 Soonkyu Hwang
Soonkyu Hwang Namil Lee1
Namil Lee1 Bernhard Palsson
Bernhard Palsson Byung-Kwan Cho
Byung-Kwan Cho- 1Systems and Synthetic Biology Laboratory, Department of Biological Sciences and KI for the BioCentury, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
- 2Department of Bioengineering, University of California, San Diego, La Jolla, CA, United States
- 3Department of Pediatrics, University of California, San Diego, La Jolla, CA, United States
- 4The Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Lyngby, Denmark
- 5Intelligent Synthetic Biology Center, Daejeon, South Korea
In nature, various enzymes govern diverse biochemical reactions through their specific three-dimensional structures, which have been harnessed to produce many useful bioactive compounds including clinical agents and commodity chemicals. Polyketide synthases (PKSs) and non-ribosomal peptide synthetases (NRPSs) are particularly unique multifunctional enzymes that display modular organization. Individual modules incorporate their own specific substrates and collaborate to assemble complex polyketides or non-ribosomal polypeptides in a linear fashion. Due to the modular properties of PKSs and NRPSs, they have been attractive rational engineering targets for novel chemical production through the predictable modification of each moiety of the complex chemical through engineering of the cognate module. Thus, individual reactions of each module could be separated as a retro-biosynthetic biopart and repurposed to new biosynthetic pathways for the production of biofuels or commodity chemicals. Despite these potentials, repurposing attempts have often failed owing to impaired catalytic activity or the production of unintended products due to incompatible protein–protein interactions between the modules and structural perturbation of the enzyme. Recent advances in the structural, computational, and synthetic tools provide more opportunities for successful repurposing. In this review, we focused on the representative strategies and examples for the repurposing of modular PKSs and NRPSs, along with their advantages and current limitations. Thereafter, synthetic biology tools and perspectives were suggested for potential further advancement, including the rational and large-scale high-throughput approaches. Ultimately, the potential diverse reactions from modular PKSs and NRPSs would be leveraged to expand the reservoir of useful chemicals.
Introduction
Enzymes are biosynthetic protein machineries that recognize specific substrates through unique three-dimensional structures, and catalyze the conversion of these substrates into new biomolecules (Agarwal, 2006). Harnessing their diverse biochemical reactions has led to the production of many bioactive-compounds as clinical agents and commodity chemicals (Tibrewal and Tang, 2014). In addition, engineering such enzymes and repurposing their reactions into new pathways enhances the biocatalytic properties and the diversity of the natural products, respectively (Tibrewal and Tang, 2014). Biosynthesis of organic compounds has several advantages compared to classical chemical synthesis methods (Wenda et al., 2011). First, enzymes are not environmentally harmful, they act as non-toxic catalysts. The reaction conditions for the production of diverse chemicals are generally moderate in terms of temperature, pressure, and pH, while the classical chemical synthesis often requires extreme conditions. High selectivity of enzymes yields high purities with specific stereochemistry of the product and reduce undesired by-products and toxic intermediates. In nature, interestingly, the mechanisms underlying a large number of enzyme reactions have not been discovered yet. For example, the recent genome mining efforts on bacteria, fungi, and plants have revealed the richness of secondary metabolite biosynthetic gene clusters (smBGCs) including many unidentified smBGCs (Rutledge and Challis, 2015). Thus, novel non-natural chemicals and pathways have been constructed by the reprogramming of the multi-enzyme complex encoded by smBGCs (Bernhardt and O’Connor, 2009; Chevrette et al., 2019).
Type I modular polyketide synthases (PKSs) and non-ribosomal peptide synthetases (NRPSs) are prominent engineering targets due to their modular properties of enzyme assembly (Ladner and Williams, 2016). Type I modular PKS comprises several modules, each responsible for the incorporation and modification of one acyl-CoA substrate to synthesize the polyketide product, such as erythromycin (antibiotic), rapamycin (immunosuppressant), amphotericin B (antifungal), and other potential clinical agents (Staunton and Weissman, 2001). Likewise, NRPS comprises numerous modules, with each of them responsible for the incorporation and modification of one amino acid substrate to extend the polypeptide product, such as daptomycin (antibiotic), actinomycin D (antitumor), cyclosporine A (immunosuppressant), and other potential clinical agents (Walsh, 2016; Sussmuth and Mainz, 2017). As the organization and the order of these modules are co-linearly correlated with each unit of the final polyketide and polypeptide products, the target modules of rational engineering could be predicted for the production of novel derivatives having the modified unit at a specific position (Alanjary et al., 2019). Using this modular biosynthetic logic of type I modular PKS and NRPS, each reaction of a module was capable of being separated from the original assembly reactions and repurposed to construct de novo biosynthetic pathway for other chemicals (Pang et al., 2019). This approach is favorable in terms of (i) the potential diversity of available synthetic parts governing unique chemical reactions, (ii) enabling retro-biosynthesis by combinatorial assembly of domains and modules, and (iii) the relative ease of engineering, owing to avoidance of the structural perturbation compared to the engineering within the multi-enzyme complex.
In this review, we briefly introduced the structure and mechanism of the modular PKS and NRPS, and thereafter focused on their repurposing examples, along with their advantages and limitations. Finally, tools in the design-build-test-learn cycle of synthetic biology and the future perspectives of the repurposing strategies were discussed.
Modular PKS and NRPS Architecture and Mechanism
Polyketide synthases are categorized into three types, namely, types I, II, and III, according to their organization and catalytic mechanisms (Yu et al., 2012). Among them, type I modular PKS has a hierarchical organization in which, the entire enzyme complex is composed of several subunits, each subunit is composed of several modules, and a module is composed of several domains (Bayly and Yadav, 2017) (Figure 1A). A minimal elongation module includes three domains; (i) an acyltransferase (AT) domain for loading the chain extender unit (typically malonyl- or methylmalonyl-CoA), (ii) an acyl carrier protein (ACP) for tethering and shuttling the extender unit or the polyketide intermediate, and (iii) a ketosynthase (KS) domain for catalyzing the condensation reaction between the extender unit of the downstream ACP domain and the polyketide intermediate attached at the KS active site which is translocated from the ACP domain of upstream module. Addition of other domains to this minimal elongation module modifies a polyketide backbone.
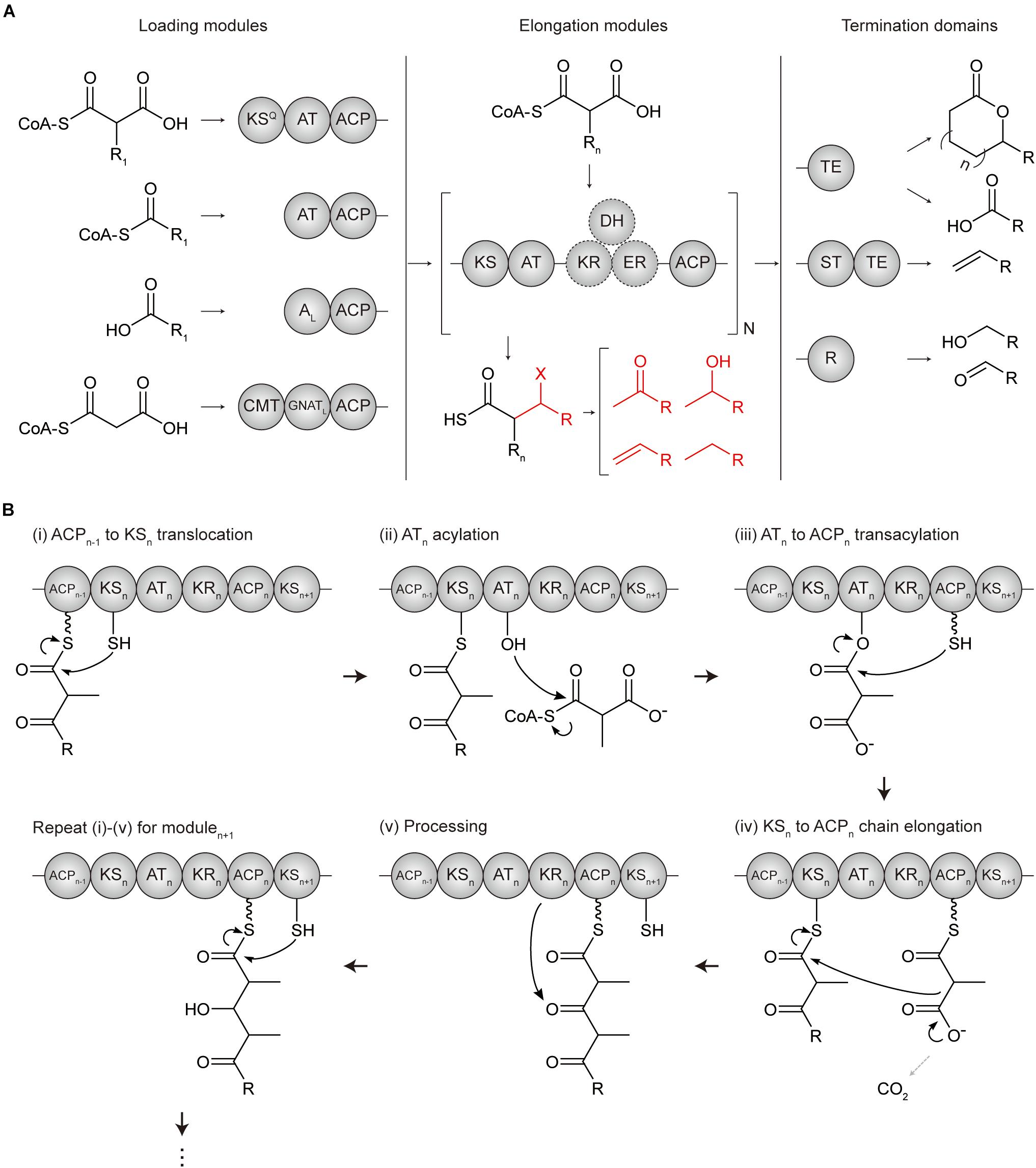
Figure 1. Domain architectures and mechanisms of polyketide chain extension in modular PKS. (A) Overall flow scheme of polyketide biosynthesis with different domain architectures of modules. Four types of loading modules load the different substrates according to involved domains (chemical examples were indicated). Next, the extender unit is selected and condensed to the growing chain one by one per elongation module for N cycles. Optional reductive domains (dashed circles) reduce the β-carbon group resulting in different X groups (indicated in red). Finally, the growing polyketide chain is cleaved by three different types of offloading domains in termination modules producing different products, including linear carboxylic acids, macrocyclic acids, olefins, aldehydes, and primary alcohols. (B) Mechanism of polyketide chain extension for the elongation modulen. (i) ACPn–1 to KSn translocation; the active site cysteine moiety of KSn receives the growing polyketide chain of ACPn–1. (ii) ATn acylation; the cognate acyl unit is incorporated into the active site serine moiety of ATn to form the acyl-O-AT intermediate. (iii) ATn to ACPn transacylation; the acyl group of ATn is transacylated to the ACPn. (iv) KSn to ACPn chain elongation; KSn catalyzes a decarboxylative Claisen condensation between the growing polyketide chain and the acyl extender unit of ACPn for the chain extension. (v) Processing; the extender units of ACPn are modified by a reductive loop or other additional domains. ACP, acyl carrier protein; AL, CoA ligase-type domain; AT, acyltransferase; CMT, C-methyltransferase; GNATL, GCN5 N-acetyltransferase-like domain; DH, dehydratase; ER, enoylreductase; KR, ketoreductase; KS, ketosynthase; KSQ, condensation-incompetent ketosynthase; R, reductive domain; ST, sulfotransferase; TE, thioesterase.
In the N- to C-terminus of a whole enzyme complex, loading, elongation, and termination modules are localized to catalyze the serial polyketide production (Barajas et al., 2017). Loading module (LM) initiates the chain formation from a broad range of priming starter units by acylation to the AT domain and transacylation to the ACP domain. According to the configuration of the domains, LMs are divided into subtype A, B, C, and D (Kornfuehrer and Eustáquio, 2019). In addition to AT-ACP didomain, type A LMs involve the condensation-incompetent KS domain that decarboxylates malonyl- or methylmalonyl-CoA to yield acetyl- or propionyl starter units, respectively. Type B LM consist of only the AT-ACP didomain and has a much broader range of substrates. Type C LM has a CoA-ligase domain rather than AT domain to incorporate carboxylic acid substrate in an ATP-dependent manner. Lastly, type D LM has a GCN5 N-acetyltransferase-like domain rather than AT domain that recently repurposed to catalyze decarboxylation (Skiba et al., 2020). A common characteristic of LMs is the absence of the condensation domain, which results a more flexible substrate specificity of the AT domain than those of downstream modules. Elongation modules consist of KS, AT, ACP, and other additional domains to serially catalyze the growth of the polyketide chain (Barajas et al., 2017; Zargar et al., 2018; Kornfuehrer and Eustáquio, 2019) (Figure 1B). Termination module contains a thioesterase (TE) domain or reductive (R) domain as an offloading domain following the ACP domain to release the ACP-bound polyketide intermediate and complete polyketide biosynthesis. TE domain performs; (i) hydrolysis to yield the linear polyketide, or (ii) cyclization to yield the macrocyclic polyketide, or (iii) sulfonate transfer and decarboxylation with sulfotransferase (ST) domain to yield the terminal olefin. Alternatively, R domain catalyzes NADPH dependent two-electron reduction, yielding the aldehyde product.
Non-ribosomal peptide synthetases are categorized into two types according to their organizations and catalytic mechanisms, which are type I modular NRPS and type II standalone NRPS (Jaremko et al., 2019). Type I modular NRPS a resemblance to type I modular PKS, with a hierarchical organization of the enzyme complex-subunit-module-domain (Winn et al., 2016) (Figure 2A). A minimal elongation module includes three domains which are, an adenylation (A) domain for loading the amino acid extender unit (both proteinogenic and non-proteinogenic), an thiolation (T or PCP) domain for tethering and shuttling the extender unit transferred from the upstream A domain or the growing polypeptide transferred from T domain of the upstream module, and a condensation (C) domain for catalyzing the peptide bond formation between the amino acid extender unit of the downstream T domain and the growing polypeptide of the T domain of the upstream module. Other cis-acting and trans-acting domains to this minimal elongation module expand the extender amino acid unit.
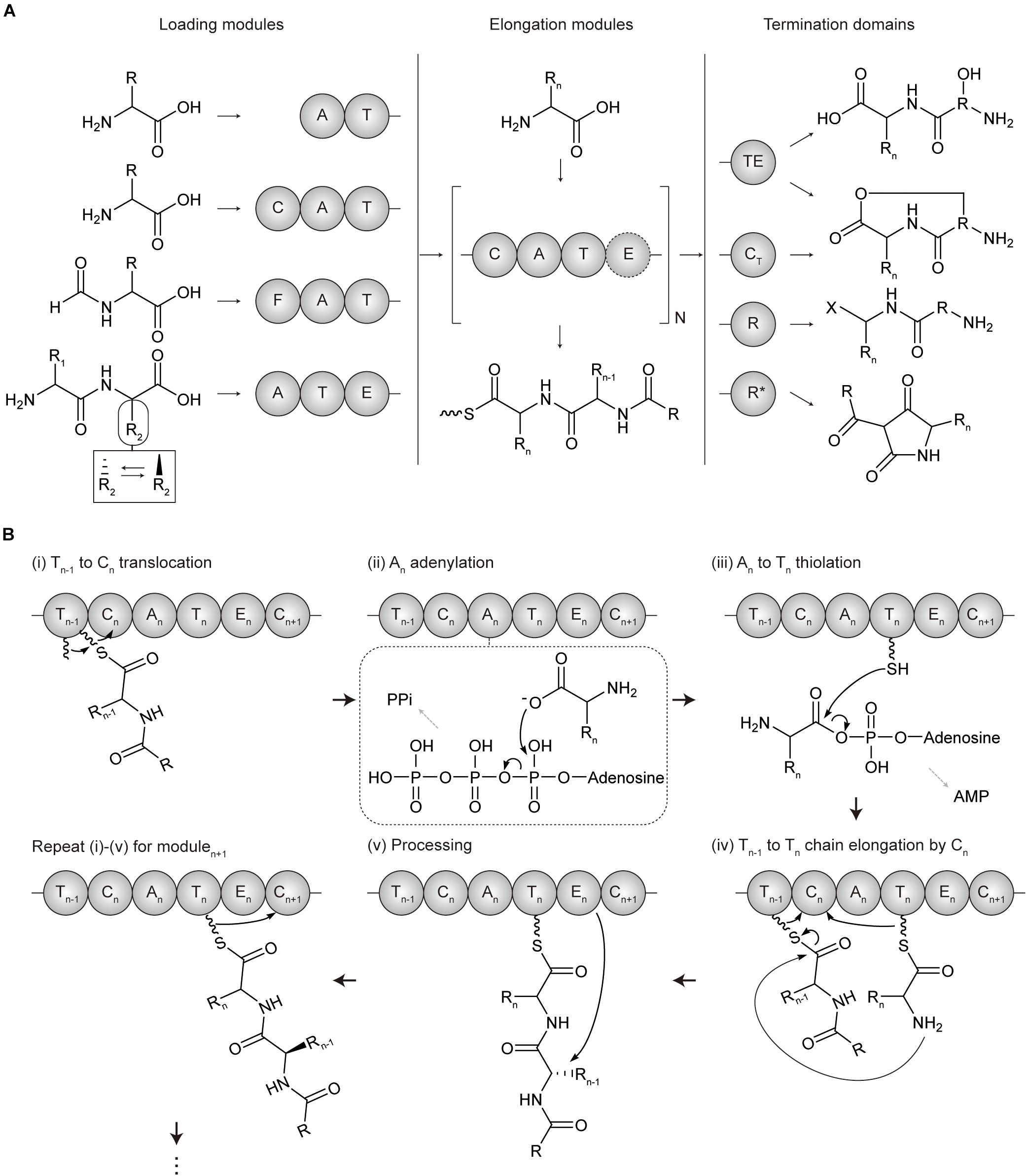
Figure 2. Domain architectures and mechanisms of non-ribosomal peptide chain extension in modular NRPS. (A) Overall flow scheme of non-ribosomal peptide biosynthesis with different domain architectures of modules. Four representative types of loading modules load the different substrates according to the involved domains (chemical examples were indicated). Next, the extender unit is selected and condensed to the growing chain one by one per elongation module for N cycles. An example of optional processing domain is indicated by the dashed circles. Finally, the growing non-ribosomal peptide chain is cleaved by four representative types of offloading domains in termination modules, producing different products including linear peptides, macrocyclic peptides, aldehydes, and tetramate moieties. The terminal X group of the product from terminal R domain includes hydroxyl group (-OH), aldehyde group (-CHO), and other aldehyde derivatives (Barajas et al., 2015; Dan et al., 2019). (B) Mechanism of polyketide chain extension for the elongation modulen. (i) Tn–1 to Cn translocation; the growing non-ribosomal peptide chain linked to Ppant arm of Tn–1 domain translocates to the solvent channel of Cn domain donor site. (ii) An adenylation; the extender amino acid unit is activated by ATP to form aminoacyl-AMP in An domain. (iii) An to Tn thiolation; the aminoacyl-AMP intermediate of An is transferred to the Ppant arm of Tn domain to form aminoacyl thioester intermediate. (iv) Tn–1 to Tn condensation at Cn; the aminoacyl thioester intermediate of Tn domain is translocated to the solvent channel of Cn domain acceptor site, and the peptide bond formation between the growing peptide of Tn–1 domain and the amino acid extender unit of Tn domain elongates by adding one amino acid to the growing peptide. (v) Processing; the extender units of ACPn are modified by an epimerase (E) domain or other additional domains. A, adenylation domain; C, condensation domain; CT, terminal condensation domain; F, formylating domain; R, reductive domain; R*, R-like domain; T, thiolation domain; TE, thioesterase.
Similar to PKS, loading, elongation, and termination module are localized to catalyze serial polypeptide production (Sussmuth and Mainz, 2017). NRPS LMs usually lack a C domain and are more variable than PKS. Many NRPS LMs carry the N-terminal modifications, which function as protection against degradation, modulating polarity, and providing specific properties such as membrane insertion. Several additional domains including formylating domain, CoA ligase domain, and other tailoring domains could be involved in LMs for acylation and formylation, among others. NRPS elongation module basically consists of three core domains C, A, and T corresponding to KS, AT, and ACP of PKS which play a role in chain elongation, substrate incorporation, and chain carrier function, respectively (Hur et al., 2012; Winn et al., 2016; Bloudoff and Schmeing, 2017; Sussmuth and Mainz, 2017) (Figure 2B). However, their overall structures and mechanisms are quite different, and there are also more various and distinct processing domains for NRPS. Examples include; epimerase (E) domain catalyzing the absolute configuration at the Cα atom, methyltransferse (MT) domain modifying the degree of C- or N-methylation, formylation (F) domain, cyclization (Cy), redox-active domain (Ox and Red), among others. These steps would be repeated between modulen and modulen + 1 till the termination module. Termination module of NRPS also contains an offloading domain such as a TE domain or reductive (R) domain after T domain to release the T-bound polypeptide intermediate for the completion of NRPS biosynthesis. The TE domain hydrolyzes or cyclize the intermediate to form a linear or cyclic polypeptide. Additionally, terminal CT domain in fungal NRPSs disconnects the oligopeptide by macrocyclization, and R∗ domain mediates Dieckmann-type cyclization of PK-NRP hybrids to obtain tetramate moieties (Sussmuth and Mainz, 2017). Diversity of NRPS is largely present in the type II NRPSs that consist of standalone or minimal domains, which have been reviewed in detail in other papers (Jaremko et al., 2019; McErlean et al., 2019).
Due to the collinear and modular biosynthetic logic, the structural diversity of the products of modular PKS and NRPS could be largely attributed to a few variables (Kornfuehrer and Eustáquio, 2019). The modules of PKS and NRPS are suitable devices for retro-biosynthesis, which starts from the target product and proceeds backward to precursors by stepwise combination of the independent module reactions (Pang et al., 2019).
Modular PKS and NRPS Engineering
As various strategies have been employed for the engineering of modular PKS and NRPS (Brown et al., 2018; Klaus and Grininger, 2018), we provided several landmark examples (Figure 3).
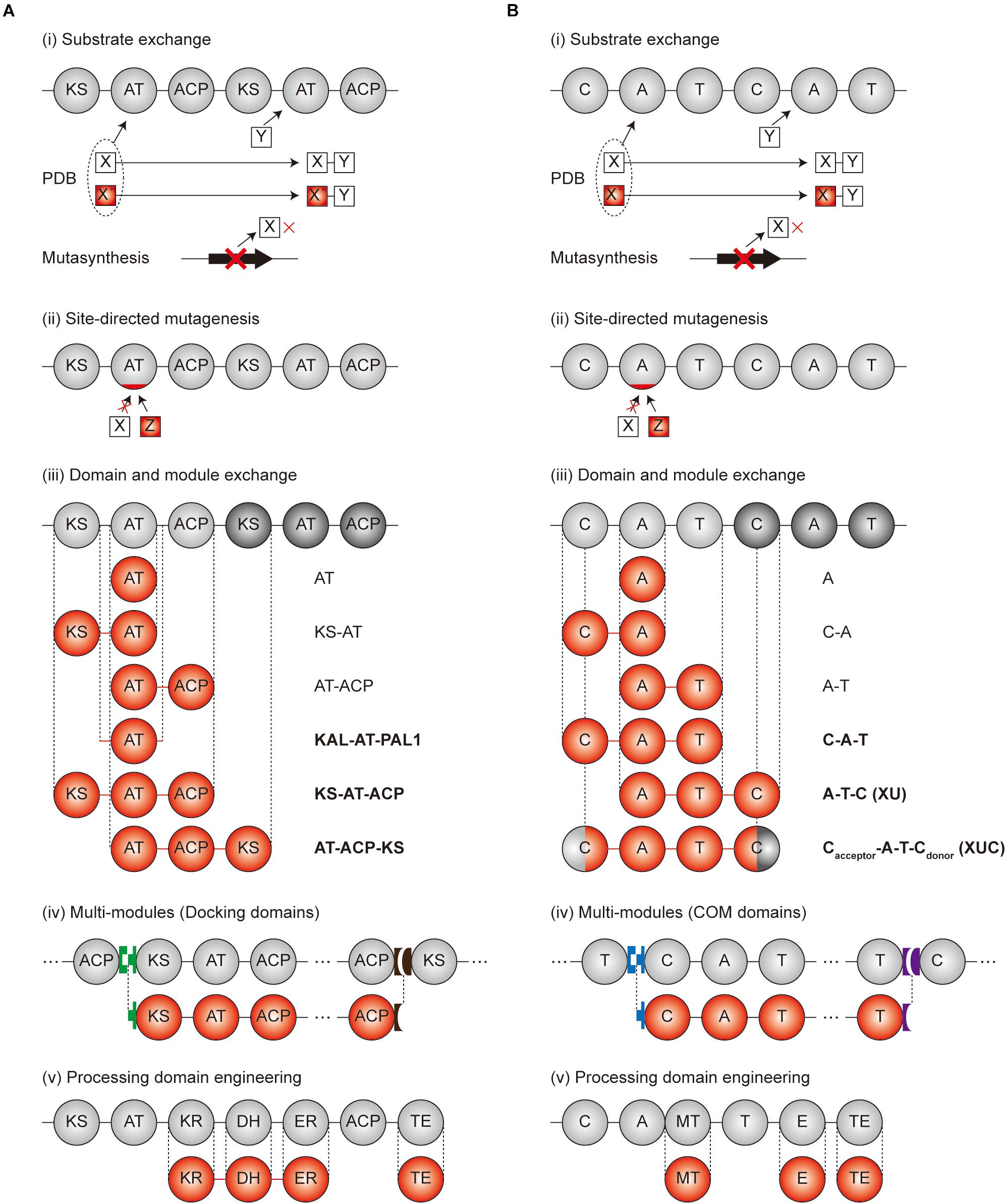
Figure 3. Engineering scheme for modular PKS and NRPS. (A) Engineering strategies of PKSs. (B) Engineering strategies of NRPSs. Gray circles and red circles indicate the original and modified domains, respectively. Green, brown, blue, and purple blocks, shaped as lock-and-key models, are the docking domains for (A) and COM domains for (B), respectively. Linkers were indicated as the lines between the domains. In case of (iii) domain and module exchange, the exchangeable units are indicated at the right of the domains. The units indicated as bold characters are currently the best exchangeable units. PDB, precursor-directed biosynthesis.
Substrate Exchange
Early attempts for the engineering of PKSs and NRPSs were conducted via precursor directed biosynthesis (PDB) and mutasynthesis (Alanjary et al., 2019) (Table 1). PDB is a strategy of creating a new product by providing an external substrate instead of the native substrate of the recognition domain (AT and A domain). This strategy is based on the native promiscuity of the recognition domains that are able to incorporate more than one kind of substrate. The advantages of this strategy are that the native enzyme could be free of any genetic engineering, and understanding of the structures and mechanisms of the enzymes is not required. This is because the promiscuity is easily confirmed by adding the target substrates and then measuring the kinetic profiles or the product formation. Despite this, native substrate and alternative substrates compete for the enzyme reaction that results in the formation of a mixture of major and minor products, hampering the yield and purity of the desired product. One of the successful examples of PDB was the incorporation of a series of 21 substrates consisting of monocyclic, polycyclic, branched aliphatic acids, benzoic acids, and heterocyclic acids to AT domain in loading module of rapamycin PKS, suggested by the previous report of relaxed substrate specificity of the domain (Lowden et al., 2004). Among the novel rapamycin analogs, one monocyclic aliphatic acid product has immunosuppressant activity comparable to the native product. In another example, native substrate L-Pro was altered by non-proteinogenic amino acids for pyreudione NRPS to generate various derivatives from pyreudione E to pyreudione K (Klapper et al., 2018). Although there were more examples for successful PDB, most of their products showed lower yield compared to the native products (Ward et al., 2007; Moran et al., 2009; Crawford et al., 2011; Xie et al., 2014).
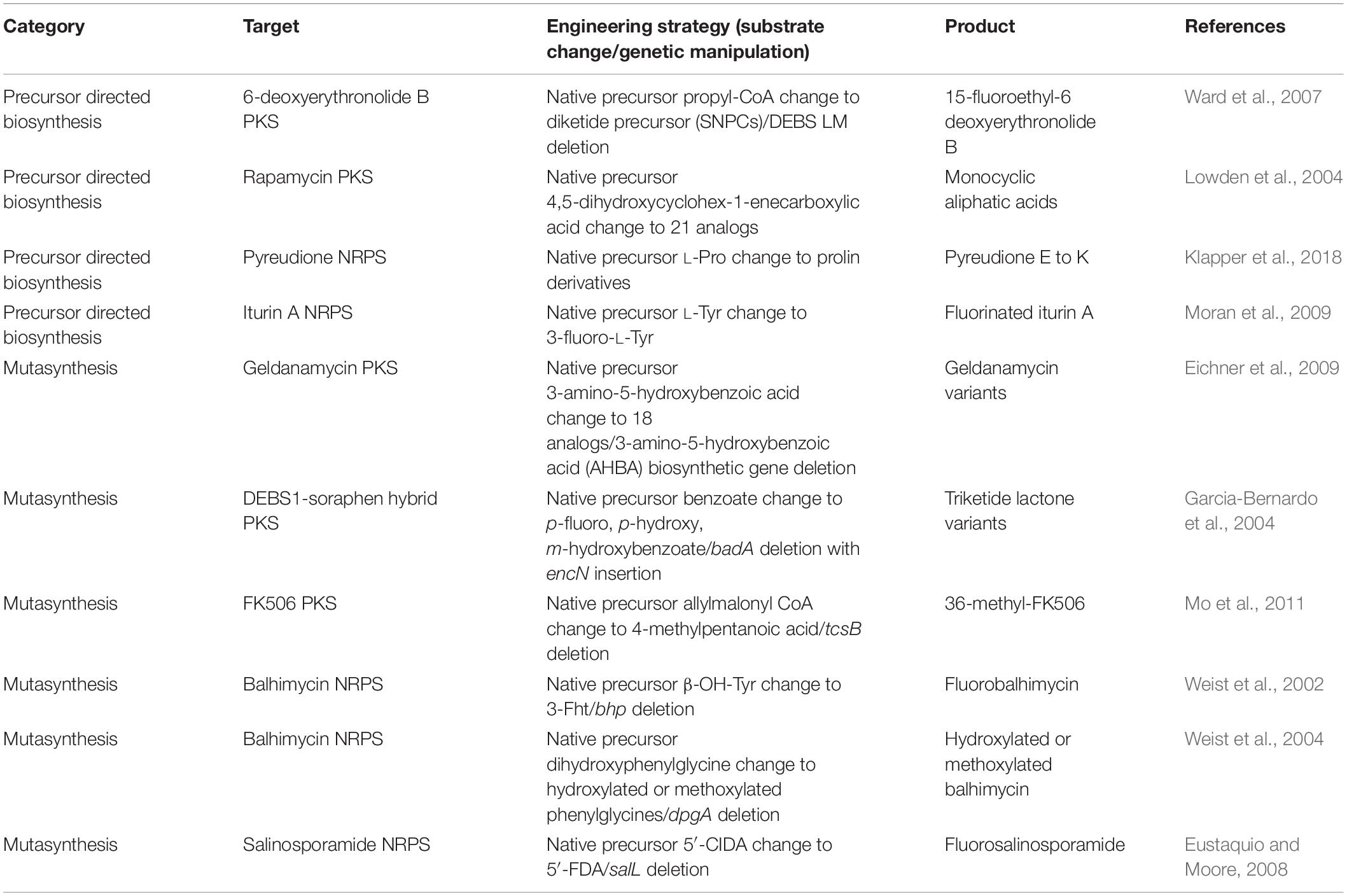
Table 1. Selected examples of the substrate exchange strategy.
To overcome the competition between native and altered substrates, mutasynthesis approaches were considered to increase the yield and purity of the desired product by feeding the alternative substrates together with the deletion of the biosynthetic genes of the native substrates in the expression host. For example, the mutasynthesis of novel FK506 derivatives was reported by the deletion of tcsB, resulting in modification of the FK506 C21 moiety (Mo et al., 2011). Utilizing the native promiscuity of AT domain of module 4 in FK506 PKS, the feeding of trans-2-hexenoic acid, 4-methylpentanoic acid, and 4-fluorocrotonic acid generated 36,37-dihydro-37-methyl-FK506, 36- methyl-FK506, and 36-fluoro-FK520, respectively. Interestingly, 36-methyl-FK506 not only has immunosuppressant function but has also improved neurite outgrowth activity. Other examples obtained various derivatives through incorporation of the non-natural units by mutasynthesis (Weist et al., 2002, 2004; Garcia-Bernardo et al., 2004; Eustaquio and Moore, 2008; Eichner et al., 2009). However, the requirement of genetic engineering and the dependency of native promiscuity are limitations of mutasynthesis. Despite these limitations, mutasynthesis is still useful when applied together with domain and module engineering in a synergistic manner. For example, non-natural precursors generated by semisynthetic or click chemistry could be fed, accompanied by the deletion of the native precursor biosynthetic genes and the mutations of PKS or NRPS enzymes to attach the pharmacophore-containing moiety or dye for novel chemical production (Alanjary et al., 2019).
Mutagenesis of Substrate Recognition Domains
The AT domain of PKS or the A domain of the NRPS have been major mutation targets for enzyme engineering (Table 2). Two directions of mutagenesis have been applied; (i) reducing or increasing the native promiscuity of the domain and (ii) creating de novo specificity of the domain (Musiol-Kroll and Wohlleben, 2018; Stanisic and Kries, 2019). Compared to domain and module exchange (see subsection “Domain and Module Exchange”), the modification of several residues in the AT or A domain could minimize the structural perturbation of entire enzyme assemblies, as well as minimally affecting the protein–protein interactions between adjacent domains and modules (Brown et al., 2018). In addition, this approach fundamentally changes the protein-substrate interaction enabling the incorporation of non-natural synthetic substrates.
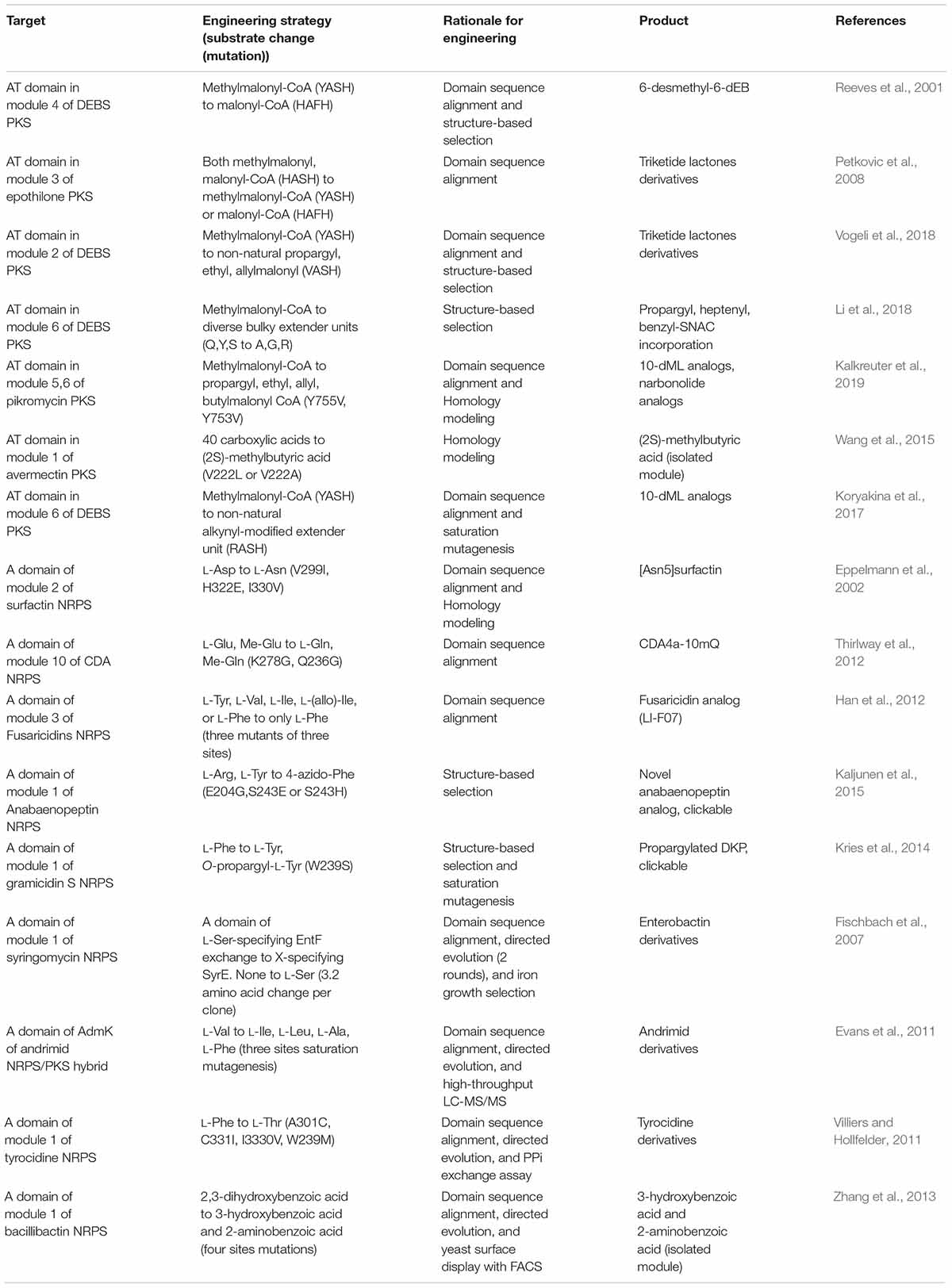
Table 2. Selected examples of the substrate recognition domain mutagenesis.
The substrate specificity of the AT domain is determined by approximately 100 residues toward the C-terminus from the active site serine (Musiol-Kroll and Wohlleben, 2018). The most common substrates of elongation AT domains are malonyl- or methylmalonyl CoA (Chan et al., 2009; Barajas et al., 2017), while at least 20 malonyl-CoA analogs have been found to be incorporated (Yuzawa et al., 2017b). The substrates of the loading AT domain are more variable (Barajas et al., 2017). Sequence alignment analysis of prototypical 6-deoxyerythronolide B synthase (DEBS) and epothilone PKS suggested the specificity code is ‘HAFH’ for malonyl-CoA, ‘YASH’ for methylmalonyl-CoA, and ‘HASH’ for the both (Reeves et al., 2001; Petkovic et al., 2008). Changing the code results in altered specificity to corresponding substrates, although there may be the mixed substrate specificity for both substrates. The specific residues for changing the promiscuous specificity toward non-natural extender units were determined based on the domain sequence alignment, X-ray crystal structure, and homology modeling (Reeves et al., 2001; Petkovic et al., 2008; Li et al., 2018; Vogeli et al., 2018; Kalkreuter et al., 2019).
The specificity conferring code of the A domain in NRPS is more variable than PKS. Early studies about X-ray structure and sequence alignment suggested the eight specificity codes, referred to as Stachelhaus codes, which were later updated to broader range of residues by computational modeling (Conti et al., 1997; Stachelhaus et al., 1999; Challis et al., 2000; Rottig et al., 2011). Based on this, the A domain in the second module of surfactin NRPS was mutated at three of the eight specificity conferring residues (V299I, H322Q, and I330V), resulting in the successful L-Asp to L-Asn specificity change (Eppelmann et al., 2002). Moreover, the corresponding code of A domain in the third module of fusaricidins NRPS was determined by aligning its residues to those of the L-Phe specific A domain of gramicidin S and tyrocidine S NRPS. These mutations resulted in increased specificity toward L-Phe to produce the fusaricidin analog (LI-F07) (Han et al., 2012). Other examples are presented in Table 2.
The limitations of the site-directed mutagenesis approach are the effects of the other residues, outside the specificity conferring code. For instance, the specialized protein–protein interactions for the different PKSs and NRPSs such as KS-AT and C-A interfaces impeded the establishment of the universal code and the accurate prediction of specificity. This is also consistent with the frequent results of the unexpected mixed substrate specificity by AT or A domain mutagenesis. To overcome this, the directed evolution method that identifies the desired clone among the random mutagenesis libraries of 104 to 106 clones by iterative selection cycles and high-throughput screening has been reported. The saturation mutagenesis of three sites in the AT domain of andrimid PKS generated a library of approximately 14,000 mutants, which were analyzed via the highly sensitive LC-MS/MS screening method (Evans et al., 2011). Consequently, mixed derivatives of adrimid containing L-Ile, L-Leu, L-Ala, and L-Phe, modified from L-Val, respectively, were obtained with improved antimicrobial activity. Together with other examples, the directed evolution approach was expected to become the universal strategy for the specificity change regardless of the kind of the enzymes (Fischbach et al., 2007; Villiers and Hollfelder, 2011; Zhang et al., 2013). However, there are still several requirements for feasible directed evolutions such as high-throughput assays, iterative cycles of mutagenesis for stability and selectivity, and effective screening. Alternatively, reducing the library size by the prediction of the residues for specificity change based on structural information and evolutionary evidence would be developed.
Domain and Module Exchange
Parallel to the site-directed mutagenesis approach, the domain or module exchange approaches have been applied to change the substrate specificity of PKS and NRPS (Sussmuth and Mainz, 2017; Kornfuehrer and Eustáquio, 2019). As the exchanging domains and modules are generally well-studied, the substrate specificity change is predictable and experimental design is more convenient. However, the correct splicing site should be precisely determined to preserve the protein–protein interactions between the acceptor and donor units, to minimize the perturbation of sophisticated conformational changes during chain elongation, and to maintain the overall structure of PKS and NRPS.
Domain or Module Exchange of PKS
In the case of PKS, successful examples were observed for the AT domain of the well-studied DEBS PKS exchange to other AT domains in different modules of DEBS PKS, or the modules from other PKSs (Oliynyk et al., 1996; Stassi et al., 1998; Hans et al., 2003; Petkovic et al., 2003) (Table 3). This AT exchange strategy is normally referred as ‘AT domain swapping.’ The splicing sites were determined as the rough boundaries of AT domains inferred by the sequence alignment between similar modules, and the AT domains were cloned by providing synonymous mutations to the boundaries for the introduction of restriction enzyme sites (Patel et al., 2004). Nevertheless, the swapping of only the AT domain was unfavorable in terms of impairment of catalytic activity owing to the non-native interactions between the swapped domains and neighboring domains such as KS, ACP, and interdomain linkers. The X-ray structural studies and mutagenesis studies of KS-AT and AT-ACP didomains revealed crucial residues in the interdomain linkers and domain interfaces for the specificity or catalytic activity, suggesting that the KS or ACP domain should be exchanged together with the AT domain (Kuhstoss et al., 1996; Marsden et al., 1998; Kim et al., 2004; Chen et al., 2007; Yuzawa et al., 2012; Miyanaga et al., 2016; Murphy et al., 2016). Recently, sequence alignments and structural studies related to module 6 of DEBS PKS and module 1 of β-lipomycin PKS provided ideal splicing sites located at interdomain linkers adjacent to AT domain despite of the protein–protein interactions with KS and ACP domains (Yuzawa et al., 2017b). The splicing sites were located at the conserved GTNAHVILE region of KS-AT linker (KAL) and conserved LPTY(A/P)FQ (H/R)xRYWL region of post AT linker 2 (PAL2) to minimize the non-native adjacent sequences, resulting in the universal KAL-AT-PAL1 unit for AT domain swapping. However, swapping by the KAL-AT-PAL1 unit is still remained to be validated for more various PKSs, because it may alter the protein–protein interactions between the domains and disrupt gatekeeping from downstream processing, which may result in incompatibility (Barajas et al., 2017).
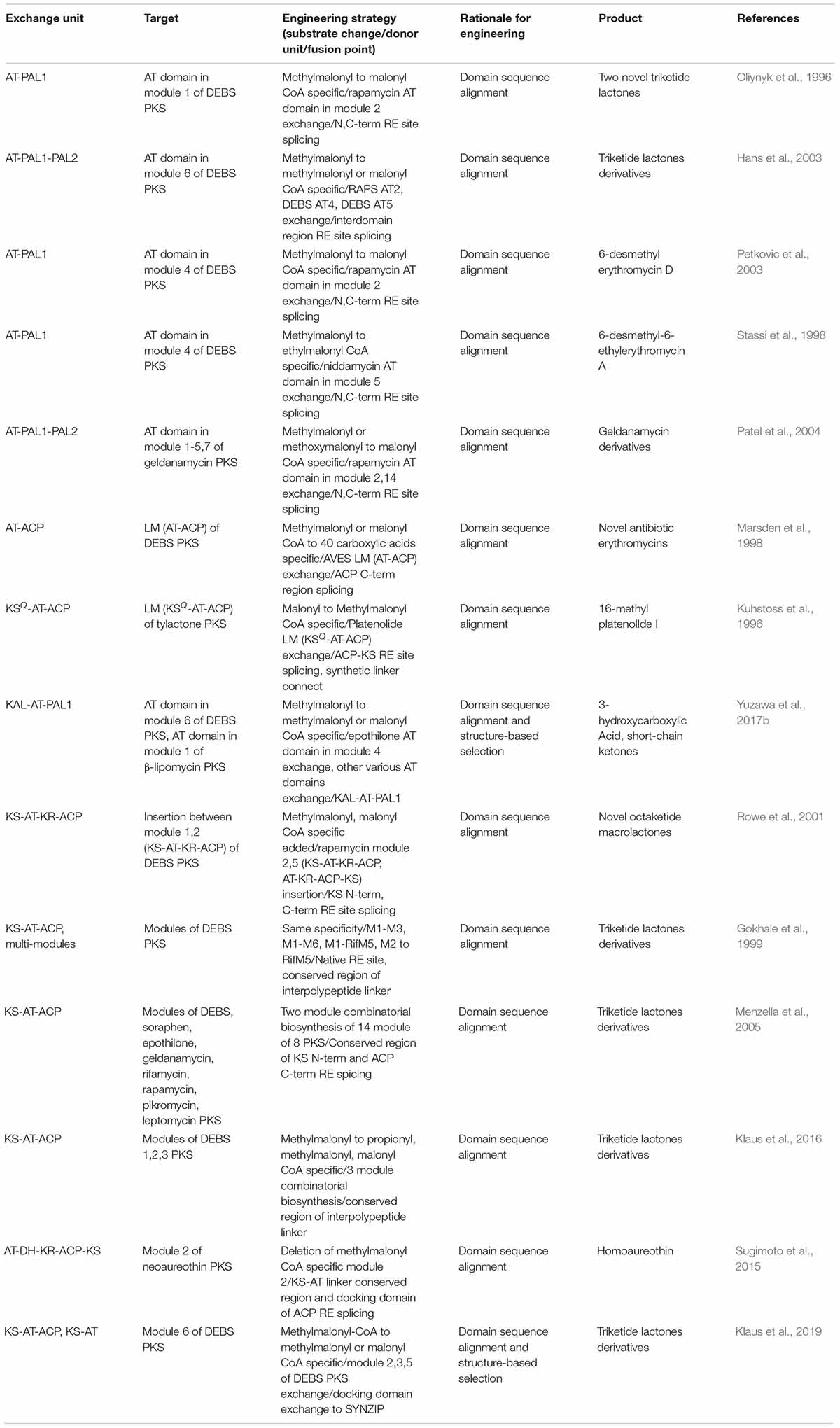
Table 3. Selected examples of domain or module exchange of modular PKS.
In addition to determination of AT domain splicing sites, the exchange of a whole module (KS-AT-ACP) could be the alternative strategy to change the specificity. In this case, the interactions between upstream ACP and downstream KS domain should be considered, including covalent interdomain linkers and non-covalent interactions between domains. By splicing at the appropriate site of the interdomain linkers, 14 KS-AT-ACP modules from 8 PKSs was successfully isolated as a functional unit, and connected to generate a total of 154 combinatorial bimodular PKSs (Gokhale et al., 1999; Menzella et al., 2005). On the other hand, recent studies suggested that the evolutionary functional module is AT-ACP-KS rather than conventional KS-AT-ACP (Sugimoto et al., 2015; Keatinge-Clay, 2017). The sequence alignment of four aminopolyol PKSs supported this unit that the higher evolutionary correlation of the sequences between processing domains and downstream KS domain compared to the upstream KS domain (Zhang et al., 2017). Also, the first half of post-AT linker sequence showed higher correlation to the AT domain than the KS-AT linker, which refers AT-(processing domains)-ACP-KS is a more evolutionarily conserved unit. Moreover, the multi-modules of PKSs with the interpolypeptide non-covalent docking domain (DD) at both ends were able to be exchanged. The DD pairs were the compatible parts for exchanging the subunits that, using heterologous DD pairs led to the successful production of diketides and triketides (Menzella et al., 2005). However, several examples showed severe impairment of catalytic activity (Klaus et al., 2016). This is because the chain translocation step from the ACP domain of upstream module to the KS domain of downstream module was the bottleneck in addition to the chain elongation step (Khosla et al., 2007; Klaus et al., 2016), emphasizing the importance of ACP-KS interaction, even when the DDs are compatible. Besides exchanging the module, incorporating the growing polypeptide with similar chain length to the native module was more successful than those of different chain length. Overall, the modular exchange strategies for PKS are diverse but the protein–protein interactions particularly for ACP-KS domains are important.
Domain or Module Exchange of NRPS
Similar to the exchange of domains and modules in PKSs, many engineering attempts for NRPSs have been reported. The swapping of only an A domain successfully resulted in the alteration of the specificity by the determination of proper splicing sites but, they were often hindered by non-native interactions between the swapping domains and neighboring domains such as C, T, and interdomain linkers (Stachelhaus et al., 1995; Doekel and Marahiel, 2000) (Table 4). The exchange of C-A didomain was more successful than the single A domain exchange in surfactin and pyoverdine NRPS (Tanovic et al., 2008; Calcott et al., 2014) but, failed in another report (Ackerley and Lamont, 2004). A-T didomain had a short interdomain linker with a conserved LPxP motif to interact with a key L-Tyr residue in the C-terminus of the A domain, and to assist the movement of the T domain during the catalytic cycle (Beer et al., 2014; Miller et al., 2014). Based on these characteristics, the A-T didomain in module 2 of actinomycin NRPS was successfully exchanged to A-T didomain in module 5 of actinomycin NRPS, but it still had low a yield (Schauwecker et al., 2000).
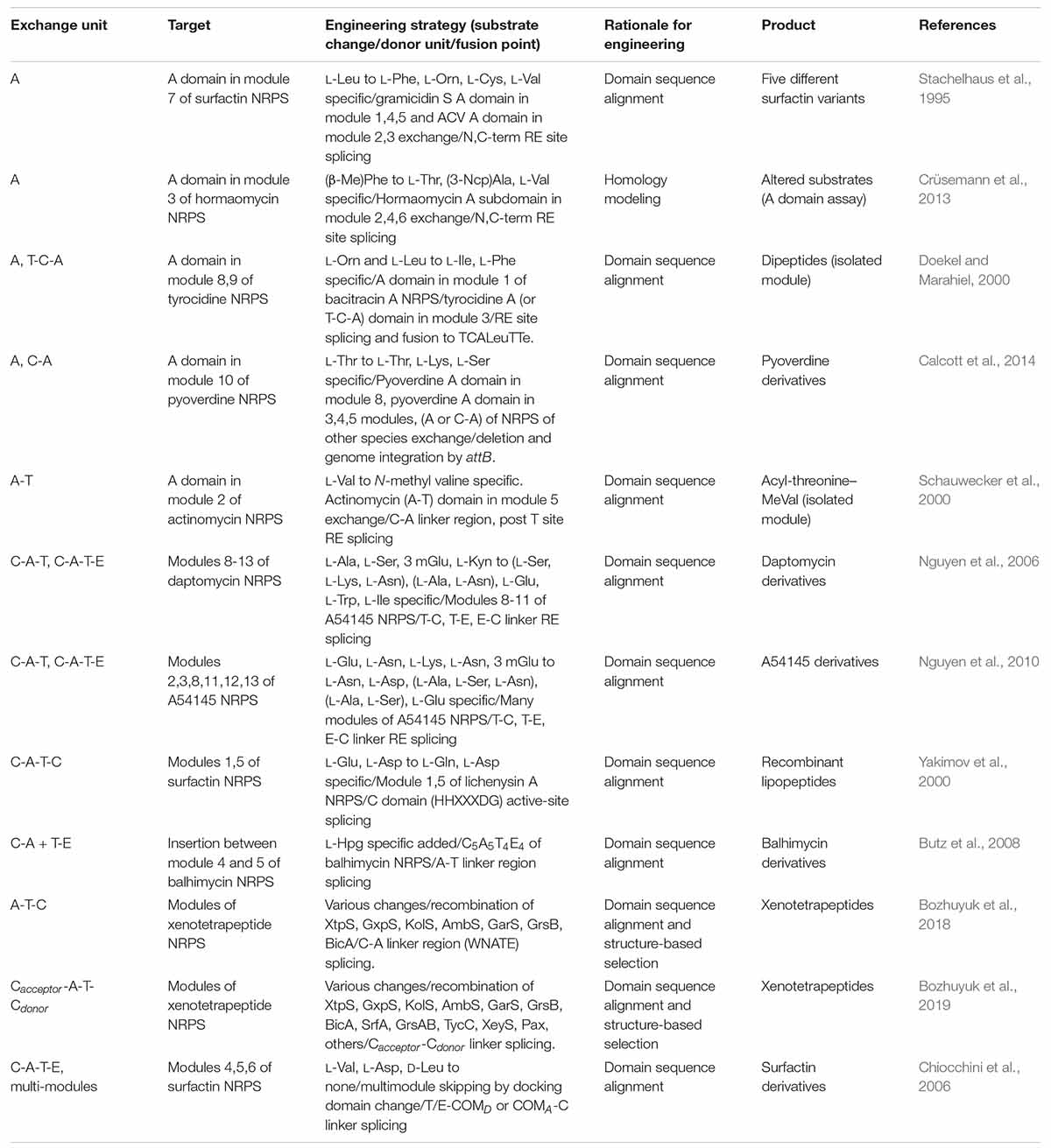
Table 4. Selected examples of domain or module exchange of modular NRPS.
Considering both interactions within the C-A and A-T didomains, C-A-T module exchange occurred in several NRPSs. The most representative examples were, the daptomycin and A54145 NRPS modules of C-A-T or C-A-T-E spliced at the interdomain linker of the T and C domain that were changed to produce the novel daptomycin and A54145 derivatives (Yakimov et al., 2000; Nguyen et al., 2006, 2010; Butz et al., 2008). But the protein–protein interactions between T and C domain, consisting of a variable interdomain linker, were disturbed resulting in impaired activity. Different from the transacylation of PKS which leads to two separated steps of upstream ACP to KS, and then KS to downstream ACP, the C domain governs the peptide bond formation by one step and has strong stereoselectivity and side-chain selectivity for both donor and acceptor peptides (Clugston et al., 2003; Samel et al., 2007; Bloudoff and Schmeing, 2017; Li et al., 2017). Due to this strong correlation between all adjacent domains and linkers, finding the optimal splicing sites for the module exchange has remained a bottleneck. Recently, a suggested exchangeable unit A-T-C (XU) that the C-A linker was thought to be a better splicing site than other linkers in view of the N-terminal conserved region, the absence of secondary structures, and the fewer interactions between other domains (Bozhuyuk et al., 2018). Nonetheless, there was a requirement for the exchange of A-T-C unit that the C domain has specificity filters for both upstream and its own module. Therefore, downstream A-T-C unit should also be exchanged together with the target A-T-C unit. To avoid the limitations of XU, the XUC exchange unit which is Cacceptor-A-T-Cdonor with the splicing site located at the intradomain conserved linker was newly suggested (Bozhuyuk et al., 2019). Through dissecting the C domain specificity for upstream and downstream module, it was theoretically ideal that the exchange unit could be fused in a combinatorial manner. However, experimental results showed that the fusion between units from different genera caused a decrease in the yield. As the overall structure disruption by the C domain was thought to be the main reason, the solution to complement this limitation would be to research and discover more exchangeable XUC units.
There were several successful multi-module exchanges produced by using communication (COM) domain pairs such as DD domain pairs of PKS (Hahn and Stachelhaus, 2004; Chiocchini et al., 2006). In addition, the specificity of COM domain pairs has the ability to be altered via modification at their key residues resulting in the non-native COM domain (Hahn and Stachelhaus, 2006). Some examples, however, displayed a low product yield due to the disruption of non-covalent protein–protein interactions between the T and C domain. Overall, the modular exchange strategies for NRPS are diverse, similar to PKS, but the protein–protein interactions between domains, particularly those involving the C domain, are commonly important considerations.
Currently, the favorable strategies of exchanging module specificity for the production of novel chemical derivatives seems to be; (i) KAL-AT-PAL1 unit, AT-ACP-KS unit, using heterologous DD pairs for PKS, and (ii) XU, XUC, using heterologous COM domain pairs for NRPS, respectively. Although numerous studies for altering specificity of PKS and NRPS have been reported, more engineering trials should be accumulated for various PKSs and NRPSs to optimize the strategies employed.
Processing and Offloading Domain Engineering
In addition to the module specificity change, the incorporated extender unit could be further modified by various processing domains. Most of the processing target residues were the α-substituent and β-keto group of acyl-ACP intermediate of PKS, and R group of amino acid intermediate of NRPS (Barajas et al., 2017; Sussmuth and Mainz, 2017) (Table 5).
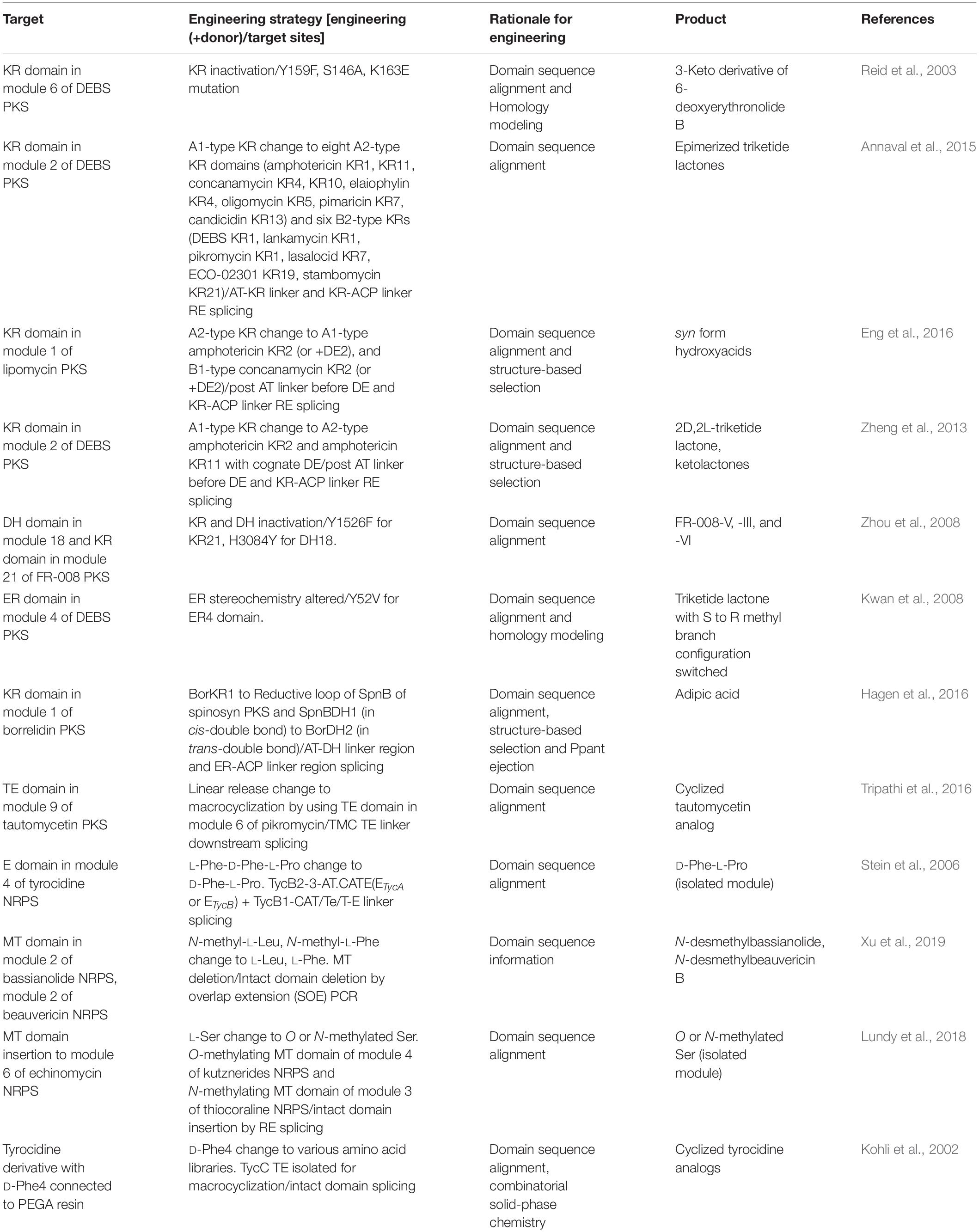
Table 5. Selected examples of processing and offloading domain engineering.
Processing and Offloading Domain Engineering of PKS
In PKS, ketoreductase (KR), dehydratase (DH), and enoyl-reductase (ER) domains are the most abundant processing domains located between the AT and ACP domain as a reductive loop, which governs the degree of β-carbonyl reduction. The KR domain performs the NADPH-mediated reduction of β-keto groups to β-hydroxyl groups and determines the stereochemistry of α-substituent and β-hydroxyl group. As the full deletion of the KR domain showed loss of specificity due to impaired protein folding and stability, the inactivation of the KR domain by its key residue mutations was more effective (Reid et al., 2003). In another approach the stereochemistry was altered by KR domain swapping, resulting in the effective stereocontrol of the product, which was generally difficult through modern synthetic methods (Annaval et al., 2015; Eng et al., 2016). Notably, substrate specificity of the KR domain has been shown to be related to its substrate size, as it tends to be less active on smaller non-native substrates (Zheng et al., 2013). Next to the KR domain, DH domain promotes the dehydration of the β-hydroxyl group to form the double bond between α- and β-carbons, and even between β- and γ-carbons in some cases. Inactivation of DH domain by single mutation at the conserved active site motif resulted the retention of the β-hydroxyl group without specificity loss of the AT domain (Zhou et al., 2008). For the insertion of a non-native DH domain, it was important to consider the stereoselectivity of DH domain to the hydroxylated product by the upstream KR domain, which means that DH domain activity is strongly dependent to cognate KR domain (Barajas et al., 2017). Additionally, DH domain was also sensitive to the length of substrates that DH domains acting on natively long chain showed impaired activity to the shorter chain substrate (Faille et al., 2017). Lastly, ER domain promotes the reduction of the unsaturated α–β double bond formed by DH domain to generate the single bond, and determines the stereochemistry of the α-carbon. As the substrate of ER domain is the product of DH domain, the specificity of ER domain is dependent to both cognate KR and DH domain. Inactivation of ER domain by single mutation at the conserved residue switched the methyl branch configuration of triketide lactone product (Kwan et al., 2008). Moreover, ER domain affected the downstream ACP arrangement by the protein–protein interactions resulting in the different stereocontrol (Zhang et al., 2018). Three domains (KR, DH, ER) could be simultaneously introduced for the exchange of the KR domain in module 1 of borrelidin PKS (Hagen et al., 2016). An offloading TE domain was abundantly fused to other modules using the splicing site at the conserved linker between the offloading domain and ACP domain to produce truncated products. Moreover, they were regio-, stereospecific, which may alter the release and cyclization nature via swapping (Pinto et al., 2012; Barajas et al., 2015; Tripathi et al., 2016). Further engineering of other processing domains, such as the methyltransferase (MT) domain, should be conducted to utilize their potential to produce novel chemicals (Yuzawa et al., 2017c).
Processing and Offloading Domain Engineering of NRPS
Among a large variety of processing domains for non-ribosomal peptides, in this review we focused on the cis-acting processing domains located in the module.
Epimerase (E) domain governs in situ epimerization of the α-carbon of the T domain tethered L-amino acid during peptide elongation to generate D-amino acid. One example was the exchange of E domain in module 4 of tyrocidine NRPS (ETycB) to E domain in loading module of tyrocidine NRPS (ETycA), that showed the epimerization of L-Phe to D-Phe (Stein et al., 2006). Methyltransferse (MT) domain is another example of the engineering that provides N-methylation of the amino acid. This domain is usually integrated inside of A domain such as MT domain of cyclosporine A NRPS (Velkov et al., 2011). A recent engineering example of the MT domain in module 2 of bassianolide synthetase showed that the deletion of this domain generated the N-desmethylbassianolide without affecting the enzyme assembly lines (Xu et al., 2019). In another study, O-methylating MT domain was inserted between the A subdomains, resulting in the incorporation of O or N-methylated serine (Lundy et al., 2018). An offloading domain of NRPS was abundantly fused to other modules, similar to PKS, using the splicing site at the conserved linker between itself and the T domain to produce truncated products. Particularly, TETycC domain of tyrocidine NRPS was fused to other modules to generate various cyclized analogs by using its macrocyclization property (Kohli et al., 2001, 2002; Trauger et al., 2001).
Cyclization (Cy), Oxidase (Ox), and Formylation (F) domain were also expected to be the favorable targets for engineering. However, there have been no examples due to the upstream substrate specificity as well as other several mechanistic problems (Miller and Walsh, 2001; Schneider et al., 2003; Schoenafinger et al., 2006; Sussmuth and Mainz, 2017). Besides, other in-trans acting tailoring enzymes have the potential to be utilized for the production of novel derivatives but, studies are lacking. Therefore, further studies and engineering about various NRPS processing domains, including both in-cis and in-trans acting, should be quantitatively increased.
To sum up, the inactivation and swapping of the individual processing domain based on homology modeling and sequence alignment was successful, however, specialized protein–protein interactions hampered the full engineering of these domains, this required the case-by-case optimization of the swapping region.
Repurposing Modular PKS and NRPS to Construct de novo Biosynthetic Pathways
As discussed above, numerous studies have repurposed modular PKSs and NRPSs via modifications and swapping of domains and modules in the original enzyme assembly lines, mainly for the production of novel derivatives. Otherwise, domains and modules were isolated and fused to elucidate the protein–protein interactions and substrate specificities of domains with the truncated product as a proof. Although there were several combinatorial examples of domain or module fusion to form the de novo enzyme assembly lines, their products were generally not the purpose of the study. Recently, technical advances of the separation and fusion of domains and modules from original enzyme assembly lines, owing to numerous engineering studies, opened the way for retro-biosynthesis. Retro-biosynthesis is a de novo pathway design that assembles reactions in a stepwise fashion in the reverse direction of synthesis, for the desired product (Birmingham et al., 2014) (Figure 4). In this approach, the final product formation is the sole selection criterion for each reaction step that reduced the screening effort for the high-throughput combinatorial experiments. By leveraging the potential diversity of the reactions from modular PKSs and NRPSs, the potential range of chemicals produced would be countless. Furthermore, this new pathway may have advantages in terms of reaction thermodynamics and economics. Despite these potentials, recently there have only been a few examples of repurposing PKSs and NRPSs due to experimental difficulties. In this section, we highlight recent efforts of repurposing PKS and NRPS domains and modules for the production of non-natural commodity chemicals or specialty chemicals by de novo pathway construction.
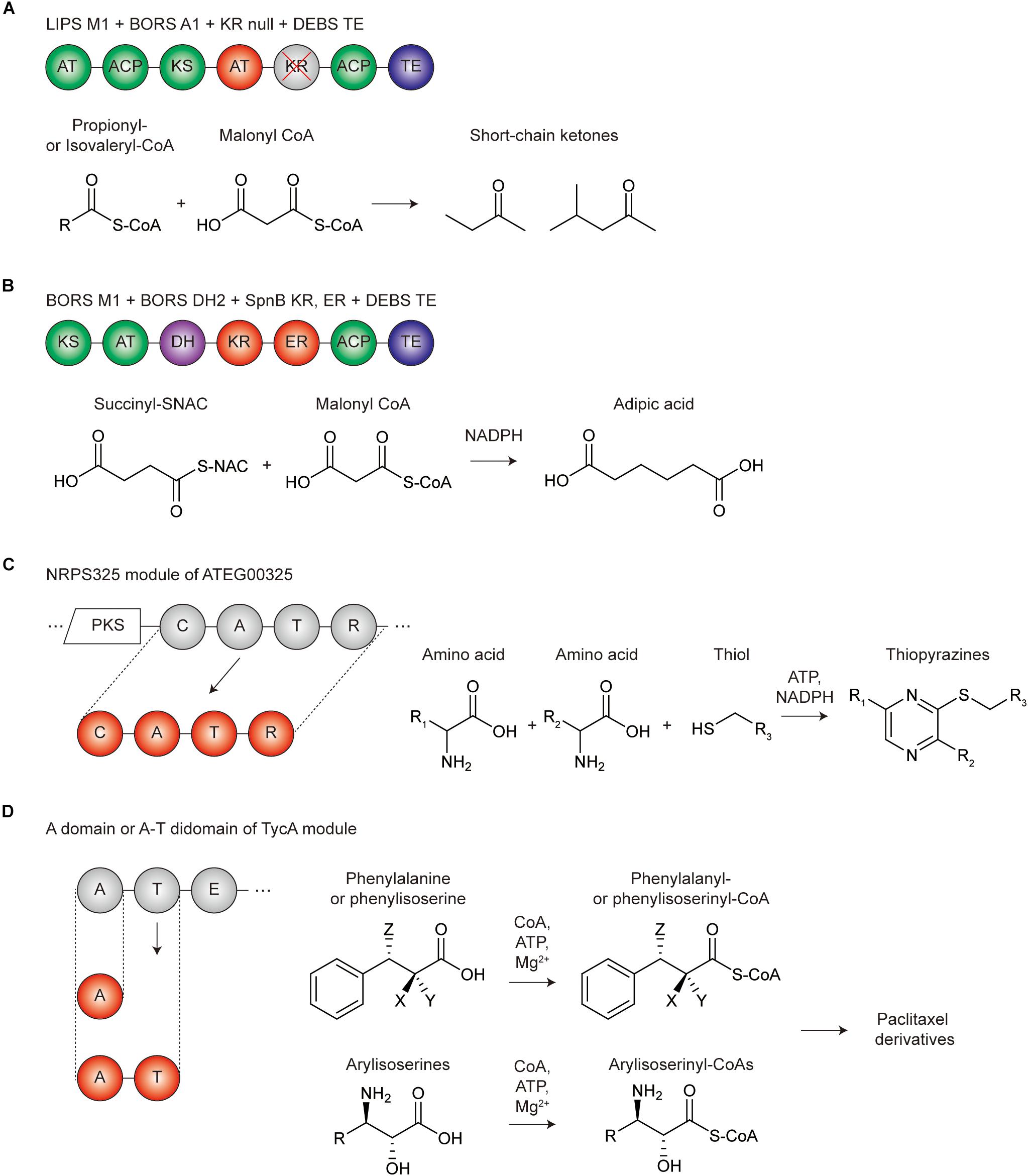
Figure 4. Representative repurposing examples of modular PKS and NRPS for de novo biosynthetic pathways. (A) Repurposing the PKS domains and modules for the production of short-chain ketones. Green circles are the domains in module 1 of β-lipomycin PKS (LIPS M1), red circles are the AT domains in module 1 of borrelidin PKS (BORS A1), gray circles with the red crossed line are the inactivated KR domain (KR null), and the blue circles are the TE domain of DEBS PKS. (B) Repurposing the PKS domains and modules for the production of adipic acid. Green circles are the domains in module 1 of borrelidin PKS (BORS M1), red circles are the KR and ER domain in SpnB module of spinosyn PKS (SpnB KR, ER), and the blue circles are the TE domain of DEBS PKS. (C) Repurposing the NRPS module for the production of thiopyrazines. NRPS325 module of ATEG00325 PKS-NRPS hybrid megasynthetase was isolated (red circles) to promote the reaction for the thiopyrazine production itself. (D) Repurposing the NRPS domain for the production of paclitaxel derivatives. The A or A-T didomain in TycA module of tyrocidine A PKS was isolated (red circles) to be repurposed for the production of phenylalanyl-, phenylisoserinyl-, arylisoserinyl-CoAs, which are the precursors of the paclitaxel derivatives; X, NH2 or H; Y, H or OH; Z, NH2 or H.
Redesigning Modular PKSs for Retro-Biosynthesis
Type I modular PKSs have favorable properties that functions and order of their modular catalytic domains determine the final product in a predictable manner. Most recently, an in silico toolkit called ClusterCAD, a computational platform for designing novel multi-modular type I PKS, has been developed and applied to several PKS repurposing studies (Eng et al., 2018). However, previous studies indicated that constructing multi-modular PKSs to produce novel chemical is still challenging (Weissman, 2016; Pang et al., 2019). The relationship between PKS module structure and acyl chain passage from one module to the next is not well understood thus, when testing the multi-modular assembly line, the acyl chain extension was frequently stalled at the middle of the synthesis process. Hence, presently, only recombination of one or two PKS modules to produce simple structured chemicals has been successfully conducted. To date, representative target molecules produced by using PKSs are divided into two categories: (i) fuels and (ii) industrial chemicals (Table 6).
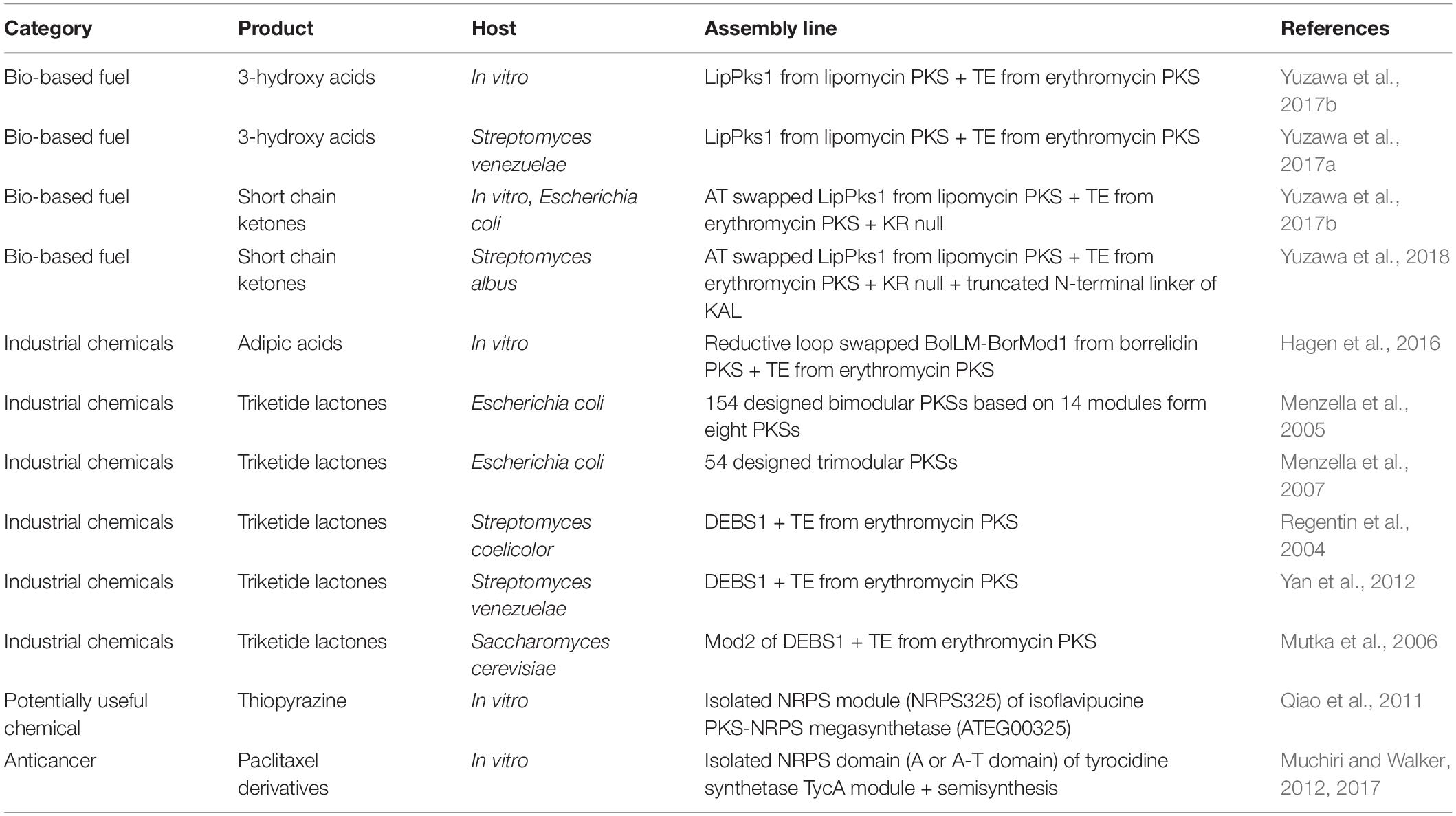
Table 6. Selected examples of modular PKS and NRPS repurposing for de novo biosynthetic pathways.
Petroleum-based fuels are mixtures of highly reduced carbons with varying chain length as in the case of gasoline, which is mixture of C5 to C8 hydrocarbons. To date, bio-based hydrocarbon production has majorly relied on utilizing enzymes involved in isoprenoid and fatty acid biosynthesis (Beller et al., 2015). However, the alkane and alkene produced from these systems were generated as a mixture (Beller et al., 2010; Sukovich et al., 2010; Howard et al., 2013). As a way to overcome this limitation, theoretically, PKS modules can be engineered to produce hydrocarbons with specific carbon length. Recently, several cases have been reported that resulted in the over-production of pentadecaheptaene (PDH) by expressing iterative PKS of enediyne biosynthesis SgcE and its cognate TE SgcE10 in Escherichia coli or Streptomyces (Van Lanen et al., 2008; Zhang et al., 2008). Furthermore, through optimizing the enzyme ratio of SgcE and SgcE10, PDH production was enhanced, followed by the additional chemical reduction to overproduce the pentadecane (PD) (Liu et al., 2015). Presently, there is no example of producing hydrocarbons using only refactored modular PKS, but chemical reduction steps such as hydrogenation are required after the precursor production. Conversely, short-chain ketones have been successfully produced by using only repurposed PKS. Due to the high octane number, short-chain ketones have the potential to be utilized as gasoline replacements or gasoline oxygenates (Yuzawa et al., 2018). In addition, short-chain ketones are widely used as flavors and fragrances. PKS for short-chain ketone production was designed by repurposing the first PKS module from the lipomycin PKS and linking TE from erythromycin PKS (LipPks1 + TE). Originally, LipPks1 + TE was constructed for production of various 3-hydroxy acids depending on the starter acyl-CoAs (Yuzawa et al., 2017a,b). Engineering the same PKS by changing active site serine residue of KR domain to alanine deactivated the KR function and resulted in the production of various short-chain ketones (Yuzawa et al., 2017b).
A representative industrial chemical produced by re-designed PKS is adipic acid, a monomer component used to prepare the polymer Nylon 6,6. Conventionally, bio-based production of adipic acid has been conducted by producing cis,cis-muconic acid, followed by chemical hydrogenation (Niu et al., 2002), or constructing β-oxidation reversal and ω-oxidation pathways, which synthesized the dicarboxylic acid from glucose or glycerol (Clomburg et al., 2015). For PKS based C6 adipic acid synthesis, a novel condensation strategy was proposed, which condenses C4 succinyl-CoA as the starter unit and C2 malonyl-CoA as the extender unit. Loading AT domain of the borrelidin PKS (BolLM) originally incorporates a trans-1,2-cyclo-pentanedicarboxylic acid CoA (CPDA-CoA) as the starter unit, but it has promiscuity to successfully recognize succinyl-CoA (Hagen et al., 2014, 2016). Furthermore, the first extension module of the borrelidin PKS (BorMod1) naturally incorporates malonyl-CoA. Thus, BolLM-BorMod1 linked with the TE domain from erythromycin produced 3-hydroxyadipic acid (Hagen et al., 2014). To convert 3-hydroxyadipic acid to adipic acid, further β-carbonyl processing governed by the DH and ER domains’ is required, however, BorMod1 only contains a KR domain and lacks the DH and ER domains. Therefore, the reductive loop of BorMod1 was replaced with reductive loop from other PKSs containing full tridomains, resulted in successful synthesis of adipic acid (Hagen et al., 2016). Nevertheless, further studies are still needed for in vivo adipic acid production using PKS, as the starter unit, succinyl-CoA, is essential for the growth of the producing host and the promiscuity of the PKS modules will decrease the titer.
As fossil resources are depleted, the bio-based eco-friendly production of transport fuels and commodity chemicals, previously been produced from these fossil resources, is becoming more common. We believe that repurposing PKS will play a large role in this field. Additionally, further mechanistic understanding of PKS domains is expected to enable the design and implementation of PKS capable of producing even chemicals that do not occur naturally.
Redesigning Modular NRPSs for Retro-Biosynthesis
Non-ribosomal peptides are the most widely spread and structurally diverse secondary metabolites. NRPS modules are much more versatile than PKS modules in terms of the number of available substrate chemicals, including non-proteinogenic amino acids, thus the potential of producing novel chemicals by repurposing the module seems to be much higher. Nonetheless, current examples of NRPS repurposing are limited, compared to PKS, this may be due to the structural and mechanistic complexity of NRPS. In the case of PKS repurposing examples, the de novo pathway constructions for the useful chemical production were done by the combination of only unrelated PKS modules themselves. On the other hand, NRPS repurposing examples used a single NRPS domain or module reaction alone or combined with other non-NRPS enzymes. Thus, selected examples were discussed that showed the potential of further NRPS module repurposing.
The first example is a case that confirmed unexpected chemical production when a single NRPS module was isolated and expressed from the PKS-NRPS hybrid assembly line. Previously, novel PKS-NRPS assembly line was designed to produce tryptophan-containing preaspyridone analog by replacing the NRPS domain of PKS-NRPS megasynthetase (ApdA) of Aspergillus nidulans, which originally synthesizes tetramic acid preaspyridone, with the NRPS domain of cyclopiazonic acid synthetase (CpaS) of Aspergillus flavus (Liu and Walsh, 2009). Based on these results, the same group dissected and tested the function of a single NRPS module (NRPS325) of PKS-NRPS megasynthetase (ATEG00325) from Aspergillus terreus, which produces isoflavipucine. Unexpectedly, the NRPS module can synthesize thiopyrazine in vitro, which is largely different from the original role of the module in the parent enzyme (Qiao et al., 2011). This showed that in a multi-modular assembly line, a single NRPS module is capable of producing potentially useful chemicals and can be utilized as a part to construct de novo biosynthetic pathways for non-natural compound production.
Another example was the repurposing of a tyrocidine NRPS domain (A domain or A-T didomain) to the paclitaxel biosynthesis pathway to produce various paclitaxel derivatives (Muchiri and Walker, 2012, 2017). Antimitotic, anticancer paclitaxel was originally produced from Taxus brevifolia or the semisynthetic method, which suffered from low yields and environmentally harmful reactions, respectively. Moreover, the synthesis of the most essential precursor, amino phenylpropanoyl CoA substrates, was a major limitation as, it required protection at their amino groups before synthetic thioesterification. The authors hypothesized that A domain in tyrocidine NRPS module TycA could be used as the potential chemoselective carboxylate CoA ligase that originally had phenylalanine specificity but showed potential promiscuity. As a result, α-, β-phenylalanyl, and (2R,3S)-phenylisoserinyl CoA were successfully generated by the A domain (Muchiri and Walker, 2012). By employing 16 substituted phenylisoserines, the A-T didomain of TycA converted them to their corresponding isoserinyl CoAs, resulting in the production of docetaxel, milataxel, and various other analogs (Muchiri and Walker, 2017). These examples showed that a single NRPS domain could be integrated to other biosynthetic or semisynthetic pathways and utilized as a part for the construction of de novo pathways for non-natural compound production.
Owing to the natural diversity of the amino acid substrates other than acyl CoA substrates, the substrate promiscuity of the NRPS modules seems to be broader than PKS modules. Although these aspects of NRPS hindered the understanding of their reactions, further advances would increase the potential of NRPS to be repurposed for the construction of de novo biosynthesis pathways of useful chemicals, by fusing the NRPS modules.
The Roadmap for Repurposing Modular PKS and NRPS
As shown above, modular PKS and NRPS have been studied with the aim of (i) engineering for the production of novel chemicals, and (ii) repurposing the domain and module reactions toward de novo biosynthetic pathways for the production of useful chemicals. To overcome previous limitations of these approaches for the expansion of the diversity of their products and optimization of their productivity, the systematic strategy such as the design-build-test-learn cycle should be applied. The brief explanations, applications, and perspectives of the tools for each step are discussed below.
Design Tools
Design tools include structural, kinetic, mechanistic, and sequence-based techniques to obtain fundamental information for the experimental design of the repurposing. Specifically, the structural and kinetic knowledge is collected and sorted to the database, and bioinformatics tools are used for domain/module selection and boundary identification based on the database. First, structural biology tools were used for the modular PKS and NRPS, which are macromolecules with numerous protein–protein interactions and dynamic conformational change during biosynthesis. Thus, the structures and interactions of the domains and one or two modules have been reported for high-resolution and dynamic scale. X-ray crystallography was the most commonly used technique that provided high-resolution images at the angstrom level (Conti et al., 1997; Keatinge-Clay and Stroud, 2006; Tang et al., 2006; Buchholz et al., 2009). However, it would require the protein to crystallize which can be difficult for large structures. Additionally, the dynamic conformations and protein–protein interactions could not be inferred. To complement these limitations, conventional electron microscopy was utilized but it required additional staining, and suffered from relatively low resolution due to the deformation of flexible particles (Hoppert et al., 2001). Also, NMR spectroscopy was predominantly practiced in the structural determination of ACP and docking domains, and their conformation dynamics (Broadhurst et al., 2003; Richter et al., 2008; Lim et al., 2012). However, the size limitation of this technique hindered to be utilized for the whole module structure. Other alternative techniques also reported were the integrated utilization of X-ray crystallography and electron microscopy (Tarry et al., 2017), and Small-angle X-ray scattering (SAXS) or spectroscopic methods for the study of time-resolved conformations (Edwards et al., 2014; Alfermann et al., 2017). Recently, cryo-EM technique was applied to obtain the high-resolution and dynamic structural information of module 5 of pikromycin PKS and pentaketide-bound form of the module, but the homogeneous sample should be prepared carefully (Dutta et al., 2014; Whicher et al., 2014). By harnessing the rapid technical developments, the dynamic structures of the larger enzyme assemblies including multi-module level could be further elucidated.
In addition to the structural efforts, kinetic profiling of the modular PKS and NRPS has been vigorously utilized to measure the substrate specificity of AT and A domain, and to understand the protein–protein interactions between various domains or modules. This information is essential for the determination of the target and method for engineering. In case of the substrate specificity and affinity assays, integrated UV assay with NADPH consumption for acyltransferase activity in PKS (Lowry et al., 2013), and pyrophosphate exchange assay, pyrophosphate release assay, and hydroxylamine quenching assay for adenylation activity in NRPS have been performed (Villiers and Hollfelder, 2009; Wilson and Aldrich, 2010; Katano et al., 2013; Stanisic and Kries, 2019). The substrate specificity of AT, A, KS, and C domains in the elongation module toward growing polyketide chains or polypeptide tethered to phosphopantetheine (Ppant) arm of ACP and T domain was studied by using the mimicking molecule such as acyl- or aminoacyl-N-acetylcysteamine thioesters (acyl- or aminoacyl-SNACs) (Ehmann et al., 2000; Jensen et al., 2012). The studies of the protein–protein interactions commonly exploited the mechanism-based crosslinkers, inhibitors, and probes. Most of them covalently attached to the Ppant arm or other active sites of ACP or T domain. There were many examples of PKSs including phosphopantetheine analogs, photo-crosslinking benzophenone, and azide-alkyne click chemistry linkers for the understanding of KS-AT, AT-ACP, and ACP-KS interactions (Worthington et al., 2006; Ye and Williams, 2014; Ye et al., 2014; Ladner and Williams, 2016). The interactions were quantified by means of radioisotopic transfer assay, thermodynamics heat using calorimetry, and fluorescence, or MS-based techniques. For NRPS, mechanism-based inhibitors such as 5′-O-sulfamoyladenosine (AMS) and adenosine vinylsulfonamide (AVS), azide-alkyne click chemistry linkers, and biotin probes were mainly used for the studies of NRPS A-T interactions (Finking et al., 2003; Sundlov et al., 2012; Ishikawa and Kakeya, 2014; Stanisic and Kries, 2019). These crosslinkers were versatile tools for the pretreatment of structural crystallization (Mitchell et al., 2012). As various tools have been used for the kinetic profiling, it might be better to compare between them for the reproducibility, or to establish the universal standard or tool.
Based on the information obtained from the structural and kinetic studies, computational approaches and bioinformatics would be required to design the detailed processes of the engineering of PKS and NRPS enzymes, or pathways (Medema and Fischbach, 2015; Alanjary et al., 2019). For example, the protein or DNA sequence boundaries of the domain, module, and linker for the swapping could be determined from the database of genome, protein, and smBGC (Marchler-Bauer et al., 2015; Blin et al., 2019; Sayers et al., 2020). In the case of the absence of the structural and kinetic profiling data, comparative analysis method was mainly used including sequence alignment tools, database search tools, and evolutionary analysis tools (Navarro-Munoz et al., 2020). Homology modeling tools such as I-TASSER, MODELLER, and SWISS-MODEL were frequently used to predict the structure of PKS or NRPS by comparative analysis with the reported similar structures for the engineering design (Roy et al., 2010; Waterhouse et al., 2018; Bitencourt-Ferreira and de Azevedo, 2019). Lastly, the integration of the enzyme reactions with their substrates, products, protein sequence, and gene sequence was applied to the automated pathway design pipeline tool of the combinations of reactions for the final products (Carbonell et al., 2014; Eng et al., 2018). Along with the structural and kinetic profiling tools, bioinformatic tools should be integrated and continuously updated to provide the universal, reproducible, and versatile strategy to involve the full pipeline of database search, comparative analysis, homology modeling, and experimental design.
Build Tools
Build tools are technical tools for preparing DNA parts, proteins, and hosts to test the experimental design from above. In recent decades synthetic biology tools have been rapidly developed, enabling large-scale, combinatorial, and high-throughput engineering (Winn et al., 2016; Lee et al., 2019). Despite these advanced tools, they are not frequently used for PKS and NRPS engineering as yet. In case of the genetic manipulation tools, the DNA sequence fragments of the domain and module for the engineering were obtained from the native source, mostly by traditional PCR amplification and restriction enzyme digestion. The rapid and inexpensive cost of DNA synthesis and the highly efficient, accurate DNA digestion tools such as CRISPR-Cas9 would replace the conventional restriction enzyme-based cloning method (Lee et al., 2015; Hughes and Ellington, 2017). Moreover, homologous recombination based DNA assembly tools such as Gibson assembly, linear-linear homologous recombination (LLHR), and yeast TAR cloning would be more efficient for the large size and numbers of DNA fragments than traditional ligation methods (Fu et al., 2012; Jiang et al., 2015; Lee et al., 2015). Site-directed mutagenesis for the PKS and NRPS enzymes would be also easier by exploiting CRISPR-Cas9 system and the base-editors, even for in vivo genome editing (Komor et al., 2016). Directed evolution of synthetic libraries for the modular PKS and NRPS are feasible with these genetic manipulation tools. Furthermore, in vitro expression techniques such as protein purification tools and precursor synthesis tools as well as in vivo expression techniques in the heterologous host, such as promoter refactoring and metabolic pathway engineering, would simultaneously be developed with genetic manipulation tools (Chen et al., 2007; Weber et al., 2015; Winn et al., 2016).
Test Tools
Test tools were used to quantitatively measure the novel derivatives from engineering, or the products from novel pathways, to analyze the chemical structure of the products and to screen the desired clone from the libraries. The most widely applied tools for the quantitative measurement of the products were high-throughput mass spectrometry techniques that were also used for the screening and chemical analysis (Krug and Muller, 2014; Boiteau et al., 2018). By integration with other techniques such as liquid chromatography, the separation and evaluation of exact molecular weight, amount, and chemical moieties for the product could be interrogated. In many cases, the screening step is the bottleneck, which requires rapid and accurate selection of the desired clone among the extensive libraries. Leveraging the biological activity of the product for screening was the traditional screening assay logic that involves an inhibition zone assay for antibacterial activity, colorimetric assay for pigment product, and growth assay for the auxotrophic strain of the product such as siderophore (Balouiri et al., 2016; Cleto and Lu, 2017; Wehrs et al., 2019). As these methods were limited to specific products and the small size of the libraries, the universal and high-throughput screening strategies such as yeast surface display combined with FACS (Zhang et al., 2013), Ppant ejection strategy detecting the intermediate of the rate-limiting step by protease (Meluzzi et al., 2008), and biosensor development for the PKS and NRPS product such as the macrolide biosensor MphR (Kasey et al., 2018), along with mass spectrometry, would be a more favorable method.
Design-Build-Test-Learn (DBTL) Cycle for Modular PKS and NRPS Repurposing
Learning from the design-build-test steps of modular PKS and NRPS repurposing would be; (i) the updated information of the protein–protein interactions and the kinetic profiles to the substrate of the enzymes, (ii) the appropriate splicing or engineering sites for the construction of synthetic parts, (iii) the rationale for the selection of compatible sets, (iv) the integration of the information of cognate reactions, sequences, substrate, and products, and (v) the reversible connections between the combination of reactions and the final product for retro-biosynthesis (Bayly and Yadav, 2017). Based on the current understanding, iterative DBTL cycles would ultimately achieve the goal of novel chemical productions as well as novel pathway generation (Figure 5).
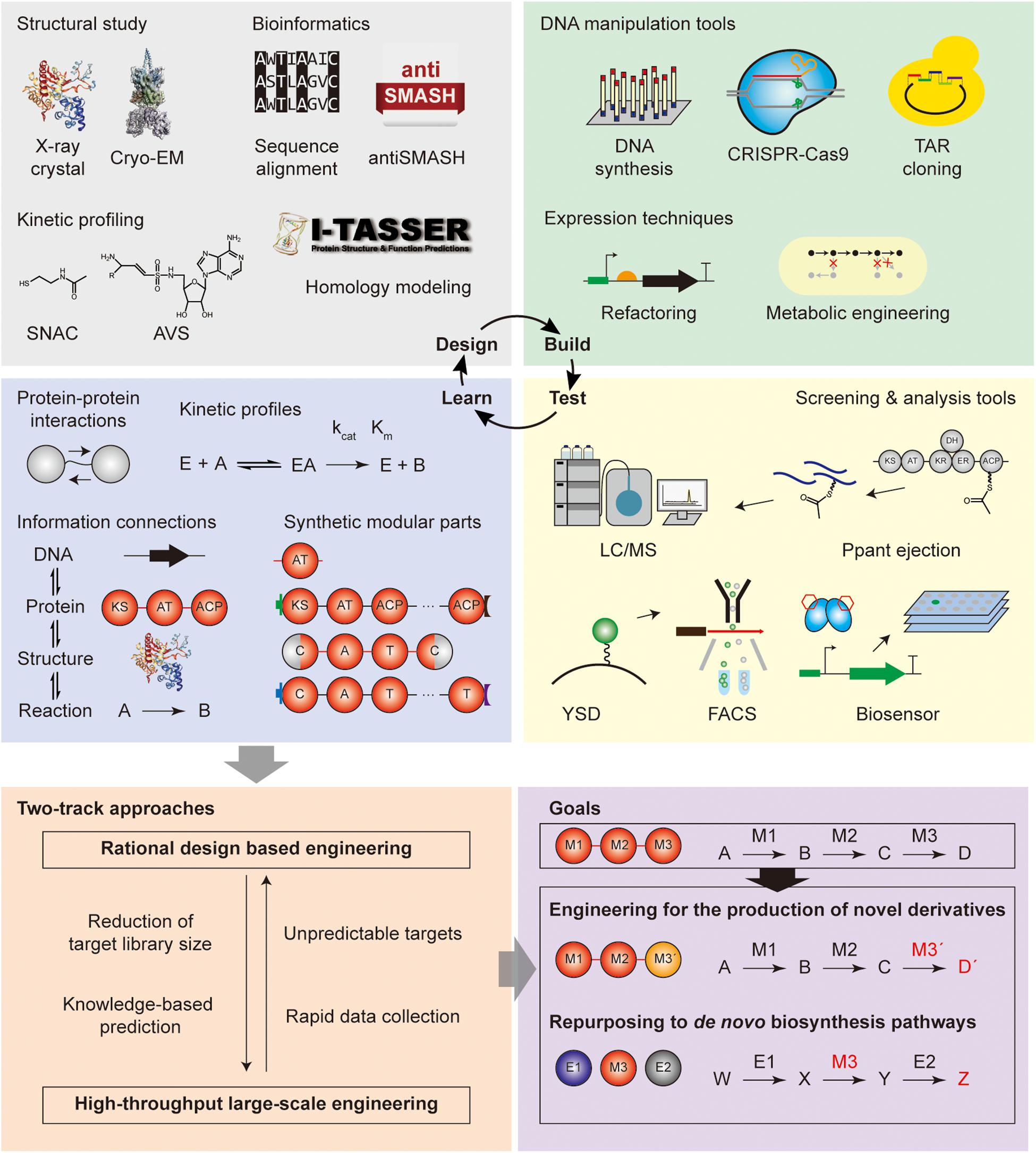
Figure 5. Roadmap for repurposing modular PKS and NRPS. Design-build-test-learn cycle with the tools for each step was illustrated.
Conclusion
We reviewed selected examples of modular PKS and NRPS repurposing for the generations of novel chemicals and pathways, followed by the roadmap in view of the synthetic biological DBTL cycle. The most important lesson from the repurposing examples was the requirement of careful considerations for dissecting the complex protein–protein interactions, despite their functionality in modular fashion. Therefore, the rational engineering design should continuously be improved by the characterization of the modular PKS and NRPS from the large amount of structural, kinetic, and genetic studies, to predict and understand the results. In parallel, the massive and rapid approaches, such as directed evolution and combinatorial strategy along with high-throughput screening, should be widely adopted to find the unpredictable factors from rational engineering. Technical advances for build tools could overcome the previous limitations for the identification of the exchangeable unit and the case-by-case optimization. Ultimately, the two approaches would complement each other by the reducing library size by rational design and the reflection of the updated information learned from the non-rational large-scale strategy. An automated pipeline of the modular PKS and NRPS repurposing for retro-biosynthesis is expected to greatly expand the reservoir of the bio-active compounds.
Author Contributions
B-KC and SH conceived the study. SH, NL, and B-KC drafted the manuscript. SH, NL, SC, BP, and B-KC contributed to the final version of the manuscript.
Funding
This work was funded by the Novo Nordisk Foundation (NNF10CC1016517 to BP), as well as the Bio & Medical Technology Development Program (2018M3A9F3079664 to B-KC), and the Basic Science Research Program (2018R1A1A3A04079196 to SC) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT). Funding for open access charge: Novo Nordisk Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ackerley, D. F., and Lamont, I. L. (2004). Characterization and genetic manipulation of peptide synthetases in Pseudomonas aeruginosa PAO1 in order to generate novel pyoverdines. Chem. Biol. 11, 971–980. doi: 10.1016/j.chembiol.2004.04.014
Agarwal, P. K. (2006). Enzymes: an integrated view of structure, dynamics and function. Microb. Cell Fact. 5:2. doi: 10.1186/1475-2859-5-2
Alanjary, M., Cano-Prieto, C., Gross, H., and Medema, M. H. (2019). Computer-aided re-engineering of nonribosomal peptide and polyketide biosynthetic assembly lines. Nat. Prod. Rep. 36, 1249–1261. doi: 10.1039/c9np00021f
Alfermann, J., Sun, X., Mayerthaler, F., Morrell, T. E., Dehling, E., Volkmann, G., et al. (2017). FRET monitoring of a nonribosomal peptide synthetase. Nat. Chem. Biol. 13, 1009–1015. doi: 10.1038/nchembio.2435
Annaval, T., Paris, C., Leadlay, P. F., Jacob, C., and Weissman, K. J. (2015). Evaluating ketoreductase exchanges as a means of rationally altering polyketide stereochemistry. Chembiochem 16, 1357–1364. doi: 10.1002/cbic.201500113
Balouiri, M., Sadiki, M., and Ibnsouda, S. K. (2016). Methods for in vitro evaluating antimicrobial activity: a review. J. Pharm. Anal. 6, 71–79. doi: 10.1016/j.jpha.2015.11.005
Barajas, J. F., Blake-Hedges, J. M., Bailey, C. B., Curran, S., and Keasling, J. D. (2017). Engineered polyketides: synergy between protein and host level engineering. Synth. Syst. Biotechnol. 2, 147–166. doi: 10.1016/j.synbio.2017.08.005
Barajas, J. F., Phelan, R. M., Schaub, A. J., Kliewer, J. T., Kelly, P. J., Jackson, D. R., et al. (2015). Comprehensive structural and biochemical analysis of the terminal myxalamid reductase domain for the engineered production of primary alcohols. Chem. Biol. 22, 1018–1029. doi: 10.1016/j.chembiol.2015.06.022
Bayly, C. L., and Yadav, V. G. (2017). Towards precision engineering of canonical polyketide synthase domains: recent advances and future prospects. Molecules 22:235. doi: 10.3390/molecules22020235
Beer, R., Herbst, K., Ignatiadis, N., Kats, I., Adlung, L., Meyer, H., et al. (2014). Creating functional engineered variants of the single-module non-ribosomal peptide synthetase IndC by T domain exchange. Mol. Biosyst. 10, 1709–1718. doi: 10.1039/c3mb70594c
Beller, H. R., Goh, E. B., and Keasling, J. D. (2010). Genes involved in long-chain alkene biosynthesis in Micrococcus luteus. Appl. Environ. Microbiol. 76, 1212–1223. doi: 10.1128/AEM.02312-09
Beller, H. R., Lee, T. S., and Katz, L. (2015). Natural products as biofuels and bio-based chemicals: fatty acids and isoprenoids. Nat. Prod. Rep. 32, 1508–1526. doi: 10.1039/c5np00068h
Bernhardt, P., and O’Connor, S. E. (2009). Opportunities for enzyme engineering in natural product biosynthesis. Curr. Opin. Chem. Biol. 13, 35–42. doi: 10.1016/j.cbpa.2009.01.005
Birmingham, W. R., Starbird, C. A., Panosian, T. D., Nannemann, D. P., Iverson, T. M., and Bachmann, B. O. (2014). Bioretrosynthetic construction of a didanosine biosynthetic pathway. Nat. Chem. Biol. 10, 392–399. doi: 10.1038/nchembio.1494
Bitencourt-Ferreira, G., and de Azevedo, W. F. Jr. (2019). Homology modeling of protein targets with MODELLER. Methods Mol. Biol. 2053, 231–249. doi: 10.1007/978-1-4939-9752-7_15
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Bloudoff, K., and Schmeing, T. M. (2017). Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: discovery, dissection and diversity. Biochim. Biophys. Acta 1865(11 Pt B), 1587–1604. doi: 10.1016/j.bbapap.2017.05.010
Boiteau, R. M., Hoyt, D. W., Nicora, C. D., Kinmonth-Schultz, H. A., Ward, J. K., and Bingol, K. (2018). Structure elucidation of unknown metabolites in metabolomics by combined NMR and MS/MS prediction. Metabolites 8:8. doi: 10.3390/metabo8010008
Bozhuyuk, K. A. J., Fleischhacker, F., Linck, A., Wesche, F., Tietze, A., Niesert, C. P., et al. (2018). De novo design and engineering of non-ribosomal peptide synthetases. Nat. Chem. 10, 275–281. doi: 10.1038/nchem.2890
Bozhuyuk, K. A. J., Linck, A., Tietze, A., Kranz, J., Wesche, F., Nowak, S., et al. (2019). Modification and de novo design of non-ribosomal peptide synthetases using specific assembly points within condensation domains. Nat. Chem. 11, 653–661. doi: 10.1038/s41557-019-0276-z
Broadhurst, R. W., Nietlispach, D., Wheatcroft, M. P., Leadlay, P. F., and Weissman, K. J. (2003). The structure of docking domains in modular polyketide synthases. Chem. Biol. 10, 723–731. doi: 10.1016/s1074-5521(03)00156-x
Brown, A. S., Calcott, M. J., Owen, J. G., and Ackerley, D. F. (2018). Structural, functional and evolutionary perspectives on effective re-engineering of non-ribosomal peptide synthetase assembly lines. Nat. Prod. Rep. 35, 1210–1228. doi: 10.1039/c8np00036k
Buchholz, T. J., Geders, T. W., Bartley, F. E., Reynolds, K. A., Smith, J. L., and Sherman, D. H. (2009). Structural basis for binding specificity between subclasses of modular polyketide synthase docking domains. ACS Chem. Biol. 4, 41–52. doi: 10.1021/cb8002607
Butz, D., Schmiederer, T., Hadatsch, B., Wohlleben, W., Weber, T., and Sussmuth, R. D. (2008). Module extension of a non-ribosomal peptide synthetase of the glycopeptide antibiotic balhimycin produced by Amycolatopsis balhimycina. Chembiochem 9, 1195–1200. doi: 10.1002/cbic.200800068
Calcott, M. J., Owen, J. G., Lamont, I. L., and Ackerley, D. F. (2014). Biosynthesis of novel pyoverdines by domain substitution in a nonribosomal peptide synthetase of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 80, 5723–5731. doi: 10.1128/AEM.01453-14
Carbonell, P., Parutto, P., Baudier, C., Junot, C., and Faulon, J. L. (2014). Retropath: automated pipeline for embedded metabolic circuits. ACS Synth. Biol. 3, 565–577. doi: 10.1021/sb4001273
Challis, G. L., Ravel, J., and Townsend, C. A. (2000). Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 7, 211–224. doi: 10.1016/s1074-5521(00)00091-0
Chan, Y. A., Podevels, A. M., Kevany, B. M., and Thomas, M. G. (2009). Biosynthesis of polyketide synthase extender units. Nat. Prod. Rep. 26, 90–114. doi: 10.1039/b801658p
Chen, A. Y., Cane, D. E., and Khosla, C. (2007). Structure-based dissociation of a type I polyketide synthase module. Chem. Biol. 14, 784–792. doi: 10.1016/j.chembiol.2007.05.015
Chevrette, M. G., Gutierrez-Garcia, K., Selem-Mojica, N., Aguilar-Martinez, C., Yanez-Olvera, A., Ramos-Aboites, H. E., et al. (2019). Evolutionary dynamics of natural product biosynthesis in bacteria. Nat. Prod. Rep. doi: 10.1039/c9np00048h [Epub ahead of print].
Chiocchini, C., Linne, U., and Stachelhaus, T. (2006). In vivo biocombinatorial synthesis of lipopeptides by COM domain-mediated reprogramming of the surfactin biosynthetic complex. Chem. Biol. 13, 899–908. doi: 10.1016/j.chembiol.2006.06.015
Cleto, S., and Lu, T. K. (2017). An engineered synthetic pathway for discovering nonnatural nonribosomal peptides in Escherichia coli. mBio 8:e01474-17. doi: 10.1128/mBio.01474-17
Clomburg, J. M., Blankschien, M. D., Vick, J. E., Chou, A., Kim, S., and Gonzalez, R. (2015). Integrated engineering of beta-oxidation reversal and omega-oxidation pathways for the synthesis of medium chain omega-functionalized carboxylic acids. Metab. Eng. 28, 202–212. doi: 10.1016/j.ymben.2015.01.007
Clugston, S. L., Sieber, S. A., Marahiel, M. A., and Walsh, C. T. (2003). Chirality of peptide bond-forming condensation domains in nonribosomal peptide synthetases: the C5 domain of tyrocidine synthetase is a DCL catalyst. Biochemistry 42, 12095–12104. doi: 10.1021/bi035090+
Conti, E., Stachelhaus, T., Marahiel, M. A., and Brick, P. (1997). Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J. 16, 4174–4183. doi: 10.1093/emboj/16.14.4174
Crawford, J. M., Portmann, C., Kontnik, R., Walsh, C. T., and Clardy, J. (2011). NRPS substrate promiscuity diversifies the xenematides. Org. Lett. 13, 5144–5147. doi: 10.1021/ol2020237
Crüsemann, M., Kohlhaas, C., and Piel, J. (2013). Evolution-guided engineering of nonribosomal peptide synthetase adenylation domains. Chem. Sci. 4, 1041–1045. doi: 10.1039/C2SC21722H
Dan, Q., Newmister, S. A., Klas, K. R., Fraley, A. E., McAfoos, T. J., Somoza, A. D., et al. (2019). Fungal indole alkaloid biogenesis through evolution of a bifunctional reductase/Diels-Alderase. Nat. Chem. 11, 972–980. doi: 10.1038/s41557-019-0326-6
Doekel, S., and Marahiel, M. A. (2000). Dipeptide formation on engineered hybrid peptide synthetases. Chem. Biol. 7, 373–384. doi: 10.1016/s1074-5521(00)00118-6
Dutta, S., Whicher, J. R., Hansen, D. A., Hale, W. A., Chemler, J. A., Congdon, G. R., et al. (2014). Structure of a modular polyketide synthase. Nature 510, 512–517. doi: 10.1038/nature13423
Edwards, A. L., Matsui, T., Weiss, T. M., and Khosla, C. (2014). Architectures of whole-module and bimodular proteins from the 6-deoxyerythronolide B synthase. J. Mol. Biol. 426, 2229–2245. doi: 10.1016/j.jmb.2014.03.015
Ehmann, D. E., Trauger, J. W., Stachelhaus, T., and Walsh, C. T. (2000). Aminoacyl-SNACs as small-molecule substrates for the condensation domains of nonribosomal peptide synthetases. Chem. Biol. 7, 765–772. doi: 10.1016/s1074-5521(00)00022-3
Eichner, S., Floss, H. G., Sasse, F., and Kirschning, A. (2009). New, highly active nonbenzoquinone geldanamycin derivatives by using mutasynthesis. Chembiochem 10, 1801–1805. doi: 10.1002/cbic.200900246
Eng, C. H., Backman, T. W. H., Bailey, C. B., Magnan, C., Garcia Martin, H., Katz, L., et al. (2018). ClusterCAD: a computational platform for type I modular polyketide synthase design. Nucleic Acids Res. 46, D509–D515. doi: 10.1093/nar/gkx893
Eng, C. H., Yuzawa, S., Wang, G., Baidoo, E. E., Katz, L., and Keasling, J. D. (2016). Alteration of polyketide stereochemistry from anti to syn by a ketoreductase domain exchange in a type I modular polyketide synthase subunit. Biochemistry 55, 1677–1680. doi: 10.1021/acs.biochem.6b00129
Eppelmann, K., Stachelhaus, T., and Marahiel, M. A. (2002). Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry 41, 9718–9726. doi: 10.1021/bi0259406
Eustaquio, A. S., and Moore, B. S. (2008). Mutasynthesis of fluorosalinosporamide, a potent and reversible inhibitor of the proteasome. Angew. Chem. Int. Ed. Engl. 47, 3936–3938. doi: 10.1002/anie.200800177
Evans, B. S., Chen, Y., Metcalf, W. W., Zhao, H., and Kelleher, N. L. (2011). Directed evolution of the nonribosomal peptide synthetase AdmK generates new andrimid derivatives in vivo. Chem. Biol. 18, 601–607. doi: 10.1016/j.chembiol.2011.03.008
Faille, A., Gavalda, S., Slama, N., Lherbet, C., Maveyraud, L., Guillet, V., et al. (2017). Insights into substrate modification by dehydratases from type I polyketide synthases. J. Mol. Biol. 429, 1554–1569. doi: 10.1016/j.jmb.2017.03.026
Finking, R., Neumuller, A., Solsbacher, J., Konz, D., Kretzschmar, G., Schweitzer, M., et al. (2003). Aminoacyl adenylate substrate analogues for the inhibition of adenylation domains of nonribosomal peptide synthetases. Chembiochem 4, 903–906. doi: 10.1002/cbic.200300666
Fischbach, M. A., Lai, J. R., Roche, E. D., Walsh, C. T., and Liu, D. R. (2007). Directed evolution can rapidly improve the activity of chimeric assembly-line enzymes. Proc. Natl. Acad. Sci. U.S.A. 104, 11951–11956. doi: 10.1073/pnas.0705348104
Fu, J., Bian, X., Hu, S., Wang, H., Huang, F., Seibert, P. M., et al. (2012). Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 30, 440–446. doi: 10.1038/nbt.2183
Garcia-Bernardo, J., Xiang, L., Hong, H., Moore, B. S., and Leadlay, P. F. (2004). Engineered biosynthesis of phenyl-substituted polyketides. Chembiochem 5, 1129–1131. doi: 10.1002/cbic.200400007
Gokhale, R. S., Tsuji, S. Y., Cane, D. E., and Khosla, C. (1999). Dissecting and exploiting intermodular communication in polyketide synthases. Science 284, 482–485. doi: 10.1126/science.284.5413.482
Hagen, A., Poust, S., de Rond, T., Yuzawa, S., Katz, L., Adams, P. D., et al. (2014). In vitro analysis of carboxyacyl substrate tolerance in the loading and first extension modules of borrelidin polyketide synthase. Biochemistry 53, 5975–5977. doi: 10.1021/bi500951c
Hagen, A., Poust, S., Rond, T., Fortman, J. L., Katz, L., Petzold, C. J., et al. (2016). Engineering a polyketide synthase for in vitro production of adipic acid. ACS Synth. Biol. 5, 21–27. doi: 10.1021/acssynbio.5b00153
Hahn, M., and Stachelhaus, T. (2004). Selective interaction between nonribosomal peptide synthetases is facilitated by short communication-mediating domains. Proc. Natl. Acad. Sci. U.S.A. 101, 15585–15590. doi: 10.1073/pnas.0404932101
Hahn, M., and Stachelhaus, T. (2006). Harnessing the potential of communication-mediating domains for the biocombinatorial synthesis of nonribosomal peptides. Proc. Natl. Acad. Sci. U.S.A. 103, 275–280. doi: 10.1073/pnas.0508409103
Han, J. W., Kim, E. Y., Lee, J. M., Kim, Y. S., Bang, E., and Kim, B. S. (2012). Site-directed modification of the adenylation domain of the fusaricidin nonribosomal peptide synthetase for enhanced production of fusaricidin analogs. Biotechnol. Lett. 34, 1327–1334. doi: 10.1007/s10529-012-0913-8
Hans, M., Hornung, A., Dziarnowski, A., Cane, D. E., and Khosla, C. (2003). Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J. Am. Chem. Soc. 125, 5366–5374. doi: 10.1021/ja029539i
Hoppert, M., Gentzsch, C., and Schorgendorfer, K. (2001). Structure and localization of cyclosporin synthetase, the key enzyme of cyclosporin biosynthesis in Tolypocladium inflatum. Arch. Microbiol. 176, 285–293. doi: 10.1007/s002030100324
Howard, T. P., Middelhaufe, S., Moore, K., Edner, C., Kolak, D. M., Taylor, G. N., et al. (2013). Synthesis of customized petroleum-replica fuel molecules by targeted modification of free fatty acid pools in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 7636–7641. doi: 10.1073/pnas.1215966110
Hughes, R. A., and Ellington, A. D. (2017). Synthetic DNA synthesis and assembly: putting the synthetic in synthetic biology. Cold Spring Harb. Perspect. Biol. 9:a023812. doi: 10.1101/cshperspect.a023812
Hur, G. H., Vickery, C. R., and Burkart, M. D. (2012). Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat. Prod. Rep. 29, 1074–1098. doi: 10.1039/c2np20025b
Ishikawa, F., and Kakeya, H. (2014). Specific enrichment of nonribosomal peptide synthetase module by an affinity probe for adenylation domains. Bioorg. Med. Chem. Lett. 24, 865–869. doi: 10.1016/j.bmcl.2013.12.082
Jaremko, M. J., Davis, T. D., Corpuz, J. C., and Burkart, M. D. (2019). Type II non-ribosomal peptide synthetase proteins: structure, mechanism, and protein-protein interactions. Nat. Prod. Rep. 37, 355–379. doi: 10.1039/c9np00047j
Jensen, K., Niederkruger, H., Zimmermann, K., Vagstad, A. L., Moldenhauer, J., Brendel, N., et al. (2012). Polyketide proofreading by an acyltransferase-like enzyme. Chem. Biol. 19, 329–339. doi: 10.1016/j.chembiol.2012.01.005
Jiang, W., Zhao, X., Gabrieli, T., Lou, C., Ebenstein, Y., and Zhu, T. F. (2015). Cas9-Assisted Targeting of CHromosome segments CATCH enables one-step targeted cloning of large gene clusters. Nat. Commun. 6:8101. doi: 10.1038/ncomms9101
Kaljunen, H., Schiefelbein, S. H. H., Stummer, D., Kozak, S., Meijers, R., Christiansen, G., et al. (2015). Structural elucidation of the bispecificity of A domains as a basis for activating non-natural amino acids. Angew. Chem. Int. Ed. Engl. 54, 8833–8836. doi: 10.1002/anie.201503275
Kalkreuter, E., CroweTipton, J. M., Lowell, A. N., Sherman, D. H., and Williams, G. J. (2019). Engineering the substrate specificity of a modular polyketide synthase for installation of consecutive non-natural extender units. J. Am. Chem. Soc. 141, 1961–1969. doi: 10.1021/jacs.8b10521
Kasey, C. M., Zerrad, M., Li, Y., Cropp, T. A., and Williams, G. J. (2018). Development of transcription factor-based designer macrolide biosensors for metabolic engineering and synthetic biology. ACS Synth. Biol. 7, 227–239. doi: 10.1021/acssynbio.7b00287
Katano, H., Watanabe, H., Takakuwa, M., Maruyama, C., and Hamano, Y. (2013). Colorimetric determination of pyrophosphate anion and its application to adenylation enzyme assay. Anal. Sci. 29, 1095–1098. doi: 10.2116/analsci.29.1095
Keatinge-Clay, A. T. (2017). Polyketide synthase modules redefined. Angew. Chem. Int. Ed. Engl. 56, 4658–4660. doi: 10.1002/anie.201701281
Keatinge-Clay, A. T., and Stroud, R. M. (2006). The structure of a ketoreductase determines the organization of the β-carbon processing enzymes of modular polyketide synthases. Structure 14, 737–748. doi: 10.1016/j.str.2006.01.009
Khosla, C., Tang, Y., Chen, A. Y., Schnarr, N. A., and Cane, D. E. (2007). Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem. 76, 195–221. doi: 10.1146/annurev.biochem.76.053105.093515
Kim, C. Y., Alekseyev, V. Y., Chen, A. Y., Tang, Y., Cane, D. E., and Khosla, C. (2004). Reconstituting modular activity from separated domains of 6-deoxyerythronolide B synthase. Biochemistry 43, 13892–13898. doi: 10.1021/bi048418n
Klapper, M., Braga, D., Lackner, G., Herbst, R., and Stallforth, P. (2018). Bacterial alkaloid biosynthesis: structural diversity via a minimalistic nonribosomal peptide synthetase. Cell Chem. Biol. 25, doi: 10.1016/j.chembiol.2018.02.013
Klaus, M., D’Souza, A. D., Nivina, A., Khosla, C., and Grininger, M. (2019). Engineering of chimeric polyketide synthases using SYNZIP docking domains. ACS Chem. Biol. 14, 426–433. doi: 10.1021/acschembio.8b01060
Klaus, M., and Grininger, M. (2018). Engineering strategies for rational polyketide synthase design. Nat. Prod. Rep. 35, 1070–1081. doi: 10.1039/c8np00030a
Klaus, M., Ostrowski, M. P., Austerjost, J., Robbins, T., Lowry, B., Cane, D. E., et al. (2016). Protein-protein interactions, not substrate recognition, dominate the turnover of chimeric assembly line polyketide synthases. J. Biol. Chem. 291, 16404–16415. doi: 10.1074/jbc.M116.730531
Kohli, R. M., Trauger, J. W., Schwarzer, D., Marahiel, M. A., and Walsh, C. T. (2001). Generality of peptide cyclization catalyzed by isolated thioesterase domains of nonribosomal peptide synthetases. Biochemistry 40, 7099–7108. doi: 10.1021/bi010036j
Kohli, R. M., Walsh, C. T., and Burkart, M. D. (2002). Biomimetic synthesis and optimization of cyclic peptide antibiotics. Nature 418, 658–661. doi: 10.1038/nature00907
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., and Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. doi: 10.1038/nature17946
Kornfuehrer, T., and Eustáquio, A. S. (2019). Diversification of polyketide structures via synthase engineering. MedChemComm 10, 1256–1272. doi: 10.1039/C9MD00141G
Koryakina, I., Kasey, C., McArthur, J. B., Lowell, A. N., Chemler, J. A., Li, S., et al. (2017). Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering. ACS Chem. Biol. 12, 114–123. doi: 10.1021/acschembio.6b00732
Kries, H., Wachtel, R., Pabst, A., Wanner, B., Niquille, D., and Hilvert, D. (2014). Reprogramming nonribosomal peptide synthetases for “clickable” amino acids. Angew. Chem. Int. Ed. Engl. 53, 10105–10108. doi: 10.1002/anie.201405281
Krug, D., and Muller, R. (2014). Secondary metabolomics: the impact of mass spectrometry-based approaches on the discovery and characterization of microbial natural products. Nat. Prod. Rep. 31, 768–783. doi: 10.1039/c3np70127a
Kuhstoss, S., Huber, M., Turner, J. R., Paschal, J. W., and Rao, R. N. (1996). Production of a novel polyketide through the construction of a hybrid polyketide synthase. Gene 183, 231–236. doi: 10.1016/s0378-1119(96)00565-3
Kwan, D. H., Sun, Y., Schulz, F., Hong, H., Popovic, B., Sim-Stark, J. C., et al. (2008). Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem. Biol. 15, 1231–1240. doi: 10.1016/j.chembiol.2008.09.012
Ladner, C. C., and Williams, G. J. (2016). Harnessing natural product assembly lines: structure, promiscuity, and engineering. J. Ind. Microbiol. Biotechnol. 43, 371–387. doi: 10.1007/s10295-015-1704-8
Lee, N., Hwang, S., Lee, Y., Cho, S., Palsson, B., and Cho, B. K. (2019). Synthetic biology tools for novel secondary metabolite discovery in Streptomyces. J. Microbiol. Biotechnol. 29, 667–686. doi: 10.4014/jmb.1904.04015
Lee, N. C., Larionov, V., and Kouprina, N. (2015). Highly efficient CRISPR/Cas9-mediated TAR cloning of genes and chromosomal loci from complex genomes in yeast. Nucleic Acids Res. 43:e55. doi: 10.1093/nar/gkv112
Li, R., Oliver, R. A., and Townsend, C. A. (2017). Identification and characterization of the sulfazecin monobactam biosynthetic gene cluster. Cell Chem. Biol. 24, 24–34. doi: 10.1016/j.chembiol.2016.11.010
Li, Y., Zhang, W., Zhang, H., Tian, W., Wu, L., Wang, S., et al. (2018). Structural basis of a broadly selective acyltransferase from the polyketide synthase of splenocin. Angew. Chem. Int. Ed. Engl. 57, 5823–5827. doi: 10.1002/anie.201802805
Lim, J., Sun, H., Fan, J. S., Hameed, I. F., Lescar, J., Liang, Z. X., et al. (2012). Rigidifying acyl carrier protein domain in iterative type I PKS CalE8 does not affect its function. Biophys. J. 103, 1037–1044. doi: 10.1016/j.bpj.2012.08.006
Liu, Q., Wu, K., Cheng, Y., Lu, L., Xiao, E., Zhang, Y., et al. (2015). Engineering an iterative polyketide pathway in Escherichia coli results in single-form alkene and alkane overproduction. Metab. Eng. 28, 82–90. doi: 10.1016/j.ymben.2014.12.004
Liu, X., and Walsh, C. T. (2009). Cyclopiazonic acid biosynthesis in Aspergillus sp.: characterization of a reductase-like R∗ domain in cyclopiazonate synthetase that forms and releases cyclo-acetoacetyl-L-tryptophan. Biochemistry 48, 8746–8757. doi: 10.1021/bi901123r
Lowden, P. A., Bohm, G. A., Metcalfe, S., Staunton, J., and Leadlay, P. F. (2004). New rapamycin derivatives by precursor-directed biosynthesis. Chembiochem 5, 535–538. doi: 10.1002/cbic.200300758
Lowry, B., Robbins, T., Weng, C. H., O’Brien, R. V., Cane, D. E., and Khosla, C. (2013). In vitro reconstitution and analysis of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 135, 16809–16812. doi: 10.1021/ja409048k
Lundy, T. A., Mori, S., and Garneau-Tsodikova, S. (2018). Engineering bifunctional enzymes capable of adenylating and selectively methylating the side chain or core of amino acids. ACS Synth. Biol. 7, 399–404. doi: 10.1021/acssynbio.7b00426
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S., Chitsaz, F., Geer, L. Y., et al. (2015). CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Marsden, A. F. A., Wilkinson, B., Cortés, J., Dunster, N. J., Staunton, J., and Leadlay, P. F. (1998). Engineering broader specificity into an antibiotic-producing polyketide synthase. Science 279, 199–202. doi: 10.1126/science.279.5348.199
McErlean, M., Overbay, J., and Van Lanen, S. (2019). Refining and expanding nonribosomal peptide synthetase function and mechanism. J. Ind. Microbiol. Biotechnol. 46, 493–513. doi: 10.1007/s10295-018-02130-w
Medema, M. H., and Fischbach, M. A. (2015). Computational approaches to natural product discovery. Nat. Chem. Biol. 11, 639–648. doi: 10.1038/nchembio.1884
Meluzzi, D., Zheng, W. H., Hensler, M., Nizet, V., and Dorrestein, P. C. (2008). Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg. Med. Chem. Lett. 18, 3107–3111. doi: 10.1016/j.bmcl.2007.10.104
Menzella, H. G., Carney, J. R., and Santi, D. V. (2007). Rational design and assembly of synthetic trimodular polyketide synthases. Chem. Biol. 14, 143–151. doi: 10.1016/j.chembiol.2006.12.002
Menzella, H. G., Reid, R., Carney, J. R., Chandran, S. S., Reisinger, S. J., Patel, K. G., et al. (2005). Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 23, 1171–1176. doi: 10.1038/nbt1128
Miller, B. R., Sundlov, J. A., Drake, E. J., Makin, T. A., and Gulick, A. M. (2014). Analysis of the linker region joining the adenylation and carrier protein domains of the modular nonribosomal peptide synthetases. Proteins 82, 2691–2702. doi: 10.1002/prot.24635
Miller, D. A., and Walsh, C. T. (2001). Yersiniabactin synthetase: probing the recognition of carrier protein domains by the catalytic heterocyclization domains, Cy1 and Cy2, in the chain-initiating HWMP2 subunit. Biochemistry 40, 5313–5321. doi: 10.1021/bi002905v
Mitchell, C. A., Shi, C., Aldrich, C. C., and Gulick, A. M. (2012). Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51, 3252–3263. doi: 10.1021/bi300112e
Miyanaga, A., Iwasawa, S., Shinohara, Y., Kudo, F., and Eguchi, T. (2016). Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 113, 1802–1807. doi: 10.1073/pnas.1520042113
Mo, S., Kim, D. H., Lee, J. H., Park, J. W., Basnet, D. B., Ban, Y. H., et al. (2011). Biosynthesis of the allylmalonyl-CoA extender unit for the FK506 polyketide synthase proceeds through a dedicated polyketide synthase and facilitates the mutasynthesis of analogues. J. Am. Chem. Soc. 133, 976–985. doi: 10.1021/ja108399b
Moran, S., Rai, D. K., Clark, B. R., and Murphy, C. D. (2009). Precursor-directed biosynthesis of fluorinated iturin A in Bacillus spp. Org. Biomol. Chem. 7, 644–646. doi: 10.1039/b816345f
Muchiri, R., and Walker, K. D. (2012). Taxol biosynthesis: tyrocidine synthetase A catalyzes the production of phenylisoserinyl CoA and other amino phenylpropanoyl thioesters. Chem. Biol. 19, 679–685. doi: 10.1016/j.chembiol.2012.05.007
Muchiri, R., and Walker, K. D. (2017). Paclitaxel biosynthesis: adenylation and thiolation domains of an NRPS TycA PheAT module produce various arylisoserine CoA thioesters. Biochemistry 56, 1415–1425. doi: 10.1021/acs.biochem.6b01188
Murphy, A. C., Hong, H., Vance, S., Broadhurst, R. W., and Leadlay, P. F. (2016). Broadening substrate specificity of a chain-extending ketosynthase through a single active-site mutation. Chem. Commun. 52, 8373–8376. doi: 10.1039/c6cc03501a
Musiol-Kroll, E. M., and Wohlleben, W. (2018). Acyltransferases as tools for polyketide synthase engineering. Antibiotics 7:62. doi: 10.3390/antibiotics7030062
Mutka, S. C., Bondi, S. M., Carney, J. R., Da Silva, N. A., and Kealey, J. T. (2006). Metabolic pathway engineering for complex polyketide biosynthesis in Saccharomyces cerevisiae. FEMS Yeast Res. 6, 40–47. doi: 10.1111/j.1567-1356.2005.00001.x
Navarro-Munoz, J. C., Selem-Mojica, N., Mullowney, M. W., Kautsar, S. A., Tryon, J. H., Parkinson, E. I., et al. (2020). A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 16, 60–68. doi: 10.1038/s41589-019-0400-9
Nguyen, K. T., He, X., Alexander, D. C., Li, C., Gu, J. Q., Mascio, C., et al. (2010). Genetically engineered lipopeptide antibiotics related to A54145 and daptomycin with improved properties. Antimicrob. Agents Chemother. 54, 1404–1413. doi: 10.1128/AAC.01307-09
Nguyen, K. T., Ritz, D., Gu, J. Q., Alexander, D., Chu, M., Miao, V., et al. (2006). Combinatorial biosynthesis of novel antibiotics related to daptomycin. Proc. Natl. Acad. Sci. U.S.A. 103, 17462–17467. doi: 10.1073/pnas.0608589103
Niu, W., Draths, K. M., and Frost, J. W. (2002). Benzene-free synthesis of adipic acid. Biotechnol. Prog. 18, 201–211. doi: 10.1021/bp010179x
Oliynyk, M., Brown, M. J., Cortes, J., Staunton, J., and Leadlay, P. F. (1996). A hybrid modular polyketide synthase obtained by domain swapping. Chem. Biol. 3, 833–839. doi: 10.1016/s1074-5521(96)90069-1
Pang, B., Valencia, L. E., Wang, J., Wan, Y., Lal, R., Zargar, A., et al. (2019). Technical advances to accelerate modular type I polyketide synthase engineering towards a retro-biosynthetic platform. Biotechnol. Bioprocess Eng. 24, 413–423.
Patel, K., Piagentini, M., Rascher, A., Tian, Z. Q., Buchanan, G. O., Regentin, R., et al. (2004). Engineered biosynthesis of geldanamycin analogs for Hsp90 inhibition. Chem. Biol. 11, 1625–1633. doi: 10.1016/j.chembiol.2004.09.012
Petkovic, H., Lill, R. E., Sheridan, R. M., Wilkinson, B., McCormick, E. L., McArthur, H. A., et al. (2003). A novel erythromycin, 6-desmethyl erythromycin D, made by substituting an acyltransferase domain of the erythromycin polyketide synthase. J. Antibiot. 56, 543–551. doi: 10.7164/antibiotics.56.543
Petkovic, H., Sandmann, A., Challis, I. R., Hecht, H. J., Silakowski, B., Low, L., et al. (2008). Substrate specificity of the acyl transferase domains of EpoC from the epothilone polyketide synthase. Org. Biomol. Chem. 6, 500–506. doi: 10.1039/b714804f
Pinto, A., Wang, M., Horsman, M., and Boddy, C. N. (2012). 6-Deoxyerythronolide B synthase thioesterase-catalyzed macrocyclization is highly stereoselective. Org. Lett. 14, 2278–2281. doi: 10.1021/ol300707j
Qiao, K., Zhou, H., Xu, W., Zhang, W., Garg, N., and Tang, Y. (2011). A fungal nonribosomal peptide synthetase module that can synthesize thiopyrazines. Org. Lett. 13, 1758–1761. doi: 10.1021/ol200288w
Reeves, C. D., Murli, S., Ashley, G. W., Piagentini, M., Hutchinson, C. R., and McDaniel, R. (2001). Alteration of the substrate specificity of a modular polyketide synthase acyltransferase domain through site-specific mutations. Biochemistry 40, 15464–15470. doi: 10.1021/bi015864r
Regentin, R., Kennedy, J., Wu, N., Carney, J. R., Licari, P., Galazzo, J., et al. (2004). Precursor-directed biosynthesis of novel triketide lactones. Biotechnol. Prog. 20, 122–127. doi: 10.1021/bp0341949
Reid, R., Piagentini, M., Rodriguez, E., Ashley, G., Viswanathan, N., Carney, J., et al. (2003). A model of structure and catalysis for ketoreductase domains in modular polyketide synthases. Biochemistry 42, 72–79. doi: 10.1021/bi0268706
Richter, C. D., Nietlispach, D., Broadhurst, R. W., and Weissman, K. J. (2008). Multienzyme docking in hybrid megasynthetases. Nat. Chem. Biol. 4, 75–81. doi: 10.1038/nchembio.2007.61
Rottig, M., Medema, M. H., Blin, K., Weber, T., Rausch, C., and Kohlbacher, O. (2011). NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39, W362–W367. doi: 10.1093/nar/gkr323
Rowe, C. J., Bohm, I. U., Thomas, I. P., Wilkinson, B., Rudd, B. A., Foster, G., et al. (2001). Engineering a polyketide with a longer chain by insertion of an extra module into the erythromycin-producing polyketide synthase. Chem. Biol. 8, 475–485. doi: 10.1016/s1074-5521(01)00024-2
Roy, A., Kucukural, A., and Zhang, Y. (2010). I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738. doi: 10.1038/nprot.2010.5
Rutledge, P. J., and Challis, G. L. (2015). Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 13, 509–523. doi: 10.1038/nrmicro3496
Samel, S. A., Schoenafinger, G., Knappe, T. A., Marahiel, M. A., and Essen, L. O. (2007). Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure 15, 781–792. doi: 10.1016/j.str.2007.05.008
Sayers, E. W., Cavanaugh, M., Clark, K., Ostell, J., Pruitt, K. D., and Karsch-Mizrachi, I. (2020). GenBank. Nucleic Acids Res. 48, D84–D86. doi: 10.1093/nar/gkz956
Schauwecker, F., Pfennig, F., Grammel, N., and Keller, U. (2000). Construction and in vitro analysis of a new bi-modular polypeptide synthetase for synthesis of N-methylated acyl peptides. Chem. Biol. 7, 287–297. doi: 10.1016/s1074-5521(00)00103-4
Schneider, T. L., Shen, B., and Walsh, C. T. (2003). Oxidase domains in epothilone and bleomycin biosynthesis: thiazoline to thiazole oxidation during chain elongation. Biochemistry 42, 9722–9730. doi: 10.1021/bi034792w
Schoenafinger, G., Schracke, N., Linne, U., and Marahiel, M. A. (2006). Formylation domain: an essential modifying enzyme for the nonribosomal biosynthesis of linear gramicidin. J. Am. Chem. Soc. 128, 7406–7407. doi: 10.1021/ja0611240
Skiba, M. A., Tran, C. L., Dan, Q., Sikkema, A. P., Klaver, Z., Gerwick, W. H., et al. (2020). Repurposing the GNAT Fold in the Initiation of Polyketide Biosynthesis. Structure 28, 63–74.e4. doi: 10.1016/j.str.2019.11.004
Stachelhaus, T., Mootz, H. D., and Marahiel, M. A. (1999). The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505. doi: 10.1016/S1074-5521(99)80082-9
Stachelhaus, T., Schneider, A., and Marahiel, M. A. (1995). Rational design of peptide antibiotics by targeted replacement of bacterial and fungal domains. Science 269, 69–72. doi: 10.1126/science.7604280
Stanisic, A., and Kries, H. (2019). Adenylation domains in nonribosomal peptide engineering. Chembiochem 20, 1347–1356. doi: 10.1002/cbic.201800750
Stassi, D. L., Kakavas, S. J., Reynolds, K. A., Gunawardana, G., Swanson, S., Zeidner, D., et al. (1998). Ethyl-substituted erythromycin derivatives produced by directed metabolic engineering. Proc. Natl. Acad. Sci. U.S.A. 95, 7305–7309. doi: 10.1073/pnas.95.13.7305
Staunton, J., and Weissman, K. J. (2001). Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 18, 380–416. doi: 10.1039/a909079g
Stein, D. B., Linne, U., Hahn, M., and Marahiel, M. A. (2006). Impact of epimerization domains on the intermodular transfer of enzyme-bound intermediates in nonribosomal peptide synthesis. Chembiochem 7, 1807–1814. doi: 10.1002/cbic.200600192
Sugimoto, Y., Ishida, K., Traitcheva, N., Busch, B., Dahse, H. M., and Hertweck, C. (2015). Freedom and constraint in engineered noncolinear polyketide assembly lines. Chem. Biol. 22, 229–240. doi: 10.1016/j.chembiol.2014.12.014
Sukovich, D. J., Seffernick, J. L., Richman, J. E., Gralnick, J. A., and Wackett, L. P. (2010). Widespread head-to-head hydrocarbon biosynthesis in bacteria and role of OleA. Appl. Environ. Microbiol. 76, 3850–3862. doi: 10.1128/AEM.00436-10
Sundlov, J. A., Shi, C., Wilson, D. J., Aldrich, C. C., and Gulick, A. M. (2012). Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 19, 188–198. doi: 10.1016/j.chembiol.2011.11.013
Sussmuth, R. D., and Mainz, A. (2017). Nonribosomal peptide synthesis-Principles and prospects. Angew. Chem. Int. Ed. Engl. 56, 3770–3821. doi: 10.1002/anie.201609079
Tang, Y., Kim, C. Y., Mathews, I. I., Cane, D. E., and Khosla, C. (2006). The 2.7-Angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc. Natl. Acad. Sci. U.S.A. 103, 11124–11129. doi: 10.1073/pnas.0601924103
Tanovic, A., Samel, S. A., Essen, L. O., and Marahiel, M. A. (2008). Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321, 659–663. doi: 10.1126/science.1159850
Tarry, M. J., Haque, A. S., Bui, K. H., and Schmeing, T. M. (2017). X-ray crystallography and electron microscopy of cross- and multi-module nonribosomal peptide synthetase proteins reveal a flexible architecture. Structure 25, 78, 783–793.e4. doi: 10.1016/j.str.2017.03.014
Thirlway, J., Lewis, R., Nunns, L., Al Nakeeb, M., Styles, M., Struck, A.-W., et al. (2012). Introduction of a non-natural amino acid into a nonribosomal peptide antibiotic by modification of adenylation domain specificity. Angew. Chem. Int. Ed. Engl. 51, 7181–7184. doi: 10.1002/anie.201202043
Tibrewal, N., and Tang, Y. (2014). Biocatalysts for natural product biosynthesis. Annu. Rev. Chem. Biomol. Eng. 5, 347–366. doi: 10.1146/annurev-chembioeng-060713-040008
Trauger, J. W., Kohli, R. M., and Walsh, C. T. (2001). Cyclization of backbone-substituted peptides catalyzed by the thioesterase domain from the tyrocidine nonribosomal peptide synthetase. Biochemistry 40, 7092–7098. doi: 10.1021/bi010035r
Tripathi, A., Choi, S. S., Sherman, D. H., and Kim, E. S. (2016). Thioesterase domain swapping of a linear polyketide tautomycetin with a macrocyclic polyketide pikromycin in Streptomyces sp. CK4412. J. Ind. Microbiol. Biotechnol. 43, 1189–1193. doi: 10.1007/s10295-016-1790-2
Van Lanen, S. G., Lin, S., and Shen, B. (2008). Biosynthesis of the enediyne antitumor antibiotic C-1027 involves a new branching point in chorismate metabolism. Proc. Natl. Acad. Sci. U.S.A. 105, 494–499. doi: 10.1073/pnas.0708750105
Velkov, T., Horne, J., Scanlon, M. J., Capuano, B., Yuriev, E., and Lawen, A. (2011). Characterization of the N-methyltransferase activities of the multifunctional polypeptide cyclosporin synthetase. Chem. Biol. 18, 464–475. doi: 10.1016/j.chembiol.2011.01.017
Villiers, B., and Hollfelder, F. (2011). Directed evolution of a gatekeeper domain in nonribosomal peptide synthesis. Chem. Biol. 18, 1290–1299. doi: 10.1016/j.chembiol.2011.06.014
Villiers, B. R., and Hollfelder, F. (2009). Mapping the limits of substrate specificity of the adenylation domain of TycA. Chembiochem 10, 671–682. doi: 10.1002/cbic.200800553
Vogeli, B., Geyer, K., Gerlinger, P. D., Benkstein, S., Cortina, N. S., and Erb, T. J. (2018). Combining promiscuous acyl-CoA oxidase and enoyl-CoA carboxylase/reductases for atypical polyketide extender unit biosynthesis. Cell Chem. Biol. 25, 833–839.e4. doi: 10.1016/j.chembiol.2018.04.009
Walsh, C. T. (2016). Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 33, 127–135. doi: 10.1039/c5np00035a
Wang, F., Wang, Y., Ji, J., Zhou, Z., Yu, J., Zhu, H., et al. (2015). Structural and functional analysis of the loading acyltransferase from avermectin modular polyketide synthase. ACS Chem. Biol. 10, 1017–1025. doi: 10.1021/cb500873k
Ward, S. L., Desai, R. P., Hu, Z., Gramajo, H., and Katz, L. (2007). Precursor-directed biosynthesis of 6-deoxyerythronolide B analogues is improved by removal of the initial catalytic sites of the polyketide synthase. J. Ind. Microbiol. Biotechnol. 34, 9–15. doi: 10.1007/s10295-006-0156-6
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi: 10.1093/nar/gky427
Weber, T., Charusanti, P., Musiol-Kroll, E. M., Jiang, X., Tong, Y., Kim, H. U., et al. (2015). Metabolic engineering of antibiotic factories: new tools for antibiotic production in actinomycetes. Trends Biotechnol. 33, 15–26. doi: 10.1016/j.tibtech.2014.10.009
Wehrs, M., Gladden, J. M., Liu, Y., Platz, L., Prahl, J.-P., Moon, J., et al. (2019). Sustainable bioproduction of the blue pigment indigoidine: expanding the range of heterologous products in R. toruloides to include non-ribosomal peptides. Green Chem. 21, 3394–3406. doi: 10.1039/C9GC00920E
Weissman, K. J. (2016). Genetic engineering of modular PKSs: from combinatorial biosynthesis to synthetic biology. Nat. Prod. Rep. 33, 203–230. doi: 10.1039/c5np00109a
Weist, S., Bister, B., Puk, O., Bischoff, D., Pelzer, S., Nicholson, G. J., et al. (2002). Fluorobalhimycin–a new chapter in glycopeptide antibiotic research. Angew. Chem. Int. Ed. Engl. 41, 3383–3385. doi: 10.1002/1521-3773(20020916)41:18<3383::AID-ANIE3383>3.0.CO;2-R
Weist, S., Kittel, C., Bischoff, D., Bister, B., Pfeifer, V., Nicholson, G. J., et al. (2004). Mutasynthesis of glycopeptide antibiotics: variations of vancomycin’s AB-ring amino acid 3,5-dihydroxyphenylglycine. J. Am. Chem. Soc. 126, 5942–5943. doi: 10.1021/ja0499389
Wenda, S., Illner, S., Mell, A., and Kragl, U. (2011). Industrial biotechnology—the future of green chemistry? Green Chem. 13, 3007–3047. doi: 10.1039/C1GC15579B
Whicher, J. R., Dutta, S., Hansen, D. A., Hale, W. A., Chemler, J. A., Dosey, A. M., et al. (2014). Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature 510, 560–564. doi: 10.1038/nature13409
Wilson, D. J., and Aldrich, C. C. (2010). A continuous kinetic assay for adenylation enzyme activity and inhibition. Anal. Biochem. 404, 56–63. doi: 10.1016/j.ab.2010.04.033
Winn, M., Fyans, J. K., Zhuo, Y., and Micklefield, J. (2016). Recent advances in engineering nonribosomal peptide assembly lines. Nat. Prod. Rep. 33, 317–347. doi: 10.1039/c5np00099h
Worthington, A. S., Rivera, H., Torpey, J. W., Alexander, M. D., and Burkart, M. D. (2006). Mechanism-based protein cross-linking probes to investigate carrier protein-mediated biosynthesis. ACS Chem. Biol. 1, 687–691. doi: 10.1021/cb6003965
Xie, Y., Cai, Q., Ren, H., Wang, L., Xu, H., Hong, B., et al. (2014). NRPS substrate promiscuity leads to more potent antitubercular sansanmycin analogues. J. Nat. Prod. 77, 1744–1748. doi: 10.1021/np5001494
Xu, F., Butler, R., May, K., Rexhepaj, M., Yu, D., Zi, J., et al. (2019). Modified substrate specificity of a methyltransferase domain by protein insertion into an adenylation domain of the bassianolide synthetase. J. Biol. Eng. 13:65. doi: 10.1186/s13036-019-0195-y
Yakimov, M. M., Giuliano, L., Timmis, K. N., and Golyshin, P. N. (2000). Recombinant acylheptapeptide lichenysin: high level of production by Bacillus subtilis cells. J. Mol. Microbiol. Biotechnol. 2, 217–224.
Yan, J., Hazzard, C., Bonnett, S. A., and Reynolds, K. A. (2012). Functional modular dissection of DEBS1-TE changes triketide lactone ratios and provides insight into Acyl group loading, hydrolysis, and ACP transfer. Biochemistry 51, 9333–9341. doi: 10.1021/bi300830q
Ye, Z., Musiol, E. M., Weber, T., and Williams, G. J. (2014). Reprogramming acyl carrier protein interactions of an Acyl-CoA promiscuous trans-acyltransferase. Chem. Biol. 21, 636–646. doi: 10.1016/j.chembiol.2014.02.019
Ye, Z., and Williams, G. J. (2014). Mapping a ketosynthase:acyl carrier protein binding interface via unnatural amino acid-mediated photo-cross-linking. Biochemistry 53, 7494–7502. doi: 10.1021/bi500936u
Yu, D., Xu, F., Zeng, J., and Zhan, J. (2012). Type III polyketide synthases in natural product biosynthesis. IUBMB Life 64, 285–295. doi: 10.1002/iub.1005
Yuzawa, S., Bailey, C. B., Fujii, T., Jocic, R., Barajas, J. F., Benites, V. T., et al. (2017a). Heterologous gene expression of N-terminally truncated variants of LipPks1 suggests a functionally critical structural motif in the N-terminus of modular polyketide synthase. ACS Chem. Biol. 12, 2725–2729. doi: 10.1021/acschembio.7b00714
Yuzawa, S., Deng, K., Wang, G., Baidoo, E. E., Northen, T. R., Adams, P. D., et al. (2017b). Comprehensive in vitro analysis of acyltransferase domain Exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth. Biol. 6, 139–147. doi: 10.1021/acssynbio.6b00176
Yuzawa, S., Kapur, S., Cane, D. E., and Khosla, C. (2012). Role of a conserved arginine residue in linkers between the ketosynthase and acyltransferase domains of multimodular polyketide synthases. Biochemistry 51, 3708–3710. doi: 10.1021/bi300399u
Yuzawa, S., Keasling, J. D., and Katz, L. (2017c). Bio-based production of fuels and industrial chemicals by repurposing antibiotic-producing type I modular polyketide synthases: opportunities and challenges. J. Antibiot. 70, 378–385. doi: 10.1038/ja.2016.136
Yuzawa, S., Mirsiaghi, M., Jocic, R., Fujii, T., Masson, F., Benites, V. T., et al. (2018). Short-chain ketone production by engineered polyketide synthases in Streptomyces albus. Nat. Commun. 9:4569. doi: 10.1038/s41467-018-07040-0
Zargar, A., Barajas, J. F., Lal, R., and Keasling, J. D. (2018). Polyketide synthases as a platform for chemical product design. AIChE J. 64, 4201–4207. doi: 10.1002/aic.16351
Zhang, J., Van Lanen, S. G., Ju, J., Liu, W., Dorrestein, P. C., Li, W., et al. (2008). A phosphopantetheinylating polyketide synthase producing a linear polyene to initiate enediyne antitumor antibiotic biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 105, 1460–1465. doi: 10.1073/pnas.0711625105
Zhang, K., Nelson, K. M., Bhuripanyo, K., Grimes, K. D., Zhao, B., Aldrich, C. C., et al. (2013). Engineering the substrate specificity of the DhbE adenylation domain by yeast cell surface display. Chem. Biol. 20, 92–101. doi: 10.1016/j.chembiol.2012.10.020
Zhang, L., Hashimoto, T., Qin, B., Hashimoto, J., Kozone, I., Kawahara, T., et al. (2017). Characterization of giant modular PKSs provides insight into genetic mechanism for structural diversification of aminopolyol polyketides. Angew. Chem. Int. Ed. Engl. 56, 1740–1745. doi: 10.1002/anie.201611371
Zhang, L., Ji, J., Yuan, M., Feng, Y., Wang, L., Deng, Z., et al. (2018). Stereospecificity of enoylreductase domains from modular polyketide synthases. ACS Chem. Biol. 13, 871–875. doi: 10.1021/acschembio.7b00982
Zheng, J., Piasecki, S. K., and Keatinge-Clay, A. T. (2013). Structural studies of an A2-type modular polyketide synthase ketoreductase reveal features controlling α-substituent stereochemistry. ACS Chem. Biol. 8, 1964–1971. doi: 10.1021/cb400161g
Keywords: polyketide synthase, non-ribosomal peptide synthetase, domain, module, repurposing
Citation: Hwang S, Lee N, Cho S, Palsson B and Cho B-K (2020) Repurposing Modular Polyketide Synthases and Non-ribosomal Peptide Synthetases for Novel Chemical Biosynthesis. Front. Mol. Biosci. 7:87. doi: 10.3389/fmolb.2020.00087
Received: 06 March 2020; Accepted: 16 April 2020;
Published: 15 May 2020.
Edited by:
Ki Duk Park, Korea Institute of Science and Technology (KIST), South KoreaReviewed by:
Satoshi Yuzawa, The University of Tokyo, JapanDong-Woo Lee, Yonsei University, South Korea
Jay D. Keasling, University of California, Berkeley, United States
Copyright © 2020 Hwang, Lee, Cho, Palsson and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Byung-Kwan Cho, YmNob0BrYWlzdC5hYy5rcg==