 María Álvarez-Satta1,2,3
María Álvarez-Satta1,2,3 Diana Valverde
Diana Valverde- 1Grupo de Biomarcadores Moleculares, Departamento de Bioquímica, Genética e Inmunología, Facultad de Biología, Universidad de Vigo, Vigo, Spain
- 2Grupo de Investigación en Enfermedades Raras y Medicina Pediátrica, Instituto de Investigación Sanitaria Galicia Sur (IIS Galicia Sur), SERGAS-UVIGO, Vigo, Spain
- 3Centro de Investigaciones Biomédicas (Centro Singular de Investigación de Galicia 2016–2019), Universidad de Vigo, Vigo, Spain
Bardet-Biedl syndrome (BBS) is a rare genetic disorder that belongs to the group of ciliopathies, defined as diseases caused by defects in cilia structure and/or function. The six diagnostic features considered for this syndrome include retinal dystrophy, obesity, polydactyly, cognitive impairment and renal and urogenital anomalies. Furthermore, three of the 21 genes currently known to be involved in BBS encode chaperonin-like proteins (MKKS/BBS6, BBS10, and BBS12), so BBS can be also considered a member of the growing group of chaperonopathies. Remarkably, up to 50% of clinically-diagnosed BBS families can harbor disease-causing variants in these three genes, which highlights the importance of chaperone defects as pathogenic factors even for genetically heterogeneous syndromes such as BBS. In addition, it is interesting to note that BBS families with deleterious variants in MKKS/BBS6, BBS10 or BBS12 genes generally display more severe phenotypes than families with changes in other BBS genes. The chaperonin-like BBS proteins have structural homology to the CCT family of group II chaperonins, although they are believed to conserve neither the ATP-dependent folding activity of canonical CCT chaperonins nor the ability to form CCT-like oligomeric complexes. Thus, they play an important role in the initial steps of assembly of the BBSome, which is a multiprotein complex essential for mediating the ciliary trafficking activity. In this review, we present a comprehensive review of those genetic, functional and evolutionary aspects concerning chaperonin-like BBS proteins, trying to provide a new perspective that expands the classical conception of BBS only from a ciliary point of view.
Bardet-Biedl Syndrome in Context
The Bardet-Biedl syndrome (BBS; MIM#209900) is a multisystem, rare genetic disorder belonging to the group of ciliopathies, which encompasses several diseases that are caused by defects in cilia structure and/or function, especially affecting the primary cilium (reviewed in Hildebrandt et al., 2011; Mitchison and Valente, 2017). This highly conserved and dynamic organelle is considered the sensorial antennae of the cell and also a central processing unit, since it captures and integrates all the extracellular signals with the cell cycle and metabolism (reviewed in Malicki and Johnson, 2017). Thus, primary cilia play a key role in coordinating the different cellular signaling pathways (reviewed in Cardenas-Rodriguez and Badano, 2009; Christensen et al., 2017), giving rise to biological responses related to the control of cell cycle, development and differentiation processes, migration and polarity, stimuli transduction or proliferation and maintenance of stem cells. Ciliopathies represent an expanding group of human inherited disorders that are valuable models to study several common conditions such as obesity, retinal dystrophy or renal cysts, considering their pleiotropic nature. Remarkably, more than 50 genes have been involved in ciliopathies (Mitchison and Valente, 2017), a number that continues to grow due to the new discoveries on ciliary proteome and ciliogenesis regulation (Mick et al., 2015; Wheway et al., 2015; Boldt et al., 2016), as well as the increasingly implementation of high-throughput sequencing technologies to ciliary disorders. Furthermore, it is important to highlight that ciliopathies are complex clinical entities with extensive genetic heterogeneity and also high phenotypic and genetic overlap among them. This, together with the progressive development of nearly all clinical features related to them, usually makes an early and specific diagnosis very difficult to establish.
BBS is considered a model disease to study the biology of the primary cilium, and is characterized by progressive retinal dystrophy, obesity, postaxial polydactyly, cognitive impairment and renal and urogenital anomalies as primary diagnostic features (reviewed in Forsythe and Beales, 2013). Furthermore, BBS is a genetically heterogeneous disorder with up to 21 genes (commonly known as BBS genes) described to date (Bujakowska et al., 2015; Heon et al., 2016; Khan et al., 2016 and references within). Intriguingly, although BBS is primarily inherited as an autosomal recessive disorder, a more complex model of oligogenic inheritance considering modifier loci and epistatic effects has been proposed for some families, trying to explain the high clinical variability reported for BBS patients (Katsanis, 2004; Badano et al., 2006). Regarding the functions of BBS proteins (reviewed in Novas et al., 2015), eight of them form a multimeric complex called BBSome, which plays a key role in mediating molecular/vesicular transport in and out of the primary cilium, and also in intraciliary trafficking as part of the intraflagellar transport machinery (Nachury et al., 2007; Loktev et al., 2008; Wei et al., 2012). Moreover, most of the remaining BBS proteins have functions related to BBSome assembly, cilia targeting of BBSome and proper recognition of BBSome cargoes, besides several extra-ciliary roles (Novas et al., 2015).
Bardet-Biedl Syndrome as a Chaperonopathy
Three of the main BBS genes, MKKS/BBS6 (MIM*604896), BBS10 (MIM*610148) and BBS12 (MIM*610683), encode chaperonin-like proteins that localize to centrosomes and ciliary basal bodies (Kim et al., 2005; Marion et al., 2009). This implies that BBS would also be part of the emerging group of diseases called chaperonopathies, which are produced by defects in molecular chaperones or any other protein resembling their structure. In this regard, it is noteworthy that chaperonin-like BBS proteins, as will be explained later, are unlikely to display a folding activity but they have functions specifically related to the assembly of the BBSome.
Chaperonopathies represent an interesting subset of disorders that have so far received little attention, although they can provide useful models to better understand some of the molecular mechanisms necessary to maintain protein homeostasis (extensively reviewed in Macario and Conway de Macario, 2005, 2007a,b; Macario et al., 2005). Chaperonopathies often manifest themselves as complex phenotypes affecting multiple organs, possibly due to the ubiquitous localization of most chaperones, and may be of genetic or acquired origin. In this latter case, defects in chaperone post-translational modifications, distribution or quantity, together with other phenomena such as generation of antichaperone autoantibodies or aggregation of chaperones with deposits of abnormal proteins, all of them usually related to aging, could be the trigger rather than mutational events. Importantly, research on chaperones and their role in disease is opening a new field of therapeutic options (termed “chaperonotherapy”) with interesting applications not only in chaperonopathies, but also in some processes such as cancer whereby chaperones may modulate the immune response against tumors (reviewed in Binder, 2008; Graner et al., 2015).
Contribution of MKKS/BBS6, BBS10 and BBS12 Genes to Bardet-Biedl Syndrome
Among ciliopathies, BBS represents a special case since as far as we know no other ciliopathy except the related McKusick-Kaufman syndrome (MKKS; MIM#236700) is caused by genetic defects in chaperone genes. At this point, it is appropriate to mention that MKKS is a monogenic ciliopathy caused by mutations in the MKKS gene leading to postaxial polydactyly, genital malformations (typically hydrometrocolpos in females) and also congenital heart disease (Schaefer et al., 2011). Furthermore, BBS is also a particular member of the chaperonopathies with regard to the very specific functions carried out by chaperonin-like BBS proteins within a ciliary context (explained in detail in the next section). In this sense, BBS constitutes a clear example of the great importance of chaperone defects as determinant pathogenic factors, taking into account that up to 50% of families clinically diagnosed with BBS can harbor pathogenic variants in MKKS/BBS6, BBS10 and BBS12 genes (Billingsley et al., 2010; Muller et al., 2010; Deveault et al., 2011). This data is even more relevant considering the high genetic heterogeneity of BBS with 21 genes currently identified, a number that is expected to grow as 20–30% of patients suspected to suffer BBS do not yet have molecular confirmation of their clinical diagnosis (Mitchison and Valente, 2017).
Chaperonin-like BBS genes are characterized by a relatively simple gene structure (Figure 1), with a low number of coding exons (one in BBS12, two in the case of BBS10 and four exons in MKKS/BBS6), which make them ideal candidates for a mutational screening previously to perform more complex and expensive analyses. Furthermore, a broad distribution of pathogenic variants throughout the coding sequence of chaperonin-like BBS genes has been reported. A brief summary of the most relevant genetic findings concerning each gene is presented below (see also Table 1).
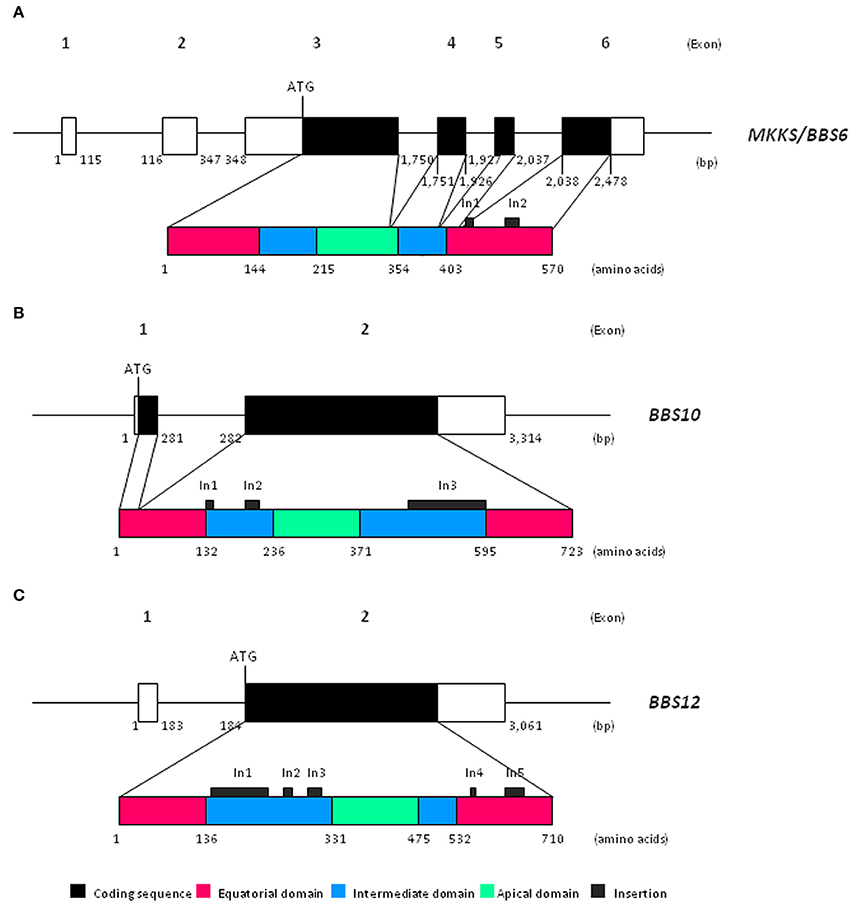
Figure 1. Schematic view of both gene and protein structure of chaperonin-like BBS proteins. (A) Representation of MKKS/BBS6 (reference transcript ENST00000347364.7); (B) Representation of BBS10 (reference transcript ENST00000393262.3); (C) Representation of BBS12 (reference transcript ENST00000314218.7). bp, base pairs.
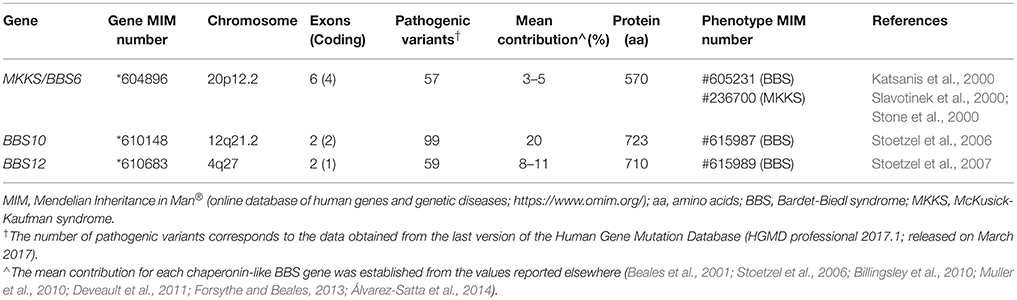
Table 1. Summary of the main features related to chaperonin-like BBS genes.
MKKS/BBS6 (chromosome 20p12.2) was the first gene coding a putative chaperonin to be associated with a human inherited disorder, the MKKS (Stone et al., 2000), being also involved in BBS shortly after (Katsanis et al., 2000; Slavotinek et al., 2000). To date, more than 50 deleterious variants have been described, predominantly missense and nonsense changes (Human Gene Mutation Database; Stenson et al., 2017). Regarding its contribution to the total load of BBS, MKKS/BBS6 is a minor contributor with 3–5% of families harboring two disease-causing variants in the multiethnic cohorts reported worldwide (Beales et al., 2001; Muller et al., 2010; Deveault et al., 2011). Interestingly, the vast majority of causal variants described in this gene have been identified in BBS patients, so it has been proposed that both syndromes MKKS and BBS are different allelic forms of the same clinical entity (Katsanis et al., 2000; Schaefer et al., 2011). Thus, MKKS phenotypes would be linked to very rare, possibly hypomorphic alleles found in MKKS/BBS6 gene.
The BBS10 gene (chromosome 12q21.2) was first identified by Stoetzel et al. (2006) in a consanguineous pedigree of Lebanese origin. It is, together with BBS1, the major contributor to BBS accounting for 20% of all cases (Stoetzel et al., 2006; Forsythe and Beales, 2013), with remarkable exceptions in ethnically homogeneous groups such as Danish (43%; Hjortshøj et al., 2010) or Spanish BBS cohorts (8.3%; Álvarez-Satta et al., 2014). About 100 different disease-causing changes have been reported elsewhere (Human Gene Mutation Database; Stenson et al., 2017), of which the p.Cys91Leufs*5 allele represents a recurrent deleterious variant in BBS cohorts of European descent, reaching 26–48% of BBS10 mutational load (Stoetzel et al., 2006; Billingsley et al., 2010; Muller et al., 2010).
Moreover, the BBS12 gene (chromosome 4q27) was linked to BBS phenotypes a decade ago (Stoetzel et al., 2007). Its contribution to BBS has grown in importance over recent years, accounting for 8–11% of the total cases in most of the cohorts reported (Billingsley et al., 2010; Muller et al., 2010; Deveault et al., 2011; Álvarez-Satta et al., 2014). About 60 pathogenic variants have been currently identified in BBS12 patients (Human Gene Mutation Database; Stenson et al., 2017), among which the nonsense change p.(Phe372*) could represent up to 20% of the mutated alleles found in this gene (Stoetzel et al., 2007).
Finally, it is also important to highlight some trends regarding the BBS phenotypes linked to changes in chaperonin-like BBS genes. Thus, there is a general consensus that BBS patients with pathogenic variants in MKKS/BBS6, BBS10 and BBS12 genes develop a more severe phenotype than those with changes affecting BBSome components such as BBS1 (Billingsley et al., 2010; Imhoff et al., 2011; Castro-Sánchez et al., 2015). In detail, they show an earlier disease onset (especially noted in BBS10 patients), greater prevalence of all BBS primary diagnostic features and also a higher frequency of overlapping features with other ciliopathies, mainly MKKS and also Alström syndrome (MIM#203800), which is a closely related ciliopathy produced by mutations in the ALMS1 gene and characterized by retinal dystrophy, sensorineural hearing loss, early-onset obesity with severe type 2 diabetes mellitus and metabolic syndrome, dilated cardiomyopathy and renal, hepatic and pulmonary injury with widespread fibrosis (reviewed in Marshall et al., 2011). One could hypothesize that differences in the severity of clinical presentation could be due to the distinct functional roles of chaperonin-like BBS genes when compared with the BBS proteins taking part of the BBSome. Thus, deleterious variants in some components of the BBSome might lead to the accumulation of intermediate complexes that maintain a residual or gain-of-function activity as compensating mechanism (Zhang et al., 2012), whereas the chaperonin-like BBS proteins are essential for the initial step of BBSome assembly (see below) so no functional complexes are formed if this subset of proteins is affected (Seo et al., 2010).
Structure and Function of Chaperonin-Like BBS Proteins: Comparison with Canonical CCT Chaperonins
The three chaperonin-like BBS proteins define a particular branch of proteins that have sequence homology to the chaperonin containing t-complex protein 1, CCT (also known as TRiC) family of group II chaperonins (Kim et al., 2005; Stoetzel et al., 2006, 2007). CCT proteins are the eukaryotic cytosolic chaperonins of type II and play key roles in the folding of a wide range of newly translated proteins in an ATP-dependent manner, mainly soluble proteins related to cytoskeleton (actin and tubulin are the quantitative major substrates) (reviewed in Dunn et al., 2001; Spiess et al., 2004). Typically, they form a functional hetero-oligomeric complex of 16 subunits that consists of two stacked rings, each composed of eight CCT monomers radially arranged (CCT1-8). With regard to their specific roles in cilia, CCT subunits are required for ciliary assembly and maintenance of cilia tip integrity, as well as cytoskeleton structure, in the ciliate Tetrahymena (Seixas et al., 2010). In addition, it has been recently reported that CCT chaperonins are essential for the biogenesis of vertebrate photoreceptors' outer segment by mediating the BBSome assembly (Sinha et al., 2014).
Recent phylogenetic analyses have revealed that chaperonin-like BBS proteins represent a highly diverged, monophyletic group derived from a duplication event in the CCT8 gene (Mukherjee et al., 2010). Remarkably, although MKKS/BBS6, BBS10, and BBS12 genes were originally considered as vertebrate-specific, the finding of several orthologs in ancient eukaryotes clearly points to an earlier evolution (Mukherjee and Brocchieri, 2013). The high rate of divergence observed for chaperonin-like BBS proteins compared with those canonical CCT chaperonins is not reflected by their primary structure, which is mostly conserved. Thus, the typical chaperonin domain architecture consisting of apical, intermediate and equatorial domains is conserved in chaperonin-like BBS proteins (Figure 1); however, they have additional specific insertions (two in MKKS/BBS6, three for BBS10 and up to five in the BBS12 sequence) that are restricted to intermediate and equatorial domains (Kim et al., 2005; Stoetzel et al., 2006, 2007). Interestingly, the three insertions located in BBS10, as well as the insertions 1 and 3 of BBS12, protrude from the same face of the intermediate domain, which suggests they constitute an additional domain maybe with specific roles (Stoetzel et al., 2006, 2007). Furthermore, BBS12 seems to be the most divergent member since more differences in several secondary-structure motifs and also in the ATP-hydrolysis motif have been identified (Stoetzel et al., 2007; Mukherjee et al., 2010).
Despite structural similarities, solid evidences point out that chaperonin-like BBS proteins neither perform folding activity nor are able to form chaperonin oligomeric complexes like canonical CCT proteins do. Thus, the ATP hydrolysis motif in the equatorial domain (highly conserved in Group I and II chaperonins) is significantly different in MKKS/BBS6 and, above all, in BBS12 protein (Kim et al., 2005; Stoetzel et al., 2007), which suggests that the catalytic activity required for protein folding is missing; conversely, it would be conserved in BBS10 (Stoetzel et al., 2006). In addition, the existence of specific insertions in chaperonin-like BBS proteins covering potential monomer-monomer contact regions makes it unlikely that they can assemble in a functional CCT-like complex (Kim et al., 2005; Stoetzel et al., 2006, 2007; Mukherjee et al., 2010).
All these data suggest that the roles of chaperonin-like BBS proteins may differ from direct protein folding. Thus, recent work has demonstrated that MKKS/BBS6, BBS10 and BBS12 play a key role in the initial steps of BBSome assembly by stabilizing BBS7 (the first component to be incorporated) and mediating its interaction with six canonical CCT chaperonins (CCT1-5 and CCT8), which would actually accomplish the folding activity (Seo et al., 2010). This means that chaperonin-like BBS proteins act as an intermediate for the binding of CCT complex to its substrates, as part of the transient BBS/CCT/TRiC-chaperonin complex. Remarkably, BBS10 is not a structural member of this complex, but it regulates the interaction of the BBS6-BBS12-BBS7 intermediate with CCT proteins to form the BBS/CCT/TRiC-chaperonin complex (Zhang et al., 2012). It is also important to note that the second step in BBSome assembly, that is, the interaction between BBS2 and the stabilized BBS7 protein, is coupled with the release of BBS6-BBS12 from the complex, and that CCT/TRiC proteins are also released after the BBSome core complex (BBS2-BBS7-BBS9) is formed (Zhang et al., 2012). Accordingly, the BBS/CCT/TRiC-chaperonin complex would assist BBSome assembly only in the first steps, so the formation of mature BBSome complexes is finally completed by intrinsic protein-protein interactions among the BBSome components, which are known to contain β-propeller, tetratricopeptide repeats and pleckstrin homology domains that typically mediate these interactions (Zhang et al., 2012). Despite the significant progress made in deciphering the specific roles of chaperonin-like BBS proteins, details on how the BBS/CCT/TRiC-chaperonin complex is formed and completes the transition of BBS7 to BBSome remain to be elucidated.
Role of Other BBS Proteins in Protein Homeostasis
The cellular network for protein-quality control necessary to maintain protein homeostasis includes besides the chaperone machinery, which ensures proper protein folding and recognition of misfolded proteins (reviewed in Hartl et al., 2011), also two proteolytic machineries, the ubiquitin-proteasome system and the autophagy pathway, which play essential roles in removing irreversibly misfolded proteins (reviewed in Chen et al., 2011). In this regard, it is appropriate to remark on some findings involving several BBS proteins and their possible role in this field.
Several BBSome components such as BBS1-2, BBS4, and BBS7, as well as the BBS6 chaperonin-like protein, interact with proteasomal subunits and could be involved in the regulation of signaling pathways coordinated by the primary cilium (reviewed in Novas et al., 2015). It has been also speculated that TRIM32/BBS11 (MIM*602290) would be the putative E3-ubiquitin ligase that targets free BBS2 to be degraded by the ubiquitin-proteasome pathway (Zhang et al., 2012). Finally, there is also evidence that the unfolded protein response (UPR) of the endoplasmic reticulum can be a pathogenic mechanism related to BBS, as the UPR is triggered by protein accumulation in the photoreceptors of Bbs12-deficient models leading to apoptosis and subsequent retinal degeneration (Mockel et al., 2012). Interestingly, the light detection ability was restored by pharmacological modulation of the UPR, which highlights both the importance of identifying disease mechanisms that involve proteostasis network components and their potential to develop new therapeutic strategies.
Perspectives
BBS is considered a model ciliopathy to study molecular mechanisms potentially involved in common disorders such as obesity, and also represents a singular component of the group of chaperonopathies. Unlike many of these clinical entities, the molecular basis underlying BBS is fairly well-known, just like the particular role of most BBS proteins in the primary cilium and also the cellular basis of several BBS phenotypes (reviewed in Novas et al., 2015). However, many mechanistic aspects remain to be uncovered, especially those concerning the particular molecular processes involved in initialization of BBsome assembly and also the role of protein degradation systems in BBS proteins turnover. In this sense, the use of prokaryotic chaperonins as models to investigate the impact of deleterious variants in chaperonin structure and function, as well as potential therapeutic strategies (Conway de Macario et al., 2017), could represent a promising tool not explored until now to further characterize chaperonin-like BBS proteins.
Moreover, a deeper understanding of the molecular mechanisms involving chaperonin-like BBS proteins could provide more opportunities to explore new therapies for BBS patients, currently unavailable. Thus, some BBSome components are found in monomeric form or aggregated with unidentified proteins in Bbs6 null mice (Seo et al., 2010), which might suggest potential therapeutic targets related to the modulation of chaperone activity. In addition, identifying the specific chaperones and partners involved in the folding of BBSome components, not yet defined, could have a great impact on the development of new strategies in this field.
Author Contributions
MA conceptualized the study, and drafted, reviewed and edited the manuscript. SC drafted and reviewed the manuscript. DV conceived of the study, critically revised the manuscript, acquired funds and supervised the work. All authors read and approved the final manuscript.
Funding
This work was supported by grants from Fondo de Investigación Sanitaria del Instituto de Salud Carlos III-FEDER (PI12/01853 and PI15/00049). MA (FPU12/01442) and SC (FPU13/01835) received graduate studentship awards (FPU fellowship) from the Spanish Ministry of Education, Culture and Sports.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Registro Español de los Síndromes de Wolfram, Bardet-Biedl y Alström (REWBA), the European Union Rare Diseases Registry for Wolfram syndrome, Alström syndrome, Bardet–Biedl syndrome and other rare diabetes syndromes (EURO-WABB), the Asociación Nacional de Ciliopatías (ANASBABI) and also to BIOCAPS Project (from European Commission under the 7th Framework Programme, FP-7-REGPOT 2012-2013-1, grant agreement no. FP7- 316265).
References
Álvarez-Satta, M., Castro-Sánchez, S., Pereiro, I., Piñeiro-Gallego, T., Baiget, M., Ayuso, C., et al. (2014). Overview of Bardet-Biedl syndrome in Spain: identification of novel mutations in BBS1, BBS10 and BBS12 genes. Clin. Genet. 86, 601–602. doi: 10.1111/cge.12334
Badano, J. L., Leitch, C. C., Ansley, S. J., May-Simera, H., Lawson, S., Lewis, R. A., et al. (2006). Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 439, 326–330. doi: 10.10938/nature04370
Beales, P. L., Katsanis, N., Lewis, R. A., Ansley, S. J., Elcioglu, N., Raza, J., et al. (2001). Genetic and mutational analyses of a large multiethnic Bardet-Biedl cohort reveal a minor involvement of BBS6 and delineate the critical intervals of other loci. Am. J. Hum. Genet. 68, 606–616. doi: 10.1086/318794
Billingsley, G., Bin, J., Fieggen, K. J., Duncan, J. L., Gerth, C., Ogata, K., et al. (2010). Mutations in chaperonin-like BBS genes are a major contributor to disease development in a multiethnic Bardet-Biedl syndrome patient population. J. Med. Genet. 47, 453–463. doi: 10.1136/jmg.2009.073205
Binder, R. J. (2008). Heat-shock protein-based vaccines for cancer and infectious disease. Expert. Rev. Vaccines 7, 383–393. doi: 10.1586/14760584.7.3.383
Boldt, K., van Reeuwijk, J., Lu, Q., Koutroumpas, K., Nguyen, T. M., Texier, Y., et al. (2016). An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 7:11491. doi: 10.1038/ncomms11491
Bujakowska, K. M., Zhang, Q., Siemiatkowska, A. M., Liu, Q., Place, E., Falk, M. J., et al. (2015). Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum. Mol. Genet. 24, 230–242. doi: 10.1093/hmg/ddu441
Cardenas-Rodriguez, M., and Badano, J. L. (2009). Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am. J. Med. Genet. C Semin. Med. Genet. 151C, 263–280. doi: 10.1002/ajmg.c.30227
Castro-Sánchez, S., Álvarez-Satta, M., Cortón, M., Guillén, E., Ayuso, C., and Valverde, D. (2015). Exploring genotype-phenotype relationships in Bardet-Biedl syndrome families. J. Med. Genet. 52, 503–513. doi: 10.1136/jmedgenet-2015-103099
Chen, B., Retzlaff, M., Roos, T., and Frydman, J. (2011). Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 3:a004374. doi: 10.1101/cshperspect.a004374
Christensen, S. T., Morthorst, S. K., Mogensen, J. B., and Pedersen, L. B. (2017). Primary Cilia and Coordination of Receptor Tyrosine Kinase (RTK) and Transforming Growth Factor β (TGF-β) Signaling. Cold Spring Harb. Perspect. Biol. 9:a028167. doi: 10.1101/cshperspect.a028167
Conway de Macario, E., Robb, F. T., and Macario, A. J. (2017). Prokaryotic chaperonins as experimental models for elucidating structure-function abnormalities of human pathogenic mutant counterparts. Front. Mol. Biosci. 3:84. doi: 10.3389/fmolb.2016.00084
Deveault, C., Billingsley, G., Duncan, J. L., Bin, J., Theal, R., Vincent, A., et al. (2011). BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum. Mutat. 32, 610–619. doi: 10.1002/humu.21480
Dunn, A. Y., Melville, M. W., and Frydman, J. (2001). Review: cellular substrates of the eukaryotic chaperonin TRiC/CCT. J. Struct. Biol. 135, 176–184. doi: 10.1006/jsbi.2001.4380
Forsythe, E., and Beales, P. L. (2013). Bardet-Biedl syndrome. Eur. J. Hum. Genet. 21, 8–13. doi: 10.1038/ejhg.2012.115
Graner, M. W., Lillehei, K. O., and Katsanis, E. (2015). Endoplasmic reticulum chaperones and their roles in the immunogenicity of cancer vaccines. Front. Oncol. 4:379. doi: 10.3389/fonc.2014.00379
Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. doi: 10.1038/nature10317
Heon, E., Kim, G., Qin, S., Garrison, J. E., Tavares, E., Vincent, A., et al. (2016). Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum. Mol. Genet. 25, 2283–2294. doi: 10.1093/hmg/ddw096
Hildebrandt, F., Benzing, T., and Katsanis, N. (2011). Ciliopathies. N. Engl. J. Med. 364, 1533–1543. doi: 10.1056/NEJMra1010172
Hjortshøj, T. D., Grønskov, K., Philp, A. R., Nishimura, D. Y., Riise, R., Sheffield, V. C., et al. (2010). Bardet-Biedl syndrome in Denmark-report of 13 novel sequence variations in six genes. Hum. Mutat. 31, 429–436. doi: 10.1002/humu.21204
Imhoff, O., Marion, V., Stoetzel, C., Durand, M., Holder, M., Sigaudy, S., et al. (2011). Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin. J. Am. Soc. Nephrol. 6, 22–29. doi: 10.2215/CJN.03320410
Katsanis, N. (2004). The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13, R65–R71. doi: 10.1093/hmg/ddh092
Katsanis, N., Beales, P. L., Woods, M. O., Lewis, R. A., Green, J. S., Parfrey, P. S., et al. (2000). Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat. Genet. 26, 67–70. doi: 10.1038/79201
Khan, S. A., Muhammad, N., Khan, M. A., Kamal, A., Rehman, Z. U., and Khan, S. (2016). Genetics of human Bardet-Biedl syndrome, an updates. Clin. Genet. 90, 3–15. doi: 10.1111/cge.12737
Kim, J. C., Ou, Y. Y., Badano, J. L., Esmail, M. A., Leitch, C. C., Fiedrich, E., et al. (2005). MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet-Biedl syndrome, is a novel centrosomal component required for cytokinesis. J. Cell Sci. 118, 1007–1020. doi: 10.1242/jcs.01676
Loktev, A. V., Zhang, Q., Beck, J. S., Searby, C. C., Scheetz, T. E., Bazan, J. F., et al. (2008). A BBSome subunit links ciliogenesis, microtubule stability, and acetylation. Dev. Cell 15, 854–865. doi: 10.1016/j.devcel.2008.11.001
Macario, A. J., and Conway de Macario, E. (2005). Sick chaperones, cellular stress, and disease. N. Engl. J. Med. 353, 1489–1501. doi: 10.1056/NEJMra050111
Macario, A. J., and Conway de Macario, E. (2007a). Chaperonopathies by defect, excess, or mistake. Ann. N.Y. Acad. Sci. 1113, 178–191. doi: 10.1196/annals.1391.009
Macario, A. J., and Conway de Macario, E. (2007b). Chaperonopathies and chaperonotherapy. FEBS Lett. 581, 3681–3688. doi: 10.1016/j.febslet.2007.04.030
Macario, A. J., Grippo, T. M., and Conway de Macario, E. (2005). Genetic disorders involving molecular-chaperone genes: a perspective. Genet. Med. 7, 3–12.
Malicki, J. J., and Johnson, C. A. (2017). The cilium: cellular antenna and central processing unit. Trends Cell Biol. 27, 126–140. doi: 10.1016/j.tcb.2016.08.002
Marion, V., Stoetzel, C., Schlicht, D., Messaddeq, N., Koch, M., Flori, E., et al. (2009). Transient ciliogenesis involving Bardet-Biedl syndrome proteins is a fundamental characteristic of adipogenic differentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 1820–1825. doi: 10.1073/pnas.0812518106
Marshall, J. D., Maffei, P., Collin, G. B., and Naggert, J. K. (2011). Alström syndrome: genetics and clinical overview. Curr. Genomics 12, 225–235. doi: 10.2174/138920211795677912
Mick, D. U., Rodrigues, R. B., Leib, R. D., Adams, C. M., Chien, A. S., Gygi, S. P., et al. (2015). Proteomics of primary cilia by proximity labeling. Dev. Cell. 35, 497–512. doi: 10.1016/j.devcel.2015.10.015
Mitchison, H. M., and Valente, E. M. (2017). Motile and non-motile cilia in human pathology: from function to phenotypes. J. Pathol. 241, 294–309. doi: 10.1002/path.4843
Mockel, A., Obringer, C., Hakvoort, T. B., Seeliger, M., Lamers, W. H., Stoetzel, C., et al. (2012). Pharmacological modulation of the retinal unfolded protein response in Bardet-Biedl syndrome reduces apoptosis and preserves light detection ability. J. Biol. Chem. 287, 37483–37494. doi: 10.1074/jbc.M112.386821
Mukherjee, K., and Brocchieri, L. (2013). Ancient origin of chaperonin gene paralogs involved in ciliopathies. J. Phylogenetics Evol. Biol. 1:107. doi: 10.4172/2329-9002.1000107
Mukherjee, K., Conway de Macario, E., Macario, A. J., and Brocchieri, L. (2010). Chaperonin genes on the rise: new divergent classes and intense duplication in human and other vertebrate genomes. BMC Evol. Biol. 10:64. doi: 10.1186/1471-2148-10-64
Muller, J., Stoetzel, C., Vincent, M. C., Leitch, C. C., Laurier, V., Danse, J. M., et al. (2010). Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum. Genet. 127, 583–593. doi: 10.1007/s00439-010-0804-9
Nachury, M. V., Loktev, A. V., Zhang, Q., Westlake, C. J., Peränen, J., Merdes, A., et al. (2007). A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129, 1201–1213. doi: 10.1016/j.cell.2007.03.053
Novas, R., Cardenas-Rodriguez, M., Irigoín, F., and Badano, J. L. (2015). Bardet-Biedl syndrome: is it only cilia dysfunction? FEBS Lett. 589, 3479–3491. doi: 10.1016/j.febslet.2015.07.031
Schaefer, E., Durand, M., Stoetzel, C., Doray, B., Viville, B., Hellé, S., et al. (2011). Molecular diagnosis reveals genetic heterogeneity for the overlapping MKKS and BBS phenotypes. Eur. J. Med. Genet. 54, 157–160. doi: 10.1016/j.ejmg.2010.10.004
Seixas, C., Cruto, T., Tavares, A., Gaertig, J., and Soares, H. (2010). CCTalpha and CCTdelta chaperonin subunits are essential and required for cilia assembly and maintenance in Tetrahymena. PLoS ONE 5:e10704. doi: 10.1371/journal.pone.0010704
Seo, S., Baye, L. M., Schulz, N. P., Beck, J. S., Zhang, Q., Slusarski, D. C., et al. (2010). BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc. Natl. Acad. Sci. U.S.A. 107, 1488–1493. doi: 10.1073/pnas.0910268107
Sinha, S., Belcastro, M., Datta, P., Seo, S., and Sokolov, M. (2014). Essential role of the chaperonin CCT in rod outer segment biogenesis. Invest. Ophthalmol. Vis. Sci. 55, 3775–3785. doi: 10.1167/iovs.14-13889
Slavotinek, A. M., Stone, E. M., Mykytyn, K., Heckenlively, J. R., Green, J. S., Heon, E., et al. (2000). Mutations in MKKS cause Bardet-Biedl syndrome. Nat. Genet. 26, 15–16. doi: 10.1038/79116
Spiess, C., Meyer, A. S., Reissmann, S., and Frydman, J. (2004). Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 14, 598–604. doi: 10.1016/j.tcb.2004.09.015
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. doi: 10.1007/s00439-017-1779-6
Stoetzel, C., Laurier, V., Davis, E. E., Muller, J., Rix, S., Badano, J. L., et al. (2006). BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat. Genet. 38, 521–524. doi: 10.1038/ng1771
Stoetzel, C., Muller, J., Laurier, V., Davis, E. E., Zaghloul, N. A., Vicaire, S., et al. (2007). Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am. J. Hum. Genet. 80, 1–11. doi: 10.1086/510256
Stone, D. L., Slavotinek, A., Bouffard, G. G., Banerjee-Basu, S., Baxevanis, A. D., Barr, M., et al. (2000). Mutation of a gene encoding a putative chaperonin causes McKusick-Kaufman syndrome. Nat. Genet. 25, 79–82. doi: 10.1038/75637
Wei, Q., Zhang, Y., Li, Y., Zhang, Q., Ling, K., and Hu, J. (2012). The BBSome controls IFT assembly and turnaround in cilia. Nat. Cell. Biol. 14, 950–957. doi: 10.1038/ncb2560
Wheway, G., Schmidts, M., Mans, D. A., Szymanska, K., Nguyen, T. M., Racher, H., et al. (2015). An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell. Biol. 17, 1074–1087. doi: 10.1038/ncb3201
Keywords: ciliopathies, chaperonopathies, Bardet-Biedl syndrome, chaperonin-like BBS proteins, MKKS/BBS6, BBS10, BBS12
Citation: Álvarez-Satta M, Castro-Sánchez S and Valverde D (2017) Bardet-Biedl Syndrome as a Chaperonopathy: Dissecting the Major Role of Chaperonin-Like BBS Proteins (BBS6-BBS10-BBS12). Front. Mol. Biosci. 4:55. doi: 10.3389/fmolb.2017.00055
Received: 25 May 2017; Accepted: 13 July 2017;
Published: 31 July 2017.
Edited by:
Alberto J. L. Macario, University of Maryland at Baltimore and Institute of Marine and Environmental Technology, United States; Istituto Euro-Mediterraneo di Scienza e Tecnologia, ItalyReviewed by:
Luciano Brocchieri, TB-SEQ, Inc., United StatesAdam Keith Walker, Macquarie University, Australia
Copyright © 2017 Álvarez-Satta, Castro-Sánchez and Valverde. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Valverde, ZGlhbmF2YWxAdXZpZ28uZXM=