107 M−1 in comparison to endothermic binding to the non-specific sequence with a binding constant of 105 M−2 (2:1 binding ratio; Goessweiner-Mohr et al., 2014b).
The Crystal Structure of the TraN-DNA Complex has been Solved to High Resolution
Recently, we solved the 1.9 Å co-crystal structure of TraN bound to its specific 34-bp binding site upstream of the oriTpIP501nic-site, described above (Goessweiner-Mohr et al., in preparation). The binding mode postulated in Goessweiner-Mohr et al. (2014b) could be confirmed: “The recognition helices of the two mirrored HTH motifs enter two adjacent major grooves of the dsDNA binding site.” Furthermore, the tip of the loops between strands 2 and 3 as well as strands 5 and 6, which are close to the internal 2-fold axis, are interacting with the minor groove. While tied to its binding site, TraN slightly bends the dsDNA oligonucleotide used in the crystallization setup (Goessweiner-Mohr et al., in preparation).
TraN is Not an Essential T4SS Protein
Very recently we generated a markerless E. faecalis JH2-2 (pIP501ΔtraN) mutant by applying a two-step recombination technique developed for construction of mutants in E. faecalis (Kristich et al., 2007). Surprisingly, in standard in vitro mating tests we could demonstrate that TraN is not an essential T4SS protein but contrary to expectations, traN deletion resulted in an increase of pIP501 transfer efficiency.
TraN Shows Structural Homology with Transcriptional Regulators: Potential Role of TraN in the T4S Process
In searches for proteins structurally similar to TraN we only found hits that resemble one half of the protein. Amongst others, the TraN fold resembles that of the N-terminal domain of transcriptional regulators of the MerR family (Goessweiner-Mohr et al., 2014b), for example a transcriptional activator from Bacillus thuringiensis (PDB entry 3gpv; New York SGX Research Center for Structural Genomics). Transcriptional activators of the MerR family comprise an N-terminal winged-helix DNA-binding domain and recognize the specific DNA site as a dimer where the recognition helices of the HTH motifs are inserted into two adjacent major grooves.
The dimerization motif of MerR proteins is completely distinct from the internal dimer configuration of TraN, which requires hydrophobic interactions within a barrel-like motif in its center. Contrarily to MerR family proteins, which contain a C-terminal effector-binding region (Brown et al., 2003), neither in TraN nor TraN-like proteins of related T4SSs such a C-terminal extension was found. All TraN-like proteins found are of enterococcal origin (from conjugative E. faecalis plasmids, pRE25 and pAMβ1, E. faecium plasmid pVEF3 and two genomically located TraN-like proteins from an E. faecalis and Enterococcus italicus strain), and their sequence is highly similar to that of TraN. All other proteins found (transposon or bacteriophage-encoded excisionases and MerR family proteins) have only a single TraN-like domain (Goessweiner-Mohr et al., 2014b).
Due to the structural similarity of TraN with MerR-like transcriptional regulators and the fact that traN deletion resulted in a 2 log increase of pIP501 transfer efficiency, we postulate that TraN could repress pIP501 transfer by regulating either expression of the pIP501 tra operon or TraA activity.
Although MerR-like proteins show only similarity to the fold of a single TraN domain, binding to DNA requires the formation of a homodimer (PDB entry 3gpv) which binds to two adjacent major grooves of dsDNA, as postulated for TraN. Expression of the pIP501 tra genes is already autoregulated by TraA relaxase (Kurenbach et al., 2006), TraA binds to the two left half repeats of IR1-1 and IR1-2 (Kurenbach et al., 2006) which overlap with the −10 and −35 box of the Ptra promoter respectively. Specific DNA recognition and binding is required for TraA-mediated site-specific nicking at the 5′-GpC-3′ dinucleotide in the nic-site (nucleotide position 1262/1263, Acc. Nr. L39769) which thus will be accessible to the enzymatic activity of TraA (Kurenbach et al., 2006; see also Figure 5). TraN could act as an additional repressor of the tra operon by specifically binding to a 34-bp sequence located 55 bp upstream of the nic-site thereby inhibiting RNA polymerase from efficient transcription of the tra operon. We hypothesize that this negative regulation could be relieved by binding of putative interaction partners, e.g., TraE or TraJ (Abajy et al., 2007) to TraN, possibly as response to (i) the presence of potential recipient cells/mating partners or (ii) an assembled putative pIP501 T4SS core complex. Experimental studies on the mechanism of TraN regulation are in progress.
Speculations on Control of pIP501 Transfer Gene Expression
Tight control of tra gene expression is a general feature of mobile genetic elements from Gram-negative and Gram-positive bacteria alike, presumably to ensure that costly—referring to bacterial fitness—expression of multiple Tra proteins only takes place when the effort is worthwhile because potential recipients are present or more generally speaking the environmental conditions allow efficient plasmid transfer. Different modes of controlling conjugative transfer are known: The well-characterized Gram-negative conjugative broad-host-range IncP plasmids, F plasmid and F-related plasmids, such as R1 and R100, have a very complex regulation system controlling expression of Tra factors at transcriptional and translational level involving not only plasmid-encoded factors but also host-factors (Zatyka et al., 1994, 1997; Taki et al., 1998; Adamczyk and Jagura-Burdzy, 2003; Will and Frost, 2006; Wong et al., 2012). Additionally, in case of TraJ from plasmid R1 and F-related plasmids, regulation of the transfer operon via a sense/antisense RNA system has been shown (Koraimann et al., 1991, 1996; Mark Glover et al., 2015). For Gram-positive bacteria, the sex-pheromone-responsive enterococcal plasmids, particularly pCF10, are those with the best studied regulatory processes controlling conjugative transfer (Tanimoto et al., 1996; Muscholl-Silberhorn, 2000; Dunny, 2007, 2013; Folli et al., 2008).
None of the known conjugation control systems fits to what we have observed for broad-host-range plasmid pIP501. pIP501 tra gene expression seems to be always on, independent of the growth phase of the host (Kurenbach et al., 2006) and presence of potential recipients, presumably at a low basic level. tra gene expression was shown to be controlled by the transfer initiator protein, TraA, which regulates its own synthesis and that of the other Tra factors by binding to the Ptra promoter overlapping with oriT (Kurenbach et al., 2006; see also Figure 5).
Recently, we detected binding of another Tra factor, TraN, to a region 55 bp upstream of the oriT nic-site (Goessweiner-Mohr et al., 2014b; Figure 5 in this article). This TraN-binding site is located only 35 bp upstream of the −35 region of the Ptra promoter. Thus, we postulate that the tra operon might be negatively controlled by two proteins, the TraA relaxase binding to the −10 and −35 region of the promoter thereby leaving the oriT nic-site accessible for specific TraA cleavage and by the winged-helix-turn-helix DNA-binding protein TraN, binding to a unique operator site (present only once on the pIP501 genome) upstream of Ptra promoter. We postulate that TraA activity is blocked by binding of TraN upstream of the oriT nic-site. Either by (i) receiving environmentals signals which could include the presence or already the contact of the donor cell with a potential recipient cell and/or (ii) by interaction of TraN with T4S key components, such as TraE, TraG or TraJ (binding to these proteins has been observed in the yeast two-hybrid system Abajy et al., 2007), TraN would be released from the DNA, likely resulting in a conformational change of the DNA in the vicinity of the TraA binding site triggering nic-cleavage by TraA. Our working model of pIP501 tra operon regulation is depicted in Figure 6.
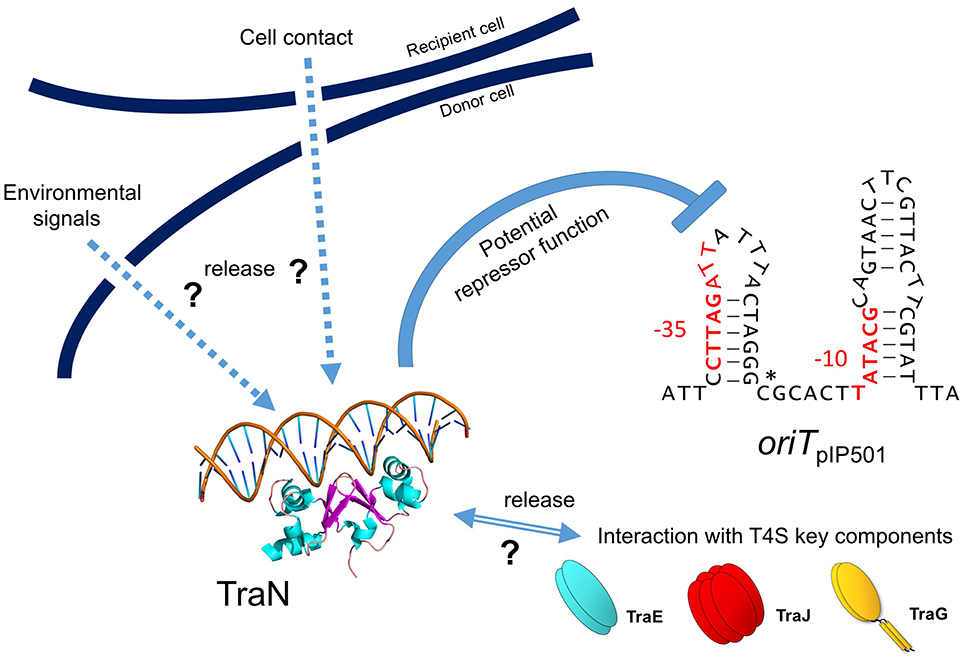
Figure 6. Model for negative tra operon regulation by potential transcriptional repressor TraN. TraN is shown docked to a DNA strand, with two helices reaching into two adjacent major grooves and two loops in the center of TraN extending into the minor groove in between the major grooves (adapted from Goessweiner-Mohr et al., 2014b; Figure S6). oriTpIP501 is depicted as a stem-loop structure as potentially generated by the two IR sequences (see also Figure 5). Putative −10 and −35 tra promoter regions are colored in red; oriT nic-site is marked by a star. Potential TraN repressor function is indicated; hypothetical release signals are denoted by arrows (environmental signals, recipient cell contact, interaction with T4S key components—TraE, TraJ, TraG).
A putative winged-helix-turn-helix DNA-binding protein, RctA from the symbiotic rhizobial megaplasmids, has been demonstrated to repress transcription of conjugative transfer genes of pRetCFN42d, the symbiotic plasmid (pSym) of Rhizobium etli (Pérez-Mendoza et al., 2005; Sepúlveda et al., 2008; Nogales et al., 2013).
The negative regulation of pIP501 tra gene expression exerted by two (putative) transcriptional regulators would be in agreement with the generally low transfer frequencies of pIP501, in the range of (2 − 8) × 10−5 transconjugants per recipient for intraspecies E. faecalis matings (Arends et al., 2013; Fercher et al., 2016).
Conclusions and Perspectives
Conjugative transfer of diverse genetic traits, such as antibiotic or heavy metal resistance genes, virulence genes or genes conferring specific metabolic capabilities such as degradation of anthropogenic compounds is a natural process going on everywhere in nature at diverse transfer rates, as these naturally depend on the plasmids involved and on the habitat. Availability of nutrients and water, or in other words, good physiological conditions of donor and recipients, are generally accepted as conditions favoring horizontal plasmid transfer. Availability of colonizable surfaces for the microorganisms is another very important feature, as the close proximity of microorganisms in surface-associated communities, the so-called biofilms, increases the chances of horizontal gene exchange (Hausner and Wuertz, 1999; Sørensen et al., 2005; Madsen et al., 2012).
The observation that tra gene expression seems to be a tightly controlled process not only holds true for plasmids of the Inc18 group, but seems to be a general feature of self-transmissible plasmids of diverse origin. In particular, the expression of relaxosome components seems to be tightly regulated, in many plasmids it is under autoregulatory control by the relaxase or relaxase accessory proteins. One of the most complex regulatory circuits controlling the production of relaxosome proteins has been deciphered in the prototype Gram-negative broad-host-range plasmid RK2. Zatyka and coworkers argued that the complex regulatory circuits involved in regulation of IncPα plasmid RK2 provide an autoregulatory way of ensuring production of enough relaxosome proteins without overburdening the host (Zatyka et al., 1994). Expression of the tra genes of F-plasmid is also tightly controlled by a number of factors, including among others, a plasmid-encoded activator and two autoregulators. One of them, TraM, is a component of the F relaxosome (Will and Frost, 2006). In all these plasmids the level and stringency of the regulatory processes appear to be in good balance with the transfer potential of the host in order to reduce its fitness costs.
Detailed knowledge of these regulatory processes in Gram-positive bacteria is still scarce, thus challenging tasks of the coming years will be to unravel the internal as well as external environmental signals triggering plasmid transfer on the molecular level to develop more efficient interference strategies to efficiently reduce conjugative spread of antibiotic resistance genes.
With regard to replication of pIP501, the identification of a third (upper) regulatory level would be required to unravel why deletion of both regulatory components, CopR and RNAIII, does not show an additive effect. In the related plasmid pSM19035, ϖ protein was found to be this central regulator (de la Hoz et al., 2000). However, in pIP501, no promoter has been detected so far preceding orf ϖ.
Thus, for both, pIP501 replication and transfer, the most urgent questions to answer concern global regulatory processes governing the success of pIP501-like multiple antibiotic resistance replicons in terms of maintenance and wide spread, particularly, in hospital environments.
Author Contributions
EG, NG, and SB contributed to writing of the manuscript. NG, SB designed the figures, all authors approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GS and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We apologize for not having mentioned all valuable contributions from colleagues in the field due to space restrictions. We thank K. Arends and C. Fercher for critical reading of the manuscript. Work in the Grohmann lab was supported by DLR grants 50WB1166 and 50WB1466 from Deutsches Zentrum für Luft und Raumfahrt, work in the Brantl lab was supported by grants BR1552/4-1 to 4-3, 6-1 to 6-3, and BR1552/8-1 from Deutsche Forschungsgemeinschaft.
References
Abajy, M. Y., Kopec, J., Schiwon, K., Burzynski, M., Döring, M., Bohn, C., et al. (2007). A type IV-secretion-like system is required for conjugative DNA transport of broad-host-range plasmid pIP501 in gram-positive bacteria. J. Bacteriol. 189, 2487–2496. doi: 10.1128/JB.01491-06
Adamczyk, M., and Jagura-Burdzy, G. (2003). Spread and survival of promiscuous IncP-1 plasmids. Acta Biochim. Pol. 50, 425–453. Available online at: http://www.actabp.pl/pdf/2_2003/425.pdf
Arends, K. (2010). Entwicklung von gfp-Basierten Monitoring Tools zur Verfolgung von Horizontalem Gentransfer und Studien zum T4SLS des Konjugativen Plasmids pIP501 aus Enterococcus faecalis. Ph.D. thesis, Technical University, Berlin.
Arends, K., Celik, E.-K., Probst, I., Goessweiner-Mohr, N., Fercher, C., Grumet, L., et al. (2013). TraG encoded by the pIP501 type IV secretion system is a two-domain peptidoglycan-degrading enzyme essential for conjugative transfer. J. Bacteriol. 195, 4436–4444. doi: 10.1128/JB.02263-12
Bhatty, M., Laverde Gomez, J. A., and Christie, P. J. (2013). The expanding bacterial type IV secretion lexicon. Res. Microbiol. 164, 620–639. doi: 10.1016/j.resmic.2013.03.012
Brantl, S. (1994). The copR gene product of plasmid pIP501 acts as a transcriptional repressor at the essential repR promoter. Mol. Microbiol. 14, 473–483. doi: 10.1111/j.1365-2958.1994.tb02182.x
Brantl, S. (2014). Plasmid replication control by Antisense RNAs. Microbiol. Spectr. 2:PLAS–0001–2013. doi: 10.1128/microbiolspec.PLAS-0001-2013
Brantl, S. (2015). Antisense-RNA mediated control of plasmid replication - pIP501 revisited. Plasmid 78, 4–16. doi: 10.1016/j.plasmid.2014.07.004
Brantl, S., and Behnke, D. (1992a). Characterization of the minimal origin required for replication of the streptococcal plasmid pIP501 in Bacillus subtilis. Mol. Microbiol. 6, 3501–3510. doi: 10.1111/j.1365-2958.1992.tb01785.x
Brantl, S., and Behnke, D. (1992b). Copy number control of the streptococcal plasmid pIP501 occurs at three levels. Nucleic Acids Res. 20, 395–400. doi: 10.1093/nar/20.3.395
Brantl, S., and Behnke, D. (1992c). The amount of RepR protein determines the copy number of plasmid pIP501 in Bacillus subtilis. J. Bacteriol. 174, 5475–5478.
Brantl, S., Behnke, D., and Alonso, J. C. (1990). Molecular analysis of the replication region of the conjugative Streptococcus agalactiae plasmid pIP501 in Bacillus subtilis. Comparison with plasmids pAMβ1 and pSM 19035. Nucleic Acids Res. 18, 4783–4790. doi: 10.1093/nar/18.16.4783
Brantl, S., Birch-Hirschfeld, E., and Behnke, D. (1993). RepR protein expression on plasmid pIP501 is controlled by an antisense RNA-mediated transcription attenuation mechanism. J. Bacteriol. 175, 4052–4061.
Brantl, S., Kummer, C., and Behnke, D. (1994). Complete nucleotide sequence of plasmid pGB3631, a derivative of the Streptococcus agalactiae plasmid pIP501. Gene 142, 155–156. doi: 10.1016/0378-1119(94)90372-7
Brantl, S., Nowak, A., Behnke, D., and Alonso, J. C. (1989). Revision of the nucleotide sequence of the Streptococcus pyogenes plasmid pSM19035 repS gene. Nucleic Acids Res. 17:10110. doi: 10.1093/nar/17.23.10110
Brantl, S., and Wagner, E. G. (1994). Antisense RNA-mediated transcriptional attenuation occurs faster than stable antisense/target RNA pairing: an in vitro study of plasmid pIP501. EMBO J. 13, 3599–3607.
Brantl, S., and Wagner, E. G. (1996). An unusually long-lived antisense RNA in plasmid copy number control: in vivo RNAs encoded by the streptococcal plasmid pIP501. J. Mol. Biol. 255, 275–288. doi: 10.1006/jmbi.1996.0023
Brantl, S., and Wagner, E. G. (1997). Dual function of the copR gene product of plasmid pIP501. J. Bacteriol. 179, 7016–7024.
Brown, N. L., Stoyanov, J. V., Kidd, S. P., and Hobman, J. L. (2003). The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 27, 145–163. doi: 10.1016/S0168-6445(03)00051-2
Bruand, C., Le Chatelier, E., Ehrlich, S. D., and Jannière, L. (1993). A fourth class of theta-replicating plasmids: the pAM beta 1 family from gram-positive bacteria. Proc. Natl. Acad. Sci. U.S.A. 90, 11668–11672. doi: 10.1073/pnas.90.24.11668
Brzozowska, I., Brzozowska, K., and Zielenkiewicz, U. (2012). Functioning of the TA cassette of streptococcal plasmid pSM19035 in various gram-positive bacteria. Plasmid 68, 51–60. doi: 10.1016/j.plasmid.2012.01.010
Ceglowski, P., Lurz, R., and Alonso, J. C. (1993). Functional analysis of pSM19035-derived replicons in Bacillus subtilis. Gene 136, 1–12. doi: 10.1016/0378-1119(93)90441-5
Çelik, E. K. (2011). Studien zur Aufklärung der Rolle von Proteinkomponenten für die Assemblierung Eines Konjugativen Typ IV Sekretionssystems in Gram-Positiven Bakterien Sowie Nachweis Konjugativer Plasmide und Antibiotika-Resistenzgene in Bodenproben. Ph.D. thesis, Karl-Franzens-University Graz, Graz.
de la Hoz, A. B., Ayora, S., Sitkiewicz, I., Fernández, S., Pankiewicz, R., Alonso, J. C., et al. (2000). Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc. Natl. Acad. Sci. U.S.A. 97, 728–733. doi: 10.1073/pnas.97.2.728
Dunny, G. M. (2007). The peptide pheromone-inducible conjugation system of Enterococcus faecalis plasmid pCF10: cell-cell signalling, gene transfer, complexity and evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 362, 1185–1193. doi: 10.1098/rstb.2007.2043
Dunny, G. M. (2013). Enterococcal sex pheromones: signaling, social behavior, and evolution. Annu. Rev. Genet. 47, 457–482. doi: 10.1146/annurev-genet-111212-133449
Evans, R. P. J. R., and Macrina, F. L. (1983). Streptococcal R plasmid pIP501: endonuclease site map, resistance determinant location, and construction of novel derivatives. J. Bacteriol. 154, 1347–1355.
Farías, M. E., Grohmann, E., and Espinosa, M. (1999). Expression of the mobM gene of the streptococcal plasmid pMV158 in Lactococcus lactis subsp. lactis. FEMS Microbiol. Lett. 176, 403–410. doi: 10.1016/S0378-1097(99)00265-7
Fercher, C., Probst, I., Kohler, V., Goessweiner-Mohr, N., Arends, K., Grohmann, E., et al. (2016). VirB8-like protein TraH is crucial for DNA transfer in Enterococcus faecalis. Sci. Rep. 6:24643. doi: 10.1038/srep24643
Folli, C., Mangiarotti, L., Folloni, S., Alfieri, B., Gobbo, M., Berni, R., et al. (2008). Specificity of the TraA-DNA interaction in the regulation of the pPD1-encoded sex pheromone response in Enterococcus faecalis. J. Mol. Biol. 380, 932–945. doi: 10.1016/j.jmb.2008.05.058
Freede, P., and Brantl, S. (2004). Transcriptional Repressor CopR: use of SELEX to study the copR operator indicates that evolution was directed at maximal binding affinity. J. Bacteriol. 186, 6254–6264. doi: 10.1128/JB.186.18.6254-6264.2004
Frost, L. S., and Manchak, J. (1998). F- phenocopies: characterization of expression of the F transfer region in stationary phase. Microbiology 144(Pt 9), 2579–2587. doi: 10.1099/00221287-144-9-2579
Goessweiner-Mohr, N., Arends, K., Keller, W., and Grohmann, E. (2013). Conjugative type IV secretion systems in gram-positive bacteria. Plasmid 70, 289–302. doi: 10.1016/j.plasmid.2013.09.005
Goessweiner-Mohr, N., Arends, K., Keller, W., and Grohmann, E. (2014a). Conjugation in gram-positive bacteria. Microbiol. Spectr. 2:PLAS-0004-2013. doi: 10.1128/microbiolspec.PLAS-0004-2013
Goessweiner-Mohr, N., Eder, M., Hofer, G., Fercher, C., Arends, K., Birner-Gruenberger, R., et al. (2014b). Structure of the double-stranded DNA-binding type IV secretion protein TraN from Enterococcus. Acta Crystallogr. D Biol. Crystallogr. 70, 2376–2389. doi: 10.1107/S1399004714014187
Goessweiner-Mohr, N., Fercher, C., Abajy, M. Y., Grohmann, E., and Keller, W. (2012). Crystallization and first data collection of the putative transfer protein TraN from the Gram-positive conjugative plasmid pIP501. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1402–1405. doi: 10.1107/S174430911204184X
Grohmann, E., Guzmán, L. M., and Espinosa, M. (1999). Mobilisation of the streptococcal plasmid pMV158: interactions of MobM protein with its cognate oriT DNA region. Mol. Gen. Genet. 261, 707–715. doi: 10.1007/s004380050014
Guzman, L. M., and Espinosa, M. (1997). The mobilization protein, MobM, of the streptococcal plasmid pMV158 specifically cleaves supercoiled DNA at the plasmid oriT. J. Mol. Biol. 266, 688–702. doi: 10.1006/jmbi.1996.0824
Hausner, M., and Wuertz, S. (1999). High rates of conjugation in bacterial biofilms as determined by quantitative in situ analysis. Appl. Environ. Microbiol. 65, 3710–3713.
Hayes, W. (1964). The Genetics of Bacteria and their Viruses. Studies in Basic Genetics and Molecular Biology. Oxford: Blackwell Scientific Publications.
Heidrich, N., and Brantl, S. (2003). Antisense-RNA mediated transcriptional attenuation: importance of a U-turn loop structure in the target RNA of plasmid pIP501 for efficient inhibition by the antisense RNA. J. Mol. Biol. 333, 917–929. doi: 10.1016/j.jmb.2003.09.020
Heidrich, N., and Brantl, S. (2007). Antisense RNA-mediated transcriptional attenuation in plasmid pIP501: the simultaneous interaction between two complementary loop pairs is required for efficient inhibition by the antisense RNA. Microbiology 153, 420–427. doi: 10.1099/mic.0.2006/002329-0
Henderson, D., and Meyer, R. J. (1996). The primase of broad-host-range plasmid R1162 is active in conjugal transfer. J. Bacteriol. 178, 6888–6894.
Kopec, J., Bergmann, A., Fritz, G., Grohmann, E., and Keller, W. (2005). TraA and its N-terminal relaxase domain of the Gram-positive plasmid pIP501 show specific oriT binding and behave as dimers in solution. Biochem. J. 387, 401–409. doi: 10.1042/BJ20041178
Koraimann, G., Koraimann, C., Koronakis, V., Schlager, S., and Högenauer, G. (1991). Repression and derepression of conjugation of plasmid R1 by wild-type and mutated finP antisense RNA. Mol. Microbiol. 5, 77–87. doi: 10.1111/j.1365-2958.1991.tb01828.x
Koraimann, G., Teferle, K., Markolin, G., Woger, W., and Högenauer, G. (1996). The FinOP repressor system of plasmid R1: analysis of the antisense RNA control of traJ expression and conjugative DNA transfer. Mol. Microbiol. 21, 811–821. doi: 10.1046/j.1365-2958.1996.361401.x
Kristich, C. J., Chandler, J. R., and Dunny, G. M. (2007). Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57, 131–144. doi: 10.1016/j.plasmid.2006.08.003
Kuhn, K., Steinmetzer, K., and Brantl, S. (2000). Transcriptional repressor CopR: the structured acidic C terminus is important for protein stability. J. Mol. Biol. 300, 1021–1031. doi: 10.1006/jmbi.2000.3929
Kuhn, K., Steinmetzer, K., and Brantl, S. (2001). Transcriptional repressor CopR: dissection of stabilizing motifs within the C terminus. Microbiology 147, 3387–3392. doi: 10.1099/00221287-147-12-3387
Kurenbach, B., Bohn, C., Prabhu, J., Abudukerim, M., Szewzyk, U., and Grohmann, E. (2003). Intergeneric transfer of the Enterococcus faecalis plasmid pIP501 to Escherichia coli and Streptomyces lividans and sequence analysis of its tra region. Plasmid 50, 86–93. doi: 10.1016/S0147-619X(03)00044-1
Kurenbach, B., Grothe, D., Farias, M. E., Szewzyk, U., and Grohmann, E. (2002). The tra region of the conjugative plasmid pIP501 is organized in an operon with the first gene encoding the relaxase. J. Bacteriol. 184, 1801–1805. doi: 10.1128/JB.184.6.1801-1805.2002
Kurenbach, B., Kopeć, J., Mägdefrau, M., Andreas, K., Keller, W., Bohn, C., et al. (2006). The TraA relaxase autoregulates the putative type IV secretion-like system encoded by the broad-host-range Streptococcus agalactiae plasmid pIP501. Microbiology 152, 637–645. doi: 10.1099/mic.0.28468-0
Le Chatelier, E., Ehrlich, S. D., and Jannière, L. (1994). The pAM beta 1 CopF repressor regulates plasmid copy number by controlling transcription of the repE gene. Mol. Microbiol. 14, 463–471. doi: 10.1111/j.1365-2958.1994.tb02181.x
Le Chatelier, E., Jannière, L., Ehrlich, S. D., and Canceill, D. (2001). The RepE initiator is a double-stranded and single-stranded DNA-binding protein that forms an atypical open complex at the onset of replication of plasmid pAMbeta 1 from Gram-positive bacteria. J. Biol. Chem. 276, 10234–10246. doi: 10.1074/jbc.M010118200
Licht, A., Freede, P., and Brantl, S. (2011). Transcriptional repressor CopR acts by inhibiting RNA polymerase binding. Microbiology 157, 1000–1008. doi: 10.1099/mic.0.047209-0
Llosa, M., Gomis-Ruth, F. X., Coll, M., and de la Cruz, F. (2002). Bacterial conjugation: a two-step mechanism for DNA transport. Mol. Microbiol. 45, 1–8. doi: 10.1046/j.1365-2958.2002.03014.x
Llosa, M., Grandoso, G., and de la Cruz, F. (1995). Nicking activity of TrwC directed against the origin of transfer of the IncW plasmid R388. J. Mol. Biol. 246, 54–62. doi: 10.1006/jmbi.1994.0065
Madsen, J. S., Burmølle, M., Hansen, L. H., and Sørensen, S. J. (2012). The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol. Med. Microbiol. 65, 183–195. doi: 10.1111/j.1574-695X.2012.00960.x
Mark Glover, J. N., Chaulk, S. G., Edwards, R. A., Arthur, D., Lu, J., and Frost, L. S. (2015). The FinO family of bacterial RNA chaperones. Plasmid 78, 79–87. doi: 10.1016/j.plasmid.2014.07.003
Matson, S. W., and Morton, B. S. (1991). Escherichia coli DNA helicase I catalyzes a site- and strand-specific nicking reaction at the F plasmid oriT. J. Biol. Chem. 266, 16232–16237.
Muscholl-Silberhorn, A. B. (2000). Pheromone-regulated expression of sex pheromone plasmid pAD1-encoded aggregation substance depends on at least six upstream genes and a cis-acting, orientation-dependent factor. J. Bacteriol. 182, 3816–3825. doi: 10.1128/JB.182.13.3816-3825.2000
Nogales, J., Blanca-Ordóñez, H., Olivares, J., and Sanjuán, J. (2013). Conjugal transfer of the Sinorhizobium meliloti 1021 symbiotic plasmid is governed through the concerted action of one- and two-component signal transduction regulators. Environ. Microbiol. 15, 811–821. doi: 10.1111/1462-2920.12073
Pansegrau, W., Balzer, D., Kruft, V., Lurz, R., and Lanka, E. (1990). In vitro assembly of relaxosomes at the transfer origin of plasmid RP4. Proc. Natl. Acad. Sci. U.S.A. 87, 6555–6559. doi: 10.1073/pnas.87.17.6555
Pérez-Mendoza, D., Sepúlveda, E., Pando, V., Muñoz, S., Nogales, J., Olivares, J., et al. (2005). Identification of the rctA gene, which is required for repression of conjugative transfer of rhizobial symbiotic megaplasmids. J. Bacteriol. 187, 7341–7350. doi: 10.1128/JB.187.21.7341-7350.2005
Ramesh, V., and Nagaraja, V. (1996). Sequence-specific DNA binding of the phage Mu C protein: footprinting analysis reveals altered DNA conformation upon protein binding. J. Mol. Biol. 260, 22–33. doi: 10.1006/jmbi.1996.0379
Rosvoll, T. C. S., Pedersen, T., Sletvold, H., Johnsen, P. J., Sollid, J. E., Simonsen, G. S., et al. (2010). PCR-based plasmid typing in Enterococcus faecium strains reveals widely distributed pRE25-, pRUM-, pIP501- and pHTbeta-related replicons associated with glycopeptide resistance and stabilizing toxin-antitoxin systems. FEMS Immunol. Med. Microbiol. 58, 254–268. doi: 10.1111/j.1574-695X.2009.00633.x
Scherzinger, E., Haring, V., Lurz, R., and Otto, S. (1991). Plasmid RSF1010 DNA replication in vitro promoted by purified RSF1010 RepA, RepB and RepC proteins. Nucleic Acids Res. 19, 1203–1211. doi: 10.1093/nar/19.6.1203
Scherzinger, E., Lurz, R., Otto, S., and Dobrinski, B. (1992). In vitro cleavage of double- and single-stranded DNA by plasmid RSF1010-encoded mobilization proteins. Nucleic Acids Res. 20, 41–48. doi: 10.1093/nar/20.1.41
Sepúlveda, E., Pérez-Mendoza, D., Ramírez-Romero, M. A., Soto, M. J., López-Lara, I. M., Geiger, O., et al. (2008). Transcriptional interference and repression modulate the conjugative ability of the symbiotic plasmid of Rhizobium etli. J. Bacteriol. 190, 4189–4197. doi: 10.1128/JB.00041-08
Sørensen, S. J., Bailey, M., Hansen, L. H., Kroer, N., and Wuertz, S. (2005). Studying plasmid horizontal transfer in situ: a critical review. Nat. Rev. Microbiol. 3, 700–710. doi: 10.1038/nrmicro1232
Steinmetzer, K., Behlke, J., and Brantl, S. (1998). Plasmid pIP501 encoded transcriptional repressor CopR binds to its target DNA as a dimer. J. Mol. Biol. 283, 595–603. doi: 10.1006/jmbi.1998.2122
Steinmetzer, K., Behlke, J., Brantl, S., and Lorenz, M. (2002a). CopR binds and bends its target DNA: a footprinting and fluorescence resonance energy transfer study. Nucleic Acids Res. 30, 2052–2060. doi: 10.1093/nar/30.9.2052
Steinmetzer, K., and Brantl, S. (1997). Plasmid pIP501 encoded transcriptional repressor CopR binds asymmetrically at two consecutive major grooves of the DNA. J. Mol. Biol. 269, 684–693. doi: 10.1006/jmbi.1997.1083
Steinmetzer, K., Hillisch, A., Behlke, J., and Brantl, S. (2000a). Transcriptional repressor CopR: amino acids involved in forming the dimeric interface. Proteins 39, 408–416. doi: 10.1002/(SICI)1097-0134(20000601)39:4<408::AID-PROT130>3.0.CO;2-0
Steinmetzer, K., Hillisch, A., Behlke, J., and Brantl, S. (2000b). Transcriptional repressor CopR: structure model-based localization of the deoxyribonucleic acid binding motif. Proteins 38, 393–406. doi: 10.1002/(SICI)1097-0134(20000301)38:4<393::AID-PROT5>3.0.CO;2-H
Steinmetzer, K., Kuhn, K., Behlke, J., Golbik, R., and Brantl, S. (2002b). Plasmid pIP501 encoded transcriptional repressor CopR: single amino acids involved in dimerization are also important for folding of the monomer. Plasmid 47, 201–209. doi: 10.1016/S0147-619X(02)00002-1
Swinfield, T. J., Oultram, J. D., Thompson, D. E., Brehm, J. K., and Minton, N. P. (1990). Physical characterisation of the replication region of the Streptococcus faecalis plasmid pAM beta 1. Gene 87, 79–90.
Szpirer, C. Y., Faelen, M., and Couturier, M. (2001). Mobilization function of the pBHR1 plasmid, a derivative of the broad-host-range plasmid pBBR1. J. Bacteriol. 183, 2101–2110. doi: 10.1128/JB.183.6.2101-2110.2001
Taki, K., Abo, T., and Ohtsubo, E. (1998). Regulatory mechanisms in expression of the traY-I operon of sex factor plasmid R100: involvement of traJ and traY gene products. Genes Cells 3, 331–345. doi: 10.1046/j.1365-2443.1998.00194.x
Tanimoto, K., Tomita, H., and Ike, Y. (1996). The traA gene of the Enterococcus faecalis conjugative plasmid pPD1 encodes a negative regulator for the pheromone response. Plasmid 36, 55–61. doi: 10.1006/plas.1996.0032
Tullius, T. D., and Dombroski, B. A. (1986). Hydroxyl radical “footprinting”: high-resolution information about DNA-protein contacts and application to lambda repressor and Cro protein. Proc. Natl. Acad. Sci. U.S.A. 83, 5469–5473. doi: 10.1073/pnas.83.15.5469
Tzou, W. S., and Hwang, M. J. (1999). Modeling helix-turn-helix protein-induced DNA bending with knowledge-based distance restraints. Biophys. J. 77, 1191–1205. doi: 10.1016/S0006-3495(99)76971-7
van der Lelie, D., Wösten, H. A., Bron, S., Oskam, L., and Venema, G. (1990). Conjugal mobilization of streptococcal plasmid pMV158 between strains of Lactococcus lactis subsp. lactis. J. Bacteriol. 172, 47–52.
Van Kaer, L., Van Montagu, M., and Dhaese, P. (1989). Purification and in vitro DNA-binding specificity of the Bacillus subtilis phage phi 105 repressor. J. Biol. Chem. 264, 14784–14791.
Wang, A., and Macrina, F. L. (1995). Streptococcal plasmid pIP501 has a functional oriT site. J. Bacteriol. 177, 4199–4206.
Will, W. R., and Frost, L. S. (2006). Hfq is a regulator of F-plasmid TraJ and TraM synthesis in Escherichia coli. J. Bacteriol. 188, 124–131. doi: 10.1128/JB.188.1.124-131.2006
Wong, J. J. W., Lu, J., and Glover, J. N. M. (2012). Relaxosome function and conjugation regulation in F-like plasmids - a structural biology perspective. Mol. Microbiol. 85, 602–617. doi: 10.1111/j.1365-2958.2012.08131.x
Zatyka, M., Jagura-Burdzy, G., and Thomas, C. M. (1994). Regulation of transfer genes of promiscuous IncP alpha plasmid RK2: repression of Tra1 region transcription both by relaxosome proteins and by the Tra2 regulator TrbA. Microbiology 140(Pt 1), 2981–2990. doi: 10.1099/13500872-140-11-2981
Zatyka, M., Jagura-Burdzy, G., and Thomas, C. M. (1997). Transcriptional and translational control of the genes for the mating pair formation apparatus of promiscuous IncP plasmids. J. Bacteriol. 179, 7201–7209.
Keywords: conjugative plasmid, replication, copy number control, type IV secretion, broad-host-range, transfer control
Citation: Grohmann E, Goessweiner-Mohr N and Brantl S (2016) DNA-Binding Proteins Regulating pIP501 Transfer and Replication. Front. Mol. Biosci. 3:42. doi: 10.3389/fmolb.2016.00042
Received: 24 June 2016; Accepted: 29 July 2016;
Published: 11 August 2016.
Edited by:
Manuel Espinosa, Spanish National Research Council, SpainReviewed by:
Itziar Alkorta, University of the Basque Country, SpainGloria Del Solar, Spanish National Research Council, Spain
Copyright © 2016 Grohmann, Goessweiner-Mohr and Brantl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabeth Grohmann, ZWxpc2FiZXRoLmdyb2htYW5uQGJldXRoLWhvY2hzY2h1bGUuZGU=
Nikolaus Goessweiner-Mohr, bmlrb2xhdXMuZ29lc3N3ZWluZXJAaW1iYS5vZWF3LmFjLmF0
Sabine Brantl, c2FiaW5lLmJyYW50bEB1bmktamVuYS5kZQ==