 Francesca Pagliari1†
Francesca Pagliari1† Maria Grazia Marafioti1†
Maria Grazia Marafioti1† Geraldine Genard1
Geraldine Genard1 Patrizio Candeloro2
Patrizio Candeloro2 Giuseppe Viglietto3
Giuseppe Viglietto3 Joao Seco1,4
Joao Seco1,4 Luca Tirinato1,2*
Luca Tirinato1,2*- 1Biomedical Physics in Radiation Oncology, German Cancer Research Center, Heidelberg, Germany
- 2BioNEM Laboratory, Department of Experimental and Clinical Medicine, Magna Graecia University, Catanzaro, Italy
- 3Department of Experimental and Clinical Medicine, Magna Graecia University, Catanzaro, Italy
- 4Department of Physics and Astronomy, Heidelberg University, Heidelberg, Germany
Since its appearance, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has immediately alarmed the World Health Organization for its very high contagiousness and the complexity of patient clinical profiles. The worldwide scientific community is today gathered in a massive effort in order to develop safe vaccines and effective therapies in the shortest possible time. Every day, new pieces of SARS-CoV-2 infective puzzle are disclosed. Based on knowledge gained with other related coronaviruses and, more in general, on single-strand RNA viruses, we highlight underexplored molecular routes in which lipids and lipid droplets (LDs) might serve essential functions in viral infections. In fact, both lipid homeostasis and the pathways connected to lipids seem to be fundamental in all phases of the coronavirus infection. This review aims at describing potential roles for lipid and LDs in host–virus interactions and suggesting LDs as new and central cellular organelles to be investigated as potential targets against SARS-CoV-2 infection.
Introduction
The rapid and global spread of a novel coronavirus (CoV) called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in Hubei Province in China (Zhang and Holmes, 2020), has immediately alarmed the World Health Organization (WHO) for the high contagiousness and the complexity of patient clinical profiles associated with the disease, referred to as coronavirus disease 2019 (COVID-19). This has induced the WHO to declare COVID-19 a pandemic in March 2020 (World Health Organization [WHO], 2020). To date (August 2020), the total confirmed cases are more than 24 million according to data provided by the Center for Systems Science and Engineering at Johns Hopkins University1.
Such a situation forced almost all governments to take strong and resolute decisions in some cases culminating in the country lockdown with enormous health, social, and economic effects, the consequences of which will be visible for a long time and nowadays are perhaps difficult to quantify. This has prompted the scientific community to make a considerable effort to quickly gain knowledge about not only SARS-CoV-2–induced pathophysiology but also its biology in terms of virus–host biomolecular and biochemical interactions to identify potential targets of intervention.
Currently, SARS-CoV-2 treatments rely on knowledge gained from other human pathogenic CoV and, more in general, single-strand RNA (ssRNA) virus infections. Although specific signatures characterize each group, ssRNA viruses share similar biological mechanisms regarding their replication and the strategies to exploit and subvert host metabolic pathways, and for some of them, similar pathologies are also triggered (De Albuquerque et al., 2006; Josset et al., 2013; Menachery et al., 2014; Pombo and Sanyal, 2018). Interestingly, modulation of the host lipid metabolism represents a common feature of several viruses (Chukkapalli et al., 2012). Lipids are also involved as immune-metabolic mediators in the innate immune responses, whose functions are crucial in determining the extent of virulence and its outcomes, as they represent the first line of host defenses against invading pathogens (Frieman et al., 2008). Moreover, host lipid reprogramming can also be a host protective response to counteract viral infections.
In light of this, after a brief overview of β-CoVs, here we have reviewed most of the knowledge linking mainly positive-sense (+)ssRNA viruses, especially CoVs, to the host lipid rearrangements, with a highlight on small multifunctional organelles named lipid droplets (LDs) as potential and underexplored targets for future, but not limited to, COVID-19 studies. Moreover, the possible interactions of lipid metabolism with the innate immune responses are also discussed. We refer readers to many excellent and comprehensive reviews and papers disseminated throughout the text providing the mechanistic details of the mentioned processes. In fact, the overall goal of this review is to draw insights and offer hints to expand knowledge on aspects that might play a role in SARS-CoV-2 virulence and other potential future pandemics. This may be helpful for developing new combined approaches interfering with specific steps of the viral life cycle.
The need to discover new strategies for SARS-CoV-2 treatment is reinforced by the fact that ssRNA viruses can rapidly mutate, and even though not all mutations are biologically and pathogenically relevant, this creates concerns for the successful realization of a vaccine in a short period.
To deepen knowledge and understanding of CoV biological interactions with the host cells, including the lipid metabolic rearrangements, will allow shedding light on the mechanisms of infection at multiple levels and will help to improve the current treatment options and to design more effective and promptly available strategies, reducing the impact on public health and supporting the development of safe and effective vaccines.
Overview of Coronaviruses
CoVs are a group of viruses belonging to the family of Coronaviridae and including four genera: α-, β-, γ-, and δ-CoVs. They share some structural and molecular features, such as the envelope, the (+)ssRNA molecule of approximately 30 kilobases, and an endoribonuclease activity (Fehr et al., 2016). CoVs can infect avian and mammalian hosts with the ability to cross the interspecies barriers and for zoonotic transmission causing respiratory, hepatic, gastrointestinal, renal, blood, and central nervous system diseases (Puelles et al., 2020). Currently, seven CoVs have been identified as being able to infect humans. In particular, three of them (all belonging to β-CoVs) gained intense attention as they were recognized as the causative agents of the outbreaks of the severe acute respiratory syndrome (SARS) that emerged in 2002 and called SARS-CoV and of Middle East respiratory syndrome (MERS) that occurred in 2012 and named MERS-CoV. More recently, in December 2019, a novel β-CoV has been identified in China and named SARS-CoV-2 because of its genetic similarity with SARS-CoV (Zhang and Holmes, 2020). Genome-phylogenetic analysis suggests that these three closely related CoVs (∼79% and 50% sequence identity between SARS-CoV-2 and SARS-CoV, and SARS-CoV-2 and MERS-CoV, respectively) (Lu et al., 2020) are most likely derived from bats with an intermediate “amplifying” host that has been potentially identified in masked palm civets and camels for SARS-CoV and MERS-CoV respectively (Zhang and Holmes, 2020) and in Malayan pangolins for SARS-CoV-2 (Xiao et al., 2020). Of note, current data on SARS-CoV-2 do not allow to exclude the possibility for a silent human-to-human transmission earlier than the evidence of the outbreak in China (Zhang and Holmes, 2020).
These highly pathogenic human CoVs induce dysfunction in host immune responses associated with respiratory diseases, which can evolve, in more severe cases, in acute respiratory distress syndrome (ARDS) and can be associated with cytokine storm and ultimately can lead to case fatality (Petrosillo et al., 2020).
The overall case fatality rate reported is ∼34% for MERS, followed by SARS ∼10%, while it is still uncertain for the ongoing pandemic of COVID-19. However, current estimates suggest that it might be lower than the other two, more male gender and age associated (Jin et al., 2020; Petrosillo et al., 2020). However, epidemiologic analyses reveal that SARS-CoV-2 is more infectious than SARS- and MERS-CoV, showing higher transmissibility among individuals (Petrosillo et al., 2020).
In general, CoVs are structurally composed by a nucleocapsid (N), which includes the (+)ssRNA and N proteins, surrounded by a lipid bilayer (envelope) containing envelope (E), spike (S), and membrane (M) structural proteins (NSPs), required to promote and drive the viral internalization and cytoplasmic assembly (Figure 1) (Bartlam et al., 2005). In particular, the S glycoprotein forms spike complexes protruding from the envelope and conferring a typical corona-like appearance (Wrapp et al., 2020). It shows a strong affinity to host cell receptor angiotensin-converting enzyme 2 (ACE2), highly expressed on the lung cell surface, and recently it has been demonstrated that SARS-CoV and SARS-CoV-2 bind the same host receptor. Nevertheless, the two S proteins differ in a few key amino acid residues localized in the receptor-binding domain, which might explain the difference in the ability of infection (Lu et al., 2020).
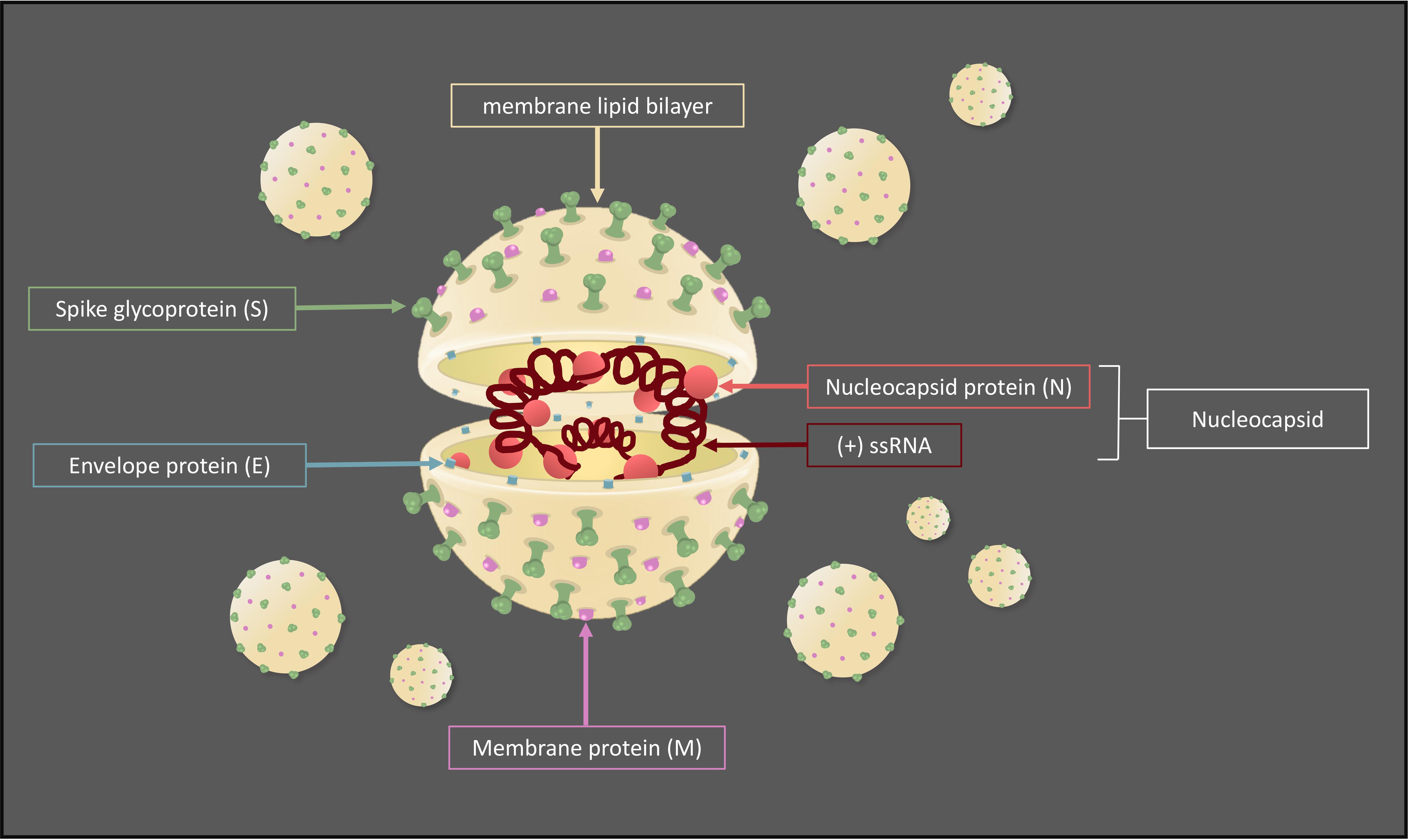
Figure 1. Typical coronavirus structure.
CoV RNA genome is made of a 5′ cap structure and a 3′ poly (A) tail that contains a replicase–transcriptase gene, occupying the largest portion of the genome and encoding for NSPs with multiple enzymatic functions and structural and accessories genes (Lu et al., 2020).
Once inside the host cells, CoVs redirect and exploit the host cell machinery for their proper replicative cycle. As a consequence, cellular signaling and metabolic pathways are rewired to support the infection process and to escape host immune responses.
In this context, based on previous studies on ssRNA viruses sharing several similarities (Table 1), the viral life cycle strongly depends on lipid metabolism, which can be considered a cellular hallmark of the infection-associated changes, thus suggesting a possible target for potential antiviral therapies (Heaton and Randall, 2011; Pombo and Sanyal, 2018).
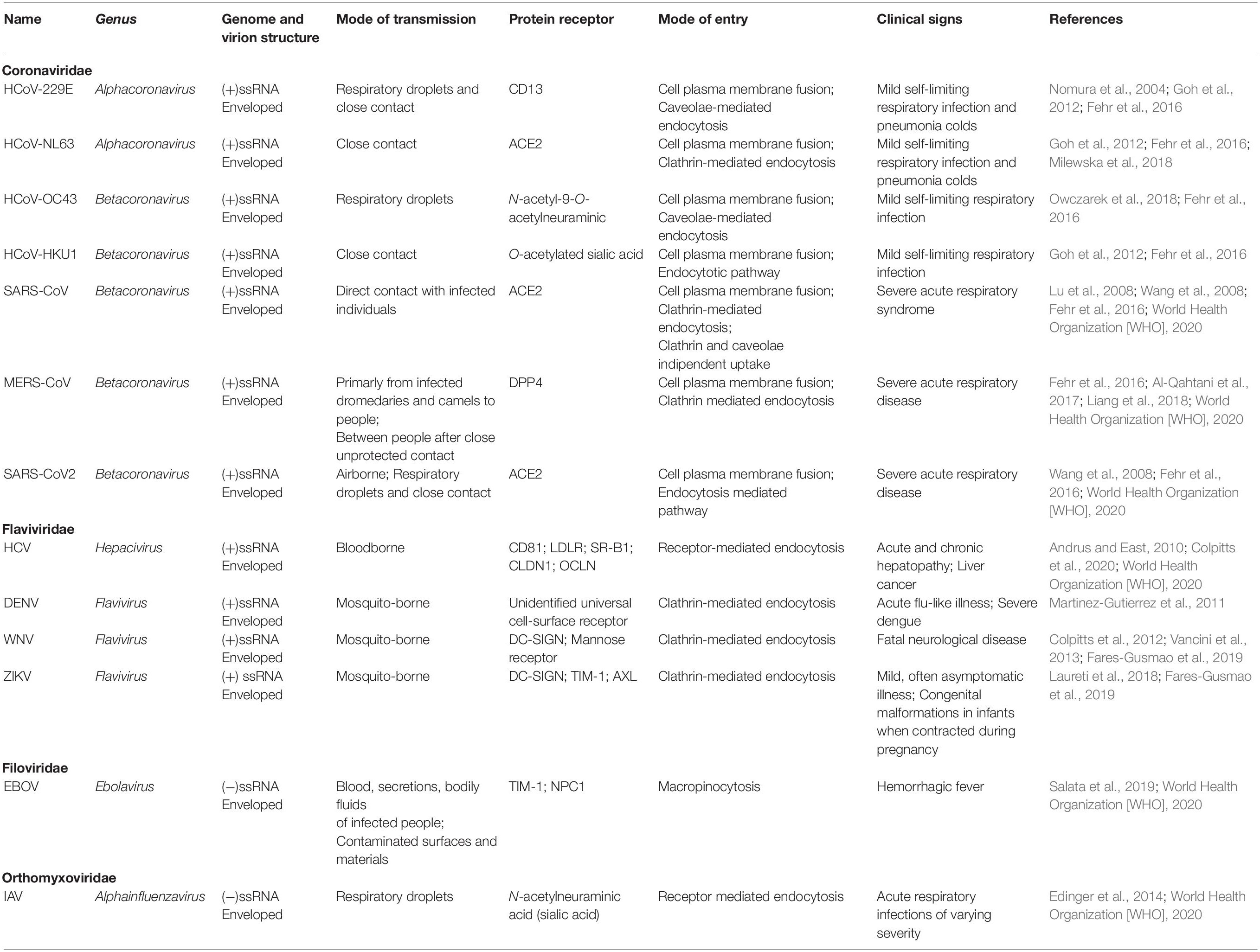
Table 1. Genetic characteristics, transmission, and clinical signs of ssRNA viruses.
To date, no specific antiviral treatments or vaccines have yet been developed against SARS-CoV and MERS-CoV, and also for SARS-CoV-2, this will require time. However, public health concerns are globally and tremendously increasing, calling for urgent solutions dictated by the high burden of these emerging infectious diseases.
Cellular Lipid Modulation Induced by Virus Infection
(+)ssRNA virus infection is a complex of well-coordinated mechanisms starting with the interaction of the virus with the host cell membrane and culminating in viral particle release, for which virus relies on cellular machinery and a rearrangement of cell metabolism. Among the adaptive metabolic responses, a considerable role is given to lipid metabolism, which has been established to be involved in all the viral infection stages (Stapleford and Miller, 2010; Heaton and Randall, 2011; Pombo and Sanyal, 2018). Alterations in host lipid metabolism, mainly fatty acid (FA) metabolism, sterol biosynthesis, synthesis of specific phosphoinositides, and utilization of lipid stores, serve to provide energy and substrates for virus replication and to dampen host antiviral responses (Mackenzie et al., 2007; Pombo and Sanyal, 2018).
Evidence has shown that members of the Coronaviridae family use lipids in different steps of their cycle, although the detailed mechanisms by which they modulate lipid metabolism are not resolved yet (Wang et al., 2008; Romero-Brey and Bartenschlager, 2014).
The binding and internalization of CoVs are mainly receptor-mediated endocytic processes, in which the envelope-located proteins, such as S glycoprotein, recognize and bind specific receptors on the plasma membrane of the host cell, thus allowing also for a synergistic interaction between the viral and the host lipid bilayers (Wang et al., 2008; Lorizate and Krausslich, 2011; Burkard et al., 2014).
The fundamental lipid component of cell membranes is cholesterol, which contributes to membrane fluidity, permeability, and, in general, to the execution of its functions (Zimmerberg and Gawrisch, 2006). It is present in both the viral envelope and the host cell membrane, where it is specifically accumulated in dynamic microdomains known as lipid rafts considered entry sites for some CoVs (Li et al., 2007; Wang et al., 2008). During the first steps of infection, cholesterol role is particularly important. In fact, it has been shown that the α-CoV human coronavirus 229E (HCoV-229E) interaction with the receptor CD13 on human fibroblast caused a specific accumulation of the receptor in lipid rafts. Moreover, the use of the methyl-β-cyclodextrin (MβCD), a plasma membrane cholesterol-removing agent, significantly reduced virus infection (Nomura et al., 2004). Similar results were reported for a pseudotyped SARS-CoV, which required intact lipid rafts, where ACE2 receptors were also sequestered, in order to produce efficient viral infectivity (Lu et al., 2008). Although controversy remains regarding ACE2 association with lipid rafts (Li et al., 2007; Wang et al., 2008), these cholesterol-enriched domains are commonly recognized as essential platforms for entry promotion of SARS-CoV and more in general of (+)ssRNA viruses. Down-regulation of cholesterol synthesis has been linked to reduced susceptibility to (+)ssRNA virus–induced fusion and increased activation of antiviral immunity and, in CoV-infected cells, also type I interferon (IFN)–mediated inflammation (Mackenzie et al., 2007; York et al., 2015; Pombo and Sanyal, 2018).
For this reason, in addition to MβCD, other cholesterol-interfering drugs are now drawing the attention of the scientific community for their potential antiviral actions, such as phytosterols and β-sitosterol (Baglivo et al., 2020). Also, statins (3-hydroxy-3-methylglutaryl CoA reductase inhibitors) cholesterol-lowering molecules are used and debated as antiviral treatments (Fedson, 2006). They have been already used in patients infected with hepatitis C virus (HCV) (Andrus and East, 2010), dengue (DENV) (Martinez-Gutierrez et al., 2011), Ebola (Fedson and Rordam, 2015), and influenza viruses (Mehrbod et al., 2014). Their pleiotropic effects, which include the capability to attenuate the proinflammatory response of cytokines, such as interleukin 6 (IL-6), and to decrease neutrophil influx, as well as to improve the cardiovascular complication, make them potential and intriguing candidates for CoV treatment, particularly for the new COVID-19 outbreak treatment (Weitz-Schmidt, 2002; Fedson et al., 2020). The long-term benefits of these safe, cheap, and widely prescribed drugs, also in combination therapy, for global public health, could be valuable (Fedson et al., 2020).
However, it should also be kept in mind that a tight feedback mechanism regulates cholesterol homeostasis; thus, low cholesterol levels might foster host-compensating mechanisms of which viruses might subsequently take advantage. Indeed, in the setting of antiviral strategies, timely administration can determine the efficacy of treatments, further stressing for the need for intense research for elucidating the modality and the underlying mechanisms of infection.
Upon binding, CoVs are internalized through endosomes, followed by RNA genome release into the cytosol. Intracellular viral replication and assembly usually occur in close association with cellular structures and membrane lipids of several organelles, such as endoplasmic reticulum (ER), Golgi complex, mitochondria, LDs, and lysosomes (Stapleford and Miller, 2010; Heaton and Randall, 2011). It has been reported that the interaction of viral proteins with specific lipids mediates viral genome replication and assembly. Lipids might serve as scaffolds and provide specific signals for viral protein functions as well as localization, thus allowing for proper virus replication (Stapleford and Miller, 2010).
These interactions, which also involve NSP subunits, lead to the extensive reorganization of host intracellular membranes and the formation of donor double-lipid membranes called double-membrane vesicles (DMVs) containing viral dsRNA (Miller and Krijnse-Locker, 2008; Romero-Brey and Bartenschlager, 2014; Lee et al., 2019) (Figure 2). DMVs in SARS-CoVs do not exist as free structures in the cytoplasm but are tightly connected to ER where they derive from, through a complex membrane network. Sometimes DMVs are referred to as “viral replication factories” because they were believed to be the sites for the synthesis of new RNA and compartments where viral components can be concentrated and protected by sensors of host innate immune surveillance (Knoops et al., 2008; Miller and Krijnse-Locker, 2008).
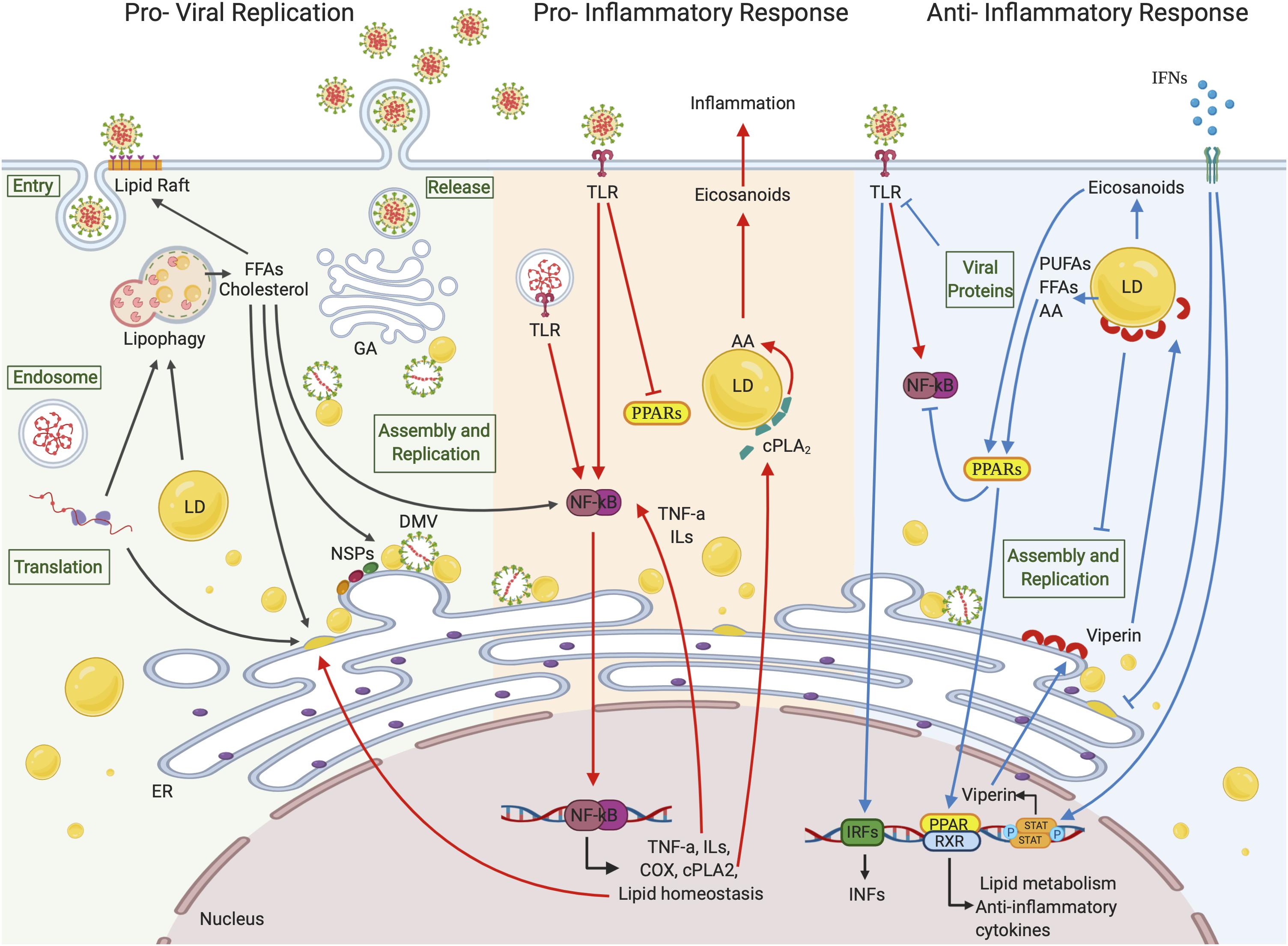
Figure 2. Lipid droplets involvement in (+)ssRNA virus infections. LDs seem to be involved in all the (+)ssRNA virus infection stages. During the first steps of a virus life cycle, thanks to a tight connection with ER vesicles and DMVs, LDs can serve as a platform for virus replication and assembly. LDs are also involved in the inflammation responses. Once viral signatures activate TLRs, an immune signaling cascade is initiated through the activation of the NF-κB pathway leading to the expression of downstream target genes. FFAs generated from LD metabolism rearrangement can also function as activating stimuli to NF-κB itself. cPLA2α localizes on the LD surface where it releases AA, which is converted in eicosanoids supporting inflammation. On the other side, LDs can hinder inflammation by activating PPAR anti-inflammatory signaling pathways or blocking virus replication and assembly through viperin, which localizes on LD surface. Created with BioRender.com.
To sustain de novo membrane formation or to recycle host cell plasma membrane, (+)ssRNA viruses subvert the lipid metabolism pathways and the autophagic flux of the infected cell. To these ends, they do need to finely, and most likely temporarily, tune the processes. In fact, if, on the one hand, lipid rearrangement involves an increase in FAs and cholesterol synthesis (Heaton et al., 2010; Lorizate and Krausslich, 2011; Ketter and Randall, 2019), on the other hand, FA oxidation, cholesterol catabolism, and autophagic membrane degradation are also crucial in order to fulfill high substrate and energy demand for viral replication (Fahmy and Labonte, 2017; Pombo and Sanyal, 2018; Ketter and Randall, 2019). Recently, the lipidomic profiling of HCoV-229E–infected cells showed the up-regulation of FA synthesis and the reprogramming of linoleic acid (LA) to arachidonic acid (AA) metabolism (Yan et al., 2019). Also, infection of lung cells with MERS-CoV produced perturbations of host lipid homeostasis, with steroid biosynthesis being the most up-regulated pathway (Yuan et al., 2019). The importance of cholesterol in (+)ssRNA viral cycle is also demonstrated by the up-regulation of the key enzyme for cholesterol synthesis, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), observed in kidney cells infected with West Nile virus (WNV). Besides, in these cells, a concurrent promotion of intracellular membrane formation and inactivation of the IFN signaling pathway, most likely due to the disruption of lipid rafts after infection, occurred (Mackenzie et al., 2007). Further, it has been shown that cytosolic phospholipase A2α (cPLA2α) which releases free FAs (FFAs), such as AA [an ω-6 polyunsaturated FA (PUFA)] from membrane-associated glycerophospholipids, was critical for CoV replication, in part owing to the production of lysophospholipids required to form DMVs. In fact, cPLA2α involvement in ER-derived membrane web formation and structure has been documented (Brown et al., 2003), and its inhibition by pyrrolidine-2 negatively impacted viral RNA accumulation (Muller et al., 2018).
Taken together, these observations point out that host lipid metabolism is a key target of viral regulation in (+)ssRNA viruses and that a specific lipid profile is most likely requested to ensure productive infection. Therefore, pharmacological perturbations of this altered cellular environment may hinder infection processes by various (+)ssRNA viruses, including the novel SARS-CoV-2.
Lipid Droplets Involvement in (+)ssRNA Virus Infection
As discussed previously, genome replication events of (+)ssRNA viruses occur in tight association with intracellular membranes on specific organelles. Among them, LDs have received particular attention, although direct studies in CoVs are still poor.
Considered only as lipid storage for a long time, their functions are increasingly being expanded. Currently, they are deemed central hubs of the lipid homeostasis, also involved in virus infection and the immune responses (Heaton and Randall, 2011; Saka and Valdivia, 2012; Zhang et al., 2017; Pereira-Dutra et al., 2019). In fact, LD formation is a frequently observed event occurring in cells from the immune system during viral infection (Saka and Valdivia, 2012; Pereira-Dutra et al., 2019).
Lipid droplets are made up of neutral lipids, including triacylglycerols (TAGs), with unsaturated and saturated chains, and cholesteryl esters (CEs), and are surrounded by a phospholipid monolayer containing several different proteins (Cermelli et al., 2006; Farese and Walther, 2009; Tirinato et al., 2017). TAGs and CEs are mainly synthesized by enzymes of glycerol phosphate and mevalonate pathways, respectively, mostly located on the membrane of ER. Among all the enzymes involved, LD generation is strongly promoted by the activity of some of them. In particular, FA synthase (FASN) yields FAs (i.e., palmitate), which are in turn elongated and converted in diacylglycerols (DAGs) and then in TAGs by diacylglycerol o-acyltransferase 1 (DGAT1) and 2 (DGAT2). Key enzymes involved in the cholesterol synthesis and its esters are HMGCR and acyl-CoA cholesterol acyltransferase 1 and 2 (ACAT 1 and 2). When needed, lipids are mobilized and hydrolyzed by specific lipases, such as adipose triglyceride lipase, to yield intermediary metabolites (such as DAGs, FFAs, phosphatidic acid, and sterols) necessary for the membrane synthesis, energy production through β-oxidation, and other anabolic reactions (Walther and Farese, 2012; Pol et al., 2014; Zhang et al., 2017). Cholesterol and FA depletion influences LD size and surface, which is a binding site of different proteins and enzymes, some of them involved in lipid synthesis and mobilization, such as DGAT2, cPLA2α, and other lipases (Yu et al., 1998; Kuerschner et al., 2008; Tirinato et al., 2017; Pereira-Dutra et al., 2019). Recent works have also suggested that HMGCR degradation, which generally occurs in response to high cholesterol levels, takes place on the LD surface (Hartman et al., 2010). Additionally, LDs can be used for protein storage as well as for the production of proinflammatory molecules, such as eicosanoids, deriving from the AA internalized within them (Cermelli et al., 2006; Accioly et al., 2008; Jarc and Petan, 2020).
Lipid droplets are considered to originate from ER and their composition, as well as number and size, can change depending on cell type and stimuli. Accumulation of cytoplasmic LDs in various (+)ssRNA virus–infected cells has been widely reported, and interestingly LDs have been recently proposed as platforms for virus replication and assembly (Miyanari et al., 2007; Samsa et al., 2009; Heaton and Randall, 2011) (Figure 2). In this regard, the targeting of mature capsid proteins to LDs is a common feature shared by some enveloped (+)ssRNA viruses, and it represents a critical step for RNA genome amplification (Miyanari et al., 2007; Samsa et al., 2009; Martins et al., 2019). As an example, it has been shown that capsid (core) protein of HCV localized on LD surface, together with some NSPs, and allowed replication complex-LD association, fundamental to produce infectious viral particles (Miyanari et al., 2007). Associations between clusters of membrane vesicles, DMVs, and LDs were also reported during HCV infection by Ferraris and colleagues (Ferraris et al., 2013).
Moreover, in HCV packaging, DGAT1 activity fostered the association of core protein with LDs, and treatments with DGAT1 inhibitor reduced such association and, consequently, the infectious viral particle production in hepatoma cells. DGAT1 is critical for LD biogenesis; hence, it may represent a druggable host target for antiviral interventions (Herker et al., 2010). Based on the fact that core protein–LD association is observed in different (+)ssRNA viruses, it might be interesting to evaluate and deepen if such a process also occurs in CoV- and SARS-CoV-2–infected cells, and if this is the case, this might open intriguing possibilities for developing broad-spectrum antiviral drugs.
Lipid droplet functions and their cross-talk with other cellular compartments and viral components in infectious diseases are only beginning to be investigated. Beyond being a structural scaffold for viral RNA and particle assembly, the readily available pool of lipids in LDs most likely provides host substrates needed for membrane materials and ATP production by regulating the balance between lipid accumulation (lipogenesis), storage, and mobilization (lipolysis). This regulation is influenced by viruses, and understanding the underlying mechanisms would help to control their replication cycle. For instance, HCV viral infection is able to trigger the sterol regulatory element-binding protein (SREBP) pathway, which induces de novo lipogenesis (Horton et al., 2002), which in turn controls the LD biogenesis, and stimulates core protein–LD formation (Li et al., 2013). Similarly, MERS-CoV caused accumulation of LDs and cholesterol, with SREBP1 and 2 being essential for viral replication. Interestingly, treatment with AM580, a retinoid derivative, inhibited SREBP-mediated lipogenesis and DMV formation (Yuan et al., 2019).
In another study, Rab18, a host protein located on LDs, was essential for DENV replication and for mediating the interaction of FASN with DENV NS3 protein, as well as FASN recruitment on LDs, in order to promote FA synthesis (Tang et al., 2014). Moreover, the inhibition of FASN by means of cerulenin or C75 (trans-4-carboxy-5-octyl-3-methylenebutyrolactone), which also reduced LD content, blocked DENV and HCV replication, indicating that FA production at the site of viral replication is an important requirement for viral particle production (Yang et al., 2008; Samsa et al., 2009; Heaton et al., 2010).
Although LD accumulation associated with viral replication is widely demonstrated, LD degradation is less reported, but equally considered an essential mechanism to establish a favorable microenvironment for infectious progeny virion production. Lipid mobilization from LDs serves not only to sustain viral energy requirements but also to provide viruses with specific lipids for membrane synthesis, and active lipid molecules might also be recruited in major signaling pathways activated or modified by viruses. For example, cPLA2α, which is associated with LDs and is also involved in their biogenesis, hydrolyzes membrane phospholipids, preferentially carrying AA of which LDs are rich deposits, thus releasing FFAs (Figure 2) (Yu et al., 1998; Gubern et al., 2008; Wymann and Schneiter, 2008).
Even though the hydrolytic activity of cPLA2α on LDs has not been proved yet, this suggests that impairment of CoV replication following cPLA2α inhibition, which in turn reduces DMV formation, might be mediated by LDs (Muller et al., 2018). Therefore, cPLA2α-regulated LD production may be investigated as a potential target for managing CoV infection. Moreover, free AA, generated by cPLA2α, might be essential in the membrane reorganization after CoV infection. It is known that AA further stimulates cPLA2α activity, which can favor viral replication. Further, it is also a biologically relevant FA converted into highly bioactive signaling molecules (eicosanoids) involved in inflammation with the aim of recruiting leukocytes and immune cells (Harizi et al., 2008; Ruiperez et al., 2009; Jarc and Petan, 2020). This suggests that by modulating the extent of cPLA2α activation, multiple pathways can be affected, and different effects may be obtained.
Lipid droplet catabolism can also take place through autophagy. In this process named lipophagy, LDs are delivered to the lytic compartments where lipases and hydrolyses mobilize FAs and cholesterol, which are either directed toward β-oxidation or their replenishment in LDs (Liu and Czaja, 2013) (Figure 2). Autophagic processes are interconnected with host cell defense systems, functioning as modulators of inflammation and both innate and adaptive immunity (Delgado et al., 2008; Deretic and Levine, 2009). However, several (+)ssRNA viruses, including CoVs, have evolved the ability either to escape or even exploit the autophagic machinery to promote their survival and sustain their expansion (Prentice et al., 2004; Cottam et al., 2011; Zhang et al., 2017). In a recent work, MERS-CoV infection reduced autophagy, most likely by suppressing the formation of autolysosomes (Gassen et al., 2019). Instead, lipophagy correlated with a reduction in LD size and an increase in β-oxidation in DENV-infected hepatoma cells. This resulted in efficient DENV replication, a process that was in turn affected by autophagy inhibition (Heaton and Randall, 2010).
The dual-effect (proviral or antiviral) of autophagic flux demonstrates the extreme complexity of the regulative mechanisms in the interaction virus–host cell.
To date, the role of LD biogenesis and lipophagy in β-CoV infection is largely underexplored. Whether and how the new SARS-CoV-2 benefits or is disadvantaged from these mechanisms may represent a new research field that would merit to be explored in order to add knowledge and potentially open new possibilities of intervention.
The Innate Immune Response: Toll-Like Receptors and Links With Lipids and Lipid Droplets
As a first line of defense, infected cell respond to viral entry by activating the innate immunity, which represents a fast, although not specific, response mechanism (Akira et al., 2006). Accumulating evidence shows a strong interaction between innate immune signaling pathways and lipid metabolism regulation (Wu et al., 2016; Pombo and Sanyal, 2018; Zhang et al., 2018). Nevertheless, the interdependence of innate immune responses with lipid metabolism in general, and LD specifically, has been little investigated in CoVs.
An important role in influencing the development of the innate antiviral immunity is played by the Toll-like receptors (TLRs). TLRs are a class of proteins (collectively known as pattern recognition receptors) expressed on the leukocyte cell surface, such as macrophages, T and B cells, as well as on non-immune cells and endosomes. TLRs are able to recognize pathogen signatures (referred to as pathogen-associated molecular patterns such as viral nucleic acids and envelope proteins) and initiate the innate immune signaling cascade in order to hinder viral replication and to promote the adaptive immunity (Akira et al., 2006; Kawai and Akira, 2010). Activated downstream pathways of TLRs involve, among others, the activation of nuclear factor-κ light chain-enhancer of activated B cells (NF-κB) signaling, which culminates in the modulation both of key proinflammatory cytokines, such as tumor necrosis factor (TNF-α), ILs, and IFNs (this latter also regulated through interferon regulatory factors), and of lipid homeostasis (Alexopoulou et al., 2001; Akira and Takeda, 2004) (Figure 2). In fact, TAG synthesis and CE accumulation upon TLR activation have been reported, with FASN, DGAT1 and DGAT2, and ACAT1 and ACAT2 being the enzymes more expressed (Pereira-Dutra et al., 2019). Further, distinct membrane lipids can influence TLR signaling, and activated TLRs can be recruited into lipid rafts, highlighting a critical role of these lipid microdomains in participating in cellular immune responses (Bonham et al., 2014; Ruysschaert and Lonez, 2015; Koberlin et al., 2016).
Evidence shows that cytokines can have an indirect antiviral activity by modulating FA and cholesterol metabolism (Zhang et al., 2018). For example, type I IFNs participate in lipid metabolism regulation, on the one hand, by down-regulating cholesterol biosynthetic pathway and inducing FA oxidation to limit the availability of substrates for viral replication, reinforce IFN responses, and establish a hostile microenvironment. On the other hand, type I IFNs can also promote cholesterol import, for example, to stimulate foam cell production (York et al., 2015; Wu et al., 2016; Pombo and Sanyal, 2018). These observations emphasize the tight link between the IFN system and lipid regulation as a possible mechanism that contributes to the metabolic changes occurring upon pathogen invasion (Figure 2).
Moreover, autophagic processes, which remove viral components, can also stimulate antiviral type I IFN-mediated responses by activating endosomal TLRs, and in turn, TLRs and cytokines are autophagy modulators (Delgado et al., 2008; Deretic and Levine, 2009; Li et al., 2012).
Conversely, (+)ssRNA viruses have developed multiple strategies to subvert the host innate immune responses at various levels, including (i) the suppression or the induction of TLR activation and its signaling, (ii) the inhibition of IFN system, and (iii) the damping of autophagy-dependent activation of immunity (Munoz-Jordan et al., 2003; Li et al., 2005; Abe et al., 2007; Deretic and Levine, 2009; Al-Qahtani et al., 2017). For instance, to favor their replication cycle while eluding the immune system, SARS-CoV can encode proteins that inhibit TLR signaling. In fact, although exogenous treatment with type I IFNs can reduce SARS-CoV and HCoV-229E infections, viral strategies, mediated by viral NSP3 macrodomain, or the open reading frame (ORF) 3b, ORF 6, and N proteins, are also activated to suppress the effect of IFNs and promote rapid spreading (Kopecky-Bromberg et al., 2007; Kuri et al., 2011; Fehr et al., 2016). Also, it has been reported that, in MERS-CoV–infected macrophages, a crucial role in immunosuppression was played by the S glycoprotein, which blocked TNF-α and IL-6 production, while down-regulating TLR signaling (Al-Qahtani et al., 2017).
However, it has been shown that SARS-CoV could also stimulate TLR3 and TRL4 expression, which resulted in protective effects in a mouse model (Totura et al., 2015). Also, protection was observed in the early phase of infection, thanks to the production of type I IFN by plasmacytoid dendritic cells (pDCs), via TRL activation, in SARS-infected donor cells (Cervantes-Barragan et al., 2007). Conversely, a delay in type I IFN signaling correlated with high levels of proinflammatory cytokines and chemokines and lethality in SARS-CoV–infected mice (Channappanavar et al., 2016). Of note, conflicting reports from both SARS and COVID-19 patients, as well as from various cell culture systems productively infected, show differences in IFN and cytokine induction, which may reflect differences in patient cohorts/cell lines, as well as in temporal analysis and assay sensitivity (Frieman et al., 2008; Guo et al., 2020). While a low type I IFN activation can benefit to viral replication, an overactivation of this pathway can be harmful for the infected patients. Actually, dysregulated IFN-mediated immune responses and aberrant cytokine release are phenomena frequently observed, although not fully characterized yet, in severe SARS patients (Peiris et al., 2004; Wang et al., 2007) and, more recently, in severe COVID-19 patients usually correlated with poor prognosis (Huang et al., 2020).
Overall, these observations indicate that properly regulated TLR activation and IFN responses represent a critical aspect of the CoV-related immunopathogenesis.
In this regard, although LDs are supposed to be a platform for viral replication, they might also contribute in antiviral signaling events, suggesting a double role for these organelles, which makes the scenario more complex and less trivial than anticipated (Monson et al., 2018). In fact, LD biogenesis is a well-established event occurring in cells of the immune system following pathogen infections and inflammation. (+)ssRNA viruses are able to trigger LD accumulation and enlargement in various leukocytes as a marker of cell activation (Bozza et al., 2009; Syed et al., 2010). Further, it has been shown that, in HCV-infected human hepatoma cell lines, LDs were the colocalization site of viperin (also known as RSAD2) and HCV NSP 5A, an event resulting in limited HCV replication (Helbig et al., 2011). In fact, viperin is a protein coded by one of the interferon-stimulated genes (ISGs) either after binding of IFNs to their receptors or via an IFN-independent pathway (Seo et al., 2011). Its antiviral properties have been documented, as well as its accumulation in LDs (Hinson and Cresswell, 2009; Helbig et al., 2011; Seo et al., 2011). Saitoh et al. (2011) have shown that viperin localized to LDs and promoted the activation of the TLR 7 and 9 pathways in pDCs, and they have proposed LDs as transit points for the consequent type I IFN production. In another study, Monson and coworkers (Monson et al., 2018) highlighted how low LD content in human cell lines challenged with dsRNA viral mimics reduced IFN production, viperin expression, thus delaying the host immune response.
As reported in the previous paragraph, cPLA2α releases AA from LDs. Of note, cPLA2α is activated by several factors, including TLRs (Pindado et al., 2007; Ruiperez et al., 2009). Once produced, AA is the substrate for the synthesis of eicosanoids involved in the initiation of inflammatory cascades and in the recruitment and infiltration of neutrophils (Harizi et al., 2008; Jarc and Petan, 2020). It was shown that AA metabolic pathway was up-regulated in HCoV-229E–infected cells and that treatment with exogenous AA inhibited both HCoV-229E and MERS-CoV replication (Yan et al., 2019). This suggests another potential link between LDs in CoVs and immune responses.
Although the connection between LDs, viral infection, and the immune response has been underexploited and is only starting to attract interest from the scientific community, current knowledge supports the idea that, through TLR activation, LDs may also act as mediators participating in the induction of innate immune responses upon CoV infection. The regulatory processes involved in a proviral or antiviral role for LDs also remain to be explored.
Linking NF-κB–Mediated Responses With Lipid Metabolism in (+)ssRNA Virus Infections
The signaling cascade initiated by TLRs leads to the activation of NF-κB pathway, which usually represents a common feature of many viral infections (Akira and Takeda, 2004; Kawai and Akira, 2006). NF-κB is a ubiquitous transcriptional factor that regulates the expression of hundreds of target genes, many of which participate in immune and inflammatory processes. NF-κB family includes five members of inducible transcription factors: NF-κB1 (also named P50), NF-κB2 (or P52), RelA (or P65), RelB, and c-Rel (Akira et al., 2006; Liu et al., 2017). Their association with inhibitory proteins, belonging to the inhibitor of κB (IκB) protein family, sequesters NF-κB factors in the cytoplasmic compartment, making them inactive. Several stimuli can induce NF-κB activation, such as exposure to cytokines (ILs and TNF) and viral infections. This results in the degradation of IκB members, followed by NF-κB dimer release and translocation to the nucleus, where it binds to DNA-specific sequences of proinflammatory genes, including a subset of ISGs (Barnes and Karin, 1997; Akira and Takeda, 2004; Pfeffer, 2011) (Figure 2). Interestingly, it has been demonstrated that viperin, which is a ISG, is involved in type I IFN production through an NF-κB–orchestrated mechanism in carcinoma cells (Moschonas et al., 2012). It may be of relevance to evaluate whether such a mechanism also occurs upon SARS-CoV-2 infection and whether LDs are entailed to some extent in this axis.
Of note, IFNs and FFAs, derived from lipid rearrangements, also lead to NF-κB signaling activation by favoring the dissociation of IκB-α, thereby initiating the proinflammatory responses (Yang et al., 2000; Baker et al., 2011). To this regard, possible links between NF-κB and lipid metabolism have been described in different cell types. For instance, it has been reported that NF-κB activation induced SREBP-1a expression in macrophages, as the SREBP1 promoter region contains an NF-κB response element, and subsequently stimulated lipogenesis and IL-1β production (Im et al., 2011). Similarly, in liver cells, exogenous FAs induced NF-κB pathway and SREBP-1c expression with LD formation (Jung et al., 2013). These observations further reinforce the connection between the innate immune responses and the lipid metabolism and LDs also through NF-κB–dependent mechanisms.
Many inflammatory lung diseases, such as asthma, are characterized by a constitutive NF-κB activation, which leads to the release of cytokines and adhesion molecules. This effect helps to recruit activated immune and inflammatory cells to the site of interest, thus amplifying and, in some cases, even exacerbating the inflammatory state (Barnes and Karin, 1997; Schuliga, 2015). Similarly, viral infections can trigger NF-κB–mediated immune and proinflammatory responses as a host defense mechanism and, at the same time, viruses can develop counter-strategies to actively modulate these networks for their purposes. In fact, prolonged NF-κB activation has been shown to be fundamental for the inflammatory immunopathology induced by β-CoVs (Smits et al., 2010; DeDiego et al., 2014). In particular, in vitro studies showed that SARS-CoV S protein triggered NF-κB pathway causing a marked up-regulation of it in macrophages compared to control with a simultaneous massive release of IL-6 and TNF-α (Wang et al., 2007; Dosch et al., 2009). Similar effects were induced by SARS-CoV E protein in in vitro and in vivo studies (DeDiego et al., 2014). Remarkably, these two cytokines can, in turn, further stimulate NF-κB through a positive feedback loop and increase cytokine levels, thus amplifying the responses and leading to aberrant inflammation (Barnes and Karin, 1997). Additionally, secreted ILs and TNF-α exhibit an intense regulatory action on lipid homeostasis. In fact, TNF-α is able to promote both de novo lipogenesis and to induce lipolysis, this latter effect also induced by IL-6, as well as to inhibit the activity of different lipid-related enzymes and to control cholesterol metabolism (Chen et al., 2009; Lehrskov and Christensen, 2019). However, other studies have shown that SARS-CoV N protein was able to either dramatically inhibit NF-κB, which in part could explain the observed inhibition of IFN synthesis or activate it. These findings suggest that NF-κB induction and its signaling might be cell context-specific and may influence distinct aspects of immune function (Kopecky-Bromberg et al., 2007; Zhang et al., 2007). Nevertheless, although accumulating data highlight a central of NF-κB as modulator of antiviral responses in β-CoVs, its involvement in lipid metabolism has not yet been fully investigated in the context of CoV infections.
As the central driver of the inflammation and innate immunity triggered by viruses, NF-κB is considered as a potential therapeutic target, and hundreds of inhibitors have been studied (Karin et al., 2004; Miller et al., 2010). Among them, it has been reported that statins reduce the levels of proinflammatory cytokines by inhibiting NF-κB activity (Hilgendorff et al., 2003). Moreover, treatments with NF-κB inhibitors, caffeic acid phenethyl ester (CAPE) and parthenolide, reduced inflammation and lung disease and increased survival after SARS-CoV infection in in vitro and in vivo studies (DeDiego et al., 2014). Another therapeutic approach explored against an α-CoV was the combination of specific NF-κB signaling inhibitors together with small molecules, called tylophorine, which resulted in interfering with the virus replication and reducing proinflammatory responses (Yang et al., 2017). However, it is worth to note that NF-κB pathway is involved in multiple cellular events and is physiologically required for the maintenance of normal immune response and cell survival; therefore, before using NF-κB–modulating drugs as a potential strategy for COVID-19 patients, it is fundamental to carefully evaluate the balance between efficacy and safety. This aspect requires intense investigations of the influenced pathways. In this respect, future integrated data from genomic, proteomic, and lipidomic analyses will help to decipher the overall functional effects of NF-κB inhibition in vitro and in vivo and to design more specific combined strategies.
Peroxisome Proliferators Activated Receptors and Lipid Metabolism in Viral Infection
Immune and inflammation reactions are also modulated by lipid molecules through another class of receptors, called peroxisome proliferator-activated receptors (PPARs), which interfere with NF-κB signaling. PPARs represent a family of nuclear hormone transcriptional factors activated by lipid ligands, such as oxidized FAs, eicosanoids, and lysophosphatidic acid, with a significant role exerted by PUFAs (Harmon et al., 2011; Varga et al., 2011). After heterodimerization with the retinoid X receptor, they recognize and bind the peroxisome proliferator–responsive elements (PPREs) present on the promoter of target genes, thus activating them (Harmon et al., 2011; Poulsen et al., 2012).
By now, three isoforms of this family have been discovered, each of them modulating lipid homeostasis in distinct ways: PPARα, PPARβ/δ, and PPARγ (Harmon et al., 2011; Varga et al., 2011). PPARα activates FA oxidation and is highly expressed in the liver, heart, and brown adipose tissue. PPARβ/δ is mainly involved in FA catabolism in several tissues but especially in skeletal muscle, whereas PPARγ regulates adipogenesis, lipid metabolism, and storage in all tissues, mostly adipose tissue (Di Paola and Cuzzocrea, 2007; Poulsen et al., 2012).
Considered as lipid sensors able to sense cellular lipid milieu, PPARs positively regulate the expression of genes whose products are involved at different stages of FA and cholesterol metabolism, such as FA transporter proteins and acyl-CoA synthetase, which catalyzes the FA conversion in ester compounds (acyl-CoAs) (Gervois et al., 2000). As such, activated FAs are substrates for de novo lipid synthesis, β-oxidation, AA production, and membrane remodeling (Grevengoed et al., 2014).
Moreover, PPARs also participate in LD biogenesis (de la Rosa Rodriguez and Kersten, 2017; Souza-Moreira et al., 2019). In particular, PPARγ is proven to up-regulate the expression of genes involved in TAG synthesis and LD-associated proteins, such as perilipin (PLIN) family members. In fact, the presence of PPRE sequences in the promoter region of some PLINs has been demonstrated (Nagai et al., 2004; de la Rosa Rodriguez and Kersten, 2017; Pereira-Dutra et al., 2019).
Peroxisome proliferator-activated receptors activity is positively modulated by intracellular concentrations of ligands, and in the absence of the ligands, the association of the heterodimers with corepressors suppresses target gene transcription. This highlights the ability of these nuclear receptors to activate positive or negative regulatory responses depending on the cellular context (Ricote and Glass, 2007; Glass and Saijo, 2010).
Peroxisome proliferator-activated receptors role upon (+)ssRNA viral infection is poorly investigated, and information is mainly obtained from studies with HCV-infected cells. In general, altered production of lipid ligands induced by viral replication may influence nuclear receptor expression and activity. Nevertheless, PPAR activation and modulation of the lipid microenvironment may interfere with viral responses. In this context, an impaired expression of PPARα, most likely influenced by HCV core protein, has been reported during the infection in patient-derived liver cells (Dharancy et al., 2005). However, further studies showed that, in chronic infection of various liver cells, treatment with a PPARα antagonist, which also down-regulates hydroxymethylglutaryl-CoA (HMG-CoA) synthase gene, was able to reduce HCV replication and resulted in increased lipid and LD accumulation likely, at least in part, owing to the role exhibited by PPARα in lipid catabolism (Rakic et al., 2006). Inhibition of the PPAR signaling pathway was also observed in DENG, and WNV-infected asymptomatic donors (Fares-Gusmao et al., 2019). By contrast, a recent study from Al-Qahtani et al. (2017) reported the induction of PPARγ in MERS-infected cells, which occurred after 24 h and not at earlier stages of stimulation. PPARγ activation was mediated by S glycoprotein with concurrent inhibition of macrophage responses and suppression of proinflammatory cytokines. In this regard, PPARs can also be stimulated by AA-derived metabolites (prostaglandins and leukotrienes), that, as previously reported, are mediators of the inflammatory responses (Devchand et al., 1996; Ricote et al., 1998; Lee et al., 2003; Varga et al., 2011). Accumulating data support the interconnection of PPAR signaling with inflammation and immunity, as already well-established in the pathogenesis of some diseases, also in the context of viral infection. In fact, PPARs are expressed in vascular and immune cells, such as in dendritic cells (DCs), macrophages, and lymphocytes (Szatmari et al., 2007; Glass and Saijo, 2010; Varga et al., 2011).
Notably, PPARγ appears to be a critical regulator involved in the immune responses, and more particularly in activated macrophages, where its expression is markedly up-regulated. PPARγ up-regulation and its ability to antagonize the activities of the transcription factors, such as NF-κB (via the up-regulation of IκBα), result in the repression of genes encoding for proinflammatory cytokines, such as TNFα, IL-6, IL-8, and IL-1β, and in the induction of anti-inflammatory cytokines, such as IL-10 and transforming growth factor (Ricote et al., 1998; Croasdell et al., 2015) (Figure 2). In addition, PPAR activation can inhibit IFN production, and concurrently cytokines can modulate PPAR expression in immune cells (Ricote and Glass, 2007).
Because of the inhibition mechanism exerted by PPARγ on NF-κB signaling, it is reasonable to argue that an interplay between PPARs and TLRs exists. Indeed, although the underlying mechanisms remain elusive, it has been proposed that TLRs/NF-κB signaling may block PPARγ activation in various cell types (Dana et al., 2019). Additionally, in vitro and in vivo studies showed that upon influenza virus infection, the expression of PPARγ in alveolar macrophages was down-regulated via type I IFNs, a strategy that most likely helps to respond to viral entry quickly. However, PPARγ complete loss produced increased proinflammatory cytokine release, which led to the worsening of the pathology (Huang et al., 2019). In fact, it is believed that activated PPARs counteract the amplification of the TLR/NF-κB pathway. In this regard, it has been shown that the activation of DCs, stimulated by synthetic analogs of dsRNA, which binds TLR3, could be inhibited by PPARγ agonists and resulted in the down-regulation of NF-κB pathway (Appel et al., 2005).
These observations, although far to be conclusive, support the idea that PPARs might participate in attenuating the inflammatory and immune responses after their induction following viral infection. This event requires to be temporarily and finely regulated in an attempt to limit the excessive production of inflammatory mediators, while not wholly abolishing them to allow an adequate antiviral reaction and to mount the adaptive immunity (Bassaganya-Riera et al., 2010). Interestingly, Szatmari et al. (2007) reported that PPARγ activation in DCs resulted in the up-regulation of lipid metabolism genes and reduction in LD content in the early stages. In contrast, genes related to immune responses were modulated at later time points, thus showing a temporal link between lipid and immune regulation (Szatmari et al., 2007).
Furthermore, based on the role of cPLA2α in AA metabolism, it was shown that PPARγ activity in airway epithelial cells could be stimulated by the overexpression of cPLA2α, which consequently increased PPARγ-mediated gene transcription through induction of PPRE reporter activity, in particular IL-8 and cyclooxygenase-2 (COX-2) expression (Pawliczak et al., 2002, 2004). Indeed, evidence shows that PPARs participate in the modulation of inflammation occurring during lung inflammation and ARDS (Di Paola and Cuzzocrea, 2007; Bassaganya-Riera et al., 2010), and activated PPARs can reduce the fibrotic responses in models of lung injury (Lakatos et al., 2007), which can also occur in severe CoV and, more specifically, COVID-19 patients (Hui et al., 2005; Spagnolo et al., 2020). In this regard, PPARγ agonists have been suggested as possible antiviral therapy for pulmonary virus-induced diseases (Bassaganya-Riera et al., 2010).
Lastly, evidence shows that a gender-related expression of PPARs in cells of the immune systems exists. In fact, while androgen hormones can increase PPARα gene expression, estrogens influence PPARγ gene expression. Consequently, PPARα is usually overexpressed in male T cells, and PPARγ is more abundant in female T cells, which also exhibit a differential cytokine production. Such a hormone-specific influence on PPAR expression might influence the gender-related infection susceptibility and disease outcomes (Zhang et al., 2012; Park and Choi, 2017).
In light of these considerations, it would be appealing to elucidate the PPAR role in lipid and LD homeostasis, as well as weather and how PPARs-TLRs/NF-kB axis might regulate them, during CoV infection especially in acute phases.
Concluding Remarks
The COVID-19 sanitary emergency has shown us that it became imperative and urgent to explore and evaluate new possible biological routes that can result in rapid and effective antiviral therapies to treat SARS-CoV-2 infection. To this purpose, the integration of clinical with molecular data and the creation of shared databases are crucial and useful to better deal with the current and the future outbreaks. This requires intense and robust research that allows unveiling the specific mechanisms of infection and identifying precise and potentially combined points of intervention.
Based on a growing body of knowledge, deregulated host lipid metabolism is a typical signature observed in many (+)ssRNA virus infections, CoVs included, as a proviral or antiviral mechanism, also having profound effects on innate immune responses. In this scenario, LDs seem to be active players at the crossroad of different ways by providing support as well as key metabolites, according to the renewed view as multifunction organelles also involved in viral replication. This evidence drove us to wonder whether a putative role for lipids and LDs in SARS-CoV-2 can also exist, and if this is the case, they might be direct or indirect druggable targets.
While waiting for safe vaccines, host-directed antiviral therapies seem to be a more promising approach against infectious diseases than pathogen-directed strategies, due to the possibility to target multiple aspects at the same time, reduce the impact of drug resistance, and develop broad-spectrum antiviral drugs able to treat different, but still similar, virus strains. Of course, this option imposes the need for a careful evaluation of the benefits, while minimizing the adverse effects for the host. Currently, several safe and cheap drugs interfering with lipids and LDs, which are host factors requested for the viral life cycle, exist and are already on the market. These could be considered and promptly tested in order to evaluate their effectiveness in SARS-CoV infections.
To date, multiple approaches have been tested on patients based on previous and limited studies, also in absence of clear data on the potential effects. This has been dictated, and intrinsically approved, by the need to rapidly manage and respond in the initial phases of this pandemic. However, now it is important both to get data from large randomized trials and to stimulate research in order to define safe therapeutic strategies only after accurate and validated studies.
Author Contributions
LT conceived the idea. FP, MM, and LT studied the topic and wrote the first draft. GG and PC organized the literature database and contributed to draw the table and figures. GV and JS provided the ideas contributing to conception and design of the manuscript. FP and LT supervised the project. All the authors contributed to manuscript revision, read, and approved the submitted version.
Funding
LT has received funding from AIRC and from the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 800924. This work was also supported by the Department of Biomedical Physics in Radiation Oncology (E041 Division) at the DKFZ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Abe, T., Kaname, Y., Hamamoto, I., Tsuda, Y., Wen, X. Y., Taguwa, S., et al. (2007). Hepatitis C virus non-structural protein 5A modulates the toll-like receptor-MyD88-dependent signaling pathway in macrophage cell lines. J. Virol. 81, 8953–8966. doi: 10.1128/Jvi.00649-07
Accioly, M. T., Pacheco, P., Maya-Monteiro, C. M., Carrossini, N., Robbs, B. K., Oliveira, S. S., et al. (2008). Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 68, 1732–1740. doi: 10.1158/0008-5472.CAN-07-1999
Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511. doi: 10.1038/nri1391
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. doi: 10.1038/35099560
Al-Qahtani, A. A., Lyroni, K., Aznaourova, M., Tseliou, M., Al-Anazi, M. R., Al-Ahdal, M. N., et al. (2017). Middle east respiratory syndrome corona virus spike glycoprotein suppresses macrophage responses via DPP4-mediated induction of IRAK-M and PPAR gamma. Oncotarget 8, 9053–9066. doi: 10.18632/oncotarget.14754
Andrus, M. R., and East, J. (2010). Use of statins in patients with chronic hepatitis C. S. Med. J. 103, 1018–1022. doi: 10.1097/SMJ.0b013e3181f0c6b4
Appel, S., Mirakaj, V., Bringmann, A., Weck, M. M., Grünebach, F., and Brossart, P. (2005). PPAR-γ agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-κB pathways. Blood 106, 3888–3894. doi: 10.1182/blood-2004-12-4709
Baglivo, M., Baronio, M., Natalini, G., Beccari, T., Chiurazzi, P., Fulcheri, E., et al. (2020). Natural small molecules as inhibitors of coronavirus lipid-dependent attachment to host cells: a possible strategy for reducing SARS-COV-2 infectivity? Acta Biomed. 91, 161–164. doi: 10.23750/abm.v91i1.9402
Baker, R. G., Hayden, M. S., and Ghosh, S. (2011). NF-κB, inflammation, and metabolic disease. Cell Metab. 13, 11–22. doi: 10.1016/j.cmet.2010.12.008
Barnes, P. J., and Karin, M. (1997). Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071. doi: 10.1056/NEJM199704103361506
Bartlam, M., Yang, H. T., and Rao, Z. H. (2005). Structural insights into SARS coronavirus proteins. Curr. Opin. Struct. Biol. 15, 664–672. doi: 10.1016/j.sbi.2005.10.004
Bassaganya-Riera, J., Song, R., Roberts, P. C., and Hontecillas, R. (2010). PPAR-gamma activation as an anti-inflammatory therapy for respiratory virus infections. Viral Immunol. 23, 343–352. doi: 10.1089/vim.2010.0016
Bonham, K. S., Orzalli, M. H., Hayashi, K., Wolf, A. I., Glanemann, C., Weninger, W., et al. (2014). A promiscuous lipid-binding protein diversifies the subcellular sites of Toll-like receptor signal transduction. Cell 156, 705–716. doi: 10.1016/j.cell.2014.01.019
Bozza, P. T., Magalhaes, K. G., and Weller, P. F. (2009). Leukocyte lipid bodies - biogenesis and functions in inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1791, 540–551. doi: 10.1016/j.bbalip.2009.01.005
Brown, W. J., Chambers, K., and Doody, A. (2003). Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 4, 214–221. doi: 10.1034/j.1600-0854.2003.00078.x
Burkard, C., Verheije, M. H., Wicht, O., van Kasteren, S. I., van Kuppeveld, F. J., Haagmans, B. L., et al. (2014). Coronavirus cell entry occurs through the Endo-/Lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 10:e1004502. doi: 10.1371/journal.ppat.1004502
Cermelli, S., Guo, Y., Gross, S. P., and Welte, M. A. (2006). The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr. Biol. 16, 1783–1795. doi: 10.1016/j.cub.2006.07.062
Cervantes-Barragan, L., Zust, R., Weber, F., Spiegel, M., Lang, K. S., Akira, S., et al. (2007). Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 109, 1131–1137. doi: 10.1182/blood-2006-05-023770
Channappanavar, R., Fehr, A. R., Vijay, R., Mack, M., Zhao, J., Meyerholz, D. K., et al. (2016). Dysregulated Type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microb. 19, 181–193. doi: 10.1016/j.chom.2016.01.007
Chen, X., Xun, K., Chen, L., and Wang, Y. (2009). TNF-α, a potent lipid metabolism regulator. Cell Biochem. Funct. 27, 407–416. doi: 10.1002/cbf.1596
Chukkapalli, V., Heaton, N. S., and Randall, G. (2012). Lipids at the interface of virus-host interactions. Curr. Opin. Microbiol. 15, 512–518. doi: 10.1016/j.mib.2012.05.013
Colpitts, C. C., Tsai, P. L., and Zeisel, M. B. (2020). Hepatitis C virus entry: an intriguingly complex and highly regulated process. Int. J. Mol. Sci. 21:2019. doi: 10.3390/ijms21062091
Colpitts, T. M., Conway, M. J., Montgomery, R. R., and Fikrig, E. (2012). West nile virus: biology, transmission, and human infection. Clin. Microbiol. Rev. 25, 635–648. doi: 10.1128/CMR.00045-12
Cottam, E. M., Maier, H. J., Manifava, M., Vaux, L. C., Chandra-Schoenfelder, P., Gerner, W., et al. (2011). Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy 7, 1335–1347. doi: 10.4161/auto.7.11.16642
Croasdell, A., Duffney, P. F., Kim, N., Lacy, S. H., Sime, P. J., and Phipps, R. P. (2015). PPARgamma and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015:549691. doi: 10.1155/2015/549691
Dana, N., Vaseghi, G., and Haghjooy Javanmard, S. (2019). Crosstalk between peroxisome proliferator-activated receptors and toll-like receptors: a systematic review. Adv. Pharm. Bull. 9, 12–21. doi: 10.15171/apb.2019.003
De Albuquerque, N., Baig, E., Ma, X., Zhang, J., He, W., Rowe, A., et al. (2006). Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 80, 10382–10394. doi: 10.1128/jvi.00747-06
de la Rosa Rodriguez, M. A., and Kersten, S. (2017). Regulation of lipid droplet-associated proteins by peroxisome proliferator-activated receptors. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862(10 Pt B), 1212–1220. doi: 10.1016/j.bbalip.2017.07.007
DeDiego, M. L., Nieto-Torres, J. L., Regla-Nava, J. A., Jimenez-Guardeño, J. M., and Fernandez-Delgado, R. (2014). Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 88, 913–924. doi: 10.1128/jvi.02576-13
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121. doi: 10.1038/emboj.2008.31
Deretic, V., and Levine, B. (2009). Autophagy, immunity, and microbial adaptations. Cell Host Microb. 5, 527–549. doi: 10.1016/j.chom.2009.05.016
Devchand, P. R., Keller, H., Peters, J. M., Vazquez, M., Gonzalez, F. J., and Wahli, W. (1996). The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 384, 39–43. doi: 10.1038/384039a0
Dharancy, S., Malapel, M., Perlemuter, G., Roskams, T., Cheng, Y., Dubuquoy, L., et al. (2005). Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology 128, 334–342. doi: 10.1053/j.gastro.2004.11.016
Di Paola, R., and Cuzzocrea, S. (2007). Peroxisome proliferator-activated receptors and acute lung injury. PPAR Res. 2007:63745. doi: 10.1155/2007/63745
Dosch, S. F., Mahajan, S. D., and Collins, A. R. (2009). SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 142, 19–27. doi: 10.1016/j.virusres.2009.01.005
Edinger, T. O., Pohl, M. O., and Stertz, S. (2014). Entry of influenza A virus: host factors and antiviral targets. J. Gen. Virol. 95, 263–277. doi: 10.1099/vir.0.059477-0
Fahmy, A. M., and Labonte, P. (2017). The autophagy elongation complex (ATG5-12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci. Rep. 7:40351. doi: 10.1038/srep40351
Farese, R. V., and Walther, T. C. (2009). Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 139, 855–860. doi: 10.1016/j.cell.2009.11.005
Fares-Gusmao, R., Rocha, B. C., Sippert, E., Lanteri, M. C., Anez, G., and Rios, M. (2019). Differential pattern of soluble immune markers in asymptomatic dengue, West Nile and Zika Virus Infections. Sci. Rep. 9:17172. doi: 10.1038/s41598-019-53645-w
Fedson, D. S. (2006). Pandemic influenza: a potential role for statins in treatment and prophylaxis. Clin. Infect. Dis. 43, 199–205. doi: 10.1086/505116
Fedson, D. S., Opal, S. M., and Rordam, O. M. (2020). Hiding in plain sight: an approach to treating patients with severe COVID-19 infection. mBio 11:e0398-20. doi: 10.1128/mBio.00398-20
Fedson, D. S., and Rordam, O. M. (2015). Treating Ebola patients: a ‘bottom up’ approach using generic statins and angiotensin receptor blockers. Intern. J. Infect. Dis. 36, 80–84. doi: 10.1016/j.ijid.2015.04.019
Fehr, A. R., Channappanavar, R., Jankevicius, G., Fett, C., Zhao, J., Athmer, J., et al. (2016). The Conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome Coronavirus Infection. mBio 7:e01721-16. doi: 10.1128/mBio.01721-16
Ferraris, P., Beaumont, E., Uzbekov, R., Brand, D., Gaillard, J., Blanchard, E., et al. (2013). Sequential biogenesis of host cell membrane rearrangements induced by hepatitis C virus infection. Cell. Mol. Life Sci. 70, 1297–1306. doi: 10.1007/s00018-012-1213-0
Frieman, M., Heise, M., and Baric, R. (2008). SARS coronavirus and innate immunity. Virus Res. 133, 101–112. doi: 10.1016/j.virusres.2007.03.015
Gassen, N. C., Niemeyer, D., Muth, D., Corman, V. M., Martinelli, S., Gassen, A., et al. (2019). SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 10:5770. doi: 10.1038/s41467-019-13659-4
Gervois, P., Torra, I. P., Fruchart, J. C., and Staels, B. (2000). Regulation of lipid and lipoprotein metabolism by PPAR activators. Clin. Chem. Lab. Med. 38, 3–11. doi: 10.1515/CCLM.2000.002
Glass, C. K., and Saijo, K. (2010). Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat. Rev. Immunol. 10, 365–376. doi: 10.1038/nri2748
Goh, G. K., Dunker, A. K., and Uversky, V. N. (2012). Understanding viral transmission behavior via protein intrinsic disorder prediction: coronaviruses. J. Pathog. 2012:738590. doi: 10.1155/2012/738590
Grevengoed, T. J., Klett, E. L., and Coleman, R. A. (2014). Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 34, 1–30. doi: 10.1146/annurev-nutr-071813-105541
Gubern, A., Casas, J., Barceló-Torns, M., Barneda, D., de la Rosa, X., Masgrau, R., et al. (2008). Group IVA phospholipase A2 is necessary for the biogenesis of lipid droplets. J. Biol. Chem. 283, 27369–27382. doi: 10.1074/jbc.M800696200
Guo, Y. R., Cao, Q. D., Hong, Z. S., Tan, Y. Y., Chen, S. D., Jin, H. J., et al. (2020). The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil. Med. Res. 7:11. doi: 10.1186/s40779-020-00240-0
Harizi, H., Corcuff, J. B., and Gualde, N. (2008). Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol. Med. 14, 461–469. doi: 10.1016/j.molmed.2008.08.005
Harmon, G. S., Lam, M. T., and Glass, C. K. (2011). PPARs and lipid ligands in inflammation and metabolism. Chem. Rev. 111, 6321–6340. doi: 10.1021/cr2001355
Hartman, I. Z., Liu, P., Zehmer, J. K., Luby-Phelps, K., Jo, Y., Anderson, R. G. W., et al. (2010). Sterol-induced dislocation of 3-Hydroxy-3-methylglutaryl Coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J. Biol. Chem. 285, 19288–19298. doi: 10.1074/jbc.M110.134213
Heaton, N. S., Perera, R., Berger, K. L., Khadka, S., LaCount, D. J., Kuhn, R. J., et al. (2010). Dengue virus non-structural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 17345–17350. doi: 10.1073/pnas.1010811107
Heaton, N. S., and Randall, G. (2010). Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microb. 8, 422–432. doi: 10.1016/j.chom.2010.10.006
Heaton, N. S., and Randall, G. (2011). Multifaceted roles for lipids in viral infection. Trends Microbiol. 19, 368–375. doi: 10.1016/j.tim.2011.03.007
Helbig, K. J., Eyre, N. S., Yip, E., Narayana, S., Li, K., Fiches, G., et al. (2011). The antiviral protein viperin inhibits Hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 54, 1506–1517. doi: 10.1002/hep.24542
Herker, E., Harris, C., Hernandez, C., Carpentier, A., Kaehlcke, K., Rosenberg, A. R., et al. (2010). Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16, 1295–1298. doi: 10.1038/nm.2238
Hilgendorff, A., Muth, H., Parviz, B., Staubitz, A., Haberbosch, W., Tillmanns, H., et al. (2003). Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int. J. Clin. Pharmacol. Ther. 41, 397–401. doi: 10.5414/cpp41397
Hinson, E. R., and Cresswell, P. (2009). The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc. Natl. Acad. Sci. U.S.A. 106, 20452–20457. doi: 10.1073/pnas.0911679106
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131. doi: 10.1172/Jci200215593
Huang, C., Wang, Y., and Li, X. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 496–496. doi: 10.1016/S0140-6736(20)30252-X
Huang, S., Zhu, B., Cheon, I. S., Goplen, N. P., Jiang, L., Zhang, R., et al. (2019). PPAR-γ in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J. Virol. 93:jvi.00030-19. doi: 10.1128/jvi.00030-19
Hui, D. S., Joynt, G. M., Wong, K. T., Gomersall, C. D., Li, T. S., Antonio, G., et al. (2005). Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity and quality of life in a cohort of survivors. Thorax 60, 401–409. doi: 10.1136/thx.2004.030205
Im, S. S., Yousef, L., Blaschitz, C., Liu, J. Z., Edwards, R. A., Young, S. G., et al. (2011). Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 13, 540–549. doi: 10.1016/j.cmet.2011.04.001
Jarc, E., and Petan, T. (2020). A twist of FATe: lipid droplets and inflammatory lipid mediators. Biochimie 169, 69–87. doi: 10.1016/j.biochi.2019.11.016
Jin, J.-M., Bai, P., He, W., Wu, F., Liu, X.-F., Han, D.-M., et al. (2020). Gender differences in patients with COVID-19: focus on severity and mortality. Front. Public Health 8:152. doi: 10.3389/fpubh.2020.00152
Josset, L., Menachery, V. D., Gralinski, L. E., Agnihothram, S., Sova, P., Carter, V. S., et al. (2013). Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio 4:e0165-13. doi: 10.1128/mBio.00165-13
Jung, T. W., Youn, B. S., Choi, H. Y., Lee, S. Y., Hong, H. C., Yang, S. J., et al. (2013). Salsalate and adiponectin ameliorate hepatic steatosis by inhibition of the hepatokine fetuin-A. Biochem. Pharmacol. 86, 960–969. doi: 10.1016/j.bcp.2013.07.034
Karin, M., Yamamoto, Y., and Wang, Q. M. (2004). The IKK NF-kappa B system: a treasure trove for drug development. Nat. Rev. Drug Discov. 3, 17–26. doi: 10.1038/nrd1279
Kawai, T., and Akira, S. (2006). TLR signaling. Cell Death Differ. 13, 816–825. doi: 10.1038/sj.cdd.4401850
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Ketter, E., and Randall, G. (2019). Virus impact on lipids and membranes. Annu. Rev. Virol. 6, 319–340. doi: 10.1146/annurev-virology-092818-015748
Knoops, K., Kikkert, M., van den Worm, S. H. E., Zevenhoven-Dobbe, J. C., van der Meer, Y., Koster, A. J., et al. (2008). SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6:e226. doi: 10.1371/journal.pbio.0060226
Koberlin, M. S., Heinz, L. X., and Superti-Furga, G. (2016). Functional crosstalk between membrane lipids and TLR biology. Curr. Opin. Cell Biol. 39, 28–36. doi: 10.1016/j.ceb.2016.01.010
Kopecky-Bromberg, S. A., Martínez-Sobrido, L., Frieman, M., Baric, R. A., and Palese, P. (2007). Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81, 548–557. doi: 10.1128/jvi.01782-06
Kuerschner, L., Moessinger, C., and Thiele, C. (2008). Imaging of lipid biosynthesis: how a neutral lipid enters lipid droplets. Traffic 9, 338–352. doi: 10.1111/j.1600-0854.2007.00689.x
Kuri, T., Eriksson, K. K., Putics, A., Zust, R., Snijder, E. J., Davidson, A. D., et al. (2011). The ADP-ribose-1”-monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J. Gen. Virol. 92(Pt 8), 1899–1905. doi: 10.1099/vir.0.031856-0
Lakatos, H. F., Thatcher, T. H., Kottmann, R. M., Garcia, T. M., Phipps, R. P., and Sime, P. J. (2007). The role of PPARs in lung fibrosis. PPAR Res. 2007:71323. doi: 10.1155/2007/71323
Laureti, M., Narayanan, D., Rodriguez-Andres, J., Fazakerley, J. K., and Kedzierski, L. (2018). Flavivirus receptors: diversity, identity, and cell entry. Front. Immunol. 9:2180. doi: 10.3389/fimmu.2018.02180
Lee, C. H., Chawla, A., Urbiztondo, N., Liao, D., Boisvert, W. A., Evans, R. M., et al. (2003). Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science 302, 453–457. doi: 10.1126/science.1087344
Lee, J. Y., Cortese, M., Haselmann, U., Tabata, K., Romero-Brey, I., Funaya, C., et al. (2019). Spatiotemporal coupling of the Hepatitis C virus replication cycle by creating a lipid droplet-proximal membranous replication compartment. Cell Rep. 27, 3602–3617. doi: 10.1016/j.celrep.2019.05.063
Lehrskov, L. L., and Christensen, R. H. (2019). The role of interleukin-6 in glucose homeostasis and lipid metabolism. Semin. Immunopathol. 41, 491–499. doi: 10.1007/s00281-019-00747-2
Li, G. M., Li, Y. G., Yamate, M., Li, S. M., and Ikuta, K. (2007). Lipid rafts play an important role in the early stage of severe acute respiratory syndrome-coronavirus life cycle. Microb. Infect. 9, 96–102. doi: 10.1016/j.micinf.2006.10.015
Li, K., Foy, E., Ferreon, J. C., Nakamura, M., Ferreon, A. C., Ikeda, M., et al. (2005). Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. U.S.A. 102, 2992–2997. doi: 10.1073/pnas.0408824102
Li, K., Li, N. L., Wei, D. H., Pfeffer, S. R., Fan, M. Y., and Pfeffer, L. M. (2012). Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology 55, 666–675. doi: 10.1002/hep.24763
Li, Q. S., Pene, V., Krishnamurthy, S., Cha, H., and Liang, T. J. (2013). Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat. Med. 19, 722–729. doi: 10.1038/nm.3190
Liang, R., Wang, L., Zhang, N., Deng, X., Su, M., Su, Y., et al. (2018). Development of small-molecule MERS-CoV inhibitors. Viruses 10:721. doi: 10.3390/v10120721
Liu, K., and Czaja, M. J. (2013). Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20, 3–11. doi: 10.1038/cdd.2012.63
Liu, T., Zhang, L., Joo, D., and Sun, S.-C. (2017). NF-κB signaling in inflammation. Signal Transd. Target. Therapy 2:17023. doi: 10.1038/sigtrans.2017.23
Lorizate, M., and Krausslich, H. G. (2011). Role of lipids in virus replication. Cold Spring Harbor Perspect. Biol. 3:a004820. doi: 10.1101/cshperspect.a004820
Lu, R. J., Zhao, X., Li, J., Niu, P. H., Yang, B., Wu, H. L., et al. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395, 565–574. doi: 10.1016/S0140-6736(20)30251-8
Lu, Y. N., Liu, D. X., and Tam, J. P. (2008). Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 369, 344–349. doi: 10.1016/j.bbrc.2008.02.023
Mackenzie, J. M., Khromykh, A. A., and Parton, R. G. (2007). Cholesterol manipulation by west nile virus perturbs the cellular immune response. Cell Host Microb. 2, 229–239. doi: 10.1016/j.chom.2007.09.003
Martinez-Gutierrez, M., Castellanos, J. E., and Gallego-Gomez, J. C. (2011). Statins reduce dengue virus production via decreased virion assembly. Intervirology 54, 202–216. doi: 10.1159/000321892
Martins, A. S., Carvalho, F. A., Faustino, A. F., Martins, I. C., and Santos, N. C. (2019). West nile virus capsid protein interacts with biologically relevant host lipid systems. Front. Cell. Infect. Microbiol. 9:8. doi: 10.3389/fcimb.2019.00008
Mehrbod, P., Omar, A. R., Hair-Bejo, M., Haghani, A., and Ideris, A. (2014). Mechanisms of action and efficacy of statins against influenza. Biomed. Res. Int. 2014:872370. doi: 10.1155/2014/872370
Menachery, V. D., Eisfeld, A. J., Schäfer, A., Josset, L., Sims, A. C., Proll, S., et al. (2014). Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 5:e001174-14. doi: 10.1128/mBio.01174-14
Milewska, A., Nowak, P., Owczarek, K., Szczepanski, A., Zarebski, M., Hoang, A., et al. (2018). Entry of human Coronavirus NL63 into the cell. J. Virol. 92:jvi.01933-17. doi: 10.1128/jvi.01933-17
Miller, S., and Krijnse-Locker, J. (2008). Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 6, 363–374. doi: 10.1038/nrmicro1890
Miller, S. C., Huang, R., Sakamuru, S., Shukla, S. J., Attene-Ramos, M. S., Shinn, P., et al. (2010). Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. Pharmacol. 79, 1272–1280. doi: 10.1016/j.bcp.2009.12.021
Miyanari, Y., Atsuzawa, K., Usuda, N., Watashi, K., Hishiki, T., Zayas, M., et al. (2007). The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–U74. doi: 10.1038/ncb1631
Monson, E. A., Crosse, K. M., Das, M., and Helbig, K. J. (2018). Lipid droplet density alters the early innate immune response to viral infection. PLoS One 13:e00190597. doi: 10.1371/journal.pone.0190597
Moschonas, A., Ioannou, M., and Eliopoulos, A. G. (2012). CD40 stimulates a “feed-forward” NF-κB-driven molecular pathway that regulates IFN-β expression in carcinoma cells. J. Immunol. 188, 5521–5527. doi: 10.4049/jimmunol.1200133
Muller, C., Hardt, M., Schwudke, D., Neuman, B. W., Pleschka, S., and Ziebuhr, J. (2018). Inhibition of cytosolic phospholipase A(2)alpha impairs an early step of coronavirus replication in cell culture. J. Virol. 92:e01463-17. doi: 10.1128/JVI.01463-17
Munoz-Jordan, J. L., Sanchez-Burgos, G. G., Laurent-Rolle, M., and Garcia-Sastre, A. (2003). Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U.S.A. 100, 14333–14338. doi: 10.1073/pnas.2335168100
Nagai, S., Shimizu, C., Umetsu, M., Taniguchi, S., Endo, M., Miyoshi, H., et al. (2004). Identification of a functional peroxisome proliferator-activated receptor responsive element within the murine perilipin gene. Endocrinology 145, 2346–2356. doi: 10.1210/en.2003-1180
Nomura, R., Kiyota, A., Suzaki, E., Kataoka, K., Ohe, Y., Miyamoto, K., et al. (2004). Human coronavirus 229E binds to CD13 in rafts and enters the cell through caveolae. J. Virol. 78, 8701–8708. doi: 10.1128/Jvi.78.16.8701-8708.2004
Owczarek, K., Szczepanski, A., Milewska, A., Baster, Z., Rajfur, Z., Sarna, M., et al. (2018). Early events during human coronavirus OC43 entry to the cell. Sci. Rep. 8:7124. doi: 10.1038/s41598-018-25640-0
Park, H. J., and Choi, J. M. (2017). Sex-specific regulation of immune responses by PPARs. Exp. Mol. Med. 49:e364. doi: 10.1038/emm.2017.102
Pawliczak, R., Han, C., Huang, X. L., Demetris, A. J., Shelhamer, J. H., and Wu, T. (2002). 85-kDa cytosolic phospholipase A2 mediates peroxisome proliferator-activated receptor gamma activation in human lung epithelial cells. J. Biol. Chem. 277, 33153–33163. doi: 10.1074/jbc.M200246200
Pawliczak, R., Logun, C., Madara, P., Lawrence, M., Woszczek, G., Ptasinska, A., et al. (2004). Cytosolic phospholipase A2 Group IValpha but not secreted phospholipase A2 Group IIA, V, or X induces interleukin-8 and cyclooxygenase-2 gene and protein expression through peroxisome proliferator-activated receptors gamma 1 and 2 in human lung cells. J. Biol. Chem. 279, 48550–48561. doi: 10.1074/jbc.M408926200
Peiris, J. S. M., Guan, Y., and Yuen, K. Y. (2004). Severe acute respiratory syndrome. Nat. Med. 10, S88–S97. doi: 10.1038/nm1143
Pereira-Dutra, F. S., Teixeira, L., de Souza Costa, M. F., and Bozza, P. T. (2019). Fat, fight, and beyond: the multiple roles of lipid droplets in infections and inflammation. J. Leukoc. Biol. 106, 563–580. doi: 10.1002/JLB.4MR0119-035R
Petrosillo, N., Viceconte, G., Ergonul, O., Ippolito, G., and Petersen, E. (2020). COVID-19, SARS and MERS: are they closely related? Clin. Microbiol. Infect. 26, 729–734. doi: 10.1016/j.cmi.2020.03.026
Pfeffer, L. M. (2011). The role of nuclear factor κB in the interferon response. J. Interf. Cytokine Res. 31, 553–559. doi: 10.1089/jir.2011.0028
Pindado, J., Balsinde, J., and Balboa, M. A. (2007). TLR3-dependent induction of nitric oxide synthase in RAW 264.7 macrophage-like cells via a cytosolic phospholipase A(2)/cyclooxygenase-2 pathway. J. Immunol. 179, 4821–4828. doi: 10.4049/jimmunol.179.7.4821
Pol, A., Gross, S. P., and Parton, R. G. (2014). Biogenesis of the multifunctional lipid droplet: lipids, proteins, and sites. J. Cell Biol. 204, 635–646. doi: 10.1083/jcb.201311051
Pombo, J. P., and Sanyal, S. (2018). Perturbation of intracellular cholesterol and fatty acid homeostasis during flavivirus infections. Front. Immunol. 9:1276. doi: 10.3389/fimmu.2018.01276
Poulsen, L., Siersbæk, M., and Mandrup, S. (2012). PPARs: fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 23, 631–639. doi: 10.1016/j.semcdb.2012.01.003
Prentice, E., Jerome, W. G., Yoshimori, T., Mizushima, N., and Denison, M. R. (2004). Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 279, 10136–10141. doi: 10.1074/jbc.M306124200
Puelles, V. G., Lütgehetmann, M., Lindenmeyer, M. T., Sperhake, J. P., Wong, M. N., Allweiss, L., et al. (2020). Multiorgan and renal tropism of SARS-CoV-2. New Engl. J. Med. 383, 590–592. doi: 10.1056/NEJMc2011400
Rakic, B., Sagan, S. M., Noestheden, M., Belanger, S., Nan, X., Evans, C. L., et al. (2006). Peroxisome proliferator-activated receptor alpha antagonism inhibits hepatitis C virus replication. Chem. Biol. 13, 23–30. doi: 10.1016/j.chembiol.2005.10.006
Ricote, M., and Glass, C. K. (2007). PPARs and molecular mechanisms of transrepression. Biochim. Biophys. Acta 1771, 926–935. doi: 10.1016/j.bbalip.2007.02.013
Ricote, M., Li, A. C., Willson, T. M., Kelly, C. J., and Glass, C. K. (1998). The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 391, 79–82. doi: 10.1038/34178
Romero-Brey, I., and Bartenschlager, R. (2014). Membranous replication factories induced by plus-strand RNA viruses. Viruses Basel 6, 2826–2857. doi: 10.3390/v6072826
Ruiperez, V., Astudillo, A. M., Balboa, M. A., and Balsinde, J. (2009). Coordinate regulation of TLR-mediated arachidonic acid mobilization in macrophages by group IVA and group V phospholipase A(2)s. J. Immunol. 182, 3877–3883. doi: 10.4049/jimmunol.0804003
Ruysschaert, J. M., and Lonez, C. (2015). Role of lipid microdomains in TLR-mediated signalling. Biochim. Biophys. Acta Biomemb. 1848, 1860–1867. doi: 10.1016/j.bbamem.2015.03.014
Saitoh, T., Satoh, T., Yamamoto, N., Uematsu, S., Takeuchi, O., Kawai, T., et al. (2011). Antiviral protein viperin promotes toll-like receptor 7- and toll-like receptor 9-mediated Type I interferon production in plasmacytoid dendritic cells. Immunity 34, 352–363. doi: 10.1016/j.immuni.2011.03.010
Saka, H. A., and Valdivia, R. (2012). Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 28, 411–437. doi: 10.1146/annurev-cellbio-092910-153958
Salata, C., Calistri, A., Alvisi, G., Celestino, M., Parolin, C., and Palù, G. (2019). Ebola virus entry: from molecular characterization to drug discovery. Viruses 11:274. doi: 10.3390/v11030274
Samsa, M. M., Mondotte, J. A., Iglesias, N. G., Assuncao-Miranda, I., Barbosa-Lima, G., Da Poian, A. T., et al. (2009). Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5:1000632. doi: 10.1371/journal.ppat.1000632
Schuliga, M. (2015). NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules 5, 1266–1283. doi: 10.3390/biom5031266
Seo, J. Y., Yaneva, R., and Cresswell, P. (2011). Viperin: a multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microb. 10, 534–539. doi: 10.1016/j.chom.2011.11.004
Smits, S. L., de Lang, A., van den Brand, J. M., Leijten, L. M., van, I. W. F., Eijkemans, M. J., et al. (2010). Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 6:e1000756. doi: 10.1371/journal.ppat.1000756
Souza-Moreira, L., Soares, V. C., Dias, S., and Bozza, P. T. (2019). Adipose-derived mesenchymal stromal cells modulate lipid metabolism and lipid droplet biogenesis via AKT/mTOR -ppargamma signalling in macrophages. Sci. Rep. 9:20304. doi: 10.1038/s41598-019-56835-8
Spagnolo, P., Balestro, E., Aliberti, S., Cocconcelli, E., Biondini, D., Casa, G. D., et al. (2020). Pulmonary fibrosis secondary to COVID-19: a call to arms? Lancet Respir. Med. 8, 750–752. doi: 10.1016/S2213-2600(20)30222-8
Stapleford, K. A., and Miller, D. J. (2010). Role of cellular lipids in positive-sense RNA virus replication complex assembly and function. Viruses 2, 1055–1068. doi: 10.3390/v2051055
Syed, G. H., Amako, Y., and Siddiqui, A. (2010). Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol. Metab. 21, 33–40. doi: 10.1016/j.tem.2009.07.005
Szatmari, I., Töröcsik, D., Agostini, M., Nagy, T., Gurnell, M., Barta, E., et al. (2007). PPARγ regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood 110, 3271–3280. doi: 10.1182/blood-2007-06-096222
Tang, W. C., Lin, R. J., Liao, C. L., and Lin, Y. L. (2014). Rab18 facilitates dengue virus infection by targeting fatty acid synthase to sites of viral replication. J. Virol. 88, 6793–6804. doi: 10.1128/Jvi.00045-14
Tirinato, L., Pagliari, F., Limongi, T., Marini, M., Falqui, A., Seco, J., et al. (2017). An overview of lipid droplets in cancer and cancer stem cells. Stem Cells Intern. 2017:1656053. doi: 10.1155/2017/1656053
Totura, A. L., Whitmore, A., Agnihothram, S., Schafer, A., Katze, M. G., Heise, M. T., et al. (2015). Toll-like receptor 3 signaling via TRIF contributes to a protective innate immune response to severe acute respiratory syndrome coronavirus infection. mBio 6:e0638-15. doi: 10.1128/mBio.00638-15
Vancini, R., Kramer, L. D., Ribeiro, M., Hernandez, R., and Brown, D. (2013). Flavivirus infection from mosquitoes in vitro reveals cell entry at the plasma membrane. Virology 435, 406–414. doi: 10.1016/j.virol.2012.10.013
Varga, T., Czimmerer, Z., and Nagy, L. (2011). PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 1812, 1007–1022. doi: 10.1016/j.bbadis.2011.02.014
Walther, T. C., and Farese, R. V. (2012). Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714. doi: 10.1146/annurev-biochem-061009-102430
Wang, H. L., Yang, P., Liu, K. T., Guo, F., Zhang, Y. L., Zhang, G. Y., et al. (2008). SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 18, 290–301. doi: 10.1038/cr.2008.15
Wang, W., Ye, L., Ye, L., Li, B., Gao, B., Zeng, Y., et al. (2007). Up-regulation of IL-6 and TNF-alpha induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaB pathway. Virus Res. 128, 1–8. doi: 10.1016/j.virusres.2007.02.007
Weitz-Schmidt, G. (2002). Statins as anti-inflammatory agents. Trends Pharmacol. Sci. 23, 482–486. doi: 10.1016/S0165-6147(02)02077-1
World Health Organization [WHO] (2020). Coronavirus Disease 2019 (COVID-19) Situation Report - 51. Geneva: World Health Organization. Available online at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200311-sitrep-51-covid-19.pdf?sfvrsn=1ba62e57_10
Wrapp, D., Wang, N. S., Corbett, K. S., Goldsmith, J. A., Hsieh, C. L., Abiona, O., et al. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263. doi: 10.1126/science.abb2507
Wu, D. J., Sanin, D. E., Everts, B., Chen, Q. Y., Qiu, J., Buck, M. D., et al. (2016). Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity 44, 1325–1336. doi: 10.1016/j.immuni.2016.06.006
Wymann, M. P., and Schneiter, R. (2008). Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 9, 162–176. doi: 10.1038/nrm2335
Xiao, K., Zhai, J., Feng, Y., Zhou, N., Zhang, X., Zou, J.-J., et al. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 583, 286–289. doi: 10.1038/s41586-020-2313-x
Yan, B. P., Chu, H., Yang, D., Sze, K. H., Lai, P. M., Yuan, S. F., et al. (2019). Characterization of the lipidomic profile of human coronavirus-infected cells: implications for lipid metabolism remodeling upon Coronavirus replication. Viruses Basel 11:73. doi: 10.3390/v11010073
Yang, C. H., Murti, A., Pfeffer, S. R., Basu, L., Kim, J. G., and Pfeffer, L. M. (2000). IFNalpha/beta promotes cell survival by activating NF-kappa B. Proc. Natl. Acad. Sci. U.S.A. 97, 13631–13636. doi: 10.1073/pnas.250477397
Yang, C.-W., Lee, Y.-Z., Hsu, H.-Y., Shih, C., Chao, Y.-S., Chang, H.-Y., et al. (2017). Targeting Coronaviral replication and cellular JAK2 mediated dominant NF-κB activation for comprehensive and ultimate inhibition of coronaviral activity. Sci. Rep. 7:4105. doi: 10.1038/s41598-017-04203-9
Yang, W., Hood, B. L., Chadwick, S. L., Liu, S. F., Watkins, S. C., Luo, G. X., et al. (2008). Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates Hepatitis C virus entry and production. Hepatology 48, 1396–1403. doi: 10.1002/hep.22508
York, A. G., Williams, K. J., Argus, J. P., Zhou, Q. D., Brar, G., Vergnes, L., et al. (2015). Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 163, 1716–1729. doi: 10.1016/j.cell.2015.11.045
Yu, W., Bozza, P. T., Tzizik, D. M., Gray, J. P., Cassara, J., Dvorak, A. M., et al. (1998). Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am. J. Pathol. 152, 759–769.
Yuan, S. F., Chu, H., Chan, J. F. W., Ye, Z. W., Wen, L., Yan, B. P., et al. (2019). SREBP-dependent lipidomic reprogramming as a broad-spectrum antiviral target. Nat. Commun. 10:120. doi: 10.1038/s41467-018-08015-x
Zhang, J. S., Lan, Y., and Sanyal, S. (2017). Modulation of lipid droplet metabolism-a potential target for therapeutic intervention in flaviviridae infections. Front. Microbiol. 8:2286. doi: 10.3389/fmicb.2017.02286
Zhang, M. A., Rego, D., Moshkova, M., Kebir, H., Chruscinski, A., Nguyen, H., et al. (2012). Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc. Natl. Acad. Sci. U.S.A. 109, 9505–9510. doi: 10.1073/pnas.1118458109
Zhang, S., Carriere, J., Lin, X. X., Xie, N., and Feng, P. H. (2018). Interplay between cellular metabolism and cytokine responses during viral infection. Viruses Basel 10:521. doi: 10.3390/v10100521
Zhang, X., Wu, K., Wang, D., Yue, X., Song, D., Zhu, Y., et al. (2007). Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-kappaB. Virology 365, 324–335. doi: 10.1016/j.virol.2007.04.009
Zhang, Y. Z., and Holmes, E. C. (2020). A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 181, 223–227. doi: 10.1016/j.cell.2020.03.035
Keywords: SARS-CoV-2, COVID-19, ssRNA virus, lipids, lipid droplets, NF-κB, TLRs, PPAR
Citation: Pagliari F, Marafioti MG, Genard G, Candeloro P, Viglietto G, Seco J and Tirinato L (2020) ssRNA Virus and Host Lipid Rearrangements: Is There a Role for Lipid Droplets in SARS-CoV-2 Infection? Front. Mol. Biosci. 7:578964. doi: 10.3389/fmolb.2020.578964
Received: 01 July 2020; Accepted: 03 September 2020;
Published: 08 October 2020.
Edited by:
Paola Patrignani, D’Annunzio University of Chieti–Pescara, ItalyReviewed by:
Francesco Taus, University of Verona, ItalyAnnalisa Trenti, University of Padua, Italy
Copyright © 2020 Pagliari, Marafioti, Genard, Candeloro, Viglietto, Seco and Tirinato. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Tirinato, dGlyaW5hdG9AdW5pY3ouaXQ=; dGlyaW5hdG9sdWNhQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work