 Jakub Luštinec
Jakub Luštinec Tomáš Koubský
Tomáš Koubský Ladislav Kalvoda
Ladislav Kalvoda- Department of Solid State Engineering, Faculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague, Prague, Czechia
Diglycolamides (DGA) form one of the most promising groups of organic ligands used in bio-inspired solvent extraction processes of lanthanide and actinide ions. Continuous experimental and theoretical research is still performed in order to further improve their application properties including their chemical stability in the real extraction environment. This work provides results of our theoretical approach focused on inclusion of an acid influence on the DGAs chemical structure, treated in frame of the density functional theory. Three different models describing the acid action are proposed and investigated in attempt to increase the resulting accuracy of the chemical stability predictions based on verified theoretical descriptors. The procedure is applied and tested on the set of selected hydrophilic DGA representatives. Comparison of the model results obtained with and without acid action shows that two types of protection effects may occur: a ‘direct’ protection, accompanied by an explicit change of the ligand stability indicators, and an ‘indirect’ one consisting in reaction of acid molecules with radicals preceding the contact of latter with the extracting ligands. The possibility of the direct acid protection route is supported by the significant decrease of the Fukui charges found with the acid models included. On the other hand, there is in general no significant difference of trends in the calculated chemical stability descriptors suggesting that an indirect mechanism must be also considered in order to explain the experimentally observed protective role of acids on the chemical stability of investigated DGA derivatives.
Introduction
Closing the nuclear fuel cycle and the maximal usage of uranium contained in the nuclear fuel are one of the biggest recent challenges related to a sustainable nuclear power plants operation. The spent nuclear fuel contains fission products, unused U, and minor actinides (MAs) which are, together with Pu, also responsible for the long term radiotoxicity of the nuclear waste. One of the possibilities for the spent nuclear fuel treatment is its partitioning and transmutation (Veliscek-Carolan, 2016). Partitioning consists of co-extraction of MAs and lanthanides from a liquid solution of nuclear waste and the subsequent separation of these components from each other. After partitioning, the transmutation of MAs is performed in order to reduce their long radiative decay lifetime and toxicity. In the result, the radiotoxicity of MAs and remaining waste can be significantly reduced (Veliscek-Carolan, 2016).
Among other organic molecules used in solvent extraction procedures, one of the most promising groups is formed by diglycolamides (DGAs). DGAs are especially well applicable for extraction of trivalent actinide and lanthanide ions, mimicking thus some of the processes observed in biological systems (Mattocks and Cotruvo, 2020). The solubility of DGAs is determined by the optional presence and length of specific N-alkyl substituents. The DGA derivatives containing short N-alkyls are water soluble, e.g., tertamethyldiglycolamide (TMDGA) or tetraethyldiglycolamide (TEDGA). These hydrophilic molecules are used as aqueous stripping and back-holding agents (Sasaki et al., 2007) in ALSEP process (Actinide Lanthanide Separation) (Lumetta et al., 2014) or EXAm process (Extraction of Americium) (Rostaing et al., 2012).
In this work, we study the following DGAs representatives (Figure 1).
• TMDGA (N,N,N′,N′-tetramethyl-diglycolamide; 2,2'_oxybis (N,N-dimethylacetamide))
• TEDGA (N,N,N′,N′-tetraethyl-diglycolamide; 2,2′-oxybis (N,N-diethylacetamide))
• Me-TEDGA (2-(2-(diethylamino)-2-oxoethoxy)-N,N-diethylpropanamide)
• Me2-TEDGA (2,2′-oxybis (N,N-diethylpropanamide)).
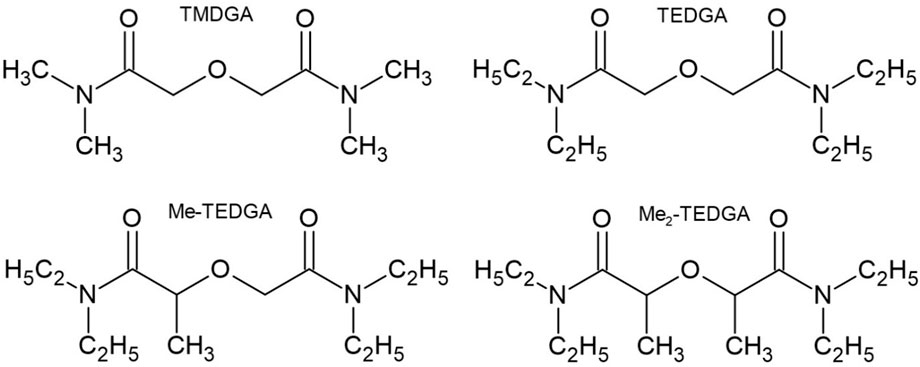
FIGURE 1. Studied hydrophilic DGA derivatives.
As indicated by experimental results (Wilden et al., 2018; Horne et al., 2019), addition of methyl group in Me-TEDGA and Me2-TEDGA results in remarkable radiolytic stability enhancement compared to the un-methylated TEDGA.
In our previous work (Koubský and Luštinec, 2018), the quantum mechanical indicators of radiolytic stability of the four above mentioned hydrophilic DGA derivatives were already evaluated and analysed in the environment of pure water. Especially, radical Fukui function, Fukui charges (condensed Fukui functions), and bond orders were found to be of key importance for the related radiolytic stability predictions. This work extends the theoretical treatment on the problem of acid influence implementation into the DFT calculations in order to improve the stability predictions. In particular, above mentioned verified stability indicators are calculated using three different acid inclusion models, and the obtained results compared and juxtaposed with the former acid-free results (Koubský and Luštinec, 2018). In addition, the condensed dual descriptor (CDD)
Reference experimental studies
Two key experimental radiolytic stability studies of the hydrophilic DGA derivatives shown in Figure 1 were performed by Wilden et al. (2018) and Horne et al. (2019).
In the first study, Wilden and co-workers tested solutions of DGA derivatives in a warmed nitric acid environment and compared the results with the behaviour in a pure water environment. In addition to the steady state measurements, the pulsed radiolysis method followed by kinetics measurements and mass spectroscopy was also applied, in order to get a deeper insight into the radiolysis process of the hydrophilic DGA derivatives. High rate constants for DGAs reactions with the hydroxyl radical are found, suggesting the important role of this radical in the radiolytic degradation mechanism in water environment. Observed decrease in dose constants with the increasing molecular weight of DGAs suggests, together with the measured rate constants, an electron transfer as the mechanism of the radical reaction. The radiolytic stability found for studied molecules follows the trend TMDGA < TEDGA < Me-TEDGA < Me2-TEDGA, i.e., growths with the molecular weight of the tested derivative (Wilden et al., 2018).
The second experimental study performed by Horne et al. (2019) (using the setup similar to the setup used by Wilden and co-workers) deals with a neutral pH concentrated aqueous nitrate solutions of the four selected hydrophilic DGA derivatives. The authors conclude that the studied hydrophilic DGAs undergo a first-order decay; the observed degradation product distributions are similar to those found in Wilden et al. (2018) under pure water conditions (except for the additional appearance of NOx adducts), and the radiolysis is driven by hydroxyl and nitrate radical oxidation chemistry, the latter then likely moderated by some secondary reactions scavenging the degradation products (Horne et al., 2019). The radiolysis rate of hydrophilic DGA representatives in aqueous nitrate solutions is found to be significantly reduced and less structurally sensitive compared to the acid-free solutions, similarly to the situation already observed for lipophilic DGA derivatives (Galan et al., 2015).
Degradation reaction mechanisms
As in our previous work (Koubský and Luštinec, 2018), we generally consider indirect radiolysis mechanisms to prevail, because of the low actual concentration of ligands used in extraction solutions (Wilden et al., 2018). Such indirect process consists in the primary radiolysis of solvent molecules, followed then by reactions of the radiolysis products with the ligand molecules. These water radiolysis products are represented by OH• and H• radicals. The overall reaction conditions anticipated in this work follow the experiments performed by Wilden et al. (2018), adding to the pure water environment [considered in the previous study (Koubský and Luštinec, 2018)] the influence of nitric acid via the proposed acid models.
The first degradation reaction mechanism considered here is supposed to start with the hydrogen abstraction followed by rupture of the ether bond C-O, in analogy with the lipophilic DGAs (Koubský et al., 2017). In the previous works (Koubský et al., 2017; Koubský and Luštinec, 2018), it was concluded that the hydrogen abstraction is more probable to occur on the ether group than on the side chains. Therefore, the ether group is mainly investigated in this work. It is worth to notice that the methylation of the ether carbons that promotes the higher molecular stability, lowers also the number of ether hydrogens: four in case of the two non-methylated studied DGA derivatives TMDGA and TEDGA, three in case of Me-TEDGA, and two for Me2-TEDGA.
The second possible degradation reaction mechanism follows the findings of Wilden et al. (2018). This mechanism is based on the known oxidation nature of the hydroxyl radical OH• that could cause the electron transfer from the amide group producing the DGA radical cation [DGA]•+. Afterwards, the rupture of the ether C-O bonds or the amide C-N bonds occurs (Wilden et al., 2018).
Methods and computational settings
Applied acid models
The acid influence is implemented into DFT simulations of the selected hydrophilic DGA derivatives by setting up three different testing models. In the first two of them, the interaction of ligand with dissociated acid molecules is assumed. These two models are independent of the particular acid type used in the experiments. In the first, simplest model, the hydrogen cation H+ is added to the calculated system in order to create a complex with the extraction molecule. In the second tested model, H3O+ cation (also applied in (Matveev et al., 2017)) is included in calculations instead of H+. Finally, in the third model, the interaction with undissociated HNO3 molecule is tested. The latter model is also relevant since the DGAs extractants are commonly dissolved in nitric acid solutions concentrated enough to contain a significant amount of undissociated HNO3 molecules (Wilden et al., 2018). This model explicitly includes the specific acid, in contrary to the first two models using general acid representations. The particular issue complicating mutual comparison of the results obtained by these models follows from the different total charge of the studied systems: it is equal to +1 for the first two models, and to zero for the last one. Thus, the behaviour of valence electrons and the related stability descriptors are affected by this difference.
Calculated stability indicators
The Frontier orbital theory of Fukui (1982) relates the molecule reactivity to the charge density
Where
Another possible implementation of the Fukui theory is the condensed Fukui function giving Fukui charges. Fukui charges are calculated from the atomic charges
Here
The CDD (Morell et al., 2005; Morell et al., 2006) for atom A is defined as follows
The sign of CDD indicates the vulnerability of the atomic site to the particular type of radical attack: the negative sign relates to electrophilic attack, the positive sign then to nucleophilic one.
Wiberg bond indices
Here
Computational settings
The DFT calculations were performed with DMol3 module from Materials Studio 8.0 (Delley, 1990; Delley, 2000) and Gaussian09 code (Frisch et al., 2013). The conformation analysis of the models combining extractants with the selected acid representation was performed using Gaussian09 code; the initial optimised extractants conformations were taken from the previous work (Koubský and Luštinec, 2018) where the geometry optimization was performed firstly with BLYP and subsequently with B3LYP functional.
The H+ and H3O+ cations were then added in vicinity of the atoms possessing negative partial charge, as calculated in Koubský and Luštinec (2018). In the case of the third model employing the undissociated nitric acid molecule, eight different initial conformations were generated for each of the tested extractants, with the HNO3 molecule placed gradually into eight different positions in vicinity of the carbonyl oxygens, ether oxygen and the two amide nitrogen atoms, which are all likely to create hydrogen bonds with the HNO3 molecule.
Geometry optimization of the initial conformations was performed using Gaussian09 code with the following settings: 6–31G (d,p) basis set (Petersson et al., 1988; Petersson and Al-Laham, 1991), PCM solvent model (Miertuš et al., 1981; Tomasi et al., 2005) with water taken as the solvent, GD3BJ dispersion correction (Grimme et al., 2010; Grimme et al., 2011), and B3LYP exchange and correlation functional (Beck, 1993). Gaussian09 code with NBO 6.0 (Glendening et al., 2013) was then used for the Natural population analysis and the Wiberg bond indices (Wiberg, 1968) calculations (the latter providing bond orders discussed in Section 3.4) with the same settings as the ones used in the conformation analysis.
Fukui functions, Fukui charges, and CDDs were calculated with DMol3 code using the following settings: DNP basis set (Delley, 1990), COSMO solvent model (Klamt and Schüürmann, 1993; Tomasi and Persico, 1994), GD2 dispersion correction (Grimme, 2006), and B3LYP exchange and correlation functional (Beck, 1993). The differences between PCM and COSMO implicit solvent models consists in the particular way in which the cavity containing the studied system is created. The specific choice of the implicit solvent model in the performed calculations was conditioned by the capabilities of the used software tool.
Results and discussion
At first, the geometrical optimization of the studied acid model systems was performed, leading to optimized conformations, indicating a localized direct interaction of the acid representatives with the extractant molecule, mediated by hydrogen atoms. These optimized conformation were then used in subsequent calculations of the studied stability indicators. The atomic denotation used in the calculations and discussion of the results achieved for the studied structures is shown in Figure 2.
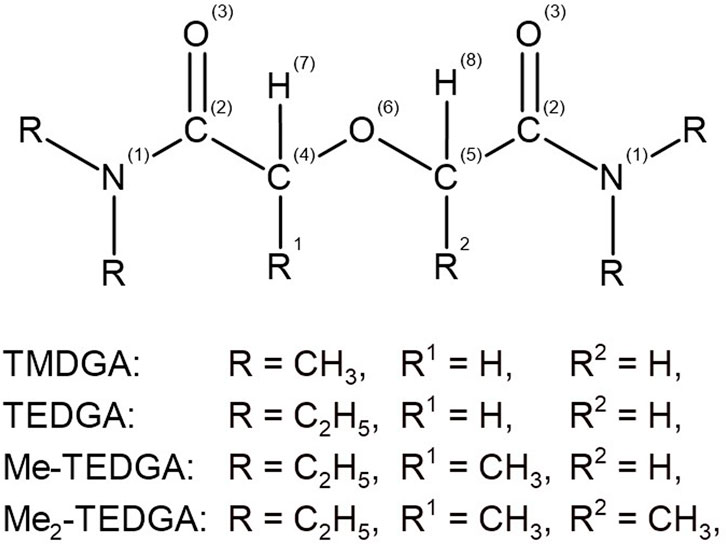
FIGURE 2. General chemical structure of the studied DGA derivatives with the atom labels indicated.
As in the previous work (Koubský and Luštinec, 2018), arithmetic averages are used for the symmetrically equivalent atoms to simplify the stability indicators analysis and also to reduce the conformation dependence of the results. Structure of Me-TEDGA is unsymmetrical due to the methyl group bonded on one of the ether carbons C (4,5). For this reason, the atoms C (4,5) are considered as inequivalent; the remaining atoms symmetrical against oxygen O (6) are analyzed as being equivalent ones.
Radical Fukui function
In our previous studies (Koubský et al., 2017; Koubský and Luštinec, 2018), the radical Fukui function (FF) (Yang and Parr, 1985) has proved to be a radical stability descriptor relevant for the investigated hydrophilic and lipophilic DGA derivatives. Therefore, the radical FF is also evaluated for the proposed DGAs acid models. The calculated values of radical FF are mapped on electron density iso-surface, and the maxima identified. Similar to the previous study (Koubský and Luštinec, 2018), the main maxima appear in a close vicinity of the ether hydrogens and the amide groups. This trend is found for all tested combinations of DGA derivatives and acid representations. The results obtained for TEDGA are given in Figure 3 and Figure 4 as examples.
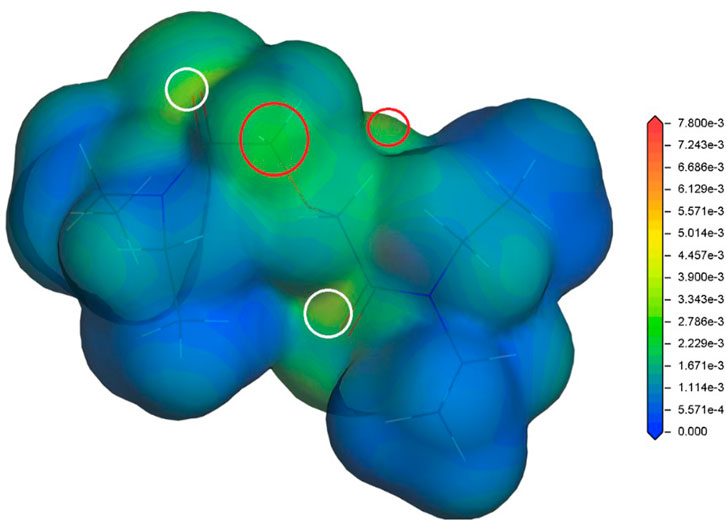
FIGURE 3. Radical Fukui function for TEDGA/acid-free model (Koubský and Luštinec, 2018), mapped on the electron density iso-surface 0.017 eÅ−3; the red circles mark the FF maxima on hydrogens adjacent to ether group, the white circles identify maxima on amide groups (Dmol3, DNP, B3LYP).

FIGURE 4. Radical Fukui function obtained for TEDGA/HNO3 acid model, mapped on the electron density iso-surface 0.017 eÅ−3; the red circles mark the FF maxima on hydrogens adjacent to ether group, the white circles identify the maxima on amide groups (Dmol3, DNP, B3LYP).
The trend of radical FF maxima located in vicinity of ether hydrogens (Table 1) agrees well with the expectations based on experimentally observed radiolytic stabilities of the derivatives. For all acid models, the calculated values of radical FF maxima decrease with the molecular weight of the derivative growing, the trend being even more pronounced than in the case of pure water environment considered in (Koubský and Luštinec, 2018). Within the three acid models considered here, the mentioned trend seems to be stronger for the H+ and H3O+ models than for the HNO3 one. However, as already stated, a direct quantitative comparison of the radical FF values obtained for the different acid models is not straightforward due to the different total charge present in the model systems.
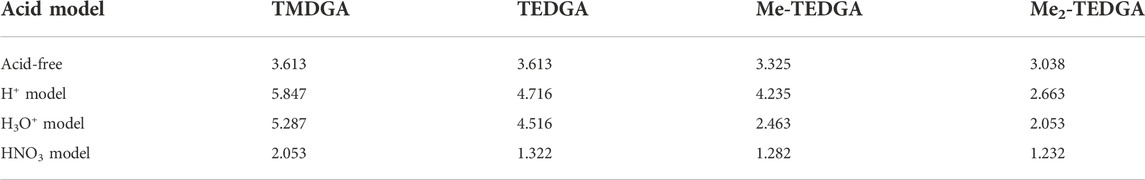
TABLE 1. Values of the radical Fukui function in 10−3 eÅ−3 close to the ether-neighboring hydrogen atoms (red circles in Figure 3 and Figure 4); results for the acid-free model taken from (Koubský and Luštinec, 2018) (DMol3, B3LYP, COSMO).
Similar to the acid-free situation (Koubský and Luštinec, 2018), the maxima of radical FF on the amide groups are also observed for all the tested acid models (Table 2). The values obtained for the HNO3 acid model are reduced as a whole compared to the acid-free model suggesting that the presence of nitric acid molecule decreases the ability of DGA derivatives to react with radicals. The values obtained for different DGA derivatives show then a gradual descend with the molecular weight increasing, supporting thus the experimentally observed stability trend. For the H+ and H3O+ models, the radical FF values on amide groups fluctuate and are dependent on the location of bonding model species. Also, the influence of different total charge of the model systems must be taken into account.
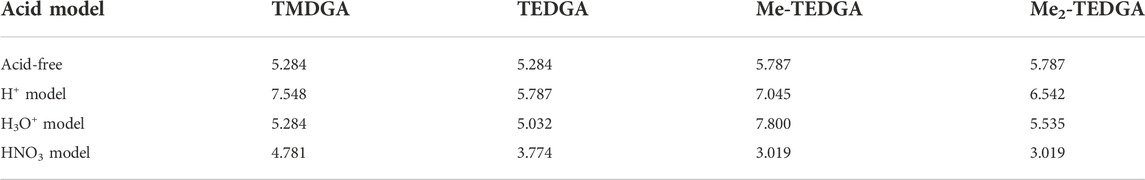
TABLE 2. Values of the radical Fukui function in 10−3 eÅ−3 close to one of the amide groups (white circles in Figure 3 and Figure 4); results for the acid-free model taken from (Koubský and Luštinec, 2018) (DMol3, B3LYP, COSMO)
Radical Fukui charges and CDD
In order to simplify the analysis and discussion of the calculated results, the volumetric radical FF can be assigned to individual atoms; the obtained condensed values are called atomic Fukui charges. As Table 3 shows, inclusion of acid does not significantly modify the trends found for the acid-free situation (Koubský and Luštinec, 2018). Since also the main features of results obtained with all tested acid models are similar, we discuss them on the example of H3O+ acid model. (Although some minor differences can be identified in the case of H+ acid model, the arguments remain the same.) The HNO3 acid model shows then a significant reduction of all calculated atomic Fukui charges (similar to the situation observed for the radical FF values, cf. Table 1 and Table 2) making analysis and a straightforward quantitative comparison with the other two models difficult.
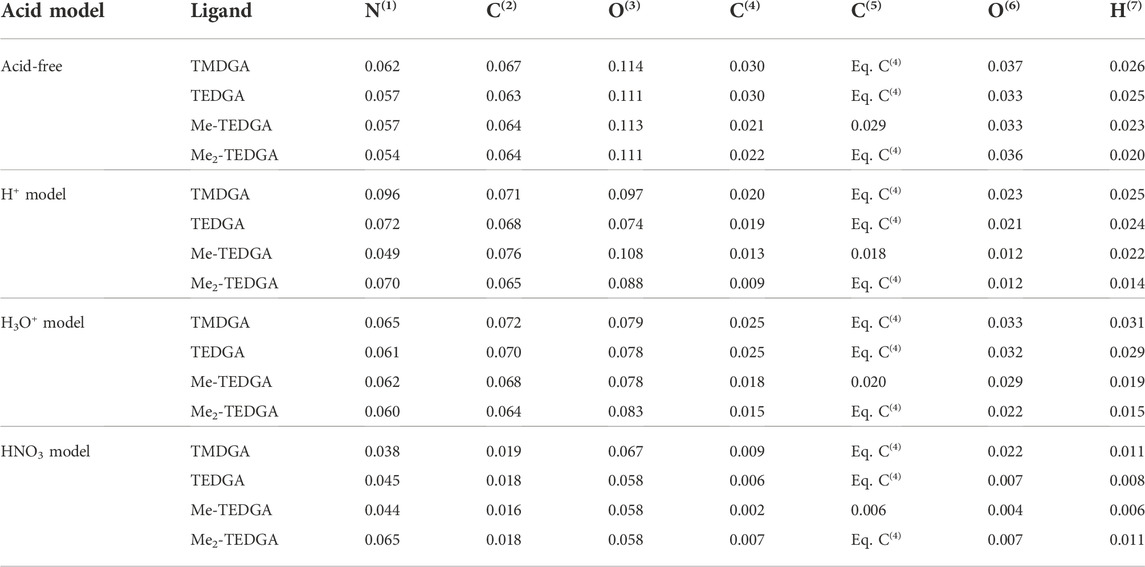
TABLE 3. Atomic radical Fukui charges based on Hirshfeld population analysis obtained for studied DGA derivatives and proposed acid models; results for the acid-free model taken from (Koubský and Luštinec, 2018); values for H8, H(R1) and H(R2) are identical to the value obtained for H7 where relevant (DMol3, B3LYP, COSMO).
Due to its high structural fragility, the ether group is the weakest part of DGA molecules when a radical attack is considered. In case of acid-free environment (Koubský and Luštinec, 2018), the values of radical Fukui charges located on the ether group (C(4,5), O(6), H(7), H(8), R1 and R2 chains) support with the greater (C(4,5), H(7)) or lesser (O(6)) extend the experimentally observed stability trend. With acid included, the analogical behaviour is observed for C(4,5), H(7) and the equivalent hydrogen atoms. For the ether oxygen O(6), the tendency of atomic Fukui charge to decrease with the ligand molecular weight is remarkably enhanced, suggesting a possible positive influence of acid presence on the tested DGAs derivatives stabilization.
The maxima of radical FF on amide group are also reproduced in the radical Fukui charges located on atoms N(1), C(2), and O(3). Again, the same trends are observed as for the ether group in case of the acid-free model (Koubský and Luštinec, 2018), and remain qualitatively unchanged after the inclusion of acid. The values of charges calculated with the HNO3 model are all reduced compared to the acid-free results.
In order to get a deeper insight into the possible reaction mechanism, the CDD indicator values are calculated and evaluated. The calculated results are summarized in (Table 4).

TABLE 4. Values of atomic CDD based on Hirshfeld population analysis calculated for the studied DGA derivatives and proposed acid models; results for the acid-free model taken from (Koubský and Luštinec, 2018); values for H8, H(R1) and H(R2) equal to the value obtained for H7 where relevant (DMol3, B3LYP, COSMO).
In general, for the acid-free model, the CDD absolute values on almost all atoms show a decreasing trend with the ligand weight growing, conformal with the experimental stability trend. The signs then indicate that atoms N(1) and O(3) are likely the only ones that are susceptible to an electrophilic attack. The remaining atoms might be vulnerable to a nucleophilic attack. This result supports the reaction degradation mechanism proposed by Wilden et al. (2018) based on the electron transfer from the amide group. In addition, in the case of H3O+ acid model, the position O(6) is predicted to be sensitive to an electrophilic attack. The similar behaviour is observed for TMDGA and TEDGA in frame of the H+ acid model, for Me2-TEDGA in the acid-free model, and for all tested derivatives in the case of the HNO3 acid model. Thus, apparently, selection of a particular acid model affects significantly the afterward obtained CDD values.
Bond orders
For all tested ligands and acid models, the order of C(4)-O(6) bond is found to be the lowest one of all bonds. The calculated C(4)-O(6) bond order values are summarized in Table 5. Similar character of the C(4)-O(6) bond was also identified for the lipophilic DGA derivatives (Koubský et al., 2017). For all tested models (with and without acid), presence of the methyl group(s) taking place in Me-TEDGA and Me2-TEDGA derivatives causes then a small drop in C(4)-O(6) bond order. Again, such effect is also observed for the analogically modified lipophilic DGA representatives (Koubský et al., 2017).
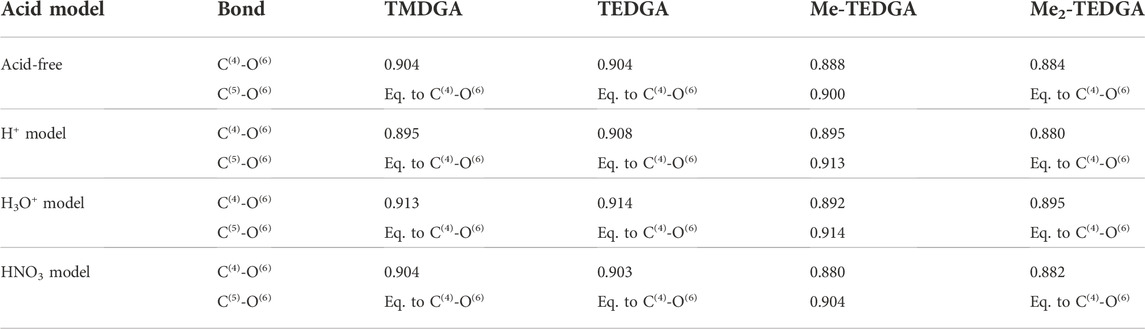
TABLE 5. Calculated bond order (Wiberg bond indices) of the C(4)-O(6) and equivalent C(5)-O(6) bond; results for the acid-free model taken from (Koubský and Luštinec, 2018) (Gaussian,B3LYP,PCM,NBO).
Bond orders closely relate to the other frequently used partitioning quantity of electron density - atomic partial charges. However, the partial charges obtained with the here tested acid models are found to be strongly dependent on the particular total charge included in the model, and show also a high sensitivity to the particular position in the ligand where the acid-representing group is attached (as a result of the geometrical optimization of the system); no reliable interpretation of the partial charges thus can be achieved and their values are not discussed here.
Conclusion
Three simplified models of acid influence on the radiolytic stability of hydrophilic DGA representatives are proposed and applied in calculations of the selected chemical stability indicators: radical FF, radical Fukui charges, CDD, and bond orders. The results obtained for the individual acid models are compared and juxtaposed with the results obtained for models with no acid influence considered. The newly tested CDD indicator shows absolute values that are generally in agreement with the experimentally observed radiolytic stability trend (TMDGA < TEDGA < Me-TEDGA < Me2-TEDGA (Wilden et al., 2018)). For different acid models however, the CDD signs significantly vary, and the CCD indicator fails to provide a reliable stability description.
All the tested acid models provide results similar to the results of the acid-free model, with the main trends remaining unaffected. However, values of the tested indicators as a whole drop down with the acid models applied, suggesting that in addition to the direct protection reflected by the local variations of calculated indicators, some indirect protection mechanism may also originate from acid presence, originating in a general decrease in the chemical reactivity of the ligands in the presence of acid.
Arguments for a direct acid protection effects follows from the two following obtained results: 1) the faster stabilizing reduction of the atomic Fukui charge values at the weakest atomic site of the tested DGA derivatives–the ether oxygen atom - found with the all acid models, and 2) the decrease of radical FF maxima on ether hydrogens and amide groups encountered with the HNO3 acid model. The latter effect indicates that undissociated acid molecules may reduce the reactivity of the studied DGA derivatives with radicals, the final protection effect being then dependent on the specific nitric acid concentration applied.
Considering the similarity between the basic trends of radical FF and Fukui charges found for the acid models and the acid-free model, the obtained results in any case do not contradict the proposition made by Horne and co-workers (Wilden et al., 2018) that the significant indirect acid protection effect consists in the preferential reaction of acid with the products of solvent radiolysis, decreasing thus the subsequent direct radical attack rate of the solvent radiolysis products on the extractant molecules.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
The calculations set up and performed by JL. All authors (JL, TK, LK) participated in evaluation, discussion and interpretation of the achieved results.
Funding
This research was funded by MEYSCR grant CZ.02.1.01/0.0/0.0/16_019/0000778 (JL and LK), MEYS CR institutional grant RVO14000 (JL, LK, and TK) and Grant Agency of the Czech Technical University in Prague, Grant No. SGS22/183/OHK4/3T/14 (JL). Support to the methodical part of the work (LK) from the CSF grant GA21-05259S is also acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Beck, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98 (7), 5648–5652. doi:10.1063/1.464913
Delley, B. (1990). An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92 (1), 508–517. doi:10.1063/1.458452
Delley, B. (2000). From molecules to solids with the DMol 3 approach. J. Chem. Phys. 113 (18), 7756–7764. doi:10.1063/1.1316015
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2013). Gaussian 09, revision D.01. Wallingford, CT: Gaussian, Inc.
Fukui, K. (1982). Role of frontier orbitals in chemical reactions. Science 218 (4574), 747–754. doi:10.1126/science.218.4574.747
Galan, H., Zarzana, C. A., Wilden, A., Nunez, A., Schmidt, H., Egberink, R. J. M., et al. (2015). Gamma-radiolytic stability of new methylated TODGA derivatives for minor actinide recycling. Dalton Trans. 44, 18049–18056. doi:10.1039/c5dt02484f
Glendening, E., Badenhoop, J., Reed, A., Carpenter, J., Bohmann, J., Morales, C., et al. (2013). Natural bond orbital analysis program: NBO 6.0. Madison, WI: Theoretical Chemistry Institute, University of Wisconsin.
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132 (15), 154104. doi:10.1063/1.3382344
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32 (7), 1456–1465. doi:10.1002/jcc.21759
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27 (15), 1787–1799. doi:10.1002/jcc.20495
Horne, G. P., Wilden, A., Mezyk, S. P., Twight, L., Hupert, M., Stärk, A., et al. (2019). Gamma radiolysis of hydrophilic diglycolamide ligands in concentrated aqueous nitrate solution. Dalton Trans. 48 (45), 17005–17013. doi:10.1039/c9dt03918j
Klamt, A., and Schüürmann, G. (1993). COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 (5), 799–805. doi:10.1039/p29930000799
Koubský, T., and Luštinec, J. (2018). Application of quantum mechanical simulations for studying the radiolytic stability of prospective extractants in the nuclear fuel cycle. J. Radioanal. Nucl. Chem. 318 (3), 2407–2413. doi:10.1007/s10967-018-6225-2
Koubský, T., Fojtíková, J., and Kalvoda, L. (2017). Radical degradation stability of ether linkage in N, N, N′, N′-tetraoctyldiglycolamide and related organic extractants: A density functional study. Prog. Nucl. Energy 94, 208–215. doi:10.1016/j.pnucene.2016.07.010
Lumetta, G. J., Gelis, A. V., Carter, J. C., Niver, C. M., and Smoot, M. R. (2014). The actinide-lanthanide separation concept. Solvent Extr. Ion Exch. 32 (4), 333–347. doi:10.1080/07366299.2014.895638
Mattocks, J. A., and Cotruvo, J. A. (2020). Biological, biomolecular, and bio-inspired strategies for detection, extraction, and separations of lanthanides and actinides. Chem. Soc. Rev. 49, 8315–8334. doi:10.1039/d0cs00653j
Matveev, P., Mitrofanov, A., Petrov, V., Zhokhov, S., Smirnova, A., Ustynyuk, Y. A., et al. (2017). Testing a simple approach for theoretical evaluation of radiolysis products in extraction systems. A case of N, O-donor ligands for Am/Eu separation. RSC Adv. 7 (87), 55441–55449. doi:10.1039/c7ra11622e
Miertuš, S., Scrocco, E., and Tomasi, J. (1981). Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55 (1), 117–129. doi:10.1016/0301-0104(81)85090-2
Morell, C., Grand, A., and Toro-Labbe, A. (2005). New dual descriptor for chemical reactivity. J. Phys. Chem. A 109 (1), 205–212. doi:10.1021/jp046577a
Morell, C., Grand, A., and Toro-Labbé, A. (2006). Theoretical support for using the Δf(r) descriptor. Chem. Phys. Lett. 425 (4-6), 342–346. doi:10.1016/j.cplett.2006.05.003
Parr, R. G., and Yang, W. (1989). Density functional theory of atoms and molecules, sv. 1. New York: Oxford University Press.
Petersson, G. A., and Al-Laham, M. A. (1991). A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 94 (9), 6081–6090. doi:10.1063/1.460447
Petersson, G. A., Bennett, A., Tensfeldt, T. G., Al-Laham, M. A., Shirley, W. A., and Mantzaris, J. (1988). A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 89 (4), 2193–2218. doi:10.1063/1.455064
Rostaing, C., Poinssot, C., Warin, D., Baron, P., and Lorraina, B. (2012). Development and validation of the EXAm separation process for single Am recycling. Procedia Chem. 7, 367–373. doi:10.1016/j.proche.2012.10.057
Sasaki, Y., Sugo, Y., Kitatsuji, Y., Kirishima, A., Kimura, T., and Choppin, G. R. (2007). Complexation and back extraction of various metals by water-soluble diglycolamide. Anal. Sci. 23 (6), 727–731. doi:10.2116/analsci.23.727
Smirnova, A., Mitrofanov, A., Matveev, P., Baygildiev, T., and Petrov, V. (2020). A search of a quantitative quantum-chemical approach for radiolytic stability prediction. Phys. Chem. Chem. Phys. 22 (26), 14992–14997. doi:10.1039/d0cp01786h
Tomasi, J., and Persico, M. (1994). Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem. Rev. 94 (7), 2027–2094. doi:10.1021/cr00031a013
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105 (8), 2999–3093. doi:10.1021/cr9904009
Veliscek-Carolan, J. (2016). Separation of actinides from spent nuclear fuel: A review. J. Hazard. Mater. 318, 266–281. doi:10.1016/j.jhazmat.2016.07.027
Wiberg, K. B. (1968). Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24 (3), 1083–1096. doi:10.1016/0040-4020(68)88057-3
Wilden, A., Mincher, B. J., Mezyk, S. P., Twight, L., Rosciolo-Johnson, K. M., Zarzana, C. A., et al. (2018). Radiolytic and hydrolytic degradation of the hydrophilic diglycolamides. Solvent Extr. Ion Exch. 36 (4), 347–359. doi:10.1080/07366299.2018.1495384
Keywords: diglycolamides, hydrophilic DGA, density functional theory, radiolytic stability, acid influence
Citation: Luštinec J, Koubský T and Kalvoda L (2022) Ab-initio evaluation of acid influence on chemical stability of hydrophilic diglycolamides. Front. Mol. Biosci. 9:1063022. doi: 10.3389/fmolb.2022.1063022
Received: 06 October 2022; Accepted: 18 November 2022;
Published: 07 December 2022.
Edited by:
Emilia Pedone, Department of Biomedical Sciences, National Research Council (CNR), ItalyReviewed by:
Chengwen Liu, The University of Texas at Austin, United StatesRamar Vanajothi, Bharathidasan University, India
Copyright © 2022 Luštinec, Koubský and Kalvoda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ladislav Kalvoda, bGFkaXNsYXYua2Fsdm9kYUBmamZpLmN2dXQuY3o=