 Deepti Nigam
Deepti Nigam Swati Saxena
Swati Saxena G. Ramakrishna
G. Ramakrishna Archana Singh
Archana Singh N. K. Singh
N. K. Singh Kishor Gaikwad
Kishor Gaikwad- 1Indian Council of Agricultural Research-National Research Centre on Plant Biotechnology, New Delhi, India
- 2Division of Biochemistry, Indian Council of Agricultural Research-Indian Agricultural Research Institute, New Delhi, India
Pigeonpea [Cajanus cajan (L.) Millsp.] is a heat and drought resilient legume crop grown mostly in Asia and Africa. Pigeonpea is affected by various biotic (diseases and insect pests) and abiotic stresses (salinity and water logging) which limit the yield potential of this crop. However, resistance to all these constraints is not readily available in the cultivated genotypes and some of the wild relatives have been found to withstand these resistances. Thus, the utilization of crop wild relatives (CWR) in pigeonpea breeding has been effective in conferring resistance, quality and breeding efficiency traits to this crop. Bud and leaf tissue of Cajanus scarabaeoides, a wild relative of pigeon pea were used for transcriptome profiling. Approximately 30 million clean reads filtered from raw reads by removal of adaptors, ambiguous reads and low-quality reads (3.02 gigabase pairs) were generated by Illumina paired-end RNA-seq technology. All of these clean reads were pooled and assembled de novo into 1,17,007 transcripts using the Trinity. Finally, a total of 98,664 unigenes were derived with mean length of 396 bp and N50 values of 1393. The assembly produced significant mapping results (73.68%) in BLASTN searches of the Glycine max CDS sequence database (Ensembl). Further, uniprot database of Viridiplantae was used for unigene annotation; 81,799 of 98,664 (82.90%) unigenes were finally annotated with gene descriptions or conserved protein domains. Further, a total of 23,475 SSRs were identified in 27,321 unigenes. This data will provide useful information for mining of functionally important genes and SSR markers for pigeonpea improvement.
Introduction
To keep pace with the growing human population and dietary requirements, food production must double in the next 25 years (McCouch et al., 2013). The intensifying food demand, climate change, soil degradation, water and land shortages are putting more pressure on the productivity of the current crop-production system (Kastner et al., 2012). In order to achieve gain in food availability, concentrated efforts to intensify food production and making it sustainable are required (Foley et al., 2011). Answers to meet these challenges lies in utilization of novel genetic diversity for production of crop varieties comprising attributes such as heat, cold and drought tolerance, disease and pest resistance (Esquinas-Alcázar, 2005). Domestication and concentration of human population has resulted in severe loss of crop genetic diversity (Olsenand and Gross, 2008). Interestingly, the wild relatives of the cultivated plant species known as Crop wild relatives (CWR) possess much broader spectrum of genetic variations in comparison to the cultivated species (Guarino and Lobell, 2011). The wild relatives have evolved in nature and show adaptation to extreme environmental changes (heat, cold, drought, salinity) and have the capability to withstand damage by insect pest and diseases (Dempewolf et al., 2014). However their utilization is hampered by limited availability of genomic resources.
Cajanus scarabaeoides (L.) Thouars. is most widely distributed wild species of pigeonpea, belongs to the secondary gene pool and is native to tropical and temperate regions of India, Sri Lanka, Australia, Africa, China etc. (Saxena, 2008). It is a creeper climber, the stem and seeds are very nutritive and can be used for feed and food purposes respectively and it is known to exhibit antibiosis as well as mechanical resistance to pod borers (Van der Maesen, 1985). It is highly resistant to Helicoverpa armigera (pod borer) due to presence of short non-glandular trichomes on the pods (Sharma et al., 2009). The flower initiation is known to be very early in C. scarabaeoides as compared to popular pigeonpea cultivars. This trait can be utilized in breeding programs to reduce harvest time to avoid drastic effects of changing climate. The somatic chromosome features of C. scarabaeoides are similar to the cultivated pigeonpea (Hari D Upadhyaya, 2006). It has many desirable attributes and is cross-compatible with cultivated pigeonpea, and interspecific gene transfer is possible through conventional hybridization. The phenomenon of heterosis has been effectively used for genetic improvement in pigeonpea as it shows considerable extent of natural outcrossing. A number of cytoplasmic male sterile (CMS) systems are available in pigeonpea but CMS lines derived from C. scarabaeoides (A2 cytoplasm) has been widely utilized to develop promising commercial hybrids (Choudhary and Singh, 2015). Unfortunately, very narrow genomic information is available for C. scarabaeoides.
Keeping in mind the decisive importance of this species, we performed Illumina paired-end sequencing to generate the bud and leaf transcriptome of C. scarabaeoides. This is the primary report of transcriptome sequencing and de novo assembly of C. scarabaeoides transcriptome. The transcriptome database will provide a valuable resource for gene discovery and marker development linked with superior traits.
Materials and Methods
Plant Material and RNA Isolation
Seeds of C. scarabaeoides (Accession N0- ICPW87) were obtained from the International Crops Research Institute for Semi-Arid Tropics (ICRISAT), Patancheru, India. The seeds were sown in pots containing 1 kg of soil with 34°C temperature at day and 25°C at night with relative humidity approximately 68% and a photoperiod of 16/8 h. The plants were maintained in the experimental green house at NRCPB, New Delhi, India. Buds and leaves of were used as plant material in the present study. Fresh leaves and buds were harvested and immediately frozen in liquid nitrogen and stored in −80°C until further use. Total RNA from the buds and leaves was extracted using Spectrum Plant Total RNA Kit (Sigma) following the manufacturer's protocol. RNA was quality was checked by Agilent 2100 Bioanalyzer RNA Nanochip (Agilent Technologies) and quantity was checked by Nanodrop spectrophotometer (Thermo scientific). Total RNA at concentration of ≥500 ng/ul, OD 260/280 = 1.8~2.2, RNA Integrity Number (RIN) ≥ 7 were used for RNA library preparation.
Library Preparation and Illumina Sequencing
A Truseq RNA Sample Prep Kit (Illumina) was employed in poly-A based mRNA purification and cDNA library construction was carried out according to the manufacturer's instructions. 1 ug of total RNA sample of bud and leaf tissue were used for poly-A mRNA selection using streptavidin-coated magnetic beads. Two rounds of enrichment for poly-A mRNA was carried followed by mRNA fragmentation. cDNA was synthesized from enriched and fragmented RNA using reverse transcriptase (Super Script II). Double stranded Illumina libraries were obtained by 15 cycles of PCR amplification using Illumina PCR primers. The size and purity of the libraries were checked by Agilent 2100 Bioanalyzer. The cDNA libraries of average fragment length 360 bp, approximately were constructed. Finally, both the libraries were sequenced using paired end protocol on the Illumina MiSeq system. The raw sequence data generated in present study have been deposited at NCBI in the Short Read Archive (SRA) database under the accession number SRA548291 (experiment accession numbers SRX2661106 and SRX2661107 for bud and leaf tissue, respectively).
De novo Transcriptome Assembly
The base quality of the raw paired end reads from bud (17,388,560) and leaf (13,146,615) respectively of 100 bp were assessed by FastQC v0.11.2 (Andrews, 2010). Raw sequenced reads were processed using Trimmomatic software version 0.36 (Bolger et al., 2014) for trimming and quality filtering. In this step, raw reads containing adapter sequence, reads with poly-N (≥10%) and low quality (sQ ≤ 5) were removed to obtain high quality de novo transcriptome sequence data. Simultaneously, Q20, Q30, GC-content and sequence duplication level of the reads were calculated and the downstream analyses was performed on the filtered clean data.
The high quality filtered reads for each library were then de novo assembled using Trinity software package version v2.4.0 (Grabherr et al., 2011), all parameters set to default. The assembly validation was performed using Bowtie2 aligner where the filtered reads were mapped back to the assembled transcripts. Further, unigenes were retrieved with the aid of CD-HIT-EST software (http://weizhongli-lab.org/cd-hit/) that clustered the transcripts with identity parameter of 95%. Unigenes were compared with the Glycine max coding sequence as well as uniprot databases of Viridiplantae by using blastx with a typical cut-off E-value of 1e−5 and similarity level > = 90% to search for homologs. Further, to calculate the abundance estimation for each unigene, reads from each sample were then mapped as separately onto the assembled transcripts i.e. unigenes and the read count for each unigene was obtained. The expression quantity of each gene (fragments per kilobase of exon model per million mapped fragments, FPKM) was estimated using RSEM (Li and Dewey, 2011) provided within the Trinity package. Potential simple sequence repeats (SSRs) were detected using MISA (MIcroSAtellite, http://pgrc.ipk-gatersleben.de/misa/) (Thiel et al., 2003). In present study, repeats of one to six nucleotides in the core motif were considered. Mononucleotide repeats ≥ 8 bases, dinucleotides ≥ 10 bases (five repeats) and trinucleotides and tetranucleotides ≥ 12 bases (four and three repeats respectively), pentanucleotide ≥ 15 bases (3 repeats) and hexanucleotides ≥ 18 bases (3 repeats).
Results
Data Quality Assessment
Next generation sequencing technology has been extensively applied to characterize the transcriptome profiles in multiple non-model plants. Equal quantity of total RNA extracted from the bud and leaf tissue of C. scarabaeoides was used to construct cDNA library which was then sequenced on Illumina MiSeq platform. Initially, the base quality of the sequenced reads was evaluated. A total of 30,535,175 raw paired-ends read of ~145 bp were obtained. The raw data has been deposited to NCBI with accession number SRA548291. Clean reads were filtered from raw reads by removal of adaptors, ambiguous reads and low-quality reads. After quality checking and pre-processing approximately 30,531,601 clean reads (7.69 GB) were retained Table 1. The GC content was 45.10% and the Q20 and Q30 were 94.243 and 90.12% respectively.
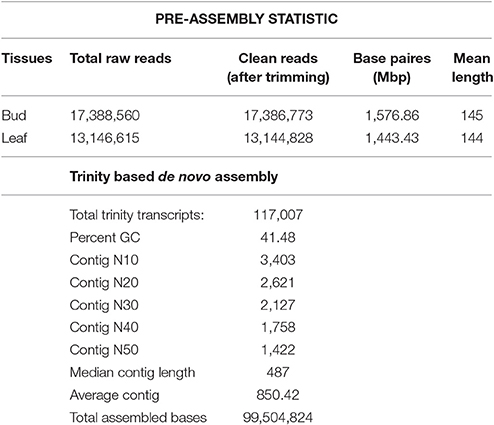
Table 1. Pre and post assembly statistics.
De novo Assembly and Validation
All the clean reads were de novo assembled by Trinity program (Grabherr et al., 2011), which generated a total of 1,17,007 transcripts with an average length of 487 bp and an N50 of 1,422 bp (Table 1). These transcripts then fed to Cluster Database at High Identity with Tolerance-EST i.e., CD-HIT-EST software (Nakasugi et al., 2013) with percent identity 95% to remove redundancy at sequence level producing non-redundant' (nr) representative sequences as output. In this way, number of assembled transcripts got reduced from a total of 1,17,007 to 98,664 transcripts, and defined as unigenes with a mean length of 396 bp and N50 of 1,393. Among the unigenes, the shortest and longest unigenes are 201 and 13,448 bp, respectively. Moreover, 26,876 unigenes were within the 200–400 bp, and 9,444 unigenes were within the 400–1,000 bp. In the meanwhile, we observed that part of unigenes (62,344) were over 1000 bp, which were very helpful to further annotation and functional analysis of unigenes. Assessment of transcriptome assembly was performed with the transcripts (obtained from Trinity assembler). First of all, Bowtie2 was used to align the reads to the constructed transcriptome from Trinity and then we counted the number of proper pairs, improper or orphan read alignments to capture the read alignment statistics. In our case, 93.75% of the mapped fragments were found mapped as proper pairs (yielding concordant alignments 1 or more times to the reconstructed transcriptome) suggesting our proper assembly. The remaining unassembled reads likely correspond to lowly expressed transcripts with unsatisfactory coverage, or are of low quality or aberrant reads. A better de novo assembly was obtained in current sequencing data and the results are shown in Supplementary Material, File 1.
Again at this level, the quality assessment of transcriptome assembly was done through the examination of the full-length or nearly full-length transcripts numbers. Therefore we performed blastx analysis of the assembled transcripts i.e. unigenes against Glycine max coding sequence (CDS) and uniprot database of whole Viridiplantae with the stringent E-value of 1e−5 and percent identity ≥ 90% resulted in 72,703 (73.68%) and 81,799 (82.90%) uniquely mapped unigenes (Table 2; Supplementary Material, Files 2, 3).

Table 2. Unigenes mapping results with Glycine max coding sequence (Ensembl).
Further, a total of 23,475 SSR loci were identified in 27,321 unigene sequences of which 6128 sequences contained more than one SSR (Supplementary Material, File 4). For identification of different types of SSRs, only the perfect repeats were used. Among them, mononucleotide was the most abundant repeat unit i.e. 12613, followed by trinucleotide which was 4402, 3027 dinucleotide, 1,139 tetranucleotide, 1,356 hexanucleotide and 938 pentanucleotide.
Conclusion
This is the first report of a transcriptome database Cajanus scarabaeoides which is a closest wild relative to C. cajan, and one of the easiest wild species that can crossed with pigeonpea cultivars. Next-generation RNA sequencing was performed on mRNAs obtained from bud and leaf tissue. Unigenes and SSR makers identified in present study can be used be used for improvement of cultivated pigeon pea with specific traits, however extended deep researches are required to confirm these findings. Incorporation of different facts at transcriptome, metabolome and proteome approaches would lead to comprehensive research programs focus on engineering C. scarabaeoides.
Author Contributions
KG and NS conceived this study. KG designed the experimental plan. SS and GR participated in sample collection, RNA sequencing. DN analyzed the sequence data. DN and SS wrote the manuscript. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the financial support received from ICAR-National Research Centre on Plant Biotechnology.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmolb.2017.00048/full#supplementary-material
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinforma. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Choudhary, A. K., and Singh, I. P. (2015). A study on comparative fertility restoration in A2 and A4 cytoplasms and its implication in breeding Hybrid Pigeonpea [Cajanus cajan (L.) Millspaugh]. Am. J. Plant Sci. 6, 385. doi: 10.4236/ajps.2015.62044
Dempewolf, H., Eastwood, R. J., Guarino, L., Khoury, C. K., Müller, J. V., and Toll, J. (2014). Adapting agriculture to climate change: a global initiative to collect, conserve, and use crop wild relatives. Agroecol. Sustainable Food Syst. 38, 369–377. doi: 10.1080/21683565.2013.870629
Esquinas-Alcázar, J. (2005). Protecting crop genetic diversity for food security: political, ethical and technical challenges. Nat. Rev. Genet. 6, 946–953. doi: 10.1038/nrg1729
Foley, J. A., Ramankutty, N., Brauman, K. A., Cassidy, E. S., Gerber, J. S., Johnston, M., et al. (2011). Solutions for a cultivated planet. Nature 478, 337–342. doi: 10.1038/nature10452
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guarino, L., and Lobell, D. B. (2011). A walk on the wild side. Nat. Climate Change 1:374. doi: 10.1038/nclimate1272
Kastner, T., Rivas, M. J. I., Koch, W., and Nonhebel, S. (2012). Global changes in diets and the consequences for land requirements for food. Proc. Natl. Acad. Sci. U.S.A. 109, 6868–6872. doi: 10.1073/pnas.1117054109
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
McCouch, S., Baute, G. J., Bradeen, J., Bramel, P., Bretting, P. K., Buckler, E., et al. (2013). Agriculture: feeding the future. Nature 499, 23–24. doi: 10.1038/499023a
Nakasugi, K., Crowhurst, R. N., Bally, J., Wood, C. C., Hellens, R. P., and Waterhouse, P. M. (2013). De novo transcriptome sequence assembly and analysis of RNA silencing genes of Nicotiana benthamiana. PLoS ONE 8:e59534. doi: 10.1371/journal.pone.0059534
Olsenand, K. M., and Gross, B. L. (2008). Detecting multiple origins of domesticated crops. Proc. Natl. Acad. Sci. U.S.A. 105, 13701–13702. doi: 10.1073/pnas.0807439105
Saxena, K. B. (2008). Genetic improvement of pigeon pea-a review. Trop. Plant Biol. 1, 159–178. doi: 10.1007/s12042-008-9014-1
Sharma, H. C., Sujana, G., and Manohar Rao, D. (2009). Morphological and chemical components of resistance to pod borer, Helicoverpa armigera in wild relatives of pigeonpea. Arthropod Plant Interact. 3, 151–161. doi: 10.1007/s11829-009-9068-5
Keywords: Cajanus scarabaeoides (L.) Thouars, cultivated wild relatives (CWR), de novo assembly, illumina sequencing, SSR's
Citation: Nigam D, Saxena S, Ramakrishna G, Singh A, Singh NK and Gaikwad K (2017) De novo Assembly and Characterization of Cajanus scarabaeoides (L.) Thouars Transcriptome by Paired-End Sequencing. Front. Mol. Biosci. 4:48. doi: 10.3389/fmolb.2017.00048
Received: 29 March 2017; Accepted: 26 June 2017;
Published: 12 July 2017.
Edited by:
Manja Marz, University of Jena, GermanyReviewed by:
Thiruvarangan Ramaraj, National Center for Genome Resources, United StatesAureliano Bombarely, Virginia Tech, United States
Alejandro Sanchez-Flores, National Autonomous University of Mexico, Mexico
Copyright © 2017 Nigam, Saxena, Ramakrishna, Singh, Singh and Gaikwad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kishor Gaikwad, a2lzaDIwMTJAbnJjcGIub3Jn
†These authors have contributed equally to this work.