 Aga Syed Sameer1*
Aga Syed Sameer1* Saniya Nissar2
Saniya Nissar2- 1Department of Basic Medical Sciences, College of Medicine, King Saud bin Abdulaziz University for Health Sciences, Jeddah, Saudi Arabia
- 2Department of Biochemistry, Kashmir University, Srinagar, India
In mammals the bulky DNA adduct lesions known to result in deleterious phenotypes are acted upon and removed from the genomic DNA by nucleotide excision repair (NER) pathway. TFIIH multi-protein complex with its important helicase–Xeroderma Pigmentosum Protein (XPD) serves as the pivotal factor for opening up of the damaged lesion DNA site and carry out the repair process. The initial damage verification step of the TFIIH is in part dependent upon the helicase activity of XPD. Besides, XPD is also actively involved in the initiation steps of transcription and in the regulation of the cell cycle and apoptosis. In this review, we will be exploring the new insights in scientific research on the functioning of the NER pathway, the role of TFIIH as the central complex of NER, the pivotal helicase XPD as the lynchpin of NER and the effects of various single nucleotide polymorphisms (SNPs) of XPD on its functioning and their consequent role in colorectal carcinogenesis.
Introduction
Colorectal carcinogenesis is a multifactorial and multigene process that is controlled by various gatekeeper and caretaker genes via definite pathway, referred to as the adenoma–carcinoma sequence/model (Vogelstein et al., 1988). The development of colorectal cancer (CRC) has been shown to occur because of cumulative accumulation of mutations in tumor-suppressor genes and oncogenes. The process of transformationinvolves several genetic changes that are essential for cancer initiation and progression (Migliore et al., 2011). On the basis of molecular profiles, CRC is now classified into three specific phenotypes (Cunningham et al., 2010); involving three major genetic instability pathways: chromosomal instability (CIN), microsatellite instability (MSI) and CpG island methylator phenotype (CIMP) (Ogino et al., 2011; Sameer, 2013).
The CIN pathway is the major mediator of colorectal tumorigenesis and is responsible for approximately 80–85% of all colorectal cancers (Goel et al., 2003; Grady, 2004). The CIN pathway involves the dynamic and continued loss or gain of whole chromosomes or chromosomal regions (due to sub-chromosomal genomic deletion and amplification respectively) that carry genes of critical importance to the process of colorectal tumorigenesis. The major initiating events in the CIN pathway are the induced mutations in two classes of critical gate-keeper genes, oncogenes and tumor suppressor genes (McGranahan et al., 2012), which in turn lead to the dysregulation of several critical signaling pathways that characterize the CIN pathway.
Microsatellite instability (MSI) usually comprises the length alterations of oligonucleotide repeat sequences (microsatellites) that occur somatically in many human tumors. The microsatellites are highly prone to replication errors due to higher susceptibility of DNA polymerase to induce mismatches during replication of long or short repetitive DNA sequences including MSs and the mutations in the form of insertions or deletions often termed as an insertion-deletion loop leads to length alterations and result in MSI. Mutations in DNA mismatch repair (MMR) genes result in a failure to repair errors that occur during DNA replication in microsatellites, resulting in an accumulation of frameshift mutations in genes that contain microsatellites (Boland and Goel, 2010). The MMR system in humans consists of several identified proteins, MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, PMS2, and Exo1 with MLH1 and MSH2 being the important members (Sameer et al., 2014). MSI is also found in 12–15% of sporadic CRCs. MIN tumors are more frequently right-sided and poorly differentiated, and more often display unusual histological type (mucinous), and marked peri-tumoral and intra-tumoral lymphocytic infiltration (Benatti et al., 2005). MSI also occurs in patients with ulcerative colitis and is fairly common in premalignant (dysplastic) and malignant lesions (21 and 19% respectively; Kerr et al., 2001).
CpG island methylator phenotype (CIMP), also referred to as methylator phenotype of CRC tumorigenesis involves gene silencing or transcriptional inactivation by CpG island methylation in tumor suppressor gene promoters. It plays a major role in about 35% of CRCs (Sporadic as well as Hereditary) (Cunningham et al., 2010; Sameer and Nissar, 2016). Thus, under epigenetic point of view, classically CRC has been divided essentially into CIMP positive (CIMP+) and non-CIMP (CIMP negative) tumors (Toyota et al., 1999). The genes implicated in colorectal tumorigenesis that are silenced by hypermethylation include DNA repair genes such as methylguanine methyltransferase (MGMT) and MLH1, tumor suppressors (p16, APC, insulin-like growth factor 2, and HIC1), cell cycle regulatory genes (Mutated In Colorectal Cancers/MCC) and Wnt signaling antagonists known as SFRPs (secreted frizzled-related proteins). The silencing of MLH1 gene by hypermethylation is a frequent event in sporadic MSI-High colorectal tumors (Kim et al., 2010).
One of the main causes of genetic instability leading to tumorigenesis is the inefficient repair of DNA lesions that creep in the cellular genome via various DNA damaging agents affecting the normal cellular functioning.
DNA Repair Mechanisms
The eukaryotic genome is always under a constant threat of wide variety of DNA-damaging agents (physical & chemical) which affect the structure and function of the genes. To prevent the effect of DNA modifying agents on the functionality of the genome and hence to allow the smooth functioning of the fundamental cellular processes, cells have a varied arsenal of a number of repair molecules and pathways that are specifically used to identify and remove the several specific types of DNA lesions which may otherwise lead to various diseases primary among them-cancers (Michailidi et al., 2012). However, each of the DNA repair pathway is specific to the particular type of DNA lesion. Hence the mechanism of restoration of intact DNA depends on various factors like the cell cycle phase, type of lesion and other environmental factors. But, one thing that is common to all repair mechanisms is that all of them use the cell's own intact complementary DNA strand as a template to restore the original strand. The various repair mechanisms operating within the confines of the cell can be divided broadly into two types: direct and indirect; depending upon the molecules/proteins used for the repair process and also the time period of the repair mechanisms (Michailidi et al., 2012; Sameer et al., 2014).
Direct repair mechanisms are carried out during the replication process itself, when the daughter DNA is being synthesized within the cell. These mechanism are mostly mediated by direct DNA interacting enzymes which may be either DNA polymerases themselves or O6-methylguanine DNA methyltransferase (MGMT) (Hoeijmakers, 2001; Michailidi et al., 2012; Iyama and Wilson, 2013; Sameer et al., 2014). Indirect repair is carried out after the synthesis process and mostly involves the repair of those lesions that occurred during the synthesis of DNA. Being essentially a post-replication process it serves to the inability of the direct repair to fix all lesions during the synthesis process (Sameer et al., 2014). Thus, essentially, it is a post synthesis process of correcting the DNA lesions, and is usually assisted by various proteins which are involved in DNA replication as well.
Indirect DNA repair mechanisms are further classified into three categories: excision repair (ER), recombination repair (RR), and mismatch repair (MMR). ER has two subcategories, namely, base excision repair (BER) for excision of abnormal bases such as uracil and breaks found only in one DNA strand, and nucleotide excision repair (NER) for the removal of bulky adducts (Michailidi et al., 2012; Iyama and Wilson, 2013; Sameer et al., 2014).
Recombination repair (RR), also called as homologous recombination (HR) comprises a series of interrelated pathways that function in the repair of DNA double-stranded breaks (DSBs) and interstrand crosslinks (ICLs) using a homologous DNA (Jasin and Rodney, 2013). MMR repairs the small loops within the duplex DNA that arise from nucleotide misincorporations—either by base—base mismatches or by insertion/deletion loops (Sameer et al., 2014).
NER: Structure and Function
The NER system is one of the principle mechanisms that cells employ in protecting themselves against genotoxic damage such as that induced by UV-irradiation or exposure to chemical carcinogens which are known to incorporate DNA lesions in the cellular genome (Benhamou and Sarasin, 2000, 2002). The most common DNA lesions which serve as the substrates of NER system are bulky covalent adducts in DNA double strands. They are produced from nitrogenous bases of DNA when exposed to strong mutagens like UV light, ionizing radiations, electrophilic chemical mutagens, drugs, and chemically active endogenous metabolites (Petruseva et al., 2014). The inability of BER to remove the bulky lesions from the duplex DNA is dealt upon by NER. In general, NER corrects the helix-distorting base lesions within the DNA that arise because of exposure to sunlight or bulky chemicals such as benzo[a]pyrene, benzo[c]anthracene, diol-epoxide, aromatic amines such as acetyl-aminofluorene, aflatoxin, nitrosamines such as MNNG, and 4-nitroquinoline oxide (Nouspikel, 2009; Michailidi et al., 2012; Petruseva et al., 2014).
The NER pathway repairs the damaged strand by excising about 24–32nt DNA fragments containing the DNA lesion (Petruseva et al., 2014). The repair of the damaged DNA strand involves five main steps; it begins with the damage recognition step, opening of the double helix at the lesion site, demarcation of the actual DNA lesion and assembly of a pre-incision complex, followed by excision of the lesion containing damaged strand and synthesis of the DNA in the gap (Benhamou and Sarasin, 2000, 2002; Hoeijmakers, 2001). Each of these steps requires the functioning of the specialized protein complexes to carry out the repair efficiently and specifically (Spivak, 2015). Till date about 30 different polypeptides have been identified which play a pivotal role in one or more steps of the NER pathway (Petruseva et al., 2014).
Based upon the initial damage recognition mechanisms, NER pathway has two functionally distinct branches for dealing with DNA lesion which distort its helical structure. One operates to repair bulky lesions throughout the entire genome (i.e., global genome NER, GG-NER) including the untranscribed regions and silent chromatin. The second works in cooperation with the transcription machinery to remove damage from actively transcribed regions of genes (i.e., transcription-coupled NER, TC-NER) (Petruseva et al., 2014; Spivak, 2015).
GG-NER is controlled by the specialized protein called XPC (Xeroderma Pigmentosum C) which senses and recognizes the helix distorting DNA lesion in the genome to start the repair process. However, some lesions like cyclobutane pyrimidine dimers (CPD) which are too small to destabilize the DNA helical structure are recognized first by damage-specific DNA binding protein 1 (DDB1) and DDB2/XPE complex (Spivak, 2015). XPC is functional as a hetero-trimer in complex with two other proteins - human RAD23 homolog B (hRAD23B /HR23B) and centrin 2 (CETN2). HR23B stabilizes the complex, protects it against proteasome degradation, and stimulates the DNA-binding activity of XPC. It plays a pivotal role in the recruitment of other repair proteins into the GG-NER process (Nouspikel, 2009; Petruseva et al., 2014; Spivak, 2015).
TC-NER is contrarily dependent upon the transcription machinery for its initial recognition of the DNA lesion. In TC-NER, DNA damage is detected by the elongating RNA polymerase II (RNAPII) when it encounters a bulky DNA lesion within the coding region of the gene to be transcribed (de Laat et al., 1999). So the blocking of the RNAPII by the damaged DNA constitutes the first step for the damage repair via TC-NER (Spivak, 2015). The arrested elongation complex then recruits CSB (ERCC6), a transcription elongation factor that translocates along template DNA with RNAPII. CSB acts as a master recruiter then by recruiting complexes of proteins required for repair mechanism like the CSA complex, NER factors (not including the GGR recognition factors XPC and XPE) and p300 to sites of arrested RNAPII. Both branches of the NER–TC-NER and GG-NER then converge with the recruitment of transcription factor II H (TFIIH) to the repair site (Spivak, 2015; Table 1).
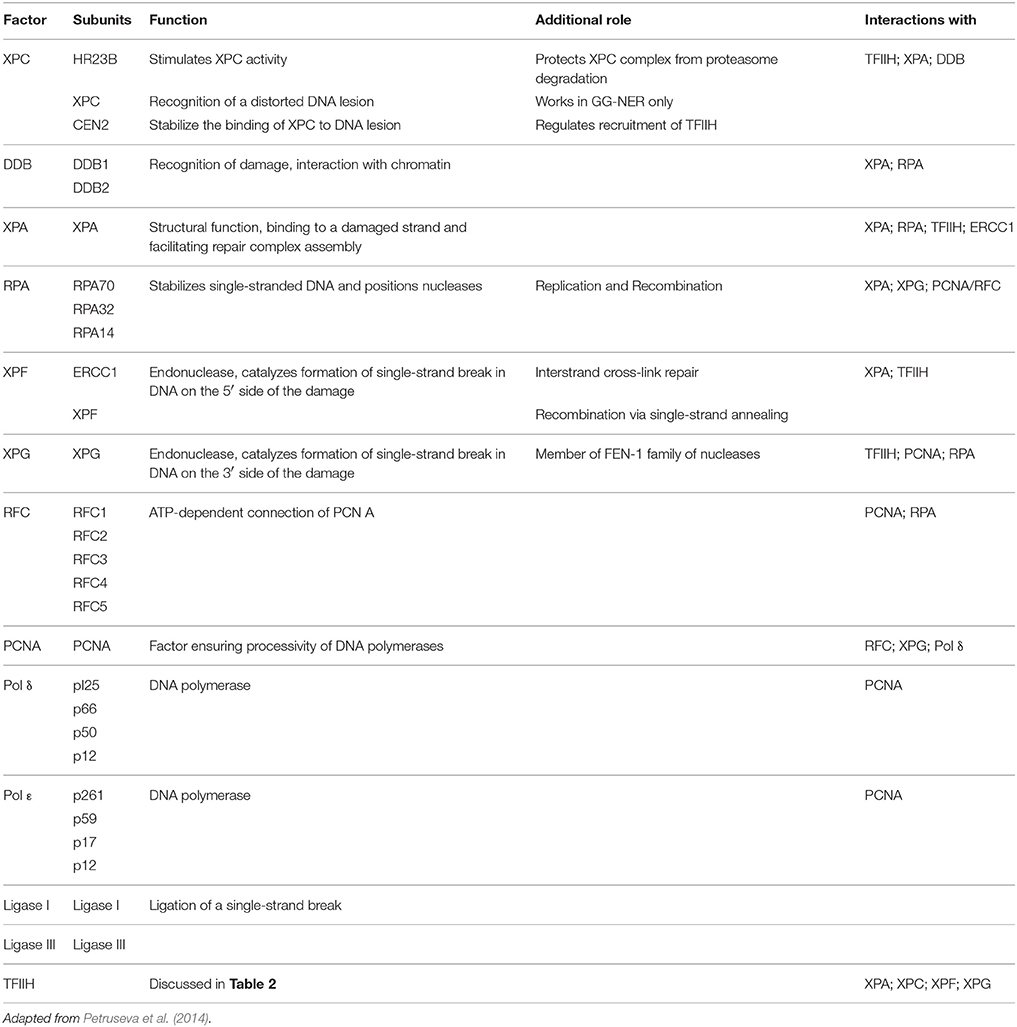
Table 1. Composition of the human NER system.
Defects in NER usually results in UV-sensitive and high-carcinogenis pathologies; and have been shown to cause at least three human genetic disorders: xeroderma pigmentosum (XP), Cockayne's syndrome (CS), and trichothiodystrophy (TTD); in addition to neurodegenerative manifestations (Iyama and Wilson, 2013; Petruseva et al., 2014; Spivak, 2015).
TFIIH: Structure and Function
TFIIH is a remarkable dual function multisubunit protein complex that not only plays a fundamental role in the transcription of protein-coding genes but also plays an important role in the NER system (Oksenych and Coin, 2010; Compe and Egly, 2012). In transcription, TFIIH is essentially required for two purposes - first for the proper binding of the RNA polymerase I & II at their specific promoter regions just upstream of the gene and second for the promoter clearance of polymerase to proceed into elongation phase of transcription via its C-terminal domain (CTD) phosphorylation (Mydlikova et al., 2010; Compe and Egly, 2012). In TC-NER, TFIIH forms a part of the core incision machinery without which NER system would not function (Egly and Coin, 2011).
The TFIIH complex is composed of two sub-complexes: core complex and cyclin-dependent kinase (CDK)-activating kinase (CAK) complex. The 3D structure of TFIIH is organized into ring-like core from which the CAK module protrudes out (Chang and Kornberg, 2000). Core complex consists of seven subunit core (XPB, p62, p52, p44, p34, and TTD-A) associated with a three subunit CAK module by the XPD helicase (Oksenych and Coin, 2010; Table 2, Figure 1). CAK module is composed of CDK7 (p40), cyclin H (p34) and menage á trois 1 (MAT1; p32). Three important enzymatic subunits are present within the confines of TFIIH complex, two ATP-dependent DNA helicases: XPB and XPD, and the kinase CDK7 (Mydlikova et al., 2010; Zhovmer et al., 2010). Proteins p62, p52, p44 and p34 previously regarded just as “structural” subunits have been shown to contain regulatory functions within TFIIH. Protein p52 modulates XPB activity by upregulating its ATPase activity through a direct XPB/p52 interaction and also anchors XPB to the TFIIH; while as protein p44 regulates XPD via direct p44/XPD interaction and also functions as ubiquitin ligase and protein p62 has been demonstrated to interact with thyroid hormone receptor TRβ (Mydlikova et al., 2010; Egly and Coin, 2011; Compe and Egly, 2012).
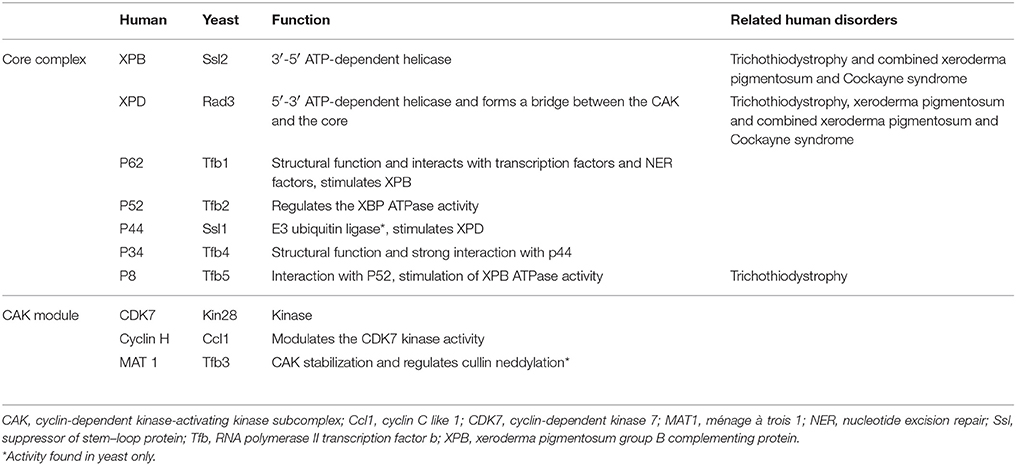
Table 2. Composition of the human TFIIH complex.
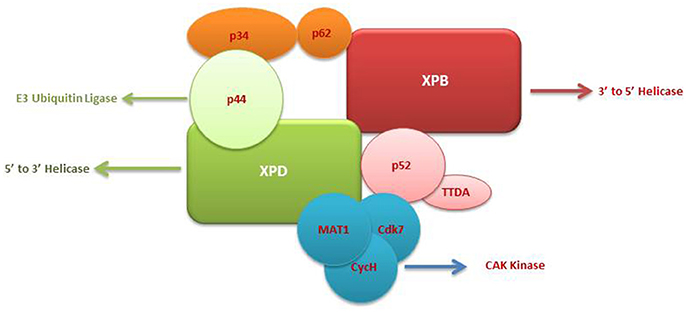
Figure 1. A multisubunit functional complex of TFIIH. The TFIIH complex is composed of eleven subunits, with XPB making its one face and XPD another. The complex also contains CAK kinase domain of three subunits (Blue). Functonally TFIIH possess four enzymatic activities; XPB and XPD has helicase activity, Cdk7 has kinase and p44 serves as E3 ubiquitin ligase.
The isolated XPB and XPD subunits of TFIIH are both ATP-dependent DNA helicases with 3′ → 5′ and 5′ → 3′ DBA helicase activity respectively (Mydlikova et al., 2010). XPB and XPD with opposite polarities have been suggested to cooperate in the opening of the damaged DNA helix on opposite sides of a lesion, with XPB working on the 3′ side of the lesion and XPD working from the 5′side (Oksenych and Coin, 2010; Fuss and Tainer, 2011; Compe and Egly, 2012). However, the differential role play by both proteins has been shown to toggle between transcription functions and NER functions (Oksenych and Coin, 2010); wherein XPB ATPase activity is essentially required for DNA opening in both NER and transcription but its helicase activity is dedicated to only promoter escape in transcription process. In contrast, helicase activity of XPD plays a minor role in transcription process but it is pivotal in NER system for the removal of DNA lesions (Tirode et al., 1999; Winkler et al., 2000; Coin et al., 2007; Richards et al., 2008; Oksenych and Coin, 2010). The opening of the DNA helix around the lesion by these two helicases in an ATP-dependents manner is the first catalytic reaction of NER system which in turn leads to the conformational changes that allows the recruitment of additional NER proteins (Wolski et al., 2010).
Within the core complex of TFIIH XPB constitutes the biggest subunit containing seven helicase motifs and belongs to helicase superfamily 2 (SF2); while as XPD contains a RecA-like fold that belongs to the SF2 family of helicases with distinct 4Fe4S (FeS) cluster that is essential for its function as a helicase differentiating into two helicase domains, HD1 and HD2 (Zurita and Merino, 2003; Rudolf et al., 2006; Mydlikova et al., 2010; Wolski et al., 2010).
In CAK domain, CDK7 protein is the biggest subunit with bifunctional activity - one phosphorylase via which CDKs participate in the cell cycle, and second as a component of the TFIIH, which is essential for CTD phosphorylation of the largest subunit of RNA polymerase II (Mydlikova et al., 2010). MAT1 protein functions to link CAK to the core TFIIH in a complex interaction which is facilitated also by both XPD and XPB helicases. MAT1 interacts with the CDK7-cyclin H complex and stimulates the CDK7 kinase activity (Busso et al., 2000).
TFIIH role in transcription is via its joining the other general transcription factors like TFIIA, TFIIB, TFIID, TFIIE, and TFIIF to form the preinitiation complex (of more than 30 polypeptides) together with central RNPII at the promoter region of the gene to be transcribed. Promoter recognition is initially carried out by TFIID which in turn recruits TFIIA and TFIIB eventually TFIIH entry is mediated via TFIIF/E (Mydlikova et al., 2010; Compe and Egly, 2012). In transcriptional process, TFIIH plays wide variety of roles; it is involved in initiation, promoter escape, and early elongation stages, to transcription reinitiation and formation of gene loops (Zhovmer et al., 2010). TFIIH controls the initiation of transcription and enhances the association of the RNPII CTD with the 7-methylguanosine (m7G) RNA capping machinery (Serizawa et al., 1993). This TFIIH kinase activity toward the CTD of Pol II can be modulated by different factors, including MAT1 (ménage à trois 1) and cyclin H, which are two binding partners of CDK7 within the CAK subcomplex (Komarnitsky et al., 2000). It is also plays important role in the RNPI transcription of ribosomal genes (Iben et al., 2002).
As already mentioned, depending upon the branches of the NER whether GG-NER or TC-NER, the recruitment of TFIIH is mediated by either XPC or by the stalled RNPII to the site of the lesion in the DNA. After recruitment, it functions to open the DNA around the lesion and thereby allow the excision of the string of 24–32nt DNA fragments containing the lesion and its consequent replacement by a new DNA fragment (Oksenych and Coin, 2010; Egly and Coin, 2011; Compe and Egly, 2012). In GG-NER, TFIIH is a part of the dual incision complex composed of XPC-HR23B, centrin2, XPA, replication protein A (RPA), XPG, and excision repair cross-complementation group 1 (ERCC1)-XPF, and is involved in the opening of the DNA around the lesion (Figure 2). Because of its high sensitivity for the recognition of damages sites; XPC not only rapidly detects the various DNA lesions but it also promotes the kinks in DNA helix forming a transient recognition intermediate; allowing the other proteins of NER to be recruited to the site (Compe and Egly, 2012). XPA is known as a scaffold protein without enzymatic activity that nevertheless shows preferential association to damaged DNA and is indispensable for DNA incision (Missura et al., 2001). After correctly seated at the damaged DNA, TFIIH then mediates the excision of the DNA lesion with the help of XPB and XPD ATPase/helicase activities (Oksenych et al., 2009).
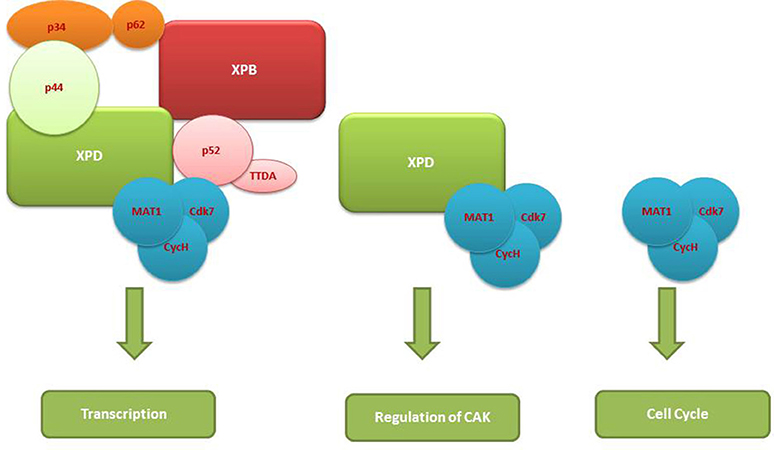
Figure 2. TFIIH complexes vs. non-TFIIH complexes and differences in their functions.
XPD: Structure and Function
XPD (also known as ERCC2) is a helicase protein of 761 amino acids with a molecular weight of 86.9kDa (Benhamou and Sarasin, 2002). XPD is one of the two pivotal ATPase/Helicase of the core unit of THIIF molecular assembly. It seems to form a bridge between the TFIIH core complex and the CAK module–which otherwise also exists as a free trimeric complex with its own distinct functions (Chen and Suter, 2003; Cameroni et al., 2010). XPD belongs to an ATP-dependent 5′-3′ superfamily 2 (SF2) helicases, which are characterized by seven “helicase motifs” (walker motif I, Ia, II, III, IV, V, and VI) constituted of highly conserved amino-acid sequences (Oksenych and Coin, 2010). Interestingly, the XPD protein also constitutes a 4Fe4S (FeS) cluster that has been demonstrated to be essential for its helicase activity. Because of this cluster XPD becomes a founding member of a family of related SF2 helicases (Rudolf et al., 2006, 2010; Liu et al., 2008). SF2 family helicases also comprises various important family members like bacterial DinG (damage-inducible G) and the eukaryotic XPD paralogs FancJ (Fanconi's anemia complementation group J), RTEL (regular of telomere length) and Chl1 (chromatid cohesion in yeast) (Wolski et al., 2010). The exact function of the FeS cluster is not known but a number of explanations as to its role have been given like–a purely structural role and providing stabilization to the FeS domain; direct interaction with the damaged DNA substrate and acting as a damage sensor and acting as a regulatory center for XPD helicase (Rudolf et al., 2006; Fan et al., 2008; Wolski et al., 2008, 2010; Houten et al., 2016).
Furthermore, XPD serves as the authenticator of the DNA lesion initially sensed by XPC-HR23B which preludes the binding of TFIIH at the site of lesion (Oksenych and Coin, 2010). The opening of the DNA duplex at the site of lesion requires the dual ATPase function of both XPB and XPD, but the helicase activity of XPD plays a critical role in the opening of the DNA. The biochemical data vividly demonstrated that mutations in the motif I (containing ATPase activity) of either XPB or XPD inhibits the formation of DNA bubble at the lesion site but the mutations in the motif III and IV (containing helicase activities) of XPB impairs its functionality but does not inhibit NER in vivo (Coin et al., 2007). However, some specific mutations in both XPB and XPD can completely prevent opening and dual incision of the DNA lesions site in NER (Evans et al., 1997). Additionally, it has also been demonstrated that binding of N-terminal p44 subunit with XPD stimulates its helicase activity by almost 10-fold. Furthermore, mutations in the C-terminal domain (CTD) of XPD prevents the interactions with p44 resulting not only in decrease in the overall TFIIH helicase activity but also modulates TFIIH composition and contributing to further transcription defects (Coin et al., 1998, 1999). XPD has also been demonstrated to control the cell cycle via its interaction with CAK domain of the TFIIH complex. Downregulation of XPD as happens at the beginning of the mitoses initiates the disengagement of CAK module from TFIIH complex and its eventual role as regulator of cell cycle independent of TFIIH core complex (Chen et al., 2003).
XPD Gene SNPs, DNA Repair Capacity and CRC
XPD gene is located at chromosome 19q13.3 and comprises of 23 exons which span around ~54.3 kb in length; cDNA of this gene is about 2,400 nt (Benhamou and Sarasin, 2002). Point mutations in the human XPD protein play a causative role in DNA repair-deficiency diseases (xeroderma pigmentosum, trichothiodystrophy, and Cockayne syndrome), which are characterized by high ultraviolet-light hypersensitivity, a high mutation frequency, and cancer-proneness, as well as some mental and growth retardation and probably aging. Till date almost 100 different mutations have been in the XPD gene (Itin et al., 2001; Fan et al., 2008) (most important are given in Table 3). Most of the mutations affecting XPD are clustered in the C-terminal domain of the protein, which is the pivotal interaction domain of XPD for p44, as already pointed out in the above section (Coin et al., 1998, 1999; Fan et al., 2008; Liu et al., 2008; Wolski et al., 2008, 2010).
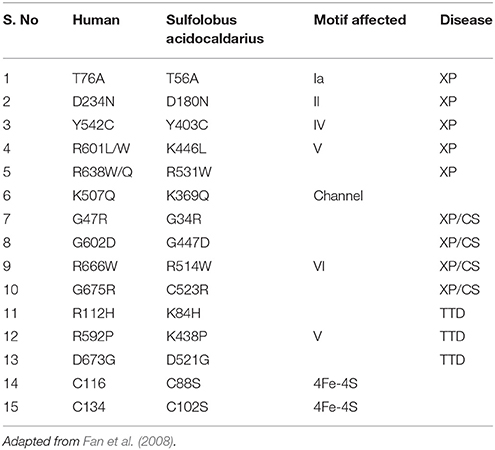
Table 3. Most common mutations affecting XPD protein.
In addition to the deleterious point mutations, a number of single nucleotide polymorphisms (SNPs) have been found in the XPD gene affecting both exonic and intronic regions of the gene). Till date researchers have defined 17 different SNPs in the XPG gene in his seminal study; seven of which affected the coding regions of the gene (exons 6, 8, 10, 17, 22, and 23) and hence affected XPD enzymatic activity (Shen et al., 1998; Mohrenweiser et al., 2002). Among all, four SNPs result in amino acid changes: isoleucine to methionine in codon 199 (C > G), histidine to tyrosine in codon 201 (C > T), aspartic acid to asparagine in codon 312 (G > A) and lysine to glutamine in codon 751 (A > C). Out of these four; only two are the most commonly occurring ones—codon 312 and 751 while as other two—codon 199 and 201 are rare (Shen et al., 1998; Benhamou and Sarasin, 2002; Gdowicz-Klosok et al., 2013).
Among all the reported SNPs of XPD, most of the population based case control studies have focused on studying the effects of SNPs affecting codons 156, 312, and 751 only, partly because of their high occurring frequency and partly because of their effects on XPD helicase activity (Coin et al., 1998; Shen et al., 1998; Benhamou and Sarasin, 2002).
XPD Asp312Asn and Lys751Gln two of the most common SNPs located within the exon 23 of the XPD gene which affects the C-terminal domain of XPD helicase that is known to interact with p44 protein of TFIIH complex, thereby stimulating XPD helicase activity (Coin et al., 1999). Thus, these two SNPs may therefore affect different protein interactions; diminish the activity of TFIIH complexes (Shen et al., 1998). In addition, XPD Lys751Gln SNP is also known to reduce the XPD protein expression by decreasing the mRNA stability (Moisan et al., 2012).
Lunn et al. (2000) was the first to report the reduced repair of X-ray induced DNA damage by XPD Lys751Lys genotype. It was reported that Individuals with the XPD 751 Lys/Lys genotype had a higher number of chromatid aberrations than those having a 751Gln allele. Possessing a Lys/Lys751 genotype increased the risk of sub-optimal DNA repair by almost 7 folds, suggesting that the Lys751 (common) allele may alter the XPD protein product resulting in sub-optimal repair of X-ray-induced DNA damage.
Furthermore, it has been also reported that the XPD Lys751 allele is associated with a high level of UVC-induced formation of DNA strand breaks (Møller et al., 2000). Also, Lunn et al. (2000) suggested that XPD Lys751 may alter the XPD protein product resulting in the suboptimal repair of X-ray induced DNA damage. However in contrast, two studies reported that the cells containing the homozygous Lys/Lys XPD protein had the elevated repair capacity than the cell containing XPD protein with Gln in either of the two forms (Spitz et al., 2001; Qiao et al., 2002).
A large number of epidemiological studies have been carried out recently to understand the effects and role of XPD SNPs on the modulation of risk of CRC; while some studies found a significant association between the two (Skjelbred et al., 2006; Gan et al., 2012; Huang et al., 2013; Procopciuc and Osian, 2013; Rezaei et al., 2013; Paszkowska-Szczur et al., 2015) others failed to link them (Yeh et al., 2005; Zhang et al., 2011, 2014; Du et al., 2014; Moghtit et al., 2014).
Two important recent meta-analyses - one by Zhang et al. (2014) on 11 case-control studies (including a total of 3 2,961 cases and 4,539 controls) and another by Zhang et al. (2011) on 15 case–control studies (including a total of 3,042 cases and 4,627 controls) did not found any evidence of a link between the XPD Lys751Gln polymorphism and risk of CRC. A recent study by Moghtit et al. (2014) on Western Algerian CRC patients (consisting of 129 cases and 148 controls) reported no association of the XPD Lys751Gln with CRC risk. Furthermore, Sliwinski et al. (2009) in their study on polish CRC cases did not find any significant association between any genotype of XPD 751 codon SNP and the occurrence of CRC; they also did not observe any relationship between XPD 751 SNP and any of the clinicopathological parameters.
Paszkowska-Szczur et al. (2015) in their study on polish CRC patients observed a significant association of XPD 312 SNP with the risk of developing CRC and strongly in men. Also, the study of Rezaei et al. (2013) in their study on Iranian CRC cases observed that individuals with heterozygous variant (Lys/Gln) SNP of XPD gene may have an increased susceptibility to CRC compared to other SNPs (Lys/Lys and Gln/Gln). Furthermore, they observed that heterozygous variant (Lys/Gln) was more frequent in CRC patients than in the control group. Similar results were also reported previously by Skjelbred et al. (2006) and Moreno et al. (2006) in their own respective populations. Also, Stern et al. (2006) have demonstrated lower risk of developing CRC in homozygous (Lys/Lys) SNP carriers. Contrarily, the study of Wang et al. (2010) on Indian CRC patients found that XPD 751Gln allele demonstrated the 3.5 times increased risk of rectal cancer.
However, meta-analysis by Mandal et al. (2014) of 13 case-control studies (including 3,087 cases and 3,599 controls) reported the likely association of the XPD Lys751Gln polymorphism with the risk of development of cancer in Indian population. Their meta-analysis concluded that XPD Lys/Gln and XPD Gln/Gln genotypes had had 1.3- and 1.6-fold increased risk of developing cancer as compared with the wild XPD Lys/Lys genotype, respectively. Similarly, another meta-analysis of 37 case-control studies (including 9,027 cases and 16,072 controls) by Du et al. (2014) suggested that the XPD 751Gln/Gln genotype was a low-penetrate risk factor for developing digestive tract cancers, especially in Asian populations.
Conclusion
Since XPD is one of the major molecules which connect the two essential processes of sustenance of life–NER pathway and transcription process, it is one of the most analyzed molecules of NER in various epidemiological studies carried out on CRC. However, even though a decade of research on XPD gene and its SNPs, no clear relationships between its various SNPs and the risk of CRC has been established till date. To establish a cohesive data on XPD SNPs well-designed studies with large statistical power is warranted to clarify the ambiguity associated with the current data on XPD SNPs.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Benatti, P., Gafa, R., Barana, D., Marino, M., Scarselli, A., Pedroni, M., Maestri, I., et al. (2005). Microsatellite instability and CRC prognosis. Clin. Cancer Res. 11, 8332–8340. doi: 10.1158/1078-0432.CCR-05-1030
Benhamou, S., and Sarasin, A. (2000). Variability in nucleotide excision repair and cancer risk: a review. Mutat. Res. 462, 149–158. doi: 10.1016/S1383-5742(00)00032-6
Benhamou, S., and Sarasin, A. (2002). ERCC2/XPD gene polymorphisms and cancer risk. Mutagenesis 17, 463–469. doi: 10.1093/mutage/17.6.463
Boland, C. R., and Goel, A. (2010). Microsatellite instability in colorectal cancer. Gastroenterology 138, 2073.e3-2087.e3. doi: 10.1053/j.gastro.2009.12.064
Busso, D., Keriel, A., Sandrock, B., Poterszman, A., Gileadi, O., and Egly, J. M. (2000). Distinct regions of MAT1 regulate cdk7 kinase and TFIIH transcription activities. J. Biol. Chem. 275, 22815–22823. doi: 10.1074/jbc.M002578200
Cameroni, E., Stettler, K., and Suter, B. (2010). On the traces of XPD: cell cycle matters - untangling the genotype-phenotype relationship of XPD mutations. Cell Div. 5:24. doi: 10.1186/1747-1028-5-24
Chang, W. H., and Kornberg, R. D. (2000). Electron crystal structure of the transcription factor and DNA repair complex, core TFIIH. Cell 102, 609–613. doi: 10.1016/S0092-8674(00)00083-0
Chen, J., Larochelle, S., Li, X., and Suter, B. (2003). Xpd/Ercc2 regulates CAK activity and mitotic progression. Nature 424, 228–232. doi: 10.1038/nature01746
Chen, J., and Suter, B. (2003). Xpd, a structural bridge and a functional link. Cell Cycle 2, 503–506. doi: 10.4161/cc.2.6.558
Coin, F., Bergmann, E., Tremeau-Bravard, A., and Egly, J. M. (1999). Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 18, 1357–1366. doi: 10.1093/emboj/18.5.1357
Coin, F., Marinoni, J. C., Rodolfo, C., Fribourg, S., Pedrini, A. M., and Egly, J. M. (1998). Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat. Genet. 20, 184–188.
Coin, F., Oksenych, V., and Egly, J. M. (2007). Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell. 26, 245–256. doi: 10.1016/j.molcel.2007.03.009
Compe, E., and Egly, J. M. (2012). TFIIH: when transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 13, 343–354. doi: 10.1038/nrm3350
Cunningham, D., Atkin, W., Lenz, H. J., Lynch, H. T., Minsky, B., Nordlinger, B., et al. (2010). Colorectal cancer. Lancet 375, 1030–1047. doi: 10.1016/S0140-6736(10)60353-4
de Laat, W. L., Jaspers, N. G., and Hoeijmakers, J. H. (1999). Molecular mechanism of nucleotide excision repair. Genes Dev. 13, 768–785. doi: 10.1101/gad.13.7.768
Du, H., Guo, N., Shi, B., Zhang, Q., Chen, Z., Lu, K., et al. (2014). The effect of XPD polymorphisms on digestive tract cancers risk: a meta-analysis. PLoS ONE 9:e96301. doi: 10.1371/journal.pone.0096301
Egly, J. M., and Coin, F. (2011). A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 10, 714–721. doi: 10.1016/j.dnarep.2011.04.021
Evans, E., Moggs, J. G., Hwang, J. R., Egly, J. M., and Wood, R. D. (1997). Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 16, 6559–6573. doi: 10.1093/emboj/16.21.6559
Fan, L., Fuss, J. O., Cheng, Q. J., Arvai, A. S., Hammel, M., Roberts, V. A., et al. (2008). XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell 133, 789–800. doi: 10.1016/j.cell.2008.04.030
Fuss, J. O., and Tainer, J. A. (2011). XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst). 10, 697–713. doi: 10.1016/j.dnarep.2011.04.028
Gan, Y., Li, X. R., Chen, D. J., and Wu, J. H. (2012). Association between polymorphisms of XRCC1 Arg399Gln and XPD Lys751Gln genes and prognosis of colorectal cancer in a Chinese population. Asian Pac. J. Cancer Prev. 13, 5721–5724. doi: 10.7314/APJCP.2012.13.11.5721
Gdowicz-Klosok, A., Widel, M., and Rzeszowska-Wolny, J. (2013). The influence of XPD, APE1, XRCC1, and NBS1 polymorphic variants on DNA repair in cells exposed to X-rays. Mutat. Res. 755, 42–48. doi: 10.1016/j.mrgentox.2013.05.002
Goel, A., Arnold, C. N., Niedzwiecki, D., Chang, D. K., Ricciardiello, L., Carethers, J. M., et al. (2003). Characterization of sporadic colon cancer by patterns of genomic instability. Cancer Res. 63, 1608–1614.
Grady, W. M. (2004). Genomic instability and colon cancer. Cancer Metastasis Rev. 23, 11–27. doi: 10.1023/A:1025861527711
Hoeijmakers, J. H. (2001). Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374. doi: 10.1038/35077232
Houten, B. V., Kuper, J., and Kisker, C. (2016). Role of XPD in cellular functions: To TFIIH and beyond. DNA Repair (Amst) 44, 136–142. doi: 10.1016/j.dnarep.2016.05.019
Huang, M. Y., Wang, J. Y., Huang, M. L., Chang, H. J., and Lin, S. R. (2013). Polymorphisms in XPD and ERCC1 associated with colorectal cancer outcome. Int. J. Mol. Sci. 14, 4121–4134. doi: 10.3390/ijms14024121
Iben, S., Tschochner, H., Bier, M., Hoogstraten, D., Hozák, P., Egly, J. M., et al. (2002). TFIIH plays an essential role in RNA polymerase I transcription. Cell 109, 297–306. doi: 10.1016/S0092-8674(02)00729-8
Itin, P. H., Sarasin, A., and Pittelkow, M. R. (2001). Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J. Am. Acad Dermatol. 44, 891–920. doi: 10.1067/mjd.2001.114294
Iyama, T., and Wilson, D. M. III. (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst). 12, 620–636. doi: 10.1016/j.dnarep.2013.04.015
Jasin, M., and Rodney, R. (2013). Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5:a012740. doi: 10.1101/cshperspect.a012740
Kerr, D. J., Young, E. M., and Hobbs, F. D. R. (2001). ABC of Colorectal Cancer, 1st Edn. London: BMJ.
Kim, M. S., Lee, J., and Sidransky, D. (2010). DNA methylation markers in colorectal cancer. Cancer Metastasis Rev. 29, 181–206. doi: 10.1007/s10555-010-9207-6
Komarnitsky, P., Cho, E. J., and Buratowski, S. (2000). Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460. doi: 10.1101/gad.824700
Liu, H., Rudolf, J., Johnson, K. A., McMahon, S. A., Oke, M., Carter, L., et al. (2008). Structure of the DNA repair helicase XPD. Cell 133, 801–812. doi: 10.1016/j.cell.2008.04.029
Lunn, R. M., Helzlsouer, K. J., Parshad, R., Umbach, D. M., Harris, E. L., Sanford, K. K., et al. (2000). XPD polymorphisms: effects on DNA repair proficiency. Carcinogenesis 21, 551–555. doi: 10.1093/carcin/21.4.551
Mandal, R. K., Yadav, S. S., and Panda, A. K. (2014). Meta-analysis on the association of nucleotide excision repair gene XPD A751C variant and cancer susceptibility among Indian population. Mol. Biol. Rep. 41, 713–719 doi: 10.1007/s11033-013-2910-y
McGranahan, N., Burrell, R. A., Endesfelder, D., Novelli, M. R., and Swanton, C. (2012). Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 13, 528–538. doi: 10.1038/embor.2012.61
Michailidi, C., Papavassiliou, A. G., and Troungos, C. (2012). DNA repair mechanisms in colorectal carcinogenesis. Curr. Mol. Med. 12, 237–246. doi: 10.2174/156652412799218859
Migliore, L., Igheli, F., Spisni, R., and Coppede, F. (2011). Genetics, cytogenetics and epigenetics of colorectal cancer. J. Biomed. Biotechnol. 1, 1–9. doi: 10.1155/2011/792362
Missura, M., Buterin, T., Hindges, R., Hübscher, U., Kaspárková, J., Brabec, V., et al. (2001). Double check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J. 20, 3554–3564. doi: 10.1093/emboj/20.13.3554
Moghtit, F. Z., Aberkane, M. S., Le Morvan, V., Louhibi, L., Bellot, R., Bousahba, A., et al. (2014). No association between XRCC3 Thr241Met and XPD Lys751Gln polymorphisms and the risk of colorectal cancer in West Algerian population: a case-control study. Med. Oncol. 31:942. doi: 10.1007/s12032-014-0942-3
Mohrenweiser, H. W., Xi, T., Vázquez-Matías, J., and Jones, I. M. (2002). Identification of 127 amino acid substitution variants in screening 37 DNA repair genes in humans. Cancer Epidemiol. Biomarkers Prev. 11(10 Pt 1), 1054–1064.
Moisan, F., Laroche-clary, A., Auzanneau, C., Ricard, N., Pourquier, P., Robert, J., et al. (2012). Deciphering the role of the ERCC2 gene polymorphism on anticancer drug sensitivity. Carcinogenesis 33, 962–968. doi: 10.1093/carcin/bgs107
Møller, P., Wallin, H., Dybdahl, M., Frentz, G., and Nexø, B. A. (2000). Psoriasis patients with basal cell carcinoma have more repair-mediated DNA strand-breaks after UVC damage in lymphocytes than psoriasis patients without basal cell carcinoma. Cancer Lett. 151, 187–192. doi: 10.1016/S0304-3835(99)00414-0
Moreno, V., Gemignani, F., Landi, S., Gioia-Patricola, L., Chabrier, A., Blanco, I., et al. (2006). Polymorphisms in genes of nucleotide and base excision repair: risk and prognosis of colorectal cancer. Clin. Cancer Res. 12, 2101–2108. doi: 10.1158/1078-0432.CCR-05-1363
Mydlikova, Z., Gursky, J., and Pirsel, M. (2010). Transcription factor IIH - the protein complex with multiple functions. Neoplasma 57, 287–290. doi: 10.4149/neo_2010_04_287
Nouspikel, T. (2009). DNA repair in mammalian cells: nucleotide excision repair: variations on versatility. Cell. Mol. Life Sci. 66, 994–1009. doi: 10.1007/s00018-009-8737-y
Ogino, S., Chan, A. T., Fuchs, C. S., Giovannucci, E., et al. (2011). Molecular pathological epidemiology of colorectal neoplasia: an emerging trans disciplinary and interdisciplinary field. Gut 60, 397–411. doi: 10.1136/gut.2010.217182
Oksenych, V., Bernardes de Jesus, B., Zhovmer, A., Egly, J. M., and Coin, F. (2009). Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 28, 2971–2980. doi: 10.1038/emboj.2009.230
Oksenych, V., and Coin, F. (2010). The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle 9, 90–96. doi: 10.4161/cc.9.1.10267
Paszkowska-Szczur, K., Scott, R. J., Górski, B., Cybulski, C., Kurzawski, G., Dymerska, D., et al. (2015). Polymorphisms in nucleotide excision repair genes and susceptibility to colorectal cancer in the Polish population. Mol. Biol. Rep. 42, 755–764. doi: 10.1007/s11033-014-3824-z
Petruseva, I. O., Evdokimov, A. N., and Lavrik, O. I. (2014). Molecular mechanism of global genome nucleotide excision repair. Acta Naturae. 6, 23–34.
Procopciuc, L. M., and Osian, G. (2013). Lys751Gln, X. P. D., and Arg399Gln XRCC1 in Romanians. Association with sporadic colorectal cancer risk and different stages of carcinomas. Chirurgia (Bucur) 108, 711–8. doi: 10.19041/Apstract/2013/4-5/17
Qiao, Y., Spitz, M. R., Shen, H., Guo, Z., Shete, S., Hedayati, M., et al. (2002). Modulation of repair of ultraviolet damage in the host-cell reactivation assay by polymorphic XPC and XPD/ERCC2 genotypes. Carcinogenesis 23, 295–299. doi: 10.1093/carcin/23.2.295
Rezaei, H., Motovali-Bashi, M., Khodadad, K., Elahi, A., Emami, H., and Naddaffnia, H. (2013). Relationship between XPD Lys 751 Gln polymorphism and colorectal cancer risk: a case-control study in a population-based study. Gastroenterol. Hepatol. Bed. Bench. 6, 18–24.
Richards, J. D., Cubeddu, L., Roberts, J., Liu, H., and White, M. F. (2008). The archaeal XPB protein is a ssDNA-dependent ATPase with a novel partner. J. Mol. Biol. 376, 634–644. doi: 10.1016/j.jmb.2007.12.019
Rudolf, J., Makrantoni, V., Ingledew, W. J., Stark, M. J., and White, M. F. (2006). The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol. Cell 23, 801–808. doi: 10.1016/j.molcel.2006.07.019
Rudolf, J., Rouillon, C., Schwarz-Linek, U., and White, M. F. (2010). The helicase XPD unwinds bubble structures and is not stalled by DNA lesions removed by the nucleotide excision repair pathway. Nucleic Acids Res. 38, 931–941. doi: 10.1093/nar/gkp1058
Sameer, A. S. (2013). Colorectal cancer: Molecular mutations and polymorphisms. Front. Oncol. 3:114. doi: 10.3389/fonc.2013.00114
Sameer, A. S., and Nissar, S. (2016). Understanding epigenetics: an alternative mechanism of colorectal carcinogenesis. Curr. Colorectal Cancer Rep. 12, 113–122. doi: 10.1007/s11888-016-0317-6
Sameer, A. S., Nissar, S., and Fatima, K. (2014). Mismatch repair pathway: molecules, functions, and role in colorectal carcinogenesis. Eur. J. Cancer Prev. 23, 246–257. doi: 10.1097/CEJ.0000000000000019
Serizawa, H., Conaway, J. W., and Conaway, R. C. (1993). Phosphorylation of C-terminal domain of RNA polymerase II is not required in basal transcription. Nature 363, 371–374. doi: 10.1038/363371a0
Shen, M. R., Jones, I. M., and Mohrenweiser, H. (1998). Nonconservative amino acid substitution variants exist at polymorphic frequency in DNA repair genes in healthy humans. Cancer Res. 58, 604–608.
Skjelbred, C. F., Saebø, M., Wallin, H., Nexø, B. A., Hagen, P. C., Lothe, I. M., et al. (2006). Polymorphisms of the XRCC1, XRCC3 and XPD genes and risk of colorectal adenoma and carcinoma, in a Norwegian cohort: a case control study. BMC Cancer 6:67. doi: 10.1186/1471-2407-6-67
Sliwinski, T., Krupa, R., Wisniewska-Jarosinska, M., Pawlowska, E., Lech, J., Chojnacki, J., et al. (2009). Common polymorphisms in the XPD and hOGG1 genes are not associated with the risk of colorectal cancer in a Polish population. Tohoku J. Exp. Med. 218, 185–191. doi: 10.1620/tjem.218.185
Spitz, M. R., Wu, X., Wang, Y., Wang, L. E., Shete, S., Amos, C. I., et al. (2001). Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients. Cancer Res. 61, 1354–1357.
Spivak, G. (2015). Nucleotide excision repair in humans. DNA Repair (Amst) 36, 13–18. doi: 10.1016/j.dnarep.2015.09.003
Stern, M. C., Siegmund, K. D., Conti, D. V., Corral, R., and Haile, R. W. (2006). XRCC1, XRCC3, and XPD polymorphisms as modifiers of the effect of smoking and alcohol on colorectal adenoma risk. Cancer 15, 2384–2390. doi: 10.1158/1055-9965.EPI-06-0381
Tirode, F., Busso, D., Coin, F., and Egly, J. M. (1999). Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunits, XPB XPD and cdk7. Mol. Cell 3, 87–95. doi: 10.1016/S1097-2765(00)80177-X
Toyota, M., Ahuja, N., Ohe-Toyota, M., Herman, J. G., Baylin, S. B., and Issa, J. P. (1999). CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 96, 8681–8686. doi: 10.1073/pnas.96.15.8681
Vogelstein, B., Fearon, E. R., Hamilton, S. R., Kern, S. E., Preisinger, A. C., Leppert, M., et al. (1988). Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319, 525–532. doi: 10.1056/NEJM198809013190901
Wang, J., Zhao, Y., Jiang, J., Gajalakshmi, V., Kuriki, K., Nakamura, S., et al. (2010). Polymorphisms in DNA repair genes XRCC1, XRCC3 and XPD, and colorectal cancer risk: a case-control study in an Indian population. J. Cancer Res. Clin. Oncol. 136, 1517–1525. doi: 10.1007/s00432-010-0809-8
Winkler, G. S., Araújo, S. J., Fiedler, U., Vermeulen, W., Coin, F., Egly, J. M., et al. (2000). TFIIH with inactive XPD helicase functions in transcription initiation but is defective in DNA repair. J. Biol. Chem. 275, 4258–4266. doi: 10.1074/jbc.275.6.4258
Wolski, S. C., Kuper, J., Hänzelmann, P., Truglio, J. J., Croteau, D. L., Van Houten, B., et al. (2008). Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 6:e149. doi: 10.1371/journal.pbio.0060149
Wolski, S. C., Kuper, J., and Kisker, C. (2010). The XPD helicase: XPanDing archaeal XPD structures to get a grip on human DNA repair. Biol. Chem. 391, 761–765. doi: 10.1515/bc.2010.076
Yeh, C. C., Sung, F. C., Tang, R., Chang-Chieh, C. R., and Hsieh, L. L. (2005). Polymorphisms of the XRCC1, XRCC3, & XPD genes, and colorectal cancer risk: a case-control study in Taiwan. BMC Cancer. 5:12. doi: 10.1186/1471-2407-5-12
Zhang, T., Zhang, D. M., Zhao, D., Hou, X. M., Ma, S. C., and Liu, X. J. (2014). Lack of association between the XPD Lys751Gln polymorphism and colorectal cancer risk: a meta-analysis. Onco. Targets. Ther. 7, 1255–1260. doi: 10.2147/OTT.S66291
Zhang, Y., Ding, D., Wang, X., Zhu, Z., Huang, M., and He, X. (2011). Lack of association between XPD Lys751Gln and Asp312Asn polymorphisms and colorectal cancer risk: a meta-analysis of case-control studies. Int. J. Colorectal Dis. 26, 1257–1264. doi: 10.1007/s00384-011-1222-8
Zhovmer, A., Oksenych, V., and Coin, F. (2010). Two sides of the same coin: TFIIH complexes in transcription and DNA repair. Sci. World J. 10, 633–643. doi: 10.1100/tsw.2010.46
Keywords: colorectal cancer, polymorphism, RFLP, DNA repair, NER, XPD, TFIIH, xeroderma pigmentosum
Citation: Sameer AS and Nissar S (2018) XPD–The Lynchpin of NER: Molecule, Gene, Polymorphisms, and Role in Colorectal Carcinogenesis. Front. Mol. Biosci. 5:23. doi: 10.3389/fmolb.2018.00023
Received: 07 January 2018; Accepted: 27 February 2018;
Published: 20 March 2018.
Edited by:
Eleonora Napoli, University of California, Davis, United StatesReviewed by:
Kaushlendra Tripathi, University of Alabama at Birmingham, United StatesPrasun K. Datta, Temple University, United States
Copyright © 2018 Sameer and Nissar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aga Syed Sameer, YWdhc3lAbmdoYS5tZWQuc2E=; YWdhc0Brc2F1LWhzLmVkdS5zYQ==