 Michael A. Reiche
Michael A. Reiche Digby F. Warner
Digby F. Warner Valerie Mizrahi
Valerie Mizrahi- SAMRC/NHLS/UCT Molecular Mycobacteriology Research Unit, DST/NRF Centre of Excellence for Biomedical Tuberculosis Research, Department of Pathology, Institute of Infectious Disease and Molecular Medicine, Faculty of Health Sciences, University of Cape Town, Cape Town, South Africa
Mycobacterium tuberculosis is the etiological agent of tuberculosis (TB), an infectious disease which results in approximately 10 million incident cases and 1.4 million deaths globally each year, making it the leading cause of mortality from infection. An effective frontline combination chemotherapy exists for TB; however, this regimen requires the administration of four drugs in a 2 month long intensive phase followed by a continuation phase of a further 4 months with two of the original drugs, and is only effective for the treatment of drug-sensitive TB. The emergence and global spread of multidrug-resistant (MDR) as well as extensively drug-resistant (XDR) strains of M. tuberculosis, and the complications posed by co-infection with the human immunodeficiency virus (HIV) and other co-morbidities such as diabetes, have prompted urgent efforts to develop shorter regimens comprising new compounds with novel mechanisms of action. This demands that researchers re-visit cellular pathways and functions that are essential to M. tuberculosis survival and replication in the host but which are inadequately represented amongst the targets of current anti-mycobacterial agents. Here, we consider the DNA replication and repair machinery as a source of new targets for anti-TB drug development. Like most bacteria, M. tuberculosis encodes a complex array of proteins which ensure faithful and accurate replication and repair of the chromosomal DNA. Many of these are essential; so, too, are enzymes in the ancillary pathways of nucleotide biosynthesis, salvage, and re-cycling, suggesting the potential to inhibit replication and repair functions at multiple stages. To this end, we provide an update on the state of chemotherapeutic inhibition of DNA synthesis and related pathways in M. tuberculosis. Given the established links between genotoxicity and mutagenesis, we also consider the potential implications of targeting DNA metabolic pathways implicated in the development of drug resistance in M. tuberculosis, an organism which is unusual in relying exclusively on de novo mutations and chromosomal rearrangements for evolution, including the acquisition of drug resistance. In that context, we conclude by discussing the feasibility of targeting mutagenic pathways in an ancillary, “anti-evolution” strategy aimed at protecting existing and future TB drugs.
Introduction
The Need for New TB Drugs
According to the most recent WHO report, 10.4 million people developed tuberculosis (TB) and 1.8 million died from this disease in 2015 (WHO, 2016), making TB the leading cause of death from an infectious disease. The threat that TB presents to global health has been significantly heightened by the evolution and spread of drug-resistant TB: in 2015, a staggering 480,000 people across the world developed multi-drug resistant (MDR)-TB, defined as TB that is resistant to isoniazid (INH) and rifampicin (RIF), with or without resistance to other first-line anti-tubercular drugs. Of these, 9.5% had extensively drug-resistant (XDR)-TB, which is resistant to INH and RIF (i.e., MDR-TB) in addition to any fluoroquinolone and at least one of the injectable second-line drugs, kanamycin, amikacin, or capreomycin. Unfortunately, this alarming situation has continued to worsen with the ongoing evolution of XDR-TB to forms of the disease that are functionally untreatable with existing antibiotics (Dheda et al., 2014).
Drug-sensitive TB is treated with a standard “short-course” regimen comprising a 2-month intensive phase of treatment with four drugs—INH, RIF, pyrazinamide (PZA), and ethambutol (EMB)—followed by four additional months of treatment with INH and RIF in a continuation phase. Under optimal conditions, this regimen is highly effective at achieving durable cure of drug-sensitive TB. However, non-adherence to this protracted therapeutic regimen is common among TB patients and may result in the emergence of drug resistance through the acquisition of chromosomal mutations in the aetiological agent, Mycobacterium tuberculosis (M. tuberculosis), leading to prolonged infectiousness and poor treatment outcomes (Dheda et al., 2014). Drug-resistant TB is far more challenging to treat, requiring the administration of combinations of second- and third-line drugs that are more toxic, more expensive, and less efficacious. As a result, this form of the disease is associated with substantial morbidity and mortality, while consuming a disproportionate share of national budgets for TB control in disease-endemic countries—thus compromising TB control programmes (Dheda et al., 2014, 2017).
The need for new TB drugs for the treatment of drug-susceptible as well as drug-resistant TB is therefore clear and urgent. After decades of neglect, a renewed interest in TB drug development in the late 1990s, which coincided with major scientific advances including the completion of the first genome sequence of M. tuberculosis (Cole et al., 1998), has resulted in a pipeline populated with new as well as repurposed drugs and drug combinations at various stages of development (http://www.newtbdrugs.org). A number of criteria are being used to guide this process: for example, all new TB drugs should: (i) have novel mechanisms of action to permit their use in the treatment of drug-resistant forms of the disease; (ii) have significant treatment-shortening potential when combined with other agents; (iii) be safe and tolerable; (iv) simplify treatment by reducing the pill burden and dosing frequency; and, (v) be compatible with antiretroviral drugs to enable treatment of patients co-infected with HIV (Zumla et al., 2013, 2014). The ability to meet these criteria is dependent upon the quality of compounds that enter the pipeline at the lead optimization stage. The identification of high-quality leads has, in turn, been critically reliant on harnessing biological insight from studies on M. tuberculosis pathogenesis in various models of infection. A major theme emerging from this work is the biological complexity of TB at the level of both host and pathogen, with the genotypic and phenotypic heterogeneity of M. tuberculosis posing particularly onerous challenges for new TB drug discovery, as discussed below.
Approaches to TB Drug Discovery
Genome-wide mutagenesis studies in M. tuberculosis (Long et al., 2015) have identified genes that are (conditionally) essential for growth and survival of the bacillus in vitro (Sassetti et al., 2003; DeJesus et al., 2017), in macrophages (Rengarajan et al., 2005), and in animal models of infection (Sassetti and Rubin, 2003). This information has underpinned target-based drug discovery efforts aimed at crippling essential cellular functions in M. tuberculosis. However, as in other areas of antimicrobial drug discovery (Payne et al., 2007), the approach has met with very limited success in the TB field, and has been confounded by a general lack of information about target vulnerability as well as the impact of compound metabolism, permeability, and efflux on efficacy. For this reason, small molecules that potently inhibit M. tuberculosis enzymes in biochemical assays have failed to translate into leads with activity against the bacillus in vitro and/or in vivo. In contrast, phenotypic screening, in which compound libraries are screened for activity against M. tuberculosis to identify molecules with whole-cell activity, has been far more successful, and has delivered the clinically approved drugs, bedaquiline (Sirturo) and delamanid (Deltyba), a number of drug candidates that are currently in development (Mdluli et al., 2015; Singh and Mizrahi, 2017)—including griselimycin (Kling et al., 2015), PA-824 (pretomanid) (Stover et al., 2000), PBTZ169 (Makarov et al., 2014), and Q203 (Pethe et al., 2013)—and other promising leads such as the Pks13 inhibitor, TAM16 (Aggarwal et al., 2017). It is worth noting, however, that this approach, too, has its challenges as mechanisms of action (MOA) of potent molecules with whole-cell activity can be difficult to elucidate, thereby complicating the progression of individual compounds or compound series through the pipeline. Importantly, though, there are signs indicating greater integration of the two approaches: on the one hand, target-based whole-cell screening, in which hit identification from phenotypic screening is biased toward prioritized targets and pathways, has begun to gain traction (Abrahams et al., 2012) while, on the other hand, screening collections of whole-cell actives identified by phenotypic approaches against high-value M. tuberculosis targets offers the prospect of discovering new drug-target pairs as starting points for hit-to-lead (H2L) programs (Esposito et al., 2017).
Managing Biological Complexity in TB Drug Discovery
Genotypic and phenotypic heterogeneity of M. tuberculosis must be taken into account from the earliest stage of TB drug discovery. Genotypic heterogeneity is managed by screening promising hits for activity against representatives from the major strain lineages of M. tuberculosis (Coscolla and Gagneux, 2014) and against panels of drug-resistant strains (e.g., Aggarwal et al., 2017; Blondiaux et al., 2017). The other major mechanism underlying differential drug susceptibility in M. tuberculosis is phenotypic antibiotic tolerance (Aldridge et al., 2014; Brauner et al., 2016), which is thought to be the main reason why prolonged TB therapy is required in order to achieve relapse-free cure (Kester and Fortune, 2014; Gold and Nathan, 2017). Antibiotic efficacy can be influenced profoundly by the physiology, metabolic state, and growth rate of the organism, with most TB drugs showing significantly reduced efficacy against M. tuberculosis in slow- or non-growing states (Baer et al., 2015). Thus, drugs that target cellular processes required to support bacterial growth tend to have reduced efficacy against slow- or non-growing organisms (Gold and Nathan, 2017). Mycobacterium tuberculosis encounters complex, hostile environments during transmission, infection, and disease (Pai et al., 2016). As an exquisitely adapted human pathogen endowed with a rich and highly flexible metabolic repertoire (Baughn and Rhee, 2014; Warner, 2014), the bacillus is able to adapt its physiology and metabolism in response to the conditions encountered during each of these stages. These conditions include intracellular residence in macrophages and other phagocytic cells, exposure to nitrosative and oxidative stress, hypoxia, nutrient deprivation, alterations in carbon source availabilities, and low pH (Baer et al., 2015). In a single patient, therefore, M. tuberculosis infection can be characterized by mixed populations of intracellular and extracellular bacilli in a variety of metabolic states and with variable growth rates. This complicates treatment (Dartois and Barry, 2013) and has led to the suggestion that TB should be treated as a polybacterial infection (Evangelopoulos and McHugh, 2015). The problem is further complicated by the impact of lesion heterogeneity on drug pharmacokinetic/pharmacodynamic (PK/PD) parameters (Dartois, 2014). To address this complexity, assays designed to recapitulate at least some of the conditions encountered during infection have been incorporated into drug screening cascades with the aim of identifying “pan-active” compounds with the ability to kill M. tuberculosis in as wide a range of metabolic states as possible.
Major Mechanistic Classes of TB Drugs
TB drugs fall into a relatively small number of mechanistic classes. A defining characteristic of the tubercle bacillus is its unusual and highly complex cell envelope, which has a number of distinguishing features including the mycolyl-arabinogalactan-peptidoglycan complex that links the peptidoglycan to the mycobacterial outer membrane. Not surprisingly, a disproportionate number of TB drugs act on biogenesis of the cell envelope; these include INH and ethambutol (EMB), the second-line agent, D-cycloserine, and those that act on the new targets, DprE1 (e.g., PBTZ169) (Makarov et al., 2014), MmpL3 (e.g., BM212 and other chemotypes) (Xu et al., 2017), and Pks13 (TAM16) (Aggarwal et al., 2017). Other drugs target transcription (RIF), protein synthesis (e.g., linezolid), and energy metabolism (bedaquiline, Q203). Furthermore, and consistent with the formidable capacity of M. tuberculosis to metabolize xenobiotics (Awasthi and Freundlich, 2017), prodrugs are common in the TB drug arsenal and, for compounds such as PZA, delamanid, and pretomanid, the respective active metabolites have pleiotropic effects on mycobacterial metabolism (Matsumoto et al., 2006; Singh et al., 2008; Anthony et al., 2016).
An important, albeit small, category of TB drugs includes those that target DNA replication. Until recently, these have been limited exclusively to the fluoroquinolones, in particular, moxifloxacin and gatifloxacin, which inhibit DNA gyrase, and are widely used for the treatment of MDR-TB. However, another component of the DNA replication machinery has emerged as an exciting new target for TB drug development through the discovery that griselimycins target the β-clamp protein, DnaN (Kling et al., 2015). In the following sections, we consider the DNA replication and repair pathways of M. tuberculosis as potential sources of new targets for TB drug development. This terrain has been extensively reviewed recently, perhaps signaling the increasing appreciation of DNA metabolism as underrepresented among common antibiotic targets. The interested reader is encouraged to consult a number of excellent articles, both specific to M. tuberculosis (Plocinska et al., 2017) and of more general interest (Robinson et al., 2012; Sanyal and Doig, 2012; van Eijk et al., 2017).
The Mycobacterial DNA Replication Machinery
Chromosomal replication in bacteria is performed by a large, multiprotein replisome that ensures coordinated synthesis of the leading and lagging DNA strands with high efficiency and accuracy (Beattie and Reyes-Lamothe, 2015; Yao and O'Donnell, 2016a,b). Broadly, this is accomplished through the concerted action of three catalytic centers: the helicase-primase complex, the core complex, and the clamp loader complex [for comprehensive recent reviews, please refer to Ditse et al. (2017) and Plocinska et al. (2017). The helicase-primase complex comprises the DnaB helicase, which unwinds the two DNA strands, and the DnaG primase, which synthesizes short RNA primers on the lagging strand to initiate replication by the replicative DNA polymerase, Pol IIIα. Two core complexes containing Pol IIIα, the exonuclease subunit, ε, and the small subunit, θ, synthesize the new DNA strand on both leading and lagging strand templates. In elegant in vivo studies that were directed by earlier in vitro studies by Yao and O'Donnell (Yao and O'Donnell, 2016a,b) and the identification of the β-clamp (O'Donnell and Kuriyan, 2006), Reyes-Lamothe and colleagues demonstrated that these core complexes bind to the torroidal β-clamp proteins that encircle the DNA, providing a tether that enables processive synthesis and dynamic exchange of replisome components (Reyes-Lamothe et al., 2012). A 1χ1ψ1 clamp-loader complex loads the β-clamp proteins onto newly synthesized RNA primers, with the τ subunits also binding to the Pol IIIα subunits to couple leading and lagging strand biosynthesis, and the χ/ψ subunits guiding single-stranded DNA binding (SSB) proteins onto the DNA lagging strand.
The composition of the replisome is dynamic (Beattie et al., 2017; Lewis et al., 2017) and, as evident from the brief description above, the majority of the constituent proteins perform specialist functions ranging across DNA unwinding, RNA primer synthesis, clamp loading, and DNA synthesis. It is not surprising, therefore, that most of the replisome components are conserved across bacteria (Robinson et al., 2012), including M. tuberculosis (Ditse et al., 2017). So, while replisome function has been most thoroughly investigated in organisms such as E. coli and B. subtilis (Beattie and Reyes-Lamothe, 2015), the resulting models of the bacterial replication machinery are considered readily applicable to less studied systems, such as M. tuberculosis, with some notable exceptions (Ditse et al., 2017). For example, there are no clear homologs of several initiation proteins (DnaC, DnaT, PriB, and PriC) in M. tuberculosis, neither is there a holE-encoded θ subunit, nor holC- and holD-encoded χ and ψ clamp-loader subunits, respectively. Moreover, recent studies have revealed additional departures of the mycobacterial system from the classic replication models, most notably in demonstrating a dominant role for the PHP domain of the essential Pol IIIα subunit, DnaE1, in proofreading in M. tuberculosis (Rock et al., 2015; Gu et al., 2016), as discussed elsewhere (Ditse et al., 2017).
Targeting DNA Replication in M. tuberculosis
The M. tuberculosis genome comprises approximately 3950 genes (Cole et al., 1998; Wang and Chen, 2013), of which ~10% (461 genes) are absolutely required for growth and survival of the bacillus under standard aerobic growth conditions in vitro (DeJesus et al., 2017). Among the “essential” genes, 15 encode components of the DNA replication machinery; these include the DnaA replication initiator, PriA helicase loader, DnaB helicase, DnaG primase, SSB, clamp loader subunits (τ/γ, δ, δ′), DNA polymerases I and III, DnaN β-clamp, DNA ligase I, and type I and II topoisomerases (Ditse et al., 2017). It is notable that effective inhibitory agents are available for only a small number of these essential mycobacterial proteins (Table 1), with DNA gyrase representing the only clinically validated target—of the fluoroquinolones, which are used in treatment of MDR-TB. This implies considerable scope for developing new compounds targeting the other essential DNA replication components, as has been proposed recently for M. tuberculosis as well as other bacterial pathogens (Robinson et al., 2012; Sanyal and Doig, 2012; Plocinska et al., 2017; van Eijk et al., 2017). In turn, it also suggests the possible utility in investigating the potential antimycobacterial efficacies of compounds developed for use against homologous DNA replication and repair proteins in other bacteria (Table 2).
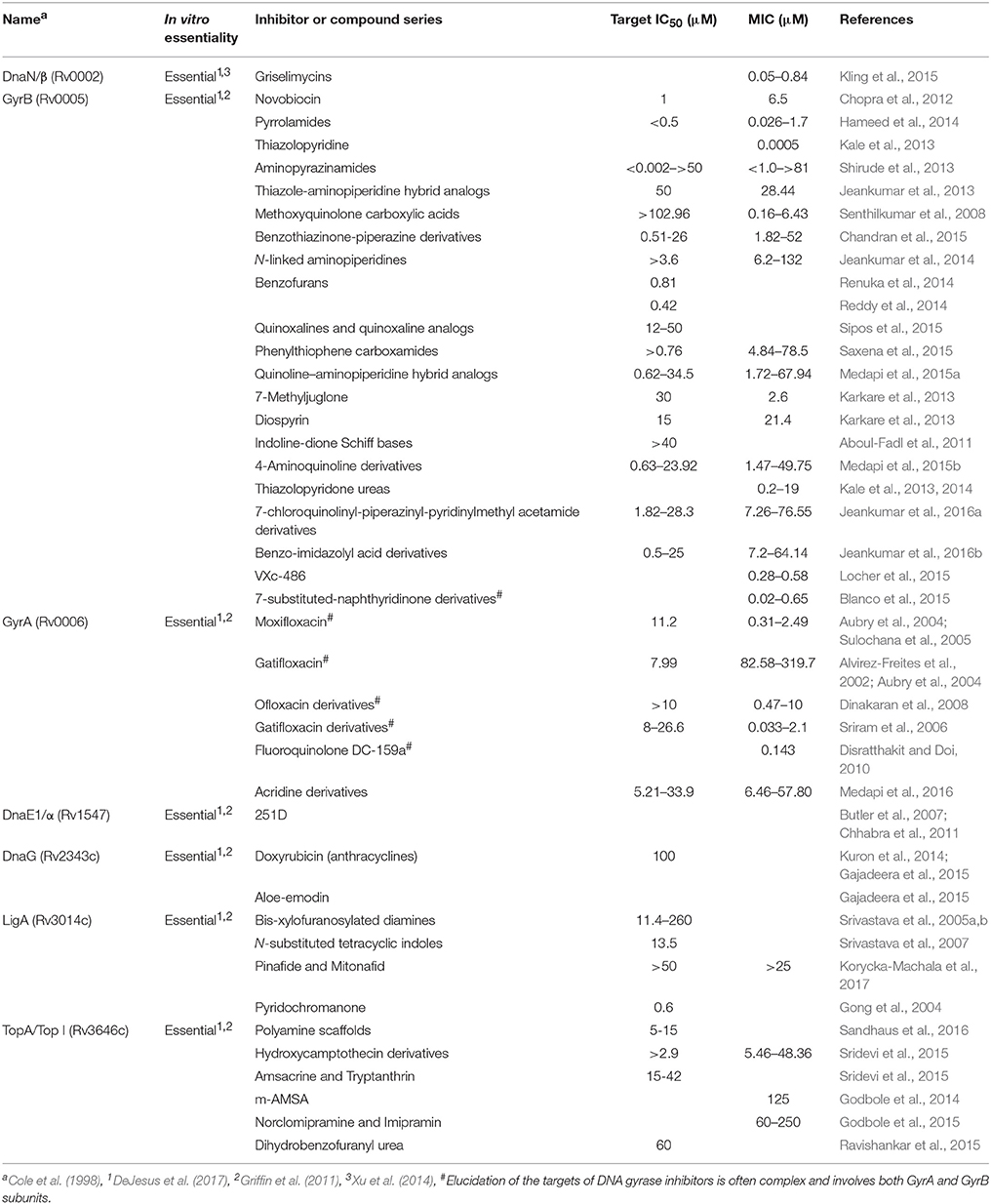
Table 1. Essential proteins involved in DNA replication targeted by anti-tubercular compounds.
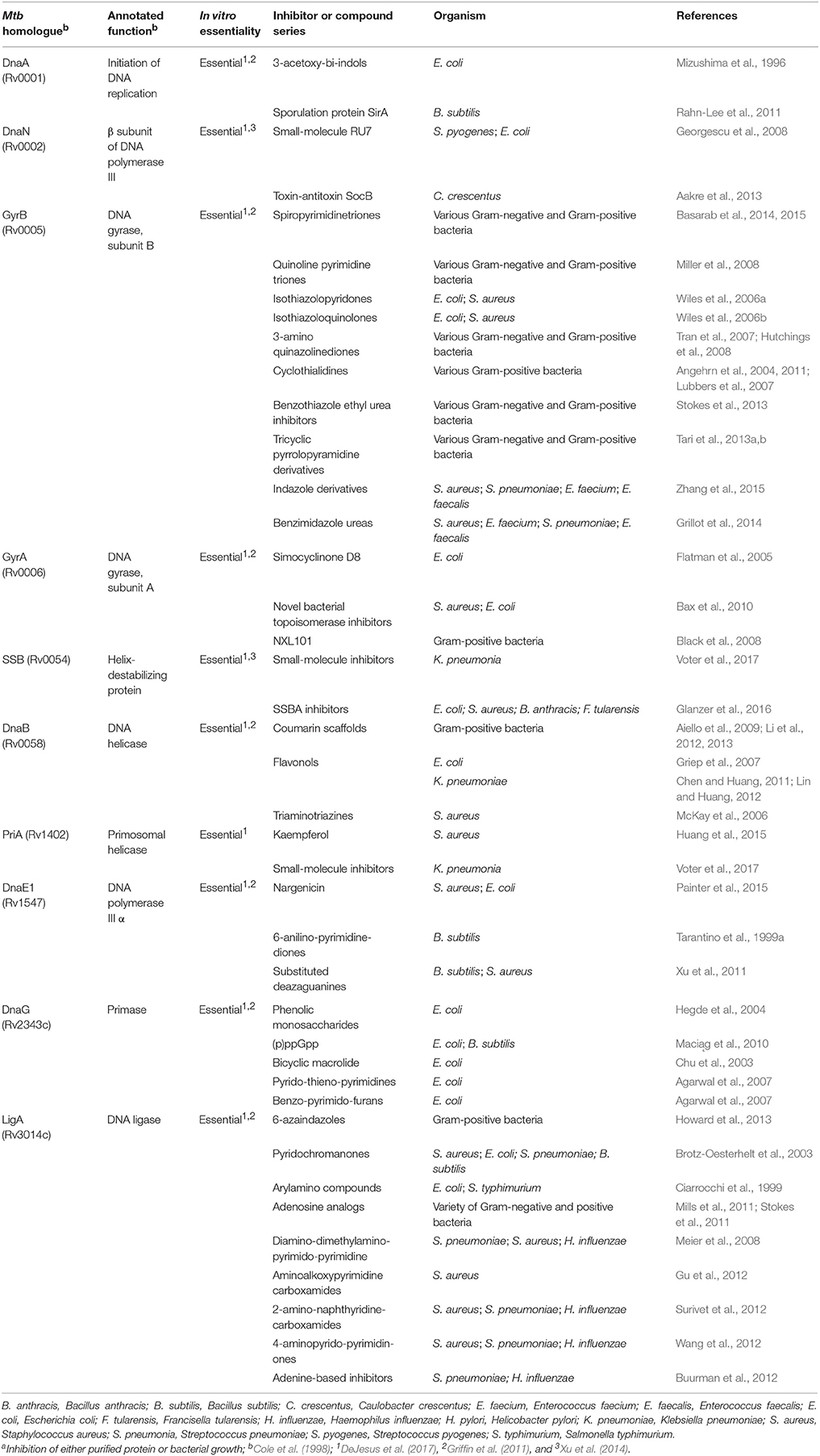
Table 2. Compounds worth investigating that inhibit homologs of essential Mtb proteins validated in other bacterial speciesa.
As applies to antibiotic development in general, overcoming the natural defenses of the target organism—in particular, the permeability barrier presented by the (myco)bacterial cell wall, and the capacity for xenobiotic extrusion via multiple efflux pumps—is often a key challenge, particularly in converting hits from biochemical assays into whole-cell actives. Avoiding compound metabolism (degradation or modification) by the target bacillus or its human host can present an additional obstacle. For DNA replication and repair specifically, the non-availability to date of purified forms of many of the mycobacterial proteins and/or reconstituted complexes has further restricted the number of in vitro screens against purified proteins, and has required that researchers rely on homology models developed using template structures from other bacteria. Importantly, this can also complicate any assessment of the druggability and ligandability of the target protein—both key additional factors in determining the success of the antibiotic development process, and which render gene essentiality alone insufficient for target validation (Hopkins and Groom, 2002; Edfeldt et al., 2011). It is pleasing to note, therefore, that several recent successes in expressing different components of the mycobacterial DNA metabolic machinery (Gong et al., 2004; Rock et al., 2015; Gu et al., 2016; Banos-Mateos et al., 2017) suggest this critical roadblock will be overcome shortly.
Targeting DNA and the array of proteins which ensure its replication and maintenance within the cell presents an additional challenge, namely ensuring specificity of the applied drug for its target organism. This can be onerous given that the proteins which interact with and modify this macromolecule have retained many key features and commonalities as they have evolved in different species. For TB, which requires lengthy treatment, the need to avoid toxicity in the human host presents an additional major challenge, and one which is likely to exclude drugs which target DNA directly, such as DNA intercalating agents (Zhang et al., 2017), and inducers of replication stress in mammalian cells (i.e., anticancer compounds). Instead, antitubercular chemotherapies need to be designed to exploit specific nuances of, and vulnerabilities within, the complement of mycobacterial DNA replication and repair proteins (Mizrahi and Huberts, 1996; Rock et al., 2015).
Despite all these challenges, there have been some exciting recent discoveries—for example, the novel DnaN-targeting natural product antibiotic, griselimycin (Kling et al., 2015), and the DnaE inhibitor, nargenicin (Young et al., 2016)—which support the potential for new drug discovery in this area, and also suggest that natural product sources are likely to offer the most promising new agents (Wright, 2017). In the ensuing sections, we discuss the very limited number of validated and experimental anti-TB drugs targeting DNA replication, and provide brief updates on recent progress suggesting the potential to develop additional experimental compounds to inhibit other components of the mycobacterial replication machinery.
Targeting the Mycobacterial Pol III Holoenzyme
Although the MOA of antifolate drugs such as sulfamethoxazole, trimethoprim, and para-aminosalicylic acid includes depletion of dNTP pools, preventing DNA replication, the impact of these agents on M. tuberculosis is polypharmacologic as it also involves inhibition of RNA and protein synthesis (Minato et al., 2015). Therefore, in its strictest sense, there are no anti-TB drugs in clinical use which directly target the DNA biosynthetic machinery in mycobacteria. That said, a handful of very exciting recent studies have established the utility of a number of compounds that prevent DNA synthesis by targeting novel Pol III holoenzyme components in M. tuberculosis.
Targeting the β Clamp, DnaN
Together with the dnaN-encoded β clamp, the Pol III* core complex (comprising α and ε subunits only, as M. tuberculosis lacks θ) and the clamp loader complex (τ/γ, δ, δ′) form the Pol III holoenzyme (Ditse et al., 2017). Griselimycin, a cyclic peptide antibiotic produced by Streptomyces spp., was originally discovered 50 years ago, but was abandoned owing to its unfavorable pharmacologic profile and the availability of other drugs such as RIF (Herrmann et al., 2017). Resurgent interest in neglected antibiotics led a team of investigators from Sanofi and Helmholtz Institute for Pharmaceutical Research Saarland to revisit this compound (Kling et al., 2015) as part of a so-called “rekindling” strategy (Herrmann et al., 2017) to identify potential anti-TB agents. In MOA studies, it was discovered that griselimycin and its metabolically more stable derivative, cyclohexylgriselimycin, bound with very high affinity (equilibrium dissociation constants of 1.0 × 10−10 and 2.0 × 10−10, respectively) to the dnaN-encoded β sliding clamp of M. tuberculosis. Importantly, the contrastingly poor binding of these compounds to the human DNA clamp protein, PNCA, results in a very high selectivity index, eliminating any concerns of general cytotoxicity.
X-ray crystallography revealed that griselimycin preferentially binds within a hydrophobic pocket located between domains II and III of DnaN—a target site known to be involved in protein-protein interactions between the β2 sliding clamp and other DNA replication and repair proteins such as the Pol IIIα replicative polymerase subunit. As such, griselimycin functions as a protein-protein interaction inhibitor and, notably, is bactericidal against mycobacteria. Moreover, resistance is rare (resistant mutants are identified at a frequency of ~5 × 10−10) and incurs a very severe fitness cost: in the non-pathogenic M. smegmatis as well as M. tuberculosis, griselimycin resistance was shown to depend on sequential amplification of the genomic region containing dnaN and the mycobacterial origin of replication (ori) site. Perhaps unsurprisingly, this resulted in a severe (slow) growth defect in vitro, and did not confer cross-resistance to other antibiotics.
From a drug development perspective, the addition of a cyclohexyl group to Proline-8 in the griselimycin backbone resulted in greater metabolic stability as well as increased lipophilicity, in turn increasing the antimycobacterial potency significantly from an initial minimum inhibitory concentration (MIC) of 1.0 μg/ml for the parental compound to 0.06 μg/ml in the derivative, all under aerobic conditions in vitro. The compound was also highly active against intracellular M. tuberculosis within macrophages, and in a mouse model—both as a single drug and in combination with the first-line drugs, RIF and PZA. These observations support the potential utility of griselimycin derivatives as anti-TB compounds, possibly to shorten therapeutic duration—though it should be noted that, under anaerobic conditions, the compound exhibited a 100-fold increase in MIC, a result which may have implications for its efficacy as a sterilizing drug. Nevertheless, griselimycin remains an exciting prospect, and is undergoing lead optimization by Sanofi and the TB Alliance (https://www.tballiance.org/portfolio/compound/cyclopeptides). Of further interest, very elegant recent work elucidating the pathway for griselimycin biosynthesis in the producer organism, Streptomyces DSM 40835, suggests the feasibility of rational modifications to the core pharmacophore (Lukat et al., 2017), thereby overcoming a common stumbling block in natural product drug development.
Targeting the Clamp Loader Complex
Mycobacterium tuberculosis possesses a restricted set of four clamp loader subunits: τ/γ, encoded by dnaX (though it must be noted that the alternative gene product, γ, has not been observed in mycobacteria), and the δ and δ′ ATPases, encoded by holA and holB, respectively (Ditse et al., 2017). Consistent with their role in loading the β clamp and co-ordinating leading and lagging strand synthesis, all four subunits are essential in M. tuberculosis (DeJesus et al., 2017); however, aside from a number of studies which have identified these components as potentially attractive targets for novel antimycobacterial agents (Anishetty et al., 2005; Kinnings et al., 2010; Xu et al., 2014), there are no reports of any experimental approaches to this effect (Plocinska et al., 2017). For this reason, these proteins are included in the small set of “non-validated, essential targets” identified as worthy of future investigation (Table 3).

Table 3. Potential, non-validated, essential Mtb targets involved in DNA replication.
Targeting the Pol IIIα Subunit, DnaE1
Mycobacterium tuberculosis encodes a single DNA Pol IIIα subunit, DnaE1, which is essential for chromosomal replication (Boshoff et al., 2003) and, therefore, a potentially attractive target for TB drug discovery (Banos-Mateos et al., 2017). Despite the fact that RNA polymerase represents a very successful therapeutic target in M. tuberculosis (Koch et al., 2014) and other pathogens (Ma et al., 2016), and that DNA polymerases have been exploited as therapeutic targets for both anti-viral and anti-cancer drugs (Lange et al., 2011), the number of compounds with demonstrated activity against bacterial replicative polymerases is very low and reduces even further when demonstrated whole-cell activity is applied as a filter (Robinson et al., 2012; van Eijk et al., 2017). There are several classes of compound known to inhibit the PolC-type polymerases: the 6-anilinouracils, which are competitive inhibitors of dGTP binding (Tarantino et al., 1999b; Wright et al., 2005); the guanine inhibitors, which are similar to the 6-anilinouracils in functioning as competitive inhibitors, but which target both PolC and DnaE (Wright et al., 2005; Xu et al., 2011); the non-nucleobase inhibitors, which include the anilinopyrimidinediones (such as 6-anilinouracils, competitive inhibitors of dGTP) (Rose et al., 2006) and the quinazolin-2-ylamino-quinazolin-4-ols (or BisQuinols), whose precise MOA remains to be elucidated but appears to involve competitive binding with the DNA template (Guiles et al., 2009); and, finally, the very recently described dicoumarin, 3,3′-(4-Nitrobenzylidene)-bis-(4-hydroxycoumarin) (Hou et al., 2015). In contrast, finding DnaE1-specific inhibitors has proved much more challenging, with some encouraging exceptions.
Very recent work has identified another natural product, nargenicin A1, as a putative DnaE1 inhibitor (Painter et al., 2015). This compound, a macrolide produced by Nocardia sp. ACC18, was shown to be active against both E. coli and S. aureus in vitro and, importantly, was effective against S. aureus in two separate mouse infection models. In S. aureus, spontaneous resistance was observed at a very low frequency (~10−9), and mapped to dnaE. This observation—in combination with in Vitro data which confirmed that nargenicin binds to the S. aureus DnaE protein in the presence of DNA, thereby inhibiting DNA replication—identified the replicative polymerase as the likely molecular target (Painter et al., 2015). However, the MOA remains to be elucidated definitively: the sole SNP in dnaE was not located in the DnaE active site, moreover nargenicin-resistant mutants displayed only low-level resistance (~4-fold over MIC). Although limited literature are available to support the potential antimycobacterial utility of nargenicin, a patent lodged by Merck claims that the compound is bactericidal against M. tuberculosis and, on that basis, under development as potential anti-TB agent (Young et al., 2016). It is assumed, therefore, that ongoing work aims to determine whether DnaE1 is the molecular target in M. tuberculosis and, furthermore, whether the bacillus is able to develop resistance—and at what cost to replicative fitness.
Compound 251D, a hybrid molecule comprising 6-(3-ethyl-4-methylanilino)uracil and fluoroquinolone moieties is another bacterial Pol IIIα inhibitor that has been identified as worthy of investigation as a potential anti-mycobacterial agent. Whether it will prove efficacious though is unclear: the target of 251D is the PolC-type replicative polymerase (Butler et al., 2007), most commonly found in low-GC Gram-positive bacteria (Timinskas et al., 2014). As noted above, M. tuberculosis encodes only the dnaE-type, which is found in both Gram-positive and Gram-negatives. Therefore, while bioinformatic analyses have predicted that the compound might be capable of docking with DnaE1, these studies utilized a model based on the replicative subunit from the Gram-negative Thermus aquaticus (Chhabra et al., 2011); inhibition of the mycobacterial DnaE1 in vitro is still to be demonstrated, so too is the activity in whole-cell assays. As noted elsewhere (Plocinska et al., 2017), an attraction of this type of hybrid compound is the potential to target both DNA gyrase and Pol IIIα with a single molecule, thereby limiting the potential for resistance development in drug-susceptible cases and retaining activity against fluoroquinolone-resistant isolates in drug-resistant TB.
Targeting the Mycobacterial Primosome
Together with nine other proteins (namely, Pol IIIα, the β2 sliding clamp, ε proofreading subunit, τ, δ, and δ′, DnaA, DNA ligase, and Pol I), the DnaB helicase, DnaG primase, and SSB constitute the basic replication module that is found across almost all sequenced bacterial genomes (McHenry, 2011; Robinson et al., 2012). DnaB and DnaG form the helicase-primase complex which, in combination with the PriA helicase loader, functions as the mycobacterial primosome: there are no identifiable mycobacterial homologs of DnaC, DnaT, PriB, or PriC. As core proteins, these represent compelling drug targets and, while much further work is required, some recent progress (summarized below) suggests that a validated clinical candidate targeting different components of the primosome is a genuine possibility.
Within any cell, ssDNA generated during DNA replication (as well as other processes, including exposure to genotoxic stress) is vulnerable to damage and prone to form secondary structures that can restrict DNA metabolic processes with potentially lethal consequences. SSB proteins have evolved to protect ssDNA, and so are essential to bacillary viability during normal replication as well as under DNA damaging conditions. Several recent high-throughput screens have been successful in identifying small-molecule inhibitors of SSB-protein interactions (Lu et al., 2010; Marceau et al., 2013; Glanzer et al., 2016). These include an attempt to identify inhibitors of SSB that might disrupt both DNA replication and SOS-mediated resistance pathways within Gram-positive and Gram-negative bacteria (Glanzer et al., 2016): following in vitro screening, six molecules were identified which successfully inhibited a broad range of bacterial SSBs, with a further four exhibiting species-specific activity—thereby establishing the potential for both broad-spectrum and species-targeted use. Notably, five of the six compounds were found to have whole-cell activity against a variety of the tested species, of which a single compound, 9-hydroxyphenylfluoron, was associated with minimal activity against the human SSB homolog. While the potential utility of this approach remains to be determined for M. tuberculosis, these results suggest the value of investigating SSB as novel anti-mycobacterial target.
An analogous approach sought to identify compounds that specifically target the eight highly-conserved residues at the C-terminus of Klebsiella pneumonia SSB with the objective of inhibiting interactions between SSB and other proteins (Voter et al., 2017). Using the interaction between SSB and the essential protein helicase, PriA, as basis for a high-throughput screen of more than 72,000 compounds, this study aimed to identify small molecules capable of inhibiting SSB interactions. Seven SSB-PriA interaction inhibitors were found to bind to SSB, with a further two binding PriA, all with IC50 values below 40 μM. No data were presented on the activity (or lack thereof) of these compounds in whole-cell assays; however, this work reinforces a common theme which suggests that protein-protein interaction inhibitors may be of specific value in inhibiting the large complex of proteins which enables DNA replication. In a similar vein, two potential PriA inhibitors, kaempferol and myricetin, were shown to inhibit the ATP hydrolysis activity of S. aureus PriA in vitro (Huang et al., 2015). While these compounds were also not validated in whole-cell assays, they too represent encouraging steps in the effort to identify antibiotics that target primosome proteins and, importantly, provide useful insight into the isolation of tractable pharmacophores for optimization against the mycobacterial homologs as part of rational structure-activity relationship (SAR) efforts.
The DnaG primase synthesizes primers for lagging strand Okazaki fragments. An early study investigating plant-derived natural products discovered two phenolic monosaccharides from Polygonum cuspidatum with low micromolar IC50 values against E. coli DnaG (Hegde et al., 2004). Similarly, another molecule from Penicillium verrucosum was shown to inhibit E. coli primase activity in biochemical assays (Chu et al., 2003). However, whole-cell activity was attainable only in a mutant E. coli strain deficient in the lipopolysaccharide layer of the cell wall as well as the AcrAB efflux system, reinforcing the potential obstacles associated with permeation and efflux as part of antibiotic discovery. This is echoed in another study which identified two classes of compounds with efficacy against E. coli DnaG in vitro and in efflux pump-deficient whole-cell assays (Agarwal et al., 2007). In that case, in vitro and whole-cell activity analyses of related pyrido-thieno-pyrimidines and benzo-pyrimido-furans identified numerous hits with attractive IC50 and MIC values, again suggesting the potential to identify novel primase inhibitors as possible anti-mycobacterial agents. In this context, it is worth noting the “natural validation” of DnaG as a suitable target for inhibiting replication: in many bacteria including M. tuberculosis, endogenous production of guanosine tetra- and penta-phosphate, (p)ppGpp, as part of the stringent response prevents the function of the replicative primase, curtailing bacterial growth (Maciąg et al., 2010).
The interaction of primase with the ssDNA template is facilitated by the replicative DNA helicase encoded by DnaB, another essential protein in M. tuberculosis (Sassetti et al., 2003; DeJesus et al., 2017). A number of flavonols have been shown to inhibit DnaB function in other bacteria (Griep et al., 2007; Lin and Huang, 2012), though no reports exist regarding the activity of these (or other) compounds in M. tuberculosis. Unusually, DnaB is among five mycobacterial proteins that contain inteins, two others of which are also involved in DNA replication: GyrA and RecA. This observation recently prompted the interesting proposal from Dziadek and colleagues (Plocinska et al., 2017) to block the protein splicing machinery as part of a polypharmacologic approach that would prevent activation of these intein-containing proteins, potentially disrupting multiple pathways simultaneously.
Targeting DNA Unwinding: DNA Gyrase and DNA Topoisomerase
Replication of the chromosomal DNA requires controlled alterations of the DNA topology to ensure processive synthesis while limiting the stresses imposed by negative supercoiling and concatenation of the double-stranded DNA molecule. The type II topoisomerase, DNA gyrase, functions to relieve torsional strain by introducing transient double-strand DNA (dsDNA) breaks which generate negative supercoils in the bacterial chromosome. Unlike those bacteria which rely on two type II topoisomerase enzymes—DNA gyrase and TopoIV—to accomplish these tasks, M. tuberculosis employs only a GyrA2B2 gyrase comprising gyrA-encoded supercoiling subunits and gyrB-encoded ATPase proteins. As a drug target, DNA gyrase represents one of the most successful in antibiotic history, primarily of the fluoroquinolones which have been used to treat both Gram-negative and Gram-positive bacterial pathogens. A series of chemical scaffolds has been employed in developing successive generations of fluoroquinolones, all of which function as topoisomerase II poisons, stabilizing the cleaved DNA-topoisomerase II complex and so resulting in a large number of double-stranded DNA breaks within the replicating bacillus which are thought to overwhelm the repair machinery, triggering a cascade of events that results in bacterial death (Dwyer et al., 2015). Fluoroquinolones are currently used as second-line anti-TB agents; however, the imperative to reduce the duration of therapy has seen several large clinical trials of novel combination regimens comprising a fluoroquinolone as frontline agent (Gillespie et al., 2014; Jindani et al., 2014; Merle et al., 2014). Although unsuccessful, these trials yielded valuable lessons about the types of preclinical data which might better inform the design of new therapies (Warner and Mizrahi, 2014), as well as the potentially critical role of drug distribution and lesion penetration in ensuring efficacy (Prideaux et al., 2015).
Other classes of gyrase inhibitors include the aminocoumarins, such as novobiocin, which were the first of many natural products found to act as gyrase inhibitors (Barreiro and Ullán, 2016). Since these compounds preferentially inhibit ATPase (GyrB) function (Lewis et al., 1996), they are less vulnerable to pre-existing resistance against the fluoroquinolones, which generally maps to mutations in gyrA (Chopra et al., 2012). Moreover, in contrast to the fluoroquinolones which result in dsDNA breaks and so upregulate the mycobacterial DNA damage response (Gillespie et al., 2005), the risk of aminocoumarin-induced mutagenesis is likely to be lower, especially in M. tuberculosis in which exposure to novobiocin does not trigger expression of the SOS regulon (Boshoff et al., 2004). However, the relatively poor penetration of aminocoumarins across cell membranes, their limited solubility, and the development of the synthetic fluoroquinolones have limited the clinical utility of this compound class (Barreiro and Ullán, 2016). In addition, issues with cytotoxicity remain a major hurdle, particularly for TB which requires extended therapeutic duration. Recent progress in the development of novel bacterial topoisomerase inhibitors (NBTIs) targeting DNA gyrase (Grillot et al., 2014; Blanco et al., 2015; Jeankumar et al., 2015a,b; Locher et al., 2015) nevertheless suggests that alternatives to the fluoroquinolones might become available in the future.
In contrast to the Type II enzymes, Type I topoisomerases have been very sparsely explored for antibiotic drug discovery. These enzymes, which cause single-stranded nicks in relaxing the DNA, perform an essential function in remodeling the chromosome for various processes including DNA replication and recombination, RNA transcription, and condensation and therefore represent an attractive target (Tse-Dinh, 2016). For this reason, Sridevi et al. conducted virtual screens of two chemical libraries for the capacity to dock with M. tuberculosis TopA (Sridevi et al., 2015). Subsequent in vitro verification of the putative hit compounds identified three with activity against purified TopA: amasacrine, tryptanthrin, and hydroxycamptothecin, a derivative of the anticancer topoisomerase inhibitor camptothecin (Wall et al., 1966). The latter hit compound was subsequently modified with terminal hydrophobic moieties to yield a library of fifteen 7-ethyl-10-hydroxycomptothecin derivatives which exhibited activity against both drug-susceptible and XDR M. tuberculosis, with MICs as low as 5.92 and 2.95 μM, respectively—a significant improvement over previous TopA inhibitors (Godbole et al., 2014, 2015). Moreover, the XDR isolates exhibited enhanced susceptibility to five of the hydroxycamptothecin derivatives relative to the drug-susceptible strains, suggesting that this might offer an attractive target in these otherwise highly resistant forms. Furthermore, four hydroxycamptothecin derivatives were identified to be more effective at inhibiting the resuscitation of non-replicating persisters in both nutrient starvation as well as oxidative and nitrosative stress models. These results, together with compounds identified in other studies and which still require validation in whole-cell assays (Ravishankar et al., 2015; Sandhaus et al., 2016), highlight the possibility of successfully and specifically inhibiting TopA as a novel therapeutic target for drug-susceptible and drug-resistant TB.
Targeting Other Functions in Chromosomal Replication
During replication, Okazaki fragments are generated which must be joined together by the bacterial NAD+-dependent DNA ligase. The enzyme is therefore essential, making it a highly attractive target for drug development. Inhibition of purified M. tuberculosis LigA has been reported numerous times (Gong et al., 2004; Srivastava et al., 2005a,b); however, very few compounds have been shown to exhibit whole-cell, micromolar-range activity against M. tuberculosis. Following the high-throughput, in silico screening of potential LigA inhibitors, Korycka-Machala et al. identified pinafide and mitonafide as attractive inhibitors of Mtb growth in vitro (Korycka-Machala et al., 2017). Both compounds exhibited an MIC of 25 μM in 7H9 liquid media and half maximal inhibitory concentration (IC50) of 50 μM. In addition, the in vitro analysis of LigA inhibition suggested that the two compounds failed to inhibit T4 ATP-dependent DNA ligase effectively and, therefore, had specificity for NAD+-dependent DNA ligase, which is not utilized by eukaryotes. Although preliminary, these results hold great promise for the development of similar compounds or analogs capable of inhibiting Mtb growth at low-micromolar concentrations in vivo through the inhibition of LigA.
Proof-of-concept Targets from Other Bacterial Systems
Sporulation Protein SirA and the SocB Toxin
Further insight into the inhibition of essential DNA replicative pathways can be obtained from natural phenomena which characterize normal bacterial physiology. Both DnaA and DnaN have been shown to be inhibited by bacterial-derived molecules, including a sporulation protein in B. subtilis and a toxin-antitoxin (TA) system found in Caulobacter crescentus. In the first example, the interaction of the sporulation protein SirA with domain I of DnaA prevents the replication initiator protein from binding to the origin of replication during the start of sporulation of B. subtilis, effectively inhibiting DNA replication initiation (Rahn-Lee et al., 2011). As noted above, DnaA is essential for initiation of DNA replication, with domain I of DnaA being required for interactions between DnaA monomers and other proteins, such as the essential dnaB-encoded helicase (Seitz et al., 2000; Abe et al., 2007). This domain of DnaA is thus an attractive target for therapeutic intervention.
In a further example, the toxin component of the atypical SocAB TA system in C. crescentus was found to inhibit DNA elongation through an interaction with the dnaN-encoded β sliding clamp (Aakre et al., 2013). Notably, mutations conferring resistance to SocB mapped to the hydrophobic Pol III-binding domain of DnaN, indicating a similar binding site to the previously mentioned antibiotic, griselimycin. The mechanism of resistance is different, though, and so cross-resistance is unlikely. In summary, these examples further validate the inhibition of novel DNA replication components and can potentially be used as a basis for the rational design of synthetic inhibitors against M. tuberculosis DnaA and DnaN in the future.
Other Mycobacterial DNA Replication and Repair Functions
In addition to the specialist DNA replication proteins detailed above (and see Figure 1), M. tuberculosis encodes a number of other DNA metabolic functions which are essential for cellular viability. For some of these, the potential to yield novel drugs and drug targets is compelling, and includes pathways and enzymes required for de novo synthesis, salvage, and recycling of dNTPs for incorporation in newly synthesized DNA, as well as during repair. A detailed discussion is beyond the scope of this review, however some examples include the mycobacterial ribonucleotide reductase (Nurbo et al., 2013; Bueno et al., 2014; Karlsson et al., 2015), thymidylate synthase (Kogler et al., 2011; Fivian-Hughes et al., 2012; Singh et al., 2015), and inosine monophosphate dehydrogenase (Park et al., 2017; Singh et al., 2017) enzymes. Notably, the roles of these and other related proteins in maintaining nucleotide homeostasis within the mycobacterial cell suggests the potential to inhibit replication and repair functions at multiple stages and, moreover, raises the possibility of disrupting indirectly other macromolecular pathways such as RNA transcription and cell wall biosynthesis owing to their convergence on many common metabolic precursors and intermediates (Singh et al., 2015, 2017).
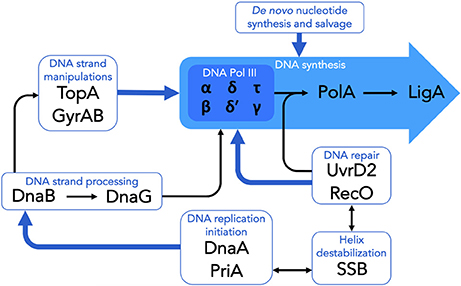
Figure 1. Essential components of DNA replication and repair in M. tuberculosis. The schematic highlights the essential DNA replication and repair functions which are targeted by existing clinical or experimental drugs, as well as those which have been identified as potential targets for the development of novel antimycobacterial compounds. See text for details.
A deeper analysis of other specialist DNA replication and repair functions reveals several more candidates such as Pol I, RecO, and UvrD2, all of which either are essential or contain essential domains (Table 3; DeJesus et al., 2017). For some of these, their perceived potential as novel drug targets requires further validation. PolA/Pol I is a DNA-dependent polymerase that possesses bi-directional exonuclease activity. Previous work identified the 5′-3′ exonuclease domain of PolA as essential to the growth of M. tuberculosis; furthermore, while the polymerase domain was shown to be dispensable in M. smegmatis, deficiency in this function was associated with DNA damage hypersensitivity (Gordhan et al., 1996; Mizrahi and Huberts, 1996). Importantly, the exonuclease domain was determined incapable of discriminating against dideoxynucleotide 5′-triphosphates, and could be inhibited by chain-terminating nucleotide analogs during DNA synthesis, suggesting the druggability of the target (Mizrahi and Huberts, 1996). Moreover, the involvement of Pol I in DNA damage tolerance has identified this protein as potential target for antimutagenesis agents (Plocinska et al., 2017), as explored further below.
Mycobacterium tuberculosis Rv2362c exhibits 28% identity with S. typhimurium RecO (Mizrahi and Andersen, 1998), a protein required in DNA repair and RecF-dependent recombination and which functions to assemble and disassemble RecA filaments at single-stranded gaps. Recently, it was reported that Rv2362c contains a domain that is essential for M. tuberculosis growth (DeJesus et al., 2017), indicating potential of targeting the under-investigated protein with therapeutic compounds in the future. Similarly, UvrD2—a component of nucleotide excision repair and methyl-directed mismatch repair pathways—is another mycobacterial protein containing an essential DNA-dependent ATPase activity implicated in DNA translocation and protein displacement, as well as a dispensable helicase activity (Kazarian et al., 2010; Williams et al., 2011). Although no compounds have been reported to inhibit either of these proteins, their implication in essential replication functions appears to warrant further investigation.
Targeting Mutagenesis
The notion of developing “anti-evolution” drugs to prevent the function of mutagenic repair pathways in M. tuberculosis has been discussed previously (Warner, 2010). This strategy seems likely to be especially appropriate for M. tuberculosis as adaptive evolution of this organism depends solely on chromosomal rearrangements and point mutations, and all drug resistance arises through spontaneous mutations in target or complementary genes (Galagan, 2014). These factors suggest that inducible mutagenic mechanisms—such as the imuA'-imuB/dnaE2 mycobacterial mutasome (Warner et al., 2010)—might drive the evolution of M. tuberculosis within its host. The limited distribution of ImuA′ and ImuB among sequenced bacterial genomes therefore identifies the mutasome as a compelling target for limiting drug resistance. In some ways, this strategy is analogous to targeting virulence factors (Liu et al., 2008) and assumes that the selective pressure to mutate to antibiotic resistance is not as great where the pathway is essential for pathogenesis but not survival (Clatworthy et al., 2007). Moreover, inhibiting mutagenesis should be effective in immune compromised individuals, and might facilitate clinical trials by identifying compounds that could supplement existing regimens without compromising efficacy.
To this end, several approaches appear worth pursuing: in the first, recent evidence suggests that selective inhibition of DnaE2 by anilinouracils might be possible (Jadaun et al., 2015), provided structural data are available to enable rational identification of compounds which target this alternative α subunit, and not the replicative DnaE1, the structure of which was recently elucidated (Banos-Mateos et al., 2017). For this reason, nargenicin may not be appropriate, though dual targeting of both DnaE proteins might nevertheless represent a profitable strategy. Secondly, targeting the PHP domain exonuclease of DnaE1 provides another attractive option as inactivation of this domain was found to render M. smegmatis hypersensitive to the chain-terminating adenosine analog, ara-A (Rock et al., 2015). The recent determination of the structure of the PHP domain, which lacks a human homolog, has created an opportunity for structure-guided design of inhibitors against this exonuclease (Banos-Mateos et al., 2017). In the third approach, the identification of griselimycin supports the potential for developing novel protein-protein interaction inhibitors designed to disrupt mutasome function. Further work is underway in our laboratory to elucidate the molecular interactions which are essential to DnaE2-dependent mutagenesis, with genetic evidence indicating that preventing ImuB from functioning as “hub” protein might collapse this pathway (Warner et al., 2010). In conclusion, the possibility of targeting replication and repair mechanisms implicated in the evolution of drug resistance seems a challenge worth tackling: if successful, it is proposed that these compounds might be co-administered with other agents in novel combination therapies designed to protect existing antibiotics.
Author Contribtions
MAR produced the tables and figure, contributed to the main text, and edited the manuscript; DFW co-developed the outline, wrote the main text, and edited the manuscript; and VM developed the outline, wrote the introduction and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Work in our laboratory on mycobacterial DNA replication and repair is funded by grants from the US National Institute of Child Health and Human Development (NICHD) U01HD085531-02 (to DFW); a Senior International Research Scholars grant from the HHMI (grant no. 55007649, to VM); the Department of Science and Technology (DST) and National Research Foundation (NRF) of South Africa (to VM; and grant no. 104683 to MAR); and the South African Medical Research Council (to VM). We gratefully acknowledge the financial assistance of the Research Council of Norway (INTPART, AMR-PART) for work on antimicrobial resistance in M. tuberculosis (to DFW).
References
Aakre, C. D., Phung, T. N., Huang, D., and Laub, M. T. (2013). A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol. Cell 52, 617–628. doi: 10.1016/j.molcel.2013.10.014
Abe, Y., Jo, T., Matsuda, Y., Matsunaga, C., Katayama, T., and Ueda, T. (2007). Structure and function of DnaA N-terminal domains: specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. J. Biol. Chem. 282, 17816–17827. doi: 10.1074/jbc.M701841200
Aboul-Fadl, T., Abdel-Aziz, H. A., Abdel-Hamid, M. K., Elsaman, T., Thanassi, J., and Pucci, M. J. (2011). Schiff bases of indoline-2, 3-dione: potential novel inhibitors of Mycobacterium tuberculosis (Mtb) DNA gyrase. Molecules 16, 7864–7879. doi: 10.3390/molecules16097864
Abrahams, G. L., Kumar, A., Savvi, S., Hung, A. W., Wen, S., Abell, C., et al. (2012). Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 19, 844–854. doi: 10.1016/j.chembiol.2012.05.020
Agarwal, A., Louise-May, S., Thanassi, J. A., Podos, S. D., Cheng, J., Thoma, C., et al. (2007). Small molecule inhibitors of E. coli primase, a novel bacterial target. Bioorg. Med. Chem. Lett. 17, 2807–2810. doi: 10.1016/j.bmcl.2007.02.056
Aggarwal, A., Parai, M. K., Shetty, N., Wallis, D., Woolhiser, L., Hastings, C., et al. (2017). Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell. 170, 249–259.e25. doi: 10.1016/j.cell.2017.06.025
Aiello, D., Barnes, M. H., Biswas, E. E., Biswas, S. B., Gu, S., Williams, J. D., et al. (2009). Discovery, characterization and comparison of inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicases. Bioorg. Med. Chem. 17, 4466–4476. doi: 10.1016/j.bmc.2009.05.014
Aldridge, B. B., Keren, I., and Fortune, S. M. (2014). The spectrum of drug susceptibility in mycobacteria. Microbiol. Spectr. 2. doi: 10.1128/microbiolspec.MGM2-0031-2013
Alvirez-Freites, E. J., Carter, J. L., and Cynamon, M. H. (2002). In vitro and in vivo activities of gatifloxacin against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 46, 1022–1025. doi: 10.1128/AAC.46.4.1022-1025.2002
Angehrn, P., Buchmann, S., Funk, C., Goetschi, E., Gmuender, H., Hebeisen, P., et al. (2004). New antibacterial agents derived from the DNA gyrase inhibitor cyclothialidine. J. Med. Chem. 47, 1487–1513. doi: 10.1021/jm0310232
Angehrn, P., Goetschi, E., Gmuender, H., Hebeisen, P., Hennig, M., Kuhn, B., et al. (2011). A new DNA gyrase inhibitor subclass of the cyclothialidine family based on a bicyclic dilactam-lactone scaffold. Synthesis and antibacterial properties. J. Med. Chem. 54, 2207–2224. doi: 10.1021/jm1014023
Anishetty, S., Pulimi, M., and Pennathur, G. (2005). Potential drug targets in Mycobacterium tuberculosis through metabolic pathway analysis. Comput. Biol. Chem. 29, 368–378. doi: 10.1016/j.compbiolchem.2005.07.001
Anthony, R. M., den Hertog, A., Mansjo, M., and Werngren, J. (2016). New insights into the mechanism of action of pyrazinamide, implications for susceptibility testing, and future regimens. Int. J. Mycobacteriol. 5 (Suppl. 1), S71–S72. doi: 10.1016/j.ijmyco.2016.08.009
Aubry, A., Pan, X. S., Fisher, L. M., Jarlier, V., and Cambau, E. (2004). Mycobacterium tuberculosis DNA gyrase: interaction with quinolones and correlation with antimycobacterial drug activity. Antimicrob. Agents Chemother. 48, 1281–1288. doi: 10.1128/AAC.48.4.1281-1288.2004
Awasthi, D., and Freundlich, J. S. (2017). Antimycobacterial metabolism: illuminating mycobacterium tuberculosis biology and drug discovery. Trends Microbiol. 25, 756–767. doi: 10.1016/j.tim.2017.05.007
Baer, C. E., Rubin, E. J., and Sassetti, C. M. (2015). New insights into TB physiology suggest untapped therapeutic opportunities. Immunol. Rev. 264, 327–343. doi: 10.1111/imr.12267
Banos-Mateos, S., van Roon, A. M., Lang, U. F., Maslen, S. L., Skehel, J. M., and Lamers, M. H. (2017). High-fidelity DNA replication in Mycobacterium tuberculosis relies on a trinuclear zinc center. Nat. Commun. 8:855. doi: 10.1038/s41467-017-00886-w
Barreiro, C., and Ullán, R. V. (2016). “DNA-synthesizing enzymes as antibacterial targets,” in New Weapons to Control Bacterial Growth, eds T. Villa and M. Vinas (Cham: Springer), 95–114. doi: 10.1007/978-3-319-28368-5_5
Basarab, G. S., Brassil, P., Doig, P., Galullo, V., Haimes, H. B., Kern, G., et al. (2014). Novel DNA gyrase inhibiting spiropyrimidinetriones with a benzisoxazole scaffold: SAR and in vivo characterization. J. Med. Chem. 57, 9078–9095. doi: 10.1021/jm501174m
Basarab, G. S., Doig, P., Galullo, V., Kern, G., Kimzey, A., Kutschke, A., et al. (2015). Discovery of novel DNA gyrase inhibiting spiropyrimidinetriones: benzisoxazole fusion with N-Linked oxazolidinone substituents leading to a clinical candidate (ETX0914). J. Med. Chem. 58, 6264–6282. doi: 10.1021/acs.jmedchem.5b00863
Baughn, A. D., and Rhee, K. Y. (2014). Metabolomics of central carbon metabolism in Mycobacterium tuberculosis. Microbiol. Spectr. 2. doi: 10.1128/microbiolspec.MGM2-0026-2013
Bax, B. D., Chan, P. F., Eggleston, D. S., Fosberry, A., Gentry, D. R., Gorrec, F., et al. (2010). Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466, 935–940. doi: 10.1038/nature09197
Beattie, T. R., and Reyes-Lamothe, R. (2015). A Replisome's journey through the bacterial chromosome. Front. Microbiol. 6:562. doi: 10.3389/fmicb.2015.00562
Beattie, T. R., Kapadia, N., Nicolas, E., Uphoff, S., Wollman, A. J., Leake, M. C., et al. (2017). Frequent exchange of the DNA polymerase during bacterial chromosome replication. Elife 6:e21763. doi: 10.7554/eLife.21763
Black, M. T., Stachyra, T., Platel, D., Girard, A. M., Claudon, M., Bruneau, J. M., et al. (2008). Mechanism of action of the antibiotic NXL101, a novel nonfluoroquinolone inhibitor of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 52, 3339–3349. doi: 10.1128/AAC.00496-08
Blanco, D., Perez-Herran, E., Cacho, M., Ballell, L., Castro, J., Gonzalez Del Rio, R., et al. (2015). Mycobacterium tuberculosis gyrase inhibitors as a new class of antitubercular drugs. Antimicrob. Agents Chemother. 59, 1868–1875. doi: 10.1128/AAC.03913-14
Blondiaux, N., Moune, M., Desroses, M., Frita, R., Flipo, M., Mathys, V., et al. (2017). Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science 355, 1206–1211. doi: 10.1126/science.aag1006
Boshoff, H. I., Myers, T. G., Copp, B. R., McNeil, M. R., Wilson, M. A., and Barry, C. E. III. (2004). The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279, 40174–40184. doi: 10.1074/jbc.M406796200
Boshoff, H. I. M., Reed, M. B., Barry, C. E. III., and Mizrahi, V. (2003). DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 113, 183–193. doi: 10.1016/S0092-8674(03)00270-8
Brauner, A., Fridman, O., Gefen, O., and Balaban, N. Q. (2016). Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 14, 320–330. doi: 10.1038/nrmicro.2016.34
Brotz-Oesterhelt, H., Knezevic, I., Bartel, S., Lampe, T., Warnecke-Eberz, U., Ziegelbauer, K., et al. (2003). Specific and potent inhibition of NAD+-dependent DNA ligase by pyridochromanones. J. Biol. Chem. 278, 39435–39442. doi: 10.1074/jbc.M306479200
Bueno, R. V., Braga, R. C., Segretti, N. D., Ferreira, E. I., Trossini, G. H., and Andrade, C. H. (2014). New tuberculostatic agents targeting nucleic acid biosynthesis: drug design using QSAR approaches. Curr. Pharm. Des. 20, 4474–4485.
Butler, M. M., Lamarr, W. A., Foster, K. A., Barnes, M. H., Skow, D. J., Lyden, P. T., et al. (2007). Antibacterial activity and mechanism of action of a novel anilinouracil-fluoroquinolone hybrid compound. Antimicrob. Agents Chemother. 51, 119–127. doi: 10.1128/AAC.01311-05
Buurman, E. T., Laganas, V. A., Liu, C. F., and Manchester, J. I. (2012). Antimicrobial Activity of Adenine-Based Inhibitors of NAD+-Dependent DNA Ligase. ACS Med. Chem. Lett. 3, 663–667. doi: 10.1021/ml300169x
Chandran, M., Renuka, J., Sridevi, J. P., Pedgaonkar, G. S., Asmitha, V., Yogeeswari, P., et al. (2015). Benzothiazinone-piperazine derivatives as efficient Mycobacterium tuberculosis DNA gyrase inhibitors. Int. J. Mycobacteriol 4, 104–115. doi: 10.1016/j.ijmyco.2015.02.002
Chen, C. C., and Huang, C. Y. (2011). Inhibition of Klebsiella pneumoniae DnaB helicase by the flavonol galangin. Protein J. 30, 59–65. doi: 10.1007/s10930-010-9302-0.
Chhabra, G., Dixit, A., and Garg, L. C. (2011). DNA polymerase III alpha subunit from Mycobacterium tuberculosis H37Rv: homology modeling and molecular docking of its inhibitor. Bioinformation 6, 69–73. doi: 10.6026/97320630006069
Chopra, S., Matsuyama, K., Tran, T., Malerich, J. P., Wan, B., Franzblau, S. G., et al. (2012). Evaluation of gyrase B as a drug target in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 67, 415–421. doi: 10.1093/jac/dkr449
Chu, M., Mierzwa, R., Xu, L., He, L., Terracciano, J., Patel, M., et al. (2003). Isolation and structure elucidation of Sch 642305, a novel bacterial DNA primase inhibitor produced by Penicillium verrucosum. J. Nat. Prod. 66, 1527–1530. doi: 10.1021/np0302302
Ciarrocchi, G., MacPhee, D. G., Deady, L. W., and Tilley, L. (1999). Specific inhibition of the eubacterial DNA ligase by arylamino compounds. Antimicrob. Agents Chemother. 43, 2766–2772.
Clatworthy, A. E., Pierson, E., and Hung, D. T. (2007). Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3, 541–548. doi: 10.1038/nchembio.2007.24
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Coscolla, M., and Gagneux, S. (2014). Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 26, 431–444. doi: 10.1016/j.smim.2014.09.012
Dartois, V. (2014). The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat. Rev. Microbiol. 12, 159–167. doi: 10.1038/nrmicro3200
Dartois, V., and Barry, C. E. III. (2013). A medicinal chemists' guide to the unique difficulties of lead optimization for tuberculosis. Bioorg. Med. Chem. Lett. 23, 4741–4750. doi: 10.1016/j.bmcl.2013.07.006
DeJesus, M. A., Gerrick, E. R., Xu, W., Park, S. W., Long, J. E., Boutte, C. C., et al. (2017). Comprehensive essentiality analysis of the Mycobacterium tuberculosis Genome via saturating transposon mutagenesis. MBio 8:e02133-16. doi: 10.1128/mBio.02133-16
Dheda, K., Gumbo, T., Gandhi, N. R., Murray, M., Theron, G., Udwadia, Z., et al. (2014). Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2, 321–338. doi: 10.1016/S2213-2600(14)70031-1.
Dheda, K., Gumbo, T., Maartens, G., Dooley, K. E., McNerney, R., Murray, M., et al. (2017). The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir. Med. 5, 291–360. doi: 10.1016/S2213-2600(17)30079-6
Dinakaran, M., Senthilkumar, P., Yogeeswari, P., China, A., Nagaraja, V., and Sriram, D. (2008). Novel ofloxacin derivatives: synthesis, antimycobacterial and toxicological evaluation. Bioorg. Med. Chem. Lett. 18, 1229–1236. doi: 10.1016/j.bmcl.2007.11.110
Disratthakit, A., and Doi, N. (2010). In vitro activities of DC-159a, a novel fluoroquinolone, against Mycobacterium species. Antimicrob. Agents Chemother. 54, 2684–2686. doi: 10.1128/AAC.01545-09
Ditse, Z., Lamers, M. H., and Warner, D. F. (2017). DNA Replication in Mycobacterium tuberculosis. Microbiol. Spectr. 5. doi: 10.1128/microbiolspec.TBTB2-0027-2016
Dwyer, D. J., Collins, J. J., and Walker, G. C. (2015). Unraveling the physiological complexities of antibiotic lethality. Annu. Rev. Pharmacol. Toxicol. 55, 313–332. doi: 10.1146/annurev-pharmtox-010814-124712
Edfeldt, F. N., Folmer, R. H., and Breeze, A. L. (2011). Fragment screening to predict druggability (ligandability) and lead discovery success. Drug Discov. Today 16, 284–287. doi: 10.1016/j.drudis.2011.02.002
Esposito, M., Szadocka, S., Degiacomi, G., Orena, B. S., Mori, G., Piano, V., et al. (2017). A phenotypic based target screening approach delivers new antitubercular CTP synthetase inhibitors. ACS Infect. Dis. 3, 428–437. doi: 10.1021/acsinfecdis.7b00006
Evangelopoulos, D., and McHugh, T. D. (2015). Improving the tuberculosis drug development pipeline. Chem. Biol. Drug Des. 86, 951–960. doi: 10.1111/cbdd.12549
Fivian-Hughes, A. S., Houghton, J., and Davis, E. O. (2012). Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiol. Sgm 158, 1388–1388. doi: 10.1099/mic.0.X00002-0
Flatman, R. H., Howells, A. J., Heide, L., Fiedler, H. P., and Maxwell, A. (2005). Simocyclinone D8, an inhibitor of DNA gyrase with a novel mode of action. Antimicrob. Agents Chemother. 49, 1093–1100. doi: 10.1128/AAC.49.3.1093-1100.2005
Gajadeera, C., Willby, M. J., Green, K. D., Shaul, P., Fridman, M., Garneau-Tsodikova, S., et al. (2015). Antimycobacterial activity of DNA intercalator inhibitors of Mycobacterium tuberculosis primase DnaG. J. Antibiot. (Tokyo) 68, 153–157. doi: 10.1038/ja.2014.131
Galagan, J. E. (2014). Genomic insights into tuberculosis. Nat. Rev. Genet. 15, 307–320. doi: 10.1038/nrg3664
Georgescu, R. E., Yurieva, O., Kim, S. S., Kuriyan, J., Kong, X. P., and O'Donnell, M. (2008). Structure of a small-molecule inhibitor of a DNA polymerase sliding clamp. Proc. Natl. Acad. Sci. U.S.A. 105, 11116–11121. doi: 10.1073/pnas.0804754105
Gillespie, S. H., Basu, S., Dickens, A. L., O'Sullivan, D. M., and McHugh, T. D. (2005). Effect of subinhibitory concentrations of ciprofloxacin on Mycobacterium fortuitum mutation rates. J. Antimicrob. Chemother. 56, 344–348. doi: 10.1093/jac/dki191
Gillespie, S. H., Crook, A. M., McHugh, T. D., Mendel, C. M., Meredith, S. K., Murray, S. R., et al. (2014). Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N. Engl. J. Med. 371, 1577–1587. doi: 10.1056/NEJMoa1407426
Glanzer, J. G., Endres, J. L., Byrne, B. M., Liu, S., Bayles, K. W., and Oakley, G. G. (2016). Identification of inhibitors for single-stranded DNA-binding proteins in eubacteria. J. Antimicrob. Chemother. 71, 3432–3440. doi: 10.1093/jac/dkw340
Godbole, A. A., Ahmed, W., Bhat, R. S., Bradley, E. K., Ekins, S., and Nagaraja, V. (2014). Inhibition of Mycobacterium tuberculosis topoisomerase I by m-AMSA, a eukaryotic type II topoisomerase poison. Biochem. Biophys. Res. Commun. 446, 916–920. doi: 10.1016/j.bbrc.2014.03.029
Godbole, A. A., Ahmed, W., Bhat, R. S., Bradley, E. K., Ekins, S., and Nagaraja, V. (2015). Targeting Mycobacterium tuberculosis topoisomerase I by small-molecule inhibitors. Antimicrob. Agents Chemother. 59, 1549–1557. doi: 10.1128/AAC.04516-14
Gold, B., and Nathan, C. (2017). Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiol. Spectr. 5. doi: 10.1128/microbiolspec.TBTB2-0031-2016
Gong, C., Martins, A., Bongiorno, P., Glickman, M., and Shuman, S. (2004). Biochemical and genetic analysis of the four DNA ligases of mycobacteria. J. Biol. Chem. 279, 20594–20606. doi: 10.1074/jbc.M401841200
Gordhan, B. G., Andersen, S. J., De Meyer, A. R., and Mizrahi, V. (1996). Construction by homologous recombination and phenotypic characterization of a DNA polymerase domain polA mutant of Mycobacterium smegmatis. Gene 178, 125–130.
Griep, M. A., Blood, S., Larson, M. A., Koepsell, S. A., and Hinrichs, S. H. (2007). Myricetin inhibits Escherichia coli DnaB helicase but not primase. Bioorg. Med. Chem. 15, 7203–7208. doi: 10.1016/j.bmc.2007.07.057
Griffin, J. E., Gawronski, J. D., Dejesus, M. A., Ioerger, T. R., Akerley, B. J., and Sassetti, C. M. (2011). High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7:e1002251. doi: 10.1371/journal.ppat.1002251
Grillot, A. L., Le Tiran, A., Shannon, D., Krueger, E., Liao, Y., O'Dowd, H., et al. (2014). Second-generation antibacterial benzimidazole ureas: discovery of a preclinical candidate with reduced metabolic liability. J. Med. Chem. 57, 8792–8816. doi: 10.1021/jm500563g
Gu, S., Li, W., Zhang, H., Fleming, J., Yang, W., Wang, S., et al. (2016). The b2 clamp in the Mycobacterium tuberculosis DNA polymerase III ab2e replicase promotes polymerization and reduces exonuclease activity. Sci. Rep. 6:18418. doi: 10.1038/srep18418
Gu, W., Wang, T., Maltais, F., Ledford, B., Kennedy, J., Wei, Y., et al. (2012). Design, synthesis and biological evaluation of potent NAD+-dependent DNA ligase inhibitors as potential antibacterial agents. Part I: aminoalkoxypyrimidine carboxamides. Bioorg. Med. Chem. Lett. 22, 3693–3698. doi: 10.1016/j.bmcl.2012.04.037
Guiles, J., Sun, X., Critchley, I. A., Ochsner, U., Tregay, M., Stone, K., et al. (2009). Quinazolin-2-ylamino-quinazolin-4-ols as novel non-nucleoside inhibitors of bacterial DNA polymerase III. Bioorg. Med. Chem. Lett. 19, 800–802. doi: 10.1016/j.bmcl.2008.12.038
Hameed, P. S., Solapure, S., Mukherjee, K., Nandi, V., Waterson, D., Shandil, R., et al. (2014). Optimization of pyrrolamides as mycobacterial GyrB ATPase inhibitors: structure-activity relationship and in vivo efficacy in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 58, 61–70. doi: 10.1128/AAC.01751-13
Hegde, V. R., Pu, H., Patel, M., Black, T., Soriano, A., Zhao, W., et al. (2004). Two new bacterial DNA primase inhibitors from the plant Polygonum cuspidatum. Bioorg. Med. Chem. Lett. 14, 2275–2277. doi: 10.1016/j.bmcl.2004.02.006
Herrmann, J., Rybniker, J., and Muller, R. (2017). Novel and revisited approaches in antituberculosis drug discovery. Curr. Opin. Biotechnol. 48, 94–101. doi: 10.1016/j.copbio.2017.03.023
Hopkins, A. L., and Groom, C. R. (2002). The druggable genome. Nat. Rev. Drug Discov. 1, 727–730. doi: 10.1038/nrd892
Hou, Z., Zhou, Y., Li, J., Zhang, X., Shi, X., Xue, X., et al. (2015). Selective in vivo and in vitro activities of 3,3′-4-nitrobenzylidene-bis-4-hydroxycoumarin against methicillin-resistant Staphylococcus aureus by inhibition of DNA polymerase III. Sci. Rep. 5:13637. doi: 10.1038/srep13637
Howard, S., Amin, N., Benowitz, A. B., Chiarparin, E., Cui, H., Deng, X., et al. (2013). Fragment-based discovery of 6-azaindazoles as inhibitors of bacterial DNA ligase. ACS Med. Chem. Lett. 4, 1208–1212. doi: 10.1021/ml4003277
Huang, Y. H., Huang, C. C., Chen, C. C., Yang, K. J., and Huang, C. Y. (2015). Inhibition of Staphylococcus aureus PriA Helicase by Flavonol Kaempferol. Protein J. 34, 169–172. doi: 10.1007/s10930-015-9609-y
Hutchings, K. M., Tran, T. P., Ellsworth, E. L., Watson, B. M., Sanchez, J. P., Showalter, H. D., et al. (2008). Synthesis and antibacterial activity of the C-7 side chain of 3-aminoquinazolinediones. Bioorg. Med. Chem. Lett. 18, 5087–5090. doi: 10.1016/j.bmcl.2008.07.117
Jadaun, A., Sudhakar, D. R., Subbarao, N., and Dixit, A. (2015). In silico screening for novel inhibitors of DNA polymerase III alpha subunit of Mycobacterium tuberculosis (MtbDnaE2, H37Rv). PLoS ONE 10:e0119760. doi: 10.1371/journal.pone.0119760
Jeankumar, V. U., Kotagiri, S., Janupally, R., Suryadevara, P., Sridevi, J. P., Medishetti, R., et al. (2015a). Exploring the gyrase ATPase domain for tailoring newer anti-tubercular drugs: hit to lead optimization of a novel class of thiazole inhibitors. Bioorg. Med. Chem. 23, 588–601. doi: 10.1016/j.bmc.2014.12.001
Jeankumar, V. U., Renuka, J., Pulla, V. K., Soni, V., Sridevi, J. P., Suryadevara, P., et al. (2014). Development of novel N-linked aminopiperidine-based mycobacterial DNA gyrase B inhibitors: scaffold hopping from known antibacterial leads. Int. J. Antimicrob. Agents 43, 269–278. doi: 10.1016/j.ijantimicag.2013.12.006.
Jeankumar, V. U., Renuka, J., Santosh, P., Soni, V., Sridevi, J. P., Suryadevara, P., et al. (2013). Thiazole-aminopiperidine hybrid analogues: design and synthesis of novel Mycobacterium tuberculosis GyrB inhibitors. Eur. J. Med. Chem. 70, 143–153. doi: 10.1016/j.ejmech.2013.09.025
Jeankumar, V. U., Reshma, R. S., Janupally, R., Saxena, S., Sridevi, J. P., Medapi, B., et al. (2015b). Enabling the (3 + 2) cycloaddition reaction in assembling newer anti-tubercular lead acting through the inhibition of the gyrase ATPase domain: lead optimization and structure activity profiling. Org. Biomol. Chem. 13, 2423–2431. doi: 10.1039/c4ob02049a
Jeankumar, V. U., Reshma, R. S., Vats, R., Janupally, R., Saxena, S., Yogeeswari, P., et al. (2016a). Engineering another class of anti-tubercular lead: Hit to lead optimization of an intriguing class of gyrase ATPase inhibitors. Eur. J. Med. Chem. 122, 216–231. doi: 10.1016/j.ejmech.2016.06.042
Jeankumar, V. U., Saxena, S., Vats, R., Reshma, R. S., Janupally, R., Kulkarni, P., et al. (2016b). Structure-guided discovery of antitubercular agents that target the gyrase ATPase domain. Chem. Med. Chem. 11, 539–548. doi: 10.1002/cmdc.201500556
Jindani, A., Harrison, T. S., Nunn, A. J., Phillips, P. P., Churchyard, G. J., Charalambous, S., et al. (2014). High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N. Engl. J. Med. 371, 1599–1608. doi: 10.1056/NEJMoa1314210
Kale, M. G., Raichurkar, A., Shahul Hameed, P., Waterson, D., McKinney, D., Manjunatha, M. R., et al. (2013). Thiazolopyridine ureas as novel antitubercular agents acting through inhibition of DNA Gyrase B. J. Med. Chem. 56, 8834–8848. doi: 10.1021/jm401268f
Kale, R. R., Kale, M. G., Waterson, D., Raichurkar, A., Hameed, S. P., Manjunatha, M. R., et al. (2014). Thiazolopyridone ureas as DNA gyrase B inhibitors: optimization of antitubercular activity and efficacy. Bioorg. Med. Chem. Lett. 24, 870–879. doi: 10.1016/j.bmcl.2013.12.080
Karkare, S., Chung, T. T., Collin, F., Mitchenall, L. A., McKay, A. R., Greive, S. J., et al. (2013). The naphthoquinone diospyrin is an inhibitor of DNA gyrase with a novel mechanism of action. J. Biol. Chem. 288, 5149–5156. doi: 10.1074/jbc.M112.419069
Karlsson, C., Blom, M., Johansson, M., Jansson, A. M., Scifo, E., Karlen, A., et al. (2015). Phototriggerable peptidomimetics for the inhibition of Mycobacterium tuberculosis ribonucleotide reductase by targeting protein-protein binding. Org. Biomol. Chem. 13, 2612–2621. doi: 10.1039/c4ob01926a.
Kazarian, K., Cassani, C., and Rizzi, M. (2010). Expression, purification and characterization of UvrD2 helicase from Mycobacterium tuberculosis. Protein Expr. Purif. 69, 215–218. doi: 10.1016/j.pep.2009.09.006
Kester, J. C., and Fortune, S. M. (2014). Persisters and beyond: mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 49, 91–101. doi: 10.3109/10409238.2013.869543
Kinnings, S. L., Xie, L., Fung, K. H., Jackson, R. M., Xie, L., and Bourne, P. E. (2010). The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLoS Comput. Biol. 6:e1000976. doi: 10.1371/journal.pcbi.1000976
Kling, A., Lukat, P., Almeida, D. V., Bauer, A., Fontaine, E., Sordello, S., et al. (2015). Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 348, 1106–1112. doi: 10.1126/science.aaa4690
Koch, A., Mizrahi, V., and Warner, D. F. (2014). The impact of drug resistance on Mycobacterium tuberculosis physiology: what can we learn from rifampicin? Emerg. Microbes Infect. 3:e17. doi: 10.1038/emi.2014.17
Kogler, M., Vanderhoydonck, B., De Jonghe, S., Rozenski, J., Van Belle, K., Herman, J., et al. (2011). Synthesis and evaluation of 5-substituted 2'-deoxyuridine monophosphate analogues as inhibitors of flavin-dependent thymidylate synthase in Mycobacterium tuberculosis. J. Med. Chem. 54, 4847–4862. doi: 10.1021/jm2004688
Korycka-Machala, M., Nowosielski, M., Kuron, A., Rykowski, S., Olejniczak, A., Hoffmann, M., et al. (2017). Naphthalimides selectively inhibit the activity of bacterial, replicative DNA Ligases and Display Bactericidal Effects against Tubercle Bacilli. Molecules 22:154. doi: 10.3390/molecules22010154
Kuron, A., Korycka-Machala, M., Brzostek, A., Nowosielski, M., Doherty, A., Dziadek, B., et al. (2014). Evaluation of DNA primase DnaG as a potential target for antibiotics. Antimicrob. Agents Chemother. 58, 1699–1706. doi: 10.1128/AAC.01721-13
Lange, S. S., Takata, K., and Wood, R. D. (2011). DNA polymerases and cancer. Nat. Rev. Cancer 11, 96–110. doi: 10.1038/nrc2998
Lewis, J. S., Spenkelink, L. M., Jergic, S., Wood, E. A., Monachino, E., Horan, N. P., et al. (2017). Single-molecule visualization of fast polymerase turnover in the bacterial replisome. Elife 6:e23932. doi: 10.7554/eLife.23932
Lewis, R. J., Singh, O. M., Smith, C. V., Skarzynski, T., Maxwell, A., Wonacott, A. J., et al. (1996). The nature of inhibition of DNA gyrase by the coumarins and the cyclothialidines revealed by X-ray crystallography. EMBO J. 15, 1412–1420.
Li, B., Pai, R., Aiello, D., Di, M., Barnes, M. H., Peet, N. P., et al. (2013). Optimization of a novel potent and selective bacterial DNA helicase inhibitor scaffold from a high throughput screening hit. Bioorg. Med. Chem. Lett. 23, 3481–3486. doi: 10.1016/j.bmcl.2013.04.055
Li, B., Pai, R., Di, M., Aiello, D., Barnes, M. H., Butler, M. M., et al. (2012). Coumarin-based inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicase: chemical optimization, biological evaluation, and antibacterial activities. J. Med. Chem. 55, 10896–10908. doi: 10.1021/jm300922h
Lin, H. H., and Huang, C. Y. (2012). Characterization of flavonol inhibition of DnaB helicase: real-time monitoring, structural modeling, and proposed mechanism. J. Biomed. Biotechnol. 2012:735368. doi: 10.1155/2012/735368
Liu, C. I., Liu, G. Y., Song, Y., Yin, F., Hensler, M. E., Jeng, W. Y., et al. (2008). A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319, 1391–1394. doi: 10.1126/science.1153018
Locher, C. P., Jones, S. M., Hanzelka, B. L., Perola, E., Shoen, C. M., Cynamon, M. H., et al. (2015). A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob. Agents Chemother. 59, 1455–1465. doi: 10.1128/AAC.04347-14
Long, J. E., DeJesus, M., Ward, D., Baker, R. E., Ioerger, T., and Sassetti, C. M. (2015). Identifying essential genes in Mycobacterium tuberculosis by global phenotypic profiling. Methods Mol. Biol. 1279, 79–95. doi: 10.1007/978-1-4939-2398-4_6
Lu, D., Bernstein, D. A., Satyshur, K. A., and Keck, J. L. (2010). Small-molecule tools for dissecting the roles of SSB/protein interactions in genome maintenance. Proc. Natl. Acad. Sci. U.S.A. 107, 633–638. doi: 10.1073/pnas.0909191107
Lubbers, T., Angehrn, P., Gmunder, H., and Herzig, S. (2007). Design, synthesis, and structure-activity relationship studies of new phenolic DNA gyrase inhibitors. Bioorg. Med. Chem. Lett. 17, 4708–4714. doi: 10.1016/j.bmcl.2006.12.065
Lukat, P., Katsuyama, Y., Wenzel, S., Binz, T., König, C., Blankenfeldt, W., et al. (2017). Biosynthesis of methyl-proline containing griselimycins, natural products with anti-tuberculosis activity. Chem. Sci. 8, 7521–7527. doi: 10.1039/C7SC02622F
Ma, C., Yang, X., and Lewis, P. J. (2016). Bacterial transcription as a target for antibacterial drug development. Microbiol. Mol. Biol. Rev. 80, 139–160. doi: 10.1128/MMBR.00055-15
Maciąg, M., Kochanowska, M., Łyżeń, R., Węgrzyn, G., and Szalewska-Pałasz, A. (2010). ppGpp inhibits the activity of Escherichia coli DnaG primase. Plasmid 63, 61–67. doi: 10.1016/j.plasmid.2009.11.002
Makarov, V., Lechartier, B., Zhang, M., Neres, J., van der Sar, A. M., Raadsen, S. A., et al. (2014). Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 6, 372–383. doi: 10.1002/emmm.201303575
Marceau, A. H., Bernstein, D. A., Walsh, B. W., Shapiro, W., Simmons, L. A., and Keck, J. L. (2013). Protein interactions in genome maintenance as novel antibacterial targets. PLoS ONE 8:e58765. doi: 10.1371/journal.pone.0058765
Matsumoto, M., Hashizume, H., Tomishige, T., Kawasaki, M., Tsubouchi, H., Sasaki, H., et al. (2006). OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3:e466. doi: 10.1371/journal.pmed.0030466
McHenry, C. S. (2011). DNA replicases from a bacterial perspective. Annu. Rev. Biochem. 80, 403–436. doi: 10.1146/annurev-biochem-061208-091655
McKay, G. A., Reddy, R., Arhin, F., Belley, A., Lehoux, D., Moeck, G., et al. (2006). Triaminotriazine DNA helicase inhibitors with antibacterial activity. Bioorg. Med. Chem. Lett. 16, 1286–1290. doi: 10.1016/j.bmcl.2005.11.076
Mdluli, K., Kaneko, T., and Upton, A. (2015). The tuberculosis drug discovery and development pipeline and emerging drug targets. Cold Spring Harb. Perspect. Med. 5:a021154. doi: 10.1101/cshperspect.a021154
Medapi, B., Meda, N., Kulkarni, P., Yogeeswari, P., and Sriram, D. (2016). Development of acridine derivatives as selective Mycobacterium tuberculosis DNA gyrase inhibitors. Bioorg. Med. Chem. 24, 877–885. doi: 10.1016/j.bmc.2016.01.011
Medapi, B., Renuka, J., Saxena, S., Sridevi, J. P., Medishetti, R., Kulkarni, P., et al. (2015a). Design and synthesis of novel quinoline-aminopiperidine hybrid analogues as Mycobacterium tuberculosis DNA gyraseB inhibitors. Bioorg. Med. Chem. 23, 2062–2078. doi: 10.1016/j.bmc.2015.03.004
Medapi, B., Suryadevara, P., Renuka, J., Sridevi, J. P., Yogeeswari, P., and Sriram, D. (2015b). 4-Aminoquinoline derivatives as novel Mycobacterium tuberculosis GyrB inhibitors: structural optimization, synthesis and biological evaluation. Eur. J. Med. Chem. 103, 1–16. doi: 10.1016/j.ejmech.2015.06.032
Meier, T. I., Yan, D., Peery, R. B., McAllister, K. A., Zook, C., Peng, S. B., et al. (2008). Identification and characterization of an inhibitor specific to bacterial NAD+-dependent DNA ligases. FEBS J. 275, 5258–5271. doi: 10.1111/j.1742-4658.2008.06652.x
Merle, C. S., Fielding, K., Sow, O. B., Gninafon, M., Lo, M. B., Mthiyane, T., et al. (2014). A four-month gatifloxacin-containing regimen for treating tuberculosis. N. Engl. J. Med. 371, 1588–1598. doi: 10.1056/NEJMoa1315817
Miller, A. A., Bundy, G. L., Mott, J. E., Skepner, J. E., Boyle, T. P., Harris, D. W., et al. (2008). Discovery and characterization of QPT-1, the progenitor of a new class of bacterial topoisomerase inhibitors. Antimicrob. Agents Chemother. 52, 2806–2812. doi: 10.1128/AAC.00247-08
Mills, S. D., Eakin, A. E., Buurman, E. T., Newman, J. V., Gao, N., Huynh, H., et al. (2011). Novel bacterial NAD+-dependent DNA ligase inhibitors with broad-spectrum activity and antibacterial efficacy in vivo. Antimicrob. Agents Chemother. 55, 1088–1096. doi: 10.1128/AAC.01181-10
Minato, Y., Thiede, J. M., Kordus, S. L., McKlveen, E. J., Turman, B. J., and Baughn, A. D. (2015). Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob. Agents Chemother. 59, 5097–5106. doi: 10.1128/AAC.00647-15
Mizrahi, V., and Andersen, S. J. (1998). DNA repair in Mycobacterium tuberculosis. What have we learnt from the genome sequence? Mol. Microbiol. 29, 1331–1339.
Mizrahi, V., and Huberts, P. (1996). Deoxy- and dideoxynucleotide discrimination and identification of critical 5′ nuclease domain residues of the DNA polymerase I from Mycobacterium tuberculosis. Nucleic Acids Res. 24, 4845–4852.
Mizushima, T., Sasaki, S., Ohishi, H., Kobayashi, M., Katayama, T., Miki, T., et al. (1996). Molecular design of inhibitors of in vitro oriC DNA replication based on the potential to block the ATP binding of DnaA protein. J. Biol. Chem. 271, 25178–25183.
Nurbo, J., Ericsson, D. J., Rosenstrom, U., Muthas, D., Jansson, A. M., Lindeberg, G., et al. (2013). Novel pseudopeptides incorporating a benzodiazepine-based turn mimetic–targeting Mycobacterium tuberculosis ribonucleotide reductase. Bioorg. Med. Chem. 21, 1992–2000. doi: 10.1016/j.bmc.2013.01.020
O'Donnell, M., and Kuriyan, J. (2006). Clamp loaders and replication initiation. Curr. Opin. Struct. Biol. 16, 35–41. doi: 10.1016/j.sbi.2005.12.004
Pai, M., Behr, M. A., Dowdy, D., Dheda, K., Divangahi, M., Boehme, C. C., et al. (2016). Tuberculosis. Nat. Rev. Dis. Primers 2:16076. doi: 10.1038/nrdp.2016.76
Painter, R. E., Adam, G. C., Arocho, M., DiNunzio, E., Donald, R. G., Dorso, K., et al. (2015). Elucidation of DnaE as the antibacterial target of the natural Product, Nargenicin. Chem. Biol. 22, 1362–1373. doi: 10.1016/j.chembiol.2015.08.015
Park, Y., Pacitto, A., Bayliss, T., Cleghorn, L. A., Wang, Z., Hartman, T., et al. (2017). Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect. Dis 3, 18–33. doi: 10.1021/acsinfecdis.6b00103
Payne, D. J., Gwynn, M. N., Holmes, D. J., and Pompliano, D. L. (2007). Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6, 29–40. doi: 10.1038/nrd2201
Pethe, K., Bifani, P., Jang, J., Kang, S., Park, S., Ahn, S., et al. (2013). Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19, 1157–1160. doi: 10.1038/nm.3262
Plocinska, R., Korycka-Machala, M., Plocinski, P., and Dziadek, J. (2017). Mycobacterial DNA replication as a target for antituberculosis drug discovery. Curr. Top. Med. Chem. 17, 2129–2142. doi: 10.2174/1568026617666170130114342
Prideaux, B., Via, L. E., Zimmerman, M. D., Eum, S., Sarathy, J., O'Brien, P., et al. (2015). The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med. 21, 1223–1227. doi: 10.1038/nm.3937
Rahn-Lee, L., Merrikh, H., Grossman, A. D., and Losick, R. (2011). The sporulation protein SirA inhibits the binding of DnaA to the origin of replication by contacting a patch of clustered amino acids. J. Bacteriol. 193, 1302–1307. doi: 10.1128/JB.01390-10.
Ravishankar, S., Ambady, A., Awasthy, D., Mudugal, N. V., Menasinakai, S., Jatheendranath, S., et al. (2015). Genetic and chemical validation identifies Mycobacterium tuberculosis topoisomerase I as an attractive anti-tubercular target. Tuberculosis (Edinb). 95, 589–598. doi: 10.1016/j.tube.2015.05.004.
Reddy, K. I., Srihari, K., Renuka, J., Sree, K. S., Chuppala, A., Jeankumar, V. U., et al. (2014). An efficient synthesis and biological screening of benzofuran and benzo[d]isothiazole derivatives for Mycobacterium tuberculosis DNA GyrB inhibition. Bioorg. Med. Chem. 22, 6552–6563. doi: 10.1016/j.bmc.2014.10.016
Rengarajan, J., Bloom, B. R., and Rubin, E. J. (2005). Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc. Natl. Acad. Sci. U.S.A. 102, 8327–8332. doi: 10.1073/pnas.0503272102
Renuka, J., Reddy, K. I., Srihari, K., Jeankumar, V. U., Shravan, M., Sridevi, J. P., et al. (2014). Design, synthesis, biological evaluation of substituted benzofurans as DNA gyraseB inhibitors of Mycobacterium tuberculosis. Bioorg. Med. Chem. 22, 4924–4934. doi: 10.1016/j.bmc.2014.06.041
Reyes-Lamothe, R., Nicolas, E., and Sherratt, D. J. (2012). Chromosome replication and segregation in bacteria. Annu. Rev. Genet. 46, 121–143. doi: 10.1146/annurev-genet-110711-155421.
Robinson, A., Causer, R. J., and Dixon, N. E. (2012). Architecture and conservation of the bacterial DNA replication machinery, an underexploited drug target. Curr. Drug Targets 13, 352–372. doi: 10.2174/138945012799424598
Rock, J. M., Lang, U. F., Chase, M. R., Ford, C. B., Gerrick, E. R., Gawande, R., et al. (2015). DNA replication fidelity in Mycobacterium tuberculosis is mediated by an ancestral prokaryotic proofreader. Nat. Genet. 47, 677–681. doi: 10.1038/ng.3269
Rose, Y., Ciblat, S., Reddy, R., Belley, A. C., Dietrich, E., Lehoux, D., et al. (2006). Novel non-nucleobase inhibitors of Staphylococcus aureus DNA polymerase IIIC. Bioorg. Med. Chem. Lett. 16, 891–896. doi: 10.1016/j.bmcl.2005.11.009
Sandhaus, S., Annamalai, T., Welmaker, G., Houghten, R. A., Paz, C., Garcia, P. K., et al. (2016). Small-molecule inhibitors targeting topoisomerase I as novel antituberculosis agents. Antimicrob. Agents Chemother. 60, 4028–4036. doi: 10.1128/AAC.00288-16
Sanyal, G., and Doig, P. (2012). Bacterial DNA replication enzymes as targets for antibacterial drug discovery. Exp. Opin. Drug Discov. 7, 327–339. doi: 10.1517/17460441.2012.660478
Sassetti, C. M., and Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994. doi: 10.1073/pnas.2134250100
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Saxena, S., Samala, G., Renuka, J., Sridevi, J. P., Yogeeswari, P., and Sriram, D. (2015). Development of 2-amino-5-phenylthiophene-3-carboxamide derivatives as novel inhibitors of Mycobacterium tuberculosis DNA GyrB domain. Bioorg. Med. Chem. 23, 1402–1412. doi: 10.1016/j.bmc.2015.02.032
Seitz, H., Weigel, C., and Messer, W. (2000). The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Mol. Microbiol. 37, 1270–1279. doi: 10.1046/j.1365-2958.2000.02096.x
Senthilkumar, P., Dinakaran, M., Banerjee, D., Devakaram, R. V., Yogeeswari, P., China, A., et al. (2008). Synthesis and antimycobacterial evaluation of newer 1-cyclopropyl-1, 4-dihydro-6-fluoro-7-(substituted secondary amino)-8-methoxy-5-(sub)-4-oxoquinoline-3-carboxylic acids. Bioorg. Med. Chem. 16, 2558–2569. doi: 10.1016/j.bmc.2007.11.050
Shirude, P. S., Madhavapeddi, P., Tucker, J. A., Murugan, K., Patil, V., Basavarajappa, H., et al. (2013). Aminopyrazinamides: novel and specific GyrB inhibitors that kill replicating and nonreplicating Mycobacterium tuberculosis. ACS Chem. Biol. 8, 519–523. doi: 10.1021/cb300510w
Singh, A., Bhagavat, R., Vijayan, M., and Chandra, N. (2016). A comparative analysis of the DNA recombination repair pathway in mycobacterial genomes. Tuberculosis (Edinb). 99, 109–119. doi: 10.1016/j.tube.2016.04.011
Singh, R., Manjunatha, U., Boshoff, H. I., Ha, Y. H., Niyomrattanakit, P., Ledwidge, R., et al. (2008). PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395. doi: 10.1126/science.1164571
Singh, V., and Mizrahi, V. (2017). Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov. Today 22, 503–509. doi: 10.1016/j.drudis.2016.09.010
Singh, V., Brecik, M., Mukherjee, R., Evans, J. C., Svetlikova, Z., Blasko, J., et al. (2015). The complex mechanism of antimycobacterial action of 5-Fluorouracil. Chem. Biol. 22, 63–75. doi: 10.1016/j.chembiol.2014.11.006
Singh, V., Donini, S., Pacitto, A., Sala, C., Hartkoorn, R. C., Dhar, N., et al. (2017). The Inosine Monophosphate Dehydrogenase, GuaB2, is a vulnerable new bactericidal drug target for tuberculosis. ACS Infect. Dis 3, 5–17. doi: 10.1021/acsinfecdis.6b00102
Sipos, A., Pato, J., Szekely, R., Hartkoorn, R. C., Kekesi, L., Orfi, L., et al. (2015). Lead selection and characterization of antitubercular compounds using the Nested Chemical Library. Tuberculosis (Edinb) 95 (Suppl. 1), S200–S206. doi: 10.1016/j.tube.2015.02.028
Sridevi, J. P., Suryadevara, P., Janupally, R., Sridhar, J., Soni, V., Anantaraju, H. S., et al. (2015). Identification of potential Mycobacterium tuberculosis topoisomerase I inhibitors: a study against active, dormant and resistant tuberculosis. Eur. J. Pharm. Sci. 72, 81–92. doi: 10.1016/j.ejps.2015.02.017
Sriram, D., Aubry, A., Yogeeswari, P., and Fisher, L. M. (2006). Gatifloxacin derivatives: synthesis, antimycobacterial activities, and inhibition of Mycobacterium tuberculosis DNA gyrase. Bioorg. Med. Chem. Lett. 16, 2982–2985. doi: 10.1016/j.bmcl.2006.02.065
Srivastava, S. K., Dube, D., Kukshal, V., Jha, A. K., Hajela, K., and Ramachandran, R. (2007). NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis: novel structure-function relationship and identification of a specific inhibitor. Proteins 69, 97–111. doi: 10.1002/prot.21457
Srivastava, S. K., Dube, D., Tewari, N., Dwivedi, N., Tripathi, R. P., and Ramachandran, R. (2005a). Mycobacterium tuberculosis NAD+-dependent DNA ligase is selectively inhibited by glycosylamines compared with human DNA ligase I. Nucleic Acids Res. 33, 7090–7101. doi: 10.1093/nar/gki1006.
Srivastava, S. K., Tripathi, R. P., and Ramachandran, R. (2005b). NAD+-dependent DNA Ligase (Rv3014c) from Mycobacterium tuberculosis. Crystal structure of the adenylation domain and identification of novel inhibitors. J. Biol. Chem. 280, 30273–30281. doi: 10.1074/jbc.M503780200
Stokes, N. R., Thomaides-Brears, H. B., Barker, S., Bennett, J. M., Berry, J., Collins, I., et al. (2013). Biological evaluation of benzothiazole ethyl urea inhibitors of bacterial type II topoisomerases. Antimicrob. Agents Chemother. 57, 5977–5986. doi: 10.1128/AAC.00719-13
Stokes, S. S., Huynh, H., Gowravaram, M., Albert, R., Cavero-Tomas, M., Chen, B., et al. (2011). Discovery of bacterial NAD+-dependent DNA ligase inhibitors: optimization of antibacterial activity. Bioorg. Med. Chem. Lett. 21, 4556–4560. doi: 10.1016/j.bmcl.2011.05.128
Stover, C. K., Warrener, P., VanDevanter, D. R., Sherman, D. R., Arain, T. M., Langhorne, M. H., et al. (2000). A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966. doi: 10.1038/35016103
Sulochana, S., Rahman, F., and Paramasivan, C. N. (2005). In vitro activity of fluoroquinolones against Mycobacterium tuberculosis. J. Chemother. 17, 169–173. doi: 10.1179/joc.2005.17.2.169
Surivet, J. P., Lange, R., Hubschwerlen, C., Keck, W., Specklin, J. L., Ritz, D., et al. (2012). Structure-guided design, synthesis and biological evaluation of novel DNA ligase inhibitors with in vitro and in vivo anti-staphylococcal activity. Bioorg. Med. Chem. Lett. 22, 6705–6711. doi: 10.1016/j.bmcl.2012.08.094
Tarantino, P. M. Jr., Zhi, C., Gambino, J. J., Wright, G. E., and Brown, N. C. (1999a). 6-Anilinouracil-based inhibitors of Bacillus subtilis DNA polymerase III: antipolymerase and antimicrobial structure-activity relationships based on substitution at uracil N3. J. Med. Chem. 42, 2035–2040. doi: 10.1021/jm980693i
Tarantino, P. M. Jr., Zhi, C., Wright, G. E., and Brown, N. C. (1999b). Inhibitors of DNA polymerase III as novel antimicrobial agents against gram-positive eubacteria. Antimicrob. Agents Chemother. 43, 1982–1987.
Tari, L. W., Li, X., Trzoss, M., Bensen, D. C., Chen, Z., Lam, T., et al. (2013a). Tricyclic GyrB/ParE (TriBE) inhibitors: a new class of broad-spectrum dual-targeting antibacterial agents. PLoS ONE 8:e84409. doi: 10.1371/journal.pone.0084409.
Tari, L. W., Trzoss, M., Bensen, D. C., Li, X., Chen, Z., Lam, T., et al. (2013b). Pyrrolopyrimidine inhibitors of DNA gyrase B (GyrB) and topoisomerase IV (ParE). Part I: Structure guided discovery and optimization of dual targeting agents with potent, broad-spectrum enzymatic activity. Bioorg. Med. Chem. Lett. 23, 1529–1536. doi: 10.1016/j.bmcl.2012.11.032
Timinskas, K., Balvociute, M., Timinskas, A., and Venclovas, C. (2014). Comprehensive analysis of DNA polymerase III alpha subunits and their homologs in bacterial genomes. Nucleic Acids Res. 42, 1393–1413. doi: 10.1093/nar/gkt900
Tran, T. P., Ellsworth, E. L., Sanchez, J. P., Watson, B. M., Stier, M. A., Showalter, H. D., et al. (2007). Structure-activity relationships of 3-aminoquinazolinediones, a new class of bacterial type-2 topoisomerase (DNA gyrase and topo IV) inhibitors. Bioorg. Med. Chem. Lett. 17, 1312–1320. doi: 10.1016/j.bmcl.2006.12.005.
Tse-Dinh, Y. C. (2016). Targeting bacterial topoisomerases: how to counter mechanisms of resistance. Future Med. Chem. 8, 1085–1100. doi: 10.4155/fmc-2016-0042
van Eijk, E., Wittekoek, B., Kuijper, E. J., and Smits, W. K. (2017). DNA replication proteins as potential targets for antimicrobials in drug-resistant bacterial pathogens. J. Antimicrob. Chemother. 72, 1275–1284. doi: 10.1093/jac/dkw548
Voter, A. F., Killoran, M. P., Ananiev, G. E., Wildman, S. A., Hoffmann, F. M., and Keck, J. L. (2017). A High-Throughput Screening Strategy to Identify Inhibitors of SSB Protein-Protein Interactions in an Academic Screening Facility. SLAS Discov. 2472555217712001. doi: 10.1177/2472555217712001
Wall, M. E., Wani, M. C., Cook, C. E., Palmer, K. H., McPhail, K. A., and Sim, G. A. (1966). Plat antitumor agents. I. The isolation and structure of camptothecin, anovel alkaloidal leukemia and tumor inhibitor from camptotheca acuminata. J. Am. Chem. Soc. 88, 3888–3890.
Wang, T., Duncan, L., Gu, W., O'Dowd, H., Wei, Y., Perola, E., et al. (2012). Design, synthesis and biological evaluation of potent NAD+-dependent DNA ligase inhibitors as potential antibacterial agents. Part 2: 4-amino-pyrido[2, 3-d]pyrimidin-5(8H)-ones. Bioorg. Med. Chem. Lett. 22, 3699–3703. doi: 10.1016/j.bmcl.2012.04.038
Wang, T. C., and Chen, F. C. (2013). The evolutionary landscape of the Mycobacterium tuberculosis genome. Gene 518, 187–193. doi: 10.1016/j.gene.2012.11.033
Warner, D. F. (2010). The role of DNA repair in M. tuberculosis pathogenesis. Drug Disc. Tod. 7, e5–e11. doi: 10.1016/j.ddmec.2010.08.002
Warner, D. F. (2014). Mycobacterium tuberculosis metabolism. Cold Spring Harb. Perspect. Med. 5:a021121. doi: 10.1101/cshperspect.a021121
Warner, D. F., and Mizrahi, V. (2014). Shortening treatment for tuberculosis–to basics. N. Engl. J. Med. 371, 1642–1643. doi: 10.1056/NEJMe1410977
Warner, D. F., Ndwandwe, D. E., Abrahams, G. L., Kana, B. D., Machowski, E. E., Venclovas, C., et al. (2010). Essential roles for imuA'- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 107, 13093–13098. doi: 10.1073/pnas.1002614107
Wiles, J. A., Hashimoto, A., Thanassi, J. A., Cheng, J., Incarvito, C. D., Deshpande, M., et al. (2006a). Isothiazolopyridones: synthesis, structure, and biological activity of a new class of antibacterial agents. J. Med. Chem. 49, 39–42. doi: 10.1021/jm051066d
Wiles, J. A., Wang, Q., Lucien, E., Hashimoto, A., Song, Y., Cheng, J., et al. (2006b). Isothiazoloquinolones containing functionalized aromatic hydrocarbons at the 7-position: synthesis and in vitro activity of a series of potent antibacterial agents with diminished cytotoxicity in human cells. Bioorg. Med. Chem. Lett. 16, 1272–1276. doi: 10.1016/j.bmcl.2005.11.065
Williams, A., Guthlein, C., Beresford, N., Bottger, E. C., Springer, B., and Davis, E. O. (2011). UvrD2 is essential in Mycobacterium tuberculosis, but its helicase activity is not required. J. Bacteriol. 193, 4487–4494. doi: 10.1128/JB.00302-11
Wright, G. D. (2017). Opportunities for natural products in 21st century antibiotic discovery. Nat. Prod. Rep. 34, 694–701. doi: 10.1039/c7np00019g
Wright, G. E., Brown, N. C., Xu, W. C., Long, Z. Y., Zhi, C., Gambino, J. J., et al. (2005). Active site directed inhibitors of replication-specific bacterial DNA polymerases. Bioorg. Med. Chem. Lett. 15, 729–732. doi: 10.1016/j.bmcl.2004.11.016
Xu, G., Ni, Z., Shi, Y., Sun, X., Wang, H., Wei, C., et al. (2014). Screening essential genes of Mycobacterium tuberculosis with the pathway enrichment method. Mol. Biol. Rep. 41, 7639–7644. doi: 10.1007/s11033-014-3654-z
Xu, W. C., Wright, G. E., Brown, N. C., Long, Z. Y., Zhi, C. X., Dvoskin, S., et al. (2011). 7-Alkyl-N(2)-substituted-3-deazaguanines. Synthesis, DNA polymerase III inhibition and antibacterial activity. Bioorg. Med. Chem. Lett. 21, 4197–4202. doi: 10.1016/j.bmcl.2011.05.093
Xu, Z., Meshcheryakov, V. A., Poce, G., and Chng, S. S. (2017). MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. U.S.A. 114, 7993–7998. doi: 10.1073/pnas.1700062114
Yao, N., and O'Donnell, M. (2016a). Bacterial and eukaryotic replisome machines. JSM Biochem. Mol. Biol. 3:1013.
Yao, N. Y., and O'Donnell, M. E. (2016b). Evolution of replication machines. Crit. Rev. Biochem. Mol. Biol. 51, 135–149. doi: 10.3109/10409238.2015.1125845
Young, K., Olsen, D. B., Singh, S. B., Su, J., Wilkening, R. R., Apgar, J. M., et al. (2016). Nargenicin Compounds and Uses thereof as Antibacterial Agents. Google Patents.
Zhang, H. Z., He, S. C., Peng, Y. J., Zhang, H. J., Gopala, L., Tangadanchu, V. K.R., et al. (2017). Design, synthesis and antimicrobial evaluation of novel benzimidazole-incorporated sulfonamide analogues. Eur. J. Med. Chem. 136, 165–183. doi: 10.1016/j.ejmech.2017.04.077
Zhang, J., Yang, Q., Romero, J. A., Cross, J., Wang, B., Poutsiaka, K. M., et al. (2015). Discovery of indazole derivatives as a novel class of Bacterial Gyrase B Inhibitors. ACS Med. Chem. Lett. 6, 1080–1085. doi: 10.1021/acsmedchemlett.5b00266
Zumla, A., Nahid, P., and Cole, S. T. (2013). Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 12, 388–404. doi: 10.1038/nrd4001
Keywords: DNA replication, Tuberculosis, bacteria, drug resistance, drug targets
Citation: Reiche MA, Warner DF and Mizrahi V (2017) Targeting DNA Replication and Repair for the Development of Novel Therapeutics against Tuberculosis. Front. Mol. Biosci. 4:75. doi: 10.3389/fmolb.2017.00075
Received: 02 October 2017; Accepted: 31 October 2017;
Published: 14 November 2017.
Edited by:
Zvi Kelman, National Institute of Standards and Technology, United StatesReviewed by:
Michael O'Donnell, Rockefeller University, United StatesKaushlendra Tripathi, University of Alabama at Birmingham, United States
Copyright © 2017 Reiche, Warner and Mizrahi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Digby F. Warner, ZGlnYnkud2FybmVyQHVjdC5hYy56YQ==
Valerie Mizrahi, dmFsZXJpZS5taXpyYWhpQHVjdC5hYy56YQ==