 Sangeeta Kakoti1,2Hiro Sato2*Siddhartha Laskar3Takaaki Yasuhara4,5Atsushi Shibata1*
Sangeeta Kakoti1,2Hiro Sato2*Siddhartha Laskar3Takaaki Yasuhara4,5Atsushi Shibata1*- 1Signal Transduction Program, Gunma University Initiative for Advanced Research (GIAR), Maebashi, Japan
- 2Department of Radiation Oncology, Gunma University, Maebashi, Japan
- 3Department of Radiation Oncology, Tata Memorial Centre, Mumbai, India
- 4Laboratory of Molecular Radiology, Center for Disease Biology and Integrative Medicine, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan
- 5Massachusetts General Hospital Cancer Center, Harvard Medical School, Charlestown, MA, United States
Cancer therapy using immune checkpoint inhibitors (ICIs) is a promising clinical strategy for patients with multiple types of cancer. The expression of programmed cell death ligand-1 (PD-L1), an immune-suppressor ligand, in cancer cells is a factor that influences the efficacy of ICI therapy, particularly in the anti-programmed cell death protein-1 (PD-1)/PD-L1 antibody therapy. PD-L1 expression in cancer cells are associated with tumor mutation burden including microsatellite instability because the accumulation of mutations in the cancer genome can produce abnormal proteins via mutant mRNAs, resulting in neoantigen production and HLA-neoantigen complex presentation in cancer cells. HLA-neoantigen presentation promotes immune activity within tumor environment; therefore, known as hot tumor. Thus, as the fidelity of DNA repair affects the generation of genomic mutations, the status of DNA repair and signaling in cancer cells can be considered prior to ICI therapy. The Cancer Genome Atlas (TCGA) and The Cancer Immunome Atlas (TCIA) database analysis showed that tumor samples harboring mutations in any non-homologous end joining, homologous recombination, or DNA damage signaling genes exhibit high neoantigen levels. Alternatively, an urgent task is to understand how the DNA damage-associated cancer treatments change the status of immune activity in patients because multiple clinical trials on combination therapy are ongoing. Recent studies demonstrated that multiple pathways regulate PD-L1 expression in cancer cells. Here, we summarize the regulation of the immune response to ICI therapy, including PD-L1 expression, and also discuss the potential strategies to improve the efficacy of ICI therapy for poor responders from the viewpoint of DNA damage response before or after DNA damage-associated cancer treatment.
Introduction
The clinical application of immune checkpoint inhibitors (ICIs), such as anti-programmed cell death protein-1 (PD-1)/programmed cell death ligand-1 (PD-L1) and anti-cytotoxic T lymphocyte-associated protein 4 (CTLA-4) antibodies, has improved the clinical outcome of patients with various malignancies. Practically, anti-PD-1/PD-L1 antibodies have been widely used at the clinical level. However, the development of a new clinical strategy remains essential because only a limited number of patients respond to anti-PD-1/PD-L1 therapy (Sharma et al., 2017a). For the selection of responders or the improvement of the therapeutic efficacy in non-responders, recent studies have actively sought to identify an optimal biomarker for the prediction of treatment response to ICI therapy (Galon and Bruni, 2019). PD-L1, a ligand that is expressed on cancer cell surface, binds to PD-1 that is a T-cell surface receptor. The binding stimulates a signal transduction within T cell, which subsequently suppresses the immune activity and proliferation of T cells (Iwai et al., 2002). Thus, the presence of PD-L1 on cancer cell surface influences the overall immune activity in tumor. The mechanistic basis for ICI therapy is that the PD-L1 expression in cells within tumor environments is also considered to be essential because anti-PD-1/PD-L1 antibodies target and inhibit the interaction between PD-1 and PD-L1, restoring immune activity in the tumor environment. As another biomarker for anti-PD-1/PD-L1 therapy, microsatellite instability (MSI) is widely used. The accumulation of mutations in tumor genome, also known as tumor mutation burden (TMB), is related to the formation of abnormal proteins, because mutations in genes at transcriptionally active loci produce mutant mRNAs that subsequently form HLA-neoantigen complex following the generation of peptides by proteasome-dependent degradation of abnormal proteins (Schumacher and Schreiber, 2015). The presentation of HLA-neoantigen on the surface of cancer cells promotes immune activity and transforms cold tumors into hot tumors. Despite the upregulation of the HLA-neoantigen-dependent immune activation, it is still not sufficient to overcome the progression of cancers under the immune-suppressive environment. Such hot tumors are considered to be sensitive to ICIs. The alleviation of immune suppression by ICIs will switch on the immune activity. Thus, the status of DNA repair in cancer cells might be important because the fidelity of repair influences the amount of TMB including MSI.
To date, studies have shown that the PD-L1 expression in tumors is influenced by DNA repair and signaling via multiple pathways. Prior to cancer treatment, endogenous DNA damage in tumors could be persistently generated due to oxidative stress or abnormal cell cycling. Under such circumstances, DNA damage responses (DDR) may upregulate immunological signaling. However, the immune activity under the situation without additional exogenous DNA damage, e.g., prior to radiotherapy (RT)/chemotherapy, is not completely able to overcome cancers. This situation will be likely in case of low TMB/MSI tumors. In contrast, recent studies have shown that multiple immunological responses including the release of interferons (IFNs, immune positive response) and PD-L1 upregulation (immune negative response) are induced after DNA damage-associated cancer treatments, such as RT and chemotherapy (McLaughlin et al., 2020). After RT/chemotherapy, both immune positive (HLA-neoantigen) and negative responses (PD-L1 upregulation) are activated, and the negative responses can be cancelled by ICIs. Thus, the introduction of DNA damage can be a trigger to transform tumors from cold to hot, irrespective of the TMB/MSI status. Here, we review the molecular linkage between immune-response and DDR, including the latest findings in the field.
Mechanism of Immune-Mediated Cancer Cell Killing
Tumor cells often harbor many “passenger” mutations, in addition to the carcinogenic “driver mutations” (McFarland et al., 2017). Among passenger mutations, non-synonymous mutations produce mRNAs containing mutations that alter the amino acid sequence of proteins, and these abnormal proteins get proteolytically degraded into short peptides. The endogenous Major Histocompatibility Complex class I (MHC-I, HLA in human) recognizes these peptides as neoantigens, which are then transferred to and presented at the tumor cell surface. This HLA-neoantigen complex then activates cytotoxic T cells by binding to the T-cell receptor (TCR) either directly or via the cross-presentation by professional antigen-presenting cells (APCs), such as dendritic cells (Garbi and Kreutzberg, 2012). Activated T cells subsequently eliminate tumor cells by a series of cytolytic events that mainly involve cytokine release (e.g., perforin and granzyme) (Martinez-Lostao et al., 2015). Thus, non-synonymous mutations in tumor cells eventually upregulate the immune activity in the tumor environment and kill cancer cells via the immune system. Recently, a phase I clinical trial in which a neoantigen vaccine was administered to glioblastoma patients showed that CD4+ and CD8+ T cells are activated in a neoantigen-dependent manner (Keskin et al., 2019). This result supports the notion that neoantigen is a key factor in immune-dependent cancer cell killing effect.
The overt manifestation of clinical malignant tumors is usually the result of “escape” from the immune-mediated “elimination” of tumor cells described above (the steps involving elimination, equilibrium, and escape are collectively termed as “Cancer Immunoediting”) (Dunn et al., 2002). Broadly, this may occur due to defects or dysfunction in any of the steps involved in elimination, such as the lack of immunogenic neoantigens on tumors, dysfunctional APCs that cannot effectively prime the neoantigens, dysfunctional cytotoxic T cells that are unable to become activated or secrete cytokines, and impaired activity of cytokines, including their receptor or ligand inactivity (Figure 1, left) (Matsushita et al., 2012; Schumacher and Schreiber, 2015; Sharma et al., 2017a). Importantly, these immune escape mechanisms also promote the resistance for immunotherapy (O’Donnell et al., 2019). Among the various mechanisms involved in the immunological escape of tumor cells, activated immune checkpoint pathways, including the PD-1/PD-L1 and CTLA-4 axis, are the central suppressors amenable to therapeutic inhibition (Figure 1, left). Specifically, PD-L1 protein expressed on tumor cells binds to PD-1 receptors on cytotoxic T cells, preventing T cells from being activated by the HLA-neoantigen complex (immune exhaustion) (Thommen and Schumacher, 2018). CTLA-4 is a member of the CD28 family of receptors that is constitutively expressed on regulatory T (Treg) cells, a type of T cell, which secrete inhibitory cytokines, such as interleukin (IL)-10, IL-35, and transforming growth factor (TGF)-β, or directly contact effector T cells, subsequently suppressing T-cell responses.
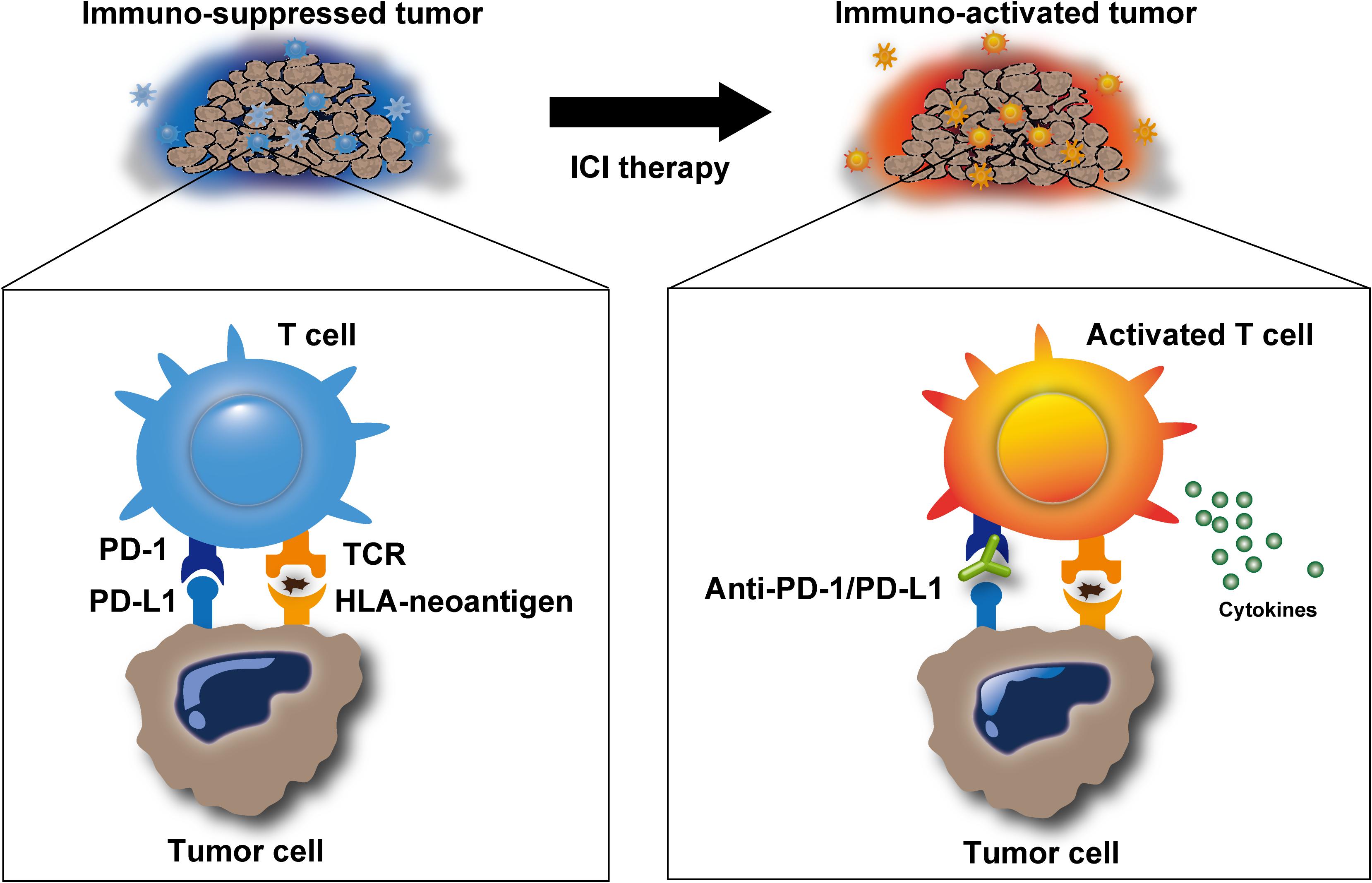
Figure 1. Release of immune suppression by immune checkpoint inhibitors. Immune checkpoint inhibitors, such as anti-programmed cell death protein-1 (PD-1)/programmed cell death ligand-1 (PD-L1) antibodies, inhibit immunosuppression in the tumor microenvironment and promote T-cell activation. Activated T cells can release cytotoxic molecules, including perforin, granzyme, and IFNγ, to eliminate target cells.
The history of the use of immunotherapy to overcome the immune-mediated escape of tumor cells began with a stimulation of host immunity. The administration of attenuated bacteria (Coley’s toxin) alone in inoperable sarcomas and carcinomas showed unprecedented results (5-year overall survival rates up to 79 and 73%, respectively) in the early 1900s (Hoption Cann et al., 2003). The ex vivo activation of cytotoxic T cells, followed by infusion back into the patients’ body (a procedure termed adoptive T-cell therapy) also showed promising results, especially in the treatment of advanced melanoma (Rosenberg et al., 2011). Alternatively, to date, ICIs, which release the patients’ intrinsic immune response from the suppressed state, have provided a significant leap in the evolution of anticancer immunotherapy (Figure 1). ICI treatment exerts antitumor activity by restoring the intrinsic immune response, as described above, in patients who are suppressed by these immune checkpoints. Anti-PD-1/PD-L1 antibodies aim to block the undesired “T-cell-exhausting” communication between PD-L1 on tumor cells and its receptor PD-1 on cytotoxic T cells. Based on this mechanism, PD-1/PD-L1 blockade has resulted in considerable improvements in the outcomes of patients with advanced malignancies across a wide range of tumor sites (Brahmer et al., 2015; Sharma et al., 2017b). Similarly, anti-CTLA-4 antibody therapy has shown impressive results in metastatic melanoma patients (Margolin et al., 2012). CTLA-4 is expressed on activated T cells, whereas CD80/86 is expressed on APCs, including tumor cells. Anti-CTLA-4 antibodies block the interaction between CTLA-4 and CD80/86, resulting in immune activity restoration. Anti-PD-1/PD-L1 blockade primarily affects the immune activity in tumor environment, whereas anti-CTLA-4 therapy restores the immune activity throughout the lymph tissues. Thus, each therapy differently targets the area of patient body; therefore, synergistic effect can be obtained using a combination of both inhibitors. A combination of inhibitors targeting both of them has the potential to improve patient outcomes compared with either one of them alone (Larkin et al., 2015; Hellmann et al., 2018; Overman et al., 2018).
Although such elegant mechanisms underlie the basis of ICI therapy, only 5% of patients are categorized as high responders, indicating that approximately 95% patients may not be effectively cured. Thus, present efforts in the field of immunotherapy are focused on identifying suitable biomarkers to predict candidates who benefit from ICI treatment. High TMB is thought to be associated with the generation of increased immunogenic neoantigens and better response rates to ICIs (Rizvi et al., 2015). In fact, a high MSI score, which is induced by defects in mismatch repair, is highly related to ICI efficacy. Therefore, MSI is currently used as one of the most reliable predictive biomarkers for ICI therapy (Le et al., 2017; Marabelle et al., 2020). Several studies have shown that base excision repair (BER) defects, chromatin remodeling, and DNA replication also contribute to MSI (Garcia-Sanz et al., 2017; Matsuno et al., 2019) (see further discussion below). In addition to the TMB/MSI/neoantigen axis, high PD-L1 expression level on the tumor cell surface is also found to be a favorable predictive biomarker (Topalian et al., 2016). In the next section, we discuss how DDRs associate with neoantigen production.
Potential Regulation of Neoantigen Production in DNA Damage Response Defective Cancers
DNA damage responses (both repair and signaling) are vital to ensure genome stability; thus, DDR defective status is often associated with somatic mutations (Jackson and Bartek, 2009) that frequently lead to frameshift errors and abnormal protein synthesis following transcription and translation. Peptides, presented as HLA-neoantigen, are produced by immunoproteasomes. Immunogenic neoantigens are transported to the endoplasmic reticulum (ER), and the HLA-neoantigen complex is formed followed by its transfer to the cell surface (Figure 2). The presented HLA-neoantigen interacts with TCR on T cells, and this interaction promotes T-cell activity enhancing the tumor cell killing effect (Schumacher et al., 2019). In contrast to the positive effect by HLA-neoantigen dependent immune stimulation, the release of IFNs from the activated T cells promotes the expression of PD-L1 in tumor environment (Garcia-Diaz et al., 2017).
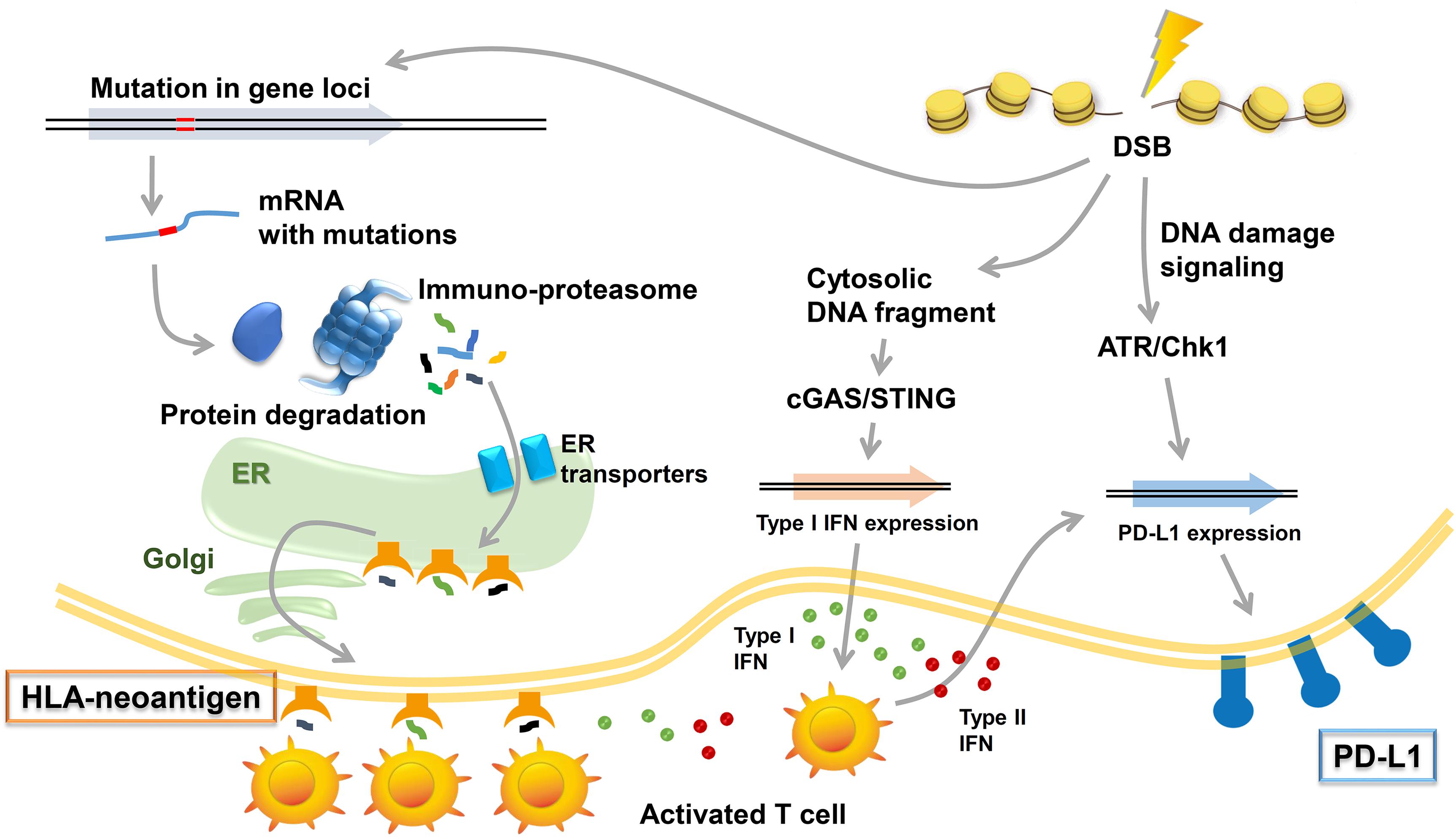
Figure 2. Immune responses induced by DNA damage response (DDR). Radiotherapy (RT)/chemotherapy change the immune microenvironment to be advantageous in combination with immune checkpoint inhibitors. Three types of immune responses are included in these responses: (1) neoantigen production induced by mutations in gene loci and the induction of HLA class I (HLA)-neoantigen complex expression, (2) PD-L1 upregulation via DNA damage signals, and (3) the accumulation of cytoplasmic DNA and activation of the cGAS/STING pathway.
The Cancer Genome Atlas (TCGA) and The Cancer Immunome Atlas (TCIA),a publicly accessible genomic dataset of clinical specimens, are a useful tool to analyze gene expressions, TMB and neoantigen production predicted by genomic mutations (Hutter and Zenklusen, 2018). Defect of mismatch repair (MMR) activity is considered to be a major cause of generation of TMB and neoantigen production in cancer cells. Mutations in MMR genes are associated with significantly increased neoantigen production and distinct immunological characteristics in lung squamous cell carcinoma (Chae et al., 2019). We previously reported that tumors with mutations in any of the double-strand break (DSB) repair genes show enhanced PD-L1 expression (Sato et al., 2017). Further, tumors with mutations in any of the BER genes exhibit statistically significant increases in neoantigen production and PD-L1 expression (Permata et al., 2019). Other dataset analyses have shown that high-grade serous ovarian cancer patients with homologous recombination (HR) deficiency have a higher neoantigen load and an increase in PD-1 and PD-L1 expression compared with HR proficient patients (Strickland et al., 2016). Another database study reported that patients with simultaneous mutations in HR and MMR, or HR and BER are associated with increased TMB and neoantigen production (Wang et al., 2018). Importantly, patients with high TMB/neoantigen in HR-MMR- or HR-BER-defective groups show a favorable clinical benefit from ICI therapy, suggesting that these DNA repair factors also can be supportive biomarkers for ICI therapy. Thus, DDR-induced neoantigen production, and subsequent immune-stimulation and PD-L1 expression, are being clarified. In this section, we present our analysis of neoantigen production in relation to the status of DDR, which is categorized by non-homologous end joining (NHEJ), HR, or DNA damage signaling by using TCGA and TCIA. To investigate the impact of NHEJ, HR, or DNA damage signaling on neoantigen production derived from genomic mutations, the neoantigen levels in patients with various tumor sites were analyzed (Figures 3A–C; representative genes of NHEJ, HR, and DNA damage signaling are summarized in Table 1). Importantly, we found increased levels of neoantigen production in tumors with mutations in either NHEJ, HR, or DNA damage signaling pathways. Neoantigen production increase was particularly evident in uterine corpus endometrial carcinoma (UCEC), stomach adenocarcinoma (STAD), and colon adenocarcinoma (COAD) across multiple genes (Figures 3A–C). We further analyzed the impact of the mutations in combination with NHEJ, HR, and DNA damage signaling on neoantigen production (Figure 4). Notably, a combination of mutations in different pathways (NHEJ + HR, NHEJ + DNA damage signaling, and HR + DNA damage signaling) showed higher levels of neoantigen production compared with mutations in one of the pathways (Figures 4A–C), which might be attributed to a compromised availability of alternate repair pathways, resulting in a higher frequency of neoantigen production when genes of more than one pathway are mutated. In combination with DNA damage signaling mutant, the NHEJ or HR mutant may cause further mutations in genes producing neoantigen under the deficiency of cell-cycle checkpoint, which arrests cell cycle at the G1/S or G2/M boundary (see below about cell-cycle checkpoint arrest). The produced neoantigen should be bound to HLA to form the HLA-neoantigen complex, even if not all, and neoantigen production increase under DDR deficiency will activate T cells, leading to the release of immune cytokines and enhanced total immune activity in the tumor environment. However, this signaling also upregulates PD-L1 expression, and ICI treatment will be effective in this situation. The result of the database analysis is supported by a recent study by Cheng’s group, which reported that the analysis of tumor samples from patients with non-small cell lung cancer treated with anti-PD-1/PD-L1 antibody using next-generation sequencing revealed that patients with DDR mutation (the most commonly mutated DDRs were ATM, ATR, BRCA2, POLQ, and RAD50 in the analysis) exhibited high TMB. The patients harboring mutations in DDR genes had longer progression-free survival and overall survival under an ICI treatment (Ricciuti et al., 2020).
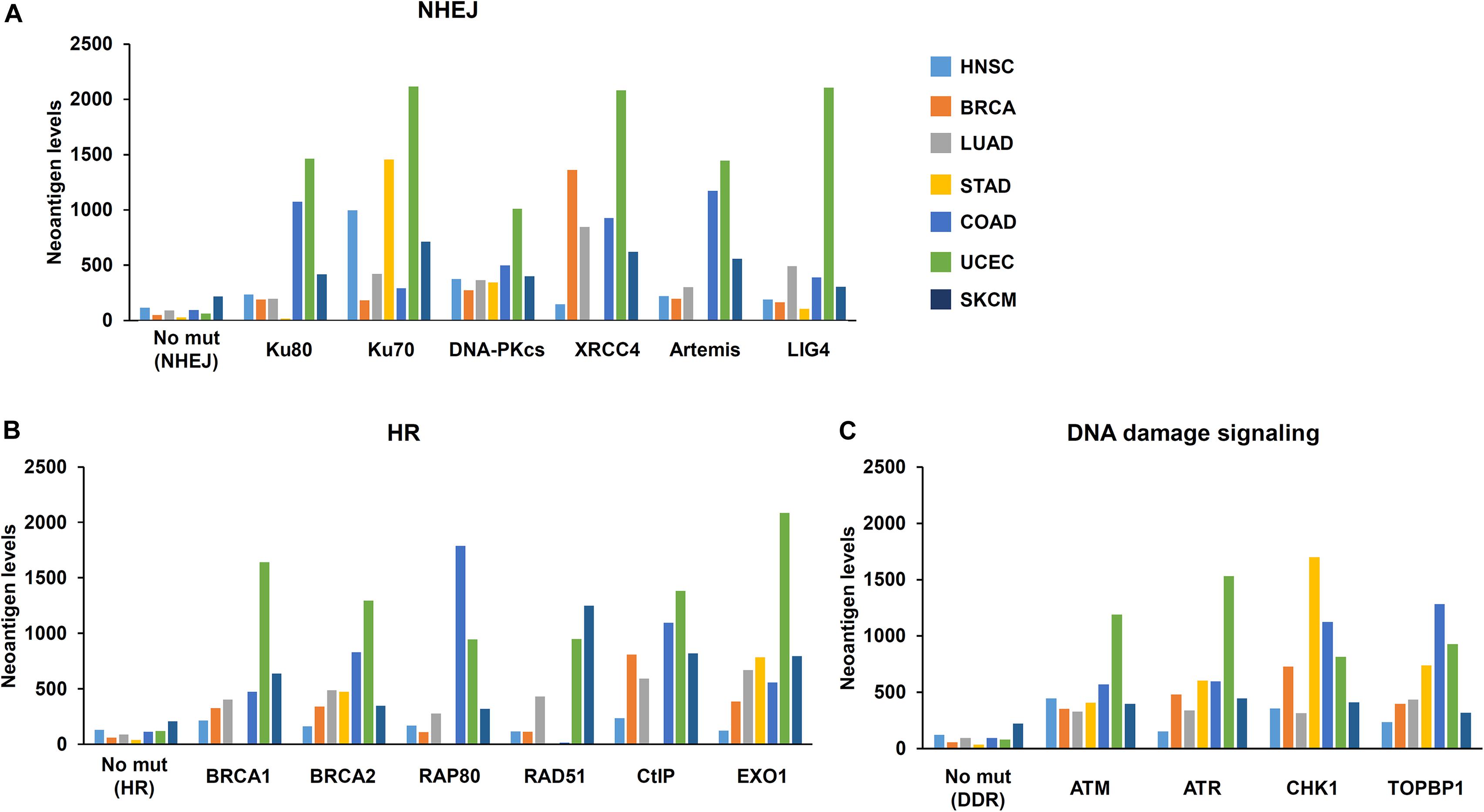
Figure 3. Mutations in DNA repair pathways and neoantigen production level. The correlation between mutations in each DNA repair pathway and the level of neoantigen production was analyzed. Mutation statuses provided by TCGA project were downloaded from the Genomic Data Commons Data Portal. The neoantigen data were obtained from The Cancer Immunome Atlas (TCIA)1. Levels of neoantigen in samples harboring mutations, including indels and point mutations in NHEJ, HR, or DNA damage signaling genes, are shown. Neoantigen levels in the y axes represent the average number of neoantigens per tumor sample in each group. The number of samples (N) in this study is listed in Table 1. (A) Genes involved in non-homologous end joining (NHEJ). (B) Genes involved in homologous recombination (HR). (C) Genes involved in the DNA damage signaling. HNSC, head and neck squamous cell carcinoma; BRCA, breast invasive carcinoma; LUAD, lung adenocarcinoma; STAD, stomach adenocarcinoma; COAD, colon adenocarcinoma; UCEC, uterine corpus endometrial carcinoma; SKCM, skin cutaneous melanoma.
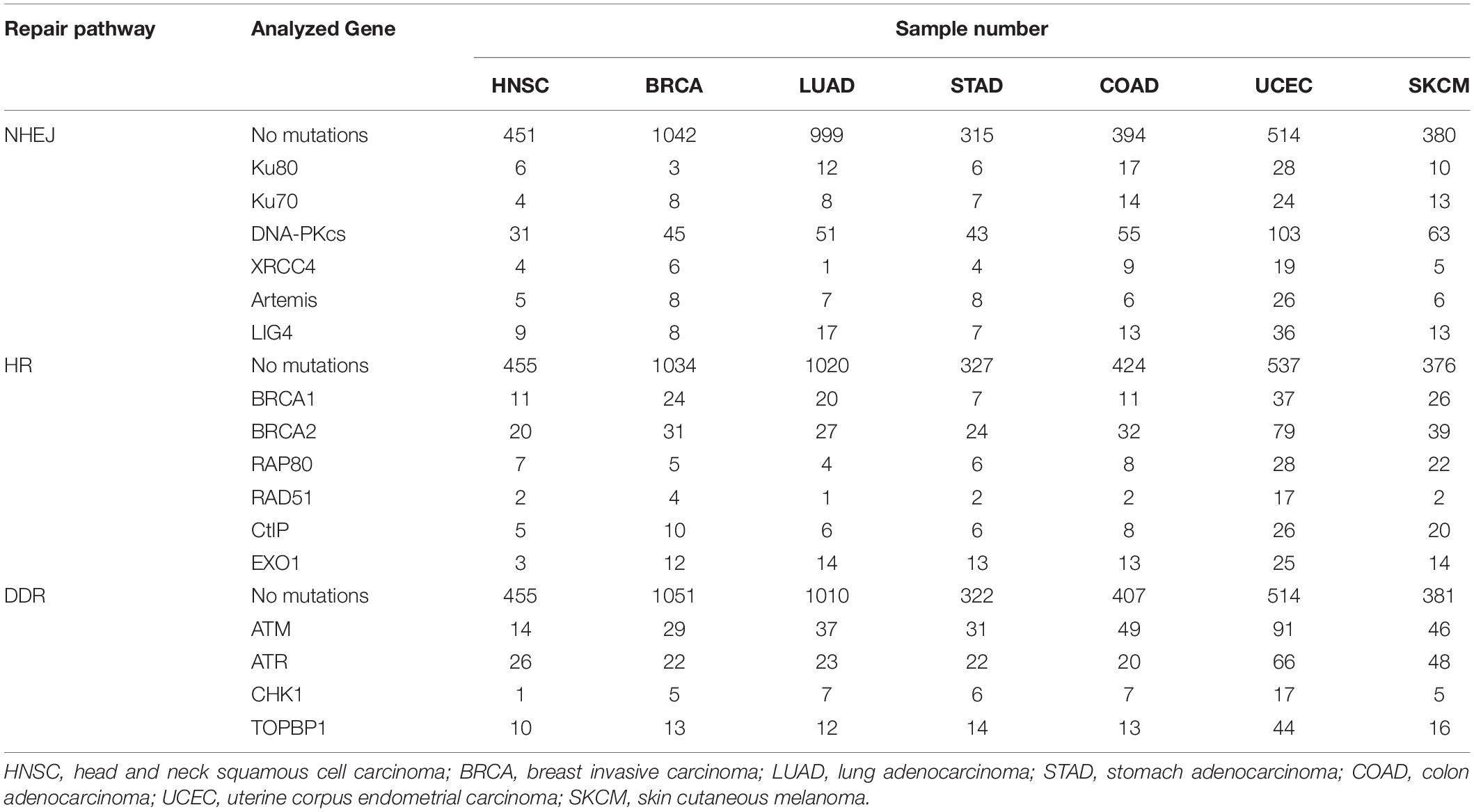
Table 1. Analysis of NHEJ, HR, and DDR pathway mutation and neoantigen production by TCGA dataset.
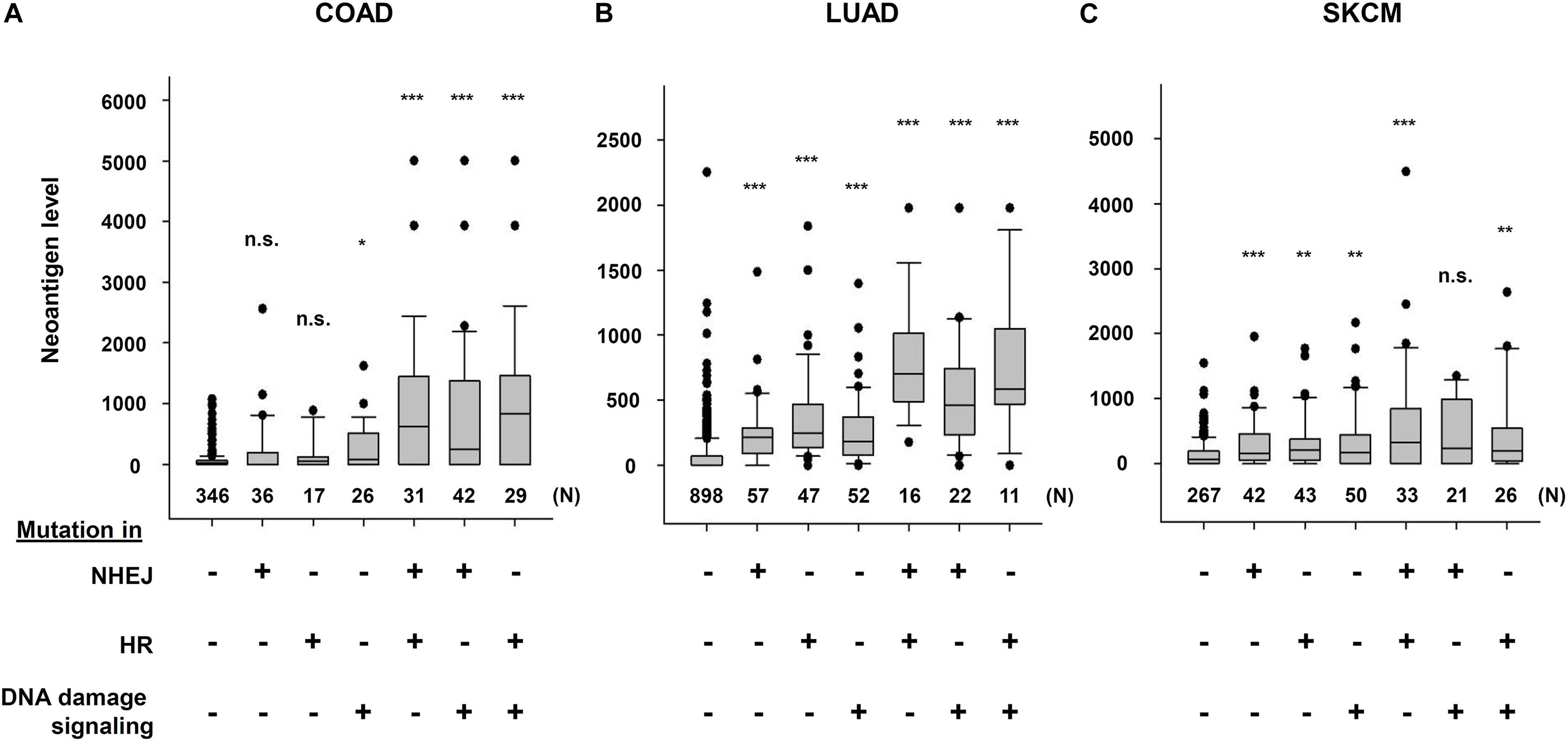
Figure 4. Multiple DNA repair pathway mutations and neoantigen production level. The correlation between mutations in multiple DNA repair pathways and neoantigen production levels using The Cancer Genome Atlas (TCGA) and The Cancer Immunome Atlas (TCIA) dataset. The distribution of the number of neoantigens in each group is shown by box plots. (A) Colon adenocarcinoma (COAD). (B) Lung adenocarcinoma (LUAD). (C) Skin cutaneous melanoma (SKCM). The analysis of neoantigen levels are described in the legend of Figure 3. Box plots were created using SigmaPlot 12.0. Statistical analysis was performed by Student’s two-tailed t-test by SigmaPlot. *p < 0.05, **p < 0.01, ***p < 0.001.
As described above, neoantigens, which are presented by HLA on the cell surface, are derived from peptides following the degradation of abnormal proteins, although not all peptides are recognized by HLA. Thus, the generation of mutations within the coding regions of gene loci is the origin of neoantigen production. MMR deficiency and DNA replication errors cause genome-wide mutations, including mutations in gene loci. A defect in other DNA repair mechanisms, such as NHEJ, HR, BER, or nucleotide excision repair, can also lead to the generation of mutations when exogenous DNA damage is induced at transcriptionally active loci. Defects in factors required for transcription-associated DNA repair at gene loci may further increase the mutation rate in the neoantigen production. Taken together, DDR status is a potential prognostic biomarker in ICI therapy.
Regulation of PD-L1 Expression in the Context of DNA Damage Response
RT and DNA damage-associated chemotherapy, such as platinum-based drugs, are widely used anticancer treatments, which act primarily by generating lethal DNA damage leading to cell death and restricted cell proliferation. DSBs are the most lethal type of DNA damage. The failure of DSB repair causes lethal mutations or cell death. In human cells, DSBs are repaired by NHEJ or HR pathways (Shibata and Jeggo, 2014). In addition to the two major pathways, alternative end joining, which is a deleterious repair pathway due to the use of a microhomology sequence for the rejoining following DSB end resection, is also used in cancer cells (Trenner and Sartori, 2019). This pathway is activated when the expression of a core NHEJ component, such as Ku70/80, is reduced. The use of NHEJ and HR is regulated by the cell cycle. For example, NHEJ functions throughout cell-cycle phases, whereas HR repairs DSBs during the S/G2 phase (Figure 5). Therefore, in proliferating cancer cells, drugs targeting and inhibiting HR in the S/G2 phase have been developed for cancer treatment (Bai et al., 2017). During repair, cells need to arrest cycling until DSBs are repaired. This cell-cycle checkpoint arrest is activated by a DNA damage signaling pathway, which is initiated at the site of DSBs. Ataxia-telangiectasia-mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) are central regulators in cell-cycle checkpoint arrest. These two kinases are distinctly activated in each cell-cycle phase. Because ATM is preferentially activated at the ends of DSBs during NHEJ, ATM greatly contributes to G1/S checkpoint arrest via the checkpoint kinase 2 (Chk2)/p53 pathway. In contrast, ATR is activated during HR and contributes to G2/M checkpoint arrest. In general, G1/S checkpoint arrest is downregulated due to the absence of the p53 pathway in cancer cells. Therefore, DNA damaged cancer cells often accumulate in the G2 phase by ATR/Chk1-dependent checkpoint arrest.
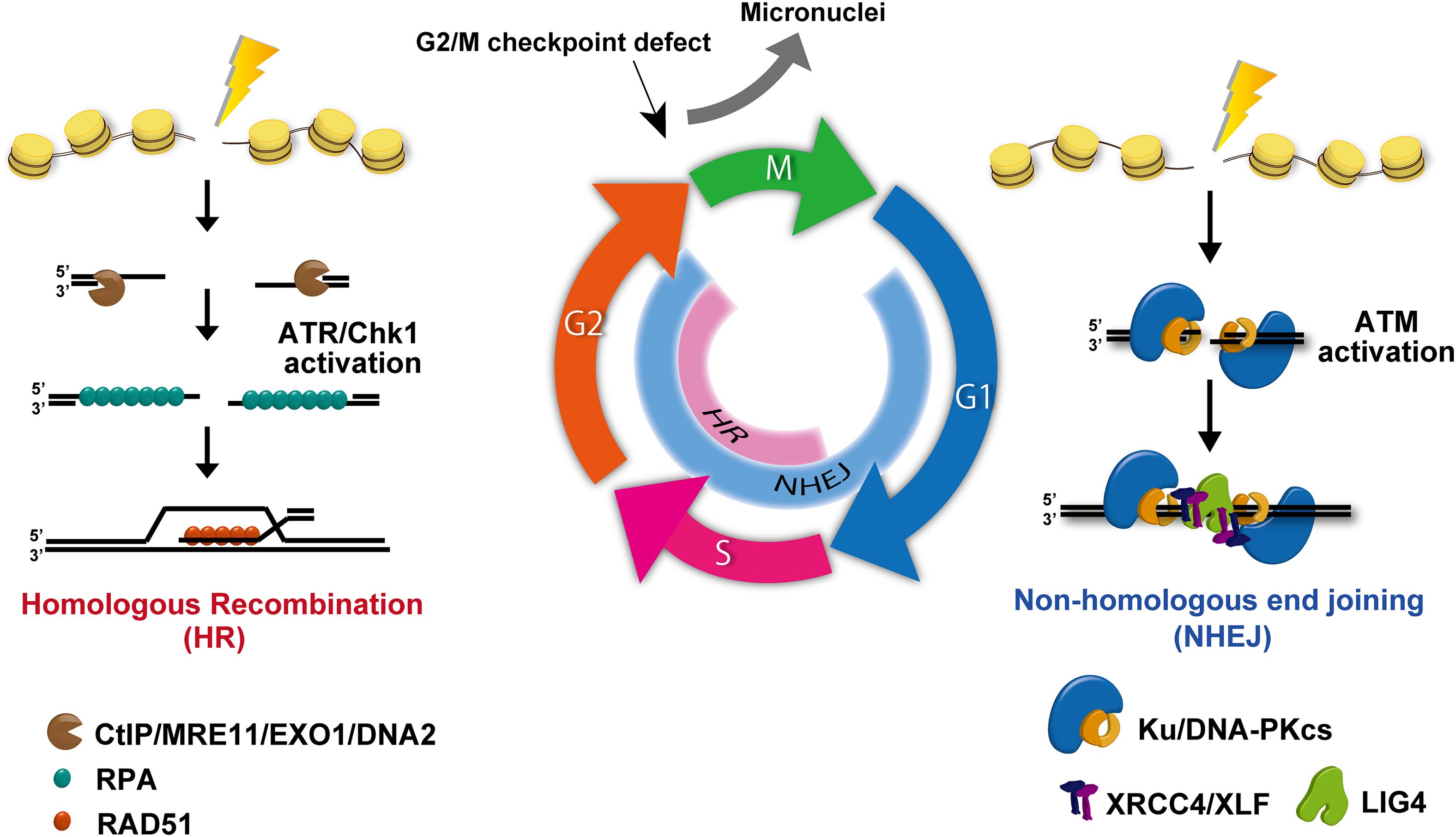
Figure 5. DNA double-strand break (DSB) repair and signal activation. Non-homologous end joining (NHEJ) functions throughout all cell-cycle phases except mitosis. Homologous recombination (HR) becomes active and repairs DSBs only in the S/G2 phase. During repair, ataxia-telangiectasia-mutated (ATM) is activated at un-resected DSB ends, i.e., at the breaks undergoing NHEJ. In addition, ataxia telangiectasia and Rad3-related (ATR) is effectively activated at ssDNA following DSB end resection. Because DSB ends are resected during HR, ATR activation occurs in the S/G2 phase.
Several reports demonstrate that PD-L1 expression is upregulated by RT or chemotherapy (Lim et al., 2016, 2017; Kelly et al., 2018; Yoneda et al., 2019). Recently, we reported that DSBs upregulate PD-L1 expression in a transcription-dependent manner via the STAT-IRF1 pathway (Figure 2; Sato et al., 2017). In this scenario, ATM, ATR, and Chk1 were found to be important for DSB-induced PD-L1 upregulation. More importantly, the depletion of DNA repair proteins, including Ku80 and breast invasive carcinoma (BRCA) 2, was found to enhance PD-L1 upregulation after DSBs, suggesting that patients with mutations of these proteins may highly express PD-L1 and thus benefit more from consolidative PD-1/PD-L1 blockade after RT or chemotherapy. Similar to the response to DSBs, oxidative stress also upregulates PD-L1 expression via the ATR/Chk1/STAT/IRF1 pathway, which is further enhanced by the depletion of BER factors (Permata et al., 2019). Furthermore, ATR inhibition downregulates PD-L1 expression in tumor cells by destabilizing PD-L1 in a proteasome-dependent manner and has resulted in enhanced immune cell killing (Sun et al., 2018). In addition, ATR inhibition attenuates irradiation-induced PD-L1 upregulation and decreases the number of tumor-infiltrating Tregs in mouse models (Vendetti et al., 2018). In contrast to these responses in cancer cells, DNA damage-induced upregulation of PD-L1 was not observed in normal human dermal fibroblasts (Hagiwara et al., 2018).
Another mechanism that regulates PD-L1 expression in response to DNA damage is the cyclic-GMP-AMP synthase (cGAS)/stimulator of IFN genes (STING) pathway. In general, cancer cells can activate G2/M checkpoint arrest, whereas G1/S checkpoint arrest is frequently lost due to the lack of p53 pathway. Despite the activation of G2/M checkpoint signaling, G2/M checkpoint machinery in human cells is not perfectly able to sustain checkpoint arrest, even if all DSBs in G2 cells have not been repaired (Deckbar et al., 2007). Indeed, premature checkpoint release can occur even in normal human cells (Deckbar et al., 2007). Once G2 cells with DSBs progress into mitosis, these cells can generate micronuclei. Additionally, micronuclei can be generated during mitosis if chromosomal aberrations such as dicentric or acentric are formed by misrepair (Figure 5). The loosely bound DNA inside micronuclei with ruptured nuclear envelopes recruit the DNA sensor protein cGAS, which then transduces the signaling toward STING (Ablasser et al., 2013; Harding et al., 2017; Mackenzie et al., 2017). cGAS/STING pathway activation eventually induces the mRNA expression of type-I IFN via an IRF3/NFκB-dependent transcriptional pathway. The produced type-I IFNs is released and incorporated into cells via IFN receptors, subsequently upregulating PD-L1 in cancer cells. Thus, cGAS/STING pathway is considered to contribute to PD-L1 regulation as well as other cytokines. Supporting this notion, DNA damage-induced activation of the cGAS-STING-type-I pathway is also implicated in the induction of PD-L1 expression in mouse models (Grabosch et al., 2019). Therefore, micronuclei and the cGAS/STING cascade are involved in PD-L1 upregulation when cells are released from G2/M checkpoint arrest after DNA damage. Importantly, in addition to cell-cycle checkpoint factors, the status of DNA repair factors also influences the generation of micronuclei. DNA repair-deficient breast cancer cells derived from patient samples were found to contain increased cytosolic DNA (specific to S phase DNA damage), followed by constitutive PD-L1 expression via the cGAS-STING-type-I IFN pathway (Parkes et al., 2017). Specifically, NHEJ deficiency causes a reduction in the number of irradiation-induced micronuclei due to prolonged G2/M checkpoint arrest (Harding et al., 2017), whereas depleting HR proteins have an opposite effect, suggesting that HR deficiency causes mitotic abnormalities via DNA replication (Feng and Jasin, 2017). In fact, although HR is known to repair DSBs in the S/G2 phase, the majority of DSB repair in G2 phase is carried out by NHEJ (Figure 5; Shibata, 2017). Therefore, the status of NHEJ activity also influences micronuclei formation.
PARP inhibitors, which cause DNA replication-associated DNA damage, induces PD-L1 upregulation (Jiao et al., 2017). Another study shows that this upregulation is dependent on the cGAS/STING pathway and the activation of cGAS/STING pathway is mediated via cytoplasmic DNA (Shen et al., 2019). Recent early clinical trials reported the result of a combination of PARP inhibitor and ICIs (Friedlander et al., 2019; Zimmer et al., 2019; Lampert et al., 2020). These are promising approaches; however, further investigation is required to clarify the clinical benefit of this combination therapy. The contribution of DNA fragment generated at stalled replication fork has been also reported (Coquel et al., 2018). In normal cells, MRE11 exonuclease in association with phosphorylated SAMHD1 digests nascent DNA strands at the stalled DNA replication fork, whose fragment is not recognized by the cGAS/STING pathway. However, in the absence of SAMHD1, MRE11 endonuclease, but not exonuclease, creates a nick at the nascent DNA strand, generating a larger DNA fragment, which is recognized by cGAS following transport into the cytosol and this promotes STING-dependent IFN release. Other DNA repair-/replication-related factors that activate the cGAS/STING pathway are cytosolic RNA:DNA hybrids (Mankan et al., 2014) and telomere erosion (Chen et al., 2017). As an alternative pathway, ATM activates STING in a cGAS-independent manner (Dunphy et al., 2018). Thus, the status of DNA repair and signaling is also an important factor influencing immune activities, indicating that it has the potential to be a predictive biomarker for guiding ICI therapy, especially in combination with DNA damage-dependent cancer treatments. As another micronuclei-independent pathway, cGAS/STING pathway is also controlled by the levels of TREX1, a cytosolic nuclease (Stetson et al., 2008; Vanpouille-Box et al., 2017). The fractionated radiation, e.g., 3 × 8 Gy, does not upregulate TREX1; therefore, the cytosolic DNA fragments generated in response to radiation are able to activate cGAS/STING pathway. On the other hand, >20 Gy per fraction induces the expression of TREX1, which cleans up the desired immunogenic cytosolic DNA fragments. Therefore, a high dose irradiation, >20 Gy, does not effectively activate cGAS/STING pathway due to TREX1 upregulation (Vanpouille-Box et al., 2017). When >20 Gy are used in a clinical setting, the efficacy might be improved by TREX1 inhibition to maximize the irradiation-induced immunogenicity (Yamazaki and Galluzzi, 2017). The studies above suggest that the introduction of DNA damage by cancer therapy is involved in the activation of the immune response. Alternatively, Sen et al. (2019) showed that Chk1 inhibition activates the STING-TBK1-IRF3 pathway, increases PD-L1 expression, and, importantly, also augments cytotoxic T-cell infiltration. Another report showed that ATM deficiency increases the release of type-I IFN in a TBK1- and SRC-dependent but cGAS/STING-independent manner. Subsequently, the release of IFN leads to PD-L1 upregulation (Zhang et al., 2019).
Thus, ATM-ATR-Chk1-dependent DNA damage signaling within damaged tumor cells is involved in regulating PD-L1 expression in response to DNA damage. In parallel, micronuclei formation due to defects in repair and/or G2/M checkpoint arrest and DNA fragment formation activate the cGAS/STING pathway, releasing IFNs within the tumor environment, and its signaling upregulates PD-L1 expression via paracrine and autocrine pathways.
Perspective for Precision Medicine for Immune Checkpoint Therapy in Combination With RT/Chemotherapy by Targeting DNA Repair and DNA Damage Signaling
In summary, evidence suggests that DNA repair and signaling are involved in the regulation of HLA-neoantigen presentation and PD-L1 expression. Therefore, the application of anti-PD-1/PD-L1 antibodies will be effective, particularly in combination with DNA damage-associated cancer treatments, such as RT and DNA-damaging chemotherapy. The mechanisms involved in the activation of the immune response after DNA damage include (1) HLA-neoantigen presentation to TCRs, (2) the ATM/ATR/Chk1 signal cascade, and (3) micronucleus/DNA fragment-dependent cGAS/STING activation. The signaling from cGAS/STING and HLA-neoantigens leads to both immune-activating and immune-suppressing responses. Under normal conditions without exogenous DNA damage (i.e., without cancer treatment), the deficiency of DDR may not sufficiently activate immune signaling. Thus, the balance of immune activity might still be toward immune suppression without additional stimuli although the presence of excessive mutations in genome may upregulate HLA-neoantigen pathway. In contrast, after DNA damage-associated cancer treatment, the immune environment may be changed from “cold tumors” to “hot tumors,” irrespective of the presence of mutations in tumors, enabling the modification of the overall immune status, which is sensitive to ICI therapy. Recent preclinical studies have shown that treatments targeting DDR alone or in combination with RT have immunostimulatory potential through micronuclei formation and type-I IFN downstream production (McLaughlin et al., 2020). Thus, DNA damage can be a powerful stimulus for immune activation. Despite the upregulation of PD-L1 expression following DNA damage, the immune-suppressive effect can be effectively inhibited by ICI therapy. Regarding the selection of combination therapy, RT might have an advantage in terms of the protection of immune cells in patients. A recent study elegantly demonstrated that a large proportion of T cells survive clinically relevant doses of radiation using longitudinal imaging of mice (Arina et al., 2019). Importantly, CD8 + tumor-infiltrating lymphocytes were recruited into the irradiated field after RT from the non-irradiated area. Further, the survived T cells after RT maintained cancer cell killing activities, such as the mobility and production of IFNs, even though T cells are generally radiosensitive.
To date, several studies have shown that such a combination exhibits a synergistic effect at the preclinical level. However, cancer is highly diverse; thus, it is essential to select the optimal approach for each patient based on the concept of precision medicine because some tumors may not express a key factor in a particular signaling pathway. For example, cGAS/STING is downregulated in some tumors. Regarding biomarkers for combination therapy, DDR status may be important for patients who receive DNA damage-associated cancer therapy. However, the timing of the balance of immune responses can be changed at any moment after RT and chemotherapy; thus, a biomarker may be appropriately selected based on the timing of ICI treatment after/during RT and chemotherapy. In the era of precision medicine, the elucidation of the mechanisms underlying the immune response in the context of DDR status will be important for considering the next generation of cancer therapy.
Data Availability Statement
All datasets presented in this study are included in the article/supplementary material.
Author Contributions
SK, HS, SL, and AS wrote the manuscript. TY developed the system of the TCGA and TCIA analysis. SK performed the TCGA analysis. AS designed the figures and HS made the table. All authors read and approved the final manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP17H04713 to AS, JP19K08195 to HS, the Takeda Science Foundation, The Uehara Memorial Foundation, the Astellas Foundation for Research on Metabolic Disorders, The Kanae Foundation for the Promotion of Medical Science, the Yasuda Memorial Medicine Foundation, the Nakajima Foundation, the Sumitomo Foundation, and Suntory foundation for Life Sciences Bioorganic Research Institute. This work was also supported by the Program of the network-type Joint Usage/Research Center for Radiation Disaster Medical Science of Hiroshima University, Nagasaki University and Fukushima Medical University. A part of this study was conducted through the Joint Usage/Research Center Program of the Radiation Biology Center, Kyoto University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Akiko Shibata, Yoko Hayashi, Yasuyo Sekiguchi, Hiroko Iino, Naho Takashima, Yukihiko Yoshimatsu, and Itaru Sato for assisting with the laboratory work. We greatly appreciate the support of TCGA analysis by Shiho Matsumoto and Naomi Kamada.
Footnotes
References
Ablasser, A., Goldeck, M., Cavlar, T., Deimling, T., Witte, G., Rohl, I., et al. (2013). cGAS produces a 2 ‘-5 ’-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. doi: 10.1038/nature12306
Arina, A., Beckett, M., Fernandez, C., Zheng, W., Pitroda, S., Chmura, S. J., et al. (2019). Tumor-reprogrammed resident T cells resist radiation to control tumors. Nat. Commun. 10:3959.
Bai, J., Li, Y., and Zhang, G. (2017). Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 14, 348–362. doi: 10.20892/j.issn.2095-3941.2017.0033
Brahmer, J., Reckamp, K. L., Baas, P., Crino, L., Eberhardt, W. E. E., Poddubskaya, E., et al. (2015). Nivolumab versus docetaxel in advanced squamous-Cell non-small-cell lung cancer. New Engl. J. Med. 373, 123–135.
Chae, Y. K., Anker, J. F., Oh, M. S., Bais, P., Namburi, S., Agte, S., et al. (2019). Mutations in DNA repair genes are associated with increased neoantigen burden and a distinct immunophenotype in lung squamous cell carcinoma. Sci. Rep. 9:3235.
Chen, Y. A., Shen, Y. L., Hsia, H. Y., Tiang, Y. P., Sung, T. L., and Chen, L. Y. (2017). Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 24, 1124–1131. doi: 10.1038/nsmb.3498
Coquel, F., Silva, M. J., Techer, H., Zadorozhny, K., Sharma, S., Nieminuszczy, J., et al. (2018). SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. doi: 10.1038/s41586-018-0050-1
Deckbar, D., Birraux, J., Krempler, A., Tchouandong, L., Beucher, A., Walker, S., et al. (2007). Chromosome breakage after G2 checkpoint release. J. Cell Biol. 176, 749–755. doi: 10.1083/jcb.200612047
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998. doi: 10.1038/ni1102-991
Dunphy, G., Flannery, S. M., Almine, J. F., Connolly, D. J., Paulus, C., Jonsson, K. L., et al. (2018). Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappaB signaling after nuclear DNA damage. Mol. Cell 71, 745–760.e745.
Feng, W., and Jasin, M. (2017). BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nat. Commun. 8:525.
Friedlander, M., Meniawy, T., Markman, B., Mileshkin, L., Harnett, P., Millward, M., et al. (2019). Pamiparib in combination with tislelizumab in patients with advanced solid tumours: results from the dose-escalation stage of a multicentre, open-label, phase 1a/b trial. Lancet Oncol. 20, 1306–1315. doi: 10.1016/s1470-2045(19)30396-1
Galon, J., and Bruni, D. (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 18, 197–218. doi: 10.1038/s41573-018-0007-y
Garbi, N., and Kreutzberg, T. (2012). Dendritic cells enhance the antigen sensitivity of T cells. Front. Immunol. 3:389. doi: 10.3389/fimmu.2012.00389
Garcia-Diaz, A., Shin, D. S., Moreno, B. H., Saco, J., Escuin-Ordinas, H., Rodriguez, G. A., et al. (2017). Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 19, 1189–1201. doi: 10.1016/j.celrep.2017.04.031
Garcia-Sanz, P., Trivino, J. C., Mota, A., Perez Lopez, M., Colas, E., Rojo-Sebastian, A., et al. (2017). Chromatin remodelling and DNA repair genes are frequently mutated in endometrioid endometrial carcinoma. Int. J. Cancer 140, 1551–1563. doi: 10.1002/ijc.30573
Grabosch, S., Bulatovic, M., Zeng, F., Ma, T., Zhang, L., Ross, M., et al. (2019). Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 38, 2380–2393. doi: 10.1038/s41388-018-0581-9
Hagiwara, Y., Sato, H., Permata, T. B. M., Niimi, A., Yamauchi, M., Oike, T., et al. (2018). Analysis of programmed death-ligand 1 expression in primary normal human dermal fibroblasts after DNA damage. Hum. Immunol. 79, 627–631. doi: 10.1016/j.humimm.2018.05.008
Harding, S. M., Benci, J. L., Irianto, J., Discher, D. E., Minn, A. J., and Greenberg, R. A. (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. doi: 10.1038/nature23470
Hellmann, M. D., Ciuleanu, T. E., Pluzanski, A., Lee, J. S., Otterson, G. A., Audigier-Valette, C., et al. (2018). Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 378, 2093–2104.
Hoption Cann, S. A., Van Netten, J. P., and Van Netten, C. (2003). Dr William Coley and tumour regression: a place in history or in the future. Postgrad. Med. J. 79, 672–680.
Hutter, C., and Zenklusen, J. C. (2018). The cancer genome atlas: creating lasting value beyond its data. Cell 173, 283–285. doi: 10.1016/j.cell.2018.03.042
Iwai, Y., Ishida, M., Tanaka, Y., Okazaki, T., Honjo, T., and Minato, N. (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. U.S.A. 99, 12293–12297. doi: 10.1073/pnas.192461099
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Jiao, S., Xia, W., Yamaguchi, H., Wei, Y., Chen, M. K., Hsu, J. M., et al. (2017). PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin. Cancer Res. 23, 3711–3720. doi: 10.1158/1078-0432.ccr-16-3215
Kelly, R. J., Zaidi, A. H., Smith, M. A., Omstead, A. N., Kosovec, J. E., Matsui, D., et al. (2018). The dynamic and transient immune microenvironment in locally advanced esophageal adenocarcinoma post chemoradiation. Ann. Surg. 268, 992–999. doi: 10.1097/sla.0000000000002410
Keskin, D. B., Anandappa, A. J., Sun, J., Tirosh, I., Mathewson, N. D., Li, S., et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. doi: 10.1038/s41586-018-0792-9
Lampert, E. J., Zimmer, A., Padget, M., Cimino-Mathews, A., Nair, J. R., Liu, Y., et al. (2020). Combination of PARP inhibitor olaparib, and PD-L1 inhibitor durvalumab, in recurrent ovarian cancer: a proof-of-concept phase II study. Clin. Cancer Res. 26:56.
Larkin, J., Hodi, F. S., and Wolchok, J. D. (2015). Combined nivolumab and ipilimumab or monotherapy in untreated melanoma Reply. New Engl. J. Med. 373, 1270–1271. doi: 10.1056/nejmc1509660
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413.
Lim, S. H., Hong, M., Ahn, S., Choi, Y. L., Kim, K. M., Oh, D., et al. (2016). Changes in tumour expression of programmed death-ligand 1 after neoadjuvant concurrent chemoradiotherapy in patients with squamous oesophageal cancer. Eur. J. Cancer 52, 1–9. doi: 10.1016/j.ejca.2015.09.019
Lim, Y. J., Koh, J., Kim, S., Jeon, S. R., Chie, E. K., Kim, K., et al. (2017). Chemoradiation-Induced alteration of programmed death-Ligand 1 and CD8(+) tumor-infiltrating lymphocytes identified patients with poor prognosis in rectal cancer: a matched comparison analysis. Int. J. Radiat. Oncol. Biol. Phys. 99, 1216–1224. doi: 10.1016/j.ijrobp.2017.07.004
Mackenzie, K. J., Carroll, P., Martin, C. A., Murina, O., Fluteau, A., Simpson, D. J., et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. doi: 10.1038/nature23449
Mankan, A. K., Schmidt, T., Chauhan, D., Goldeck, M., Honing, K., Gaidt, M., et al. (2014). Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 33, 2937–2946. doi: 10.15252/embj.201488726
Marabelle, A., Le, D. T., Ascierto, P. A., Di Giacomo, A. M., De Jesus-Acosta, A., Delord, J. P., et al. (2020). Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10. doi: 10.1200/jco.19.02105
Margolin, K., Ernstoff, M. S., Hamid, O., Lawrence, D., Mcdermott, D., Puzanov, I., et al. (2012). Ipilimumab in patients with melanoma and brain metastases: an open-label, phase 2 trial. Lancet Oncol. 13, 459–465. doi: 10.1016/s1470-2045(12)70090-6
Martinez-Lostao, L., Anel, A., and Pardo, J. (2015). How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 21, 5047–5056. doi: 10.1158/1078-0432.ccr-15-0685
Matsuno, Y., Atsumi, Y., Shimizu, A., Katayama, K., Fujimori, H., Hyodo, M., et al. (2019). Replication stress triggers microsatellite destabilization and hypermutation leading to clonal expansion in vitro. Nat. Commun. 10:3925.
Matsushita, H., Vesely, M. D., Koboldt, D. C., Rickert, C. G., Uppaluri, R., Magrini, V. J., et al. (2012). Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404. doi: 10.1038/nature10755
McFarland, C. D., Yaglom, J. A., Wojtkowiak, J. W., Scott, J. G., Morse, D. L., Sherman, M. Y., et al. (2017). The damaging effect of passenger mutations on cancer progression. Cancer Res. 77, 4763–4772. doi: 10.1158/0008-5472.can-15-3283-t
McLaughlin, M., Patin, E. C., Pedersen, M., Wilkins, A., Dillon, M. T., Melcher, A. A., et al. (2020). Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 20, 203–217. doi: 10.1038/s41568-020-0246-1
O’Donnell, J. S., Teng, M. W. L., and Smyth, M. J. (2019). Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 16, 151–167. doi: 10.1038/s41571-018-0142-8
Overman, M. J., Lonardi, S., Wong, K. Y. M., Lenz, H. J., Gelsomino, F., Aglietta, M., et al. (2018). Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 36, 773–779. doi: 10.1200/jco.2017.76.9901
Parkes, E. E., Walker, S. M., Taggart, L. E., Mccabe, N., Knight, L. A., Wilkinson, R., et al. (2017). Activation of STING-dependent innate immune signaling by S-phase-specific DNA damage in breast cancer. JNCI J. Natl. Cancer Instit. 109:djw199. doi: 10.1093/jnci/djw199
Permata, T. B. M., Hagiwara, Y., Sato, H., Yasuhara, T., Oike, T., Gondhowiardjo, S., et al. (2019). Base excision repair regulates PD-L1 expression in cancer cells. Oncogene 38, 4452–4466. doi: 10.1038/s41388-019-0733-6
Ricciuti, B., Recondo, G., Spurr, L. F., Li, Y. Y., Lamberti, G., Venkatraman, D., et al. (2020). Impact of DNA damage response and repair (DDR) gene mutations on efficacy of PD-(L)1 immune checkpoint inhibition in non-small cell lung cancer. Clin. Cancer Res. 26:529.
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128.
Rosenberg, S. A., Yang, J. C., Sherry, R. M., Kammula, U. S., Hughes, M. S., Phan, G. Q., et al. (2011). Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557. doi: 10.1158/1078-0432.ccr-11-0116
Sato, H., Niimi, A., Yasuhara, T., Permata, T. B. M., Hagiwara, Y., Isono, M., et al. (2017). DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 8:1751.
Schumacher, T. N., Scheper, W., and Kvistborg, P. (2019). Cancer neoantigens. Annu. Rev. Immunol. 37, 173–200.
Schumacher, T. N., and Schreiber, R. D. (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74.
Sen, T., Rodriguez, B. L., Chen, L., Corte, C. M. D., Morikawa, N., Fujimoto, J., et al. (2019). Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 9, 646–661. doi: 10.1158/2159-8290.cd-18-1020
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., and Ribas, A. (2017a). Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723. doi: 10.1016/j.cell.2017.01.017
Sharma, P., Retz, M., Siefker-Radtke, A., Baron, A., Necchi, A., Bedke, J., et al. (2017b). Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 18, 312–322. doi: 10.1016/s1470-2045(17)30065-7
Shen, J., Zhao, W., Ju, Z., Wang, L., Peng, Y., Labrie, M., et al. (2019). PARPi Triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 79, 311–319. doi: 10.1158/0008-5472.can-18-1003
Shibata, A. (2017). Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. 803–805, 51–55. doi: 10.1016/j.mrfmmm.2017.07.011
Shibata, A., and Jeggo, P. A. (2014). DNA double-strand break repair in a cellular context. Clin. Oncol. 26, 243–249. doi: 10.1016/j.clon.2014.02.004
Stetson, D. B., Ko, J. S., Heidmann, T., and Medzhitov, R. (2008). Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. doi: 10.1016/j.cell.2008.06.032
Strickland, K. C., Howitt, B. E., Shukla, S. A., Rodig, S., Ritterhouse, L. L., Liu, J. F., et al. (2016). Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 7, 13587–13598. doi: 10.18632/oncotarget.7277
Sun, L. L., Yang, R. Y., Li, C. W., Chen, M. K., Shao, B., Hsu, J. M., et al. (2018). Inhibition of ATR downregulates PD-L1 and sensitizes tumor cells to T cell-mediated killing. Am. J. Cancer Res. 8, 1307–1316.
Thommen, D. S., and Schumacher, T. N. (2018). T cell dysfunction in cancer. Cancer Cell 33, 547–562. doi: 10.1016/j.ccell.2018.03.012
Topalian, S. L., Taube, J. M., Anders, R. A., and Pardoll, D. M. (2016). Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287.
Trenner, A., and Sartori, A. A. (2019). Harnessing DNA double-strand break repair for cancer treatment. Front. Oncol. 9:1388. doi: 10.3389/fimmu.2012.01388
Vanpouille-Box, C., Alard, A., Aryankalayil, M. J., Sarfraz, Y., Diamond, J. M., Schneider, R. J., et al. (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 8:15618.
Vendetti, F. P., Karukonda, P., Clump, D. A., Teo, T., Lalonde, R., Nugent, K., et al. (2018). ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Invest. 128, 3926–3940.
Wang, Z., Zhao, J., Wang, G., Zhang, F., Zhang, Z., Zhang, F., et al. (2018). Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 78, 6486–6496.
Yamazaki, T., and Galluzzi, L. (2017). TREX1 cuts down on cancer immunogenicity. Trends Cell Biol. 27, 543–545.
Yoneda, K., Kuwata, T., Kanayama, M., Mori, M., Kawanami, T., Yatera, K., et al. (2019). Alteration in tumoural PD-L1 expression and stromal CD8-positive tumour-infiltrating lymphocytes after concurrent chemo-radiotherapy for non-small cell lung cancer. Br. J. Cancer 121, 490–496.
Zhang, Q., Green, M. D., Lang, X., Lazarus, J., Parsels, J. D., Wei, S., et al. (2019). Inhibition of ATM increases interferon signaling and sensitizes pancreatic cancer to immune checkpoint blockade therapy. Cancer Res. 79, 3940–3951.
Zimmer, A. S., Nichols, E., Cimino-Mathews, A., Peer, C., Cao, L., Lee, M. J., et al. (2019). A phase I study of the PD-L1 inhibitor, durvalumab, in combination with a PARP inhibitor, olaparib, and a VEGFR1-3 inhibitor, cediranib, in recurrent women’s cancers with biomarker analyses. J. Immunother. Cancer 7:197.
Keywords: DNA repair, non-homologous end joining, homologous recombination, ataxia-telangiectasia-mutated, ataxia telangiectasia and Rad3-related, immune checkpoint inhibitor
Citation: Kakoti S, Sato H, Laskar S, Yasuhara T and Shibata A (2020) DNA Repair and Signaling in Immune-Related Cancer Therapy. Front. Mol. Biosci. 7:205. doi: 10.3389/fmolb.2020.00205
Received: 12 May 2020; Accepted: 28 July 2020;
Published: 08 September 2020.
Edited by:
Emma Bolderson, Queensland University of Technology, AustraliaReviewed by:
Jessica Downs, The Institute of Cancer Research (ICR), United KingdomRichard Chahwan, University of Zurich, Switzerland
Copyright © 2020 Kakoti, Sato, Laskar, Yasuhara and Shibata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiro Sato, aGlyby5zYXRvQGd1bm1hLXUuYWMuanA=; Atsushi Shibata, c2hpYmF0YS5hdEBndW5tYS11LmFjLmpw