 Kecheng Zhou
Kecheng Zhou Minxia Liu1,2†
Minxia Liu1,2†- 1Laboratory of Molecular and Experimental Pathology, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
- 2Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming, China
Tumorigenesis is a multi-step and complex process with multi-factors involved. Deregulated oncogenes and tumor suppressor genes (TSGs) induced by genetic and epigenetic factors are considered as the driving force in the development and progression of cancer. Besides, microRNAs (miRNAs) act vital roles in tumorigenesis through regulating some oncogenes and TSGs. Interestingly, miRNAs are also regulated by oncogenes and TSGs. Considering the entangled regulation, here we propose a new insight into these regulation relationships in cancer: oncogene–miRNA–TSG network, which further emphasizes roles of miRNA, as well as highlights the network regulation among oncogene, miRNA, and TSG during tumorigenesis. The oncogene–miRNA–TSG network demonstrates that oncogenes and TSGs not only show functional synergy, but also there are regulatory relationships among oncogenes and TSGs during tumorigenesis, which could be mediated by miRNAs. In view of the oncogene–miRNA–TSG network involved in many oncogenes, miRNAs, and TSGs, as well as occurring in various tumor types, the anomaly of this network may be a common event in cancers and participates in tumorigenesis. This hypothesis broadens horizons of molecular mechanisms underlying tumorigenesis, and may provide a new promising venue for the prediction, diagnosis, and even therapy of cancer.
Introduction
Tumorigenesis is a complicated process, induced by multi-factors, such as environment, genetics, and epigenetics. Mechanisms underlying tumorigenesis are involved in gene mutation, chromosome stability, DNA repair, epigenetic changes, and cell growth, differentiation, movement, apoptosis, autophagy, and so on. Abnormal expressions of oncogene and tumor suppressor gene (TSG) are regarded as the key driving force promoting the cell malignant transformation. For instance, loss of p53 (an important TSG) promotes cell proliferation (Drosten et al., 2014), and disturbs p53-dependent apoptosis (Vazquez et al., 2008); meanwhile, activation of p53 often occurs in the response of DNA damage (Lakin and Jackson, 1999; Smith and Seo, 2002); activated Myc (an important oncogene) was involved in dysfunction of several important cell processes, including growth control (Schmidt, 1999), apoptosis (Hoffman and Liebermann, 2008), and DNA damage response and repair (Campaner and Amati, 2012; Li et al., 2012).
MicroRNAs (miRNAs), a post-transcriptional level regulator, have fundamental activities in cancer development and progression by modulating oncogene and/or TSG through specifically targeting messenger RNA (mRNA) 3′-untranslated region (UTR), either degrading targeted mRNA or blocking translation. Numerous studies indicated that miRNAs act as oncogenic miRNA (by targeting TSG) or tumor suppressive miRNA (by targeting oncogene). Interestingly, some oncogenes or tumor suppressor genes (TSGs) are capable of activating miRNA transcription by binding to promoter regions of miRNA host genes. MiRNA expressions are also regulated by oncogene and/or TSG in addition to mutations and epigenetic changes (mainly methylation) of miRNA host genes. Therefore, miRNAs are involved in the construction of convoluted signal transduction pathways together with oncogene and TSG. Here, we propose oncogene–miRNA–TSG network hypothesis, which highlights miRNAs may function as mediators among oncogenes as well as TSGs, and as key nodes of the collaborated regulation network. Furthermore, the hypothesis provides other potential regulation relationships among oncogenes and TSGs.
Functions and Functional Synergy of Oncogene and TSG
Many factors are involved in tumorigenesis and promote the cell malignant transformation, consequences of the characters or the hallmarks of cancer (Hanahan and Weinberg, 2011). Functional alterations of oncogene and TSG are one of the most important mechanisms underlying tumorigenesis (Figure 1A). Gene mutation can induce functional changes of gene. Many environmental factors, for instance, chemical carcinogens (e.g., benzopyrene from smoking and air pollution; Hecht, 1999), physical exposure (such as ultraviolet irradiation and radiation exposure), as well as pathogenic bacteria and viruses (including human hepatitis B and C virus, and human papilloma virus), induce somatic mutation (Minamoto et al., 1999). Random mutations during DNA replication in stem cells are also an important source of somatic mutations (Tomasetti and Vogelstein, 2015; Tomasetti et al., 2017). Germline mutations arise from parent heredity. A sequence of oncogene and TSG mutations including somatic mutations and germline mutations could result in the cell malignant transformation (Luzzatto, 2011). Genetic instability and mutations are not only the hallmark of cancer, but also central to the genesis, development, and evolution of cancer (Loeb and Loeb, 2000). The gene mutation leads to either the active function, like EGFR in non-small cell lung cancer (NSCLC) (Lynch et al., 2004), or the inactive function, like p53 mutation (Muller and Vousden, 2013). The gain-of-function of oncogene and loss-of-function of TSG play key roles during tumorigenesis, and also provide potential therapeutic targets (Paez et al., 2004; Hong et al., 2014), even though current situation is still challenging (Yu et al., 2014).
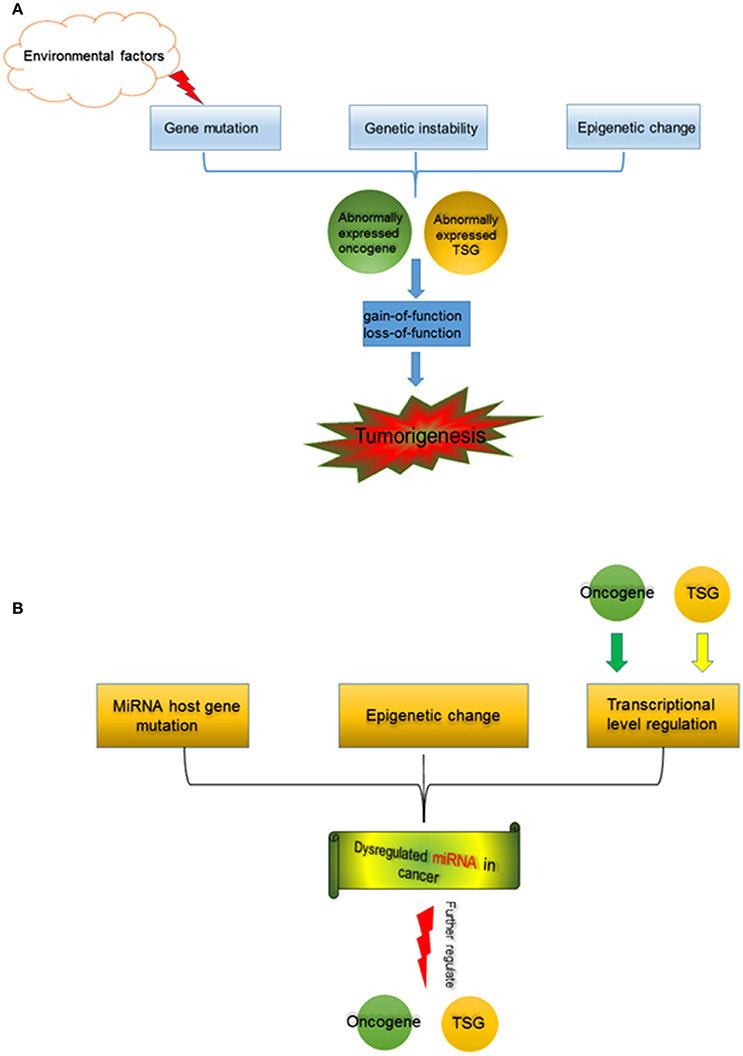
Figure 1. (A) Tumorigenesis is a multi-factor induced process. Gene mutation, which could be induced by environmental factors (such as chemical carcinogens, physical carcinogens, pathogenic bacteria, and viruses), and genetic instability, as well as alterations in epigenetics cause abnormal expression of oncogene and TSG. Functional changes of oncogene and TSG, separately or jointly drive tumorigenesis. (B) miRNA in cancer. MiRNA host gene mutation, epigenetic change (including aberrant DNA demethylation, aberrant modification of histone deacetylase, etc.), and the transcription regulation by oncogene and TSG contribute to miRNA dysregulation in cancer. The abnormally expressed miRNAs further regulate oncogene and TSG during tumorigenesis.
Epigenetic changes, including DNA methylation (Worm et al., 2002), histone modifications (Cohen et al., 2011), chromatin remodeling (Wolffe, 2001), as well as miRNAs (Kala et al., 2013), etc. also induce dysfunction of oncogene and TSG, which are observed in kinds of cancers (Grønbæk et al., 2007; Singh et al., 2016; Wijetunga et al., 2016). Many studies showed that the collaboration of epigenetic and genetic changes is critically vital to drive the cancer development and progression (Baylin and Jones, 2016).
It has been noted that multi-factors and series cell processes emerge during tumorigenesis. By preforming whole-genome-sequencing studies of 3281 tumors from 12 cancer types, more than 600,000 somatic mutations were observed in cancer cells (Kandoth et al., 2013). Those mutations occur in plenty of oncogenes and TSGs, further induce either gain-of-function or loss-of-function. Eventually, altered oncogenes and TSGs collaboratively drive the cancer development and progression.
MiRNA Roles in Cancer
MiRNA is a vital factor contributing to the epigenetic regulation. After its first discovery (Lee et al., 1993), miRNAs were quickly shown with a conserved mechanism and broad functional significance (Bartel, 2004). Moreover, abnormally expressed miRNAs are useful biomarkers for cancer diagnosis, and even promising targets for cancer therapy (Hayes et al., 2014).
Origins of dysregulated miRNAs are various in cancer cells (Figure 1B). MiRNA host gene codes miRNA, and it can regulate miRNA expression at the transcription level. Some mutations within miRNA host genes induced incorrect miRNA expressions at the DNA level. MiRNA dysregulation induced by gene mutation was observed in cancers and other diseases (Meola et al., 2009). Interestingly, in colorectal cancers from Kashmiri population, mutations/SNPs within miRNA genes or their binding sites in 3′-UTR are infrequent events, indicating that miRNA gene mutations may not be as common as mutations of protein coding genes during tumorigenesis (Maqbool et al., 2014). Epigenetic changes are also vital factors contributing abnormal miRNA expressions in cancer. A research revealed that the 17 miRNAs were up-regulated by simultaneous treatment with the chromatin-modifying drugs 5-aza-2′-deoxycytidine (5-Aaza-Ccdr; a DNA methyltransferase inhibitor) and 4-phenylbutyric acid (PBA; a histone deacetylase inhibitor), demonstrating that epigenetic mechanisms (DNA demethylation and histone acetylation) could alter miRNA expressions (Saito et al., 2006). In addition, 5-Aaza-Ccdr and PBA treatments activated the miR-512-5p expression in gastric cancer cells, which further caused the MCL1 suppression and resulted in the cell apoptosis (Saito et al., 2009). These confirmed that chromatin remodeling induced by the epigenetic treatment was capable to directly affect specific miRNA expression.
Apart from genetic and epigenetic mechanisms, transcription factors can also bind to promoter regions of miRNA host genes, and regulate miRNA expression. Transcription factor binding sites were highly enriched within miRNA precursor sequences, suggesting that transcription factors may directly bind to primary miRNA gene transcripts or small hairpin miRNA precursors, thus, regulate their processing (Piriyapongsa et al., 2011). Moreover, some well-known oncogenes and TSGs regulate miRNA expression at the transcription level. For instance, c-Myc up-regulated miR-17 ~ 92 and miR-106a ~ 363 expressions in cultured human trophoblasts (Kumar et al., 2013). Jackstadt et al carried out genome-wide analyses of c-Myc-regulated mRNAs and miRNAs, collaborating an accelerated and comprehensive understanding of c-Myc function (Jackstadt et al., 2013). Strikingly, c-Myc also transactivated drosha expression though directly binding to the E-box of the drosha promoter (Wang et al., 2013), which imposed on board and general regulation of miRNAs. P53 could activate miR-34 family, such as miR-34a (Chang et al., 2007), miR-34b, and miR-34c (Corney et al., 2007), further affected cell apoptosis, proliferation, and adhesion-independent growth.
Interestingly, oncogene and TSG are also targets of miRNAs (Figure 1B). MiRNA let-7a down-regulated Myc, and thus reverted the Myc-Induced cell growth in burkitt lymphoma cells (Sampson et al., 2007); miR-34a modulated c-Myc transcriptional complexes, suppressing malignancy in human prostate cancer cells (Yamamura et al., 2012). MiR-504 acted as a negative regulator of p53 by directly binding to the 3′-UTR, therefore, decreased the p53 mediated-apoptosis and cell-cycle stress (Hu et al., 2010). Additionally, miR-125b was a novel regulator of p53, further affects the p53-induced apoptosis during development and stress response (Le et al., 2009). Besides c-Myc and p53, other oncogenes and TSGs were regulated by miRNAs as well, and involved in the development and progression of cancer. For instance, miR-144 targeted oncogene ZEB1, and a decreased miR-144 expression resulted in the increased Zeb1 expression and epithelial-mesenchymal transition (EMT) (Pan et al., 2015). Another study identified three differentially expressed miRNA in metastases as key drivers of EMT though targeting SIAH1, SETD2, ZEB2, and FOXN3 (Mudduluru et al., 2015). Currently, we found miR-203a regulated the expression of ERGIC3 (a candidate oncogene), and further affected cell proliferation in lung cancer (Lin et al., 2015).
It is worthy to note about several characters of miRNA regulation and related problems in miRNA researches. (1) MiRNA regulation is a dynamic process. By using Affymetrix microarray platform, Kumari et al profiled miRNA and mRNA expressions at multiple time points, and revealed the dynamic changes in global miRNAome and transcriptome (Kumari et al., 2016). A previous study also suggested the dynamic modeling of miRNA regulation during the mesenchymal stem cell differentiation (Weber et al., 2013). Thus, the dynamic character implies that miRNA study needs to consider the effects of time and state on results. (2) One miRNA can target multiple mRNAs, thence, one miRNA has different functions. For example, miR-21 affected cell survival by targeting PCPD4 (programmed cell death protein 4) (Jiao et al., 2015), regulated cell proliferation by targeting PTEN (Zhang et al., 2010), and meanwhile was involved in cancer metastasis by targeting Maspin (Zhu et al., 2008). Sometimes, opposite functions were observed for one miRNA in diverse cases, because it targeted different mRNAs in specific cell types or cell states. For instance, one study suggested miR-944 functioned as a novel oncogenic miRNA and regulated the chemoresistance in breast cancer (He et al., 2016), meanwhile another study reported that the miR-944 expression was severely repressed in breast cancer cells and clinical specimens, and suppressed the cell migration by targeting SIAH1 (Flores-Pérez et al., 2016). Interestingly, our recent study revealed miR-944 was significantly down-regulated in NSCLC by utilizing small RNA deep sequencing, and found that miR-944 inhibited cellular proliferation through targeting EPHA7 in NSCLC, which may offer a new mechanism underlying the development and progression of NSCLC (Liu et al., 2016). Therefore, when studying miRNA functions, it is necessary to declare detail mechanisms and targets. Since one specific miRNA could possess various functions, physiological roles of miRNA should be further investigated by animal models, such as knockout and transgenic animals. (3) One mRNA is regulated by multiple miRNAs simultaneously, which may be involved in the complexity of miRNA synergy and may be one reason of practical problems in miRNA studies. Utilizing miRNA mimic and miRNA inhibitor to study the miRNA's target and examine its related function is a classical and established strategy. However, in practice, effects of miRNA mimic and inhibitor are not always at opposite ends, some discrepancies results by using miRNA mimics and antisense inhibitors were observed (Thomson et al., 2013). Occasionally, transfected small RNA may compete with endogenous miRNA for the RNA-induced silencing complex (RISC) or other machinery, thus, could result in artificial readout (Khan et al., 2009). Meanwhile, miRNA inhibitors may not be sufficiently specific to separate different members from the same miRNA family harboring similar sequences (Thomson et al., 2011). Another potential explanation may be due to neutralization and compensation mechanisms. Briefly, after one miRNA is modulated through mimic or inhibitor treatments, other miRNAs which target the same mRNA like this miRNA, may change their expressions to neutralize or compensate the biological effects.
Oncogene–miRNA–TSG Network Hypothesis
In early studies, we screened novel cancer-related genes and found that microspherule protein 1 (MCRS1) was overexpressed in lung cancer (Liang et al., 2013). Subsequently, we confirmed the overexpression of MCRS1 was associated with the cellular proliferation, EMT, and metastasis (Liu et al., 2014). MCRS1 was a candidate oncogene. Furthermore, MCRS1 transactivated the miR-155 expression by directly binding to its promoter region (Liu et al., 2014). Interestingly, in the continuing study, we established that MCRS1 promoted cancer cell growth via miR-155 targeting Rb1 (Liu et al., 2015). Thus, there is the MCRS1–miR-155–Rb1 pathway in lung cancer. MiR-155 mediated the regulation relationship between MCRS1 (a candidate oncogene) and Rb1 (a famous TSG), suggesting miRNAs could potentially act as a mediator between oncogene and TSG. Additionally, a recent study demonstrated that miR-200 mediated the regulation relationship between p53 (a well-established TSG) and ZEB1/BMI1 (a well-known oncogene), and p53–miR-200–ZEB1/BMI1 contributed to EMT in breast cancer cells (Chang et al., 2011). Meanwhile, MCRS1 and Rb1, p53 and ZEB1/BMI1 are simultaneously regulated by other miRNAs, such as miR-129* (targeting MCRS1) and miR-125b (targeting p53), respectively. Taking those regulation pathways into consideration, we thought that oncogenes/TSGs can regulate miRNAs expression by binding to promoter regions; and the disrupted miRNAs, in turn, continue to regulate other oncogenes/TSGs. Here, we propose a new hypothesis, oncogene–miRNAs–TSG network (Figure 2). MiRNAs act as transductor/mediator among oncogenes and/or TSGs. In one case, miRNAs regulated by oncogenes further target TSGs, to collaborate and accelerate effects of oncogene (the miRNA upstream). In another case, if the miRNA regulated by one oncogene further aims at another oncogene, this oncogene 1–miRNA–oncogene 2 feedback will alleviate the effect of oncogene 1, suggesting that there may be precise regulation relationships among oncogenes. For examples, oncogenic KRAS regulated miR-200c (Tsunoda et al., 2011), while miR-200c targeted ZEB1/ZEB2 (Korpal et al., 2008) (ZEB1/ZEB2 functions as an oncogene to promote the cancer metastasis). Interestingly, miR-200c was also shown to target KRAS (Kopp et al., 2014). Here, miR-200c mediated the regulation relationship among oncogenes (KRAS, ZEB1/ZEB2), and acted the feedback regulation of one oncogene (KRAS). Considering the regulation relationships involved in many oncogenes, miRNAs, and TSGs, we extend the hypothesis to oncogene/TSG–miRNA–oncogene/TSG network (in order to express the convenience and simplicity, we still used “oncogene–miRNA–TSG network” in this article), which addresses the function of miRNA, and the complexity of the regulation network as well.
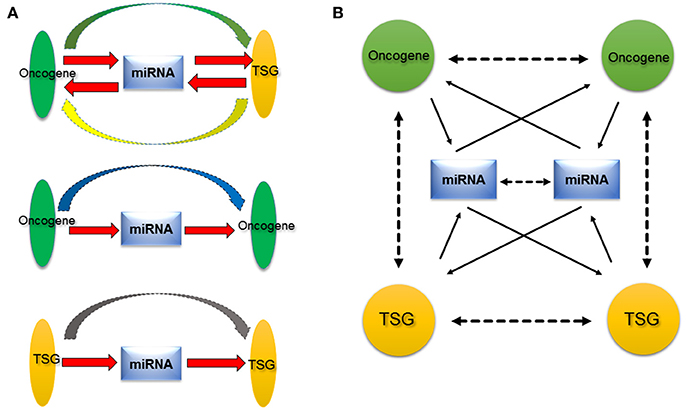
Figure 2. Proposed oncogene-miRNA-TSG regulation network. (A) MiRNA acts a central role among oncogene or TSG. On one hand, miRNA regulates oncogene or TSG, on the other hand, miRNA is regulated by oncogene or TSG (red arrow). Therefore, miRNA may serve as transductor/mediator among oncogene and TSG. Besides the functional synergy of oncogene and TSG, there are regulatory relationships between oncogene and TSG (green and yellow arrow), between oncogenes (blue arrow), and between TSGs (gray arrow). (B) Because the existence of numerous oncogenes, miRNAs, and TSGs, their relationships are presented as a complex network, the oncogene–miRNA–TSG network. The network provides a new insight into roles of miRNAs, oncogene, and TSG in cancer. Solid arrows indicate direct relationships that have been verified with experiments at the molecular level, and dotted arrows denote indirect relationships that are supported by experiments at the cellular level.
We think that this oncogene–miRNA–TSG network may be prevalent in normal cells too, and it generates various regulation relationships and possesses important biological functions, which is extremely essential to keep intermolecular homeostasis for the normal status of cells. In diseases, particularly in cancers, dysregulations of certain oncogenes, miRNAs, and TSGs that were induced by genetic mutations or epigenetic changes etc., disrupt the balance of this molecular network, and transduct mislead signals to more downstream pathway molecules (including oncogene, miRNA, and TSG), and arouse domino effects, therefore, promote the cell malignant transformation. The oncogene–miRNA–TSG network is involved in many oncogenes, miRNAs, and TSGs, as well as occurs in various tumor types, hence it may participate in the development and progression of a wide range of tumors. The anomaly of this network may be a common event in cancers. In addition, the oncogene–miRNA–TSG network hypothesis demonstrated that oncogenes and TSGs not only show functional synergy, but also there are regulatory relationships among oncogenes and TSGs during tumorigenesis, which could be mediated by miRNAs. Finally, this hypothesis also addresses that tumorigenesis may be a result of the loss of the intermolecular homeostasis, scilicet, the disruption of entire oncogene–miRNA–TSG network rather than single dysregulation of oncogene or TSG. MiRNAs can be released through exosome from cancer cells into body fluids, such as blood, urine, milk, sputum, saliva. These miRNAs can serve as biomarkers for diagnosis (Tran, 2016). Recognizing the significances of oncogene–miRNA–TSG network may help better understanding oncogenes, TSGs, and miRNAs roles during the development and progression of cancer, and may provide a new promising venue of prediction, diagnosis, and therapy of cancer.
Author Contributions
KZ, ML, and YC jointly wrote this review. KZ drafted the manuscript. ML participated in in-depth discussions and made constructive suggestions and comments. YC developed the structure, design the scope of the review, and helped to draft the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Natural Science Foundation of China (81272617).
References
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Baylin, S. B., and Jones, P. A. (2016). Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 8:a019505. doi: 10.1101/cshperspect.a019505
Campaner, S., and Amati, B. (2012). Two sides of the Myc-induced DNA damage response: from tumor suppression to tumor maintenance. Cell Div. 7:6. doi: 10.1186/1747-1028-7-6
Chang, C.-J., Chao, C.-H., Xia, W., Yang, J.-Y., Xiong, Y., Li, C.-W., et al. (2011). p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol. 13, U317–U296. doi: 10.1038/ncb2173
Chang, TC., Wentzel, EA., Kent, OA., Ramachandran, K., Mullendore, M., Lee, KH., et al. (2007). Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 26, 745–752. doi: 10.1016/j.molcel.2007.05.010
Cohen, I., Poręba, E., Kamieniarz, K., and Schneider, R. (2011). Histone modifiers in cancer: friends or foes? Genes Cancer 2, 631–647. doi: 10.1177/1947601911417176
Corney, D. C., Flesken-Nikitin, A., Godwin, A. K., Wang, W., and Nikitin, A. Y. (2007). MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 67, 8433–8438. doi: 10.1158/0008-5472.CAN-07-1585
Drosten, M., Sum, E. Y. M., Lechuga, C. G., Simón-Carrasco, L., Jacob, H. K. C., García-Medina, R., et al. (2014). Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 111, 15155–15160. doi: 10.1073/pnas.1417549111
Flores-Pérez, A., Marchat, L. A., Rodríguez-Cuevas, S., Bautista, V. P., Fuentes-Mera, L., Romero-Zamora, D., et al. (2016). Suppression of cell migration is promoted by miR-944 through targeting of SIAH1 and PTP4A1 in breast cancer cells. BMC Cancer 16:379. doi: 10.1186/s12885-016-2470-3
Grønbæk, K., Hother, C., and Jones, P. A. (2007). Epigenetic changes in cancer. APMIS 115, 1039–1059. doi: 10.1111/j.1600-0463.2007.apm_636.xml.x
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hayes, J., Peruzzi, P. P., and Lawler, S. (2014). MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol. Med. 20, 460–469. doi: 10.1016/j.molmed.2014.06.005
He, H., Tian, W., Chen, H., and Jiang, K. (2016). MiR-944 functions as a novel oncogene and regulates the chemoresistance in breast cancer. Tumor Biol. 37, 1599–1607. doi: 10.1007/s13277-015-3844-x
Hecht, S. S. (1999). Tobacco smoke carcinogen and lung cancer. J. Natl. Cancer Inst. 91, 1194–1210. doi: 10.1093/jnci/91.14.1194
Hoffman, B., and Liebermann, D. A. (2008). Apoptotic signaling by c-MYC. Oncogene 27, 6462–6472. doi: 10.1038/onc.2008.312
Hong, B., van den Heuvel, A. P. J., Prabhu, V. V, Zhang, S., and El-Deiry, W. S. (2014). Targeting tumor suppressor p53 for cancer therapy: strategies, challenges and opportunities. Curr. Drug Targets 15, 80–89. doi: 10.2174/1389450114666140106101412
Hu, W., Chan, C. S., Wu, R., Zhang, C., Sun, Y., Song, J. S., et al. (2010). Negative regulation of tumor suppressor p53 by MicroRNA miR-504. Mol. Cell 38, 689–699. doi: 10.1016/j.molcel.2010.05.027
Jackstadt, R., Menssen, A., and Hermeking, H. (2013). Genome-wide analysis of c-MYC-regulated mRNAs and miRNAs, and c-MYC DNA binding by next-generation sequencing. Methods Mol. Biol. 1012, 145–185. doi: 10.1007/978-1-62703-429-6_11
Jiao, J., Fan, Y., and Zhang, Y. (2015). Expression and clinicopathological significance of microRNA-21 and programmed cell death 4 in malignant melanoma. J. Int. Med. Res. 43, 672–678. doi: 10.1177/0300060515583707
Kala, R., Peek, G. W., Hardy, T. M., and Tollefsbol, T. O. (2013). MicroRNAs: an emerging science in cancer epigenetics. J. Clin. Bioinforma. 3:6. doi: 10.1186/2043-9113-3-6
Kandoth, C., McLellan, M. D., Vandin, F., Ye, K., Niu, B., Lu, C., et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339. doi: 10.1038/nature12634
Khan, A. A., Betel, D., Miller, M. L., Sander, C., Leslie, C. S., and Marks, D. S. (2009). Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat. Biotechnol. 27, 549–555. doi: 10.1038/nbt0709-671a
Kopp, F., Wagner, E., and Roidl, A. (2014). The proto-oncogene KRAS is targeted by miR-200c. Oncotarget 5, 185–195. doi: 10.18632/oncotarget.1427
Korpal, M., Lee, E. S., Hu, G., and Kang, Y. (2008). The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 283, 14910–14914. doi: 10.1074/jbc.C800074200
Kumar, P., Luo, Y., Tudela, C., Alexander, J. M., and Mendelson, C. R. (2013). The c-Myc-regulated microRNA-17~92 (miR-17~92) and miR-106a~363 clusters target hCYP19A1 and hGCM1 to inhibit human trophoblast differentiation. Mol. Cell. Biol. 33, 1782–1796. doi: 10.1128/MCB.01228-12
Kumari, B., Jain, P., Das, S., Ghosal, S., Hazra, B., Trivedi, A. C., et al. (2016). Dynamic changes in global microRNAome and transcriptome reveal complex miRNA-mRNA regulated host response to Japanese encephalitis virus in microglial cells. Sci. Rep. 6:20263. doi: 10.1038/srep20263
Lakin, N. D., and Jackson, S. P. (1999). Regulation of p53 in response to DNA damage. Oncogene 18, 7644–7655. doi: 10.1038/sj.onc.1203015
Le, M. T. N., Teh, C., Shyh-Chang, N., Xie, H., Zhou, B., Korzh, V., et al. (2009). MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 23, 862–876. doi: 10.1101/gad.1767609
Lee, R. C., Feinbaum, R. L., and Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854. doi: 10.1016/0092-8674(93)90529-Y
Li, Z., Owonikoko, T. K., Sun, S.-Y., Ramalingam, S. S., Doetsch, P. W., Xiao, Z.-Q., et al. (2012). c-Myc suppression of DNA double-strand break repair. Neoplasia 14, 1190–1202. doi: 10.1593/neo.121258
Liang, Y., Liu, M., Wang, P., Ding, X., and Cao, Y. (2013). Analysis of 20 genes at chromosome band 12q13: RACGAP1 and MCRS1 overexpression in non-small-cell lung cancer. Genes Chromosom. Cancer 52, 305–315. doi: 10.1002/gcc.22030
Lin, Q. H., Zhang, K. D., Duan, H. X., Liu, M. X., Wei, W. L., and Cao, Y. (2015). ERGIC3, which is regulated by miR-203a, is a potential biomarker for non-small cell lung cancer. Cancer Sci. 106, 1463–1473. doi: 10.1111/cas.12741
Liu, M.-X., Zhou, K.-C., and Cao, Y. (2014). MCRS1 overexpression, which is specifically inhibited by miR-129*, promotes the epithelial-mesenchymal transition and metastasis in non-small cell lung cancer. Mol. Cancer 13:245. doi: 10.1186/1476-4598-13-245
Liu, M., Zhou, K., and Cao, Y. (2016). MicroRNA-944 affects cell growth by targeting EPHA7 in non-small cell lung cancer. Int. J. Mol. Sci. 17:1493. doi: 10.3390/ijms17101493
Liu, M., Zhou, K., Huang, Y., and Cao, Y. (2015). The candidate oncogene (MCRS1) promotes the growth of human lung cancer cells via the miR-155-Rb1 pathway. J. Exp. Clin. Cancer Res. 34:121. doi: 10.1186/s13046-015-0235-5
Loeb, K. R., and Loeb, L. A. (2000). Significance of multiple mutations in cancer. Carcinogenesis 21, 379–385. doi: 10.1093/carcin/21.3.379
Luzzatto, L. (2011). Somatic mutations in cancer development. Environ. Health 10(Suppl. 1), S12. doi: 10.1186/1476-069x-10-s1-s12
Lynch, T. J., Bell, D. W., Sordella, R., Gurubhagavatula, S., Okimoto, R. A., Brannigan, B. W., et al. (2004). Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 350, 2129–2139. doi: 10.1056/NEJMoa040938
Maqbool, R., Ismail, R., and Hussain, M. (2014). Mutations in MicroRNA genes and their binding sites are infrequently associated with human colorectal cancer in the kashmiri population. Microrna 2, 219–224. doi: 10.2174/2211536602666140102001007
Meola, N., Gennarino, V. A., and Banfi, S. (2009). microRNAs and genetic diseases. Pathogenetics 2:7. doi: 10.1186/1755-8417-2-7
Minamoto, T., Mai, M., and Ronai, Z. (1999). Environmental factors as regulators and effectors of multistep carcinogenesis. Carcinogenesis 20, 519–527. doi: 10.1093/carcin/20.4.519
Mudduluru, G., Abba, M., Batliner, J., Patil, N., Scharp, M., Lunavat, T. R., et al. (2015). A systematic approach to defining the microrna landscape in metastasis. Cancer Res. 75, 3010–3019. doi: 10.1158/0008-5472.CAN-15-0997
Muller, P. A. J., and Vousden, K. H. (2013). P53 mutations in cancer. Nat. Cell Biol. 15, 2–8. doi: 10.1038/ncb2641
Paez, J. G., Jänne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., et al. (2004). EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500. doi: 10.1126/science.1099314
Pan, H.-L., Wen, Z.-S., Huang, Y.-C., Cheng, X., Wang, G.-Z., Zhou, Y.-C., et al. (2015). Down-regulation of microRNA-144 in air pollution-related lung cancer. Sci. Rep. 5:14331. doi: 10.1038/srep14331
Piriyapongsa, J., Jordan, I. K., Conley, A. B., Ronan, T., and Smalheiser, N. R. (2011). Transcription factor binding sites are highly enriched within microRNA precursor sequences. Biol. Direct 6:61. doi: 10.1186/1745-6150-6-61
Saito, Y., Liang, G., Egger, G., Friedman, J. M., Chuang, J. C., Coetzee, G. A., et al. (2006). Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 9, 435–443. doi: 10.1016/j.ccr.2006.04.020
Saito, Y., Suzuki, H., Tsugawa, H., Nakagawa, I., Matsuzaki, J., Kanai, Y., et al. (2009). Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 28, 2738–2744. doi: 10.1038/onc.2009.140
Sampson, V. B., Rong, N. H., Han, J., Yang, Q., Aris, V., Soteropoulos, P., et al. (2007). MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 67, 9762–9770. doi: 10.1158/0008-5472.CAN-07-2462
Schmidt, E. V (1999). The role of c-myc in cellular growth control. Oncogene 18, 2988–2996. doi: 10.1038/sj.onc.1202751
Singh, U., Malik, M. A., Goswami, S., Shukla, S., and Kaur, J. (2016). Epigenetic regulation of human retinoblastoma. Tumor Biol. 37, 14427–14441. doi: 10.1007/s13277-016-5308-3
Smith, M. L., and Seo, Y. R. (2002). p53 regulation of DNA excision repair pathways. Mutagenesis 17, 149–156. doi: 10.1093/mutage/17.2.149
Thomson, D. W., Bracken, C. P., and Goodall, G. J. (2011). Experimental strategies for microRNA target identification. Nucleic Acids Res. 39, 6845–6853. doi: 10.1093/nar/gkr330
Thomson, D. W., Bracken, C. P., Szubert, J. M., and Goodall, G. J. (2013). On Measuring miRNAs after Transient Transfection of Mimics or Antisense Inhibitors. PLoS ONE 8:e55214. doi: 10.1371/journal.pone.0055214
Tomasetti, C., Li, L., and Vogelstein, B. (2017). Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355, 1330–1334. doi: 10.1126/science.aaf9011
Tomasetti, C., and Vogelstein, B. (2015). Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347, 78–81. doi: 10.1126/science.1260825
Tran, N., (2016). Cancer exosomes as miRNA factories. Trends Cancer 2, 329–331. doi: 10.1016/j.trecan.2016.05.008
Tsunoda, T., Takashima, Y., Yoshida, Y., Doi, K., Tanaka, Y., Fujimoto, T., et al. (2011). Oncogenic KRAS regulates miR-200c and miR-221/222 in a 3D-specific manner in colorectal cancer cells. Anticancer Res. 31, 2453–2459.
Vazquez, A., Bond, E. E., Levine, A. J., and Bond, G. L. (2008). The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 7, 979–987. doi: 10.1038/nrd2656
Wang, X., Zhao, X., Gao, P., and Wu, M. (2013). c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci. Rep. 3:1942. doi: 10.1038/srep01942
Weber, M., Sotoca, A. M., Kupfer, P., Guthke, R., and van Zoelen, E. J. (2013). Dynamic modelling of microRNA regulation during mesenchymal stem cell differentiation. BMC Syst. Biol. 7:124. doi: 10.1186/1752-0509-7-124
Wijetunga, N. A., Belbin, T. J., Burk, R. D., Whitney, K., Abadi, M., Greally, J. M., et al. (2016). Novel epigenetic changes in CDKN2A are associated with progression of cervical intraepithelial neoplasia. Gynecol. Oncol. 142, 566–573. doi: 10.1016/j.ygyno.2016.07.006
Wolffe, A. P. (2001). Chromatin remodeling: why it is important in cancer. Oncogene 20, 2988–2990. doi: 10.1038/sj.onc.1204322
Worm, J., Guldberg, P., and Article, R. (2002). DNA methylation: an epigenetic pathway to cancer and a promising target for anticancer therapy. J. Oral Pathol. Med. 31, 443–449. doi: 10.1034/j.1600-0714.2002.00034.x
Yamamura, S., Saini, S., Majid, S., Hirata, H., Ueno, K., Deng, G., et al. (2012). Microrna-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 7:e29722. doi: 10.1371/journal.pone.0029722
Yu, X., Narayanan, S., Vazquez, A., and Carpizo, D. R. (2014). Small molecule compounds targeting the p53 pathway: are we finally making progress? Apoptosis 19, 1055–1068. doi: 10.1007/s10495-014-0990-3
Zhang, J. G., Wang, J. J., Zhao, F., Liu, Q., Jiang, K., and Yang, G. H. (2010). MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 411, 846–852. doi: 10.1016/j.cca.2010.02.074
Keywords: oncogene–microRNA–tumor suppressor gene network, miRNA function, cancer, oncogenes, tumor suppressor genes
Citation: Zhou K, Liu M and Cao Y (2017) New Insight into microRNA Functions in Cancer: Oncogene–microRNA–Tumor Suppressor Gene Network. Front. Mol. Biosci. 4:46. doi: 10.3389/fmolb.2017.00046
Received: 15 April 2017; Accepted: 20 June 2017;
Published: 07 July 2017.
Edited by:
Sanjeev Kumar Srivastava, Mitchell Cancer Institute, United StatesReviewed by:
Lihua Li, University of Minnesota, United StatesVijay Kumar Prajapati, Central University of Rajasthan, India
Copyright © 2017 Zhou, Liu and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Cao, Y2FveUBtYWlsLmtpei5hYy5jbg==
†These authors have contributed equally to this work.