 Mariola J. Edelmann
Mariola J. Edelmann Gustavo H. B. Maegawa
Gustavo H. B. Maegawa- 1Department of Microbiology and Cell Science, The University of Florida's Institute of Food and Agricultural Sciences, University of Florida, Gainesville, FL, United States
- 2Department of Pediatrics, College of Medicine, University of Florida, Gainesville, FL, United States
During the past decades, several therapeutic approaches have been developed and made rapidly available for many patients afflicted with lysosomal storage disorders (LSDs), inborn organelle disorders with broad clinical manifestations secondary to the progressive accumulation of undegraded macromolecules within lysosomes. These conditions are individually rare, but, collectively, their incidence ranges from 1 in 2,315 to 7,700 live-births. Most LSDs are manifested by neurological symptoms or signs, including developmental delay, seizures, acroparesthesia, motor weakness, and extrapyramidal signs. The chronic and later-onset clinical forms are at one end of the continuum spectrum and are characterized by a subtle and slow progression of neurological symptoms. Due to its inherent physiological properties, unfortunately, the blood-brain barrier (BBB) constitutes a significant obstacle for current and upcoming therapies to achieve the central nervous system (CNS) and treat neurological problems so prevalent in these conditions. To circumvent this limitation, several strategies have been developed to make the therapeutic agent achieve the CNS. This narrative will provide an overview of current therapeutic strategies under development to permeate the BBB, and address and unmet need for treatment of the progressive neurological manifestations, which are so prevalent in these inherited lysosomal disorders.
Lysosomal Storage Diseases: Inborn Organelle Disorders Predominantly Affecting the CNS
Lysosomal storage diseases (LSDs) are inborn organelle disorders characterized by multisystemic and progressive manifestations, being most of them neurological in nature (Table 1) (Patil and Maegawa, 2013; Maegawa, 2019). Intravenous enzyme replacement therapy (ERT), the mainstay treatment for several LSDs, does not address the neurological problems, as these recombinant proteins, large molecular-weight molecules, are unable to permeate through the blood-brain barrier (BBB) effectively. Several novel therapeutic agents such as intrathecal or intracerebroventricular delivery of enzymes, fusion proteins that cross the BBB, substrate reduction therapy (SRT), pharmacological chaperones (PCs), and gene therapy, are currently being developed to treat the neurological manifestations of LSDs (Figure 1).
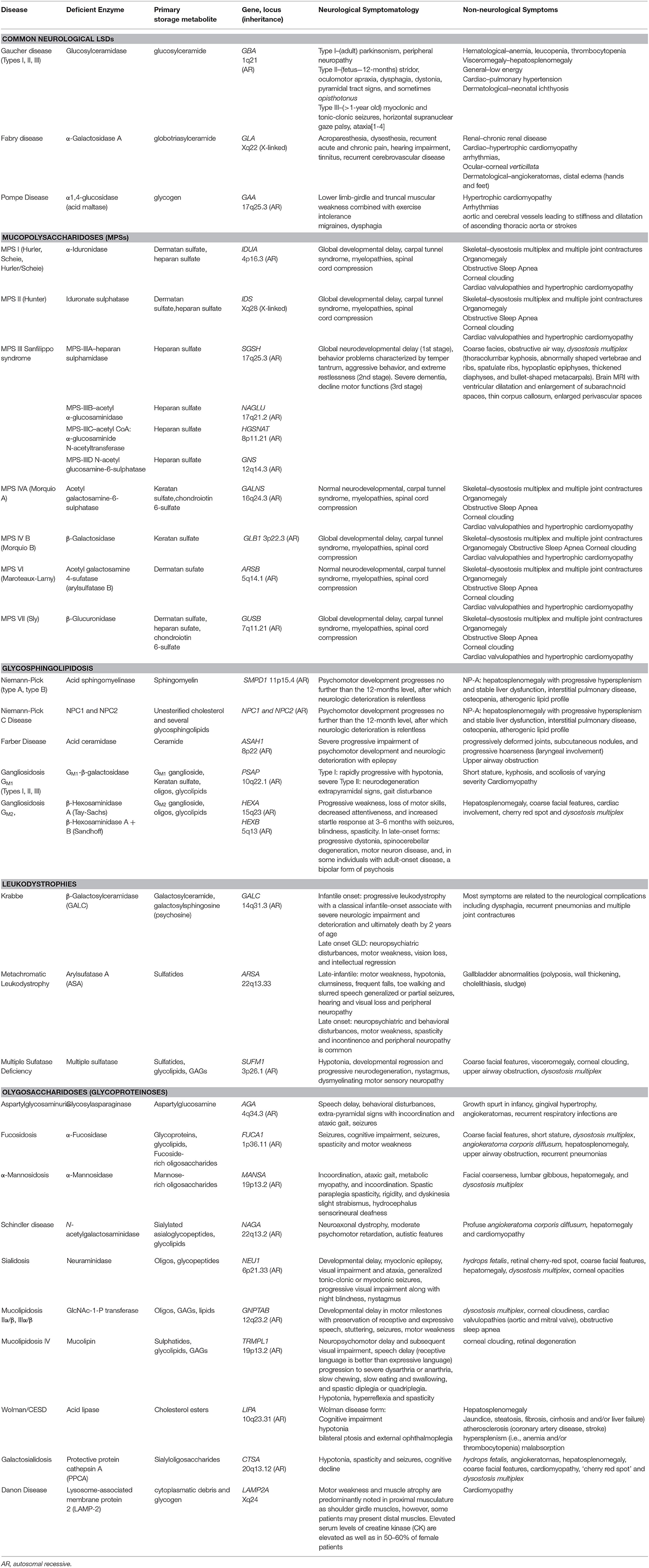
Table 1. Enzyme-deficiency LSDs, classified according to primary substrate storage.
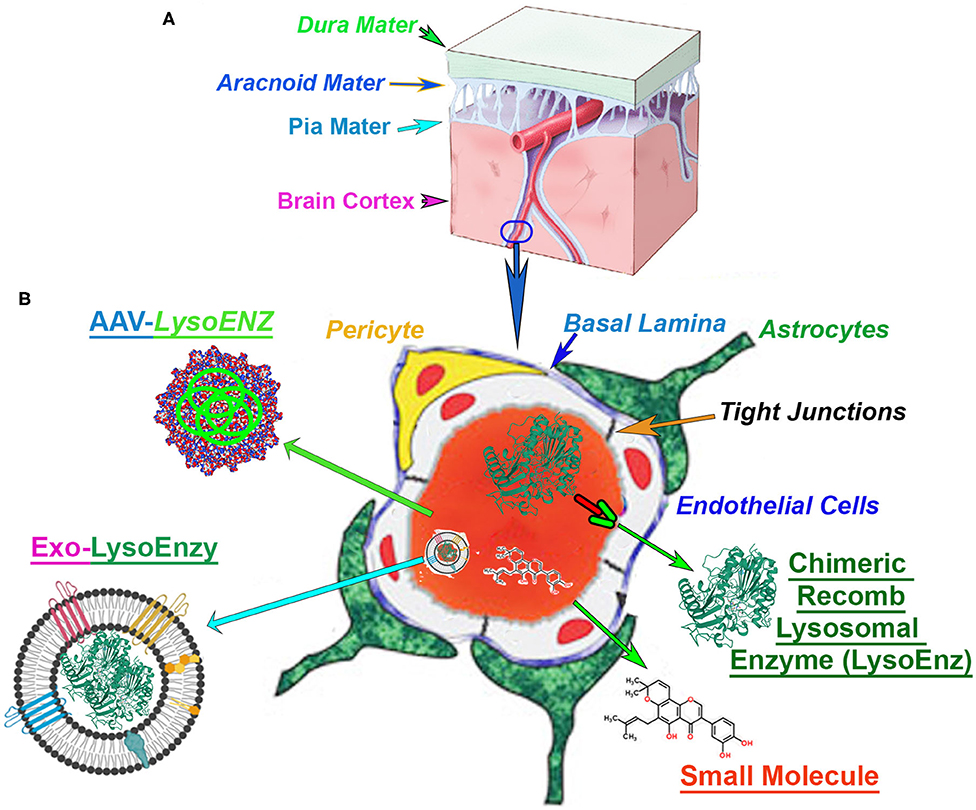
Figure 1. Blood-brain barrier (BBB) in the context of CNS-targetting therapeutics. (A) The central nervous system (CNS), and the cerebrovascular system are protected by a series of robust membranes as dura, arachnoid and pia mater. (B) The components of the BBB are established by the dynamic relationship among astrocytes endfeet, basal membrane, pericytes, endothelial cells and the tight junctions between the brain-endothelial cells. An illustration of the cross-section of a CNS-capillary shows the BBB components (italized), and current therapies under investigation. The modified chimeric lysosomal enzymes (LysoEnz), small molecules, gene therapy exploring specific AAV serotypes, and nanoparticle-based therapies (Exo-LysoEnzy) are designed to overcome the BBB and tackle the neuropathogenesis processes prevalent in many LSDs.
The unmet need for more efficacious drug delivery to the central nervous system (CNS) is currently the primary problem and often attribute to the properties of the BBB, considered the “problem behind the problem” when developing therapies for neurological conditions (Pardridge, 2005). The BBB is a term used to describe the unique properties of the microvasculature of the CNS, constituted by continuous non-fenestrated vessels, containing a series of additional “gatekeepers” tightly regulating the movement of molecules, ions, and cells between the blood and its neural cells (Figure 1) (Daneman and Prat, 2015). Neurodegenerative disorders such as Alzheimer's, Parkinson's, and multiple sclerosis, among others, constitute ~17% of total deaths globally (GBD 2016 Neurology Collaborators Group, 2017). The majority of the promising drugs with the potential to treat CNS disorders, including mostly small molecules, fall in the late pre-preclinical phase due to their failure to achieve therapeutic levels in the brain. In fact, among the current small molecule drugs approved, only 2% or less can produce significant trough levels in CNS and used therapeutically to treat different neurobehavioral manifestations. Those are mostly antipsychotics, mood stabilizers, anti-depressants, and anti-epileptics (Ghose et al., 2012). Many orphan disorders also manifest with significant progressive neurodegeneration and have served as models for the development of novel strategies to address the BBB drug delivery challenge (Begley et al., 2008). The efficacy of therapeutic options depends on the bioactivity of the therapeutic agent, but equally important is its ability to reach critical targets within tissues and cells affected by the disease processes. Intravenous administration is the simplest and least invasive method for systemic delivery of drugs by infusing molecules directly into the circulatory system. The blood supplies all cells and tissues with vital elements and nutrients. However, the delivery of macromolecules such as proteins from the circulation into adjacent cell layers is limited by a variety of biological barriers and selectivity of cell uptake mechanisms. The macromolecular transport across plasma and intracellular membranes are regulated by integral transmembrane channels or carriers. For any particular drug, the specific components required for uptake might be limited or lacking, especially in specialized tissues such as the endothelium of the CNS. Exogenous molecules interface with at least three distinct barriers that modulate transport into the brain: BBB, the blood-cerebrospinal fluid (CSF) barrier, and the arachnoid barrier (Begley et al., 2008; Abbott et al., 2010). The BBB is formed by vascular endothelial cells and other neural cells making a specialized network of capillaries with an extensive interface for blood-brain exchange with a total surface area of 12–18 m2 in humans (Pardridge, 2001, 2007a; Begley et al., 2008). Molecules in the blood gain access to the brain by exploiting selective cell surface transporters or by free diffusion if the molecule is lipophilic and has a molecular weight <400 Da (Pardridge, 2006; Sweeney et al., 2018). In response to the challenges imposed by BBB to treat neurological manifestations which are prevalent in several LSDs, a number of strategies to deliver small molecules and biological agents to the CNS include neurosurgical-based interventions, chemical-based strategies (e.g., promoting lipid solubility of the molecules) (Corraliza-Gomez et al., 2019); and biology-based strategies (e.g., vesicular mechanism regulated by endogenous BBB transporters) (Sweeney et al., 2018).
Perspectives on the CNS-Targeting Therapies and Their Limitations
Unfortunately, for the vast majority of the neurological forms of LSDs, the core of patient management is based on multidisciplinary supportive care, which includes addressing the disease-related neurological complications with neurosurgical procedures. The classical example is the decompression of the cervical spine and the placement of ventricular-peritoneal shunt for hydrocephalus (Muenzer et al., 2009b). In addition, the use of anticonvulsants for seizures and assistance for patients with learning disabilities, orthopedic interventions to alleviate spinal deformities, joint limitations, and retractions; nutritional support; cardiac surgery when involvement of cardiac valves or coronaries are part of the complex care required in management of patients diagnosed with LSDs.
Over recent years, intensive and continual efforts have been made to develop therapies to tackle the neuro-pathogenesis of these inborn organelle disorders (Alderson et al., 2019). The approaches developed to treat LSDs are based on different strategies, each directed to manipulate a specific event in the downstream pathogenic cascades. The majority of these approaches target the deficiency of lysosomal enzyme/protein and/or the accumulated substrate. The former can be achieved by administering the recombinant human lysosomal enzyme, or by taking advantage of the ability of cells to uptake the agent through the mannose 6-phosphate receptor (Ghosh et al., 2003), delivering it to the lysosomal compartment. This approach is the enzyme replacement therapy (ERT) and is currently the core therapeutic modality used in the majority of LSDs, but limited to treat non-neurological, or somatic, manifestations of these conditions (Platt, 2018).
More recently, to address the poor biodistribution of the agent to certain organs, including CNS studies on modifying the ERT agent have emerged (Condori et al., 2016) and skeletal muscle (Koeberl et al., 2011; Peng et al., 2017; Han et al., 2019). To investigate the potential of large-molecular weight compounds to treat neurological disorders, novel approaches are required to surmount the BBB. Based on the ability of the adsorptive-mediated endocytosis of plant RTB-lectin, plant-based bioproduction of a fusion enzyme of ERT agent and lectin were accomplished, preserving the lysosomal enzymatic activity (Acosta and Cramer, 2020). Two murine LSD models, IDUA−/− (MPS I), GM1 gangliosidosis mice, treated with the ERT agent-RTB-lectin showed significant normalization of substrate levels in CNS, and correction of learning and memory deficits of the mouse model (Condori et al., 2016; Ou et al., 2018). Other studies have shown that the cellular uptake of α-iduronidase (IDUA), the enzyme deficient in MPS I, is enhanced by a fusion of IgG-IDUA fusion protein, where the IgG domain is a genetically engineered monoclonal antibody (MAb) against the human insulin receptor (HIR) (Boado et al., 2008, 2013). Based on these studies on murine IDUA−/− (MPS I), a phase 1/2 trial was performed in which patients affected by MPS I received the HIRMAb–IDUA fusion protein by intravenous infusion for 52-weeks DQ, and the cortical gray matter volume of the brain were stabilized by valanafusp alpha treatment. Somatic manifestations were stabilized, or improved, based on urinary glycosaminoglycan levels, hepatic and spleen volumes, and shoulder range of motion (Giugliani et al., 2018). The adverse events were dominated by transient hypoglycemia in 6.4% of subjects enrolled (Giugliani et al., 2018). Further studies will be necessary in earlier symptomatic or presymptomatic patients with the neuronopathic predictive genotype of MPS-I, as well as, studies in other neurological lysosomal storage disorders are needed. In preclinical studies, two fusion candidates, IDUAe1 and IDUAe2, were identified and shown to be desirable receptor-mediated binding, endocytosis, and transendothelial transport as well as appropriate lysosomal enzyme trafficking and biological function (Wang et al., 2013). The increased peripheral IDUAe1 or IDUAe2 through the hepatic expression resulted in BBB capillary-depleted endothelial cells and protein delivery into non-endothelium perivascular cells, neurons, and astrocytes in the murine MPS-I model, achieving levels of 2–3% of normal brain IDUA activities (Wang et al., 2013). Using the mucopolysaccharidosis type IIIA (MPS-IIIA) murine model, a neurological MPS caused by the N-sulfoglucosamine sulfohydrolase deficiency (Table 1), studies on a chimeric sulphamidase, containing a signal peptide from Apolipoprotein B (ApoB-BD), showed a highly secreted iduronate-2-sulphatase (IDS) and efficient BBB transcytosis and restoration of sulphamidase activity in the brain of treated mice (Sorrentino et al., 2013).
Alternatively, the recombinant enzyme can be secreted by donor stem-cell-derived cells or genetically manipulated cells from the patient. This strategy is the hematopoietic stem-cell therapy (HSCT) and is efficacious for a few neurological LSDs including the neuronopathic forms of mucopolysaccharidosis type I (MPS-I) (Wiseman et al., 2013). In a recent study, a brain-targeted hematopoietic stem cell gene therapy approach using lentiviral IDS fused to ApoEII (IDS.ApoEII) compared to a lentivirus expressing wild-type (normal) IDS to standard bone marrow transplant. In mucopolysaccharidosis II mice, only IDS.ApoEII mediated complete normalization of brain pathology and behavior, providing significantly enhanced correction compared to IDS. Gleitz et al. (2018) In addition, the corrected macrophages traffic to the brain, secreting IDS/IDS.ApoEII enzyme for cross-correction. The IDS.ApoEII enzyme showed to be more active in plasma and was taken up by endothelial cells, and transcytosed across the BBB at a significantly higher rate than the native IDS via both heparan sulfate/ApoE-dependent receptors and mannose-6-phosphate receptors (Gleitz et al., 2018). Recently, lentiviral vector-mediated expression of the chimeric GALC enzymes was shown to be safe and leads to supranormal enzymatic activity in both neural and hematopoietic cells. The IDSsp.GALC showed enhanced expression and secretion in comparison to the unmodified GALC. The chimeric GALC enzymes produced by LV-transduced cells reduce intracellular galactosylceramide (GalCer) storage and effectively cross-correct GLD murine neurons and glial cells. These findings suggest that ERT of chimeric GALC can be used by the affected CNS cells and tissues, supporting the development of novel and more effective GT approaches for GLD (Ricca et al., 2020).
The gene encoding the deficient lysosomal enzyme can be delivered to the cells through the use of AAV systems. Subsequently, patients cells can synthesize the normal lysosomal enzymatic cells (Biffi, 2017a,b). This approach is known as ex-vivo gene therapy. Another approach is to restore the equilibrium between the synthesis of the accumulated, or “stored,” substrates, and their limited degradation due to the lysosomal enzymatic deficiency (Platt and Jeyakumar, 2008; Marshall et al., 2016). This strategy is called substrate reduction therapy (SRT), which is achieved by modulating the primary substrate biosynthesis with small molecule inhibitors of critical limiting steps. The “reduction of the accumulated substrate” can also be achieved by promoting the clearance of primary and secondary accumulated products from the affected cells (Gatto et al., 2017; Torra et al., 2018).
In this review, we focus on current treatment strategies for LSDs targeting the CNS, including those that hold promise for future advancements in the treatment of the progressive neurological manifestations of these devastating disorders.
Therapeutic Strategies Potentially Targeting CNS Manifestations of LSDs
Small Molecule Therapies
Small molecules are most frequently used experimentally to modulate the function of a protein target and assess the consequences. The small molecules are low-molecular-weight (<900 Da) synthetic organic compounds that have antagonistic or agonistic effects on intra and extracellular targets. By being small molecules, these therapeutic agents are more likely to cross the BBB and achieve the CNS, and ultimately the neural cells with endolysosomal network dysfunction due to the lysosomal enzyme/protein deficiency. Examples of small molecules that have been used to treat LSDs are pharmacological chaperones (PCs), proteostasis regulators, substrate reduction therapy (SRT) agents, and drugs that increase gene expression by suppressing translation termination, also known as premature termination codon (PTC) suppressing agents. Here, we describe the current classes of small molecules that have shown evidence of BBB penetration and potentially treating some neurological manifestations of some LSDs (Table 2).
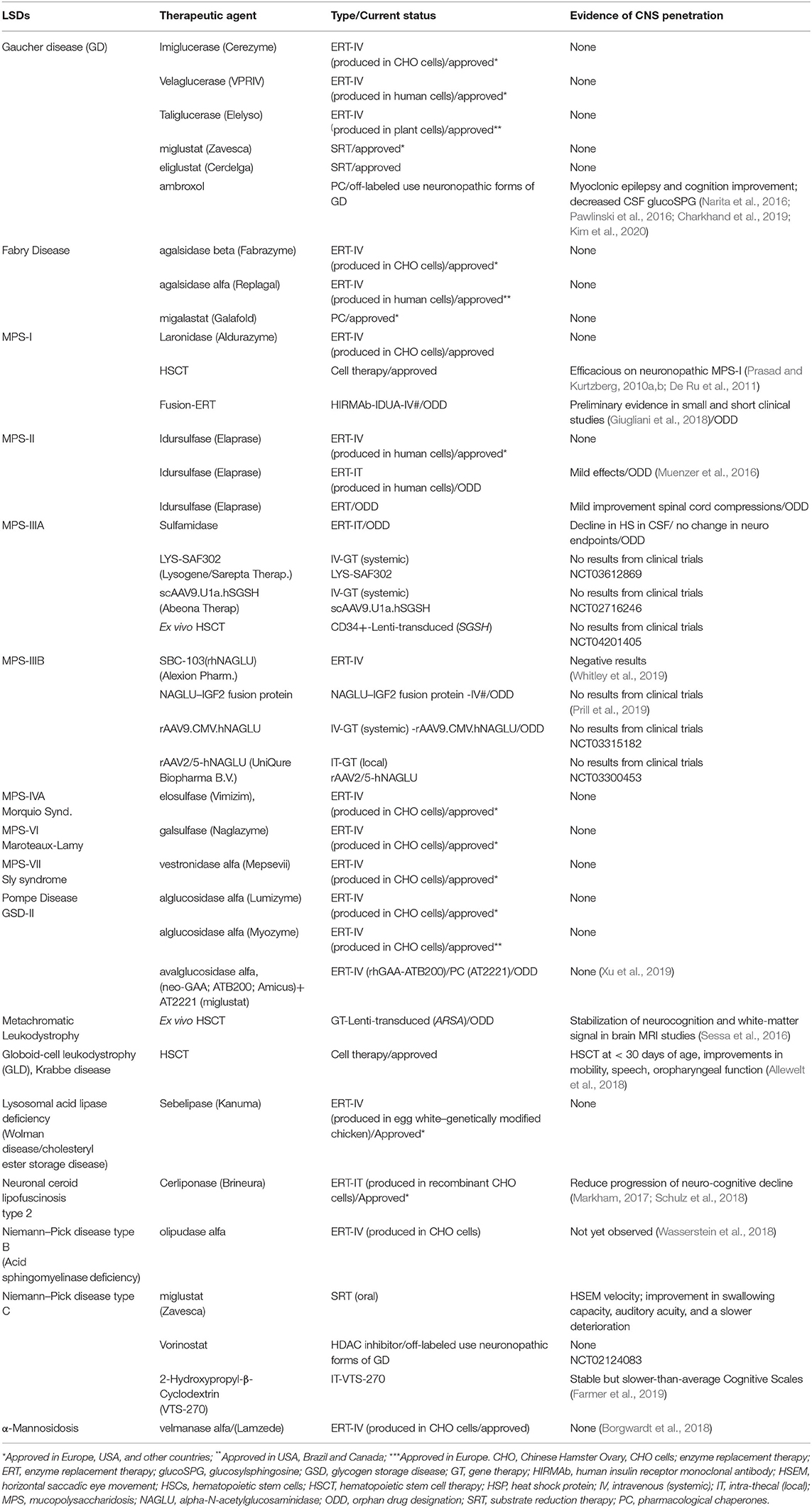
Table 2. Overview of approved, orphan drug designations, off-labeled therapies for treating lysosomal storage disorders, and examples of some products under development with an orphan drug designation.
Pharmacological Chaperones (PC)
A number of missense pathogenic gene variants encode mutant misfolded proteins, resulting in interactions with elements of the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway (Bhattacharya and Qi, 2019), and subsequent premature degradation by the endoplasmic reticulum quality control (ERQC) machinery (Maegawa et al., 2007; Balch et al., 2008; Mu et al., 2008b). Pharmacological chaperones (PCs) interact earlier with the target misfolded mutant protein in the endoplasmic reticulum, in the case of lysosomal enzymes, assisting its folding, attenuating its high-free energy (ΔG0), toward a lower ΔG0” state (Powers and Balch, 2013), passing the ERQC, and evading the ERAD (Wiseman et al., 2007). Subsequently, the properly folded mutant protein reaches the cis-Golgi network and, where specific biochemical modifications allow the interactions with mannose-6 phosphate receptors and, eventually, landing in the lysosomal compartment (Sawkar et al., 2006). Once at the lysosomes, due to its acidic environment (pH 4–5) and abundance of the undegraded and accumulated substrate, optimal PCs lose their affinity to the target mutant, leaving their “site-of-binding,” often the active-site, free for intercations with the natural enzymatic substrate (Tropak and Mahuran, 2007). Several compounds have been tested as potential PCs for the treatment of neurological symptoms of lysosomal disorders. These compounds include ambroxol (Maegawa et al., 2009b; Klionsky et al., 2016), and most recently progranulin (Jian et al., 2016) and NCGC607 for the treatment of Gaucher disease (Aflaki et al., 2016); pyrimethamine for late-onset Tay- Sachs disease (Maegawa et al., 2007; Clarke et al., 2011; Osher et al., 2011); N-octyl-epi-β-valienamine (Aflaki et al., 2016) and, recently, derivatives of 4-epi isofagomine for β-galactosidase deficiency, betaine for aspartylglucosaminuria (Lebl et al., 2017) and the current PC molecule approved for Fabry disease, migalastat (Germain et al., 2016; Hughes et al., 2017). Protein homeostasis, or proteostasis, consists of a network of interacting activities, or “interactome,” in charge of maintaining the “health” of the proteome and the organism (Balch et al., 2008; Song et al., 2013; Wang and Segatori, 2013). This network comprises several ER-resident chaperones and degradation elements as well as stress-responsive signaling pathways that detect the misfolding and/or aggregation of proteins in specific subcellular compartments using stress sensors, which respond by generating an active transcription factor (Roth and Balch, 2011; Klionsky et al., 2016; Kelly, 2020).
Proteostasis Regulators
Proteostasis regulators are defined as small molecules able to increase the residual function of lysosomal mutant enzymes by manipulating components of several folding and protein degradation signaling pathways (Kelly, 2020). By affecting and interacting with components of the interactome network, the proteostasis regulators have a broader and more pervasive outcome effect on multiple misfolded mutant proteins, which implies in a broader clinical application for several “misfolding protein disorders.” (Mu et al., 2008a; Kelly, 2020). One of the examples of proteostasis regulators is bortezomib, an approved-proteasome inhibitor approved for myeloma, which showed to decrease the levels of intracellular accumulated cholesterol in cultured cells with NPC1 deficiency from individuals with the Niemann-Pick disease type C (Macias-Vidal et al., 2014). In addition, the same proteostasis regulator showed to restore glucocerebrosidase (GCase) activity from Gaucher disease by inhibiting histone deacetylases and preventing mutant GCase degradation (Fan et al., 2011). Other histone deacetylase inhibitors, vorinostat, and panobinostat showed to decrease the cholesterol typically accumulated in human Niemann-Pick C1 fibroblasts (Pipalia et al., 2017). Also, the manipulation of ER-calcium concentrations in the ER has been investigated as a potential therapeutic approach with promising results in Gaucher disease, Niemann-Pick disease type C, and mucolipidosis type IV (Matalonga et al., 2017). However, the long-term effects of the regulation of the proteostasis cannot be predicted. The link between autophagy impairment and lysosomal disorders has led to novel approaches to modulate cellular clearance, which has been investigated in preclinical studies (Palmieri et al., 2017), but the translation to clinical trials is still premature.
Substrate Reduction Therapy
The substrate reduction therapy (SRT) agent consists of a small molecule that partially inhibits the biosynthesis of the natural substrate accumulated secondary to the enzymatic deficiency. It is an attractive approach to mitigate the load of the primary and eventually secondary substrate of the deficient lysosomal enzyme (Platt et al., 1994a,b). The first substrate reduction therapy agent developed for a LSD was miglustat, an imino-sugar deoxynojirimycin, and its alkylated derivatives, known inhibitors of the N-linked oligosaccharide processing enzymes (Platt et al., 1994b). In preclinical studies, the imino sugar N-butyldeoxynojirimycin showed also to interact with glucosyltransferases, especially inhibiting ceramide-specific glucosyltransferase that catalyzes the first step in glycosphingolipid biosynthesis (Platt et al., 1997). Miglustat was later approved for the treatment of Gaucher disease type I (Cox et al., 2000), and Niemann-Pick C disease (Patterson et al., 2007). Despite the reduction of the increasing levels of the glycosphingolipids in the CNS of murine models of gangliosidoses, in early human clinical trials in GM2 and GM1 gangliosidoses, the small molecule failed to achieve sufficient CNS levels to result in measurable clinical benefits (Maegawa et al., 2009a,c; Shapiro et al., 2009).
Studies using miglustat in combination with ERT for neuropathic Gaucher disease (types II and III) showed no significant alterations of clinical endpoints (Schiffmann et al., 2008). Despite the anecdotal reports on neurological improvement in some cases (Capablo et al., 2007), no SRT agents, a standard of care has been used with ERT. In this setting, and given the significant side effects of miglustat (Pastores et al., 2007; Giraldo et al., 2009), another ceramide analog, eliglustat, was developed by Dr. Shayman's group (Mceachern et al., 2007; Shayman, 2010, 2013), and later approved for the treatment of Gaucher disease type I (Cox et al., 2015; Mistry et al., 2015). However, eliglustat fails to permeate the BBB, limiting the therapeutic use to non-neuropathic symptoms of Gaucher disease. Small molecules that function as cholesterol-lowering agents have been identified for Niemann-Pick disease type C disease (Camargo et al., 2001;Davidson et al., 2009).
The intrathecal 2-hydroxypropyl-β-cyclodextrin has been shown to have clinical benefits or stabilization of disease progression in most of the 14 patients reported (Ory et al., 2017; Farmer et al., 2019). Further clinical trials with a larger number of patients with NPC studies are either being analyzed (NCT01747135) or ongoing (NCT03887533, NCT03893071) using the intrathecal and intravenous route, respectively. In terms of the mucopolysaccharidosis (MPS) I, II, and III (all subtypes), and their neurological manifestations, the genistein, an isoflavone, and the protein tyrosine kinase and epidermal growth factor inhibitor resulted in the reduction of glycosaminoglycan (GAG) accumulation (Malinowska et al., 2009). The early phase clinical studies showed a reduction of urinary excretion of GAGs and plasma heparan sulfate concentration in patients with MPS III (De Ruijter et al., 2012). However, the absolute reduction of these metabolites was small after a 12-months treatment period coinciding within the range as observed in untreated patients. Also, genistein showed no efficacy in the clinical endpoints when examining in 30 affected MPS-III enrolled in the study (De Ruijter et al., 2012). Evidence of another potential mechanism of action of the 2-hydroxypropyl-β-cyclodextrin has been described against cytotoxic psychosine, which is a sphingolipid found at high and cytotoxic levels in Krabbe disease due to the GALC deficiency in the CNS (Katabuchi et al., 2018).
Small Molecules Targeting Nonsense Mutations in LSDs
By definition, nonsense pathogenic genetic variants result in the premature termination codon (PTC) and subsequent translation of a truncated protein that is typically degraded by nonsense-mediated decay. An extensive meta-analysis on the Human Gene Mutation Database has revealed that approximately 11% of all described pathogenic variants resulting in human inherited disorders are caused by nonsense mutations (Mort et al., 2008; Bidou et al., 2012). Nonsense pathogenic variants are identified in several LSDs that may potentially benefit from small molecules that function as PTC suppression-stimulating agents by promoting “read-through' the premature termination or stop codon. In LSDs, chloramphenicol showed to increase the IDUA gene expression and, subsequent, α-L iduronidase (IDUA) activity in cell lines derived from patients with MPS-I (Mayer et al., 2013). Also, B84 suppressed the IDUA-W392X nonsense mutation much more efficiently than any of the other compounds tested. NB84 treatment restored enough functional α-L-iduronidase activity to partially reverse abnormal GAG accumulation and lysosomal abundance in mouse embryonic fibroblasts derived from the Idua-W392X mouse (Wang et al., 2012). In cultured oligodendrocytes from Twitcher mice (galctwi/twi), naturally occurring murine model for Krabbe disease, and patient fibroblasts with nonsense mutations in GALC gene, the nonsense-mediated mRNA decay (NMD) inhibitor 1 (NMDI1) (Durand et al., 2007) increased the levels of mRNA and rescued galactocerebrosidase (GALC) activity in a dose-dependent manner, and improve the morphology of the differentiated oligodendrocytes (Luddi et al., 2016). In infantile neuronal ceroid lipofuscinoses, using the novel Cln1(R151X) mouse model, mice receiving PTC124 (ataluren) showed enhancements of the palmitoyl-protein thioesterase 1 activity were noted, but limited to the liver and muscle tissues, alluding the challenges of biodistribution of this specific class of small molecules (Thada et al., 2016).
Cell Therapy
The basis and rationale of using hemopoietic stem cell transplantation (HSCT) to treat LSDs originated from elegant and pioneering experiments of Neufeld and cols. who demonstrated in cultured cells of patients with different mucopolysaccharidoses, cross-correction with reductions of the accumulated GAGs were observed (Fratantoni et al., 1968; Neufeld, 1989). The HSCT potential resides in the permanent biological source (donor cells) of steady functional lysosomal enzymes secretion and their internalization through the mannose-6 phosphate receptor system by surrounding “recipient” affected cells (Platt et al., 2018). The mannose 6-phosphate receptor allows the exogenous lysosomal enzyme to directly targeting the dysfunctional and enlarged lysosomes. The hematopoietic stem cells (HSCs) can also cross the BBB and differentiate into microglial cells, and populate the CNS, becoming active sources of lysosomal enzymes (Tan et al., 2019). Numerous factors may impact the efficacy of the HSCT in LSDs and other inherited metabolic disorders. These factors can be clinical and HSCT-related, including drug regimens of conditioning pre-HSCT, presence and degree of graft-vs. host disease, time of neutropenic recovery, and, ultimately, clinical status and, especially, the disease stage of the affected patient undergoing the procedure (Biffi, 2017b). From a biological standpoint, several factors can affect the level of “cross-correction” of the deficient lysosomal enzyme, including the number of donor HSCs administered, level of differentiation once in CNS, the degree of secretion of the lysosomal enzyme by the donor HSCs, and also the properties of the lysosomal enzyme to be uptaken by the recipient neural cells. The sum of these factors will determine the outcome, which is the arrest and/or improvement of neurological manifestations of LSDs. In addition, in the setting of a generalized lysosomal dysfunction, the extracellular microenvironment is altered by the presence of a number of cytokines and inflammatory response cells, which also contribute substantially to the pathogenesis of the neurodegeneration observed in LSDs (Walkley, 2009; Platt et al., 2018). If successful, with full engraftment, the HSCT can single-intervention, offering a lifelong source of secretion of “deficient' enzyme” for the recipient patient. However, the physiological demands of lysosomal levels may change during the natural history of the LSDs. The donor HSCs are allogenic donors either from family-related members, usually siblings. Given the setting of early diagnosis and also with the newborn screen for MPS-I and Krabbe disease, the Umbilical cord blood (UCB) as an alternative stem cell source has been used in HSCT (Lund, 2018). The UCB is a safe and effective source of HSCs.
In terms of limitations, only in MPS-I (iduronate deficiency), the HSCT procedure has shown to be efficacious to prevent and attenuate the neurological manifestations of the neuronopathic forms (Ballen et al., 2013). The numbers of umbilical HSCs are usually lower than those from bone marrow sources. In pediatric patients, the UCB source can provide desired doses of HSCs number/kilogram, whereas, adults undergoing HCT may suffer from inadequate cell numbers (Ballen et al., 2013). As in an adult setting, the UBC as a source of HSCs becomes limited, several approaches have been proposed, including combined infusion of 2 UCB units (Barker et al., 2001), increasing the homing efficiency of UCB cells (Cutler et al., 2013), and expanding the UCB in vitro (Klionsky et al., 2016). Recently, small molecule, carlecortemcel-L, a copper chelator known for its ability to expand UCB HSCs in vitro (Peled et al., 2004), allowed a selected CD133+ cell expansion post-selection from UCB units, followed by a 21-days expansion with cytokines and carlecortemcel-L in vitro (Stiff et al., 2018).
In sum, the significant advantages of the HSCT are dependent upon a number of factors, and a higher concentration of enzymes delivered to the CNS will result in better clinical outcomes. Therefore, it is essential that HSCs source, either the UCT- or BM-derived, to be screened for the carrier status of the specific lysosomal enzyme deficiency diagnosed in the recipient patient.
Since 1980, several patients with LSDs have been treated and a wide spectrum of outcomes reported. At present, HSCT is highly recommended for patients with MPS-I if they are younger than 24 months, as per the European Union (Clarke et al., 2017), and 36-months, as per US guidelines (De Ru et al., 2011). The MPS-I is the only LSD in which full prevention of the neuropathogenic processes is observed if the HSCT is done before the neurocognitive impairment arises (Aldenhoven et al., 2015). In addition, HSCT has been beneficial for patients with metachromatic leukodystrophy (MLD) and Krabbe disease if the transplantation is performed at the presymptomatic stage (Escolar et al., 2005; Mcgraw et al., 2005; Cartier and Aubourg, 2008; Duffner et al., 2009; Krageloh-Mann et al., 2013; Tejera et al., 2013; Groeschel et al., 2016; Van Rappard et al., 2016; Allewelt et al., 2018; Kwon et al., 2018). However, cognitive decline and neurodegenerative disease are observed in a number with early-onset Tay-Sachs disease and Sandhoff disease (Boelens et al., 2014), MPS-III and MPS-II (Mckinnis et al., 1996; Guffon et al., 2009; Muenzer et al., 2009a; Scarpa et al., 2011), who underwent HSCT. Recently, in preclinical studies, encouraging results of human neural cells in several LSD murine models, including Niemann-Pick A disease (sphingomyelinase deficiency) (Shihabuddin et al., 2004), infantile neuronal ceroid lipofuscinosis (Tamaki et al., 2009) and MLD (Biffi et al., 2004; Givogri et al., 2006). The substantial attenuation of the neuropathology in the CNS of murine models should be reproduced in a larger brain before moving this therapeutic strategy into the clinic. In addition, a well-characterized and non-tumorigenic human-derived source of stem cells will be needed.
In general, several factors, including age at diagnosis, and HSCT, the clinical status, levels of chimerism, and specific genotype can also be determinants of the final clinical outcome for each patient undergoing this therapeutic modality (Biffi, 2017b). However, mortality has been reduced to approximately 10%, with variable rates for different lysosomal disorders (Prasad and Kurtzberg, 2008; Biffi, 2017b).
CSF-Delivery of ERT Agents
Currently, ERT is considered an effective treatment for non-neurological manifestations of LSDs. It is conceivable that, to circumvent the BBB, ERT agents directly delivered into the cerebrospinal fluid (CSF) can achieve the neural cells. Several routes can be used, including intracerebroventricular (ICV) injection into the lateral ventricle (through a catheter/reservoir) or intrathecal (IT) injection into the lumbar spine or subarachnoid space at the cisterna magna lumbar puncture or an IT drug-delivery device. The first ERT agents to be CSF injected were laronidase (Aldurazyme) in a patient with MPS I- with cervical spinal cord compression (SCC), and unable to undergo necessary neurosurgery procedures (Munoz-Rojas et al., 2008). Another patient received the galsulfase ERT agent for MPS-VI (Bernal-Bayard et al., 2010). The IT-administered ERT was well-tolerated and safe, and some improvement in symptoms of SCC was observed (Klionsky et al., 2016). Later on, three-phase 1/2 studies investigating the safety (as primary outcome) and efficacy of CSF-delivery through IT injections for MPS-I, MPS-II, and MPS-III were performed (Klionsky et al., 2016; Muenzer et al., 2016; Nestrasil et al., 2017). One patient with attenuated MPS-I patient after receiving IT ERT showed signs of neurological benefits, improving brain structure, and reversing cognitive decline (Nestrasil et al., 2017). In addition, IT ERT was also assessed in conjunction with HSCT. Twenty-four patients received over 1–2 min in 4 mL of Elliott's B® solution containing recombinant ERT agent iduronidase (0.05 mg/kg) at four discrete time points: 8–12 weeks before HSCT, 2 weeks before HSCT, 100 days after HSCT, and 6 months after HSCT (Eisengart et al., 2019). All of the patients who demonstrated increases in IQ 2 years after treatment had at least a 50% reduction in CSF glycosaminoglycan non-reducing ends across the four-timepoints (Eisengart et al., 2019). The most notable limitation of this study is a lack of a comparison CSF from a group that received only intravenous ERT and HCT. In 2017, cerliponase alfa (Brineura®) was approved-based on the clinical trials showing to slow loss of ability to walk or crawl (ambulation) in symptomatic pediatric patients 3 years of age and older with late infantile neuronal ceroid lipofuscinosis type 2 (CLN2), also known as tripeptidyl peptidase 1 (TPP1) deficiency. In a clinical study of 24 patients with CLN2 receiving an intraventricular infusion of cerliponase alfa, the slope of decline in motor and language function was less than the one observed in the historical controls. Serious adverse events included the failure of the intraventricular device and device-related infections (Schulz et al., 2018).
In terms of immunogenicity, in the majority of patients who had intravenous ERT at least 6 months preceding the IT administration, no immune response or antibody was present in the CSF (Klionsky et al., 2016)]. The patients who developed antibodies against the ERT agents in the CSF did before the IT-ERT administration, and no safety issues were a concern. It is important to point out that significant ERT agent leakage occurs to the systemic circulation during IT-ERT administrations (Chung et al., 2017). For this reason, CSF-administered ERT agents can eventually result in systemic effects in non-CNS areas of patients receiving only this route as ERT administration.
Gene-Therapy
The LSDs, as single-gene disorders with well-characterized biochemical and clinical phenotypes, are ideal candidates for gene therapy approaches. Also, the critical threshold of 10–15% of residual enzymatic activity for most lysosomal enzymes, below which no disease is manifested (Conzelmann and Sandhoff, 1983; Sandhoff and Conzelmann, 1984), has shown that only a small percentage of correction is required to reinstate the physiological turnover of the primary substrate and shift the cell homeostasis (Conzelmann and Sandhoff, 1991; Schueler et al., 2004). The gene therapy approach can be accomplished in two general ways, in vivo and ex vivo, which are both discussed below.
In vivo Gene Therapy
The gene therapy (GT) approach has made significant advances in several animal models of LSDs (Platt, 2018). Based on the encouraging pre-clinical studies in animal models, early phase clinical trials have started and shown positive results. A number of approaches have been developed to administer GT agents to target the CNS. Most of them are based on the direct administration of recombinant adeno-associated vectors (rAAVs) administered systemically (mostly intravenously) or locally injected (CNS). The rAAVs are derived from small, non-enveloped, and non-integrating AAVs. The transduction efficiency and safety characteristics make the rAAVs the ideal candidate for gene delivery (Bey et al., 2017). The rAAV serotypes traditionally used in gene-transferring into CNS are the 1, 2, 5, 8, 9, and the recombinant human (rh)10. Usually, rAAV-mediated gene transfer in the brain efficiently targets neurons and scarcely astrocytes, oligodendrocytes, and microglia (Hocquemiller et al., 2016). Specific rAAV serotypes, including AAV9 and 10, can cross the BBB, allowing transduction of the CNS after systemic administration (Foust et al., 2009; Hocquemiller et al., 2016). As issues raised on the CSF-injected ERT, because of leakage, local rAAV administration into the CNS results in transduction of non-target tissues and peripheral transgene expression, particularly in the liver (Hocquemiller et al., 2016). The leakage may trigger immune responses and can mitigate the efficacy of the in vivo GT. Transient immunosuppression as induction of tolerance has been developed (Colella et al., 2018; Keeler et al., 2019). In particular, relevant results have been obtained with strategies inducing liver-mediated tolerance or neonatal AAV-mediated systemic expression of a therapeutic protein before CNS-directed in vivo GT (Hinderer et al., 2015). Based on this and other preclinical studies (Fraldi et al., 2007), a phase I/II clinical trial was initiated in patients affected by MPS type IIIA. In this study, four children aged between 2 years 8 months and 6 years received intracerebral injections of an adeno-associated virus (AAV) vector carrying the complementary DNA encoding sulfamidase and the gene for the sulfatase-modifying factor SUMF1 (Tardieu et al., 2014). The therapeutic vector was administered to the brain by stereotaxic surgery. The immunosuppressive regimen given 2 weeks before the procedure was generally well-tolerated. In two patients, the brain magnetic resonance imaging (MRI) showed brain size and neuropsychological tests were performed. The brain atrophy seemed to be stable, but tended to increase in the other two individuals. In three other patients, moderate improvements in neurocognitive areas, including behavior, attention, and sleep, were noted in neuropsychological evaluations (Tardieu et al., 2014; Marco et al., 2019). Intracerebral injections of adeno-associated viruses carrying a therapeutic gene have also been performed or are under evaluation in other LSDs and neurodegenerative diseases (Hocquemiller et al., 2016).
Ex vivo Therapy
As we described earlier, the BBB prevents circulating molecules, either small or macromolecules, from entering the CNS (Pardridge, 2007b). Hematopoietic cells permeate the BBB, where they subsequently differentiate into microglia. The HSCT in neurological LSDs may be beneficial but is associated with significant risks of GVHD host and other complications that range from 8 to 10%, even in the most advanced centers (Graf, 2017; Tan et al., 2019). In this setting, to circumvent the risks associated with the allogenic HSCT, autologous HSCs becomes attractive as it opens the opportunity to genetically modify the patient's HSCs to express the deficient lysosomal protein. The lentiviral vectors (LVs) showed strong safety and the most efficient tool to deliver genes to HSCs (Magrin et al., 2019). The lentiviral genome is stably integrated into the host cell genome after transduction, allowing a long-term and stable transgene expression over time and in the differentiated progeny of the original transduced HSPCs (Naldini, 2019). In general, to inject the lenti-transduced HSCs, a partial or full myeloablative bone marrow will be required to allow “space” and promote the novel HSCs engraftment (Biffi, 2017b; Naldini, 2019). One successful ex vivo gene therapy targeting CNS has been reported in MLD, an LSD caused by arylsulfatase A (ASA) deficiency and manifested by a severely progressive demyelination of CNS and peripheral nervous system. Based on previous studies (Lattanzi et al., 2010; Meneghini et al., 2016), nine children asymptomatically diagnosed with MLD underwent haemopoietic stem-cell gene therapy (HSC-GT) in a non-randomized, open-label, single-arm phase 1/2 trial with 3-years follow-up (Sessa et al., 2016). Initial data of some of the first subjects enrolled were published earlier (Biffi et al., 2013). After reinfusion of the genetically modified cells, high enzyme activity was found throughout haematopoietic lineages and in the cerebrospinal fluid. Eight patients, seven of whom received treatment when presymptomatic, had prevention of disease onset or halted disease progression as per clinical and instrumental assessment, compared with historical untreated control patients with early-onset disease. Gross Motor Function Measures scores for six patients up to the last follow-up showed that gross motor performance was similar to that of normally developing children. The extent of benefit appeared to be influenced by the interval between HSC-GT and the expected time of disease onset (Sessa et al., 2016). The ad-hoc results showed evidence of the safety and therapeutic benefit of ex vivo gene therapy for a neurological LSD when administered at presymptomatic or very early-symptomatic disease stages. Despite these promising results, the long-term safety and efficacy of the approach are needed to assure options for patients affected MLD and other LSDs (Sessa et al., 2016).
Nano-Vesicle-Based Delivery of Therapies for LSDs
Since the BBB forbids the translocation of large proteins into the CNS, only certain small low-molecular-weight or lipophilic molecules can pass through this natural barrier. Several types of nanovesicle-based delivery technologies have been designed to transport therapeutic agents to CNS to allow BBB delivery and membrane permeability. Amongst these approaches, polymeric nanoparticles (NPs), lipid NPs, liposomes, as well as extracellular vesicles (EVs), such as exosomes, produced by cells can be used to transport bioactive enzymes and other types of therapeutic molecules to CNS (Figure 2, Table 3). The specific characteristics that allow delivery through BBB are the size of vesicles/ NPs, their lipophilic composition, and the ability to carry a high payload of the chosen therapeutic agent. The modifications of these nanomaterials with specific ligands can open the possibility to target the nanocarriers to a specific tissue, cell, and even an organelle. In addition, these nanosystems offer a protective niche for the delivered cargo, due to increased physicochemical and biological stability of encapsulated and carried products. Intravenously injected enzymes avoid being exposed to blood and reach the body organs more efficiently if these molecules can be protected by the nanocarriers. Moreover, the delivery of therapeutic molecules to organs of interest in a more targeted manner can make the delivery more effective and control the half-life of the delivered enzymes. The qualities of these nanosystems also provide improved bioavailability, and many of these vehicles of delivery are non-toxic, biocompatible, and biodegradable (Onaca-Fischer et al., 2012; Pirooznia et al., 2012; Del Grosso et al., 2019).
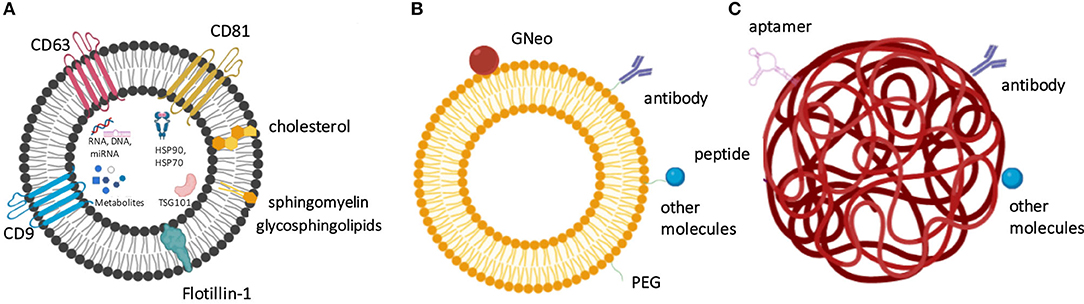
Figure 2. The nanocarrier-based delivery systems proposed for the treatment of lysosomal storage diseases include exosomes (A), liposomes (B), and nanoparticles (C). (A) Exosomes as physiological extracellular vesicles produced by cells and contain specific protein- and lipid-based markers and also carry various metabolites, nucleic acids and proteins as cargo. (B) Liposome surface can be derivatized to contain targeting molecules, such as antibodies, GNeo, and other molecules, which are often attached to the liposomes after prior coating with polyethylene glycol (PEG). (C) Nanoparticles, such as here shown polymeric nanoparticle can be modified by the presence of peptides, aptamers, antibodies, and other molecules to increase their targeting efficiency.
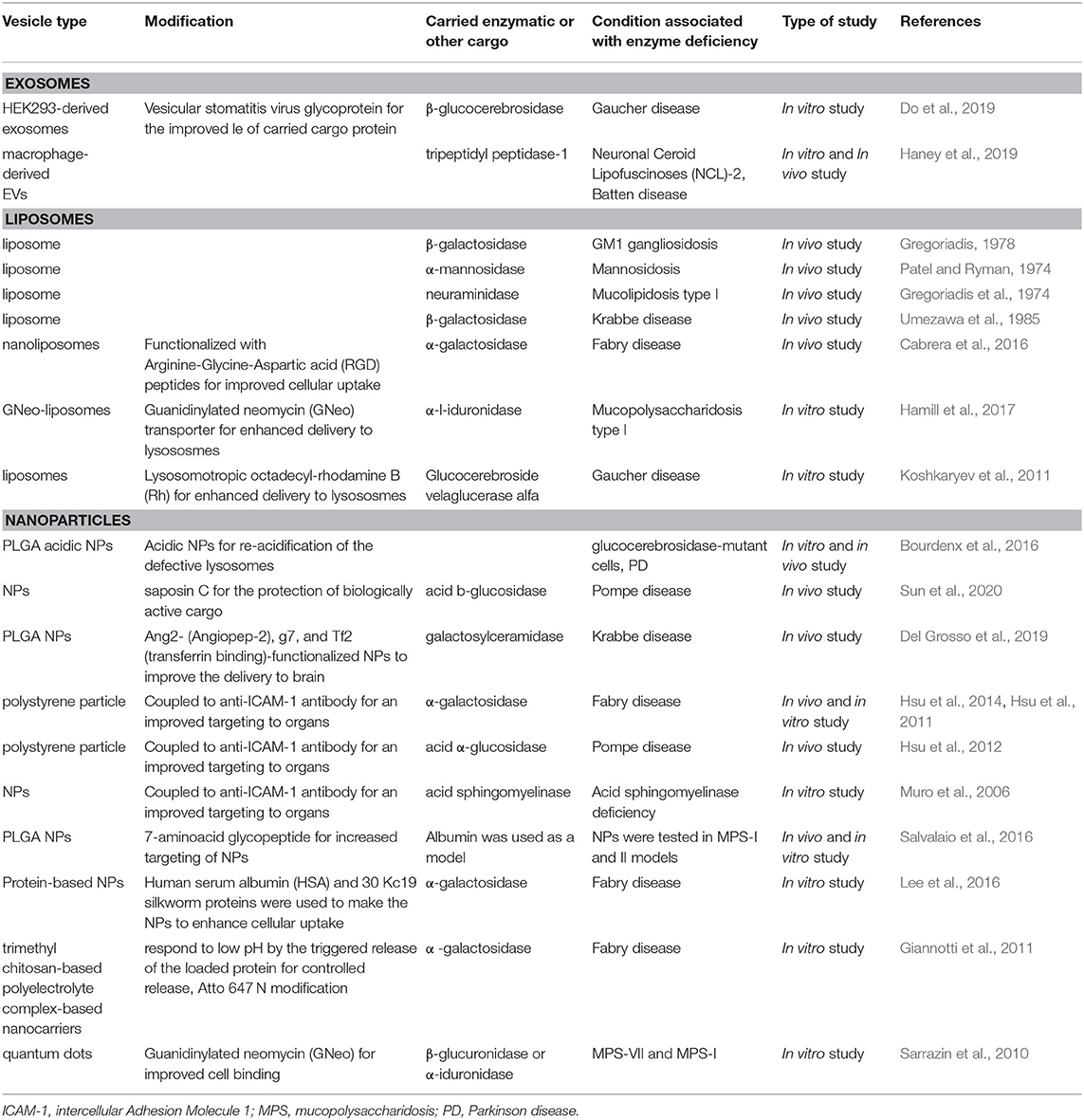
Table 3. Vesicle-based delivery systems of therapeutic molecules for the treatment of LSDs.
The EV-Mediated Delivery of Therapied to Target CNS
The heterogeneity of EVs
All eukaryotic cells secrete extracellular vesicles (EVs), which vary in function and characteristics, but they all carry specific molecules from the cell of origin to the target cells, thus performing essential roles in the cell-to-cell communication and homeostasis. EVs are a heterogeneous population of vesicles that vary in size, biogenesis, and protein markers (Raposo and Stoorvogel, 2013). The subpopulation of EVs include exosomes, microvesicles (MVs), apoptotic bodies (APOs), oncosomes (Di Vizio et al., 2009; Minciacchi et al., 2015), and exomeres (Nolte-'T Hoen et al., 2012). In particular, exosomes are small EVs, which have a size a range of 30–120 nm (Théry et al., 2002), contain tetraspanin-based biomarkers CD9, CD63, or CD81 (Raposo and Stoorvogel, 2013), and have unique biogenesis originating in the endosomal pathway (Raposo and Stoorvogel, 2013). Exosomes are capable of carrying and transmitting protein and RNA cargo, but also specific lipids and small molecules (Rajendran et al., 2006; Alvarez-Erviti et al., 2011a; Noerholm et al., 2012). This exosomal cargo can be subsequently transmitted to other cells. Because exosomes travel in physiological fluids, these vesicles can reach cells in distant organs to transmit their contents from the cells of origin (Alvarez-Erviti et al., 2011b). Since exosomes are formed by all known cell types, not surprisingly, they are also present in the CNS. Within the brain, exosomes have specific functions regulating the interactions between cells, such as neurons and glial cells, for instance by mediating and facilitating the delivery of bioactive molecules from one cell to another (Frühbeis et al., 2012; Zhang and Yang, 2018).
Brain-homing signals for exosomes
Although exosomes contain some molecules on their surface that allow them to reach specific organs, engineered exosomes containing modified membrane decorations that result in improved targeted delivery. One of the ways by which exosomes can be engineered to possess increased targeting capabilities is by a fusion of targeting peptides to the extracellular region of proteins displayed on naïve exosomes. One of such proteins with an extracellular N-terminus is Lamp2b (Alvarez-Erviti et al., 2011b), which is abundantly present on exosomal membranes. Lamp2b molecule can then fused with a peptide utilized for the targeting of specific organs (Alvarez-Erviti et al., 2011b). For the CNS-specific deliveries, rabies viral glycoprotein (RVG) peptide (YTIWMPENPRPGTPCDIFTNSRGKRASNG) can be used, which specifically binds to the acetylcholine receptor 3. Dendritic cells-derived exosomes engineered to express an exosomal membrane protein, Lamp2b, which was fused to the neuron-specific RVG (rabies virus glycoprotein) peptide were used to deliver exosomes to the brain by intravenous injections. These exosomes containing this siRNA cargo were introduced by electroporation. By using this model, exosomes were able to deliver the cargo to neurons, microglia, and oligodendrocytes in the brain, where the carried siRNA resulted in a gene knockdown (Alvarez-Erviti et al., 2011b). The RVG peptide was also used by other groups for exosome homing to the brain (Yang et al., 2017). Another type of peptide that can be fused with Lamp2b to target exosomes to the brain is T7 peptide. Antisense miRNA oligonucleotides against miR-21 (AMO-21) with potential for the treatment of glioblastoma were delivered by exosomes decorated with Lamp2b-fused T7 peptide since the transferrin receptor is overexpressed on the surface of glioblastoma cells. T7-exosomes had a higher delivery efficiency to glioblastoma cells in comparison to the unmodified exosomes or RVG-decorated exosomes in vitro. Similar observations were obtained in vivo, where the delivery of AMO-21 by exosomes via an intravenous injection led to a reduction in the miR-21 levels, followed by the reduction of tumor size (Kim et al., 2020). Finally, muscle-specific peptide (MSP) identified by in vivo phage display (ASSLNIA) can also be fused with Lamp2b for muscle-specific delivery of exosomes (Alvarez-Erviti et al., 2011b).
Exploring Therapeutic EVs to Treat Disorders Affecting CNS
Although exosomes were evaluated in terms of the therapeutic function in such CNS conditions as glioblastoma (Kim et al., 2020), these and other natural nanovesicles have not been extensively characterized as delivery vehicles of therapeutic agents for LSD. A single study focused on exosome-based delivery of a lysosomal enzyme for LSD treatment, in which lysosomal β-glucocerebrosidase (GBA) was fused to a protein that anchored it to the exosomes, termed vesicular stomatitis virus glycoprotein (VSVG), without altering the protein function, but significantly increasing the levels of β-glucocerebrosidase. This is an evidence that the lysosomal the enzyme payloads within exosomes are active. The β-glucocerebrosidase enclosed within these HEK293-derived exosomes was taken up by HEK293 recipient cells by the endocytosis pathway. Finally, such obtained exosomes were explicitly targeted to endocytic compartments and increased GBA activity in the recipient cells (Do et al., 2019).
Macrophage-derived EVs were also used for brain delivery of a soluble lysosomal enzyme tripeptidyl peptidase-1, TPP1, for the treatment of Neuronal Ceroid Lipofuscinoses 2 (CLN2) or Batten disease (Haney et al., 2019). Because macrophages are often associated with inflamed tissue, macrophage derived EVs were hypothesized to interact with inflamed tissues for enhanced targeted delivery of the therapeutic protein. In this study, TPP1-encoding plasmid DNA was transfected into macrophages, or TPP1 protein was packed into the empty EVs by sonication or permeabilization with saponin, and all these methods yielded proper incorporation of functional TPP1 into the vesicles. EVs also increased the stability of TPP1 and were able to deliver the protein to the recipient cells, where over 70% of the protein was localized to lysosomes. TPP1-containing EVs were also administered intraperitoneally and led to the accumulation of TPP1-EVs in brain tissue, improving the lifespan of the late-infantile neuronal ceroid lipofuscinosis mouse (Haney et al., 2019).
Finally, companies such as Evox Therapeutics have an interest in applying exosome-based deliveries for the improved treatment of Niemann-Pick disease type C (Zipkin, 2019). The exosomes produced by Evox Therapeutics carry a copy of the NPC protein to the neural cells by using surface molecules that bind certain, yet undisclosed, ligands (Hean et al., 2019). The EV-based therapies are promising delivery systems for the proteins, which carry large payloads of cargo, and also vectors which are used for gene therapy-based therapeutic approaches.
Liposome-Based Delivery of LSD Therapeutics
Liposomal drug delivery systems composed of biocompatible nanomaterials enhance the efficacy of ERT since these nanostructures encapsulate and protect the enzymes in biodegradable micelles. Liposomes are spherical vesicles that contain one or more lipid bilayers, and their hydrophilic character is ideal for efficient drug delivery. The bilayer components affect the properties of liposomes, including the rigidity, charge, and fluidity of the bilayer. Unsaturated phosphatidylcholine lipids obtained from natural sources can be used to manufacture liposomes, but these species of lipids increase the permeability and negatively affect the stability of these vesicles. On the other hand, saturated phospholipids containing longer acyl chains are more rigid and impermeable (Akbarzadeh et al., 2013). ERT utilizing liposomes that contain enzymes which are deficient in LDS were already proposed in the 1970s and 1980s (Patel and Ryman, 1974, Gregoriadis et al., 1974, Steger and Desnick, 1977, Gregoriadis, 1978, Umezawa et al., 1985). However, in these early studies, liposomes often failed to specifically localize the enzyme of interest to the brain (Umezawa et al., 1985). Since then, considerable effort has been made to improve the targeted delivery of therapeutic proteins to CNS by liposomes.
First, liposomes can be modified to contain homing signals to enhance liposome-mediated transport to specific organs. Immunoliposomes—or antibody-directed liposomes—are sterically stabilized with PEG, which is conjugated to a linker lipid, allowing for conjugation of thiolated antibody on the surface of the vesicle. The monoclonal antibodies are used as modifiers attached to the surface of such liposomes. Examples of antibodies that were previously used to direct the liposomes to the brain are the OX26 antibody recognizing the transferrin receptor, which increased the uptake of liposomes to rat brain and specifically to the microvascular endothelium that builds the blood-brain barrier (Huwyler et al., 1996).
Second, the liposome modification can increase the uptake of liposomes by cells. Here, modification of the liposomal surface with a peptide has enhanced the intracellular penetration of liposomes. Also, nanoliposomes functionalized with Arginine-Glycine-Aspartic acid (RGD) peptides containing α -galactosidase-a, had an enhanced enzymatic activity and improved intracellular penetration (Cabrera et al., 2016). Proteins such as apolipoprotein E can be added on the surface of liposomes for more efficient delivery of receptor-mediated uptake of the liposomes to the cells (Ansari et al., 1997).
To further improve the delivery of liposomes to lysosomes, guanidinylated neomycin (GNeo) transporter can be used. GNeo is capable of transport of bioactive cargo inside the cells, which can also be of high molecular weight. GNeo works by a process utilizing heparan sulfate proteoglycans in the cell surface. The synthesis of GNeo-lipids, which were then incorporated into the liposomes, termed GNeo-liposomes, or GNeosomes, was able to enhance the specific delivery of these particles to lysosomes, which encapsulated α-l-iduronidase (IDUA). These GNeosomes successfully restored the function in fibroblasts isolated from a patient that lacked this enzyme due to the lysosomal storage disorder mucopolysaccharidosis type I (MPS I) (Hamill et al., 2017). Finally, lysosomotropic octadecyl-rhodamine B (Rh) (Koshkaryev et al., 2011) was used to increase the delivery of the encapsulated glucocerebroside velaglucerase-α to the lysosomes in the Gaucher's fibroblasts, where a 68% increase in the targeted delivery was observed in contrast to liposomes that were not modified with the Rh (Thekkedath et al., 2013).
NP-Based Delivery of Therapeutic Molecules for the Treatment of LDS
Another particle type that can be used for the delivery is polymeric NP. Poly-(lactide-co-glycolide), PLGA is one of the preferred materials used for the production of NPs, which is a non-toxic material and thus offers increased biocompatibility (Patel et al., 2012). PLGA has been approved by the Food and Drug Administration for administration in humans. As an example, the catalytic activity of lipase was improved by immobilization of this enzyme onto a porous super magnetic polymeric microsphere carrier. The glycerol-oleic-acid-esterification activity of the immobilized lipase increased seven fold in comparison with the naked enzyme. Whereas, the activity normalized to the total protein content of the immobilized enzyme was improved by 200-fold, and the stability of the lipase was also improved by the immobilization of this enzyme to the NPs (Meng et al., 2013).
The targeting function of NPs can be enhanced by the NP modifications, where peptides, antibodies, and aptamers can be added to the surface of these structures. As an example, Ang2- (Angiopep-2) (Demeule et al., 2008), g7 (Tosi et al., 2011), and Tf2 (transferrin binding)-functionalized (Santi et al., 2017). PLGA NPs containing cross-linked enzyme aggregates were tested in the encapsulation efficiency, activity yield of encapsulated enzymes, and targeting to the brain if delivered intraperitoneally to a TWI murine model of Krabbe disease (Del Grosso et al., 2019). These NPs were able to promote recovery in the brain up to the control mice level. Specifically, the enzyme-delivering NPs lead to significant GALC activity in the organs, where both targeted and control NPs accumulated in the liver and kidneys 4 h after injection. However, only the targeted NPs were detected in the brain, displaying a GALC activity that was comparable to the activity of the heterozygous mice for GALC, which is identical to the wild-type mouse phenotype.
In contrast, the non-targeted NPs did not show any significant GALC activity increase in the brain tissues (Del Grosso et al., 2019).
Another modification of NPs is antibody coupling. One of the commonly targeted molecules is ICAM-1, which is an endothelial surface protein upregulated in many LSD pathologies, such as inflammation or metabolic imbalance (Springer, 1994). Polystyrene and PLGA polymer nanocarriers targeting ICAM-1, delivered by intravenous injection to mice, were reported not to cause lung injury or have other adverse effects, such as abnormal vascular permeability. These NPs were rapidly removed from the circulation and accumulated in various organs such as the kidney, heart, liver, spleen, or lung (Garnacho et al., 2008). Polystyrene particles coupled to an antibody specific to ICAM-1 were then used to improve targeting of α-galactosidase (αGal), which is an enzyme affected in the Fabry disease. While naked αGal intravenously delivered to animals preferentially remained in circulation and not up-taken by tissues and organs, αGal coupled to nanocarriers accumulated in the brain, kidneys, heart, liver, lungs, and spleen (Hsu et al., 2014). The nanocarriers also specifically were targeted to lysosomes and led to an enhanced globotriaosylceramide degradation, thus constituting possible improvement of drug delivery for Fabry disease (Hsu et al., 2011). The same ICAM-1 molecule was targeted for the delivery of ERT to treat Pompe disease caused by a deficiency of acid α-glucosidase (GAA). GAA was coupled to ~180 nm polystyrene bead-based nanocarriers coated with anti-ICAM-1, and this enhanced targeting of anti-ICAM/GAA NCs to organs in comparison to the naked enzyme delivery (Hsu et al., 2012). Anti-ICAM-1-coated nanocarriers have also been used to target recombinant proteins, such as human acid sphingomyelinase to ICAM-1-positive cells, including activated endothelial cells and Niemann–Pick disease patient fibroblasts. The sphingomyelinase delivery by nanocarriers was enabled by the CAM-mediated endocytosis pathway, which is distinct from the clathrin-dependent endocytosis. As a result, the bioactive proteins were successfully delivered to lysosomes, leading to a decreased accumulation of lysosomal lipids (Muro et al., 2006).
Peptides are another class of modifications that can be added to NPs to enhance their targeting to brain regions. A 7-aminoacid glycopeptide (g7) was used to decorate PLGA NPs, which were then tested in MPS I and MPS II models. The g7-modified PLGA NPs successfully delivered high molecular weight molecules across the BBB in a murine model, where a model molecule FITC-albumin was used as an example (Salvalaio et al., 2016).
Human serum albumin (HSA) and 30Kc19 silkworm protein were tested as an enhancement strategy for the delivery of α-galactosidase. HSA was used because this protein is an FDA-approved molecule, and it binds to receptors within the caveolae while the 30Kc19 silkworm protein enhances cellular uptake and stabilizes the cargo (Park et al., 2012, 2014). Using primary cultured fibroblast, 30Kc19-HSA NPs led to enhanced cellular uptake of the carried protein. The α-galactosidase carried by these NPs had improved globotriaosylceramide degradation in the fibroblasts (Lee et al., 2016).
Controlled release of proteins to the lysosomes can also be achieved by functional polyelectrolyte-based NPs, which respond to low pH by the triggered release of the loaded protein. In one study, trimethyl chitosan (TMC)-based polyelectrolyte complexes (PECs) nanocarriers were used to deliver α-Gal. These NPs released the enzyme at acidic pH, and upon their further functionalization with fluorescent group Atto 647N, PECs were shown to be further internalized by human endothelial cells and enriched in the lysosomes (Giannotti et al., 2011).
Apart from transferring molecules, NPs can be used to lower the pH within lysosomes. PLGA acidic NPs, which were transported to lysosomes, were able to restore impaired lysosomal function glucocerebrosidase-mutant cells by inducing re-acidification of the defective lysosomes. The same PLGA-acidic NP was localized to neurons following the intracerebral injection, where they were able to attenuate the neurodegeneration in vivo in the murine model of PD by rescuing the lysosomal dysfunction (Bourdenx et al., 2016).
Other modification to the nanovesicles include the addition of stabilization agents. Saposin C (SapC) is a lysosomal glycoprotein which naturally protects GCase from the degradation (Sun et al., 2003). The CNS-selective delivery system based on nanovesicles of saposin C (SapC) and dioleoylphosphatidylserine (DOPS) was used to deliver acid b-glucosidase (GCase) via the BBB. The enclosure of the enzyme within the nanovesicles improved the stability and activity of the GCase and mediated the uptake of the enzyme by a mannose receptor-independent pathway. SapC-DOPS-GCase-containing nanovesicles were able to penetrate through BBB via the surface phosphatidylserine. These vesicles also led to a reduction of GCase substrate levels, attenuating brain inflammation (Sun et al., 2020).
Quantum Dots
One more category of the nanocarriers worth mentioning are quantum dots, also known as “artificial atoms,” which are semiconductor-based nanocrystals in a size range of 2–10 nm. Guanidinylated neomycin (GNeo), which targets liposomes to lysosomes (Hamill et al., 2017), as described above, was also used to modify quantum dots, which were able to undergo endocytosis and translocate the lysosomes. The activated ester of GNeo was conjugated to β-glucuronidase or α–iduronidase enzymes, which did not affect enzymatic activity. This GNeo modification enabled efficient binding of the heparan sulfate on primary human fibroblast cells, where the GNeo quantum dots containing the enzyme led to a restoration of glycosaminoglycan turnover in the cells (Sarrazin et al., 2010).
Conclusions and Future Directions
The mainstay therapy for LSDs, ERT, and current small molecules functioning as SRT and PC agents have a very limited biodistribution to CNS, PNS, and even other organs including bone, skeletal and cardiac muscle and connective tissues (Hollak and Wijburg, 2014). A summary of the current therapies targeting the CNS are described in Table 2. In the majority of LSDs, the predominant clinical manifestations are neurological in origin and associated with progressive morbidity. Therefore, there is an urgent and unmet need for therapies to the CNS, tackling the primary defect (enzyme deficiency/substrate build-up), and the multiple secondary pathogenic cascades (Pastores and Maegawa, 2013; Giugliani et al., 2018; Platt, 2018; Platt et al., 2018). Despite remarkable advances in the understanding of neuropathogenesis of these disorders, the CNS impairment remains the major cause of disabilities, hospitalizations, and the requirement of prolonged and complex care for the affected patients. Cognizance of this unmet therapeutic need and challenges imposed by the BBB, several therapeutic modalities have been investigated, including modified ERT agents, novel small molecules, HSCT, gene therapy and innovative therapeutic-CNS delivery strategies. The advances in combinatorial chemistry, nanomolecule and chemical synthesis allowed the expansion of the chemical space, resulting in novel and unique molecules capable of permeating the BBB and tackling the neurodegenerative processes in the CNS. A better understanding of exosomes and other nanovesicles as carriers of signaling metabolites will allow us to explore the therapeutic aspect of these nanovesicles (O'loughlin et al., 2012; Van Niel et al., 2018). The advances in, now “micro,” robotic technologies, high-resolution microscopy will offer multiple and concomitant readouts in high-through screening (HTS) assays, generating more disease-relevant and precise and “hits.”(O'loughlin et al., 2012; Van Niel et al., 2018). In addition, the “tissue-chips” or “microphysiological systems,” bioengineered microsystems capable of recreating aspects of human organ physiology and function, will accelerate the validation and prioritization of the small molecule “hits” against a specific target and its mutant variants (Low et al., 2020). With the advances of mass spectrometry and next-generation sequencing (NGS), allowing both newborn screen programs and early diagnosis, respectively, an increasing number of individuals are being diagnosed at early or presymptomatic disease stages. In this setting, diverse modalities and more tailored and individualized therapeutics will be required to treat and, eventually prevent, the lysosomal, cellular, and organ damages, often irreversible by the time of diagnosis. In sum, applying the basic understanding of the neuropathogenesis to develop safer and target therapies, the prevention of the LSD manifestations throughout the life span of early diagnosed individuals, who maybe “apparently healthy,” will be achieved.
Author Contributions
GM and ME contributed to the design, writing, and review of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work was funded by NIH/NINDS: R21NS113649-01 (GM); R03AI135610-01A1 (ME).
Conflict of Interest
GM has received research funding from NINDS/NIH, U.S. Department of Defense, Pfizer Inc., Moderna Inc., Protalix Pharm. and also non-profit foundations as The Legacy of Angels Foundation. GM has received honorarium for consulting for Genzyme-Sanofi.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Acosta, W., and Cramer, C. L. (2020). Targeting macromolecules to CNS and other hard-to-treat organs using lectin-mediated delivery. Int. J. Mol. Sci. 21:971. doi: 10.3390/ijms21030971
Aflaki, E., Borger, D. K., Moaven, N., Stubblefield, B. K., Rogers, S. A., Patnaik, S., et al. (2016). A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with gaucher disease and parkinsonism. J. Neurosci. 36, 7441–7452. doi: 10.1523/JNEUROSCI.0636-16.2016
Akbarzadeh, A., Rezaei-Sadabady, R., Davaran, S., Joo, S. W., Zarghami, N., Hanifehpour, Y., et al. (2013). Liposome: classification, preparation, and applications. Nanoscale Res. Lett. 8:102. doi: 10.1186/1556-276X-8-102
Aldenhoven, M., Wynn, R. F., Orchard, P. J., O'meara, A., Veys, P., Fischer, A., et al. (2015). Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood 125, 2164–2172. doi: 10.1182/blood-2014-11-608075
Alderson, L. M., Joksaite, S. X., Kemp, J., Main, E., Watson, T., Platt, F. M., et al. (2019). Age-related gait standards for healthy children and young people: the GOS-ICH paediatric gait centiles. Arch. Dis. Child. 104, 755–760. doi: 10.1136/archdischild-2018-316311
Allewelt, H., Taskindoust, M., Troy, J., Page, K., Wood, S., Parikh, S., et al. (2018). Long-term functional outcomes after hematopoietic stem cell transplant for early infantile krabbe disease. Biol. Blood Marrow Transplant. 24, 2233–2238. doi: 10.1016/j.bbmt.2018.06.020
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J., et al. (2011a). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S., and Wood, M. J. (2011b). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 29, 341–345. doi: 10.1038/nbt.1807
Ansari, N. H., He, Q., Cook, J. D., Wen, J., and Srivastava, S. K. (1997). Delivery of liposome-sequestered hydrophobic proteins to lysosomes of normal and Batten disease cells. J. Neurosci. Res. 47, 341–347. doi: 10.1002/(SICI)1097-4547(19970201)47:3<341::AID-JNR12>3.0.CO;2-4
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Ballen, K. K., Gluckman, E., and Broxmeyer, H. E. (2013). Umbilical cord blood transplantation: the first 25 years and beyond. Blood 122, 491–498. doi: 10.1182/blood-2013-02-453175
Barker, J. N., Weisdorf, D. J., and Wagner, J. E. (2001). Creation of a double chimera after the transplantation of umbilical-cord blood from two partially matched unrelated donors. N. Engl. J. Med. 344, 1870–1871. doi: 10.1056/NEJM200106143442417
Begley, D. J., Pontikis, C. C., and Scarpa, M. (2008). Lysosomal storage diseases and the blood-brain barrier. Curr. Pharm. Des. 14, 1566–1580. doi: 10.2174/138161208784705504
Bernal-Bayard, J., Cardenal-Muñoz, E., and Ramos-Morales, F. (2010). The Salmonella type III secretion effector, salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J. Biol. Chem. 285, 16360–16368. doi: 10.1074/jbc.M110.100669
Bey, K., Ciron, C., Dubreil, L., Deniaud, J., Ledevin, M., Cristini, J., et al. (2017). Efficient CNS targeting in adult mice by intrathecal infusion of single-stranded AAV9-GFP for gene therapy of neurological disorders. Gene Ther. 24, 325–332. doi: 10.1038/gt.2017.18
Bhattacharya, A., and Qi, L. (2019). ER-associated degradation in health and disease - from substrate to organism. J. Cell Sci. 132:jcs.232850. doi: 10.1242/jcs.232850
Bidou, L., Allamand, V., Rousset, J. P., and Namy, O. (2012). Sense from nonsense: therapies for premature stop codon diseases. Trends Mol. Med. 18, 679–688. doi: 10.1016/j.molmed.2012.09.008
Biffi, A. (2017a). Hematopoietic gene therapies for metabolic and neurologic diseases. Hematol. Oncol. Clin. North Am. 31, 869–881. doi: 10.1016/j.hoc.2017.06.004
Biffi, A. (2017b). Hematopoietic stem cell gene therapy for storage disease: current and new indications. Mol. Ther. 25, 1155–1162. doi: 10.1016/j.ymthe.2017.03.025
Biffi, A., de Palma, M., Quattrini, A., Del Carro, U., Amadio, S., Visigalli, I., et al. (2004). Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J. Clin. Invest. 113, 1118–1129. doi: 10.1172/JCI200419205
Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341:1233158. doi: 10.1126/science.1233158
Boado, R. J., Hui, E. K., Lu, J. Z., and Pardridge, W. M. (2013). IgG-enzyme fusion protein: pharmacokinetics and anti-drug antibody response in rhesus monkeys. Bioconjug. Chem. 24, 97–104. doi: 10.1021/bc3005123
Boado, R. J., Zhang, Y., Zhang, Y., Xia, C. F., Wang, Y., and Pardridge, W. M. (2008). Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood-brain barrier. Biotechnol. Bioeng. 99, 475–484. doi: 10.1002/bit.21602
Boelens, J. J., Orchard, P. J., and Wynn, R. F. (2014). Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br. J. Haematol. 167, 293–303. doi: 10.1111/bjh.13059
Borgwardt, L., Guffon, N., Amraoui, Y., Dali, C. I., De Meirleir, L., Gil-Campos, M., et al. (2018). Efficacy and safety of Velmanase alfa in the treatment of patients with alpha-mannosidosis: results from the core and extension phase analysis of a phase III multicentre, double-blind, randomised, placebo-controlled trial. J. Inherit. Metab. Dis. 41, 1215–1223. doi: 10.1007/s10545-018-0185-0
Bourdenx, M., Daniel, J., Genin, E., Soria, F. N., Blanchard-Desce, M., Bezard, E., et al. (2016). Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy 12, 472–483. doi: 10.1080/15548627.2015.1136769
Cabrera, I., Abasolo, I., Corchero, J. L., Elizondo, E., Gil, P. R., Moreno, E., et al. (2016). α-galactosidase-a loaded-nanoliposomes with enhanced enzymatic activity and intracellular penetration. Adv. Healthc. Mater. 5, 829–840. doi: 10.1002/adhm.201500746
Camargo, F., Erickson, R. P., Garver, W. S., Hossain, G. S., Carbone, P. N., Heidenreich, R. A., et al. (2001). Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 70, 131–142. doi: 10.1016/S0024-3205(01)01384-4
Capablo, J. L., Franco, R., de Cabezon, A. S., Alfonso, P., Pocovi, M., and Giraldo, P. (2007). Neurologic improvement in a type 3 Gaucher disease patient treated with imiglucerase/miglustat combination. Epilepsia 48, 1406–1408. doi: 10.1111/j.1528-1167.2007.01074.x
Cartier, N., and Aubourg, P. (2008). Hematopoietic stem cell gene therapy in Hurler syndrome, globoid cell leukodystrophy, metachromatic leukodystrophy and X-adrenoleukodystrophy. Curr. Opin. Mol. Ther. 10, 471–478.
Charkhand, B., Scantlebury, M. H., Narita, A., Zimran, A., and Al-Hertani, W. (2019). Effect of Ambroxol chaperone therapy on Glucosylsphingosine (Lyso-Gb1) levels in two Canadian patients with type 3 Gaucher disease. Mol. Genet. Metab. Rep. 20:100476. doi: 10.1016/j.ymgmr.2019.100476
Chung, J. K., Pan, L., Palmieri, K., Youssef, A. S., and Mccauley, T. G. (2017). Whole body and CNS biodistribution of rhHNS in cynomolgus monkeys after intrathecal lumbar administration: treatment implications for patients with MPS IIIA. Int. J. Mol. Sci. 18:2594. doi: 10.3390/ijms18122594
Clarke, J. T., Mahuran, D. J., Sathe, S., Kolodny, E. H., Rigat, B. A., Raiman, J. A., et al. (2011). An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol. Genet. Metab. 102, 6–12. doi: 10.1016/j.ymgme.2010.09.004
Clarke, L. A., Atherton, A. M., Burton, B. K., Day-Salvatore, D. L., Kaplan, P., Leslie, N. D., et al. (2017). Mucopolysaccharidosis type I newborn screening: best practices for diagnosis and management. J. Pediatr. 182, 363–370. doi: 10.1016/j.jpeds.2016.11.036
Colella, P., Ronzitti, G., and Mingozzi, F. (2018). Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 8, 87–104. doi: 10.1016/j.omtm.2017.11.007
Condori, J., Acosta, W., Ayala, J., Katta, V., Flory, A., Martin, R., et al. (2016). Enzyme replacement for GM1-gangliosidosis: uptake, lysosomal activation, and cellular disease correction using a novel beta-galactosidase:RTB lectin fusion. Mol. Genet. Metab. 117, 199–209. doi: 10.1016/j.ymgme.2015.12.002
Conzelmann, E., and Sandhoff, K. (1983). Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev. Neurosci. 6, 58–71. doi: 10.1159/000112332
Conzelmann, E., and Sandhoff, K. (1991). Biochemical basis of late-onset neurolipidoses. Dev. Neurosci. 13, 197–204. doi: 10.1159/000112160
Corraliza-Gomez, M., Sanchez, D., and Ganfornina, M. D. (2019). Lipid-binding proteins in brain health and disease. Front. Neurol 10:1152. doi: 10.3389/fneur.2019.01152
Cox, T., Lachmann, R., Hollak, C., Aerts, J., Van Weely, S., Hrebicek, M., et al. (2000). Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet 355, 1481–1485. doi: 10.1016/S0140-6736(00)02161-9
Cox, T. M., Drelichman, G., Cravo, R., Balwani, M., Burrow, T. A., Martins, A. M., et al. (2015). Eliglustat compared with imiglucerase in patients with Gaucher's disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet 385, 2355–2362. doi: 10.1016/S0140-6736(14)61841-9
Cutler, C., Multani, P., Robbins, D., Kim, H. T., Le, T., Hoggatt, J., et al. (2013). Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood 122, 3074–3081. doi: 10.1182/blood-2013-05-503177
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb. Perspect. Biol 7:a020412. doi: 10.1101/cshperspect.a020412
Davidson, C. D., Ali, N. F., Micsenyi, M. C., Stephney, G., Renault, S., Dobrenis, K., et al. (2009). Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE 4:e6951. doi: 10.1371/journal.pone.0006951
De Ru, M. H., Boelens, J. J., Das, A. M., Jones, S. A., van Der Lee, J. H., Mahlaoui, N., et al. (2011). Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: results of a European consensus procedure. Orphanet J. Rare Dis. 6:55. doi: 10.1186/1750-1172-6-55
De Ruijter, J., Valstar, M. J., Narajczyk, M., Wegrzyn, G., Kulik, W., Ijlst, L., et al. (2012). Genistein in Sanfilippo disease: a randomized controlled crossover trial. Ann. Neurol. 71, 110–120. doi: 10.1002/ana.22643
Del Grosso, A., Galliani, M., Angella, L., Santi, M., Tonazzini, I., Parlanti, G., et al. (2019). Brain-targeted enzyme-loaded nanoparticles: a breach through the blood-brain barrier for enzyme replacement therapy in Krabbe disease. Sci. Adv. 5:eaax7462. doi: 10.1126/sciadv.aax7462
Demeule, M., Currie, J. C., Bertrand, Y., Ché, C., Nguyen, T., Régina, A., et al. (2008). Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J. Neurochem. 106, 1534–1544. doi: 10.1111/j.1471-4159.2008.05492.x
Di Vizio, D., Kim, J., Hager, M. H., Morello, M., Yang, W., Lafargue, C. J., et al. (2009). Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 69, 5601–5609. doi: 10.1158/0008-5472.CAN-08-3860
Do, M. A., Levy, D., Brown, A., Marriott, G., and Lu, B. (2019). Targeted delivery of lysosomal enzymes to the endocytic compartment in human cells using engineered extracellular vesicles. Sci. Rep. 9:17274. doi: 10.1038/s41598-019-53844-5
Duffner, P. K., Caviness, V. S. Jr., Erbe, R. W., Patterson, M. C., Schultz, K. R., Wenger, D. A., et al. (2009). The long-term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet. Med 11, 450–454. doi: 10.1097/GIM.0b013e3181a16e04
Durand, S., Cougot, N., Mahuteau-Betzer, F., Nguyen, C. H., Grierson, D. S., Bertrand, E., et al. (2007). Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol. 178, 1145–1160. doi: 10.1083/jcb.200611086
Eisengart, J. B., Pierpont, E. I., Kaizer, A. M., Rudser, K. D., King, K. E., Pasquali, M., et al. (2019). Intrathecal enzyme replacement for Hurler syndrome: biomarker association with neurocognitive outcomes. Genet. Med. 21, 2552–2560. doi: 10.1038/s41436-019-0522-1
Escolar, M. L., Poe, M. D., Provenzale, J. M., Richards, K. C., Allison, J., Wood, S., et al. (2005). Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N. Engl. J. Med. 352, 2069–2081. doi: 10.1056/NEJMoa042604
Fan, Y. H., Yu, Y., Mao, R. F., Tan, X. J., Xu, G. F., Zhang, H., et al. (2011). USP4 targets TAK1 to downregulate TNFα-induced NF-κB activation. Cell Death Differ. 18, 1547–1560. doi: 10.1038/cdd.2011.11
Farmer, C. A., Thurm, A., Farhat, N., Bianconi, S., Keener, L. A., and Porter, F. D. (2019). Long-term neuropsychological outcomes from an open-label phase I/IIa trial of 2-hydroxypropyl-beta-cyclodextrins (VTS-270) in niemann-pick disease, type C1. CNS Drugs 33, 677–683. doi: 10.1007/s40263-019-00642-2
Foust, K. D., Nurre, E., Montgomery, C. L., Hernandez, A., Chan, C. M., and Kaspar, B. K. (2009). Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 27, 59–65. doi: 10.1038/nbt.1515
Fraldi, A., Hemsley, K., Crawley, A., Lombardi, A., Lau, A., Sutherland, L., et al. (2007). Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum. Mol. Genet. 16, 2693–2702. doi: 10.1093/hmg/ddm223
Fratantoni, J. C., Hall, C. W., and Neufeld, E. F. (1968). Hurler and hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 162, 570–572. doi: 10.1126/science.162.3853.570
Frühbeis, C., Fröhlich, D., and Krämer-Albers, E. M. (2012). Emerging roles of exosomes in neuron-glia communication. Front. Physiol. 3:119. doi: 10.3389/fphys.2012.00119
Garnacho, C., Dhami, R., Simone, E., Dziubla, T., Leferovich, J., Schuchman, E. H., et al. (2008). Delivery of acid sphingomyelinase in normal and niemann-pick disease mice using intercellular adhesion molecule-1-targeted polymer nanocarriers. J. Pharmacol. Exp. Ther. 325, 400–408. doi: 10.1124/jpet.107.133298
Gatto, F., Rossi, B., Tarallo, A., Polishchuk, E., Polishchuk, R., Carrella, A., et al. (2017). AAV-mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe disease. Sci. Rep 7:15089. doi: 10.1038/s41598-017-15352-2
GBD 2016 Neurology Collaborators Group (2017). Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the global burden of disease study 2015. Lancet Neurol. 16, 877–897. doi: 10.1016/S1474-4422(17)30299-5
Germain, D. P., Hughes, D. A., Nicholls, K., Bichet, D. G., Giugliani, R., Wilcox, W. R., et al. (2016). Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 375, 545–555. doi: 10.1056/NEJMoa1510198
Ghose, A. K., Herbertz, T., Hudkins, R. L., Dorsey, B. D., and Mallamo, J. P. (2012). Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 3, 50–68. doi: 10.1021/cn200100h
Ghosh, P., Dahms, N. M., and Kornfeld, S. (2003). Mannose 6-phosphate receptors: new twists in the tale. Nat. Rev. Mol. Cell Biol. 4, 202–212. doi: 10.1038/nrm1050
Giannotti, M. I., Esteban, O., Oliva, M., García-Parajo, M. F., and Sanz, F. (2011). pH-responsive polysaccharide-based polyelectrolyte complexes as nanocarriers for lysosomal delivery of therapeutic proteins. Biomacromolecules 12, 2524–2533. doi: 10.1021/bm2003384
Giraldo, P., Alfonso, P., Atutxa, K., Fernandez-Galan, M. A., Barez, A., Franco, R., et al. (2009). Real-world clinical experience with long-term miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL project. Haematologica 94, 1771–1775. doi: 10.3324/haematol.2009.008078
Giugliani, R., Vairo, F., Kubaski, F., Poswar, F., Riegel, M., Baldo, G., et al. (2018). Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child Adolesc. Health 2, 56–68. doi: 10.1016/S2352-4642(17)30087-1
Givogri, M. I., Galbiati, F., Fasano, S., Amadio, S., Perani, L., Superchi, D., et al. (2006). Oligodendroglial progenitor cell therapy limits central neurological deficits in mice with metachromatic leukodystrophy. J. Neurosci. 26, 3109–3119. doi: 10.1523/JNEUROSCI.4366-05.2006
Gleitz, H. F., Liao, A. Y., Cook, J. R., Rowlston, S. F., Forte, G. M., D'souza, Z., et al. (2018). Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 10:e8730. doi: 10.15252/emmm.201708730
Graf, W. D. (2017). Stem cell transplantation in Krabbe disease: new truths discovered and opinions change. Neurology 89, 1318–1319. doi: 10.1212/WNL.0000000000004427
Gregoriadis, G. (1978). Liposomes in the therapy of lysosomal storage diseases. Nature 275, 695–696. doi: 10.1038/275695a0
Gregoriadis, G., Putman, D., Louis, L., and Neerunjun, D. (1974). Comparative effect and fate of non-entrapped and liposome-entrapped neuraminidase injected into rats. Biochem. J. 140, 323–330. doi: 10.1042/bj1400323
Groeschel, S., Kuhl, J. S., Bley, A. E., Kehrer, C., Weschke, B., Doring, M., et al. (2016). Long-term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol. 73, 1133–1140. doi: 10.1001/jamaneurol.2016.2067
Guffon, N., Bertrand, Y., Forest, I., Fouilhoux, A., and Froissart, R. (2009). Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J. Pediatr. 154, 733–737. doi: 10.1016/j.jpeds.2008.11.041
Hamill, K. M., Wexselblatt, E., Tong, W., Esko, J. D., and Tor, Y. (2017). Delivery of cargo to lysosomes using GNeosomes. Methods Mol. Biol. 1594, 151–163. doi: 10.1007/978-1-4939-6934-0_9
Han, S. O., Li, S., Everitt, J. I., and Koeberl, D. D. (2019). Salmeterol with liver depot gene therapy enhances the skeletal muscle response in murine pompe disease. Hum. Gene Ther. 30, 855–864. doi: 10.1089/hum.2018.197
Haney, M. J., Klyachko, N. L., Harrison, E. B., Zhao, Y., Kabanov, A. V., and Batrakova, E. V. (2019). TPP1 delivery to lysosomes with extracellular vesicles and their enhanced brain distribution in the animal model of batten disease. Adv. Healthc. Mater. 8:e1801271. doi: 10.1002/adhm.201801271
Hean, J., Mager, I., Wood, M., El Andaloussi, S., Wiklander, O., and Nordin, J. (2019). Exosomes Comprising Therapeutic Polypeptides. Oxford: Google Patents.
Hinderer, C., Bell, P., Louboutin, J. P., Zhu, Y., Yu, H., Lin, G., et al. (2015). Neonatal systemic AAV induces tolerance to CNS gene therapy in MPS I dogs and nonhuman primates. Mol. Ther. 23, 1298–1307. doi: 10.1038/mt.2015.99
Hocquemiller, M., Giersch, L., Audrain, M., Parker, S., and Cartier, N. (2016). Adeno-associated virus-based gene therapy for CNS diseases. Hum. Gene Ther. 27, 478–496. doi: 10.1089/hum.2016.087
Hollak, C. E., and Wijburg, F. A. (2014). Treatment of lysosomal storage disorders: successes and challenges. J. Inherit. Metab. Dis. 37, 587–598. doi: 10.1007/s10545-014-9718-3
Hsu, J., Bhowmick, T., Burks, S. R., Kao, J. P., and Muro, S. (2014). Enhancing biodistribution of therapeutic enzymes in vivo by modulating surface coating and concentration of ICAM-1-targeted nanocarriers. J. Biomed. Nanotechnol. 10, 345–354. doi: 10.1166/jbn.2014.1718
Hsu, J., Northrup, L., Bhowmick, T., and Muro, S. (2012). Enhanced delivery of α-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders. Nanomedicine 8, 731–739. doi: 10.1016/j.nano.2011.08.014
Hsu, J., Serrano, D., Bhowmick, T., Kumar, K., Shen, Y., Kuo, Y. C., et al. (2011). Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease. J. Control. Release 149, 323–331. doi: 10.1016/j.jconrel.2010.10.031
Hughes, D. A., Nicholls, K., Shankar, S. P., Sunder-Plassmann, G., Koeller, D., Nedd, K., et al. (2017). Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet 54, 288–296. doi: 10.1136/jmedgenet-2016-104178
Huwyler, J., Wu, D., and Pardridge, W. M. (1996). Brain drug delivery of small molecules using immunoliposomes. Proc. Natl. Acad. Sci. U.S.A. 93, 14164–14169. doi: 10.1073/pnas.93.24.14164
Jian, J., Tian, Q. Y., Hettinghouse, A., Zhao, S., Liu, H., Wei, J., et al. (2016). Progranulin recruits HSP70 to beta-glucocerebrosidase and is therapeutic against gaucher disease. EBioMedicine 13, 212–224. doi: 10.1016/j.ebiom.2016.10.010
Katabuchi, A. U., Godoy, V., Shil, P., Moser, A., and Maegawa, G. H. B. (2018). Serendipitous effects of beta-cyclodextrin on murine model of Krabbe disease. Mol Genet Metab Rep. 15, 98–99. doi: 10.1016/j.ymgmr.2018.03.002
Keeler, G. D., Markusic, D. M., and Hoffman, B. E. (2019). Liver induced transgene tolerance with AAV vectors. Cell. Immunol. 342:103728. doi: 10.1016/j.cellimm.2017.12.002
Kelly, J. W. (2020). Pharmacologic approaches for adapting proteostasis in the secretory pathway to ameliorate protein conformational diseases. Cold Spring Harb. Perspect. Biol. 12, 1–19. doi: 10.1101/cshperspect.a034108
Kim, G., Kim, M., Lee, Y., Byun, J. W., Hwang, D. W., and Lee, M. (2020). Systemic delivery of microRNA-21 antisense oligonucleotides to the brain using T7-peptide decorated exosomes. J. Control. Release 317, 273–281. doi: 10.1016/j.jconrel.2019.11.009
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Koeberl, D. D., Luo, X., Sun, B., Mcvie-Wylie, A., Dai, J., Li, S., et al. (2011). Enhanced efficacy of enzyme replacement therapy in Pompe disease through mannose-6-phosphate receptor expression in skeletal muscle. Mol. Genet. Metab. 103, 107–112. doi: 10.1016/j.ymgme.2011.02.006
Koshkaryev, A., Thekkedath, R., Pagano, C., Meerovich, I., and Torchilin, V. P. (2011). Targeting of lysosomes by liposomes modified with octadecyl-rhodamine B. J. Drug Target 19, 606–614. doi: 10.3109/1061186X.2010.550921
Krageloh-Mann, I., Groeschel, S., Kehrer, C., Opherk, K., Nagele, T., Handgretinger, R., et al. (2013). Juvenile metachromatic leukodystrophy 10 years post transplant compared with a non-transplanted cohort. Bone Marrow Transplant. 48, 369–375. doi: 10.1038/bmt.2012.155
Kwon, J. M., Matern, D., Kurtzberg, J., Wrabetz, L., Gelb, M. H., Wenger, D. A., et al. (2018). Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease. Orphanet J. Rare Dis 13:30. doi: 10.1186/s13023-018-0766-x
Lattanzi, A., Neri, M., Maderna, C., Di Girolamo, I., Martino, S., Orlacchio, A., et al. (2010). Widespread enzymatic correction of CNS tissues by a single intracerebral injection of therapeutic lentiviral vector in leukodystrophy mouse models. Hum. Mol. Genet. 19, 2208–2227. doi: 10.1093/hmg/ddq099
Lebl, R., Thonhofer, M., Tysoe, C., Pabst, B. M., Schalli, M., Weber, P., et al. (2017). A Morita-Baylis-Hillman based route to C-5a-chain-extended 4-epi-isofagomine type glycosidase inhibitors. Carbohydr. Res. 442, 31–40. doi: 10.1016/j.carres.2017.03.003
Lee, H. J., Park, H. H., Sohn, Y., Ryu, J., Park, J. H., Rhee, W. J., et al. (2016). α-Galactosidase delivery using 30Kc19-human serum albumin nanoparticles for effective treatment of Fabry disease. Appl. Microbiol. Biotechnol. 100, 10395–10402. doi: 10.1007/s00253-016-7689-z
Low, L. A., Sutherland, M., Lumelsky, N., Selimovic, S., Lundberg, M. S., and Tagle, D. A. (2020). Organs-on-a-chip. Adv. Exp. Med. Biol. 1230, 27–42. doi: 10.1007/978-3-030-36588-2_3
Luddi, A., Crifasi, L., Capaldo, A., Piomboni, P., and Costantino-Ceccarini, E. (2016). Suppression of galactocerebrosidase premature termination codon and rescue of galactocerebrosidase activity in twitcher cells. J. Neurosci. Res. 94, 1273–1283. doi: 10.1002/jnr.23790
Lund, T. C. (2018). Umbilical cord blood expansion: are we there yet? Biol. Blood Marrow Transplant. 24, 1311–1312. doi: 10.1016/j.bbmt.2018.05.002
Macias-Vidal, J., Giros, M., Guerrero, M., Gascon, P., Serratosa, J., Bachs, O., et al. (2014). The proteasome inhibitor bortezomib reduced cholesterol accumulation in fibroblasts from Niemann-Pick type C patients carrying missense mutations. FEBS J. 281, 4450–4466. doi: 10.1111/febs.12954
Maegawa, G. H., Banwell, B. L., Blaser, S., Sorge, G., Toplak, M., Ackerley, C., et al. (2009a). Substrate reduction therapy in juvenile GM2 gangliosidosis. Mol. Genet. Metab. 98, 215–224. doi: 10.1016/j.ymgme.2009.06.005
Maegawa, G. H., Tropak, M., Buttner, J., Stockley, T., Kok, F., Clarke, J. T., et al. (2007). Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 282, 9150–9161. doi: 10.1074/jbc.M609304200
Maegawa, G. H., Tropak, M. B., Buttner, J. D., Rigat, B. A., Fuller, M., Pandit, D., et al. (2009b). Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 284, 23502–23516. doi: 10.1074/jbc.M109.012393
Maegawa, G. H., Van Giersbergen, P. L., Yang, S., Banwell, B., Morgan, C. P., Dingemanse, J., et al. (2009c). Pharmacokinetics, safety and tolerability of miglustat in the treatment of pediatric patients with GM2 gangliosidosis. Mol. Genet. Metab. 97, 284–291. doi: 10.1016/j.ymgme.2009.04.013
Maegawa, G. H. B. (2019). Lysosomal leukodystrophies lysosomal storage diseases associated with white matter abnormalities. J. Child Neurol. 34, 339–358. doi: 10.1177/0883073819828587
Magrin, E., Miccio, A., and Cavazzana, M. (2019). Lentiviral and genome-editing strategies for the treatment of beta-hemoglobinopathies. Blood 134, 1203–1213. doi: 10.1182/blood.2019000949
Malinowska, M., Wilkinson, F. L., Bennett, W., Langford-Smith, K. J., O'leary, H. A., Jakobkiewicz-Banecka, J., et al. (2009). Genistein reduces lysosomal storage in peripheral tissues of mucopolysaccharide IIIB mice. Mol. Genet. Metab. 98, 235–242. doi: 10.1016/j.ymgme.2009.06.013
Marco, S., Haurigot, V., and Bosch, F. (2019). In vivo gene therapy for mucopolysaccharidosis type III (Sanfilippo Syndrome): a new treatment horizon. Hum. Gene Ther. 30, 1211–1221. doi: 10.1089/hum.2019.217
Markham, A. (2017). Cerliponase alfa: first global approval. Drugs 77, 1247–1249. doi: 10.1007/s40265-017-0771-8
Marshall, J., Sun, Y., Bangari, D. S., Budman, E., Park, H., Nietupski, J. B., et al. (2016). CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic gaucher disease. Mol. Ther. 24, 1019–1029. doi: 10.1038/mt.2016.53
Matalonga, L., Gort, L., and Ribes, A. (2017). Small molecules as therapeutic agents for inborn errors of metabolism. J. Inherit. Metab. Dis. 40, 177–193. doi: 10.1007/s10545-016-0005-3
Mayer, F. Q., Artigalas, O. A., Lagranha, V. L., Baldo, G., Schwartz, I. V., Matte, U., et al. (2013). Chloramphenicol enhances IDUA activity on fibroblasts from mucopolysaccharidosis I patients. Curr. Pharm. Biotechnol. 14, 194–198. doi: 10.2174/1389201011314020009
Mceachern, K. A., Fung, J., Komarnitsky, S., Siegel, C. S., Chuang, W. L., Hutto, E., et al. (2007). A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol. Genet. Metab. 91, 259–267. doi: 10.1016/j.ymgme.2007.04.001
Mcgraw, P., Liang, L., Escolar, M., Mukundan, S., Kurtzberg, J., and Provenzale, J. M. (2005). Krabbe disease treated with hematopoietic stem cell transplantation: serial assessment of anisotropy measurements–initial experience. Radiology 236, 221–230. doi: 10.1148/radiol.2353040716
Mckinnis, E. J., Sulzbacher, S., Rutledge, J. C., Sanders, J., and Scott, C. R. (1996). Bone marrow transplantation in Hunter syndrome. J. Pediatr. 129, 145–148. doi: 10.1016/S0022-3476(96)70202-0
Meneghini, V., Lattanzi, A., Tiradani, L., Bravo, G., Morena, F., Sanvito, F., et al. (2016). Pervasive supply of therapeutic lysosomal enzymes in the CNS of normal and Krabbe-affected non-human primates by intracerebral lentiviral gene therapy. EMBO Mol. Med. 8, 489–510. doi: 10.15252/emmm.201505850
Meng, X., Xu, G., Zhou, Q.-L., Wu, J.-P., and Yang, L.-R. (2013). Improvements of lipase performance in high-viscosity system by immobilization onto a novel kind of poly (methylmethacrylate-co-divinylbenzene) encapsulated porous magnetic microsphere carrier. J. Mol. Catal. B Enzymatic 89, 86–92. doi: 10.1016/j.molcatb.2013.01.006
Minciacchi, V. R., You, S., Spinelli, C., Morley, S., Zandian, M., Aspuria, P. J., et al. (2015). Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget 6, 11327–11341. doi: 10.18632/oncotarget.3598
Mistry, P. K., Lukina, E., Ben Turkia, H., Amato, D., Baris, H., Dasouki, M., et al. (2015). Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA 313, 695–706. doi: 10.1001/jama.2015.459
Mort, M., Ivanov, D., Cooper, D. N., and Chuzhanova, N. A. (2008). A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 29, 1037–1047. doi: 10.1002/humu.20763
Mu, T. W., Fowler, D. M., and Kelly, J. W. (2008a). Partial restoration of mutant enzyme homeostasis in three distinct lysosomal storage disease cell lines by altering calcium homeostasis. PLoS Biol. 6:e26. doi: 10.1371/journal.pbio.0060026
Mu, T. W., Ong, D. S., Wang, Y. J., Balch, W. E., Yates, J. R. 3rd, Segatori, L., and Kelly, J. W. (2008b). Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134, 769–781. doi: 10.1016/j.cell.2008.06.037
Muenzer, J., Beck, M., Eng, C. M., Escolar, M. L., Giugliani, R., Guffon, N. H., et al. (2009a). Multidisciplinary management of Hunter syndrome. Pediatrics 124, e1228–e1239. doi: 10.1542/peds.2008-0999
Muenzer, J., Hendriksz, C. J., Fan, Z., Vijayaraghavan, S., Perry, V., Santra, S., et al. (2016). A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet. Med. 18, 73–81. doi: 10.1038/gim.2015.36
Muenzer, J., Wraith, J. E., Clarke, L. A., and International Consensus Panel On M. Treatment of Mucopolysaccharidosis I. (2009b). Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 123, 19–29. doi: 10.1542/peds.2008-0416
Munoz-Rojas, M. V., Vieira, T., Costa, R., Fagondes, S., John, A., Jardim, L. B., et al. (2008). Intrathecal enzyme replacement therapy in a patient with mucopolysaccharidosis type I and symptomatic spinal cord compression. Am. J. Med. Genet. A 146A, 2538–2544. doi: 10.1002/ajmg.a.32294
Muro, S., Schuchman, E. H., and Muzykantov, V. R. (2006). Lysosomal enzyme delivery by ICAM-1-targeted nanocarriers bypassing glycosylation- and clathrin-dependent endocytosis. Mol. Ther. 13, 135–141. doi: 10.1016/j.ymthe.2005.07.687
Naldini, L. (2019). Genetic engineering of hematopoiesis: current stage of clinical translation and future perspectives. EMBO Mol. Med. 11:e9985. doi: 10.15252/emmm.201809958
Narita, A., Shirai, K., Itamura, S., Matsuda, A., Ishihara, A., Matsushita, K., et al. (2016). Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann. Clin. Transl. Neurol. 3, 200–215. doi: 10.1002/acn3.292
Nestrasil, I., Shapiro, E., Svatkova, A., Dickson, P., Chen, A., Wakumoto, A., et al. (2017). Intrathecal enzyme replacement therapy reverses cognitive decline in mucopolysaccharidosis type I. Am. J. Med. Genet. A 173, 780–783. doi: 10.1002/ajmg.a.38073
Neufeld, E. F. (1989). Natural history and inherited disorders of a lysosomal enzyme, beta-hexosaminidase. J. Biol. Chem 264, 10927–10930.
Noerholm, M., Balaj, L., Limperg, T., Salehi, A., Zhu, L. D., Hochberg, F. H., et al. (2012). RNA expression patterns in serum microvesicles from patients with glioblastoma multiforme and controls. BMC Cancer 12:22. doi: 10.1186/1471-2407-12-22
Nolte-'T Hoen, E. N., van Der Vlist, E. J., Aalberts, M., Mertens, H. C., Bosch, B. J., Bartelink, W., et al. (2012). Quantitative and qualitative flow cytometric analysis of nanosized cell-derived membrane vesicles. Nanomedicine 8, 712–720. doi: 10.1016/j.nano.2011.09.006
O'loughlin, A. J., Woffindale, C. A., and Wood, M. J. (2012). Exosomes and the emerging field of exosome-based gene therapy. Curr. Gene Ther. 12, 262–274. doi: 10.2174/156652312802083594
Onaca-Fischer, O., Liu, J., Inglin, M., and Palivan, C. G. (2012). Polymeric nanocarriers and nanoreactors: a survey of possible therapeutic applications. Curr. Pharm. Des. 18, 2622–2643. doi: 10.2174/138161212800492822
Ory, D. S., Ottinger, E. A., Farhat, N. Y., King, K. A., Jiang, X., Weissfeld, L., et al. (2017). Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1-2 trial. Lancet 390, 1758–1768. doi: 10.1016/S0140-6736(17)31465-4
Osher, E., Fattal-Valevski, A., Sagie, L., Urshanski, N., Amir-Levi, Y., Katzburg, S., et al. (2011). Pyrimethamine increases beta-hexosaminidase A activity in patients with late onset tay Sachs. Mol. Genet. Metab. 102, 356–363. doi: 10.1016/j.ymgme.2010.11.163
Ou, L., Przybilla, M. J., Koniar, B., and Whitley, C. B. (2018). RTB lectin-mediated delivery of lysosomal alpha-l-iduronidase mitigates disease manifestations systemically including the central nervous system. Mol. Genet. Metab. 123, 105–111. doi: 10.1016/j.ymgme.2017.11.013
Palmieri, M., Pal, R., Nelvagal, H. R., Lotfi, P., Stinnett, G. R., Seymour, M. L., et al. (2017). mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 8:14338. doi: 10.1038/ncomms15793
Pardridge, W. M. (2001). Crossing the blood-brain barrier: are we getting it right? Drug Discov. Today 6, 1–2. doi: 10.1016/S1359-6446(00)01583-X
Pardridge, W. M. (2005). The blood-brain barrier and neurotherapeutics. NeuroRx 2, 1–2. doi: 10.1602/neurorx.2.1.1
Pardridge, W. M. (2006). Molecular Trojan horses for blood-brain barrier drug delivery. Curr. Opin. Pharmacol 6, 494–500. doi: 10.1016/j.coph.2006.06.001
Pardridge, W. M. (2007a). Blood-brain barrier delivery. Drug Discov. Today 12, 54–61. doi: 10.1016/j.drudis.2006.10.013
Pardridge, W. M. (2007b). Blood-brain barrier delivery of protein and non-viral gene therapeutics with molecular Trojan horses. J. Control. Release 122, 345–348. doi: 10.1016/j.jconrel.2007.04.001
Park, H. H., Sohn, Y., Yeo, J. W., Park, J. H., Lee, H. J., Ryu, J., et al. (2014). Dimerization of 30Kc19 protein in the presence of amphiphilic moiety and importance of Cys-57 during cell penetration. Biotechnol. J. 9, 1582–1593. doi: 10.1002/biot.201400253
Park, J. H., Lee, J. H., Park, H. H., Rhee, W. J., Choi, S. S., and Park, T. H. (2012). A protein delivery system using 30Kc19 cell-penetrating protein originating from silkworm. Biomaterials 33, 9127–9134. doi: 10.1016/j.biomaterials.2012.08.063
Pastores, G. M., Elstein, D., Hrebicek, M., and Zimran, A. (2007). Effect of miglustat on bone disease in adults with type 1 Gaucher disease: a pooled analysis of three multinational, open-label studies. Clin. Ther. 29, 1645–1654. doi: 10.1016/j.clinthera.2007.08.006
Pastores, G. M., and Maegawa, G. H. (2013). Clinical neurogenetics: neuropathic lysosomal storage disorders. Neurol. Clin. 31, 1051–1071. doi: 10.1016/j.ncl.2013.04.007
Patel, T., Zhou, J., Piepmeier, J. M., and Saltzman, W. M. (2012). Polymeric nanoparticles for drug delivery to the central nervous system. Adv. Drug Deliv. Rev. 64, 701–705. doi: 10.1016/j.addr.2011.12.006
Patel, H., and Ryman, B. E. (1974). α-Mannosidase in Zinc-Deficient Rats: Possibility of Liposomal Therapy in Annosidosis. London: Portland Press Ltd. doi: 10.1042/bst0021014
Patil, S. A., and Maegawa, G. H. (2013). Developing therapeutic approaches for metachromatic leukodystrophy. Drug Des. Devel. Ther. 7, 729–745. doi: 10.2147/DDDT.S15467
Patterson, M. C., Vecchio, D., Prady, H., Abel, L., and Wraith, J. E. (2007). Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6, 765–772. doi: 10.1016/S1474-4422(07)70194-1
Pawlinski, L., Malecki, M. T., and Kiec-Wilk, B. (2016). The additive effect on the antiepileptic treatment of ambroxol in type 3 Gaucher patient. The early observation. Blood Cells Mol. Dis. 11, 192–193. doi: 10.1016/j.bcmd.2016.12.001
Peled, T., Landau, E., Mandel, J., Glukhman, E., Goudsmid, N. R., Nagler, A., et al. (2004). Linear polyamine copper chelator tetraethylenepentamine augments long-term ex vivo expansion of cord blood-derived CD34+ cells and increases their engraftment potential in NOD/SCID mice. Exp. Hematol. 32, 547–555. doi: 10.1016/j.exphem.2004.03.002
Peng, J., Dalton, J., Butt, M., Tracy, K., Kennedy, D., Haroldsen, P., et al. (2017). Reveglucosidase alfa (BMN 701), an IGF2-tagged rhacid alpha-glucosidase, improves respiratory functional parameters in a murine model of pompe disease. J. Pharmacol. Exp. Ther. 360, 313–323. doi: 10.1124/jpet.116.235952
Pipalia, N. H., Subramanian, K., Mao, S., Ralph, H., Hutt, D. M., Scott, S. M., et al. (2017). Histone deacetylase inhibitors correct the cholesterol storage defect in most Niemann-Pick C1 mutant cells. J. Lipid Res. 58, 695–708. doi: 10.1194/jlr.M072140
Pirooznia, N., Hasannia, S., Lotfi, A. S., and Ghanei, M. (2012). Encapsulation of alpha-1 antitrypsin in PLGA nanoparticles: in vitro characterization as an effective aerosol formulation in pulmonary diseases. J. Nanobiotechnology 10:20. doi: 10.1186/1477-3155-10-20
Platt, F. M. (2018). Emptying the stores: lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 17, 133–150. doi: 10.1038/nrd.2017.214
Platt, F. M., D'azzo, A., Davidson, B. L., Neufeld, E. F., and Tifft, C. J. (2018). Lysosomal storage diseases. Nat. Rev. Dis. Primers 4:27. doi: 10.1038/s41572-018-0037-0
Platt, F. M., and Jeyakumar, M. (2008). Substrate reduction therapy. Acta Paediatr. Suppl. 97, 88–93. doi: 10.1111/j.1651-2227.2008.00656.x
Platt, F. M., Neises, G. R., Dwek, R. A., and Butters, T. D. (1994a). N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J. Biol. Chem. 269, 8362–8365.
Platt, F. M., Neises, G. R., Karlsson, G. B., Dwek, R. A., and Butters, T. D. (1994b). N-butyldeoxygalactonojirimycin inhibits glycolipid biosynthesis but does not affect N-linked oligosaccharide processing. J. Biol. Chem. 269, 27108–27114.
Platt, F. M., Reinkensmeier, G., Dwek, R. A., and Butters, T. D. (1997). Extensive glycosphingolipid depletion in the liver and lymphoid organs of mice treated with N-butyldeoxynojirimycin. J. Biol. Chem. 272, 19365–19372. doi: 10.1074/jbc.272.31.19365
Powers, E. T., and Balch, W. E. (2013). Diversity in the origins of proteostasis networks–a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 14, 237–248. doi: 10.1038/nrm3542
Prasad, V. K., and Kurtzberg, J. (2008). Emerging trends in transplantation of inherited metabolic diseases. Bone Marrow Transplant. 41, 99–108. doi: 10.1038/sj.bmt.1705970
Prasad, V. K., and Kurtzberg, J. (2010a). Cord blood and bone marrow transplantation in inherited metabolic diseases: scientific basis, current status and future directions. Br. J. Haematol. 148, 356–372. doi: 10.1111/j.1365-2141.2009.07974.x
Prasad, V. K., and Kurtzberg, J. (2010b). Transplant outcomes in mucopolysaccharidoses. Semin. Hematol. 47, 59–69. doi: 10.1053/j.seminhematol.2009.10.008
Prill, H., Luu, A., Yip, B., Holtzinger, J., Lo, M. J., Christianson, T. M., et al. (2019). Differential uptake of NAGLU-IGF2 and unmodified NAGLU in cellular models of sanfilippo syndrome Type, B. Mol. Ther. Methods Clin. Dev. 14, 56–63. doi: 10.1016/j.omtm.2019.05.008
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U. S. A. 103, 11172–11177. doi: 10.1073/pnas.0603838103
Raposo, G., and Stoorvogel, W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J. Cell Biol. 200, 373–383. doi: 10.1083/jcb.201211138
Ricca, A., Cascino, F., Morena, F., Martino, S., and Gritti, A. (2020). In vitro validation of chimeric beta-Galactosylceramidase enzymes with improved enzymatic activity and increased secretion. Front Mol Biosci. 7:167. doi: 10.3389/fmolb.2020.00167
Roth, D. M., and Balch, W. E. (2011). Modeling general proteostasis: proteome balance in health and disease. Curr. Opin. Cell Biol. 23, 126–134. doi: 10.1016/j.ceb.2010.11.001
Salvalaio, M., Rigon, L., Belletti, D., D'avanzo, F., Pederzoli, F., Ruozi, B., et al. (2016). Targeted polymeric nanoparticles for brain delivery of high molecular weight molecules in lysosomal storage disorders. PLoS ONE 11:e0156452. doi: 10.1371/journal.pone.0156452
Sandhoff, K., and Conzelmann, E. (1984). The biochemical basis of gangliosidoses. Neuropediatrics 15 (Suppl.):85–92. doi: 10.1055/s-2008-1052387
Santi, M., Maccari, G., Mereghetti, P., Voliani, V., Rocchiccioli, S., Ucciferri, N., et al. (2017). Rational design of a transferrin-binding peptide sequence tailored to targeted nanoparticle internalization. Bioconjug. Chem. 28, 471–480. doi: 10.1021/acs.bioconjchem.6b00611
Sarrazin, S., Wilson, B., Sly, W. S., Tor, Y., and Esko, J. D. (2010). Guanidinylated neomycin mediates heparan sulfate-dependent transport of active enzymes to lysosomes. Mol. Ther. 18, 1268–1274. doi: 10.1038/mt.2010.78
Sawkar, A. R., Schmitz, M., Zimmer, K. P., Reczek, D., Edmunds, T., Balch, W. E., et al. (2006). Chemical chaperones and permissive temperatures alter localization of Gaucher disease associated glucocerebrosidase variants. ACS Chem. Biol. 1, 235–251. doi: 10.1021/cb600187q
Scarpa, M., Almassy, Z., Beck, M., Bodamer, O., Bruce, I. A., de Meirleir, L., et al. (2011). Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J. Rare Dis. 6:72. doi: 10.1186/1750-1172-6-72
Schiffmann, R., Fitzgibbon, E. J., Harris, C., Devile, C., Davies, E. H., Abel, L., et al. (2008). Randomized, controlled trial of miglustat in Gaucher's disease type 3. Ann. Neurol. 64, 514–522. doi: 10.1002/ana.21491
Schueler, U. H., Kolter, T., Kaneski, C. R., Zirzow, G. C., Sandhoff, K., and Brady, R. O. (2004). Correlation between enzyme activity and substrate storage in a cell culture model system for Gaucher disease. J. Inherit. Metab. Dis. 27, 649–658. doi: 10.1023/B:BOLI.0000042959.44318.7c
Schulz, A., Ajayi, T., Specchio, N., de Los Reyes, E., Gissen, P., Ballon, D., et al. (2018). Study of intraventricular cerliponase Alfa for CLN2 disease. N. Engl. J. Med. 378, 1898–1907. doi: 10.1056/NEJMoa1712649
Sessa, M., Lorioli, L., Fumagalli, F., Acquati, S., Redaelli, D., Baldoli, C., et al. (2016). Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 388, 476–487. doi: 10.1016/S0140-6736(16)30374-9
Shapiro, B. E., Pastores, G. M., Gianutsos, J., Luzy, C., and Kolodny, E. H. (2009). Miglustat in late-onset Tay-Sachs disease: a 12-month, randomized, controlled clinical study with 24 months of extended treatment. Genet. Med. 11, 425–433. doi: 10.1097/GIM.0b013e3181a1b5c5
Shayman, J. A. (2010). Eliglustat tartrate: glucosylceramide synthase inhibitor treatment of type 1 gaucher disease. Drugs Future 35, 613–620. doi: 10.1358/dof.2010.035.08.1505566
Shayman, J. A. (2013). The design and clinical development of inhibitors of glycosphingolipid synthesis: will invention be the mother of necessity? Trans. Am. Clin. Climatol. Assoc. 124, 46–60.
Shihabuddin, L. S., Numan, S., Huff, M. R., Dodge, J. C., Clarke, J., Macauley, S. L., et al. (2004). Intracerebral transplantation of adult mouse neural progenitor cells into the Niemann-Pick-A mouse leads to a marked decrease in lysosomal storage pathology. J. Neurosci. 24, 10642–10651. doi: 10.1523/JNEUROSCI.3584-04.2004
Song, W., Wang, F., Savini, M., Ake, A., Di Ronza, A., Sardiello, M., et al. (2013). TFEB regulates lysosomal proteostasis. Hum. Mol. Genet 22, 1994–2009. doi: 10.1093/hmg/ddt052
Sorrentino, N. C., D'orsi, L., Sambri, I., Nusco, E., Monaco, C., Spampanato, C., et al. (2013). A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol. Med. 5, 675–690. doi: 10.1002/emmm.201202083
Springer, T. A. (1994). Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell 76, 301–314. doi: 10.1016/0092-8674(94)90337-9
Steger, L. D., and Desnick, R. J. (1977). Enzyme therapy. VI: comparative in vivo fates and effects on lysosomal integrity of enzyme entrapped in negatively and positively charged liposomes. Biochim. Biophys. Acta 464, 530–546. doi: 10.1016/0005-2736(77)90028-1
Stiff, P. J., Montesinos, P., Peled, T., Landau, E., Goudsmid, N. R., Mandel, J., et al. (2018). Cohort-controlled comparison of umbilical cord blood transplantation using carlecortemcel-L, a single progenitor-enriched cord blood, to double cord blood unit transplantation. Biol. Blood Marrow Transplant. 24, 1463–1470. doi: 10.1016/j.bbmt.2018.02.012
Sun, Y., Liou, B., Chu, Z., Fannin, V., Blackwood, R., Peng, Y., et al. (2020). Systemic enzyme delivery by blood-brain barrier-penetrating SapC-DOPS nanovesicles for treatment of neuronopathic Gaucher disease. EBioMedicine 2020:102735. doi: 10.1016/j.ebiom.2020.102735
Sun, Y., Qi, X., and Grabowski, G. A. (2003). Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J. Biol. Chem. 278, 31918–31923. doi: 10.1074/jbc.M302752200
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Tamaki, S. J., Jacobs, Y., Dohse, M., Capela, A., Cooper, J. D., Reitsma, M., et al. (2009). Neuroprotection of host cells by human central nervous system stem cells in a mouse model of infantile neuronal ceroid lipofuscinosis. Cell Stem Cell 5, 310–319. doi: 10.1016/j.stem.2009.05.022
Tan, E. Y., Boelens, J. J., Jones, S. A., and Wynn, R. F. (2019). Hematopoietic stem cell transplantation in inborn errors of metabolism. Front. Pediatr. 7:433. doi: 10.3389/fped.2019.00433
Tardieu, M., Zerah, M., Husson, B., de Bournonville, S., Deiva, K., Adamsbaum, C., et al. (2014). Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum. Gene Ther. 25, 506–516. doi: 10.1089/hum.2013.238
Tejera, E., Rocha-Perugini, V., López-Martín, S., Pérez-Hernández, D., Bachir, A. I., Horwitz, A. R., et al. (2013). CD81 regulates cell migration through its association with Rac GTPase. Mol. Biol. Cell 24, 261–273. doi: 10.1091/mbc.e12-09-0642
Thada, V., Miller, J. N., Kovacs, A. D., and Pearce, D. A. (2016). Tissue-specific variation in nonsense mutant transcript level and drug-induced read-through efficiency in the Cln1(R151X) mouse model of INCL. J. Cell. Mol. Med. 20, 381–385. doi: 10.1111/jcmm.12744
Thekkedath, R., Koshkaryev, A., and Torchilin, V. P. (2013). Lysosome-targeted octadecyl-rhodamine B-liposomes enhance lysosomal accumulation of glucocerebrosidase in Gaucher's cells in vitro. Nanomedicine 8, 1055–1065. doi: 10.2217/nnm.12.138
Théry, C., Zitvogel, L., and Amigorena, S. (2002). Exosomes: composition, biogenesis and function. Nat. Rev. Immunol. 2, 569–579. doi: 10.1038/nri855
Torra, A., Parent, A., Cuadros, T., Rodriguez-Galvan, B., Ruiz-Bronchal, E., Ballabio, A., et al. (2018). Overexpression of TFEB drives a pleiotropic neurotrophic effect and prevents parkinson's disease-related neurodegeneration. Mol. Ther. 26, 1552–1567. doi: 10.1016/j.ymthe.2018.02.022
Tosi, G., Fano, R. A., Bondioli, L., Badiali, L., Benassi, R., Rivasi, F., et al. (2011). Investigation on mechanisms of glycopeptide nanoparticles for drug delivery across the blood-brain barrier. Nanomedicine 6, 423–436. doi: 10.2217/nnm.11.11
Tropak, M. B., and Mahuran, D. (2007). Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase A activity in GM2 gangliosidosis cells. FEBS J. 274, 4951–4961. doi: 10.1111/j.1742-4658.2007.06040.x
Umezawa, F., Eto, Y., Tokoro, T., Ito, F., and Maekawa, K. (1985). Enzyme replacement with liposomes containing beta-galactosidase from Charonia lumpas in murine globoid cell leukodystrophy (twitcher). Biochem. Biophys. Res. Commun. 127, 663–667. doi: 10.1016/S0006-291X(85)80212-6
Van Niel, G., D'angelo, G., and Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 19, 213–228. doi: 10.1038/nrm.2017.125
Van Rappard, D. F., Boelens, J. J., van Egmond, M. E., Kuball, J., Van Hasselt, P. M., Oostrom, K. J., et al. (2016). Efficacy of hematopoietic cell transplantation in metachromatic leukodystrophy: the Dutch experience. Blood 127, 3098–3101. doi: 10.1182/blood-2016-03-708479
Walkley, S. U. (2009). Pathogenic cascades in lysosomal disease-why so complex? J. Inherit. Metab. Dis. 32, 181–189. doi: 10.1007/s10545-008-1040-5
Wang, D., Belakhov, V., Kandasamy, J., Baasov, T., Li, S. C., Li, Y. T., et al. (2012). The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol. Genet. Metab. 105, 116–125. doi: 10.1016/j.ymgme.2011.10.005
Wang, D., El-Amouri, S. S., Dai, M., Kuan, C. Y., Hui, D. Y., Brady, R. O., et al. (2013). Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. U. S. A. 110, 2999–3004. doi: 10.1073/pnas.1222742110
Wang, F., and Segatori, L. (2013). Remodeling the proteostasis network to rescue glucocerebrosidase variants by inhibiting ER-associated degradation and enhancing ER Folding. PLoS ONE 8:e61418. doi: 10.1371/journal.pone.0061418
Wasserstein, M. P., Diaz, G. A., Lachmann, R. H., Jouvin, M. H., Nandy, I., Ji, A. J., et al. (2018). Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): safety and efficacy in adults treated for 30 months. J. Inherit. Metab. Dis. 41, 829–838. doi: 10.1007/s10545-017-0123-6
Whitley, C. B., Vijay, S., Yao, B., Pineda, M., Parker, G. J. M., Rojas-Caro, S., et al. (2019). Final results of the phase 1/2, open-label clinical study of intravenous recombinant human N-acetyl-alpha-d-glucosaminidase (SBC-103) in children with mucopolysaccharidosis IIIB. Mol. Genet. Metab. 126, 131–138. doi: 10.1016/j.ymgme.2018.12.003
Wiseman, D. H., Mercer, J., Tylee, K., Malaiya, N., Bonney, D. K., Jones, S. A., et al. (2013). Management of mucopolysaccharidosis type IH (Hurler's syndrome) presenting in infancy with severe dilated cardiomyopathy: a single institution's experience. J. Inherit. Metab. Dis. 36, 263–270. doi: 10.1007/s10545-012-9500-3
Wiseman, R. L., Powers, E. T., Buxbaum, J. N., Kelly, J. W., and Balch, W. E. (2007). An adaptable standard for protein export from the endoplasmic reticulum. Cell 131, 809–821. doi: 10.1016/j.cell.2007.10.025
Xu, S., Lun, Y., Frascella, M., Garcia, A., Soska, R., Nair, A., et al. (2019). Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight. 4:e125358. doi: 10.1172/jci.insight.125358
Yang, J., Zhang, X., Chen, X., Wang, L., and Yang, G. (2017). Exosome mediated delivery of miR-124 promotes neurogenesis after Ischemia. Mol. Ther. Nucleic Acids 7, 278–287. doi: 10.1016/j.omtn.2017.04.010
Zhang, G., and Yang, P. (2018). A novel cell-cell communication mechanism in the nervous system: exosomes. J. Neurosci. Res. 96, 45–52. doi: 10.1002/jnr.24113
Keywords: lysosomes, small molecules, therapy, enzyme replacement therapy, extracellular vesicles, exosomes, liposomes, gene therapy
Citation: Edelmann MJ and Maegawa GHB (2020) CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 7:559804. doi: 10.3389/fmolb.2020.559804
Received: 07 May 2020; Accepted: 15 September 2020;
Published: 12 November 2020.
Edited by:
Caroline Sevin, Bicêtre Hospital, FranceReviewed by:
Carlos Romá-Mateo, University of Valencia, SpainMahendra Pratap Kashyap, University of Alabama at Birmingham, United States
Copyright © 2020 Edelmann and Maegawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gustavo H. B. Maegawa, Z21hZWdhd2FAdWZsLmVkdQ==