 Victor M. Guerrero-Sanchez
Victor M. Guerrero-Sanchez Ana M. Maldonado-Alconada
Ana M. Maldonado-Alconada Francisco Amil-Ruiz
Francisco Amil-Ruiz Jesús V. Jorrin-Novo
Jesús V. Jorrin-Novo- 1Agroforestry and Plant Biochemistry, Proteomics and Systems Biology, Department Biochemistry and Molecular Biology, Universidad de Córdoba, Cordoba, Spain
- 2Servicio Central de Apoyo a la Investigación, Universidad de Córdoba, Cordoba, Spain
Introduction
Holm oak (Quercus ilex L. subsp. ballota [Desf.] Samp.) is the dominant tree species in the Mediterranean forest with great ecological and economic value (Pulido et al., 2001). It constitutes, together with cork oak (Q. suber), the “dehesa,” a typical Mediterranean agro-forestry-pastoral ecosystem, covering almost four million hectares in the western Iberian Península (Joffre et al., 1999). Besides, holm oak is widely used in reforestation programs and silvicultural practices, being their seeds, acorns, used for feed, and fatten the exclusive Iberian race pigs, whose meat is the basis of a high-quality food industry (Vicente and Alés, 2006; Cañellas et al., 2007).
Nowadays, Q. ilex forest maintenance and sustainability are facing severe problems and challenges. Those are related to agricultural practices, low natural regeneration, seed viability, which may be due to their non-orthodox seed character (Doody and O'Reilly, 2008), plant mortality in both adult trees and young plants after field transplantation resulting from adverse environmental conditions like drought, the so-called decline syndrome (Gallego et al., 1999), especially considering the current and future climate change scenario (Plieninger et al., 2004; Bates et al., 2008; Corcobado et al., 2013). Overcoming those threats could be greatly facilitated if olm oak ecophysiological behavior was better understood at the molecular level. Nowadays, multidisciplinary approaches by integrating the so-called—omic studies—transcriptomics, proteomics and metabolomics—have become indispensable to shed light on the fine-tuned molecular regulation in many biological systems/species. Thus, system biology aims to describe and interpret the full complexity of cells, tissues, organs, and organisms.
In this context, our research group has been investigating different aspects of Q. ilex biology such as natural variation, seed germination and seedling growth, physiology, biotic and abiotic stress-responses, combining classical biochemistry, and integrating those multidisciplinary “omics” analysis (Echevarría-Zomeño et al., 2009, 2012; Jorrín-Novo et al., 2009; Valero-Galván et al., 2011, 2012, 2013; Sghaier-Hammami et al., 2013, 2016; Romero-Rodríguez et al., 2014). Nevertheless, the scarce genomic information (to date) available for Q. ilex, supposes, such as for other orphan tree species (Abril et al., 2011; Jorrín-Novo et al., 2015), a notable obstacle to successfully carry out these global studies at molecular level. Driven by that need, our main aim has been to generate a reference transcriptome of Q. ilex which will support and complement future research within this species. For that purpose, as a first approach we sequenced the mRNA of a pooled plant sample containing equal amounts of homogenized tissue from acorn embryo, leaves, and roots, using an Illumina Hiseq 2500 platform. Contrasting different assembly strategies and algorithms, we present here the first de novo assembled transcriptome of the non-conventional plant Q. ilex.
The pre-processed raw reads generated by the sequencing platform, and used for the de novo assembly, have been deposited at the NCBI SRA database with accession number SRR5815058.
This new genomic resource will set the stage for ongoing and future studies to obtain a better understanding of molecular mechanisms involved in physiological processes such as seed germination, seedling establishment, drought, which are essential for selection of superior phenotypes or Candidate Plus for restoration and reforestation programs under the impending climate change in Mediterranean regions.
Materials and Methods
Plant Material
Mature acorns from Holm oak (Q. ilex L. subsp. ballota [Desf.] Samp.) were collected from a tree located in Aldea de Cuenca (province of Córdoba, Andalusia, Spain). Acorns were germinated and seedlings grew in a chamber under controlled conditions (a 12 h photoperiod, a temperature of 21 ± 1°C, a relative humidity of 60 ± 5% and an irradiance of 200 μmol m−2 s−1, Echevarría-Zomeño et al., 2009). Germinated embryo, leaves and roots from 1 year plantlets were collected separately, weighted, and individually frozen in liquid nitrogen. The plant material used for RNA sequencing experiments consisted in a pool generated by mixing equal amounts of homogenized tissue from acorn embryo, leaves, and roots.
RNA Extraction
Total RNA was extracted from 50 mg pooled plant sample according the procedures previously set up in our laboratory for Q. ilex samples (Echevarría-Zomeño et al., 2012). Contaminating genomic DNA was removed by DNase I (Ambion) treatment. Total RNA was quantified spectrophotometrically (DU 228800 Spectrophotometer, Beckman Coulter, TrayCell Hellma GmbH & Co. KG). The high quality and integrity of the RNA preparation was tested electrophoretically (Agilent 2100 Bioanalyzer). Only high-quality RNAs with RIN values > 8 and A260:A280 ratios near 2.0 were used for subsequent experiments.
Enrichment of mRNA, cDNA Synthesis, and Library Generation for Illumina HiSeq 2500 Platform. Paired-End Sequencing
The library construction of cDNA molecules was carried out using Illumina TruSeq Stranded mRNA Library Preparation Kit according to manufacturer instructions using 2 μg of total RNA followed by poly-A mRNA enrichment using streptavidin coated magnetic beads and thermal mRNA fragmentation. The cDNA was synthesized, followed by a chemical fragmentation (DNA library) and sequenced in the Illumina Hiseq 2500 platform, using 100 bp paired-end sequencing (Conesa and Götz, 2008; De Wit et al., 2012).
De novo Assembly and Analysis of High Throughput RNA Sequencing Data
The raw reads obtained from the sequencing platform were pre-processed in order to retain only high-quality sequences to be subsequently used in the assembly. Thus, each original sequence was quality trimmed considering several parameters (quality trimming based on minimum quality scores, ambiguity trimming to trim off e.g., stretches of Ns, base trim to remove specified number of bases at either 3′ or 5′ end of the reads). The pre-processing parameters used were selected as following: trimming sequences by maximum 2 ambiguous nucleotides), minimum mean quality assuming error probability < 0.01, and filtering out those sequences shorter than 30 nucleotides.
Three different assemblers were employed to de novo assemble the Q. ilex transcriptome, considering there is not a reference genome available, and further evaluated to contrast the results obtained (Figure 1).
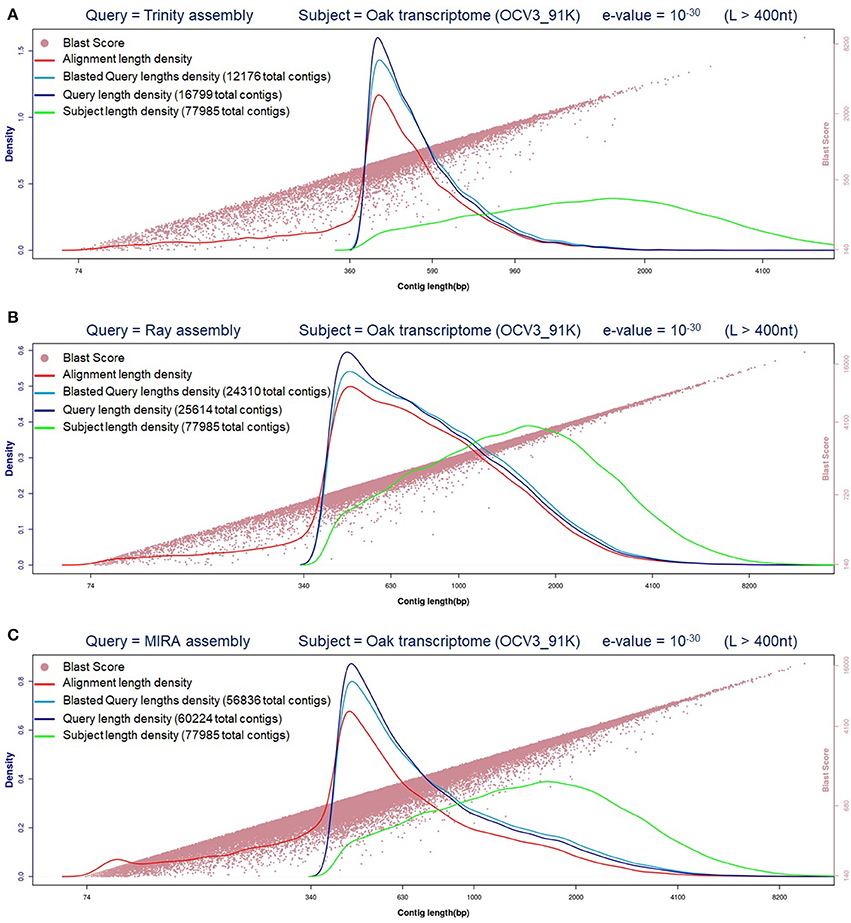
Figure 1. Evaluation of Q.ilex transcriptomes generated. Contig (longer than 400 nucleotides = L > 400 nt) length distribution and comparative evaluation against oak transcriptome (BlastN e-value = 10−30). (A) Trinity; (B) Ray; (C) MIRA.
Trinity 2.4.0. performs a de novo assembly using an algorithm based on Bruijn graphs (Grabherr et al., 2011). For the assembly, Trinity 2.4.0 was launched with a k-mer value (k = 25).
Ray 2.3.1. assembly uses de Bruijn graphs but its framework is not based on the Eulerian steps. Specific subsequences, seeds, are defined, and for each of them, the algorithm extends it to a contig. Heuristics are defined that control the extension process in such a way that the process stops if, at some point, the readings family does not clearly indicate the address of the extension (Boisvert et al., 2010). In this case we selected a k-mer value of 31.
MIRA 4.9.6 software (Chevreux et al., 1999), unlike Trinity and Ray, is based on the strategy known as Overlap /Layout/ Consensus. Following the author guidelines/recommendations for Illumina data, we used the complete raw data without a filtering process like we described previously.
Evaluation of the structure of the generated assemblies was done with the QUAST software (Gurevich et al., 2013).
The assemblies obtained using the three aforementioned softwares were blasted (e-value of 10−30) against the most accurate and nearest phylogenetic transcriptome currently available, the oak transcriptome (containing Quercus robur and Quercus petraea sequences) (Lesur et al., 2015). That transcriptome database is divided in two files OCV3_91K and OCV3_101K but OCV3_91K has a larger amount of valuable information of Quercus spp. transcriptome. So, we chose OCV3_91K as a general oak transcriptome database.
Results
Evaluation and Annotation of the Assembled Transcriptomes
There are differences between the three assembled transcriptomes in terms of transcriptome architecture/structure. Thus, the N50 value, number of contigs and the average length of the sequences generated by each algorithm differ (Table 1).
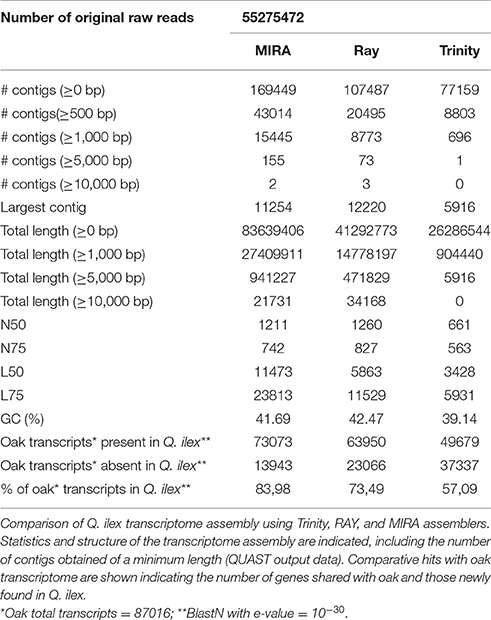
Table 1. Assembly structure and similarity with oak transcriptome.
Considering these results, we can state that MIRA generated more and longer contigs than RAY and Trinity (MIRA>RAY>Trinity), suggesting that a more robust architecture/structure is obtained by MIRA for the Q. ilex transcriptome assemby. Upon the continuous development of NGS methods, data processing, and transcript assemby remains a main challenge. Several studies have been published devoted to evaluate different de novo assemblers varying in performance and quality in terms of number and length of transcripts and computational speed (Clarke et al., 2013). Besides, it has been reported that the quality of the assembly using a given software depends on the biological sample on study (Bradnam et al., 2013). Thus, these aspects should be taken into consideration when comparing different softwares.
The comparison between the sequences generated from Q. ilex and those available from the close species, oak transcriptome, reveals that MIRA assembly was the one which shared the higher number of transcripts (73073), followed by RAY assembler (Table 1). Besides, MIRA assembly sequences blasted against oak transcriptome render the longest alignment lengths and better blast scores (Figure 1).
Taking into consideration the data and parameters evaluated (Table 1 and Figure 1), we decided to use the MIRA assembly to continue with the corresponding annotation of Q. ilex transcriptome. After blastX was completed against Uni-Prot (Swiss-Prot) curated database (e-value of 10−5), followed by the corresponding mapping process, 31973 annotated sequences were obtained by Blast2GO (Conesa and Götz, 2008).
Direct Link to Deposited Data
The pre-processed raw reads of the transcriptome assembly generated by the sequencing platform, and used for the de-novo assembly, have been deposited at the NCBI SRA database with the following accession number SRX2993508 and direct link: ftp://ftp-trace.ncbi.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR581/SRR5815058/SRR5815058.sra
Author Contributions
AM: Collected samples, performed RNA isolation. VG and FA: Bioinformatic analysis of the data. VG, FA, JJ, and AM: Wrote the manuscript. JJ: Supervised the Project and acquired funding.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the University of Cordoba and financial support from the Spanish Ministry of Economy and Competitiveness (Project BIO2015-64737-R2). The staff of the Central Service for Research Support (SCAI) at the University of Cordoba is acknowledged for its technical support in Bioinformatics data analysis.
References
Abril, N., Gion, J. M., Kerner, R., Müller-Starck, G., Cerrillo, R. M., Plomion, C., et al. (2011). Proteomics research on forest trees, the most recalcitrant and orphan plant species. Phytochemistry 72, 1219–1242. doi: 10.1016/j.phytochem.2011.01.005
Bates, B. C., Kundzewicz, Z. W., Wu, S., and Palutikof, J. P. (2008). Climate Change and Water Technical Paper of the Intergovernmental Panel on Climate Change. Geneva: IPCC Secretariat.
Boisvert, S., Laviolette, F., and Corbeil, J. (2010). Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J. Comput. Biol. 17, 1519–1533. doi: 10.1089/cmb.2009.0238
Bradnam, K. R., Fass, J. N., Alexandrov, A., Baranay, P., Bechner, M., Birol, I., et al. (2013). Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2:10. doi: 10.1186/2047-217X-2-10
Cañellas, I., Roig, S., Poblaciones, M., Gea-Izquierdo, G., and Olea, L. (2007). An approach to acorn production in Iberian dehesas. Agrofor. Syst. 70, 3–9. doi: 10.1007/s10457-007-9034-0
Chevreux, B., Wetter, T., and Suhai, S. (1999). “Genome sequence assembly using trace signals and additional sequence information,” in German Conference on Bioinformatics (Heidelberg), 45–56.
Clarke, K., Yang, Y., Marsh, R., Xie, L., and Zhang, K. K. (2013). Comparative analysis of de novo transcriptome assembly. Sci. China Life Sci. 56, 56–162. doi: 10.1007/s11427-013-4444-x
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832. doi: 10.1155/2008/619832
Corcobado, T., Cubera, E., Moreno, G., and Solla, A. (2013). Quercus ilex forests are influenced by annual variations in water table, soil water deficit and fine root loss caused by Phytophthora cinnamomi. Agric. For. Meteorol. 169, 92–99. doi: 10.1016/j.agrformet.2012.09.017
De Wit, P., Pespeni, M. H., Ladner, J. T., Barshis, D. J., Seneca, F., Jaris, H., et al. (2012). The simple fool's guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis. Mol. Ecol. Resour. 12, 1058–1067. doi: 10.1111/1755-0998.12003
Doody, C. N., and O'Reilly, C. (2008). Drying and soaking pretreatments affect germination in pedunculate oak. Ann. For. Sci. 65, 509–509. doi: 10.1051/forest:2008027
Echevarría-Zomeño, S., Abril, N., Ruiz-Laguna, J., Jorrín-Novo, J., and Maldonado-Alconada, A. M. (2012). Simple, rapid and reliable methods to obtain high quality RNA and genomic DNA from Quercus ilex L. leaves suitable for molecular biology studies. Acta Physiol. Plant. 34, 793–805. doi: 10.1007/s11738-011-0880-z
Echevarría-Zomeño, S., Ariza, D., Jorge, I., Lenz, C., Del Campo, A., Jorrín, J. V., et al. (2009). Changes in the protein profile of Quercus ilex leaves in response to drought stress and recovery. J. Plant Physiol. 166, 233–245. doi: 10.1016/j.jplph.2008.05.008
Gallego, B. F. J., de Algaba, A. P., and Fernandez-Escobar, R. (1999). Etiology of oak decline in Spain. For. Pathol. 29, 17–27. doi: 10.1046/j.1439-0329.1999.00128.x
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Joffre, R., Rambal, S., and Ratte, J. P. (1999). The dehesa system of southern Spain and Portugal as a natural ecosystem mimic. Agrofor. Syst. 45, 57–79.
Jorrín-Novo, J. V., Maldonado, A. M., Echevarría-Zomeño, S., Valledor, L., Castillejo, M. A., Curto, M., et al. (2009). Plant proteomics update (2007–2008): Second-generation proteomic techniques, an appropriate experimental design, and data analysis to fulfill MIAPE standards, increase plant proteome coverage and expand biological knowledge. J. Proteomics 72, 285–314. doi: 10.1016/j.jprot.2009.01.026
Jorrín-Novo, J. V., Pascual, J., Sánchez-Lucas, R., Romero-Rodríguez, M. C., Rodríguez-Ortega, M. J., Lenz, C., et al. (2015). Fourteen years of plant proteomics reflected in Proteomics : moving from model species and 2DE-based approaches to orphan species and gel-free platforms. PROTEOMICS 15, 1089–1112. doi: 10.1002/pmic.201400349
Lesur, I., Le Provost, G., Bento, P., Da Silva, C., Leplé, J. C., Murat, F., et al. (2015). The oak gene expression atlas: insights into Fagaceae genome evolution and the discovery of genes regulated during bud dormancy release. BMC Genomics 16:112. doi: 10.1186/s12864-015-1331-9
Plieninger, T., Pulido, F. J., and Schaich, H. (2004). Effects of land-use and landscape structure on holm oak recruitment and regeneration at farm level in Quercus ilex L. dehesas. J. Arid Environ. 57, 345–364. doi: 10.1016/S0140-1963(03)00103-4
Pulido, F. J., Diaz, M., and Hidalgo de Trucios, S. J., (2001). Size structure and regeneration of Spanish holm oak Quercus ilex forests and dehesas: effects of agroforestry use on their long-term sustainability. For. Ecol. Manag. 146, 1–13. doi: 10.1016/S0378-1127(00)00443-6
Romero-Rodríguez, M. C., Pascual, J., Valledor, L., and Jorrín-Novo, J. (2014). Improving the quality of protein identification in non-model species. Characterization of Quercus ilex seed and Pinus radiata needle proteomes by using SEQUEST and custom databases. J. Proteomics 105, 85–91. doi: 10.1016/j.jprot.2014.01.027
Sghaier-Hammami, B., Redondo-López, I., Valero-Galvàn, J., and Jorrín-Novo, J. V. (2016). Protein profile of cotyledon, tegument, and embryonic axis of mature acorns from a non-orthodox plant species: Quercus ilex. Planta 243, 369–396. doi: 10.1007/s00425-015-2404-3
Sghaier-Hammami, B., Valero-Galvàn, J., Romero-Rodríguez, M. C., Navarro Cerrillo, R. M., Abdelly, C., and Jorrín-Novo, J. (2013). Physiological and proteomics analyses of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) responses to Phytophthora cinnamomi. Plant Physiol. Biochem. 71, 191–202. doi: 10.1016/j.plaphy.2013.06.030
Valero-Galván, J., González-Fernández, R., Navarro-Cerrillo, R. M., Gil-Pelegrín, E., and Jorrín-Novo, J. V. (2013). Physiological and proteomic analyses of drought stress response in holm oak provenances. J. Proteome Res. 12, 5110–5123. doi: 10.1021/pr400591n
Valero-Galván, J., Valledor, L., Navarro Cerrillo, R. M. N., Gil Pelegrín, E. G., and Jorrín-Novo, J. V. (2011). Studies of variability in Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) through acorn protein profile analysis. J. Proteomics 74, 1244–1255. doi: 10.1016/j.jprot.2011.05.003
Valero-Galván, J., Valledor, L., González Fernandez, R., Navarro Cerrillo, R. M., and Jorrín-Novo, J. V. (2012). Proteomic analysis of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) pollen. J. Proteomics 75, 2736–2744. doi: 10.1016/j.jprot.2012.03.035
Keywords: Holm oak, Quercus ilex, RNA-sequencing, assemblers, illumina
Citation: Guerrero-Sanchez VM, Maldonado-Alconada AM, Amil-Ruiz F and Jorrin-Novo JV (2017) Holm Oak (Quercus ilex) Transcriptome. De novo Sequencing and Assembly Analysis. Front. Mol. Biosci. 4:70. doi: 10.3389/fmolb.2017.00070
Received: 27 July 2017; Accepted: 22 September 2017;
Published: 06 October 2017.
Edited by:
Sanjeev Kumar Srivastava, Mitchell Cancer Institute, United StatesReviewed by:
Kehua Wang, China Agricultural University, ChinaKishor Gaikwad, National Research Centre on Plant Biotechnology (ICAR), India
Copyright © 2017 Guerrero-Sanchez, Maldonado-Alconada, Amil-Ruiz and Jorrin-Novo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor M. Guerrero-Sanchez, YjEyZ3VzYXZAdWNvLmVz
Ana M. Maldonado-Alconada, YmIybWFhbGFAdWNvLmVz