 Giuseppina Mariano
Giuseppina Mariano Rebecca J. Farthing2
Rebecca J. Farthing2 Julien R. C. Bergeron
Julien R. C. Bergeron- 1Microbes in Health and Disease Theme, Newcastle University Biosciences Institute, Newcastle University, Newcastle upon Tyne, United Kingdom
- 2Randall Centre for Cell and Molecular Biophysics, King’s College London, London, United Kingdom
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread in humans in almost every country, causing the disease COVID-19. Since the start of the COVID-19 pandemic, research efforts have been strongly directed towards obtaining a full understanding of the biology of the viral infection, in order to develop a vaccine and therapeutic approaches. In particular, structural studies have allowed to comprehend the molecular basis underlying the role of many of the SARS-CoV-2 proteins, and to make rapid progress towards treatment and preventive therapeutics. Despite the great advances that have been provided by these studies, many knowledge gaps on the biology and molecular basis of SARS-CoV-2 infection still remain. Filling these gaps will be the key to tackle this pandemic, through development of effective treatments and specific vaccination strategies.
Introduction
Recently, a novel Coronavirus (CoV), SARS-CoV-2, has emerged in China at the end of 2019 and quickly spread nearly to every country, causing a global pandemic. SARS-CoV-2 causes severe infections of the respiratory tract, with symptoms that range from dry cough, to fever and pneumonia, and has a high mortality rate (Benvenuto et al., 2020; Huang et al., 2020; Mousavizadeh and Ghasemi, 2020). As of mid-November 2020, it has infected an estimated 60 M people word-wide, and caused > 1.4M deaths (World Health Organisation, 2020). The virus is still endemic in most countries, and no vaccine or prophylactic drugs have been approved yet to prevent its spread. Two drugs (Remdesivir and Dexamethasone) have recently been approved in its treatment, but these only help alleviate symptoms in the most severe case (National Institute of Allergy, and Infectious Diseases (NIAID), 2020; The Recovery Collaborative Group, 2020).
Coronaviruses (CoVs) belong to the family of Coronaviridae. These are enveloped viruses that contain a positive, single-stranded RNA genome, which is packaged within a capsid. The capsid consists of the nucleocapsid protein N and this is further surrounded by a membrane, that contains three proteins: the membrane protein (M) and the envelope protein (E), which are involved in the virus budding process, and the spike glycoprotein (S), which is a key player in binding host receptor and mediating membrane fusion and virus entry into host cells (Li, 2016; Cai et al., 2020).
CoVs can be divided in four genera, Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and, Deltacoronavirus (Woo et al., 2012; Cui et al., 2019). Alpha and Betacoronaviruses more commonly cause infections in humans and mammal (Tang et al., 2015) and in particular, Betacoronaviruses include severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), which caused previous pandemics in 2002 and 2012, respectively (Snijder et al., 2003; Chan et al., 2015), and the newly emerged virus SARS-CoV-2. Gammacoronavirus, and Deltacoronavirus instead prevalently infect birds and fish, but some instances were also found to infect mammals (Woo et al., 2010). The main distinctive characteristic between the 4 genera is the presence of the non-structural protein Nsp1 in Alpha and Betacoronaviruses, with Nsp1 displaying distinct protein size and sequence features between these two genera (King et al., 2012). Conversely, a Nsp1 counterpart has not been reported in Gammacoronavirus, and Deltacoronavirus (King et al., 2012). Furthermore, Alphacoronaviruses exclusively possess a common accessory gene which encodes for the multi-spanning alphacoronavirus membrane protein (αmp) (King et al., 2012). Different types of Alphacoronaviruses can possess a different number of copies of this accessory gene (King et al., 2012). Within each genus, different types of CoVs will be equipped with different types of accessory genes, determining the distinctive host-range, virulence and mortality rate of each CoV subtype.
SARS-CoV and MERS-CoV are highly virulent and caused global pandemics in 2002 and 2012, respectively, with high mortality rates (∼10% for SARS-CoV and ∼36% for MERS-CoV) (Rota et al., 2003; de Groot et al., 2013; Li, 2016). Similarly, SARS-CoV-2 shows high mortality rate (reported globally as ∼ 3.8%) (World Health Organisation, 2020). SARS-CoV-2 additionally shows a higher infection rate compared to the closely related SARS-CoV (Benvenuto et al., 2020; Huang et al., 2020; Mousavizadeh and Ghasemi, 2020).
The SARS-CoV-2 genome (Wu F. et al., 2020) shows a similar organization to other CoVs. The positive-stranded RNA genome presents a 5′-cap and a 3′-poly-A tail (Figure 1A), allowing its translation from the host translation machinery. Similarly to other CoVs, at the 5′-end of SARS-CoV-2 a frameshift between two Orfs, Orf1a and Orf1b, allows the production of two polypeptides that are then proteolytically processed to produce 16 non-structural proteins (Nsp1-16) (Mousavizadeh and Ghasemi, 2020; Figure 1A), which are involved in various processes of the virus infection cycle (Gordon et al., 2020). At the 3′-end the structural proteins S (spike glycoprotein), N (nucleocapsid protein), M (membrane protein) and E (Envelope protein) are encoded. The nucleocapsid protein binds to the viral genome, aiding its packing against the internal surface of the envelope. The viral envelope is instead constituted of the S, M and E proteins (Paules et al., 2020; Figure 1B).
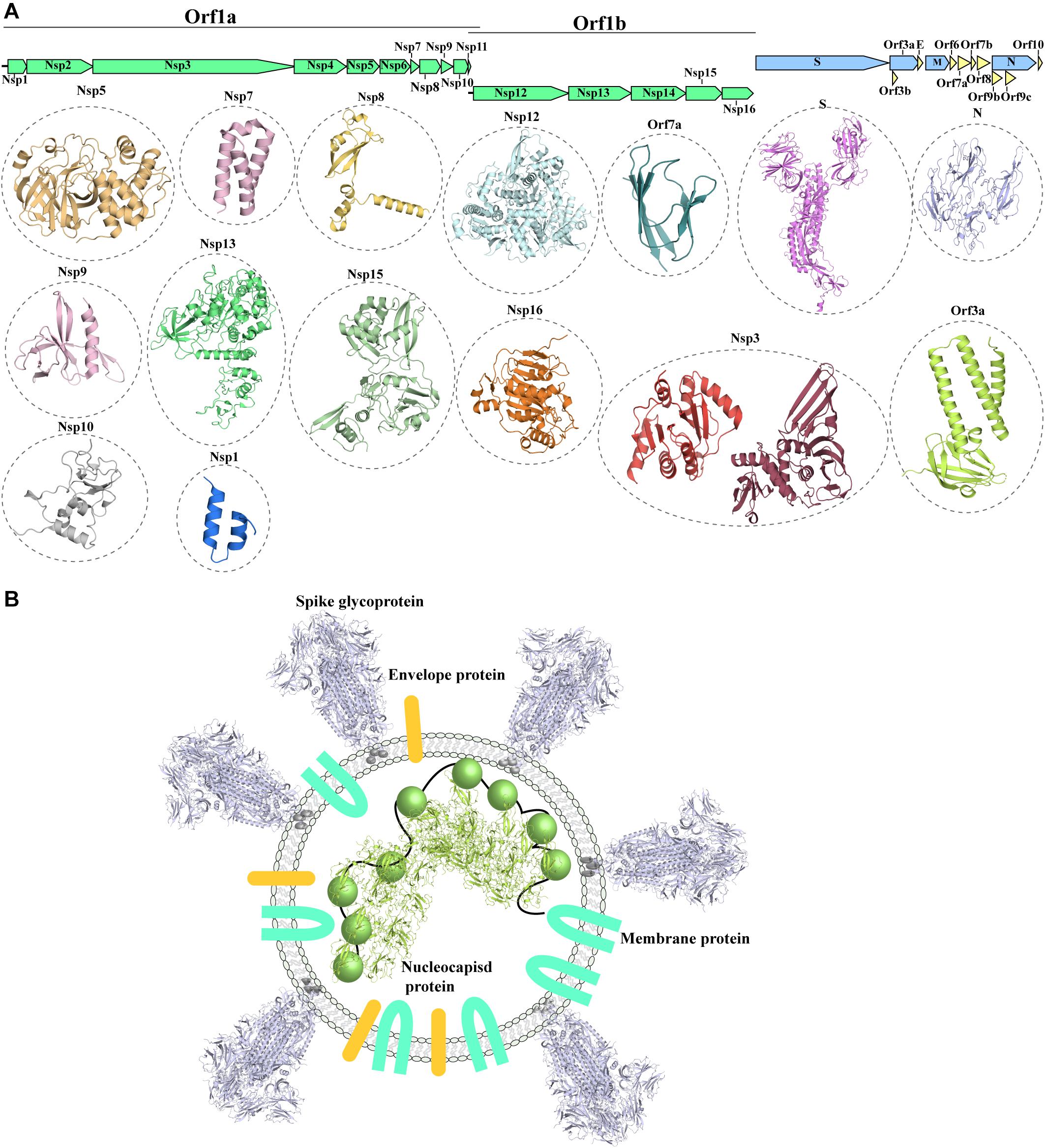
Figure 1. Known structures for SARS-CoV-2 proteins. (A) Schematic representation of genomic organization of SARS-CoV-2. Structural proteins are shown in pale blue, non-structural proteins are shown in green and accessory proteins are represented in pale yellow. Where available, a cartoon representation of the 3D structure for each protein is shown. 3D structure representations are based on PDBIDs shown in Table 1, only individual monomers are shown. (B) Schematic representation of an assembled SARS-CoV2 virus. The structural S glycoprotein is depicted through the use of a cartoon representation of its molecular structure (PDBID: 6VXX). E and M proteins are depicted with colored shapes. The nucleocapsid protein binding to viral RNA is represented by a cartoon representation of the molecular structure of its N-terminal domain (PDBID: 6M3M), while the C-terminal domain, whose structure is not available, is depicted with a colored sphere.
In addition to the 4 structural protein, the 3’-end also encodes nine accessory proteins (Orf3a, Orf3b, Orf6, Orf7a, Orf7b, Orf8, Orf9b, Orf9c, Orf10) (Figure 1A; Gordon et al., 2020). Accessory proteins were suggested to play an important role in virulence and host interaction in other coronaviruses (Liu et al., 2014). Whilst structural and non-structural proteins are shared amongst coronaviruses, the accessory proteins do not show highly similar distribution with other coronaviruses, with the exception of SARS-CoV (Liu et al., 2014). However, despite the close phylogenetical relationship between SARS-CoV and SARS-CoV-2 and their similar genomic organization, accessory proteins show decreased conservation, detectable both in lower sequence similarity between shared accessory proteins and variable content of accessory proteins between the two viruses (Table 1; Wu A. et al., 2020).
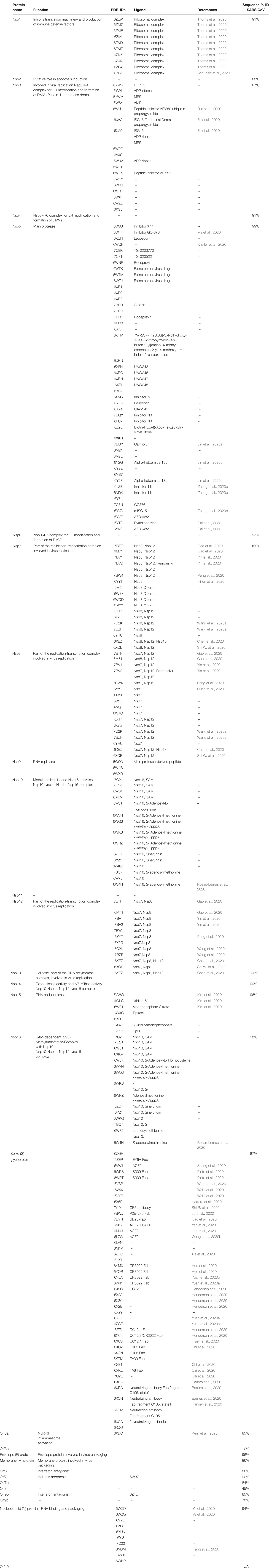
Table 1. Summary of available PDB structures of SARS-CoV-2 proteins.
Since the release of SARS-CoV-2 genome sequence, a significant effort has been directed to the investigation of the molecular mechanism of SARS-CoV-2 infection. In particular, many studies have focused on the structural characterization of SARS-CoV-2 structural, accessory and non-structural proteins (Figure 1A and Table 1) in order to elucidate in detail their involvement in the various steps of the infection process and to explore their role as effective molecular targets for therapy development.
The aim of this review is to summarize the progress towards the structural determination of SARS-CoV-2 proteins, but also to highlight the challenges that still remain and areas deserving further investigations. In particular, a more complete understanding of the network of interactions between the viral proteins, and how they cooperate in the crucial steps of the virus infection, will allow a comprehensive understanding of the infection mechanism and facilitate the identification of pivotal proteins and processes that can be exploited for structure-guided drug and vaccine design.
Structural Proteins
The Spike Glycoprotein
The first step in SARS-CoV-2 infection is the invasion of a host cell, a process that is mediated by the spike (S) glycoprotein (Figure 2; Tortorici and Veesler, 2019). The S protein is a glycosylated type I membrane protein that consists of two subunits, S1 and S2. The S protein exists in a trimeric pre-fusion form that is later cleaved by a host furin protease into the two subunits S1 and S2 (Bosch et al., 2003). The N-terminal S1 subunit contains the receptor-binding domain (RBD), which mediated binding to the host cell receptor, namely the angiotensin converting enzyme 2 (ACE2) for both SARS-CoV and SARS-CoV-2 (Bosch et al., 2003; Hoffmann et al., 2020b). Binding of RBD to ACE2, followed by additional cleavage of the S2 subunit at a second specific site by the host serine protease TMPRSS2, are fundamental to trigger the disassociation between S1 and S2, leading to the conformational changes in S2 that are responsible for the fusion of viral and host membranes and virus entry (Figure 2; Hoffmann et al., 2020b; Walls et al., 2020).
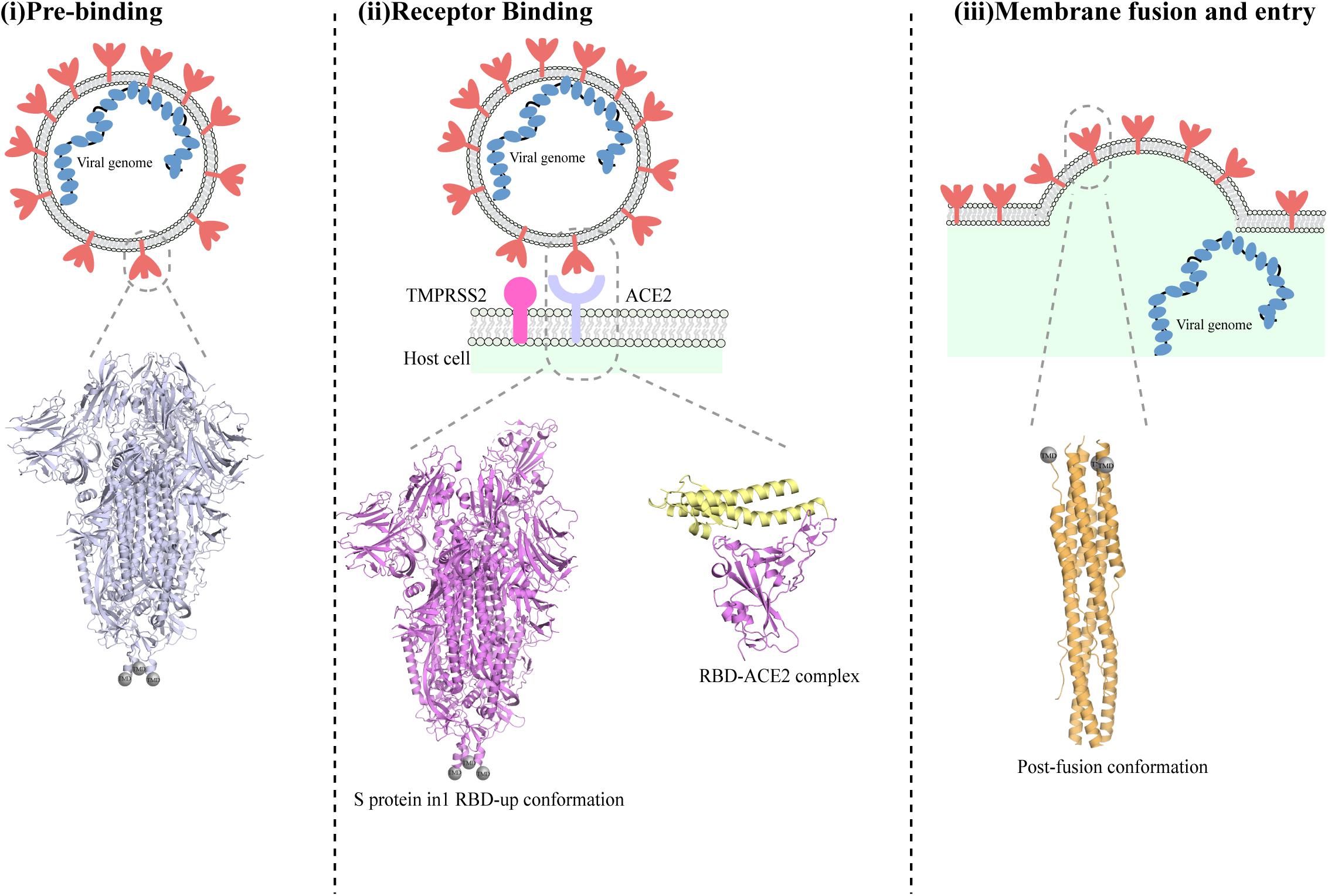
Figure 2. Different conformations of the SARS-CoV-2 spike protein S. Schematic representation of the stages of SARS-CoV-2 fusion to host cells membrane. The S trimer is exposed on the envelope of assembled viruses in its closed conformation (i). Below the schematic, a cartoon representation of the SARS-CoV-2 protein in its pre-fusion closed conformation (PDBID:6vxx) is shown. Cleavage by furin allows exposure of RBDs in their ‘up’ conformation and binding to ACE2 receptor (ii). The determined molecular structure of the S protein with one RBD open (PDBID:6vyb) is shown below. Following binding to ACE2 and further cleavage from TMPRSS2, additional conformational changes occurs that ultimately lead to the post-fusion conformation of the S protein and cause fusion between viral and host membrane (iii). A cartoon representation of the structure of the post-fusion SARS-CoV-2 trimer is shown below the schematic in part (iii).
Given its pivotal role in host recognition and invasion, the S protein has become a major target for the design of drugs and vaccine. Indeed, since SARS-CoV-2 sudden emergence, numerous studies have reported the structure of the S protein in various states (pre-fusion, post-fusion, and/or in complex with ACE2). A summary of all the available S protein structures, with their correspondent PDB ID and study is available in Table 1.
Numerous studies have reported the structure of the SARS-CoV-2 S protein by cryo-EM (Table 1) (some of them published merely a few weeks after the genome sequence of the virus was obtained, a remarkable tour-de-force). Two different approaches were used: in the first reported structures, the transmembrane (TM) domain of the protein was missing, and the ectodomain of the S protein was stabilized by introducing two prolines at the C-terminal S2 fusion domain, which stabilize the S protein soluble domain (Walls et al., 2020; Wrapp et al., 2020). Despite employing a similar strategy to yield a stable soluble domain, Wrapp et al. (2020) reported the structure of the S protein soluble domain in a partially open state (with one exposed RBD), whereas Walls et al., obtained a sample with multiple conformational states of the S protein. Through 3D classification, Walls et al. (2020) have successfully separated the two species and resolved the structure of the S protein soluble domain in a closed conformation and in its partially open state (with one exposed RBD). One representative structure for the S protein soluble domain in a closed state and in a partially open state (with one exposed RBD) are shown in Figure 2. More recently the structure of the full-length S protein has been solved using cryo-EM (Cai et al., 2020). Interestingly, Cai et al. (2020) observed that the full-length protein exists in vitro as a mixture of different pre-fusion and post-fusion forms.
In several studies, isolation of monoclonal or polyclonal antibodies from plasma from recovered COVID-19 patients has produced a plethora of potential neutralizing antibodies with diverse targeted epitopes (Chi et al., 2020; Liu et al., 2020; Piccoli et al., 2020; Robbiani et al., 2020). In particular, in a study that evaluated ∼600 plasma and serum samples obtained from symptomatic and asymptomatic patients, it emerged that the majority of the elicited antibodies specifically bind to the S trimer RBD, with a minority recognizing other sites of the S trimer or binding to quaternary epitopes (Chi et al., 2020; Liu et al., 2020; Piccoli et al., 2020). Through the determination of the structure of six mABs, Piccoli et al., were able to map sites within the RBD that are predominantly recognized by the analyzed neutralizing antibodies. The study found that the dominant neutralizing strategy adopted by these antibodies includes binding to the receptor binding motif (RBM) which is only exposed in the S trimer open state or binding to a second site, which is also exposed in the closed state (Piccoli et al., 2020).
Epitopes targeted by the isolated antibodies have been precisely identified through determination of the structure of the S protein-candidate antibody complex with cryo-EM and X-ray crystallography. Barnes et al. (2020) isolated polyclonal antibodies which bound to the RBDs in ‘up’ conformation, sterically blocking ACE2 binding. Chi et al. (2020) instead isolated antibodies that were able to bind to the N-terminal domain (NTD) of the S protein and displayed high potency. Liu et al., isolated and characterized the epitope of a group of monoclonal antibodies that can target both the RBD and the NTD. Among these monoclonal antibodies, the structure of the 2-43 Fab in complex with the S trimer highlighted that three copies of the 2-43 Fab bind to a quaternary epitope on the spike protein, which in part includes the RBD of two adjacent protomers, when they are in the closed conformation (Liu et al., 2020). Similarly, Tortorici et al. (2020) identified a potent monoclonal human antibody, S2M11, which forms electrostatic interactions with a quaternary epitope that encompasses two of the RBDs of a S trimer. Whilst S2M11 and 2-43 Fab recognize distinct quaternary epitopes (Liu et al., 2020; Tortorici et al., 2020), they ultimately lead to locking of the S trimer in a closed conformation, achieving inhibition of ACE2 binding and virus entry. The study by Tortorici et al. additionally identified the S2E12 antibody, which binds to the receptor binding motif and thus can only bind to S trimer in the open state. In both cases, S2M11 and S2E12 prevent binding of the S trimer with ACE2, through targeting of different epitopes and when used in combination display additive effects on SARS-CoV-2 neutralization (Tortorici et al., 2020).
Intriguingly, the X-ray structure of the antibody CR3022 in complex with the S trimer showed that CR3022 can bind to the pre-fusion trimer leading to its transition to a disordered and inactive state (Huo et al., 2020). A full summary of the structures of the S trimer in complex with candidate neutralizing antibodies is shown in Table 1. The increasing effort of isolating and characterizing the epitope recognized by S protein-targeting antibodies becomes highly relevant in the context of a study from Hansen et al., where the authors showed that the use of cocktails of antibodies targeting the S protein display highest rate of success and lowest probability to lead to development of SARS-CoV-2 escape mutants (Hansen et al., 2020). The cryo-EM structure of two candidate antibodies bound to S, showed that these bind different epitopes of the RBD and thereby offer high protection against SARS-CoV-2 (Hansen et al., 2020), without increasing the rate of escape mutants. A similar potential for using antibodies in combination was displayed by S2M11 and S2E12 antibodies, which showed to both bind the S protein RBD, without competing. S2M11 and S2E12 were tested against several variants of the S protein and whilst some of them showed decreased binding to S2M11, they did not affect the neutralizing activity of S2E12 (Tortorici et al., 2020). The evidence of successful combination of different and potent antibodies in cocktails with high neutralizing properties presented by Tortorici et al., and Hansen et al., becomes particularly relevant in the context of the prevalence of RBD-targeting antibodies elicited in the immune response and the high frequency of mutations rising in the RBD region in escape mutants of SARS-CoV-2 (Li Q. et al., 2020; Piccoli et al., 2020). With the same principles, gaining structural information on more S protein-candidate antibodies will allow to design more efficient therapeutic strategies, taking advantage of the best combination of antibodies that can bind multiple sites of SARS-CoV-2 without posing steric obstacles to their partners and, thus, allowing to develop treatments with a low probability to rise escape mutants of SARS-CoV-2.
The structure of the S protein pre-fusion trimer consists of the S trimer with all three molecules in the closed state (Cai et al., 2020; Walls et al., 2020) or with one RBD in the open state (hereinafter referred to as ‘open state’) (Walls et al., 2020; Wrapp et al., 2020; Figure 2 and Table 1). Despite the high similarity between the different structures of the pre-fusion conformations, several minor discrepancies have emerged (Cai et al., 2020). In particular, the proximal region to the fusion subunit was found to be disordered in pre-fusion ectodomains in their closed state and in the available open state structure. In contrast, the same region was fully ordered in the full-length S glycoprotein structure in the closed state (Cai et al., 2020). Considering the suggested role of the transition from ordered to disordered fold of this region in the conformational changes that lead to the switch from closed to fully open state (Cai et al., 2020), the inconsistencies observed between available structures of the closed state need to be further investigated.
In a subsequent report, the S trimer was purified in baculovirus-infected Hi5 insect cells, with the use of media supplemented with free fatty acids. In these conditions, the obtained cryo-EM structure highlighted the presence of a hydrophobic fatty acid binding pocket located at the RBD of the S glycoprotein, in a distal position compared to the ACE2 binding motif that displayed specific binding to linoleic acid (Toelzer et al., 2020). The authors further report that the molecular features involved in fatty acid binding are conserved in SARS-CoV and less extensively in other coronaviruses, including MERS-CoV. Considering some differences in the 3D conformations of S protein bound to linoleic acid compared to previously determined structures, it has been proposed that previous structure represent an ‘apo form’ of the S protein, in absence of a ligand. Binding to linoleic acid seems to switch the RBDs from loosely to tightly packed in the closed conformation, opening to the possibility to exploit this feature to inhibit ACE2 binding and virus spread (Toelzer et al., 2020). However, lipid-binding pocket was not reported from structural studies that focused on the S-trimers in the context of the virion membrane (Ke et al., 2020). One emerging feature from the numerous studies that focused on the S trimer structure points out that experimental and purification conditions tend to have a great influence on the predominant conformation of the S trimer. Therefore, the remaining uncertainty on the physiological relevance of S-trimer lipid binding ability and its putative predominance in certain S-trimer conformations requires further addressing. Remarkably, cryo-ET studies that have focused on the study of the S protein in its physiological context have highlighted that the number of S proteins per virion, as well as the ratio of pre- and post-fusion conformation of the S protein is dependent on the level of expression of ACE2 and TMPRSS2 in the infected cell line used (Ke et al., 2020; Klein et al., 2020).
The structure of the post-fusion S protein was also determined, consistently shown to be quite rigid across independently determined structures, with a central stranded coiled-coil and two six helix-bundle that contribute to the rigidity of the central coiled-coil (Cai et al., 2020; Xia et al., 2020; Figure 2).
The structure of the RBD-ACE2 complex has been reported in several studies, employing both X-ray crystallography and cryo-EM (Lan et al., 2020; Shang et al., 2020; Yan et al., 2020). These structures showed that the SARS-CoV-2 S protein binds to the ACE2 receptor, similarly, to the SARS-CoV S protein and provided a comprehensive map of the amino acids residues involved in the RBD-ACE2 interactions (Lan et al., 2020). Despite the overall similarity, the RBD of SARS-CoV and SARS-CoV-2 shows a higher degree of divergence compared to other proteins (Table 1; Walls et al., 2020). Indeed, the SARS-CoV-2 S protein shows unique amino acids substitutions that establish additional contacts with the ACE2 receptor, compared to SARS-CoV S protein (Lan et al., 2020).
To date great progress has been made into characterizing the interaction of the S protein with ACE2 receptor and how this mediates virus entry, whereas not much information is currently available on the details of S protein interaction with the proteases involved in its processing and activation.
More detailed information on the molecular basis of the S protein rearrangements that allow transition from pre- to post-fusion conformation were provided by the combination of cryo-ET and molecular dynamics simulations (Turoòová et al., 2020). This study showed that the stalk that connects the S protein ectodomain to its trans-membrane domain possesses three flexible hinges (Turoòová et al., 2020). The subtomogram averages also allowed to visualize a right-handed coiled-coil region at the top of the stalk region, which the authors speculate might be involved in conformational rearrangements that allow to transition from the pre- to the post-fusion states (Turoòová et al., 2020).
Human and lung and bronchial cells were observed to co-express ACE2, TMPRSS2 and furin (Lukassen et al., 2020) and consistently, the necessity for both proteases for the efficient activation and pathogenicity of SARS-CoV-2 was shown in several studies (Bestle et al., 2020; Hoffmann et al., 2020a).
TMPRSS2 protease activity was found to determine activation of fusion proteins in many viruses, including SARS-CoV, MERS CoV, influenza A virus and hepatitis C virus (Kido et al., 2008; Sakai et al., 2014; Esumi et al., 2015; Böttcher-Friebertshäuser, 2018).
In contrast to SARS-CoV, where the S1-S2 boundary is cleaved by Cathepsin L (Bosch et al., 2008), SARS-CoV-2 shows a furin cleavage site between the S1 and S2 subunit. A similar cleavage site is also found on the spike protein of MERS CoV (Millet and Whittaker, 2014).
It is speculated that furin-dependent cleavage between S1 and S2 could mediate conformational changes that ultimately lead to exposure and binding of the RBD to ACE2 and exposure of the TMPRSS2 cleavage site (Millet and Whittaker, 2015; Hoffmann et al., 2018). However, molecular details of the interaction between furin and the SARS-CoV-2 S protein are currently not available. Interestingly, a structure for human furin has been solved and exploited for designing of small protease inhibitors (Dahms et al., 2017). The availability of this structure could not only favor the design or repurposing of existing drugs, but it could aid to establish important amino acid positions for furin-S protein contacts and help to determine the consequences and importance of the furin-dependent cleavage on the S protein conformational changes.
TMPRSS2 is responsible for cleavage of the S2 subunit at a second site, which ultimately leads to production of a fusion protein that mediates fusion of virus and host membrane and virus entry. To date structure of TMPRSS2, either on its own or in complex with the SARS-CoV-2 S protein has not been determined. However, in a recent computational study the structure of TMPRSS2 was modeled, showing in detail the fold of TMPRSS2 domains, including its C-terminal catalytic domain (Hussain et al., 2020). The proposed S protein cleavage sites were employed for docking simulations that allowed to locate TMPRSS2 and S protein residues involved in the interaction, including those residues neighboring cleavage sites and the C-terminal catalytic domain of TMPRSS2 (Hussain et al., 2020). These results represent a starting point for the investigation of priming of S protein and the conformational changes that drive virus entry, especially considering the intrinsic difficulties of solving TMPRSS2 structure experimentally (Hussain et al., 2020). Nevertheless, further investigation, utilizing both experimental and computational methodologies, that takes into account also the novel findings in S protein full length structure (Cai et al., 2020) might confirm or implement this current model.
For SARS-CoV it has been reported that TMPRSS2 and human airway trypsin-like protease (HAT) cleave and prime the ACE2 receptor and this is fundamental to enhance infectivity of SARS-CoV (Heurich et al., 2014). Whether this process is crucial also for SARS-CoV-2 entry and whether it also involves the furin protease remains to be established. If both proteases play a multifaceted role in the virus entry, the hierarchy and interdependence of these processes and their molecular aspects has to be established in order to safely exploit them in drug and therapy design.
Interestingly, treatment with protease inhibitors in some studies has shown to be largely effective in inhibition of SARS-CoV-2 infection indicating that targeting of furin and TMPRSS2 activity might represent a promising route for treatment (Bestle et al., 2020; Hoffmann et al., 2020b) and further highlighting the necessity to determine of TMPRSS2- S protein and furin-S protein complexes, with the ultimate aim of providing alternative routes of inhibition for structure-guided therapeutic strategies.
Finally, none of the S protein structures in closed or open state have yielded defined densities for the trans-membrane domains (TMDs) (Figure 2) – see Box 1. Following conformational changes in the post-fusion trimers, the fusion peptide and the trans-membrane regions come in close proximity, thereby initiating the membrane fusion (Schroth-Diez et al., 2000; Schibli and Weissenhorn, 2004). In a study on SARS-CoV, systematic point mutations within the trans-membrane domain on the S protein has allowed to elucidate residues in these regions that are fundamental for successful virus entry (Corver et al., 2009). The authors of this work propose a model whereby, following transition to a post-fusion state, TMD’s tryptophan residues bind to the membrane, causing rearrangements of the TMD’s hydrophobic region and insertion in the membrane of its neighboring region. TMDs then oligomerise, ultimately leading to membrane curvature (Corver et al., 2009). This model provides a starting point in understanding the role of TMDs in the membrane fusion, albeit it requires experimental validation. Progress in this direction has been made by Ke et al., with the in situ resolution of the S trimer structure. Compared to other available structure, this study was able to obtain additional densities for TMDs proximal regions. The study further shows that the TMDs proximal regions act as hinge that allow bending on the surface of some S trimers (Ke et al., 2020). Nevertheless, the flexibility of this region still represents an obstacle in the full resolution of the membrane-associated region of the S trimer. Efforts to determine the TMD structure and placing this in context with the available molecular details of others S protein structures may lead to a more in depth understanding of virus entry process.
BOX 1. SARS-CoV-2 membrane proteins and membrane-embedded domains.
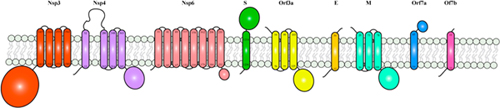
Figure | Schematic representation of SARS-CoV-2 membrane proteins. Trans-membrane helices were predicted using TMHMM Server v. 2.0. All membrane proteins were represented as monomers.
Membrane proteins exhibit significant challenges in their recombinant expression, purification, solubilisation and stability, which represent major obstacles in obtaining their structures. X-ray crystallography has a particularly low success rate with membrane proteins, as the use of detergents, protein stability and flexibility often prevents their crystallization (Rawson et al., 2016). In recent years, advances in cryo-EM have allowed to overcome some of these challenges and to increase the success rate of membrane proteins structure resolution. Detergent usage, whilst hindering preparation of cryo-grids, less frequently represents a problem in structure determination. Additional biochemical methods such as nanodiscs and amphipols, which helps with their solubilization but are not compatible with crystallization, have also contributed to successful cryo-EM analyses of membrane proteins. Nevertheless, heterogenicity of the protein sample and highly flexible regions still hamper the determination of high resolution molecular structures.
Within the protein repertoire of SARS-CoV-2, several proteins harbor one or more membrane domains. These include the structural proteins S, M and E, which are embedded in the virion membrane and permit virus fusion or budding. In addition, Nsp3-4-6, as well as the accessory proteins Orf3a, Orf7a and 7b, assemble at the plasma membrane or at the membrane of the Golgi complex or endoplasmic reticulum, during the various stages of the viral infection (von Brunn et al., 2007; Schaecher et al., 2008; Taylor et al., 2015). For Orf3a, studies conducted on SARS-CoV-2 have shown Orf3a mainly localizing at the plasma membrane (Kern et al., 2020; Ren et al., 2020).
Accordingly, cryo-EM was used in most studies focused on SARS CoV-2 proteins. One such example is represented by the two trans-membrane domains (TMDs) of the S protein. TMDs have been shown to mediate conformational changes that promotes virus entry, nevertheless despite the huge effort and the massive number of S protein structures that have been deposited during the past months (Table 1), near-atomic resolution determination of the TM region was not achieved in these analyses. Nonetheless, cryo-ET studies, in combination with molecular dynamics simulations yielded structures of the S protein in intact virions, and allowed to clearly distinguish the localization of TMDs of individual S copies (Turoòová et al., 2020). This study was not able to yield a high resolution structure of the TMDs, however, the authors were able to identify a right handed coiled-coil region at the top of a stalk region, which connects the TMDs to the ectodomain. The stalk region consists of three hinges, which provide flexibility to the S protein and lead the authors to speculate that it may contribute to drive the conformational changes that underlie the transition to a post-fusion conformation (Turoòová et al., 2020).
Similarly, the structure of the membrane channel Orf3a, determined by cryo-EM, has been reported recently. Orf3a formed dimeric and tetrameric ion channels, where tetrameric channels were found to be constituted by the juxtaposition of two dimers. Each dimer showed a novel fold, where the 3 TMHs of each protomer followed each other in a clockwise order, tracing an ellipse, ultimately forming a bifurcated channel, connected to the cytoplasm (Kern et al., 2020). Nonetheless, the structures of Nsp3, Nsp4, Nsp6, as well as E, M, and Orf7a and Orf7b, remain to be determined. Nsp3, Nsp4 and Nsp6 assemble at the endoplasmic reticulum, driving its modification into double membrane vesicles (DMVs) (Angelini et al., 2013; Hagemeijer et al., 2014). Numerous studies have reported the ability of Nsp3 to additionally interact with Nsp2, Orf3a, Orf7a and Orf9b (von Brunn et al., 2007; Neuman et al., 2008; Lei et al., 2018), which leads to the speculation that more proteins may either be involved in DMVs formation or might intervene at an earlier or later stage of the virus replication cycle to this process. Orf7a is localized at the Golgi-endoplasmic reticulum intermediary compartment, whereas reports on Orf3a localization have been contrasting, reporting this protein at the plasma membrane or Golgi complex membrane (Kern et al., 2020; Ren et al., 2020). In SARS CoV, Orf3a was found to interact with N, M and S (Tan, 2005), which are also localized at the Golgi-endoplasmic reticulum intermediary compartment. It is possible that Orf3a might play a ‘bridging’ role between the DMVs formation and later virus budding phase.
It is likely that homotypic and heterotypic interaction between TMDs will play a crucial role in formation of these complexes. It is therefore increasingly clear that much of the future understanding of SARS-CoV-2 replication cycle relies on the possibility to obtain near-atomic resolution structures of these putative complexes and to establish their network of interactions can underlying their function.
In addition to the structural details of the trans-membrane domains of the S protein trimer and its conformational changes, it still remains to establish whether other envelope proteins are involved in its conformational switches or stabilization. Indeed, cooperation of the S protein with other structural components N, E and M was shown to play a role during the membrane fusion step in other coronaviruses (Bosch et al., 2005). The lower variability of the S2 fusion subunit render this region an ideal target for development of inhibitors that may prevent virus entry and several inhibitors targeting this infection step have been identified (Walls et al., 2020; Xia et al., 2020). Consequently, additional steps need to be taken in order to determine the full structure of the S protein trimer by itself and in the context of the viral assembly, in relation to the other structural protein. These findings will shed light on additional residues involved in conformational changes within the S protein during membrane fusion and also reveal key residues involved into interactions between the S and other structural proteins, that are essential for the membrane fusion step to take place. Identification of these crucial amino acid positions may guide to design of inhibitors that prevent virus entry by targeting critical regions of the protein, less likely to mutate if the SARS-CoV-2 virus subsequently goes through genetic drift, as observed in other RNA viruses (Treanor, 2015).
The Nucleocapsid, Membrane and Envelope Proteins
Severe acute respiratory syndrome coronavirus 2structural proteins also include the nucleocapsid protein N, the membrane protein (M) and the envelope protein (E) (Figure 1B; Paules et al., 2020). Unlike the S protein, structural information on these proteins is limited.
The nucleocapsid protein N plays a multifaceted role in the infection cycle of CoVs (McBride et al., 2014). In SARS-CoV, the N protein was reported to bind to, and package the viral RNA into ribonucleoprotein RNP complexes (Figure 1B). The packaged RNPs particles are located on the internal face of the viral membrane, forming a separate layer from the envelope proteins M, E and S (Figure 1B). Furthermore, RNPs localization could be aided by the interaction between N and the C-terminus of the M protein (Chang et al., 2014).
The architecture of the RNPs within newly assembled virions has been recently elucidated with the use of cryo-ET. The authors of this study observed the RNPs form a cylindrical, semi-circular assembly of 16 nm that shows a preferred orientation. Within this assembly, multiple and aligned copies of N proteins form pillar-shaped densities that are stacked in parallel. This assembly ultimately results in the formation in two juxtaposed curved walls (Klein et al., 2020).
Additionally, in SARS-CoV, the N protein is recruited at the replication-transcription complex by Nsp3 and thus, is thought to be involved in viral genome replication (Cong et al., 2020). In particular, this interaction seems to involve the N-terminal domains of Nsp3 and the N protein and its function is to guide the viral genome to the newly assembled replication complex (Hurst et al., 2013; Lei et al., 2018).
The membrane protein M is embedded in the viral membrane (Figure 1B), through three predicted transmembrane helices. Its role is to drive the assembly of new virions within the host cells (Masters, 2006; Neuman et al., 2011). Coronaviruses M proteins were shown to oligomerize at the membrane of Golgi-endoplasmic reticulum intermediary compartment (Stertz et al., 2007) and were shown to induce apoptosis (Tsoi et al., 2014). S, N and E proteins are then recruited through interaction with the M protein. Thus, the current model suggests that the M protein acts as a scaffolding platform that recruits the other structural proteins and promotes membrane curvature during virion budding (Schoeman and Fielding, 2019).
Finally, the envelope protein E presents one trans-membrane domain and shows oligomerization properties. Indeed it was reported that E protein can self-interact, through its trans-membrane domain, to form a ion channel (Pervushin et al., 2009). Interaction between the C-terminus of E and M proteins guides E recruitment to the Golgi-endoplasmic reticulum intermediary compartment, initiating virus budding into host cells (Corse and Machamer, 2003; Masters, 2006; Schoeman and Fielding, 2019).
Additionally, several studies have suggested that the envelope protein E can also establish interactions with the nucleocapsid protein N (Tseng et al., 2014; Schoeman and Fielding, 2019). Currently it is thought that this interaction takes place independently from M-N and M-E interactions and its necessity in the assembly and release of viral particles is still unclear (Schoeman and Fielding, 2019).
So far, high-resolution structures have been reported for the N protein, but not M or E. The domain organization of coronaviruses N protein includes a RNA binding N-terminal domain, a C-terminal domain involved in dimerization (Chang et al., 2009, 2014) and a disordered serine/arginine rich linker, which plays a regulative role (Wu et al., 2009; Chang et al., 2014).
The structure of the RNA-binding domain of SARS-CoV-2 N has been determined recently, identifying the binding site and proposing a thorough characterization of the electrostatic interactions that take place between the viral RNA and the N-terminal domain (Kang et al., 2020). An independent study has instead unveiled the structure of the N protein dimerization domain, demonstrating that this domain can dimerise in a structure that is highly conserved with SARS-CoV. Interestingly, the authors showed that inclusion of an additional linker region allowed formation of homo-tetramers (Ye et al., 2020).
In the case of the E protein, only low-resolution structural details are available. Indeed, the structure of the E protein from SARS-CoV protein in lyso-myristoyl phosphatidylglycerol (LMPG) micelles, has been determined through NMR. The monomer of E consists of one trans-membrane helix, with two additional middle and C-terminal helical segments (Surya et al., 2018). Following determination of the pentameric state of E through BN-PAGE, the authors exploited the monomeric structure to dock a pentameric model, where the E pentamer displays a α-helical bundle fold, where the C-terminal tails coil around each other (Surya et al., 2018).
Conversely, to date the molecular structure of M is unavailable. M, N and E proteins were all found to be indispensable for the proper virus assembly and release in SARS-CoV and other coronaviruses (Siu et al., 2008). Definition of the missing structural details will not only help to determine the folding of these protein on their own, but it will provide also a considerable enhancement in the attempt to obtain higher resolution structural and molecular details of the RNPs and their packaging against the envelope, including the elucidation of network and hierarchy of the interactions between N,M,S,E that leads to the final viral assembly.
Non-Structural Proteins
Coronaviruses non-structural proteins are translated from the precursors proteins pp1a and pp1b. These precursors are processed by the pp1a-encoded proteases, Nsp3 and Nsp5, and the resulting non-structural proteins (Nsp1-16) are then essentials in viral replication, transcription and production of envelope proteins (Gordon et al., 2020; Mousavizadeh and Ghasemi, 2020).
The Replication-Transcription Complex
Following virus entry, replication of SARS-CoV-2 genome is operated by the cooperation of several non-structural proteins (Mousavizadeh and Ghasemi, 2020). The core RNA-dependent RNA polymerase (RdRp) consists of the protein Nsp12, which is bound to a heterodimer of Nsp7 and Nsp8. A second Nsp8 molecule interacts with Nsp12 at a different site to the Nsp7-8 heterodimer. Several groups have determined the structure of the Nsp12-7-8 subcomplex (Table 1), underlining the high conservation of the structural features of this complex between SARS-CoV and SARS-CoV-2 (Kirchdoerfer and Ward, 2019; Gao et al., 2020; Peng et al., 2020; Romano et al., 2020; Yin et al., 2020).
Interestingly, the independent studies reported an additional N-terminal β-hairpin motif that was not reported for SARS-CoV Nsp12 (Kirchdoerfer and Ward, 2019; Gao et al., 2020; Peng et al., 2020; Romano et al., 2020; Yin et al., 2020). Furthermore, the contact sites between Nsp12 and Nsp7-8 were determined and it was demonstrated that Nsp7 mediated the majority of contacts between the heterodimer and Nsp12 and that the two Nsp8 copies of this complex have differential conformations that ensure their binding to the right Nsp12 site (Table 1; Gao et al., 2020; Peng et al., 2020; Romano et al., 2020; Yin et al., 2020). Peng et al. (2020) further show that existing amino acid substitutions in SARS-CoV-2 Nsp7-8-12 overall decrease the efficiency of the RNA-dependent RNA polymerase activity and the thermostability of these proteins. The RdRp is one of the most promising drug targets identified to date. Structural details of the mechanism of inhibition of the anti-viral drug Remdesivir have been determined (Yin et al., 2020). Remdesivir is a nucleotide analog that is able to access the conserved RdRp active site and terminates elongation of the viral RNA (Yin et al., 2020). Given the high conservation of the active residues of the RdRp, Remdesivir has the potential to be employed as a broad-spectrum antiviral drug for several coronaviruses infections (Yin et al., 2020). Additionally, another independent study has solved the structure of RdRp in a post-translocation conformation, highlighting that the flexible N-terminal region of Nsp8 undergoes conformational changes during DNA elongation and these movements are then transmitted to the Nsp7-Nsp8 heterodimer causing, in turn, its conformational re-arrangements (Shi W. et al., 2020). Nsp7-Nsp8 heterodimer rearrangements are concomitant with translocation and nucleotide addition and are suggested to play a regulatory role of the polymerase activity (Shi W. et al., 2020).
The Nsp13 helicase is also part of the replication-transcription complex (RTC). The structure of Nsp13 in complex with the core RdRp complex has been determined, highlighting important protein-protein contacts between Nsp13 and Nsp7, Nsp8 and Nsp12 (Table 1). In particular, the Nsp13 zinc-binding domain interacts with the N-terminal regions of the two copies of Nsp8 that are part of RdRp, and the residues involved in these interactions are highly conserved in Betacoronaviruses (Chen et al., 2020). Additionally, this study shows that the nucleic acid-binding pocket is located directly in front of the downstream tRNA (Chen et al., 2020).
The structural details obtained for the Nsp13-RdRp complex highlighted that RdRp translocates the RNA in a 3′ – > 5′ direction, whereas Nsp13 translocates it in the opposite direction (Chen et al., 2020). For this reason Chen et al., proposed a ‘backtracking’ model, typical of proof-reading activity of polymerases complexes, whereby the active site of Nsp13 is divided into two ‘pockets’ by a β-hairpin, and backwards movement of Nsp13 generates a single stranded 3′-RNA that extrudes from the secondary pocket of Nsp13 active site. The authors further suggest that this process, could aid proof-reading during synthesis of the viral genome (Chen et al., 2020). Finally, the structural details of Nsp13-RdRp complex allowed to determine the molecular details of binding between ADP-Mg2+ and the N-terminal nidovirus RdRp-associated nucleotidyltransferase (NiRAN) domain, albeit the physiological role of this activity remains unknown.
Despite the advances achieved so far in structural characterization of SARS-CoV-2, it still remains to be established how the interaction between Nsp13 and Nsp8 N-terminal region influence the regulatory role of the Nsp8 N-terminal region and the Nsp7-Nsp8 heterodimer. Additionally, an interaction between Nsp8 and Orf6 has been reported in SARS-CoV (Kumar et al., 2007), and is likely conserved in SARS-CoV-2 because of the high sequence similarity between the proteins in the different viruses. However, the functional role of this interaction is not known, and how Orf6 and, potentially, other accessory protein can influence viral replication remain to be investigated.
Despite these studies have broadened the understanding of the mechanism of SARS-CoV-2 RNA replication, many details still remain elusive. The understanding of how other Nsp proteins cooperate to achieve efficient RNA synthesis is a crucial step in order to fully and efficiently target this process.
Nsp15 is a RNA uridylate−specific endoribonuclease (NendoU) (Deng and Baker, 2018), and forms a double−ring hexamer in isolation, with the active sites situated at the top and bottom faces of this hexamer (Table 1; Kim et al., 2020). This assembly is conserved also in SARS-CoV and MERS CoV Nsp15 (Bhardwaj et al., 2008; Zhang et al., 2018; Kim et al., 2020).
Whilst it’s not clear what the role of Nsp15 is in viral replication, it has been shown to co-localize with the RdRp complex in MHV (Shi et al., 1999).
Nsp9 instead shows a dimeric fold (Table 1; Egloff et al., 2004; Littler et al., 2020). In SARS-CoV, Nsp9 dimerisation was shown to be necessary for viral replication (Miknis et al., 2009). Littler et al., additionally showed that Nsp9 has low affinity for RNA binding. Nsp9 was also shown to co-localize with Nsp7-8-10 in MHV (Bost et al., 2000) and an interaction with Nsp7 was reported in Porcine Reproductive and Respiratory Syndrome Virus (Chen et al., 2017). The precise role of Nsp9, and of its interaction with other proteins, isn’t currently known.
Another necessary step for viral RNA production is RNA processing and capping, which protects the viral RNA from being targeted by the host cells immune system (Wang Y.-N. et al., 2020). In SARS-CoV-2, and more generally in coronaviruses, this function is accomplished by the Nsp10-14-16 subcomplex.
Nsp14 shows both an exonuclease activity, performed by its N-terminal domain, and a N7-Methyltransferase (MTase) domain at its C-terminus. Concerted activity of domains accomplishes both proof-reading and capping of the viral RNA, consistent with reports of their importance for correct viral replication (Chen et al., 2009; Eckerle et al., 2010; Bouvet et al., 2014).
The structure of SARS-CoV-2 Nsp14 has not been determined yet, but molecular details of Nsp10-14 interactions and important amino acids involved in these contacts have been determined in SARS-CoV (Chen et al., 2009; Bouvet et al., 2014).
Nsp10 plays multiple roles in RNA capping: its interaction with Nsp14 stimulates the exonuclease activity by stabilizing the N-terminal domain (Chen et al., 2009). Nsp10 can additionally interact with Nsp16, forming a hetero-dimer (Table 1), with 7-methylguanine-triphosphate-adenosine (m7GppA)-specific, 2′-O-methyltransferase activity. Following addition of the m7GppA cap to the RNA, in presence of a methyl group donor, Nsp16 catalyses the methylation of the nucleotide adjacent to the cap, at the 2′-O position (Chen et al., 2011; Rosas-Lemus et al., 2020). This interaction is responsible for activation of Nsp16 activity and completion of the second RNA capping step (Bouvet et al., 2010). The structure of the Nsp10-16 complex in SARS-CoV-2 revealed the high similarity of this complex in SARS-CoV-2, SARS-CoV and MERS CoV, underlining its conserved role (Rosas-Lemus et al., 2020).
Intriguingly, while Nsp10 is required for the function of Nsp14 and Nsp16 (Bouvet et al., 2014; Rosas-Lemus et al., 2020), no intrinsic enzymatic activity for this protein has been identified. It is likely that Nsp10 fulfills its role by simply acting as a scaffold for the recruitment of Nsp14 and Nsp16 to the replication-transcription complex and by triggering their specific activity. It remains to determine whether Nsp10 can spatially and temporally coordinate the proof-reading and capping activities of Nsp14 and Nsp16, also in relation to the timings of RNA synthesis operated by the replication-transcription complex.
Nsp3, Nsp4, Nsp6 Subcomplex
Nsp3 is the largest SARS-CoV-2 protein. In coronaviruses, Nsp3 comprises multiple domains, suggesting a pleiotropic role (Lei et al., 2018). The domain composition and sequence conservation of Nsp3 across the coronaviruses genera is variable (Neuman et al., 2011). Nsp3 encompasses one or two papain-like protease domains which are responsible for the cleavage of several Nsp proteins, including Nsp3 self-cleavage (Lei et al., 2018). Cleavage site and target specificity of papain-like protease domains in Nsp3 can greatly vary across coronaviruses (Lei et al., 2018). This is mainly due to the fact that whilst papain-like protease domain 2 is present in all coronaviruses, presence of papain-like protease domain 1 is retained in only some viruses, generating the differential cleavage specificity (Lei et al., 2018). Similarly to SARS-CoV and MERS CoV, SARS-CoV-2 only possess one papain-like protease domain that is responsible for the cleavage activity (Rut et al., 2020). Additionally, the papain-like protease cleaves poly-ubiquitin chains, hampering the host inflammation response, and impairs type I interferon response by interfering with ISG15 modifications (Box 2) (Lindner et al., 2005; Clementz et al., 2010).
BOX 2. SARS-CoV-2 proteins hijacking the ubiquitin-proteasome pathway.
The ubiquitin-proteasome system plays a central role in regulating eukaryotic cells homeostasis (Lecker et al., 2006), and influence many cellular processes, such as signaling, apoptosis, immunity and development (Gao and Luo, 2006; Randow and Lehner, 2009). It consists of several enzymes that, in a series of sequential reactions, promote the attachment of multiple ubiquitin molecules to cellular protein to be targeted for degradation (Lecker et al., 2006). Because of its versatility and ubiquity, the ubiquitin-proteasome system is frequently hijacked by viral proteins, for example to promote the degradation of key proteins involved in defense mechanisms (Gao and Luo, 2006; Randow and Lehner, 2009; Biard-Piechaczyk et al., 2012).
In SARS-CoV and SARS-CoV-2, Orf6 and Orf9b represent two such examples of proteins hijacking the ubiquitination pathway, to induce the degradation of interferon-induced antiviral proteins: Orf6 targets STAT1 (Frieman et al., 2007; Lei et al., 2020), a nuclear messenger that is transported to the nucleus through karyopherin α2 (KPNA2) and karyopherin ß1 (KPNB1), where it stimulates transcription of interferon stimulated genes and determines an antiviral state of the cell (McBride and Reich, 2003). Conversely, Orf9b induces the degradation of MAVS, TRAF3 and TRAF6 (Shi et al., 2014; Cheng et al., 2015; Sa Ribero et al., 2020). Mitochondrial antiviral signaling proteins (MAVS) are activated by retinoic acid-inducible gene I-like receptors (RIG-I) (Wu and Hur, 2015), which in response to detection of viral nucleic acids promotes oligomerisation of MAVS. MAVS form large filaments on the surface of mitochondria and recruit several members of the tumor necrosis factor receptor-associated factor (TRAF), including TRAF3 and TRAF6 (Saha et al., 2006; Liu et al., 2013). TRAF factors are E3 ligases which can lead to activation of IκB kinase ϵ and IKKα/β (Guo and Cheng, 2007). These factors in turn activate interferon regulatory factor (IRF) 3 and 7 and NF-κB, which are translocated to the nucleus, thereby inducing the expression of type I interferon and proinflammatory cytokines (Shi et al., 2015). However, the molecular details of how this is achieved are not currently understood, and accordingly, the structural basis for this activity has not been identified.
Furthermore, an interaction was also reported between the N protein and the p42 subunit of the proteasome complex, speculating that this interaction might inhibit proteasome functions (Wang et al., 2010). This in turn might reduce inflammation, by reducing antigen presentation.
However this remains largely speculative. Nonetheless, characterization of the p42-N complex may aid to understand how N binding to the proteasome can result in its inhibition and how this could benefit the viral replication cycle. It is also noteworthy that Orf10, exclusively found in SARS-CoV-2 (and not in SARS-CoV), has been shown to interact with the Cullin 2 (CUL2) RING E3 ligase complex, possibly hijacking its function. It is not known what the implications of this interaction is for virus replication, but a similar complex is recruited by other viral proteins, including HIV Vif, Adenovirus E4Orf6, EBV Bzlf and others (Mahon et al., 2014). In all cases, the virus exploits this to target an antiviral protein for proteasome degradation, and it is therefore likely the case also for Orf10. Identifying its target could potentially provide new avenues for antiviral treatment.
Finally, in recent years it has been identified that de- ubiquitination, together with the ubiquitin-proteasome pathway, also plays a role in protein turnover regulation. Accordingly, many viruses have been found to encode de-ubiquitinase enzymes, and these proteins play an important role in viral replication (Bailey-Elkin et al., 2017). In SARS-CoV-2, the papain-like protease domain of Nsp3 also shows de-ubiquitinase properties and leads to the removal of ISG15 from target proteins (Fu et al., 2020). Although the role of ISG15 in antiviral response is not completely understood, ISGylation was shown to enhance JAK/STAT-dependent antiviral response and to reduce virus-dependent IRF-3 degradation, thereby promoting the expression of type I interferon genes (Lu, 2006; Malakhova and Zhang, 2008). Nsp3-mediated ISG15 modifications removal could therefore result in suppression of IFN-I production and favor the progression of the viral infection.
Because of its broad and multi-step impact on the viral life cycle, one can speculate that the ubiquitin-proteasome system could be a promising target for the development of specific and/or wide spectrum antiviral drugs. However, a better structural understanding of the molecular details and contacts involved in viral and ubiquitin-proteasome system protein-protein interactions is required for such targeted drug development.
Several structures of the SARS-CoV-2 papain-like domain, alone or in complex with inhibitors, have been deposited in PDB (Table 1) but some are not published yet. These structures show that the papain-like protease domain has a homo-trimeric fold, conserved with its SARS-CoV and MERS CoV counterparts. Remarkably, one promising study has revealed the structure of the papain-like domain with GRL0617, a repurposed drug that can target both SARS-CoV and SARS-CoV-2. GRL0617 binds to the SARS-CoV-2 Nsp3 papain-like protease domain away from its catalytic site, but instead inhibits Nsp3 ability to interfere with ISG15 (Fu et al., 2020).
In an independent study the structure of the papain-like protease in complex with several inhibitors, which have been engineered to carry a vinylmethyl ester to increase their reactivity toward deubiquitinases enzymes, was determined (Rut et al., 2020). This approach yielded irreversible inhibitors that are specifically active against SARS-CoV and SARS-CoV-2, but not MERS-CoV (Rut et al., 2020). The structural data obtained provided the molecular basis of inhibition operated by these compounds and their potential in prevention of the viral infection (Lei et al., 2018; Rut et al., 2020), highlighting that the papain-like protease/inhibitors complexes adopt an overall very similar structure and that inhibitors binding takes place in proximity of the active site, thereby preventing substrate binding (Rut et al., 2020).
Furthermore, Nsp3 possesses two ubiquitin-like domains that fulfill distinct roles. The ubiquitin-like domain 1 is positioned at the N-terminal of the protein and it can mediate interaction with the N protein and with ssRNA (Cong et al., 2020). The role of the ubiquitin-like domain 2 is object of contrasting reports, with one study suggesting it plays a role in inhibiting IFN production, while a distinct study suggests it is involved in innate immune response (Frieman et al., 2009; Clementz et al., 2010).
Finally, Nsp3 proteins have also a macro domain that is involved in disruption of the expression of innate immunity genes (Fehr et al., 2016). Interestingly in vitro experiments have shown the ability of SARS-CoV Nsp3 macro domain to interact with Nsp12, however, the physiological relevance of this interaction in the virus life cycle remains unknown (Imbert et al., 2008). Several structures of the macro domain have been released on PDB, although are still not published (Table 1) showing high similarity to MERS CoV Nsp3 Macro domain (Lei et al., 2018).
Aside from its protease and Ubiquitin-like domains, Nsp3 is also responsible for initiation of the assembly of the replication-transcription complex in SARS-CoV, mouse hepatitis virus (MHV) and MERS-CoV-infected cells and therefore is essential for virus replication (Lei et al., 2018). Nsp3 was shown to be interacting with a number of other CoV proteins, including Nsp2, Nsp4, Nsp6, Orf3a, Orf7a and Orf9b (von Brunn et al., 2007; Neuman et al., 2008; Angelini et al., 2013; Hagemeijer et al., 2014; Lei et al., 2018), highlighting the central role of the multi-domain protein in the virus life cycle and thus, rendering it an attractive target for therapeutic development. In particular, Nsp3, Nsp4 and Nsp6 have been shown to form a complex that modifies the endoplasmic (ER) reticulum into double membrane vesicles (DMVs) (Angelini et al., 2013; Hagemeijer et al., 2014). These studies also reveal that the C-terminal domain of Nsp3 is sufficient for DMVs formation, and further identified fundamental residues in Nsp4 involved in DMVs formation (Angelini et al., 2013; Hagemeijer et al., 2014). Whilst the exact mechanism by which Nsp3-4-6 can trigger this event is still unknown, the investigation of fully assembled virions by cryo-ET by Klein and co-authors has allowed to shed light on the dynamics of DMVs assembly and fusion during virus budding. This study showed that DMVs content in RNA is relatively high and their role might involve concealment of RNA intermediates from the immune system (Klein et al., 2020). The authors were further able to prove that, similarly, to other viruses, SARS-CoV-2 DMVs can form multi-vesicular compartment through membrane fusion. Instances of multiple vesicles contained in one single outer membrane, referred as vesicle packets (VPs), were observed and in some cases, inner membrane fusion allowed inter-connection between vesicle packets (Klein et al., 2020). Although a role for multi-vesicular DMVs in virus budding has been suggested, this has still to be experimentally verified. Virus budding was observed to occur in regions with a high concentration of DMVs and the authors observed that S proteins alone are not sufficient for membrane curvature, hypothesizing that other proteins, such M, initiate this process (Klein et al., 2020). These findings provide great advance in understanding the fundamental stages of virus replication and assembly. Nevertheless, further investigation and molecular details are required to understand how the membrane curvature occurs and the role of M protein and, potentially of other proteins in this process.
The PDB IDs for the aforementioned structures of the Nsp3 macro and papain-like protease domains can be found in Table 1. Nonetheless, the structure of the full-length protein remains to be determined, and in particular it is not clear if the Nsp3 protein forms a large, defined structure, or if it is merely constituted of isolated “beads-on-a-string”-like domains.
Nsp3 plays a central role in viral replication, and therefore is an attractive target for therapeutics development. However, there are still a lot of uncharacterized domains, and the relevance to its interaction with other non-structural proteins remains to be elucidated. In particular, an effort to obtain structural information of Nsp3 in complex with other proteins could provide several drug targets to block viral replication in host cells.
Nevertheless, one of the most important pending questions is to understand how the whole replication-transcription complex elements cooperate and/or are hierarchically recruited during viral replication. In Figure 3, the available evidence of SARS-CoV-2 replication-transcription complex recruitment to date is represented. Putative localization and interactions networks have been presented based on the evidences reviewed in this manuscript.
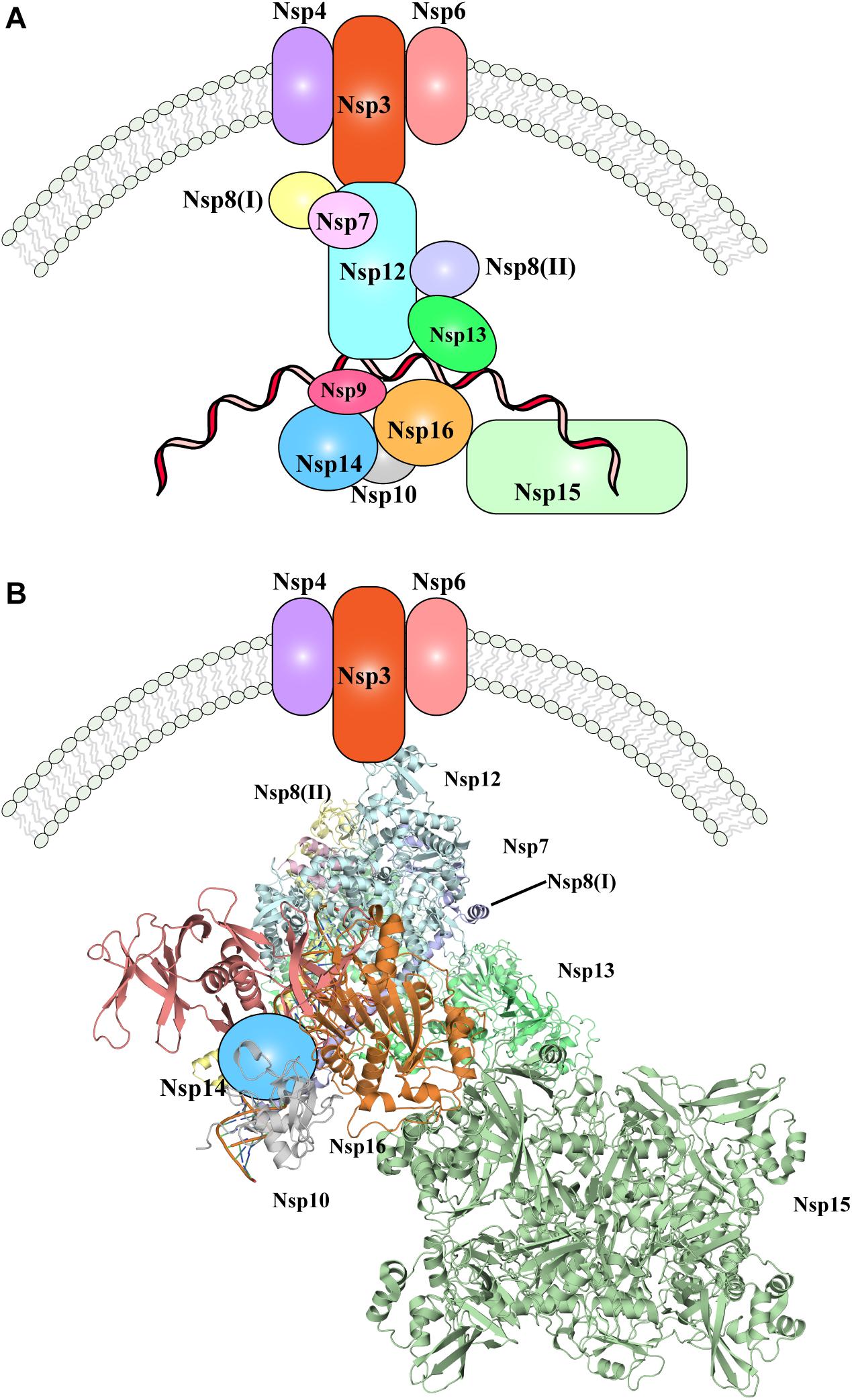
Figure 3. The SARS-CoV-2 replication-transcription complex. (A) Schematic representation of the non-structural proteins that were reported to intervene during the distinct steps of viral RNA replication. Endoplasmic reticulum rearrangements are performed by the Nsp3-5-6 subcomplex resulting in recruitment of the Nsp proteins, initiating RNA synthesis, proof-reading and capping. (B) Structural representation of the proteins involved in viral RNA replication, as shown in (A). Where protein structures have been determined, cartoon models of the molecular structures are represented, whilst proteins with no structural details are depicted as colored shapes, as in part A. Relative positions and molecular contacts between Nsp proteins represented are based on the experimental evidence reviewed in this manuscript, while putative localization have been provided for those protein whose interaction network is still not determined. PDB IDs for the structures depicted in these figures are reported in Table 1.
Following cleavage by the proteases Nsp3 and Nsp5 (presumably still on the uncleaved full polypeptide, or carried over within virus particles), the individual Nsp proteins are produced. Nsp3-4-6 initiate ER rearrangements, which in turn recruits the remaining components of the RTC complex. Nsp12-7-8, together with Nsp13 are then likely responsible for production of novel viral RNA, and further modification and proof-reading is operated by Nsp10-14-16 complex (Figure 3). The role played by Nsp9 and Nsp15 within the RTC complex and the step at which their function is executed has not been fully elucidated, but given the interactions and co-localization of Nsp9 and Nsp15 with other components of the RTC complex, they are likely involved in RNA production or processing (Figure 3). Moreover, the N protein will be recruited in situ through interaction with Nsp3, however it remains to determine at which stage of the RNA replication its role is exerted.
While it is possible to propose a tentative model of the RTC role and functions within the infected host cells, many knowledge gaps are currently present that prevent to fully understand how SARS-CoV-2 replication works. For instance, it is not clear whether the recruitment of the RTC components and the exertion of their function is simultaneous or whether this is a timely coordinated process. In the latter case, it would be of great interest to determine the full hierarchy of Nsp proteins recruitment and the inter-dependence that exists between them. In particular, in the context of the interaction network of the Nsp proteins, their relative fold and orientation within the RTC complex should be determined, as well as a full characterization of the regions that are involved in protein-protein interactions and with the nascent RNA molecules. To date, Nsp1 and Nsp2 involvement in RTC complex or the existence of a mechanism that regulates and coordinates their activities with the RTC complex has not been reported and thus it is currently not clear where the role of Nsp1-2 falls within this preliminary putative model.
Similarly, the reported interactions of Nsp3 with Orf3a, Orf7a and Orf9b (von Brunn et al., 2007; Neuman et al., 2008; Angelini et al., 2013; Hagemeijer et al., 2014; Lei et al., 2018) also pose the question on whether these interactions are conserved in SARS-CoV-2 and whether other accessory proteins are involved in the various steps of the RTC assembly and mechanism.
Efforts to answer these questions could provide a comprehensive molecular dissection of the interplay between the SARS-CoV-2 proteins during viral replication and greatly improve the understanding of RTC complex function in its physiological context by taking into account also the supportive role that the accessory proteins can provide. It will also provide new avenues for targeting these interactions by antiviral drugs.
Nsp1, Nsp2, Nsp5
Nsp1 displays high sequence divergence amongst the Betacoronavirus genus. Nevertheless, numerous reports on distinct viruses have highlighted that its function is conserved, despite the relatively low sequence similarity (Narayanan et al., 2015). In SARS-CoV, Nsp1 abrogates translation of the host cell genes, which may in turn promote enhanced translation of viral proteins (Lokugamage et al., 2012). This is achieved through two strategies; by binding of Nsp1 to the 40S ribosomal subunit and inhibiting mRNA translation at different stages (Kamitani et al., 2009; Lokugamage et al., 2012); and by endonucleolytic RNA cleavage in the 5′-UTR of host mRNAs (Kamitani et al., 2009). The translational inhibition does not apply to the viral mRNAs, however how this is achieved is unknown, but might implicate the interaction of Nsp1 with the 5′-UTR region of the viral mRNAs (Huang et al., 2011). Translational inhibition also determines lack of an efficient IFN-dependent antiviral signaling and, consequently, of efficient innate immune response (Narayanan et al., 2008). Importantly, amino acids that are crucial for Nsp1 functions have been identified and suggested as potential targets for therapy or generation of attenuated virus for vaccine development (Narayanan et al., 2008, 2015; Lokugamage et al., 2012).
Recent studies have confirmed that Nsp1 from SARS-CoV-2 plays the same roles as its counterpart in SARS-CoV during virus infection (Schubert et al., 2020; Thoms et al., 2020). In the study by Thoms et al. (2020) a complex between purified Nsp1 and 40S ribosomal subunits was reconstituted in vitro by cryo-EM. The authors found two densities that they identified as two helices from Nsp1 C-terminus. These helices are localized inside the ribosomal mRNA entry channel, blocking mRNA accommodation and thereby shutting down mRNA translation (Thoms et al., 2020). The authors further identified a globular density, which may represent the N-terminal domain of Nsp1, but were unable to confirm it (Thoms et al., 2020). Importantly, Nsp1 C-terminal densities were also observed when the protein was co-purified with 40S and 80S ribosomal complexes in vivo. Structural data from these isolated complexes highlighted that Nsp1-40S and Nsp1-80S complexes could be divided in different populations, representing different intermediate states of the translation initiation process. In all these distinct intermediates, Nsp1 was found to bind in the position and conformation to that observed in the in vitro complex, confirming its role as inhibitor of translation initiation (Thoms et al., 2020). These findings were confirmed in an independently resolved structure of Nsp1 in complex with several ribosomal components (Schubert et al., 2020).
Much less is known instead about Nsp2. A study on the SARS-CoV Nsp2 protein has shown the ability of this protein to interact with prohibitin 1 and prohibitin 2 (Cornillez-Ty et al., 2009), however, this protein was found to be dispensable for virulence of several coronaviruses (Graham et al., 2005). Prohibitin proteins play a role in apoptosis regulation, mitochondrial functions and cell cycle regulation (Wang et al., 1999; Peng et al., 2015; Signorile et al., 2019). These findings imply that Nsp2, similarly, to Nsp1, may contribute to viral replication by arresting normal host cell functions. Whilst there are no available structures for Nsp2, a bioinformatic approach allowed to predict the presence of 4 putative transmembrane helices and a region that displays structural similarity to the endosome−associated protein of the avian infectious bronchitis virus (Cornillez-Ty et al., 2009). This latter region contains an amino acid substitution, compared to SARS-CoV, which might stabilize the protein and contribute to the higher infectivity of the virus (Angeletti et al., 2020).
Nsp5 is the main protease and, together with the papain-like protease domain in Nsp3, is responsible of cleavage of Orf1ab polypeptides and release of the mature Nsp proteins (Mousavizadeh and Ghasemi, 2020). Proteases and Nsp5 in particular have been identified as one of the most attractive drug targets to treat coronaviruses infections (Anand et al., 2003). Indeed inhibition of the Nsp5-mediated cleavage would prevent Nsp proteins productions and thus, prevent viral replication. For this reason, since the start of the SARS-CoV-2 pandemic, many studies have focused on determining the structure of Nsp5, alone or in complex with potential inhibitors, and have allowed to identify a plethora of compounds that are able to antagonize Nsp5 activity, providing the molecular mechanism behind the inhibition (Jin et al., 2020a, b; Zhang et al., 2020b). The chymotrypsin-like and 3C protease-like domains (domain I and II, respectively) both adopt a six-strand antiparallel β barrell fold. Domain III shows a globular fold that consists of a group of five helices. Interaction between two domains III from two distinct Nsp5 protomers is responsible of modulating the dimerization between their respective domain I and II. SARS-CoV-2 Nsp5 dimerises with the same affinity as its SARS-CoV counterpart (Zhang et al., 2020b). Dimerization of Nsp5 protomers ultimately results in the formation of a substrate-binding catalytic site, where Cys145 and His41 from each protomer constitute the catalytic residues (Zhang et al., 2020b). These reports demonstrated that the overall structure of SARS-CoV-2 Nsp5 is similar to SARS-CoV Nsp5, albeit some minor structural differences were found. In particular, a hydrogen bond between Thr285 residues of the two domains III of SARS-CoV Nsp5 was observed (Zhang et al., 2020b). In SARS-CoV2 Nsp5, Thr285 is replaced by an Alanine residue, thereby abolishing the hydrogen bond. Absence of this bond allows the domain III from each SARS-CoV2 Nsp5 to come closer contact, but only causes a slight increase in catalytic efficiency, compared to SARS-CoV (Zhang et al., 2020b).
Interestingly, several promising drugs were already designed for other purposes, including targeting of SARS-CoV and seem to be successfully repurposed for Nsp5 inhibition (Jin et al., 2020a, b; Zhang et al., 2020a, b). For most of such compounds, these structures revealed that the molecules act as competitive inhibitors, binding in the substrate-binding pocket (Jin et al., 2020a, b; Zhang et al., 2020b).
Accessory Proteins
Severe acute respiratory syndrome coronavirus 2 accessory proteins are generally less well characterized, and their role in infection is mostly not understood. Accessory proteins display the highest degree of variability, even within very closely related coronaviruses (Wu A. et al., 2020). Indeed, Orf10 is exclusively present in SARS-CoV-2 whilst it is not encoded by the closely related SARS-CoV. Similarly, SARS-CoV encodes two Orfs, Orf8a and Orf8b, in place of the single Orf8 that is conversely found in SARS-CoV-2 (Nguyen et al., 2020). Investigation of the role of the accessory proteins and the different pathways they influence could allow to pin-point those proteins responsible for instance, of the different infectivity or mortality of SARS-CoV and SARS-CoV-2 viruses.
Orf3a displays high conservation in the Betacoronavirus subgenus Sarbecovirus, which includes SARS-CoV and SARS-CoV-2 (Kern et al., 2020). In SARS-CoV, Orf3a was shown to form an ion channel likely causing a calcium influx, involved in increased formation of DMVs (Lu et al., 2009). This pore shows modest selectivity towards K+ and Ca2+ over Na+ in vitro, however, the ion actually transported in vivo is not known. The structure of SARS-CoV-2 Orf3a in its inactive or ‘closed’ conformation has been solved with cryo-EM, highlighting that this protein adopts a novel fold, forming a large and bifurcated channel that is connected to the cytoplasm through a large cavity (Kern et al., 2020). Orf3a was found to determine apoptosis and in SARS-CoV the pro-apoptotic activity of Orf3a has been associated to its channel-forming activity (Chan et al., 2009; Ren et al., 2020).
Orf3a can additionally interact with the structural proteins N, M and S (Tan, 2005), supporting a role of Orf3a in virus budding. These observations are consistent with the reported interaction between Orf3a and Nsp3 (von Brunn et al., 2007), which initiates DMVs formation.
Nsp3 also interacts with Orf9b, suggesting that the role of these three proteins might be connected. Orf9b is a E3 ubiquitin ligase, and evidences have shown that it promotes the ubiquitination and proteasomal degradation of dynamin-like protein 1 (Box 2) (Kamerkar et al., 2018). Dynamin-like protein 1 promotes mitochondrial fission in physiological conditions. Orf9b-mediated degradation of dynamin-like protein 1 may in turn lead to elongation of mitochondria, ultimately compromising the association between mitochondria and ER, a crucial step for IFN production and signaling during innate immune response (Chatel-Chaix et al., 2016; Kim et al., 2018). Furthermore, it hinders the cell IFN response by triggering the degradation of MAVS, TRAF3 and TRAF6 (Shi et al., 2014; Sa Ribero et al., 2020). A structure for SARS-CoV-2 Orf9b has been deposited in PDB (Table 1), where it shows high conservation with SARS-CoV Orf9b dimeric, β-sheet-rich, structure (Meier et al., 2006). Additionally, a central, lipid-binding pocket was notably observed and it was suggested to mediate ORF9b localization in proximity of ER/Golgi complex (Meier et al., 2006). Furthermore, Orf9b was proposed to be involved in the interaction with Nsp3, N and S proteins (von Brunn et al., 2007; Lu et al., 2009). Binding to the ER/Golgi complex membrane, through its lipid binding pocket, would provide Orf9b with an ideal localization to accomplish these interactions and would suggest a role for this protein in virion assembly.
Despite the currently available evidence, the exact role(s) of Orf9b is not fully understood. Specifically, it is not known if the Orf9b interactions are all necessary for its Ubiquitin Ligase activity, or whether Orf9b interactions with other SARS-CoV-2 proteins indicate that it plays multiple roles in the virus cycle. Structural characterization of these sub-complexes could provide the necessary information to understand the role of Orf9b.
Like Orf3a, evidence suggests that Orf7a also might play a role in virus budding and release. In SARS-CoV, ORF7a enables virion release, by preventing the antiviral activity of the bone marrow stromal antigen 2 (BST-2/tetherin), which tethers budding virions to the host cell (Taylor et al., 2015). In agreement with this, Orf7a was also shown to interact with Nsp3 (Neuman et al., 2008). In SARS-CoV, Orf3a and Orf7a were additionally found to localize at the Golgi-endoplasmic reticulum intermediary compartment, with Nsp3 (von Brunn et al., 2007; Taylor et al., 2015), where Orf7a can additionally interact with Orf3a, which in turn interacts with M, N and S proteins (Tan, 2005). However, recent observation in SARS-CoV-2 found that Orf3a is mainly localized at the plasma membrane (Kern et al., 2020; Ren et al., 2020). The structure of SARS-CoV-2 Orf7a has been solved (PDB ID: 6W37, Table 1), and it shows high degree of conservation with the structure of SARS-CoV Orf7a, revealing that this protein possesses an Ig fold (Nelson et al., 2005). It also possesses a predicted C-terminal TM helix.
Orf8 has newly been described as a paralog of Orf7a, thought to have evolved via gene duplication. It also possesses an Ig-like fold, but Orf8 lacks the TM helix, and has an additional large insertion between its third and fourth strand. A homology model of Orf8, based on the solved Orf7a structure has suggested that this insertion in Orf8 would lead to an enhancement of the putative peptide-ligand binding activity that was reported for Orf7a (Tan et al., 2020). Orf8 binds to MHC-I, leading to decreased antigen presentation, whereas this was not observed for Orf8a and 8b. Orf8-mediated inhibition of MHC-I antigen presentation ultimately leads to inefficient viral clearing (Zhang et al., 2020c). Additionally, Orf8 was found to inhibit IFN-I signaling pathway, likely through downregulation of the Interferon Stimulation Response Element (ISRE) (Li J.-Y. et al., 2020). In SARS-CoV, Orf8b was shown to interact with Orf7a, M and E proteins (Keng and Tan, 2009), leaving open the possibility that it could additionally cooperate in DMVs formation and virus budding. It is not known if SARS-CoV-2 Orf8 possesses a similar activity. Future efforts should address this question and further clarify the basis of the different localization observed for Orf3a in SARS-CoV and SARS-CoV-2. Answers to the latter questions will further allow to shed light on whether Orf3a interaction with Orf7a and Nsp3 is retained in SARS-CoV-2 and in this case, to work towards determination of the molecular details of the interactions between Orf3a, Orf7a with Nsp3 and the other proteins found at the DMVs interface, as well as interactions with the structural proteins during virus budding, to establish the contacts that are required for virus budding and release.
Orf7b is predicted to be an integral membrane protein and its single TMH was found to be sufficient to direct localization to the Golgi complex in SARS-CoV (Schaecher et al., 2008). Furthermore, Orf7a and Orf7b favor apoptotic activity of infected cells, although their role is dispensable for SARS-CoV infections in Vero African green monkey kidney cells, CaLu-3 human lung adenocarcinoma cells and CaCo-2 human colorectal carcinoma cells (Schaecher et al., 2007). To date, research efforts focused on SARS-CoV-2 investigations have not provided many details on Orf7b functions. Given the high sequence similarity between Orf7b in the two closely related viruses, it likely that its role will be conserved. Nevertheless, a structure Orf7b from either SARS-CoV and SARS-CoV-2 has not yet been determined.
Orf3b has been recently identified as one of the accessory proteins of SARS-CoV-2 that downregulates IFN-I signaling (Konno et al., 2020) (Box 3). Importantly, SARS-CoV-2 Orf3b ability to antagonize IFN-I is significantly higher than its homologue in SARS-CoV (Konno et al., 2020), which may play a role in the higher transmission rate. This function is determined by the C-terminus of Orf3b, and specifically correlates with length of this region (Konno et al., 2020). To date, there are no studies that report a structure for Orf3b. Determination of structural details could shed light onto the nature of its fold, facilitating identification of proteins with structural similarities, which may in turn provide a better understanding on the putative roles of this protein and locate sites to target.
BOX 3. SARS-CoV-2 manipulation of the Interferon response pathway.
Figure | Schematic representation of SARS-CoV-2 proteins and their involvement in IFN hijacking. Proteins involved in IFN production pathways were represented in gray, while SARS-CoV-2 proteins are represented in color. The stage of IFN pathways which non-structural or accessory proteins are predicted to interfere with, is based on the reported studies cited within the box and the main text.
Interferons are a class of cytokines, central to the innate immune response (for extensive review of the pathway, see Muller et al., 1994; Samuel, 2001; Zhou et al., 2018). The release of IFN, a response to cell infection by a viral (or bacterial) pathogen, triggers downstream events to prevent their replication, such as suppression of viral transcription, translation and replication and prevention of entry of the virus (Samuel, 2001), which ultimately protect the host. Given the broad spectrum of IFN-dependent response, many viruses, including SARS-CoV-2, have evolved multiple effective strategies to subvert this defense (Spiegel et al., 2005; Hadjadj et al., 2020).
A number of SARS-CoV-2 proteins have been implicated in modulating the IFN response either at the level of IFN synthesis, downstream IFN-signaling, or both (Sa Ribero et al., 2020). Over-expression of Orf3b, Orf6 and N protein has been demonstrated to antagonize IFN (Yuen et al., 2020; Konno et al., 2020; Li J.-Y. et al., 2020). In SARS-CoV, these proteins were demonstrated to play a role in IFN-ß downregulation, via repression of the interferon regulatory factor-3 (IRF-3) (Kopecky-Bromberg et al., 2007). Hindering of this response results in enhanced viral replication, as IFN-ß stimulates the expression of antiviral and antiproliferative factors by neighboring cells, thereby limiting spread (Ivashkiv and Donlin, 2014). Orf6 inhibits IFN production through hindering of the of STAT1-nuclear complex formation (Frieman et al., 2007; Lei et al., 2020) by sequestering karyopherin α2 (KPNA2) and karyopherin ß1 (KPNB1) to the endoplasmic reticulum/Golgi interface, thereby preventing nuclear translocation of the messenger STAT1 and, consequently hampering the host defense against viral infection (McBride and Reich, 2003; Frieman et al., 2007).
Orf3b has also been shown to bind to karyopherins α3 and α4 (Veljkovic and Paessler, 2020). Interestingly, the same mechanism of IFN inhibition is shared by the Ebola virus, whereby VP24 binds karyopherins α1, α5 and α6 (Reid et al., 2007). Alongside Orf6 and Orf3b, Orf8 and N have also been shown to inhibit IFN-I and the NF-κB response (Li J.-Y. et al., 2020). Both Orf6 and Orf8 reduce expression of the Interferon Stimulation Response Element (ISRE), whereas N has been proposed to use an alternative mode of IFN downregulation (Li J.-Y. et al., 2020): it was shown to interact with stress granule proteins G3BP1, G3BP2 and La-related protein 1 (LARP1) (Gordon et al., 2020). G3BP1 and G3BP2 upregulate and allow accumulation of interferon-stimulated genes (ISG), whereas the role of LARP1 in ISG regulation has been object of controversial reports (Alam and Kennedy, 2019; Kim et al., 2019; Berman et al., 2020).
More recently, Nsp13, Nsp14 and Nsp15 of SARS-CoV-2 have also been implicated in the inhibition of nuclear localization of IRF-3 (Yuen et al., 2020; Gordon et al., 2020). Putative interactions between Orf9c and multiple regulators of Iκß kinase, an upstream kinase of NF-κB which activates IRF-3, have also been reported (Gordon et al., 2020). Furthermore, an interaction between Orf9b and the adaptor protein TOMM70, required for IRF-3 activation, was also observed (Gordon et al., 2020). Nevertheless, the physiological relevance of these interactions has yet to be determined. Finally, the papain-like domain on Nsp3 was also found to hamper the IFN-I response by altering the normal ISG15-modifications pathway, through cleavage of ISG15 (Fu et al., 2020).
The sensitivity of SARS-CoV to IFN treatment has been known for many years (Cinatl et al., 2003). IFN-I is therefore an emerging candidate for COVID-19 treatment, as IFN-α and IFN-ß are also able to inhibit SARS-CoV-2 replication (Hensley et al., 2004; Clementi et al., 2020; Felgenhauer et al., 2020). Yet to this date, we do not have any structural information on the mechanisms by which the virus overcomes this effect. Such data will be necessary to exploit the interferon response in the combat against COVID19.
Similar to Orf3b, Orf6 also down-regulates interferon signaling (Yuen et al., 2020). Previous immunofluorescence studies performed for SARS-CoV demonstrated that Orf6 achieves IFN-I antagonism by disrupting formation of STAT1 complex, through sequestering its components to the endoplasmic reticulum/Golgi complex (Boxes 2, 3) (Frieman et al., 2007; Lei et al., 2020). Whilst this mode of action needs to be confirmed for SARS-CoV-2, an additional interaction between Orf6 and an mRNA export factor induced by IFN-I, NUP98 RAE1, has been reported (Gordon et al., 2020). Therefore, identifying the key binding interactions between Orf6 and its binding partners by solving the complex structure could be crucial for understanding the mechanism of inhibition employed and resultant pathogenesis. In addition to IFN-I antagonism, it has been suggested that Orf6 could play a role in viral replication: In SARS-CoV, Orf6 was found to co-localize and interact with Nsp8 (Kumar et al., 2007). It is currently unclear whether Orf6 is involved in RNA synthesis or whether its interaction with Nsp8 might play a modulating role on the activity of the replication-transcription complex. Furthermore, it still needs to be investigated whether the interaction between Orf6 and Nsp8 and its physiological role in viral replication are conserved in SARS-CoV-2.
Finally, very few information are available for Orf9c and Orf10. SARS-CoV-2 Orf9c is encoded within the nucleocapsid gene, similarly, to Orf9b. Evidence in SARS-CoV suggests that it interacts with human NLRX1, F2RL1, NDFIP2, proteins, which in turn are associated with NF-kB signaling pathway (Gordon et al., 2020). Orf9c can further influence the modulation of IkB kinase (Gordon et al., 2020), thereby suggesting a role of this protein in modulation of inflammation.
Orf10 is exclusively found in SARS-CoV-2 and its use has been suggested as a strategy to identify ongoing COVID-19 infections (Koyama et al., 2020). Available details about Orf10 are very few, however, a recent analysis of the interactome of SARS-CoV-2 proteins has interestingly pointed out that that Orf10 interacts with the Cullin2 RING E3 ligase complex, in particular with the ZYG11B adapter (Gordon et al., 2020) (Box 2). The physiological role and mechanistic of this interaction is not known, but it suggests that as many other viral proteins, Orf10 is involved in hijacking the ubiquitin-proteasome complex in order to favor viral replication and inhibit the antiviral host response, with a mechanism that is yet to be determined.
Conclusive Remarks
SARS-CoV-2 first appeared in China in 2019 and has quickly become a major global threat to public health. Despite the impressive amount of research efforts that have been devoted to fully characterize the mode of infection of this novel virus and determine the details of its mechanism of hijacking on host factors, a specific treatment with high rate of success has not been yet found. Similarly, a clinically approved vaccine has not been developed yet, despite the great advances that have been made in the characterization of SARS-CoV-2 entry into host cells.
In this review we have summarized the current state of knowledge of the structural studies that have provided detailed molecular details of some of the SARS-COV-2 proteins and the basis of their biological functions during the viral infection cycle. Furthermore, several of the reported studies within this review have employed X-ray crystallography and cryo-EM approaches to not only determine the mode of action of SARS-COV-2 proteins in their physiological context but to additionally determine the basis of inhibition of their activity operated by potential inhibitors that may be developed for treatment of COVID-19.
Nevertheless, there are many aspects of the distinct phases of SARS-CoV-2 infection cycle that are not well characterized. For instance, despite the high number of structures now available for the SARS-CoV-2 S protein in its open or closed state or in complex with the ACE2 receptor, it still remains to be understood how the TMDs of the S protein effectively contribute to the structural rearrangements that are coupled with virus entry. One of the main challenges still to be addressed at the structural level, is to obtain high-resolution details of membrane domains from the other structural proteins (M and E).
Similarly, many structures of the replication-transcription complex have been determined, providing new insights into the mechanism of RNA synthesis, the basis of the proofreading activity operated by Nsp13 and the regulatory role of the conformational changes of Nsp7 and Nsp8. Nevertheless, multiple studies in SARS-CoV have indicated that accessory proteins may intervene at different stages of the replication process or in the previous phase of DMVs formation (Kumar et al., 2007; von Brunn et al., 2007; Neuman et al., 2008; Angelini et al., 2013; Hagemeijer et al., 2014; Lei et al., 2018). However, the molecular basis for this is largely not understood.
Research effort to date have provided molecular details of several subcomplexes involved in viral entry, replication and budding. However, one of the current limitations in fully understanding these processes is lack of structural and functional characterization of the full protein complexes, protein interaction networks and its hierarchy, involved in the main steps of SARS-CoV-2 infection. Integrating the available structural data on subcomplexes involved in viral replication with additional functional and biochemical studies will provide information on crucial protein-protein interactions within the full complexes, including transient interactions that play a modulatory role or mediate distinct phases of infection. In particular, accessory proteins may play multifaceted role in infection, and require further characterization. Ultimately, a more comprehensive knowledge of SARS-CoV-2 infection that these additional studies would provide will allow a more effective and specific structure-guided design of vaccination and therapeutic strategies. Structural biologists, like Wayne Gretzky, “need to skate to where the puck is going to be, not to where the puck has been”.
Author Contributions
JRCB and GM planned the overall structure of the review. GM wrote the manuscript and made the figures, with help from RJF and SLML-F for the accessory proteins section, and with edits from JRCB. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by BBSRC grant BB/R019061 (to JRCB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the other members of the Bergeron lab for help gathering information on the SARS-CoV-2 proteins, and for discussion. We apologize to those authors’ work we were not able to mention due to space restrictions.
References
Alam, U., and Kennedy, D. (2019). G3BP1 and G3BP2 regulate translation of interferon-stimulated genes: IFITM1. IFITM2 and IFITM3 in the cancer cell line MCF7. Mol. Cell. Biochem. 459, 189–204. doi: 10.1007/s11010-019-03562-3
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R., and Hilgenfeld, R. (2003). Coronavirus main proteinase (3CLpro) structure: basis for design of Anti-SARS Drugs. Science 300, 1763–1767. doi: 10.1126/science.1085658
Angeletti, S., Benvenuto, D., Bianchi, M., Giovanetti, M., Pascarella, S., and Ciccozzi, M. (2020). COVID-2019: the role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 92, 584–588. doi: 10.1002/jmv.25719
Angelini, M. M., Akhlaghpour, M., Neuman, B. W., and Buchmeier, M. J. (2013). Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio 4:e00524-13. doi: 10.1128/mBio.00524-13
Bailey-Elkin, B. A., Knaap, R. C. M., Kikkert, M., and Mark, B. L. (2017). Structure and function of viral deubiquitinating enzymes. J. Mol. Biol. 429, 3441–3470. doi: 10.1016/j.jmb.2017.06.010
Barnes, C. O., West, A. P., Huey-Tubman, K. E., Hoffmann, M. A. G., Sharaf, N. G., Hoffman, P. R., et al. (2020). Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 182, 828.e16–842.e16. doi: 10.1016/j.cell.2020.06.025
Benvenuto, D., Giovanetti, M., Ciccozzi, A., Spoto, S., Angeletti, S., and Ciccozzi, M. (2020). The 2019-new coronavirus epidemic: evidence for virus evolution. J. Med. Virol. 92, 455–459. doi: 10.1002/jmv.25688
Berman, A. J., Thoreen, C. C., Dedeic, Z., Chettle, J., Roux, P. P., and Blagden, S. P. (2020). Controversies around the function of LARP1. RNA Biol. [Epub ahead of print]. doi: 10.1080/15476286.2020.1733787
Bestle, D., Heindl, M. R., Limburg, H., Van Lam van,, T., Pilgram, O., Moulton, H., et al. (2020). TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 3:e202000786. doi: 10.26508/lsa.202000786
Bhardwaj, K., Palaninathan, S., Alcantara, J. M. O., Yi, L. L., Guarino, L., Sacchettini, J. C., et al. (2008). Structural and functional analyses of the severe acute respiratory syndrome coronavirus endoribonuclease Nsp15. J. Biol. Chem. 283, 3655–3664. doi: 10.1074/jbc.M708375200
Biard-Piechaczyk, M., Borel, S., Espert, L., de Bettignies, G., and Coux, O. (2012). HIV-1, ubiquitin and ubiquitin-like proteins: the dialectic interactions of a virus with a sophisticated network of post-translational modifications. Biol. Cell 104, 165–187. doi: 10.1111/boc.201100112
Bosch, B. J., Bartelink, W., and Rottier, P. J. M. (2008). Cathepsin L functionally cleaves the severe acute respiratory syndrome coronavirus class I fusion protein upstream of rather than adjacent to the fusion peptide. J. Virol. 82, 8887–8890. doi: 10.1128/JVI.00415-08
Bosch, B. J., de Haan, C. A. M., Smits, S. L., and Rottier, P. J. M. (2005). Spike protein assembly into the coronavirion: exploring the limits of its sequence requirements. Virology 334, 306–318. doi: 10.1016/j.virol.2005.02.001
Bosch, B. J., van der Zee, R., de Haan, C. A. M., and Rottier, P. J. M. (2003). The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811. doi: 10.1128/JVI.77.16.8801-8811.2003
Bost, A. G., Carnahan, R. H., Lu, X. T., and Denison, M. R. (2000). Four proteins processed from the replicase gene polyprotein of mouse hepatitis virus colocalize in the cell periphery and adjacent to sites of virion assembly. J. Virol. 74, 3379–3387. doi: 10.1128/JVI.74.7.3379-3387.2000
Böttcher-Friebertshäuser, E. (2018). Membrane-anchored serine proteases: host cell factors in proteolytic activation of viral glycoproteins. Act. Viruses Host Proteases 153–203. doi: 10.1007/978-3-319-75474-1_8
Bouvet, M., Debarnot, C., Imbert, I., Selisko, B., Snijder, E. J., Canard, B., et al. (2010). In Vitro Reconstitution of SARS-Coronavirus mRNA Cap Methylation. PLoS Pathog. 6:e1000863. doi: 10.1371/journal.ppat.1000863
Bouvet, M., Lugari, A., Posthuma, C. C., Zevenhoven, J. C., Bernard, S., Betzi, S., et al. (2014). Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 289, 25783–25796. doi: 10.1074/jbc.M114.577353
Cai, Y., Zhang, J., Xiao, T., Peng, H., Sterling, S. M., Walsh, R. M., et al. (2020). Distinct conformational states of SARS-CoV-2 spike protein. Science 369, 1586–1592. doi: 10.1126/science.abd4251
Cao, Y., Su, B., Guo, X., Sun, W., Deng, Y., Bao, L., et al. (2020). Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182, 73–84.e16. doi: 10.1016/j.cell.2020.05.025
Chan, C.-M., Tsoi, H., Chan, W.-M., Zhai, S., Wong, C.-O., Yao, X., et al. (2009). The ion channel activity of the SARS-coronavirus 3a protein is linked to its pro-apoptotic function. Int. J. Biochem. Cell Biol. 41, 2232–2239. doi: 10.1016/j.biocel.2009.04.019
Chan, J. F. W., Lau, S. K. P., To, K. K. W., Cheng, V. C. C., Woo, P. C. Y., and Yuen, K.-Y. (2015). Middle east respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 28, 465–522. doi: 10.1128/CMR.00102-14
Chang, C., Hou, M.-H., Chang, C.-F., Hsiao, C.-D., and Huang, T. (2014). The SARS coronavirus nucleocapsid protein – Forms and functions. Antiviral Res. 103, 39–50. doi: 10.1016/j.antiviral.2013.12.009
Chang, C.-K., Hsu, Y.-L., Chang, Y.-H., Chao, F.-A., Wu, M.-C., Huang, Y.-S., et al. (2009). Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J. Virol. 83, 2255–2264. doi: 10.1128/JVI.02001-08
Chatel-Chaix, L., Cortese, M., Romero-Brey, I., Bender, S., Neufeldt, C. J., Fischl, W., et al. (2016). Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe 20, 342–356. doi: 10.1016/j.chom.2016.07.008
Chen, J., Malone, B., Llewellyn, E., Grasso, M., Shelton, P. M. M., Olinares, P. D. B., et al. (2020). Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 182, 1560.e13–1573.e13. doi: 10.1016/j.cell.2020.07.033
Chen, J., Xu, X., Tao, H., Li, Y., Nan, H., Wang, Y., et al. (2017). Structural Analysis of Porcine Reproductive and Respiratory Syndrome Virus Non-structural Protein 7α (NSP7α) and Identification of Its Interaction with NSP9. Front. Microbiol 8:853. doi: 10.3389/fmicb.2017.00853
Chen, Y., Cai, H., Pan, J., Xiang, N., Tien, P., Ahola, T., et al. (2009). Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. U.S.A. 106, 3484–3489. doi: 10.1073/pnas.0808790106
Chen, Y., Su, C., Ke, M., Jin, X., Xu, L., Zhang, Z., et al. (2011). Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2’-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog. 7:e1002294. doi: 10.1371/journal.ppat.1002294
Cheng, W., Chen, S., Li, R., Chen, Y., Wang, M., and Guo, D. (2015). Severe acute respiratory syndrome coronavirus protein 6 mediates ubiquitin-dependent proteosomal degradation of N-Myc (and STAT) interactor. Virol. Sin. 30, 153–161. doi: 10.1007/s12250-015-3581-8
Chi, X., Yan, R., Zhang, J., Zhang, G., Zhang, Y., Hao, M., et al. (2020). A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 369, 650–655. doi: 10.1126/science.abc6952
Cinatl, J., Morgenstern, B., Bauer, G., Chandra, P., Rabenau, H., and Doerr, H. W. (2003). Treatment of SARS with human interferons. Lancet Lond. Engl. 362, 293–294. doi: 10.1016/s0140-6736(03)13973-6
Clementi, N., Ferrarese, R., Criscuolo, E., Diotti, R. A., Castelli, M., Scagnolari, C., et al. (2020). Interferon-β-1a inhibition of severe acute respiratory syndrome–coronavirus 2 in vitro when administered after virus infection. J. Infect. Dis. 222, 722–725. doi: 10.1093/infdis/jiaa350
Clementz, M. A., Chen, Z., Banach, B. S., Wang, Y., Sun, L., Ratia, K., et al. (2010). Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84, 4619–4629. doi: 10.1128/JVI.02406-09
Cong, Y., Ulasli, M., Schepers, H., Mauthe, M., V’kovski, P., Kriegenburg, F., et al. (2020). Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 94:e01925-19. doi: 10.1128/JVI.01925-19
Cornillez-Ty, C. T., Liao, L., Yates, J. R., Kuhn, P., and Buchmeier, M. J. (2009). Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 83, 10314–10318. doi: 10.1128/JVI.00842-09
Corse, E., and Machamer, C. E. (2003). The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 312, 25–34. doi: 10.1016/s0042-6822(03)00175-2
Corver, J., Broer, R., van Kasteren, P., and Spaan, W. (2009). Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: refinement of the requirements for SARS coronavirus cell entry. Virol. J. 6:230. doi: 10.1186/1743-422X-6-230
Cui, J., Li, F., and Shi, Z.-L. (2019). Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 17, 181–192. doi: 10.1038/s41579-018-0118-9
Dahms, S. O., Jiao, G.-S., and Than, M. E. (2017). Structural Studies Revealed Active Site Distortions of Human Furin by a Small Molecule Inhibitor. ACS Chem. Biol. 12, 1211–1216. doi: 10.1021/acschembio.6b01110
Dai, W., Zhang, B., Su, H., Li, J., Zhao, Y., Xie, X., et al. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335. doi: 10.1126/science.abb4489
de Groot, R. J., Baker, S. C., Baric, R. S., Brown, C. S., Drosten, C., Enjuanes, L., et al. (2013). Middle East Respiratory Syndrome Coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 87, 7790–7792. doi: 10.1128/JVI.01244-13
Deng, X., and Baker, S. C. (2018). An “Old” protein with a new story: coronavirus endoribonuclease is important for evading host antiviral defenses. Virology 517, 157–163. doi: 10.1016/j.virol.2017.12.024
Eckerle, L. D., Becker, M. M., Halpin, R. A., Li, K., Venter, E., Lu, X., et al. (2010). Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 6:e1000896. doi: 10.1371/journal.ppat.1000896
Egloff, M.-P., Ferron, F., Campanacci, V., Longhi, S., Rancurel, C., Dutartre, H., et al. (2004). The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. U.S.A. 101, 3792–3796. doi: 10.1073/pnas.0307877101
Esumi, M., Ishibashi, M., Yamaguchi, H., Nakajima, S., Tai, Y., Kikuta, S., et al. (2015). Transmembrane serine protease TMPRSS2 activates hepatitis C virus infection. Hepatol. Baltim. Md. 61, 437–446. doi: 10.1002/hep.27426
Fehr, A. R., Channappanavar, R., Jankevicius, G., Fett, C., Zhao, J., Athmer, J., et al. (2016). The conserved coronavirus macrodomain promotes virulence and suppresses the innate immune response during severe acute respiratory syndrome coronavirus infection. mBio 7:13. doi: 10.1128/mBio.01721-16
Felgenhauer, U., Schoen, A., Gad, H. H., Hartmann, R., Schaubmar, A. R., Failing, K., et al. (2020). Inhibition of SARS-CoV-2 by type I and type III interferons. J. Biol. Chem. 295, 13958–13964. doi: 10.1074/jbc.AC120.013788
Frieman, M., Ratia, K., Johnston, R. E., Mesecar, A. D., and Baric, R. S. (2009). Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83, 6689–6705. doi: 10.1128/JVI.02220-08
Frieman, M., Yount, B., Heise, M., Kopecky-Bromberg, S. A., Palese, P., and Baric, R. S. (2007). Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824. doi: 10.1128/JVI.01012-07
Fu, Z., Huang, B., Tang, J., Liu, S., Liu, M., Ye, Y., et al. (2020). Structural basis for the inhibition of the papain-like protease of SARS-CoV-2 by small molecules. bioRxiv[Preprint]. doi: 10.1101/2020.07.17.208959
Gao, G., and Luo, H. (2006). The ubiquitin-proteasome pathway in viral infections. Can. J. Physiol. Pharmacol. 84, 5–14. doi: 10.1139/y05-144
Gao, Y., Yan, L., Huang, Y., Liu, F., Zhao, Y., Cao, L., et al. (2020). Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368, 779–782. doi: 10.1126/science.abb7498
Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., et al. (2020). A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468. doi: 10.1038/s41586-020-2286-9
Graham, R. L., Sims, A. C., Brockway, S. M., Baric, R. S., and Denison, M. R. (2005). The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication. J. Virol. 79, 13399–13411. doi: 10.1128/JVI.79.21.13399-13411.2005
Guo, B., and Cheng, G. (2007). Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J. Biol. Chem. 282, 11817–11826. doi: 10.1074/jbc.M700017200
Hadjadj, J., Yatim, N., Barnabei, L., Corneau, A., Boussier, J., Smith, N., et al. (2020). Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724. doi: 10.1126/science.abc6027
Hagemeijer, M. C., Monastyrska, I., Griffith, J., van der Sluijs, P., Voortman, J., van Bergen en Henegouwen, P. M., et al. (2014). Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology 458–459, 125–135. doi: 10.1016/j.virol.2014.04.027
Hansen, J., Baum, A., Pascal, K. E., Russo, V., Giordano, S., Wloga, E., et al. (2020). Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 369, 1010–1014. doi: 10.1126/science.abd0827
Henderson, R., Edwards, R. J., Mansouri, K., Janowska, K., Stalls, V., Gobeil, S., et al. (2020). Controlling the SARS-CoV-2 spike glycoprotein conformation. bioRxiv [Preprint]. doi: 10.1101/2020.05.18.102087
Hensley, L. E., Fritz, E. A., Jahrling, P. B., Karp, C., Huggins, J. W., and Geisbert, T. W. (2004). Interferon-β 1a and SARS coronavirus replication. Emerg. Infect. Dis. 10, 317–319. doi: 10.3201/eid1002.030482
Herrera, N. G., Morano, N. C., Celikgil, A., Georgiev, G. I., Malonis, R. J., Lee, J. H., et al. (2020). Characterization of the SARS-CoV-2 S protein: biophysical, biochemical, structural, and antigenic analysis. bioRxiv [Preprint]. doi: 10.1101/2020.06.14.150607
Heurich, A., Hofmann-Winkler, H., Gierer, S., Liepold, T., Jahn, O., and Pöhlmann, S. (2014). TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 88, 1293–1307. doi: 10.1128/JVI.02202-13
Hillen, H. S., Kokic, G., Farnung, L., Dienemann, C., Tegunov, D., and Cramer, P. (2020). Structure of replicating SARS-CoV-2 polymerase. Nature 584, 154–156. doi: 10.1038/s41586-020-2368-8
Hoffmann, M., Hofmann-Winkler, H., and Pöhlmann, S. (2018). Priming time: how cellular proteases arm coronavirus spike proteins. Act. Viruses Host Proteases 16, 71–98. doi: 10.1007/978-3-319-75474-1_4
Hoffmann, M., Kleine-Weber, H., and Pöhlmann, S. (2020a). A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 78, 779.e–784.e. doi: 10.1016/j.molcel.2020.04.022
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020b). SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271.e–280.e. doi: 10.1016/j.cell.2020.02.052
Hsieh, C.-L., Goldsmith, J. A., Schaub, J. M., DiVenere, A. M., Kuo, H.-C., Javanmardi, K., et al. (2020). Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. bioRxiv [Preprint]. doi: 10.1101/2020.05.30.125484
Huang, C., Lokugamage, K. G., Rozovics, J. M., Narayanan, K., Semler, B. L., and Makino, S. (2011). SARS coronavirus nsp1 protein induces template-dependent endonucleolytic cleavage of mRNAs: viral mRNAs Are Resistant to nsp1-Induced RNA Cleavage. PLoS Pathog. 7:e1002433. doi: 10.1371/journal.ppat.1002433
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/S0140-6736(20)30183-5
Huo, J., Zhao, Y., Ren, J., Zhou, D., Duyvesteyn, H. M. E., Ginn, H. M., et al. (2020). Neutralization of SARS-CoV-2 by destruction of the prefusion spike. Cell Host Microbe 28, 445.e–454.e. doi: 10.1016/j.chom.2020.06.010
Hurst, K. R., Koetzner, C. A., and Masters, P. S. (2013). Characterization of a critical interaction between the coronavirus nucleocapsid protein and nonstructural protein 3 of the viral replicase-transcriptase complex. J. Virol. 87, 9159–9172. doi: 10.1128/JVI.01275-13
Hussain, M., Jabeen, N., Amanullah, A., Baig, A. A., Aziz, B., Shabbir, S., et al. (2020). Structural basis of SARS-CoV-2 spike protein priming by TMPRSS2. bioRxiv[Preprint]. doi: 10.1101/2020.04.21.052639
Imbert, I., Snijder, E. J., Dimitrova, M., Guillemot, J.-C., Lécine, P., and Canard, B. (2008). The SARS-Coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 133, 136–148. doi: 10.1016/j.virusres.2007.11.017
Ivashkiv, L. B., and Donlin, L. T. (2014). Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49. doi: 10.1038/nri3581
Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., et al. (2020a). Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293. doi: 10.1038/s41586-020-2223-y
Jin, Z., Zhao, Y., Sun, Y., Zhang, B., Wang, H., Wu, Y., et al. (2020b). Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 27, 529–532. doi: 10.1038/s41594-020-0440-6
Ju, B., Zhang, Q., Ge, J., Wang, R., Sun, J., Ge, X., et al. (2020). Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584, 1–5. doi: 10.1038/s41586-020-2380-z
Kamerkar, S. C., Kraus, F., Sharpe, A. J., Pucadyil, T. J., and Ryan, M. T. (2018). Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat. Commun. 9:5239. doi: 10.1038/s41467-018-07543-w
Kamitani, W., Huang, C., Narayanan, K., Lokugamage, K. G., and Makino, S. (2009). A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 16, 1134–1140. doi: 10.1038/nsmb.1680
Kang, S., Yang, M., Hong, Z., Zhang, L., Huang, Z., Chen, X., et al. (2020). Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 10, 1228–1238. doi: 10.1016/j.apsb.2020.04.009
Ke, Z., Oton, J., Qu, K., Cortese, M., Zila, V., McKeane, L., et al. (2020). Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature doi: 10.1038/s41586-020-2665-2
Keng, C.-T., and Tan, Y.-J. (2009). “Molecular and Biochemical Characterization of the SARS-CoV Accessory Proteins ORF8a, ORF8b and ORF8ab,” in Molecular Biology of the SARS-Coronavirus, ed. S. K. Lal (New York, NY: Springer Science & Business Media), 177–191. doi: 10.1007/978-3-642-03683-5_12
Kern, D. M., Sorum, B., Hoel, C. M., Sridharan, S., Remis, J. P., Toso, D. B., et al. (2020). Cryo-EM structure of the SARS-CoV-2 3a ion channel in lipid nanodiscs. bioRxiv[Preprint]. doi: 10.1101/2020.06.17.156554
Kido, H., Okumura, Y., Takahashi, E., Pan, H.-Y., Wang, S., Chida, J., et al. (2008). Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J. Mol. Genet. Med. Int. J. Biomed. Res. 3, 167–175.
Kim, S.-J., Ahn, D.-G., Syed, G. H., and Siddiqui, A. (2018). The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 41, 21–27. doi: 10.1016/j.mito.2017.11.007
Kim, S. S.-Y., Sze, L., and Lam, K.-P. (2019). The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-β response. J. Biol. Chem. 294, 6430–6438. doi: 10.1074/jbc.RA118.005868
Kim, Y., Jedrzejczak, R., Maltseva, N. I., Wilamowski, M., Endres, M., Godzik, A., et al. (2020). Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci. 29, 1596–1605. doi: 10.1002/pro.3873
King, A. M. Q., Adams, M. J., Carstens, E. B., and Lefkowitz, E. J. (eds) (2012). “Family - Coronaviridae,” in Virus Taxonomy. San Diego, CA: Elsevier, 806–828. doi: 10.1016/B978-0-12-384684-6.00068-9
Kirchdoerfer, R. N., and Ward, A. B. (2019). Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 10:2342. doi: 10.1038/s41467-019-10280-3
Klein, S., Cortese, M., Winter, S. L., Wachsmuth-Melm, M., Neufeldt, C. J., Cerikan, B., et al. (2020). SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. bioRxiv[Preprint]. doi: 10.1101/2020.06.23.167064
Kneller, D. W., Phillips, G., O’Neill, H. M., Jedrzejczak, R., Stols, L., Langan, P., et al. (2020). Structural plasticity of SARS-CoV-2 3CL M pro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 11:3202. doi: 10.1038/s41467-020-16954-7
Konno, Y., Kimura, I., Uriu, K., Fukushi, M., Irie, T., Koyanagi, Y., et al. (2020). SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 32:108185. doi: 10.1016/j.celrep.2020.108185
Kopecky-Bromberg, S. A., Martínez-Sobrido, L., Frieman, M., Baric, R. A., and Palese, P. (2007). Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 81, 548–557. doi: 10.1128/JVI.01782-06
Koyama, T., Platt, D., and Parida, L. (2020). Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ. 98, 495–504. doi: 10.2471/BLT.20.253591
Kumar, P., Gunalan, V., Liu, B., Chow, V. T. K., Druce, J., Birch, C., et al. (2007). The nonstructural protein 8 (nsp8) of the SARS coronavirus interacts with its ORF6 accessory protein. Virology 366, 293–303. doi: 10.1016/j.virol.2007.04.029
Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., et al. (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220. doi: 10.1038/s41586-020-2180-5
Lecker, S. H., Goldberg, A. L., and Mitch, W. E. (2006). Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 17, 1807–1819. doi: 10.1681/ASN.2006010083
Lei, J., Kusov, Y., and Hilgenfeld, R. (2018). Nsp3 of coronaviruses: structures and functions of a large multi-domain protein. Antiviral Res. 149, 58–74. doi: 10.1016/j.antiviral.2017.11.001
Lei, X., Dong, X., Ma, R., Wang, W., Xiao, X., Tian, Z., et al. (2020). Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 11:3810. doi: 10.1038/s41467-020-17665-9
Li, F. (2016). Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 3, 237–261. doi: 10.1146/annurev-virology-110615-042301
Li, J.-Y., Liao, C.-H., Wang, Q., Tan, Y.-J., Luo, R., Qiu, Y., et al. (2020). The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 286:198074. doi: 10.1016/j.virusres.2020.198074
Li, Q., Wu, J., Nie, J., Zhang, L., Hao, H., Liu, S., et al. (2020). The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 182, 1284.e9–1294.e9. doi: 10.1016/j.cell.2020.07.012
Lindner, H. A., Fotouhi-Ardakani, N., Lytvyn, V., Lachance, P., Sulea, T., and Ménard, R. (2005). The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 79, 15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005
Littler, D. R., Gully, B. S., Colson, R. N., and Rossjohn, J. (2020). Crystal structure of the SARS-CoV-2 non-structural protein 9, Nsp9. iScience 23:101258. doi: 10.1016/j.isci.2020.101258
Liu, D. X., Fung, T. S., Chong, K. K.-L., Shukla, A., and Hilgenfeld, R. (2014). Accessory proteins of SARS-CoV and other coronaviruses. Antiviral Res. 109, 97–109. doi: 10.1016/j.antiviral.2014.06.013
Liu, L., Wang, P., Nair, M. S., Yu, J., Rapp, M., Wang, Q., et al. (2020). Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 584, 450–456. doi: 10.1038/s41586-020-2571-7
Liu, S., Chen, J., Cai, X., Wu, J., Chen, X., Wu, Y.-T., et al. (2013). MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2:e00785. doi: 10.7554/eLife.00785
Lokugamage, K. G., Narayanan, K., Huang, C., and Makino, S. (2012). Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 86, 13598–13608. doi: 10.1128/JVI.01958-12
Lu, G. (2006). ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell. Mol. Biol. 52, 29–41.
Lu, W., Xu, K., and Sun, B. (2009). “SARS Accessory Proteins ORF3a and 9b and Their Functional Analysis,” in Mol. Biol. SARS-Coronavirus, ed. S. K. Lal (New York, NY: Springer), 167–175. doi: 10.1007/978-3-642-03683-5_11
Lukassen, S., Chua, R. L., Trefzer, T., Kahn, N. C., Schneider, M. A., Muley, T., et al. (2020). SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 39:e105114. doi: 10.15252/embj.20105114
Ma, C., Sacco, M. D., Hurst, B., Townsend, J. A., Hu, Y., Szeto, T., et al. (2020). Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692. doi: 10.1038/s41422-020-0356-z
Mahon, C., Krogan, N. J., Craik, C. S., and Pick, E. (2014). Cullin E3 ligases and their rewiring by viral factors. Biomolecules 4, 897–930. doi: 10.3390/biom4040897
Malakhova, O. A., and Zhang, D.-E. (2008). ISG15 inhibits Nedd4 Ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 283, 8783–8787. doi: 10.1074/jbc.C800030200
Masters, P. S. (2006). “The Molecular Biology of Coronaviruses,” in Advances in Virus Research. Cambridge, MA: Academic Press, 193–292. doi: 10.1016/S0065-3527(06)66005-3
McBride, K. M., and Reich, N. C. (2003). The ins and outs of STAT1 nuclear transport. Sci. STKE Signal. Transduct. Knowl. Environ. 2003:RE13. doi: 10.1126/stke.2003.195.re13
McBride, R., van Zyl, M., and Fielding, B. C. (2014). The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 6, 2991–3018. doi: 10.3390/v6082991
Meier, C., Aricescu, A. R., Assenberg, R., Aplin, R. T., Gilbert, R. J. C., Grimes, J. M., et al. (2006). The crystal structure of ORF-9b, a lipid binding protein from the SARS coronavirus. Struct. Lond. Engl. 1993, 1157–1165. doi: 10.1016/j.str.2006.05.012
Miknis, Z. J., Donaldson, E. F., Umland, T. C., Rimmer, R. A., Baric, R. S., and Schultz, L. W. (2009). Severe acute respiratory syndrome coronavirus nsp9 dimerization is essential for efficient viral growth. J. Virol. 83, 3007–3018. doi: 10.1128/JVI.01505-08
Millet, J. K., and Whittaker, G. R. (2014). Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. U.S.A. 111, 15214–15219. doi: 10.1073/pnas.1407087111
Millet, J. K., and Whittaker, G. R. (2015). Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 202, 120–134. doi: 10.1016/j.virusres.2014.11.021
Mousavizadeh, L., and Ghasemi, S. (2020). Genotype and phenotype of COVID-19: their roles in pathogenesis. J. Microbiol. Immunol. Infect. doi: 10.1016/j.jmii.2020.03.022 [Epub ahead of print],
Muller, U., Steinhoff, U., Reis, L. F., Hemmi, S., Pavlovic, J., Zinkernagel, R. M., et al. (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. doi: 10.1126/science.8009221
Narayanan, K., Huang, C., Lokugamage, K., Kamitani, W., Ikegami, T., Tseng, C.-T. K., et al. (2008). Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82, 4471–4479. doi: 10.1128/JVI.02472-07
Narayanan, K., Ramirez, S. I., Lokugamage, K. G., and Makino, S. (2015). Coronavirus nonstructural protein 1: common and distinct functions in the regulation of host and viral gene expression. Virus Res. 202, 89–100. doi: 10.1016/j.virusres.2014.11.019
National Institute of Allergy, and Infectious Diseases (NIAID) (2020). A Multicenter, Adaptive, Randomized Blinded Controlled. (Trial)of the Safety and Efficacy of Investigational Therapeutics for the Treatment of COVID-19 in Hospitalized Adults (ACTT-3). Available online at: https://clinicaltrials.gov/ct2/show/NCT04492475 (accessed October 7, 2020).
Nelson, C. A., Pekosz, A., Lee, C. A., Diamond, M. S., and Fremont, D. H. (2005). Structure and Intracellular Targeting of the SARS-Coronavirus Orf7a Accessory Protein. Structure 13, 75–85. doi: 10.1016/j.str.2004.10.010
Neuman, B. W., Joseph, J. S., Saikatendu, K. S., Serrano, P., Chatterjee, A., Johnson, M. A., et al. (2008). Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 82, 5279–5294. doi: 10.1128/JVI.02631-07
Neuman, B. W., Kiss, G., Kunding, A. H., Bhella, D., Baksh, M. F., Connelly, S., et al. (2011). A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 174, 11–22. doi: 10.1016/j.jsb.2010.11.021
Nguyen, T. M., Zhang, Y., and Pandolfi, P. P. (2020). Virus against virus: a potential treatment for 2019-nCov (SARS-CoV-2) and other RNA viruses. Cell Res. 30, 189–190. doi: 10.1038/s41422-020-0290-0
Paules, C. I., Marston, H. D., and Fauci, A. S. (2020). Coronavirus infections-more than just the common cold. JAMA 323, 707–708. doi: 10.1001/jama.2020.0757
Peng, Q., Peng, R., Yuan, B., Zhao, J., Wang, M., Wang, X., et al. (2020). Structural and biochemical characterization of the nsp12-nsp7-nsp8 core polymerase complex from SARS-CoV-2. Cell Rep. 31:107774. doi: 10.1016/j.celrep.2020.107774
Peng, Y.-T., Chen, P., Ouyang, R.-Y., and Song, L. (2015). Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis Int. J. Program. Cell Death 20, 1135–1149. doi: 10.1007/s10495-015-1143-z
Pervushin, K., Tan, E., Parthasarathy, K., Lin, X., Jiang, F. L., Yu, D., et al. (2009). Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 5:e1000511. doi: 10.1371/journal.ppat.1000511
Piccoli, L., Park, Y.-J., Tortorici, M. A., Czudnochowski, N., Walls, A. C., Beltramello, M., et al. (2020). Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell doi: 10.1016/j.cell.2020.09.037
Pinto, D., Park, Y.-J., Beltramello, M., Walls, A. C., Tortorici, M. A., Bianchi, S., et al. (2020). Structural and functional analysis of a potent sarbecovirus neutralizing antibody. bioRxiv [Preprint]. doi: 10.1101/2020.04.07.023903
Randow, F., and Lehner, P. J. (2009). Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 11, 527–534. doi: 10.1038/ncb0509-527
Rawson, S., Davies, S., Lippiat, J. D., and Muench, S. P. (2016). The changing landscape of membrane protein structural biology through developments in electron microscopy. Mol. Membr. Biol. 33, 12–22. doi: 10.1080/09687688.2016.1221533
Reid, S. P., Valmas, C., Martinez, O., Sanchez, F. M., and Basler, C. F. (2007). Ebola Virus VP24 Proteins Inhibit the Interaction of NPI-1 subfamily karyopherin α proteins with activated STAT1. J. Virol. 81, 13469–13477. doi: 10.1128/JVI.01097-07
Ren, Y., Shu, T., Wu, D., Mu, J., Wang, C., Huang, M., et al. (2020). The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol 17, 881–883. doi: 10.1038/s41423-020-0485-9
Robbiani, D. F., Gaebler, C., Muecksch, F., Lorenzi, J. C. C., Wang, Z., Cho, A., et al. (2020). Convergent antibody responses to SARS-CoV-2 infection in convalescent individuals. bioRxiv[Preprint]. doi: 10.1101/2020.05.13.092619
Romano, M., Ruggiero, A., Squeglia, F., Maga, G., and Berisio, R. (2020). A structural view of SARS-CoV-2 RNA replication machinery: RNA synthesis, proofreading and final capping. Cells 9:1267. doi: 10.3390/cells9051267
Rosas-Lemus, M., Minasov, G., Shuvalova, L., Inniss, N. L., Kiryukhina, O., Wiersum, G., et al. (2020). The crystal structure of nsp10-nsp16 heterodimer from SARS-CoV-2 in complex with S-adenosylmethionine. bioRxivp[Preprint]. doi: 10.1101/2020.04.17.047498
Rota, P. A., Oberste, M. S., Monroe, S. S., Nix, W. A., Campagnoli, R., Icenogle, J. P., et al. (2003). Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394–1399. doi: 10.1126/science.1085952
Rut, W., Lv, Z., Zmudzinski, M., Patchett, S., Nayak, D., Snipas, S. J., et al. (2020). Activity profiling and structures of inhibitor-bound SARS-CoV-2-PLpro protease provides a framework for anti-COVID-19 drug design. bioRxiv[Preprint]. doi: 10.1101/2020.04.29.068890
Sa Ribero, M., Jouvenet, N., Dreux, M., and Nisole, S. (2020). Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog 16:e1008737. doi: 10.1371/journal.ppat.1008737
Saha, S. K., Pietras, E. M., He, J. Q., Kang, J. R., Liu, S.-Y., Oganesyan, G., et al. (2006). Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J. 25, 3257–3263. doi: 10.1038/sj.emboj.7601220
Sakai, K., Ami, Y., Tahara, M., Kubota, T., Anraku, M., Abe, M., et al. (2014). The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J. Virol. 88, 5608–5616. doi: 10.1128/JVI.03677-13
Samuel, C. E. (2001). Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778–809. doi: 10.1128/CMR.14.4.778-809.2001
Schaecher, S. R., Diamond, M. S., and Pekosz, A. (2008). The transmembrane domain of the severe acute respiratory syndrome coronavirus ORF7b protein is necessary and sufficient for its retention in the golgi complex. J. Virol. 82, 9477–9491. doi: 10.1128/JVI.00784-08
Schaecher, S. R., Touchette, E., Schriewer, J., Buller, R. M., and Pekosz, A. (2007). Severe acute respiratory syndrome coronavirus gene 7 products contribute to virus-induced apoptosis. J. Virol. 81, 11054–11068. doi: 10.1128/JVI.01266-07
Schibli, D. J., and Weissenhorn, W. (2004). Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol. Membr. Biol. 21, 361–371. doi: 10.1080/09687860400017784
Schoeman, D., and Fielding, B. C. (2019). Coronavirus envelope protein: current knowledge. Virol. J. 16:69. doi: 10.1186/s12985-019-1182-0
Schroth-Diez, B., Ludwig, K., Baljinnyam, B., Kozerski, C., Huang, Q., and Herrmann, A. (2000). The role of the transmembrane and of the intraviral domain of glycoproteins in membrane fusion of enveloped viruses. Biosci. Rep. 20, 571–595. doi: 10.1023/A:1010415122234
Schubert, K., Karousis, E. D., Jomaa, A., Scaiola, A., Echeverria, B., Gurzeler, L.-A., et al. (2020). SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 27, 957-966. doi: 10.1038/s41594-020-0511-8
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224. doi: 10.1038/s41586-020-2179-y
Shi, C.-S., Qi, H.-Y., Boularan, C., Huang, N.-N., Abu-Asab, M., Shelhamer, J. H., et al. (2014). SARS-CoV ORF-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. Baltim. Md. 1950, 3080–3089. doi: 10.4049/jimmunol.1303196
Shi, R., Shan, C., Duan, X., Chen, Z., Liu, P., Song, J., et al. (2020). A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 584, 120–124. doi: 10.1038/s41586-020-2381-y
Shi, S. T., Schiller, J. J., Kanjanahaluethai, A., Baker, S. C., Oh, J.-W., and Lai, M. M. C. (1999). Colocalization and membrane association of murine hepatitis virus gene 1 products and de novo-synthesized viral RNA in infected cells. J. Virol. 73, 5957–5969. doi: 10.1128/JVI.73.7.5957-5969.1999
Shi, W., Chen, M., Yang, Y., Zhou, W., Chen, S., Yang, Y., et al. (2020). A dynamic regulatory interface on SARS-CoV-2 RNA polymerase. bioRxiv[Preprint]. doi: 10.1101/2020.07.30.229187
Shi, Z., Zhang, Z., Zhang, Z., Wang, Y., Li, C., Wang, X., et al. (2015). Structural insights into mitochondrial antiviral signaling protein (MAVS)-tumor necrosis factor receptor-associated Factor 6 (TRAF6) signaling. J. Biol. Chem. 290, 26811–26820. doi: 10.1074/jbc.M115.666578
Signorile, A., Sgaramella, G., Bellomo, F., and De Rasmo, D. (2019). Prohibitins: a critical role in mitochondrial functions and implication in diseases. Cells 8:71. doi: 10.3390/cells8010071
Siu, Y. L., Teoh, K. T., Lo, J., Chan, C. M., Kien, F., Escriou, N., et al. (2008). The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J. Virol. 82, 11318–11330. doi: 10.1128/JVI.01052-08
Snijder, E. J., Bredenbeek, P. J., Dobbe, J. C., Thiel, V., Ziebuhr, J., Poon, L. L. M., et al. (2003). Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 Lineage. J. Mol. Biol. 331, 991–1004. doi: 10.1016/S0022-2836(03)00865-9
Spiegel, M., Pichlmair, A., Martínez-Sobrido, L., Cros, J., García-Sastre, A., Haller, O., et al. (2005). Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 79, 2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005
Stertz, S., Reichelt, M., Spiegel, M., Kuri, T., Martínez-Sobrido, L., García-Sastre, A., et al. (2007). The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361, 304–315. doi: 10.1016/j.virol.2006.11.027
Surya, W., Li, Y., and Torres, J. (2018). Structural model of the SARS coronavirus E channel in LMPG micelles. Biochim. Biophys. Acta 1860, 1309–1317. doi: 10.1016/j.bbamem.2018.02.017
Tan, Y., Schneider, T., Leong, M., Aravind, L., and Zhang, D. (2020). Novel immunoglobulin domain proteins provide insights into evolution and pathogenesis of SARS-CoV-2-related viruses. mBio 11, doi: 10.1128/mBio.00760-20
Tan, Y.-J. (2005). The Severe Acute Respiratory Syndrome (SARS)-coronavirus 3a protein may function as a modulator of the trafficking properties of the spike protein. Virol. J. 2:5. doi: 10.1186/1743-422X-2-5
Tang, Q., Song, Y., Shi, M., Cheng, Y., Zhang, W., and Xia, X.-Q. (2015). Inferring the hosts of coronavirus using dual statistical models based on nucleotide composition. Sci. Rep. 5:17155. doi: 10.1038/srep17155
Taylor, J. K., Coleman, C. M., Postel, S., Sisk, J. M., Bernbaum, J. G., Venkatarajan, T., et al. (2015). SARS Coronavirus ORF7a inhibits BST-2 virion tethering through a novel mechanism of glycosylation interference. J. Virol. 89. doi: 10.1128/JVI.02274-15
The Recovery Collaborative Group (2020). Dexamethasone in hospitalized patients with Covid-19 — preliminary report. N. Engl. J. Med. doi: 10.1056/NEJMoa2021436
Thoms, M., Buschauer, R., Ameismeier, M., Koepke, L., Denk, T., Hirschenberger, M., et al. (2020). Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 369, 1249–1255. doi: 10.1126/science.abc8665
Toelzer, C., Gupta, K., Yadav, S. K. N., Borucu, U., Davidson, A. D., Williamson, M. K., et al. (2020). Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science [Epub ahead of print]. doi: 10.1126/science.abd3255
Tortorici, M. A., Beltramello, M., Lempp, F. A., Pinto, D., Dang, H. V., Rosen, L. E., et al. (2020). Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science [Epub ahead of print]. doi: 10.1126/science.abe3354
Tortorici, M. A., and Veesler, D. (2019). Structural insights into coronavirus entry. Adv. Virus Res. 105, 93–116. doi: 10.1016/bs.aivir.2019.08.002
Treanor, J. J. (2015). “167 - Influenza (Including Avian Influenza and Swine Influenza),” in Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th Edn,, eds J. E. Bennett, R. Dolin, and M. J. Blaser (Philadelphia, PA: Wiley), doi: 10.1016/B978-1-4557-4801-3.00167-3
Tseng, Y.-T., Wang, S.-M., Huang, K.-J., and Wang, C.-T. (2014). SARS-CoV envelope protein palmitoylation or nucleocapid association is not required for promoting virus-like particle production. J. Biomed. Sci. 21:34. doi: 10.1186/1423-0127-21-34
Tsoi, H., Li, L., Chen, Z. S., Lau, K.-F., Tsui, S. K. W., and Chan, H. Y. E. (2014). The SARS-coronavirus membrane protein induces apoptosis via interfering with PDK1–PKB/Akt signalling. Biochem. J. 464, 439–447. doi: 10.1042/BJ20131461
Turoòová, B., Sikora, M., Schürmann, C., Hagen, W. J. H., Welsch, S., Blanc, F. E. C., et al. (2020). In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 370, 203-208. doi: 10.1126/science.abd5223
Veljkovic, V., and Paessler, S. (2020). COVID-19 Orf3b protein: the putative biological function and the therapeutic target. Review doi: 10.21203/rs.2.24483/v1
von Brunn, A., Teepe, C., Simpson, J. C., Pepperkok, R., Friedel, C. C., Zimmer, R., et al. (2007). Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLoS One 2:e459. doi: 10.1371/journal.pone.0000459
Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., and Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181, 281.e6–292.e6. doi: 10.1016/j.cell.2020.02.058
Wang, Q., Li, C., Zhang, Q., Wang, T., Li, J., Guan, W., et al. (2010). Interactions of SARS coronavirus nucleocapsid protein with the host cell proteasome subunit p42. Virol. J. 7:99. doi: 10.1186/1743-422X-7-99
Wang, S., Nath, N., Adlam, M., and Chellappan, S. (1999). Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function. Oncogene 18, 3501–3510. doi: 10.1038/sj.onc.1202684
Wang, Y.-N., Yu, C.-Y., and Jin, H.-Z. (2020). RNA N6-methyladenosine modifications and the immune response. J. Immunol. Res. 2020:e6327614. doi: 10.1155/2020/6327614
Wang, Q., Wu, J., Wang, H., Gao, Y., Liu, Q., Mu, A., et al. (2020a). Structural basis for RNA replication by the SARS-CoV-2 polymerase. Cell 182, 417–428.e13. doi: 10.1016/j.cell.2020.05.034
Wang, Q., Zhang, Y., Wu, L., Niu, S., Song, C., Zhang, Z., et al. (2020b). Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904.e9. doi: 10.1016/j.cell.2020.03.045
Woo, P. C. Y., Huang, Y., Lau, S. K. P., and Yuen, K.-Y. (2010). Coronavirus genomics and bioinformatics analysis. Viruses 2, 1804–1820. doi: 10.3390/v2081803
Woo, P. C. Y., Lau, S. K. P., Lam, C. S. F., Lau, C. C. Y., Tsang, A. K. L., Lau, J. H. N., et al. (2012). Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86, 3995–4008. doi: 10.1128/JVI.06540-11
World Health Organisation (2020). Coronavirus Dis. COVID-19 Weekly-epi-update – 9. Available online at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20201012-weekly-epi-update-9.pdf (accessed October 11, 2020).
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C.-L., Abiona, O., et al. (2020). Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263. doi: 10.1126/science.abb2507
Wu, A., Peng, Y., Huang, B., Ding, X., Wang, X., Niu, P., et al. (2020). Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27, 325–328. doi: 10.1016/j.chom.2020.02.001
Wu, B., and Hur, S. (2015). How RIG-I like receptors activate MAVS. Curr. Opin. Virol. 12, 91–98. doi: 10.1016/j.coviro.2015.04.004
Wu, C.-H., Yeh, S.-H., Tsay, Y.-G., Shieh, Y.-H., Kao, C.-L., Chen, Y.-S., et al. (2009). Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J. Biol. Chem. 284, 5229–5239. doi: 10.1074/jbc.M805747200
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., et al. (2020). A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269. doi: 10.1038/s41586-020-2008-3
Xia, S., Liu, M., Wang, C., Xu, W., Lan, Q., Feng, S., et al. (2020). Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 30, 343–355. doi: 10.1038/s41422-020-0305-x
Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y., and Zhou, Q. (2020). Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367, 1444–1448. doi: 10.1126/science.abb2762
Ye, Q., West, A. M. V., Silletti, S., and Corbett, K. D. (2020). Architecture and self-assembly of the SARS-CoV-2 nucleocapsid protein. Prot. Sci. 29, 1890–1901. doi: 10.1002/pro.3909
Yin, W., Mao, C., Luan, X., Shen, D.-D., Shen, Q., Su, H., et al. (2020). Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 368, 1499–1504. doi: 10.1126/science.abc1560
Yuan, M., Liu, H., Wu, N. C., Lee, C.-C. D., Zhu, X., Zhao, F., et al. (2020a). Structural basis of a public antibody response to SARS-CoV-2. bioRxiv [Preprint]. doi: 10.1101/2020.06.08.141267
Yuan, M., Wu, N. C., Zhu, X., Lee, C.-C. D., So, R. T. Y., Lv, H., et al. (2020b). A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368, 630–633. doi: 10.1126/science.abb7269
Yuen, C.-K., Lam, J.-Y., Wong, W.-M., Mak, L.-F., Wang, X., Chu, H., et al. (2020). SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 9, 1418–1428. doi: 10.1080/22221751.2020.1780953
Zhang, L., Li, L., Yan, L., Ming, Z., Jia, Z., Lou, Z., et al. (2018). Structural and biochemical characterization of endoribonuclease Nsp15 encoded by middle east respiratory syndrome coronavirus. J. Virol. 92, e893–e818. doi: 10.1128/JVI.00893-18
Zhang, L., Lin, D., Kusov, Y., Nian, Y., Ma, Q., Wang, J., et al. (2020a). α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design. synthesis, and activity assessment. J. Med. Chem. 63, 4562–4578. doi: 10.1021/acs.jmedchem.9b01828
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., et al. (2020b). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412. doi: 10.1126/science.abb3405
Zhang, Y., Zhang, J., Chen, Y., Luo, B., Yuan, Y., Huang, F., et al. (2020c). The ORF8 protein of SARS-CoV-2 mediates immune evasion through potently downregulating MHC-I. bioRxiv[Preprint]. doi: 10.1101/2020.05.24.111823
Keywords: COVID19, SARS-CoV-2, coronavirus, structural biology, cryo-EM
Citation: Mariano G, Farthing RJ, Lale-Farjat SLM and Bergeron JRC (2020) Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 7:605236. doi: 10.3389/fmolb.2020.605236
Received: 11 September 2020; Accepted: 22 October 2020;
Published: 17 December 2020.
Edited by:
Yan Xiang, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Christian Cambillau, Aix-Marseille Université, FranceDimitrios P. Vlachakis, Agricultural University of Athens, Greece
Copyright © 2020 Mariano, Farthing, Lale-Farjat and Bergeron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppina Mariano, Z2l1c3kubWFyaWFub0BuY2wuYWMudWs=; Julien R. C. Bergeron, anVsaWVuLmJlcmdlcm9uQGtjbC5hYy51aw==