 Evan Mercier
Evan Mercier Marina V. Rodnina
Marina V. Rodnina- Department of Physical Biochemistry, Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany
Nascent polypeptides emerging from the ribosome during translation are rapidly scanned and processed by ribosome-associated protein biogenesis factors (RPBs). RPBs cleave the N-terminal formyl and methionine groups, assist cotranslational protein folding, and sort the proteins according to their cellular destination. Ribosomes translating inner-membrane proteins are recognized and targeted to the translocon with the help of the signal recognition particle, SRP, and SRP receptor, FtsY. The growing nascent peptide is then inserted into the phospholipid bilayer at the translocon, an inner-membrane protein complex consisting of SecY, SecE, and SecG. Folding of membrane proteins requires that transmembrane helices (TMs) attain their correct topology, the soluble domains are inserted at the correct (cytoplasmic or periplasmic) side of the membrane, and – for polytopic membrane proteins – the TMs find their interaction partner TMs in the phospholipid bilayer. This review describes the recent progress in understanding how growing nascent peptides are processed and how inner-membrane proteins are targeted to the translocon and find their correct orientation at the membrane, with the focus on biophysical approaches revealing the dynamics of the process. We describe how spontaneous fluctuations of the translocon allow diffusion of TMs into the phospholipid bilayer and argue that the ribosome orchestrates cotranslational targeting not only by providing the binding platform for the RPBs or the translocon, but also by helping the nascent chains to find their correct orientation in the membrane. Finally, we present the auxiliary role of YidC as a chaperone for inner-membrane proteins. We show how biophysical approaches provide new insights into the dynamics of membrane protein biogenesis and raise new questions as to how translation modulates protein folding.
Introduction
Membrane proteins comprise 20–30% of the cellular proteome and are critical for allowing cells to communicate with the extracellular environment. The common feature of inner-membrane proteins is the presence of transmembrane helices (TMs) which contain large patches of hydrophobic residues that are likely to misfold if not inserted into the phospholipid bilayer of the membrane. Efficient biogenesis of properly folded membrane proteins relies on multiple processes including N-terminal processing, membrane targeting, membrane insertion and folding, which are all coordinated in the cell. In bacteria like Escherichia coli, this coordination has to keep up with the rapid synthesis of new proteins by the ribosome, which churns out polypeptide chains at 10–20 amino acids per second on average. The rate of translation defines the time window in which a membrane protein emerging from the ribosome exit tunnel has to be recognized, processed, and targeted to the membrane to prevent misfolding. In this review, we summarize the main findings and current models of how bacteria have overcome this challenge by ensuring that all steps of membrane protein biogenesis are efficient and rapid in order to win the race against the clock as a new membrane protein is synthesized on the ribosome.
Recent insights into cotranslational protein biogenesis have revealed that protein folding starts early, when most of the nascent protein chain is still inside the ribosome (reviewed in (Bustamante et al., 2020; Cassaignau et al., 2020; Liutkute et al., 2020b)). Compaction of several nascent proteins has been observed inside the ribosome and, in some cases, the extent of folding can be quite substantial, with secondary structure elements such as α-helices and even small domains folding relatively deep inside the exit tunnel of the ribosome (Woolhead et al., 2004; Lu and Deutsch, 2005; Nilsson et al., 2015; Marino et al., 2016; Farías-Rico et al., 2018; Liutkute et al., 2020a; Agirrezabala et al., 2022). Larger structural elements can fold upon arrival at the vestibule region of the ribosome exit tunnel or outside the ribosome (Holtkamp et al., 2015; Bustamante et al., 2020; Liutkute et al., 2020b). Remarkably, an all-β protein has been shown to compact inside the exit tunnel of the ribosome as an α-helix, suggesting that the environment of the exit tunnel favors formation of α-helices (Agirrezabala et al., 2022). On the other hand, protein structures remain dynamic as long as the nascent protein is bound to the ribosome (Kaiser et al., 2011; Liutkute et al., 2020a; Agirrezabala et al., 2022). Together, these findings indicate not only a clear trend for nascent-protein folding inside the ribosome, but also a profound ability of the ribosome to alter the folding landscape of nascent proteins. How well the cotranslational folding trends elucidated for cytosolic proteins extend to membrane proteins is still unclear, but recent advances in biophysical techniques provide new insights into how nascent membrane proteins fold on the ribosome, and how the ribosome influences the different steps in membrane protein biogenesis.
The influence of the ribosome on cotranslational protein biogenesis does not end when the nascent protein emerges from the ribosome. Proteins called ribosome-associated protein biogenesis factors (RPBs) that facilitate nascent chain maturation and targeting bind at the tunnel exit and interact with the emerging nascent protein (Kramer et al., 2009; Giglione et al., 2015; Koubek et al., 2021). RPBs comprise peptide deformylase (PDF) and methionine aminopeptidase (MAP), which are responsible for N-terminal processing; the chaperone trigger factor (TF); and the signal recognition particle (SRP) which targets inner membrane proteins and secretory proteins to the membrane with the help of the SRP receptor (SR), FtsY (Figure 1). All RPBs scan ribosome-nascent-chain complexes (RNCs) for suitable nascent chain substrates by binding to the ribosome close to the tunnel exit where the new protein emerges (Holtkamp et al., 2012; Sandikci et al., 2013; Bornemann et al., 2014). The area near the tunnel exit, therefore, provides a platform capable of coordinating the timing and action of different RPBs. The region is also responsible for binding the SecYEG translocon, which helps nascent membrane proteins to insert into the phospholipid bilayer. The TMs of a membrane protein reach the phospholipid bilayer during ongoing translation and must attain the correct topology with respect to the membrane, and fold appropriately. We focus this review on the role of the ribosome in coordinating these different events to ensure that all processes occur in a timely manner.
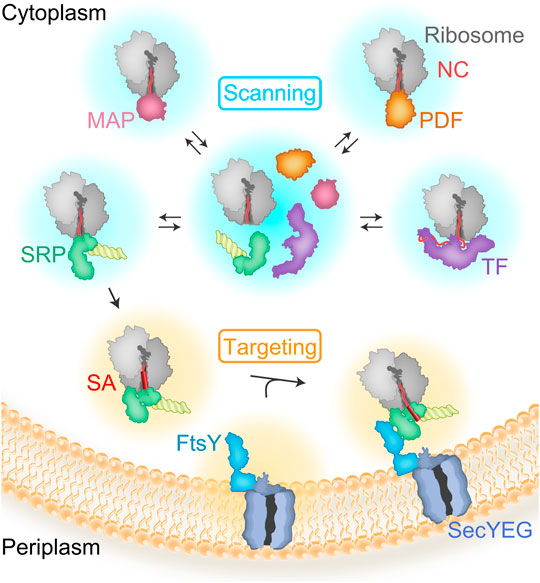
FIGURE 1. Scanning and targeting of nascent polypeptides. RBPs rapidly bind to the ribosome during ongoing translation to scan emerging nascent polypeptide chains (NC). Most of the proteins are first deformylated by PDF and then processed by MAP, which cleaves the N-terminal methionine. TF is a chaperone with preference for cytosolic or outer membrane proteins. TF and SRP compete for binding to ribosomes with short nascent chains. Nascent peptides emerging from the exit tunnel of the ribosome expose recognition motifs leading to kinetic stabilization or rejection of TF vs SRP. SRP recognizes the signal-anchor sequence (SA) and targets the RNCs to the translocon, SecYEG, with the help of the SRP receptor, FtsY.
Scanning of RNCs by RPBs
N-Terminal Processing of Nascent Cytosolic Proteins
N-terminal processing begins cotranslationally as soon as the formylated methionine at the N-terminus of a nascent chain emerges from the polypeptide exit tunnel of the ribosome (Figure 1). In the first step, the formyl group is removed by PDF, which is the prerequisite for subsequent removal of methionine by MAP. The essential protein PDF is a small metalloenzyme that carries an Fe(II) cofactor in its active site (Mazel et al., 1994; Rajagopalan et al., 1997) and contains a C-terminal helix that binds to the ribosomal protein uL22 (Chan et al., 1997; Bingel-Erlenmeyer et al., 2008; Bhakta et al., 2019; Akbar et al., 2021). Binding of PDF to the ribosome and dissociation of the complex is rapid, estimated to occur 65 times per second, which allows the enzyme to randomly scan ribosomes until it encounters an RNC carrying its reaction substrate, a formylated nascent chain (Sandikci et al., 2013; Bogeholz et al., 2021). The binding sites of PDF and MAP, which both bind at uL22, overlap, precluding simultaneous recruitment of the two proteins at their primary binding site on the ribosome (Bhakta et al., 2019). The competition between PDF and MAP enhances the scanning process by increasing the selectivity of PDF to N-formylated nascent chain (Sandikci et al., 2013). MAP interacts with the ribosome via positively charged amino acids, which allows MAP to evade to a secondary binding site in the presence of PDF (Sandikci et al., 2013; Bhakta et al., 2019). As the binding site of PDF lies in close proximity of the polypeptide exit tunnel, PDF can rapidly deformylate short peptide substrates starting at a length of about 50 amino acids (Giglione et al., 2015; Bogeholz et al., 2021). For stalled RNCs, however, a preferred length of deformylation is about 70 amino acids (Ranjan et al., 2017; Yang et al., 2019).
The cleavage of the formyl group is rapid, but the subsequent turnover of PDF is delayed by a slow conformational rearrangement most likely reflecting the release of the nascent chain from the active site of the enzyme (Bogeholz et al., 2021). Owing to this slow step, deformylation is rate-limiting for N-terminal processing, because MAP can only access the N-terminus after dissociation of PDF (Yang et al., 2019). Methionine is removed from about 50% of proteins in E. coli. There is a strong dependence on the size of the second amino acid: nascent chains with a small amino acid in this position are preferred MAP substrates (Hirel et al., 1989; Frottin et al., 2006; Xiao et al., 2010; Bienvenut et al., 2015). The retention of the nascent chain by PDF during ongoing translation on one hand and nascent-peptide folding as it moves away from the ribosome surface on the other hand, define the time window for MAP to act before it can no longer access the N-terminus (Yang et al., 2019), which may explain the incomplete methionine removal.
Lack of Deformylation of Membrane Proteins
While proteomics studies show that most cytosolic proteins are deformylated, the only proteins that retain the formyl group to an extent greater than 50% are inner-membrane proteins (Bienvenut et al., 2015). When an inner-membrane protein is synthesized on the ribosome, it displays an SRP-specific signal sequence that is recognized by SRP as soon as the N-terminus of the nascent chain emerges from the polypeptide exit tunnel. Binding of SRP inhibits deformylation by PDF (Ranjan et al., 2017) even though PDF and SRP can bind the ribosome simultaneously (Bornemann et al., 2014). The extent of the inhibitory effect, however, seems to depend on the distance between the signal sequence and the N-terminus (Ranjan et al., 2017; Yang et al., 2019). Interference with deformylation by SRP recruitment allows bypassing of the relatively slow N-terminal processing and ensures efficient targeting of inner-membrane proteins to the membrane. After insertion into the membrane, N-termini that reach into the cytosol can potentially be deformylated posttranslationally comparable to the deformylation of model peptides (Ranjan et al., 2017).
Competition Between RPBs at the Tunnel Exit
TF is another key player monitoring the emerging nascent chains. Interacting with short stretches enriched in basic and aromatic amino acids, TF acts as a chaperone mainly for cytosolic or outer-membrane proteins (Oh et al., 2011; Deckert et al., 2021; Koubek et al., 2021). TF consists of three domains. The N-terminal ribosome-binding domain interacts with the ribosome at protein uL23, which allows TF to arch over the polypeptide exit tunnel and accommodate nascent proteins (Kramer et al., 2002; Kristensen and Gajhede, 2003; Ferbitz et al., 2004). The peptidyl-prolyl isomerase domain facilitates cis/trans-isomeration of proline residues and the C-terminal domain contains the main chaperone activity (Kramer et al., 2002; Kristensen and Gajhede, 2003; Ferbitz et al., 2004; Merz et al., 2006). In the cell, TF is in two-to-three-fold excess over ribosomes (Lill et al., 1988) and as the Kd is about 0.1 µM (Raine et al., 2006; Bornemann et al., 2014), nearly all ribosomes can have one TF bound. TF dissociates rapidly from non-translating ribosomes, allowing the chaperone to scan for its substrates. On the ribosome, TF and PDF can bind at the same time due to a conformational change in TF (Bornemann et al., 2014; Bhakta et al., 2019). TF and MAP can also bind simultaneously (Bornemann et al., 2014); however, simultaneous binding of all three RPBs is only observed at very high MAP concentrations (Bhakta et al., 2019; Akbar et al., 2021). When TF encounters an RNC synthesizing one of its client proteins, TF binding on the ribosome is stabilized (Bornemann et al., 2014). Even though TF contacts the nascent chain early on (Merz et al., 2008), a stable engagement of TF starts at a nascent chain length of 100 amino acids for most proteins (Oh et al., 2011), which leaves sufficient time for N-terminal processing of proteins that are TF clients. TF and SRP can bind to the ribosome at the same time displaying partially competitive binding to the ribosome and the nascent chain (Buskiewicz et al., 2004; Eisner et al., 2006; Ullers et al., 2006; Bornemann et al., 2014). When SRP–TF–RNC complexes that carry a nascent chain displaying a signal peptide bind to the SRP receptor, FtsY, TF is displaced from the complex (Buskiewicz et al., 2004). On the other hand, TF slows down the recruitment of FtsY for suboptimal SRP substrates and reduces the affinity of SRP towards longer substrates, suggesting that if a protein has not been targeted to the membrane within a certain time, it will remain in the cytosol until it has been fully synthesized (Ariosa et al., 2015). Thus, interplay between TF and SRP leads to efficient selection of nascent chains for different pathways.
Biogenesis of Inner-Membrane Proteins
Membrane Targeting
Bacterial proteins which are destined for the inner membrane, the outer membrane, or the periplasmic space are first targeted to the inner membrane through one of three pathways. The Tat and SecA/SecB pathways help to translocate proteins across the inner membrane and act largely after synthesis of the client protein is completed (reviewed in (Frain et al., 2019)). The SecA/B pathway exports proteins in an unfolded state and these clients then go on to fold in the periplasm or insert into the bacterial outer membrane (reviewed in (Smets et al., 2019)). The Tat pathway exports folded proteins from the cytosol and tends to handle proteins which either fold rapidly in the cytosol or bind cytosolic cofactors (reviewed in (Kudva et al., 2013)). Inner-membrane proteins, on the other hand, are targeted by the SRP pathway that guides actively translating ribosomes to the Sec translocon (SecYEG in bacteria) with the assistance of FtsY.
Targeting through any of these pathways requires a hydrophobic signal sequence located near the N-terminus. Signal sequences for the Tat and SecA/B pathways include positively charged amino acids N-terminal to the hydrophobic segment, which is included in the SRRxFLK consensus sequence for Tat substrates (Kudva et al., 2013; Palmer and Stansfeld, 2020). For these posttranslational export pathways, hydrophobic signal sequences are inserted into the membrane and cleaved at a peptidase recognition motif located downstream of the hydrophobic segment. Signal peptide cleavage disconnects the exported protein from the inner membrane and allows it to find its proper place in the periplasm, outer membrane, or beyond. Inner-membrane proteins are targeted by the SRP pathway (reviewed in (Kuhn et al., 2017; Steinberg et al., 2018); signal sequences of the SRP pathway do not all contain positively charged amino acids at the N-terminus, and they tend to be more hydrophobic than those of the other export pathways (Cristóbal et al., 1999; Kudva et al., 2013). The SRP-specific signal sequence is not a conserved sequence motif, but a hydrophobic segment with high propensity to form an α-helix (Lee and Bernstein, 2001; Adams et al., 2002; Peterson et al., 2010; Robinson et al., 2012). The first transmembrane domain (TM1) of an integral membrane protein is typically used for membrane targeting and is referred to as a signal-anchor sequence (SA). Some membrane proteins are targeted by a signal sequence that is later cleaved off by leader peptidase; for these proteins, the second hydrophobic segment will become the first TM of the mature protein (e.g. CyoA (Celebi et al., 2008), FliP (Pradel et al., 2004)). During targeting, SRP interacts with the signal sequence and protects it from the cellular environment, thus helping to prevent aggregation or misfolding of the hydrophobic segment.
SRP is an RNA-protein complex which, in E. coli, comprises 4.5S RNA and a protein, Ffh. The 4.5S RNA is 114 nucleotides long and folds into a stem-loop structure, while the Ffh protein contains an NG domain which binds and hydrolyzes GTP, a methionine-rich M-domain, and a flexible linker connecting the two larger domains (Schaffitzel et al., 2006). Ffh binds near the proximal end of 4.5S RNA and the resulting SRP complex binds to the ribosome at the peptide tunnel exit. The M-domain contacts ribosomal proteins uL23 and uL29, as well as 23S rRNA in the vicinity of the exit tunnel (Figure 2). The flexible C-terminus of Ffh protrudes into the interior of the exit tunnel where it contacts uL23 (Jomaa et al., 2016; Denks et al., 2017). The affinity (Kd) of SRP to ribosomes that do not carry a nascent chain is about 100 nM, (Bornemann et al., 2008; Zhang et al., 2010)). In a translating ribosome, the growing nascent chain of about 25 aa length displaces Ffh from its uL23 contact inside the exit tunnel (Denks et al., 2017), but SRP remains bound to the ribosome in a stand-by state at the exit tunnel. This enables SRP to sense a nascent protein in the exit tunnel of the ribosome, and facilitates binding of the signal sequence to the M domain of SRP immediately upon emergence from the ribosome (Halic et al., 2006; Schaffitzel et al., 2006; Hainzl et al., 2011). Recognition of the signal sequence results in a high-affinity complex whose thermodynamic stability is somewhat sensitive to the hydrophobicity of the signal sequence (Kd = 1–10 nM, (Bornemann et al., 2008; Zhang et al., 2010; Mercier et al., 2017)). The high thermodynamic stability of SRP-ribosome and SRP-RNC complexes (Kd = 1–100 nM), along with the large cellular concentration of ribosomes in E. coli (20–45 µM (Rudorf and Lipowsky, 2015)), indicate that all SRP complexes in the cell are bound to translating ribosomes.
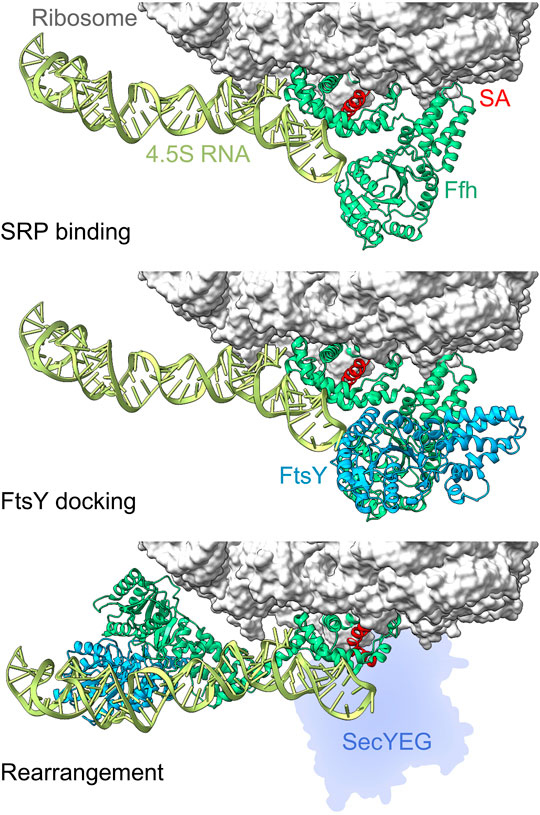
FIGURE 2. Conformational rearrangements of SRP and FtsY on the ribosome. Top panel: cryo-EM structure of the SRP binding complex at the ribosome with the M-domain of Ffh (green) recognizing the signal-anchor sequence (SA, red) emerging from the ribosome (gray). Ffh NG domain contacts the proximal end of the 4.5S RNA while the M-domain interacts with ribosomal proteins uL23 and uL29 as well as the 23S rRNA (PDB 5GAF (Jomaa et al., 2016)). Middle panel: Initial docking of FtsY on the RNC-SRP complex involves interactions between the NG domains of FtsY (blue) and Ffh (green) (PDB 5GAD (Jomaa et al., 2016)). Bottom panel, NG domains of Ffh and FtsY relocate towards the distal end of the 4.5S RNA (PDB 5NCO (Jomaa et al., 2017)), promoting handover of the RNC to the translocon (silhouette; not in the structure, inferred from PDB 5GAE (Jomaa et al., 2016)).
Because the concentration of SRP is about 400 nM in E. coli (Kudva et al., 2013), SRP can occupy only 1–2% of all ribosomes at any time, and this generates a potential predicament for SRP which has to bind hydrophobic signal sequences as they emerge from the ribosome. The solution to the problem lies in a rapid scanning function of SRP that enables it to bind and dissociate from ribosomes 5–10 times per second (Holtkamp et al., 2012; Mercier et al., 2017). Detection of a non-signal sequence-containing peptide causes SRP to reject the RNC as a substrate, while a hydrophobic signal sequence induces a switch in SRP from scanning mode to targeting mode (Holtkamp et al., 2012). The switch has been observed for actively translating ribosomes, and occurs after the ribosome has incorporated 40–50 amino acids, and likely depends on where the signal sequence is located relative to the N-terminus (Noriega et al., 2014; Schibich et al., 2016; Mercier et al., 2017). In targeting mode, the SRP-RNC complex is kinetically stabilized (koff = 0.03–0.1 s−1), which ensures that SRP remains bound until the RNC has been targeted to the membrane, and effectively protects the hydrophobic signal sequence (Holtkamp et al., 2012; Noriega et al., 2014; Saraogi et al., 2014; Mercier et al., 2017).
The SRP-RNC targeting complex is then directed to the membrane where FtsY is bound at the SecYEG translocon. FtsY contains an intrinsically disordered A domain at the N-terminus that interacts with phospholipids and SecYEG, as well as an NG domain homologous to that in SRP protein Ffh, which enables binding of FtsY to SRP (Figure 2) (Stjepanovic et al., 2011; Lakomek et al., 2016). Activation of FtsY is enhanced by anionic phospholipids (phosphatidylglycerol and cardiolipin), and binding of the A domain to the membrane-embedded translocon induces a conformational rearrangement of FtsY and effectively tethers the SRP-receptor to the translocon (de Leeuw et al., 2000; Lam et al., 2010; Stjepanovic et al., 2011; Draycheva et al., 2016). The A domain remains intrinsically disordered, thus providing a volume around the translocon where the NG domain can search for SRP-bound targeting complexes (Draycheva et al., 2016; Lakomek et al., 2016). Binding of the SRP-RNC targeting complex to the FtsY-translocon complex is driven by interaction of the NG domains in SRP and FtsY. In the complex, the NG domains of Ffh and FtsY relocate together towards the distal end of the 4.5S RNA and vacate a space in the vicinity of the tunnel exit for SecYEG (Figure 2C). This weakens SRP-RNC binding to Ffh-SecYEG, promoting handover of the RNC to the translocon (Jomaa et al., 2016; Jomaa et al., 2017; Draycheva et al., 2018). The details of handover of the nascent chain from SRP to SecYEG are unclear, but it seems to require GTP-bound SRP and FtsY, while GTP hydrolysis occurs after handover of the nascent chain in order to recycle SRP and FtsY for another round of targeting (Valent et al., 1998; Zhang et al., 2010).
Membrane Insertion
Insertion of TMs into the phospholipid bilayer takes place at the translocon. The core translocon is a ternary transmembrane protein complex composed of proteins SecY, SecE and SecG in bacteria (Denks et al., 2014). The largest component, SecY, is comprised of 10 α-helices that form a channel across the inner membrane (Van den Berg et al., 2004). The proteins SecE and SecG are both small, each comprising two α-helices, and are located at the periphery of SecY (Park et al., 2014; Tanaka et al., 2015). The translocon can open in two ways. For protein export across the inner membrane, the small plug domain in SecY that docks onto the hydrophobic ring in the middle of the pore can be displaced into periplasm (Zimmer et al., 2008; Park et al., 2014). To permit insertion of TMs into the phospholipid bilayer, the two halves of the translocon can move apart, opening the lateral gate formed by TM2 and TM7 and exposing the pore of SecY to the phospholipid bilayer (Egea and Stroud, 2010; Frauenfeld et al., 2011; Ge et al., 2014).
Upon targeting, the translocon docks at the tunnel exit of the ribosome, with its cytoplasmic loops occupying space in the vestibule of the ribosomal exit tunnel (Frauenfeld et al., 2011; Jomaa et al., 2016). In this complex, the peptide exit tunnel forms a continuous conduit with the translocon pore that allows nascent proteins to pass directly from the ribosome to the translocon. Inside this conduit, TMs can fold as soon as they pass the constriction formed by ribosomal proteins uL4 and uL22, about 25 Å away from the peptidyl transferase center (PTC) (reviewed in (Liutkute et al., 2020b)). This is supported by compaction of nascent membrane proteins observed in eukaryotic and prokaryotic ribosomes through distance-dependent FRET measurements and crosslinking to proteins in the exit tunnel (Woolhead et al., 2004; Robinson et al., 2012). The conduit formed by the ribosome-translocon complex allows TMs to pass directly from the peptide exit tunnel of the ribosome into the translocon pore. This permits a nascent membrane protein to span a distance of about 140 Å from the PTC to the periplasmic side of the translocon in an environment largely shielded from the aqueous environment of the cell. Because the translocon binds to the ribosome partially in the vestibule, the space available for folding a membrane protein in the ribosome is restricted compared to a cytosolic domain. Experiments with eukaryotic microsomes show that nascent membrane proteins remain in a compacted state upon entering the translocon, indicating that TMs insert as helices into the translocon pore (Woolhead et al., 2004). The dynamic properties of TMs inside the ribosome have not been characterized in detail, and it is possible that they exhibit breathing-type motions between compact α-helical and extended conformations (Liutkute et al., 2020b).
Membrane Protein Topology
TMs can insert into the membrane in two basic topologies: with the N-terminus pointing towards the cytosol (N-in) or towards the periplasm (N-out) (Figure 3). The principal determinant for TM topology is the location of positively charged amino acids within the TM, with regions of net positive charge retained on the cytosolic side of the membrane, known as the positive-inside rule (von Heijne, 1989; 1992). Because cotranslational deformylation of membrane proteins is inhibited by SRP during targeting, the N-terminus does not carry a charge and should not contribute to topology during insertion (Ranjan et al., 2017). Several factors influencing the efficiency of the positive-inside rule have been discussed, including strength of the positive charge, hydrophobicity of the TM, membrane potential, and phospholipid composition of the membrane (reviewed in (Higy et al., 2004; Kuhn et al., 2017; Spiess et al., 2019)). The simplest case is insertion with N-out topology, because no inversion of the TM is required. In this case, the loop or region preceding the TM is negatively charged or neutral, while the loop following the TM, which will be retained on the inside of the inner membrane, has a net positive charge. Retention of positive charges is favored by electrostatic interaction with negatively charged phospholipids in the inner membrane (Bogdanov et al., 2009; Dowhan et al., 2019), as well as an electric potential across the inner membrane (Cao et al., 1995), and, at least in eukaryotes, amino acids in the translocon (Junne et al., 2007).
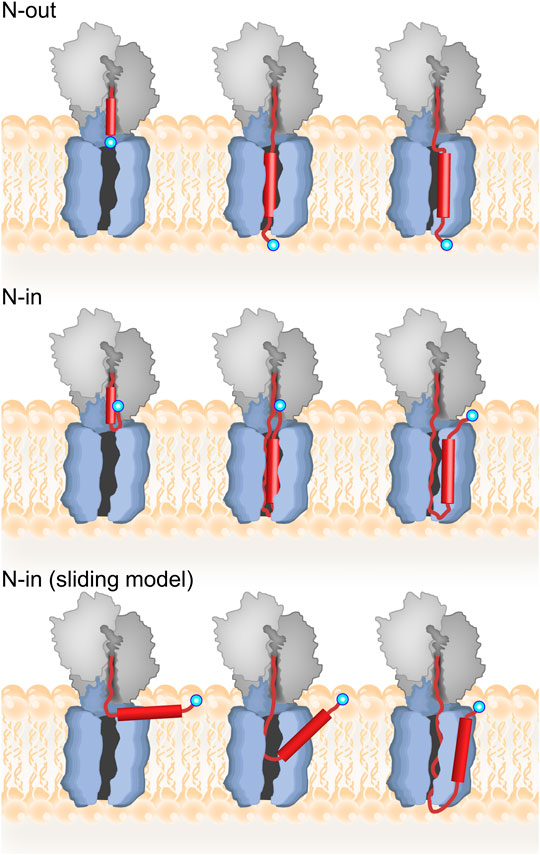
FIGURE 3. Topology of inner-membrane proteins. Top panel: model for cotranslational N-out TM insertion where the TM inserts first into the translocon prior to membrane integration. The nascent protein is depicted in red with the TM as a cylinder, and the N-terminus indicated by a blue circle. Middle panel: model for N-in TM insertion with inversion occurring in the ribosome prior to translocon insertion and then membrane integration. Bottom panel: alternative “sliding model” for N-in insertion where the TM does not insert into the translocon. For further discussion, see text.
The exact mechanism of TM insertion into the phospholipid bilayer is not clear (Cymer et al., 2015). For N-out topology, one can imagine a TM transitioning seamlessly from the ribosome into the translocon in a head-on fashion, all while the protein is being synthesized on the ribosome. This process has been monitored directly by real-time fluorescence-based measurements with high-efficiency in-vitro translation (Mercier et al., 2020). These experiments used distance-dependent FRET measurements to show that a FRET acceptor linked to the N-terminus of the N-out membrane protein LepB approaches a FRET donor positioned on the cytoplasmic side of the translocon, followed by insertion of TM1 in an N-out orientation, which causes a FRET decrease. Analysis of these results revealed that the simplistic description above is largely correct. In particular, kinetic analysis of these experiments indicated that translocon insertion of nascent membrane proteins is rate-limited by translation. Thus, no kinetic barrier restricts transition of the TM from the ribosome to the translocon pore. Interestingly, detailed analysis of translation rates revealed a strong pause in mRNA translation just prior to translocon insertion of the first TM. The cause of this pause is unclear, but it appears to be encoded in the mRNA since translation was carried out in an in-vitro system lacking any regulatory factors (Mercier et al., 2020).
Insertion of a TM with N-in topology is more complex as it requires TM inversion. In principle, inversion could result either from retention of the N-terminus on the inside of the membrane or could occur after initial insertion in an N-out orientation (Figure 3). Real-time FRET and protease protection experiments suggest that for the N-in membrane protein EmrD, the FRET acceptor linked to the N-terminus approached the FRET donor on the cytoplasmic side of the translocon to yield a high-FRET state, and then remained in this state throughout cotranslational insertion of TM1 with N-out topology (Mercier et al., 2020). Thus, insertion of TM1 with N-in topology followed retention, rather than insertion/inversion, pathway (Figure 3). That study also showed that inversion of the N-terminus occurs early, before the N-terminus of EmrD leaves the peptide exit tunnel of the ribosome. Moreover, very little FRET is observed between the cytoplasmic side of the translocon and the N-terminus of EmrD when it reaches 50 aa length, indicating a compact nascent chain that tends towards the N-in topology already at this length inside the exit tunnel of the ribosome.
The precise conformation of the inverted N-terminus inside the ribosomal exit tunnel is unclear. Since formation of the first α-helix should be possible early in the exit tunnel (see above), the simplest inverted conformation would be a looped structure with the N-terminus pointing in the direction of the PTC in the ribosome. Stabilization of the positively charged N-terminus could be accomplished by electrostatic interactions with negatively charged rRNA in the exit tunnel. This would suggest that the ribosome could also participate in establishing the positive-inside orientation and actively contribute to membrane protein topology before nascent membrane proteins emerge from the ribosome. Consistent with this idea, coarse-grained molecular dynamics simulations have revealed that electrostatic repulsion of negatively charged amino acids from the ribosome helps to drive cotranslational translocation of nascent proteins across the membrane (Niesen et al., 2018). Interestingly, cotranslational experiments revealed that during N-in insertion of EmrD, the ribosome paused during translation at or near codon 48 of the mRNA, at a length where the nascent chain is most likely attaining the inverted conformation (Mercier et al., 2020). Thus, the pause in mRNA translation, which is encoded in the mRNA and independent of the SecYEG insertion machinery, may provide additional time to stabilize the inverted N-in conformation of EmrD. It is not clear if formation of more complex inverted conformations (e.g. helix-turn-helix motifs) are possible in the exit tunnel, since the width of the exit tunnel is in the range of 10–13 Å, while two α-helices would require 15–20 Å depending on the side-chain conformers (O'Shea et al., 1991). While the ribosome exit tunnel widens significantly at the vestibule (up to 18 Å), binding of SecY in the vestibule constricts this space and the ribosome-translocon conduit remains narrow throughout (5–10 Å) (Frauenfeld et al., 2011). Thus, insertion with N-in topology would involve bending of the TM within the conduit of the ribosome-translocon complex or, conversely, involve lateral gate opening in SecY to provide the necessary space for acrobatics of a rigid TM.
Findings from real-time FRET measurements are in contrast to endpoint observations from ER insertion assays in COS-1 cells, which indicate that inversion occurs after initial N-out insertion of TM1 (Goder and Spiess, 2003). These studies take advantage of the glycosylation machinery present in the ER, which can modify specific sites in a region following the TM only if the TM is inserted into the ER membrane with N-in topology. In these experiments, inversion to an N-in topology is favored by increasing the time required for protein synthesis, either by elongating the C-terminal region after the TM, or by addition of cycloheximide, a reversible inhibitor of ribosomal protein synthesis. In addition, more hydrophobic TMs invert with lower efficiency than TMs with lower hydrophobicity. These findings point towards a mechanism where the TM first inserts in N-out configuration and inversion occurs later. This mechanism is also supported by chemical probing, crosslinking, and fluorescence quenching experiments performed with stalled RNCs on mammalian ER microsomes (Devaraneni et al., 2011). Here, exposure of the nascent-chain N-terminus on the luminal side of the membrane was observed at intermediate nascent-chain lengths, but at longer lengths luminal exposure was reduced. These results advocate for N-out insertion of the TM prior to inversion.
The two scenarios, where inversion can occur either by looping of the N-terminus while still inside the ribosome, or N-out insertion of the TM followed by flipping, are reflected in a kinetic model for TM inversion based on coarse-grained molecular dynamics simulations (Zhang and Miller, 2012). The results from these simulations indicate that both flipping mechanisms are possible, with the looping mechanism preferred at faster translation rates, while reducing the simulated rate of nascent chain elongation increased the fraction of trajectories showing N-out insertion and subsequent TM inversion. This kinetic description helps to reconcile the findings from real-time TM insertion experiments and endpoint assays by revealing that the insertion/inversion mechanism would be favored if in-vitro translation was slow, or if inversion was studied using stalled RNCs. This is also evident in real-time measurements, where an N-out intermediate was found to form posttranslationally when translation of an N-in protein was terminated after the incorporation of 85 amino acids (Mercier et al., 2020). While this intermediate could be identified as off-pathway through kinetic analysis of real-time data, this is not possible for endpoint assays. It is also noteworthy that the real-time FRET studies were performed using a translation/insertion system from E. coli, while the other studies have employed eukaryotic ribosomes, which have a somewhat different exit tunnel anatomy. The part of the peptide exit tunnel close to the vestibule is more narrow in eukaryotic than in bacterial ribosomes, so there would be less space available for looping of the nascent chain N-terminus inside the ribosome of eukaryotes (Dao Duc et al., 2019). In addition, real-time measurements were made with nascent polytopic membrane proteins, while the other studies have used exported proteins with a single TM: a model hydrophobic sequence rather than a naturally occurring amino acid sequence (Goder and Spiess, 2003; Devaraneni et al., 2011; Zhang and Miller, 2012). For exported proteins, TM inversion may be related to translocation of the protein C-terminus across the membrane, which is generally a posttranslational process, and thus it is unclear how strictly these findings should be applied for integral membrane proteins which are inserted cotranslationally.
In addition to cotranslational TM inversion, posttranslational inversion may play an important role in determining the membrane-protein topology of some integral membrane proteins. For example, the dual-topology protein EmrE has been suggested to invert after membrane insertion (Woodall et al., 2017). Examination of EmrE by in-vivo chemical probing, however, revealed that protomer topology is unaffected by the topology of the dimerization partner, indicating that protein topology is determined prior to dimerization and complete topological inversion is unlikely in vivo (Fluman et al., 2017). Interestingly, addition of a single positive charge at the C-terminal TM5 of EmrE is capable of inverting the final topology of the protein, suggesting that inversion may occur very late during protein synthesis, probably after insertion of several TMs (Seppälä et al., 2010). In-vitro measurements with proteoliposomes suggested that inversion of the membrane protein LacY occurs posttranslationally, and can be induced either by changing the lipid composition or by LacY phosphorylation (Vitrac et al., 2013; Vitrac et al., 2017). In addition, an internal TM (TM3) of human aquaporin was shown to insert with different topology if the protein was truncated after 3 TMs, or if 4, 5 or 6 TMs were translated (in Xenopus oocytes), indicating that TM topology can be influenced by elements that are translated later (Lu et al., 2000). Further work should clarify how common posttranslational TM inversion is, and whether it occurs shortly after TM insertion or awaits specific cellular signals.
TM and Translocon Dynamics
In order for a TM to transit from the pore of the translocon into the phospholipid bilayer it must pass through the lateral gate. Opening of the lateral gate has been observed upon insertion of a TM into the translocon using fluorescence quenching, indicating that lateral gate opening is induced during cotranslational insertion, likely by the TM itself (Ge et al., 2014). RNCs carrying nascent membrane proteins have been visualized in complex with translocons using cryo-EM, and demonstrate that the lateral gate can be opened to different degrees during TM insertion (Frauenfeld et al., 2011; Bischoff et al., 2014; Park et al., 2014; Kater et al., 2019). The modest resolution of these structures (6–10 Å) makes it difficult to place the substrate TMs unequivocally, but they appear to occupy positions at the lateral gate, partially in the lateral gate and partially in the phospholipid bilayer, or in the lipid bilayer at the outside of the lateral gate. Thus, the information available from cryo-EM indicates that both the lateral gate and substrate TM are dynamic during the process of membrane integration. Two groups have carried out single-molecule FRET studies to characterize lateral-gate conformations, and these studies suggest a very dynamic picture of the lateral gate (Fessl et al., 2018; Mercier et al., 2021). Specifically, in addition to open and closed conformations, at least two partly open conformations are evident, and both studies confirm that lateral gate opening occurs even in the absence of substrates. Kinetic analysis of conformational changes at the lateral gate indicated that spontaneous lateral gate opening is rapid, occurring multiple times per second (Mercier et al., 2021). This is fast enough to support cotranslational insertion of TMs, each one requiring about 2 s for synthesis by the ribosome. Thus, acceleration of lateral gate opening by the ribosome or TM is not necessary for efficient phospholipid insertion. Rather, stochastic lateral gate opening permits TMs to integrate into the phospholipid bilayer when the nascent chain has reached a sufficient length (Mercier et al., 2021). Thus, the timing of TM integration into the membrane is dictated by the ribosome and the speed at which it translates the mRNA.
An alternative model for TM insertion proposes that TMs do not enter the pore of the translocon (Cymer et al., 2015). In this model, TMs exit the ribosome and adsorb at the membrane interface, parallel to the membrane surface, consistent with the notion that TMs tend to adsorb to the membrane interface (Ulmschneider et al., 2014). Only periplasmic loops enter the translocon pore where they are translocated across the membrane as the TM integrates into the phospholipid bilayer and orients itself perpendicular to the plane of the membrane. This “sliding model” is which TMs slide along the outside of the lateral gate can be distinguished from the “translocon-insertion” model based on the length of nascent chain required for N-out versus N-in insertion. For the translocon-insertion model, N-out TM insertion should occur at shorter nascent chain lengths than N-in, because N-out insertion requires the nascent protein to traverse the membrane only once (Figure 3). For the sliding model, however, both N-out and N-in insertion should occur at similar nascent-chain lengths. Support for the sliding model is found in detailed force-profile analysis (FPA) which indicates that TMs stop generating pulling force when they are about 45 amino acids away from the PTC, regardless of orientation (N-in vs N-out; (Nicolaus et al., 2021)). This nascent chain length would mean interaction with lipids rather than TM insertion in its correct topology. However, other FPA experiments clearly demonstrate a biphasic force profile generated by TM insertion, with the first peak attributed to translocon insertion, and the second (larger) peak to membrane insertion, thus supporting the canonical translocon-insertion model (Ismail et al., 2012; Niesen et al., 2018). As mentioned above, the translocon-insertion model is also supported by real-time measurements of cotranslational insertion, which indicate that insertion monitored via FRET, and protection against protease digestion occur faster for TMs with N-out topology than N-in (Mercier et al., 2020).
It is noteworthy that the two models are not mutually exclusive. A TM could maintain continuous access to the lipid bilayer (Nicolaus et al., 2021) – as suggested by the sliding model – if rapid lateral gate opening allowed the TM to fluctuate between positions in the translocon pore and the phospholipid bilayer, thus satisfying both models. This idea would be consistent with the weak density for nascent-chain TMs near the lateral gate observed in cryo-EM (Kater et al., 2019). With the demonstration of rapid lateral gate opening comes the possibility that TMs may fluctuate between the translocon pore and the phospholipid bilayer during insertion (Mercier et al., 2021). The ability of TMs to rapidly sample multiple positions in the lipid bilayer and the translocon could have a significant impact on the cotranslational folding of polytopic membrane proteins by allowing the TMs to sample different positions in an incomplete helix bundle, before finding the correct position. Understanding potential fluctuations of the TM between the translocon pore and the phospholipid bilayer and impacts on folding will require future single-molecule measurements focusing on TM behavior.
Cotranslational Folding of Membrane Proteins
Insights into how polytopic membrane proteins start to fold became available recently through the FPA technique (Nicolaus et al., 2021). In general, elements of polytopic membrane proteins are inserted or translocated when they are about 45 amino acids away from the PTC (Mercier et al., 2020; Nicolaus et al., 2021). In some cases, however, TM insertion can be delayed (in terms of amino acid length) by positively charged amino acids that are likely retained by interactions with negative charges on the ribosome and/or phospholipids (Nicolaus et al., 2021). An important finding is that mutations at key positions in TMs decrease the propensity of the following TM to insert into the phospholipids, suggesting that TMs are stabilized in the phospholipid bilayer by TM-TM interactions (Cymer and von Heijne, 2013; Nicolaus et al., 2021). Based on the nature of the technique, these pulling forces are detected in partially synthesized proteins and the TM-TM interactions identified are, therefore, formed before the protein can be completely folded. The importance of these interactions is supported by single-molecule atomic-force microscopy (AFM) experiments, where insertion of LacY in membranes containing SecYEG follows a preferred mechanism where a single C-terminial helix inserts first, followed by neighbouring helices (Laskowski et al., 2021). Another single-molecule force spectroscopy study utilized magnetic tweezers to monitor transmembrane protein folding in bicelles, albeit in the absence of translocon (Choi et al., 2019). The results of this work suggest that folding of E. coli GlpG as well as the human ß2-adrenergic receptor proceed from the protein N-terminus to the C-terminus. A variety of pathways may, therefore, be accessible to different transmembrane proteins. The TM-TM interactions can be transient during ongoing translation. By analogy to cotranslational protein folding of cytosolic proteins, nascent TMs may remain in a dynamic state, where TM-TM interactions rearrange rapidly until enough elements have reached the correct location and stable interactions are assembled. Maintaining a nascent membrane protein in a quasi-stable structure would facilitate formation of rather complicated folds where for example, a TM interacts with TMs that do not immediately precede it, or TMs are intertwined rather than parallel. While the compaction of TMs into the their native fold in the membrane remains an open question, this TM-centered view of folding says nothing of the loops, which may remain dynamic to facilitate interactions with ligands and/or other proteins. Conversely, some loops may actually contribute to stabilizing the native folded structure of membrane proteins, perhaps being buried within other loops (Tastan et al., 2009).
While the application of FPA has given us detailed insights into critical events during polytopic membrane protein folding, it does not provide kinetic information about the folding process, as FPA relies on intermediates generated by programmed ribosome stalling. Determining the speed of TM insertion, loop translocation, and TM-TM interactions is important to understand the mechanism of folding in the membrane, and which steps occur co- vs. posttranslationally. This becomes even more important when we consider the influence of ongoing translation, where TMs and loops reach their respective locations only after they have been synthesized and traversed the peptide exit tunnel and (potentially) the translocon. Extrapolating the trend observed for real-time insertion of TM1 would suggest that translation on the ribosome is rate-limiting for all other steps in protein folding. This idea is supported by the timescales of loop motions and larger domain motions which are generally on the microsecond-millisecond timescales (Henzler-Wildman and Kern, 2007), much faster than the relatively low rate of protein synthesis. Thus, the ribosome will dictate when each new TM can join the nascent protein as it folds in the phospholipid bilayer, and folding events that may require significant time, such as formation of knots or interweaving of TMs, may be modulated by encoded regions in the mRNA that are translated slowly.
Another interesting question is where membrane protein folding occurs. Some loops are long enough to permit TMs to move away from the translocon and equilibrate in the lipid bilayer. Other loops, however, are so short that a preceding TM would have to wait at the lateral gate before both TMs can insert together. Cryo-EM structures indicates that a nascent membrane protein could fold in the vicinity of the translocon, perhaps even using TMs of SecY to chaperone folding (Frauenfeld et al., 2011; Bischoff et al., 2014; Park et al., 2014; Kater et al., 2019). Folding in the vicinity of the translocon would benefit proteins with internal soluble domains, where N-terminal TMs would have to wait for later insertion of C-terminal TMs, and the translocon would act as a chaperone to hold early TMs until arrival of late TMs. The TMs that follow the soluble domain may require an additional round of targeting by SRP, which has been observed in some membrane proteins (Schibich et al., 2016).
Membrane Protein Chaperone YidC
Cotranslational folding of membrane proteins at the translocon may be facilitated by the membrane-protein chaperone YidC. YidC is an essential protein in bacteria (Gerdes et al., 2003), with homologues in mitochondria (Oxa1), chloroplasts (Alb3), ER membrane (GET1, EMC3 and TMCO1) and archaea (Ylp1 or DUC160) (Borowska et al., 2015; Anghel et al., 2017; Kuhn and Kiefer, 2017; McDowell et al., 2021). YidC is comprised of 6 TMs and, in E. coli, a large periplasmic domain situated between TMs 1 and 2 (Kumazaki et al., 2014b). The structure of YidC is mostly solved apart from TM1, which is thought to be flexible (Figure 4). A central hydrophilic cavity which is surrounded by TMs 2–6 extends about halfway across the width of the membrane, and opens towards the cytoplasm. YidC is thought to promote TM insertion by facilitating translocation of charged loops, which bind in the hydrophilic groove. After passing a gap between TMs 2 and 4, loops are only required to traverse about half the width of the lipid bilayer (Kumazaki et al., 2014a; Wickles et al., 2014). The insertase function of YidC has been monitored by single-molecule AFM experiments, where insertion of an unfolded membrane protein attached to the AFM stylus was monitored in lipid bilayers containing YidC (Serdiuk et al., 2016; Serdiuk et al., 2017; Serdiuk et al., 2019; Laskowski et al., 2021). In these experiments, YidC alone was sufficient to enable insertion and folding of LacY, although structural elements were inserted in random order (Serdiuk et al., 2019).
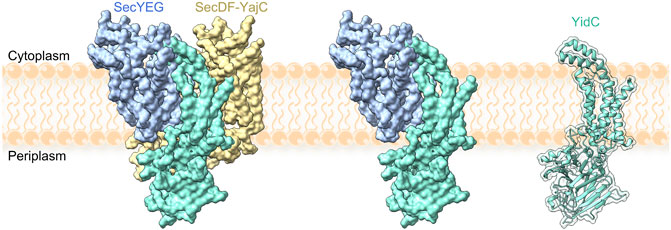
FIGURE 4. YidC in complex with the holotranslocon SecYEG–SecDF–YajC (left), with SecYEG (middle) and alone (right). Models were constructed by alignment of the holotranslocon (PDB: 5MG3 (Botte et al., 2016)) with a recent structure of YidC (PDB: 6AL2 (Tanaka et al., 2018)). TM1 of YidC is missing from all structures.
In the cell, YidC is present in excess relative to SecYEG and can function as part of the holo-translocon containing YidC, SecYEG and SecDF (Schulze et al., 2014), in complex with SecYEG (Scotti et al., 2000; Nouwen and Driessen, 2002; Sachelaru et al., 2013; Sachelaru et al., 2017) or as a stand-alone insertase (Luirink et al., 2001; Samuelson et al., 2001; Serek et al., 2004; Welte et al., 2012) (Figure 4). RNCs are targeted to YidC with the help of SRP, and YidC was reported to bind to FtsY as well as to the exit tunnel of the ribosome (Driessen, 1994; Urbanus et al., 2002; Welte et al., 2012; Li et al., 2014; Geng et al., 2015; Tanaka et al., 2018). Thus, there seems to be a potential for cotranslational membrane protein targeting and insertion by YidC without the need for SecYEG in bacteria, but the details of the pathway remain poorly understood. The presence of the YidC homologue Oxa1 in mitochondria, which lack SecY, provides an obvious precedence for such a model.
The known substrates which require YidC for insertion are listed in Table 1. It is not clear, however, which of these substrates utilize YidC-only, SecYEG–YidC, or holotranslocon pathways, or whether there is any crosstalk between pathways. Only a few substrates have been identified that utilise the YidC-only pathway for membrane insertion (Table 1 and (Samuelson et al., 2000; Chen et al., 2003)). The precise role of YidC is unclear, but it has been shown to be required for the correct folding of LacY (Nagamori et al., 2004; Serdiuk et al., 2017) and stability and complex formation for MalF (Wagner et al., 2008). Since most YidC substrates are components of membrane protein complexes, YidC may act as a chaperone during complex formation in the phospholipid bilayer. So far, only the substrate MalF is known to require YidC for complex formation, so this needs to be explored for the other YidC substrates as well.
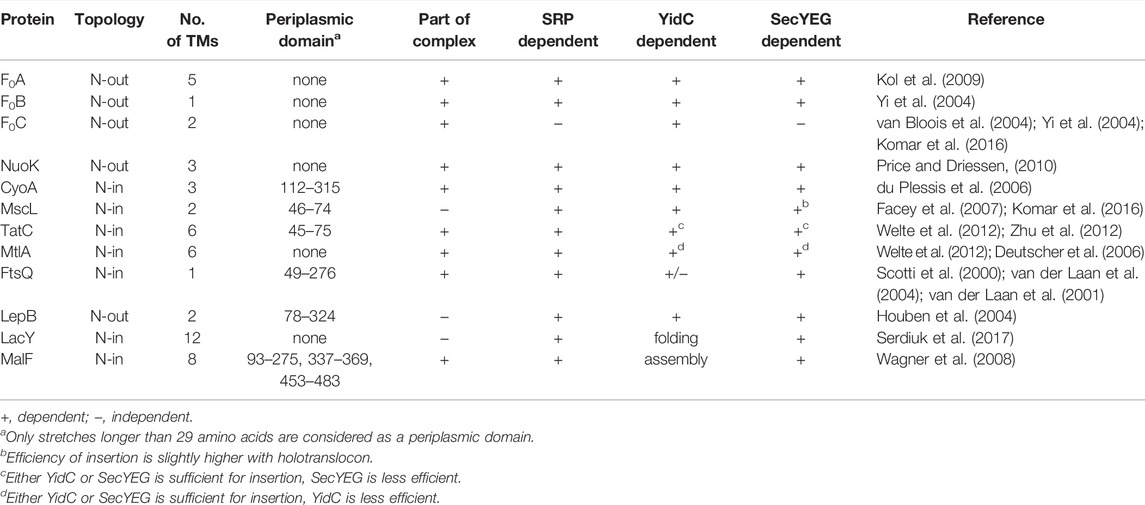
TABLE 1. YidC substrates and their attributes.
When bound to SecYEG, YidC contacts SecY at TMs 1, 2b, 3, 5 and 7, including residues inside the pore and at the lateral gate, through interactions with TM1 and loop P1 of YidC (Sachelaru et al., 2013; Petriman et al., 2018; Jauss et al., 2019). The interaction between TM1 of YidC and the SecY pore is displaced by insertion of a substrate TM, suggesting that YidC recognizes nascent protein substrates within the pore of the translocon (Sachelaru et al., 2013). Single-molecule studies of SecYEG-YidC complexes using FRET and electrophysiology measurements indicate that the complex containing YidC is more closed than SecYEG alone, although rapid fluctuations at the lateral gate are still observed by smFRET, even when YidC is present (Sachelaru et al., 2017; Mercier et al., 2021). During insertion of a membrane protein, however, YidC strongly stabilizes the open conformation of the lateral gate, although it is unclear whether YidC holds the lateral gate open, or interacts with the substrate TM in the open lateral gate (Mercier et al., 2021). In single-molecule AFM studies, insertion and folding of LacY in membranes containing SecYEG and YidC proceeds similarly to membranes containing SecYEG only, suggesting that the role of the translocon dominates over that of YidC (Serdiuk et al., 2019). The current model of YidC-dependent insertion is that TM1 of YidC occupies the channel in SecY, and is displaced by TM insertion. YidC then interacts with the lateral gate of SecY and fetches the nascent TM before it is released into the phospholipid bilayer. Any potential effects of YidC on the kinetics or specificity of membrane protein targeting await further investigations.
Concluding Remarks
The predominant theme emerging from mechanistic studies of the cotranslational biosynthesis of membrane proteins has been that individual steps are inherently fast, and rate-limited by translation on the ribosome. Rapid targeting of RNCs to the membrane via the SRP pathway permits bypassing of N-terminal processing and ensures that ribosomes reach the membrane while nascent chains are still short. Subsequent insertion of TMs into the translocon and then membrane are also rapid, occurring when the nascent chain is sufficiently long that TMs can enter. This theme reinforces the concept of vectorial folding, as TMs become available to fold in the lipid bilayer in the order in which they are synthesized on the ribosome. Despite the recent progress in understanding the mechanism of membrane protein biogenesis, there are still many outstanding questions.
For instance, a clear understanding of how cytosolic loops are handled at the ribosome-translocon complex is lacking. On the one hand, there may be enough space with or without minor rearrangement to permit cytosolic loops to exit the ribosome and enter the cytosol directly. Exit of these loops may, however, cause the ribosome to leave its position on the translocon and remain tethered to the membrane by nascent protein. Such complexes may require SRP for retargeting and while there is evidence for retargeting based on selective ribosome profiling (Schibich et al., 2016), this significantly complicates the picture of membrane protein biogenesis.
During the process of TM insertion, there is evidence from several groups to suggest that entry of some TMs is delayed, and this seems to involve positively charged amino acids (Mercier et al., 2020; Nicolaus et al., 2021). Delayed entry may permit TMs to enter the lipid bilayer in pairs, rather than one at a time but it is unclear whether this is critical for efficient folding. This is difficult to disentangle, however, since positive charges are important for maintaining a correct topology and likely slow down protein synthesis.
In addition, a great deal of work has characterized ribosome pausing during mRNA translation which influences protein folding in general (reviewed in (Samatova et al., 2020)). Translational pausing can dominate the speed at which TMs enter the membrane, and could conceivably be programmed in mRNA to permit folding of complex structures before additional TMs arrive. On the other hand, if additional TMs do not interfere with initial folding steps, then pauses may simply result from the lack of any pressure to optimize translation speed at particular locations in the mRNA. Here we need a better handle on what causes translational pauses, and how likely kinetic traps are to hinder membrane protein folding.
Finally, an important outstanding question relates to which folding events and rearrangements occur inside the phospholipid bilayer. It is possible that partially folded membrane proteins remain dynamic in the lipid bilayer to permit reorganization during continued synthesis until all TMs are present. Another interesting question is at what stage membrane protein complexes start to form. Additional real-time measurements within phospholipid bilayers will be necessary to address these questions.
Author Contributions
All authors contributed to writing the manuscript.
Funding
This work was supported by the German Science Foundation (Deutsche Forschungsgemeinschaft, DFG, in the framework of SFB1190 to MVR), the European Research Council (ERC) Advanced Investigator Grant RIBOFOLD to MVR (proposal number n° 787926), through Germany’s Excellence Strategy—EXC 2067/1- 390729940 (to MVR), and by the Max Planck Society.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adams, H., Scotti, P. A., De Cock, H., Luirink, J., and Tommassen, J. (2002). The Presence of a helix Breaker in the Hydrophobic Core of Signal Sequences of Secretory Proteins Prevents Recognition by the Signal-Recognition Particle inEscherichia Coli. Eur. J. Biochem. 269 (22), 5564–5571. doi:10.1046/j.1432-1033.2002.03262.x
Agirrezabala, X., Samatova, E., Macher, M., Liutkute, M., Maiti, M., Gil‐Carton, D., et al. (2022). A Switch from α‐Helical to β‐Strand Conformation during Co‐translational Protein Folding. EMBO J. 41, e109175. doi:10.15252/embj.2021109175
Akbar, S., Bhakta, S., and Sengupta, J. (2021). Structural Insights into the Interplay of Protein Biogenesis Factors with the 70S Ribosome. Structure 29 (7), 755–767.e4. doi:10.1016/j.str.2021.03.005
Anghel, S. A., McGilvray, P. T., Hegde, R. S., and Keenan, R. J. (2017). Identification of Oxa1 Homologs Operating in the Eukaryotic Endoplasmic Reticulum. Cel Rep. 21 (13), 3708–3716. doi:10.1016/j.celrep.2017.12.006
Ariosa, A., Lee, J. H., Wang, S., Saraogi, I., and Shan, S.-o. (2015). Regulation by a Chaperone Improves Substrate Selectivity during Cotranslational Protein Targeting. Proc. Natl. Acad. Sci. U.S.A. 112 (25), E3169–E3178. doi:10.1073/pnas.1422594112
Bhakta, S., Akbar, S., and Sengupta, J. (2019). Cryo-EM Structures Reveal Relocalization of MetAP in the Presence of Other Protein Biogenesis Factors at the Ribosomal Tunnel Exit. J. Mol. Biol. 431 (7), 1426–1439. doi:10.1016/j.jmb.2019.02.002
Bienvenut, W. V., Giglione, C., and Meinnel, T. (2015). Proteome-wide Analysis of the Amino Terminal Status ofEscherichia Coliproteins at the Steady-State and upon Deformylation Inhibition. Proteomics 15 (14), 2503–2518. doi:10.1002/pmic.201500027
Bingel-Erlenmeyer, R., Kohler, R., Kramer, G., Sandikci, A., Antolić, S., Maier, T., et al. (2008). A Peptide Deformylase-Ribosome Complex Reveals Mechanism of Nascent Chain Processing. Nature 452 (7183), 108–111. doi:10.1038/nature06683
Bischoff, L., Wickles, S., Berninghausen, O., van der Sluis, E. O., and Beckmann, R. (2014). Visualization of a Polytopic Membrane Protein during SecY-Mediated Membrane Insertion. Nat. Commun. 5, 4103. doi:10.1038/ncomms5103
Bogdanov, M., Xie, J., and Dowhan, W. (2009). Lipid-protein Interactions Drive Membrane Protein Topogenesis in Accordance with the Positive inside Rule. J. Biol. Chem. 284 (15), 9637–9641. doi:10.1074/jbc.R800081200
Bogeholz, L. A. K., Mercier, E., Wintermeyer, W., and Rodnina, M. V. (2021). Kinetic Control of Nascent Protein Biogenesis by Peptide Deformylase. Sci. Rep. 11 (1), 24457. doi:10.1038/s41598-021-03969-3
Bornemann, T., Jöckel, J., Rodnina, M. V., and Wintermeyer, W. (2008). Signal Sequence-independent Membrane Targeting of Ribosomes Containing Short Nascent Peptides within the Exit Tunnel. Nat. Struct. Mol. Biol. 15 (5), 494–499. doi:10.1038/nsmb.1402
Bornemann, T., Holtkamp, W., and Wintermeyer, W. (2014). Interplay between Trigger Factor and Other Protein Biogenesis Factors on the Ribosome. Nat. Commun. 5, 4180. doi:10.1038/ncomms5180
Borowska, M. T., Dominik, P. K., Anghel, S. A., Kossiakoff, A. A., and Keenan, R. J. (2015). A YidC-like Protein in the Archaeal Plasma Membrane. Structure 23 (9), 1715–1724. doi:10.1016/j.str.2015.06.025
Botte, M., Zaccai, N. R., Nijeholt, J. L. à., Martin, R., Knoops, K., Papai, G., et al. (2016). A central Cavity within the Holo-Translocon Suggests a Mechanism for Membrane Protein Insertion. Sci. Rep. 6, 38399. doi:10.1038/srep38399
Buskiewicz, I., Deuerling, E., Gu, S.-Q., Jöckel, J., Rodnina, M. V., Bukau, B., et al. (2004). Trigger Factor Binds to Ribosome-Signal-Recognition Particle (SRP) Complexes and Is Excluded by Binding of the SRP Receptor. Proc. Natl. Acad. Sci. U.S.A. 101 (21), 7902–7906. doi:10.1073/pnas.0402231101
Bustamante, C., Alexander, L., Maciuba, K., and Kaiser, C. M. (2020). Single-molecule Studies of Protein Folding with Optical Tweezers. Annu. Rev. Biochem. 89, 443–470. doi:10.1146/annurev-biochem-013118-111442
Cao, G., Kuhn, A., and Dalbey, R. E. (1995). The Translocation of Negatively Charged Residues across the Membrane Is Driven by the Electrochemical Potential: Evidence for an Electrophoresis-like Membrane Transfer Mechanism. EMBO J. 14 (5), 866–875. doi:10.1002/j.1460-2075.1995.tb07068.x
Cassaignau, A. M. E., Cabrita, L. D., and Christodoulou, J. (2020). How Does the Ribosome Fold the Proteome? Annu. Rev. Biochem. 89, 389–415. doi:10.1146/annurev-biochem-062917-012226
Celebi, N., Dalbey, R. E., and Yuan, J. (2008). Mechanism and Hydrophobic Forces Driving Membrane Protein Insertion of Subunit II of Cytochrome Bo3 Oxidase. J. Mol. Biol. 375 (5), 1282–1292. doi:10.1016/j.jmb.2007.11.054
Chan, M. K., Gong, W., Rajagopalan, P. T. R., Hao, B., Tsai, C. M., and Pei, D. (1997). Crystal Structure of the Escherichia coli Peptide Deformylase,. Biochemistry 36 (45), 13904–13909. doi:10.1021/bi9711543
Chen, M., Xie, K., Nouwen, N., Driessen, A. J. M., and Dalbey, R. E. (2003). Conditional Lethal Mutations Separate the M13 Procoat and Pf3 Coat Functions of YidC: Different YIDC Structural Requirements for Membrane Protein Insertion. J. Biol. Chem. 278 (26), 23295–23300. doi:10.1074/jbc.M301008200
Choi, H.-K., Min, D., Kang, H., Shon, M. J., Rah, S.-H., Kim, H. C., et al. (2019). Watching Helical Membrane Proteins Fold Reveals a Common N-To-C-Terminal Folding Pathway. Science 366 (6469), 1150–1156. doi:10.1126/science.aaw8208
Cristóbal, S., de Gier, J. W., Nielsen, H., and von Heijne, G. (1999). Competition between Sec- and TAT-dependent Protein Translocation in Escherichia coli. EMBO J. 18 (11), 2982–2990. doi:10.1093/emboj/18.11.2982
Cymer, F., and von Heijne, G. (2013). Cotranslational Folding of Membrane Proteins Probed by Arrest-Peptide-Mediated Force Measurements. Proc. Natl. Acad. Sci. U.S.A. 110 (36), 14640–14645. doi:10.1073/pnas.1306787110
Cymer, F., von Heijne, G., and White, S. H. (2015). Mechanisms of Integral Membrane Protein Insertion and Folding. J. Mol. Biol. 427 (5), 999–1022. doi:10.1016/j.jmb.2014.09.014
Dao Duc, K., Batra, S. S., Bhattacharya, N., Cate, J. H. D., and Song, Y. S. (2019). Differences in the Path to Exit the Ribosome across the Three Domains of Life. Nucleic Acids Res. 47 (8), 4198–4210. doi:10.1093/nar/gkz106
de Leeuw, E., te Kaat, K., Moser, C., Menestrina, G., Demel, R., de Kruijff, B., et al. (2000). Anionic Phospholipids Are Involved in Membrane Association of FtsY and Stimulate its GTPase Activity. Embo j 19 (4), 531–541. doi:10.1093/emboj/19.4.531
Deckert, A., Cassaignau, A. M. E., Wang, X., Włodarski, T., Chan, S. H. S., Waudby, C. A., et al. (2021). Common Sequence Motifs of Nascent Chains Engage the Ribosome Surface and Trigger Factor. Proc. Natl. Acad. Sci. U.S.A. 118 (52), e210301511. doi:10.1073/pnas.2103015118
Denks, K., Vogt, A., Sachelaru, I., Petriman, N.-A., Kudva, R., and Koch, H.-G. (2014). The Sec Translocon Mediated Protein Transport in Prokaryotes and Eukaryotes. Mol. Membr. Biol. 31 (2-3), 58–84. doi:10.3109/09687688.2014.907455
Denks, K., Sliwinski, N., Erichsen, V., Borodkina, B., Origi, A., and Koch, H.-G. (2017). The Signal Recognition Particle Contacts uL23 and Scans Substrate Translation inside the Ribosomal Tunnel. Nat. Microbiol. 2, 16265. doi:10.1038/nmicrobiol.2016.265
Deutscher, J., Francke, C., and Postma, P. W. (2006). How Phosphotransferase System-Related Protein Phosphorylation Regulates Carbohydrate Metabolism in Bacteria. Microbiol. Mol. Biol. Rev. 70 (4), 939–1031. doi:10.1128/mmbr.00024-06
Devaraneni, P. K., Conti, B., Matsumura, Y., Yang, Z., Johnson, A. E., and Skach, W. R. (2011). Stepwise Insertion and Inversion of a Type II Signal Anchor Sequence in the Ribosome-Sec61 Translocon Complex. Cell 146 (1), 134–147. doi:10.1016/j.cell.2011.06.004
Dowhan, W., Vitrac, H., and Bogdanov, M. (2019). Lipid-assisted Membrane Protein Folding and Topogenesis. Protein J. 38 (3), 274–288. doi:10.1007/s10930-019-09826-7
Draycheva, A., Bornemann, T., Ryazanov, S., Lakomek, N. A., and Wintermeyer, W. (2016). The Bacterial SRP Receptor, FtsY, Is Activated on Binding to the Translocon. Mol. Microbiol. 102 (1), 152–167. doi:10.1111/mmi.13452
Draycheva, A., Lee, S., and Wintermeyer, W. (2018). Cotranslational Protein Targeting to the Membrane: Nascent-Chain Transfer in a Quaternary Complex Formed at the Translocon. Sci. Rep. 8 (1), 9922. doi:10.1038/s41598-018-28262-8
Driessen, A. J. M. (1994). How Proteins Cross the Bacterial Cytoplasmic Membrane. J. Membarin Biol. 142 (2), 145–159. doi:10.1007/bf00234937
du Plessis, D. J. F., Nouwen, N., and Driessen, A. J. M. (2006). Subunit a of Cytochrome O Oxidase Requires Both YidC and SecYEG for Membrane Insertion. J. Biol. Chem. 281 (18), 12248–12252. doi:10.1074/jbc.M600048200
Egea, P. F., and Stroud, R. M. (2010). Lateral Opening of a Translocon upon Entry of Protein Suggests the Mechanism of Insertion into Membranes. Proc. Natl. Acad. Sci. U.S.A. 107 (40), 17182–17187. doi:10.1073/pnas.1012556107
Eisner, G., Moser, M., Schäfer, U., Beck, K., and Müller, M. (2006). Alternate Recruitment of Signal Recognition Particle and Trigger Factor to the Signal Sequence of a Growing Nascent Polypeptide. J. Biol. Chem. 281 (11), 7172–7179. doi:10.1074/jbc.M511388200
Facey, S. J., Neugebauer, S. A., Krauss, S., and Kuhn, A. (2007). The Mechanosensitive Channel Protein MscL Is Targeted by the SRP to the Novel YidC Membrane Insertion Pathway of Escherichia coli. J. Mol. Biol. 365 (4), 995–1004. doi:10.1016/j.jmb.2006.10.083
Farías-Rico, J. A., Ruud Selin, F., Myronidi, I., Frühauf, M., and von Heijne, G. (2018). Effects of Protein Size, Thermodynamic Stability, and Net Charge on Cotranslational Folding on the Ribosome. Proc. Natl. Acad. Sci. U.S.A. 115 (40), E9280–E9287. doi:10.1073/pnas.1812756115
Ferbitz, L., Maier, T., Patzelt, H., Bukau, B., Deuerling, E., and Ban, N. (2004). Trigger Factor in Complex with the Ribosome Forms a Molecular Cradle for Nascent Proteins. Nature 431 (7008), 590–596. doi:10.1038/nature02899
Fessl, T., Watkins, D., Oatley, P., Allen, W. J., Corey, R. A., Horne, J., et al. (2018). Dynamic Action of the Sec Machinery during Initiation, Protein Translocation and Termination. Elife 7, e35112. doi:10.7554/eLife.35112
Fluman, N., Tobiasson, V., and von Heijne, G. (2017). Stable Membrane Orientations of Small Dual-Topology Membrane Proteins. Proc. Natl. Acad. Sci. U.S.A. 114 (30), 7987–7992. doi:10.1073/pnas.1706905114
Frain, K. M., Robinson, C., and van Dijl, J. M. (2019). Transport of Folded Proteins by the Tat System. Protein J. 38 (4), 377–388. doi:10.1007/s10930-019-09859-y
Frauenfeld, J., Gumbart, J., Sluis, E. O. v. d., Funes, S., Gartmann, M., Beatrix, B., et al. (2011). Cryo-EM Structure of the Ribosome-SecYE Complex in the Membrane Environment. Nat. Struct. Mol. Biol. 18 (5), 614–621. doi:10.1038/nsmb.2026
Frottin, F., Martinez, A., Peynot, P., Mitra, S., Holz, R. C., Giglione, C., et al. (2006). The Proteomics of N-Terminal Methionine Cleavage. Mol. Cell Proteomics 5 (12), 2336–2349. doi:10.1074/mcp.M600225-MCP200
Ge, Y., Draycheva, A., Bornemann, T., Rodnina, M. V., and Wintermeyer, W. (2014). Lateral Opening of the Bacterial Translocon on Ribosome Binding and Signal Peptide Insertion. Nat. Commun. 5, 5263. doi:10.1038/ncomms6263
Geng, Y., Kedrov, A., Caumanns, J. J., Crevenna, A. H., Lamb, D. C., Beckmann, R., et al. (2015). Role of the Cytosolic Loop C2 and the C Terminus of YidC in Ribosome Binding and Insertion Activity. J. Biol. Chem. 290 (28), 17250–17261. doi:10.1074/jbc.M115.650309
Gerdes, S. Y., Scholle, M. D., Campbell, J. W., Balázsi, G., Ravasz, E., Daugherty, M. D., et al. (2003). Experimental Determination and System Level Analysis of Essential Genes in Escherichia coli MG1655. J. Bacteriol. 185 (19), 5673–5684. doi:10.1128/jb.185.19.5673-5684.2003
Giglione, C., Fieulaine, S., and Meinnel, T. (2015). N-terminal Protein Modifications: Bringing Back into Play the Ribosome. Biochimie 114, 134–146. doi:10.1016/j.biochi.2014.11.008
Goder, V., and Spiess, M. (2003). Molecular Mechanism of Signal Sequence Orientation in the Endoplasmic Reticulum. EMBO J. 22 (14), 3645–3653. doi:10.1093/emboj/cdg361
Hainzl, T., Huang, S., Meriläinen, G., Brännström, K., and Sauer-Eriksson, A. E. (2011). Structural Basis of Signal-Sequence Recognition by the Signal Recognition Particle. Nat. Struct. Mol. Biol. 18 (3), 389–391. doi:10.1038/nsmb.1994
Halic, M., Blau, M., Becker, T., Mielke, T., Pool, M. R., Wild, K., et al. (2006). Following the Signal Sequence from Ribosomal Tunnel Exit to Signal Recognition Particle. Nature 444 (7118), 507–511. doi:10.1038/nature05326
Henzler-Wildman, K., and Kern, D. (2007). Dynamic Personalities of Proteins. Nature 450 (7172), 964–972. doi:10.1038/nature06522
Higy, M., Junne, T., and Spiess, M. (2004). Topogenesis of Membrane Proteins at the Endoplasmic Reticulum. Biochemistry 43 (40), 12716–12722. doi:10.1021/bi048368m
Hirel, P. H., Schmitter, M. J., Dessen, P., Fayat, G., and Blanquet, S. (1989). Extent of N-Terminal Methionine Excision from Escherichia coli Proteins Is Governed by the Side-Chain Length of the Penultimate Amino Acid. Proc. Natl. Acad. Sci. U.S.A. 86 (21), 8247–8251. doi:10.1073/pnas.86.21.8247
Holtkamp, W., Lee, S., Bornemann, T., Senyushkina, T., Rodnina, M. V., and Wintermeyer, W. (2012). Dynamic Switch of the Signal Recognition Particle from Scanning to Targeting. Nat. Struct. Mol. Biol. 19 (12), 1332–1337. doi:10.1038/nsmb.2421
Holtkamp, W., Kokic, G., Jäger, M., Mittelstaet, J., Komar, A. A., and Rodnina, M. V. (2015). Cotranslational Protein Folding on the Ribosome Monitored in Real Time. Science 350 (6564), 1104–1107. doi:10.1126/science.aad0344
Houben, E. N. G., ten Hagen‐Jongman, C. M., Brunner, J., Oudega, B., and Luirink, J. (2004). The Two Membrane Segments of Leader Peptidase Partition One by One into the Lipid Bilayer via a Sec/YidC Interface. EMBO Rep. 5 (10), 970–975. doi:10.1038/sj.embor.7400261
Ismail, N., Hedman, R., Schiller, N., and von Heijne, G. (2012). A Biphasic Pulling Force Acts on Transmembrane Helices during Translocon-Mediated Membrane Integration. Nat. Struct. Mol. Biol. 19 (10), 1018–1022. doi:10.1038/nsmb.2376
Jauss, B., Petriman, N.-A., Drepper, F., Franz, L., Sachelaru, I., Welte, T., et al. (2019). Noncompetitive Binding of PpiD and YidC to the SecYEG Translocon Expands the Global View on the SecYEG Interactome in Escherichia coli. J. Biol. Chem. 294 (50), 19167–19183. doi:10.1074/jbc.RA119.010686
Jomaa, A., Boehringer, D., Leibundgut, M., and Ban, N. (2016). Structures of the E. coli Translating Ribosome with SRP and its Receptor and with the Translocon. Nat. Commun. 7, 10471. doi:10.1038/ncomms10471
Jomaa, A., Fu, Y.-H. H., Boehringer, D., Leibundgut, M., Shan, S.-o., and Ban, N. (2017). Structure of the Quaternary Complex between SRP, SR, and Translocon Bound to the Translating Ribosome. Nat. Commun. 8 (1), 15470. doi:10.1038/ncomms15470
Junne, T., Schwede, T., Goder, V., and Spiess, M. (2007). Mutations in the Sec61p Channel Affecting Signal Sequence Recognition and Membrane Protein Topology. J. Biol. Chem. 282 (45), 33201–33209. doi:10.1074/jbc.M707219200
Kaiser, C. M., Goldman, D. H., Chodera, J. D., Tinoco, I., and Bustamante, C. (2011). The Ribosome Modulates Nascent Protein Folding. Science 334 (6063), 1723–1727. doi:10.1126/science.1209740
Kater, L., Frieg, B., Berninghausen, O., Gohlke, H., Beckmann, R., and Kedrov, A. (2019). Partially Inserted Nascent Chain Unzips the Lateral Gate of the Sec Translocon. EMBO Rep. 20, e48191. doi:10.15252/embr.201948191
Kol, S., Majczak, W., Heerlien, R., van der Berg, J. P., Nouwen, N., and Driessen, A. J. M. (2009). Subunit a of the F(1)F(0) ATP Synthase Requires YidC and SecYEG for Membrane Insertion. J. Mol. Biol. 390 (5), 893–901. doi:10.1016/j.jmb.2009.05.074
Komar, J., Alvira, S., Schulze, R. J., Martin, R., Lycklama, A. N. J. A., Lee, S. C., et al. (2016). Membrane Protein Insertion and Assembly by the Bacterial Holo-Translocon SecYEG-SecDF-YajC-YidC. Biochem. J. 473 (19), 3341–3354. doi:10.1042/bcj20160545
Koubek, J., Schmitt, J., Galmozzi, C. V., and Kramer, G. (2021). Mechanisms of Cotranslational Protein Maturation in Bacteria. Front. Mol. Biosci. 8, 689755. doi:10.3389/fmolb.2021.689755
Kramer, G., Rauch, T., Rist, W., Vorderwülbecke, S., Patzelt, H., Schulze-Specking, A., et al. (2002). L23 Protein Functions as a Chaperone Docking Site on the Ribosome. Nature 419 (6903), 171–174. doi:10.1038/nature01047
Kramer, G., Boehringer, D., Ban, N., and Bukau, B. (2009). The Ribosome as a Platform for Co-translational Processing, Folding and Targeting of Newly Synthesized Proteins. Nat. Struct. Mol. Biol. 16 (6), 589–597. doi:10.1038/nsmb.1614
Kristensen, O., and Gajhede, M. (2003). Chaperone Binding at the Ribosomal Exit Tunnel. Structure 11 (12), 1547–1556. doi:10.1016/j.str.2003.11.003
Kudva, R., Denks, K., Kuhn, P., Vogt, A., Müller, M., and Koch, H.-G. (2013). Protein Translocation across the Inner Membrane of Gram-Negative Bacteria: the Sec and Tat Dependent Protein Transport Pathways. Res. Microbiol. 164 (6), 505–534. doi:10.1016/j.resmic.2013.03.016
Kuhn, A., and Kiefer, D. (2017). Membrane Protein Insertase YidC in Bacteria and Archaea. Mol. Microbiol. 103 (4), 590–594. doi:10.1111/mmi.13586
Kuhn, A., Koch, H.-G., and Dalbey, R. E. (2017). Targeting and Insertion of Membrane Proteins. EcoSal Plus 7 (2), 1–27. doi:10.1128/ecosalplus.ESP-0012-2016
Kumazaki, K., Chiba, S., Takemoto, M., Furukawa, A., Nishiyama, K.-i., Sugano, Y., et al. (2014a). Structural Basis of Sec-independent Membrane Protein Insertion by YidC. Nature 509 (7501), 516–520. doi:10.1038/nature13167
Kumazaki, K., Kishimoto, T., Furukawa, A., Mori, H., Tanaka, Y., Dohmae, N., et al. (2014b). Crystal Structure of Escherichia coli YidC, a Membrane Protein Chaperone and Insertase. Sci. Rep. 4 (1), 7299. doi:10.1038/srep07299
Lakomek, N.-A., Draycheva, A., Bornemann, T., and Wintermeyer, W. (2016). Electrostatics and Intrinsic Disorder Drive Translocon Binding of the SRP Receptor FtsY. Angew. Chem. Int. Ed. 55, 9544–9547. doi:10.1002/anie.201602905
Lam, V. Q., Akopian, D., Rome, M., Henningsen, D., and Shan, S.-o. (2010). Lipid Activation of the Signal Recognition Particle Receptor Provides Spatial Coordination of Protein Targeting. J. Cel Biol 190 (4), 623–635. doi:10.1083/jcb.201004129
Laskowski, P. R., Pluhackova, K., Haase, M., Lang, B. M., Nagler, G., Kuhn, A., et al. (2021). Monitoring the Binding and Insertion of a Single Transmembrane Protein by an Insertase. Nat. Commun. 12 (1), 7082. doi:10.1038/s41467-021-27315-3
Lee, H. C., and Bernstein, H. D. (2001). The Targeting Pathway of Escherichia coli Presecretory and Integral Membrane Proteins Is Specified by the Hydrophobicity of the Targeting Signal. Proc. Natl. Acad. Sci. U.S.A. 98 (6), 3471–3476. doi:10.1073/pnas.051484198
Li, Z., Boyd, D., Reindl, M., and Goldberg, M. B. (2014). Identification of YidC Residues that Define Interactions with the Sec Apparatus. J. Bacteriol. 196 (2), 367–377. doi:10.1128/jb.01095-13
Lill, R., Crooke, E., Guthrie, B., and Wickner, W. (1988). The "trigger Factor Cycle" Includes Ribosomes, Presecretory Proteins, and the Plasma Membrane. Cell 54 (7), 1013–1018. doi:10.1016/0092-8674(88)90116-x
Liutkute, M., Maiti, M., Samatova, E., Enderlein, J., and Rodnina, M. V. (2020a). Gradual Compaction of the Nascent Peptide during Cotranslational Folding on the Ribosome. Elife 9, e60895. doi:10.7554/eLife.60895
Liutkute, M., Samatova, E., and Rodnina, M. V. (2020b). Cotranslational Folding of Proteins on the Ribosome. Biomolecules 10 (1), 97. doi:10.3390/biom10010097
Lu, J., and Deutsch, C. (2005). Folding Zones inside the Ribosomal Exit Tunnel. Nat. Struct. Mol. Biol. 12 (12), 1123–1129. doi:10.1038/nsmb1021
Lu, Y., Turnbull, I. R., Bragin, A., Carveth, K., Verkman, A. S., and Skach, W. R. (2000). Reorientation of Aquaporin-1 Topology during Maturation in the Endoplasmic Reticulum. MBoC 11 (9), 2973–2985. doi:10.1091/mbc.11.9.2973
Luirink, J., Samuelsson, T., and de Gier, J.-W. (2001). YidC/Oxa1p/Alb3: Evolutionarily Conserved Mediators of Membrane Protein Assembly. FEBS Lett. 501 (1), 1–5. doi:10.1016/s0014-5793(01)02616-3
Marino, J., von Heijne, G., and Beckmann, R. (2016). Small Protein Domains Fold inside the Ribosome Exit Tunnel. FEBS Lett. 590 (5), 655–660. doi:10.1002/1873-3468.12098
Mazel, D., Pochet, S., and Marlière, P. (1994). Genetic Characterization of Polypeptide Deformylase, a Distinctive Enzyme of Eubacterial Translation. EMBO J. 13 (4), 914–923. doi:10.1002/j.1460-2075.1994.tb06335.x
McDowell, M. A., Heimes, M., and Sinning, I. (2021). Structural and Molecular Mechanisms for Membrane Protein Biogenesis by the Oxa1 Superfamily. Nat. Struct. Mol. Biol. 28 (3), 234–239. doi:10.1038/s41594-021-00567-9
Mercier, E., Holtkamp, W., Rodnina, M. V., and Wintermeyer, W. (2017). Signal Recognition Particle Binds to Translating Ribosomes before Emergence of a Signal Anchor Sequence. Nucl. Acids Res. 45 (20), 11858–11866. doi:10.1093/nar/gkx888
Mercier, E., Wintermeyer, W., and Rodnina, M. V. (2020). Co‐translational Insertion and Topogenesis of Bacterial Membrane Proteins Monitored in Real Time. EMBO J. 39 (15), e104054. doi:10.15252/embj.2019104054
Mercier, E., Wang, X., Maiti, M., Wintermeyer, W., and Rodnina, M. V. (2021). Lateral Gate Dynamics of the Bacterial Translocon during Cotranslational Membrane Protein Insertion. Proc. Natl. Acad. Sci. U.S.A. 118 (26), e2100474118. doi:10.1073/pnas.2100474118
Merz, F., Hoffmann, A., Rutkowska, A., Zachmann-Brand, B., Bukau, B., and Deuerling, E. (2006). The C-Terminal Domain ofEscherichia coliTrigger Factor Represents the Central Module of its Chaperone Activity. J. Biol. Chem. 281 (42), 31963–31971. doi:10.1074/jbc.M605164200
Merz, F., Boehringer, D., Schaffitzel, C., Preissler, S., Hoffmann, A., Maier, T., et al. (2008). Molecular Mechanism and Structure of Trigger Factor Bound to the Translating Ribosome. EMBO J. 27 (11), 1622–1632. doi:10.1038/emboj.2008.89
Nagamori, S., Smirnova, I. N., and Kaback, H. R. (2004). Role of YidC in Folding of Polytopic Membrane Proteins. J. Cel Biol 165 (1), 53–62. doi:10.1083/jcb.200402067
Nicolaus, F., Metola, A., Mermans, D., Liljenström, A., Krč, A., Abdullahi, S. M., et al. (2021). Residue-by-residue Analysis of Cotranslational Membrane Protein Integration In Vivo. Elife 10, e64302. doi:10.7554/eLife.64302
Niesen, M. J. M., Müller-Lucks, A., Hedman, R., von Heijne, G., and Miller, T. F. (2018). Forces on Nascent Polypeptides during Membrane Insertion and Translocation via the Sec Translocon. Biophysical J. 115 (10), 1885–1894. doi:10.1016/j.bpj.2018.10.002
Nilsson, O. B., Hedman, R., Marino, J., Wickles, S., Bischoff, L., Johansson, M., et al. (2015). Cotranslational Protein Folding inside the Ribosome Exit Tunnel. Cel Rep. 12 (10), 1533–1540. doi:10.1016/j.celrep.2015.07.065
Noriega, T. R., Chen, J., Walter, P., and Puglisi, J. D. (2014). Real-time Observation of Signal Recognition Particle Binding to Actively Translating Ribosomes. Elife 3, e04418. doi:10.7554/eLife.04418
Nouwen, N., and Driessen, A. J. M. (2002). SecDFyajC Forms a Heterotetrameric Complex with YidC. Mol. Microbiol. 44 (5), 1397–1405. doi:10.1046/j.1365-2958.2002.02972.x
Oh, E., Becker, A. H., Sandikci, A., Huber, D., Chaba, R., Gloge, F., et al. (2011). Selective Ribosome Profiling Reveals the Cotranslational Chaperone Action of Trigger Factor In Vivo. Cell 147 (6), 1295–1308. doi:10.1016/j.cell.2011.10.044
O'Shea, E. K., Klemm, J. D., Kim, P. S., and Alber, T. (1991). X-ray Structure of the GCN4 Leucine Zipper, a Two-Stranded, Parallel Coiled Coil. Science 254 (5031), 539–544. doi:10.1126/science.1948029
Palmer, T., and Stansfeld, P. J. (2020). Targeting of Proteins to the Twin‐arginine Translocation Pathway. Mol. Microbiol. 113 (5), 861–871. doi:10.1111/mmi.14461
Park, E., Ménétret, J.-F., Gumbart, J. C., Ludtke, S. J., Li, W., Whynot, A., et al. (2014). Structure of the SecY Channel during Initiation of Protein Translocation. Nature 506 (7486), 102–106. doi:10.1038/nature12720
Peterson, J. H., Woolhead, C. A., and Bernstein, H. D. (2010). The Conformation of a Nascent Polypeptide inside the Ribosome Tunnel Affects Protein Targeting and Protein Folding. Mol. Microbiol. 78 (1), 203–217. doi:10.1111/j.1365-2958.2010.07325.x
Petriman, N.-A., Jauß, B., Hufnagel, A., Franz, L., Sachelaru, I., Drepper, F., et al. (2018). The Interaction Network of the YidC Insertase with the SecYEG Translocon, SRP and the SRP Receptor FtsY. Sci. Rep. 8 (1), 578. doi:10.1038/s41598-017-19019-w
Pradel, N., Ye, C., and Wu, L.-F. (2004). A Cleavable Signal Peptide Is Required for the Full Function of the Polytopic Inner Membrane Protein FliP of Escherichia coli. Biochem. Biophys. Res. Commun. 319 (4), 1276–1280. doi:10.1016/j.bbrc.2004.05.123
Price, C. E., and Driessen, A. J. M. (2010). Conserved Negative Charges in the Transmembrane Segments of Subunit K of the NADH:ubiquinone Oxidoreductase Determine its Dependence on YidC for Membrane Insertion. J. Biol. Chem. 285 (6), 3575–3581. doi:10.1074/jbc.M109.051128
Raine, A., Lovmar, M., Wikberg, J., and Ehrenberg, M. (2006). Trigger Factor Binding to Ribosomes with Nascent Peptide Chains of Varying Lengths and Sequences. J. Biol. Chem. 281 (38), 28033–28038. doi:10.1074/jbc.M605753200
Rajagopalan, P. T. R., Yu, X. C., and Pei, D. (1997). Peptide Deformylase: A New Type of Mononuclear Iron Protein. J. Am. Chem. Soc. 119 (50), 12418–12419. doi:10.1021/ja9734096
Ranjan, A., Mercier, E., Bhatt, A., and Wintermeyer, W. (2017). Signal Recognition Particle Prevents N-Terminal Processing of Bacterial Membrane Proteins. Nat. Commun. 8, 15562. doi:10.1038/ncomms15562
Robinson, P. J., Findlay, J. E., and Woolhead, C. A. (2012). Compaction of a Prokaryotic Signal-Anchor Transmembrane Domain Begins within the Ribosome Tunnel and Is Stabilized by SRP during Targeting. J. Mol. Biol. 423 (4), 600–612. doi:10.1016/j.jmb.2012.07.023
Rudorf, S., and Lipowsky, R. (2015). Protein Synthesis in E. coli: Dependence of Codon-specific Elongation on tRNA Concentration and Codon Usage. PLoS One 10 (8), e0134994. doi:10.1371/journal.pone.0134994
Sachelaru, I., Petriman, N. A., Kudva, R., Kuhn, P., Welte, T., Knapp, B., et al. (2013). YidC Occupies the Lateral Gate of the SecYEG Translocon and Is Sequentially Displaced by a Nascent Membrane Protein. J. Biol. Chem. 288 (23), 16295–16307. doi:10.1074/jbc.M112.446583
Sachelaru, I., Winter, L., Knyazev, D. G., Zimmermann, M., Vogt, A., Kuttner, R., et al. (2017). YidC and SecYEG Form a Heterotetrameric Protein Translocation Channel. Sci. Rep. 7 (1), 101. doi:10.1038/s41598-017-00109-8
Samatova, E., Daberger, J., Liutkute, M., and Rodnina, M. V. (2020). Translational Control by Ribosome Pausing in Bacteria: How a Non-uniform Pace of Translation Affects Protein Production and Folding. Front. Microbiol. 11, 619430. doi:10.3389/fmicb.2020.619430
Samuelson, J. C., Chen, M., Jiang, F., Möller, I., Wiedmann, M., Kuhn, A., et al. (2000). YidC Mediates Membrane Protein Insertion in Bacteria. Nature 406 (6796), 637–641. doi:10.1038/35020586
Samuelson, J. C., Jiang, F., Yi, L., Chen, M., de Gier, J.-W., Kuhn, A., et al. (2001). Function of YidC for the Insertion of M13 Procoat Protein in Escherichia coli: Translocation of Mutants that Show Differences in Their Membrane Potential Dependence and Sec Requirement. J. Biol. Chem. 276 (37), 34847–34852. doi:10.1074/jbc.M105793200
Sandikci, A., Gloge, F., Martinez, M., Mayer, M. P., Wade, R., Bukau, B., et al. (2013). Dynamic Enzyme Docking to the Ribosome Coordinates N-Terminal Processing with Polypeptide Folding. Nat. Struct. Mol. Biol. 20 (7), 843–850. doi:10.1038/nsmb.2615
Saraogi, I., Akopian, D., and Shan, S.-o. (2014). Regulation of Cargo Recognition, Commitment, and Unloading Drives Cotranslational Protein Targeting. J. Cel. Biol. 205 (5), 693–706. doi:10.1083/jcb.201311028
Schaffitzel, C., Oswald, M., Berger, I., Ishikawa, T., Abrahams, J. P., Koerten, H. K., et al. (2006). Structure of the E. coli Signal Recognition Particle Bound to a Translating Ribosome. Nature 444 (7118), 503–506. doi:10.1038/nature05182
Schibich, D., Gloge, F., Pöhner, I., Björkholm, P., Wade, R. C., von Heijne, G., et al. (2016). Global Profiling of SRP Interaction with Nascent Polypeptides. Nature 536 (7615), 219–223. doi:10.1038/nature19070
Schulze, R. J., Komar, J., Botte, M., Allen, W. J., Whitehouse, S., Gold, V. A. M., et al. (2014). Membrane Protein Insertion and Proton-motive-force-dependent Secretion through the Bacterial Holo-Translocon SecYEG-SecDF-YajC-YidC. Proc. Natl. Acad. Sci. U.S.A. 111 (13), 4844–4849. doi:10.1073/pnas.1315901111
Scotti, P. A., Urbanus, M. L., Brunner, J., de Gier, J.-W. L., von Heijne, G., van der Does, C., et al. (2000). YidC, the Escherichia coli Homologue of Mitochondrial Oxa1p, Is a Component of the Sec Translocase. EMBO J. 19 (4), 542–549. doi:10.1093/emboj/19.4.542
Seppälä, S., Slusky, J. S., Lloris-Garcerá, P., Rapp, M., and von Heijne, G. (2010). Control of Membrane Protein Topology by a Single C-Terminal Residue. Science 328 (5986), 1698–1700. doi:10.1126/science.1188950
Serdiuk, T., Balasubramaniam, D., Sugihara, J., Mari, S. A., Kaback, H. R., and Müller, D. J. (2016). YidC Assists the Stepwise and Stochastic Folding of Membrane Proteins. Nat. Chem. Biol. 12 (11), 911–917. doi:10.1038/nchembio.2169
Serdiuk, T., Mari, S. A., and Müller, D. J. (2017). Pull-and-Paste of Single Transmembrane Proteins. Nano Lett. 17 (7), 4478–4488. doi:10.1021/acs.nanolett.7b01844
Serdiuk, T., Steudle, A., Mari, S. A., Manioglu, S., Kaback, H. R., Kuhn, A., et al. (2019). Insertion and Folding Pathways of Single Membrane Proteins Guided by Translocases and Insertases. Sci. Adv. 5 (1), eaau6824. doi:10.1126/sciadv.aau6824
Serek, J., Bauer-Manz, G., Struhalla, G., van den Berg, L., Kiefer, D., Dalbey, R., et al. (2004). Escherichia coli YidC Is a Membrane Insertase for Sec-independent Proteins. EMBO J. 23 (2), 294–301. doi:10.1038/sj.emboj.7600063
Smets, D., Loos, M. S., Karamanou, S., and Economou, A. (2019). Protein Transport across the Bacterial Plasma Membrane by the Sec Pathway. Protein J. 38 (3), 262–273. doi:10.1007/s10930-019-09841-8
Spiess, M., Junne, T., and Janoschke, M. (2019). Membrane Protein Integration and Topogenesis at the ER. Protein J. 38 (3), 306–316. doi:10.1007/s10930-019-09827-6
Steinberg, R., Knüpffer, L., Origi, A., Asti, R., and Koch, H.-G. (2018). Co-Translational Protein Targeting in Bacteria. FEMS Microbiol. Lett. 365 (11), 1–15. doi:10.1093/femsle/fny095
Stjepanovic, G., Kapp, K., Bange, G., Graf, C., Parlitz, R., Wild, K., et al. (2011). Lipids Trigger a Conformational Switch that Regulates Signal Recognition Particle (SRP)-mediated Protein Targeting. J. Biol. Chem. 286 (26), 23489–23497. doi:10.1074/jbc.M110.212340
Tanaka, Y., Sugano, Y., Takemoto, M., Mori, T., Furukawa, A., Kusakizako, T., et al. (2015). Crystal Structures of SecYEG in Lipidic Cubic Phase Elucidate a Precise Resting and a Peptide-Bound State. Cel Rep. 13 (8), 1561–1568. doi:10.1016/j.celrep.2015.10.025
Tanaka, Y., Izumioka, A., Abdul Hamid, A., Fujii, A., Haruyama, T., Furukawa, A., et al. (2018). 2.8-Å crystal Structure of Escherichia coli YidC Revealing All Core Regions, Including Flexible C2 Loop. Biochem. Biophysical Res. Commun. 505 (1), 141–145. doi:10.1016/j.bbrc.2018.09.043
Tastan, O., Klein-Seetharaman, J., and Meirovitch, H. (2009). The Effect of Loops on the Structural Organization of α-Helical Membrane Proteins. Biophys. J. 96 (6), 2299–2312. doi:10.1016/j.bpj.2008.12.3894
Ullers, R. S., Houben, E. N. G., Brunner, J., Oudega, B., Harms, N., and Luirink, J. (2006). Sequence-specific Interactions of Nascent Escherichia coli Polypeptides with Trigger Factor and Signal Recognition Particle. J. Biol. Chem. 281 (20), 13999–14005. doi:10.1074/jbc.M600638200
Ulmschneider, M. B., Ulmschneider, J. P., Schiller, N., Wallace, B. A., von Heijne, G., and White, S. H. (2014). Spontaneous Transmembrane helix Insertion Thermodynamically Mimics Translocon-Guided Insertion. Nat. Commun. 5, 4863. doi:10.1038/ncomms5863
Urbanus, M. L., Fröderberg, L., Drew, D., Björk, P., de Gier, J.-W. L., Brunner, J., et al. (2002). Targeting, Insertion, and Localization of Escherichia coli YidC. J. Biol. Chem. 277 (15), 12718–12723. doi:10.1074/jbc.M200311200
Valent, Q. A., Scotti, P. A., High, S., de Gier, J. W., von Heijne, G., Lentzen, G., et al. (1998). The Escherichia coli SRP and SecB Targeting Pathways Converge at the Translocon. EMBO J. 17 (9), 2504–2512. doi:10.1093/emboj/17.9.2504
van Bloois, E., Jan Haan, G., de Gier, J.-W., Oudega, B., and Luirink, J. (2004). F(1)F(0) ATP Synthase Subunit C Is Targeted by the SRP to YidC in the E. Coli Inner Membrane. FEBS Lett. 576 (1-2), 97–100. doi:10.1016/j.febslet.2004.08.069
Van den Berg, B., Clemons, W. M., Collinson, I., Modis, Y., Hartmann, E., Harrison, S. C., et al. (2004). X-ray Structure of a Protein-Conducting Channel. Nature 427 (6969), 36–44. doi:10.1038/nature02218
van der Laan, M., Houben, E. N. G., Nouwen, N., Luirink, J., and Driessen, A. J. M. (2001). Reconstitution of Sec‐dependent Membrane Protein Insertion: Nascent FtsQ Interacts with YidC in a SecYEG‐dependent Manner. EMBO Rep. 2 (6), 519–523. doi:10.1093/embo-reports/kve106
van der Laan, M., Bechtluft, P., Kol, S., Nouwen, N., and Driessen, A. J. M. (2004). F1F0 ATP Synthase Subunit C Is a Substrate of the Novel YidC Pathway for Membrane Protein Biogenesis. J. Cel. Biol. 165 (2), 213–222. doi:10.1083/jcb.200402100
Vitrac, H., Bogdanov, M., and Dowhan, W. (2013). In Vitro Reconstitution of Lipid-dependent Dual Topology and Postassembly Topological Switching of a Membrane Protein. Proc. Natl. Acad. Sci. U.S.A. 110 (23), 9338–9343. doi:10.1073/pnas.1304375110
Vitrac, H., MacLean, D. M., Karlstaedt, A., Taegtmeyer, H., Jayaraman, V., Bogdanov, M., et al. (2017). Dynamic Lipid-Dependent Modulation of Protein Topology by post-translational Phosphorylation. J. Biol. Chem. 292 (5), 1613–1624. doi:10.1074/jbc.M116.765719
von Heijne, G. (1989). Control of Topology and Mode of Assembly of a Polytopic Membrane Protein by Positively Charged Residues. Nature 341 (6241), 456–458. doi:10.1038/341456a0
von Heijne, G. (1992). Membrane Protein Structure Prediction. Hydrophobicity Analysis and the Positive-Inside Rule. J. Mol. Biol. 225 (2), 487–494. doi:10.1016/0022-2836(92)90934-c
Wagner, S., Pop, O., Haan, G.-J., Baars, L., Koningstein, G., Klepsch, M. M., et al. (2008). Biogenesis of MalF and the MalFGK(2) Maltose Transport Complex in Escherichia coli Requires YidC. J. Biol. Chem. 283 (26), 17881–17890. doi:10.1074/jbc.M801481200
Welte, T., Kudva, R., Kuhn, P., Sturm, L., Braig, D., Müller, M., et al. (2012). Promiscuous Targeting of Polytopic Membrane Proteins to SecYEG or YidC by theEscherichia Colisignal Recognition Particle. MBoC 23 (3), 464–479. doi:10.1091/mbc.E11-07-0590
Wickles, S., Singharoy, A., Andreani, J., Seemayer, S., Bischoff, L., Berninghausen, O., et al. (2014). A Structural Model of the Active Ribosome-Bound Membrane Protein Insertase YidC. Elife 3, e03035. doi:10.7554/eLife.03035
Woodall, N. B., Hadley, S., Yin, Y., and Bowie, J. U. (2017). Complete Topology Inversion Can Be Part of normal Membrane Protein Biogenesis. Protein Sci. 26 (4), 824–833. doi:10.1002/pro.3131
Woolhead, C. A., McCormick, P. J., and Johnson, A. E. (2004). Nascent Membrane and Secretory Proteins Differ in FRET-Detected Folding Far inside the Ribosome and in Their Exposure to Ribosomal Proteins. Cell 116 (5), 725–736. doi:10.1016/s0092-8674(04)00169-2
Xiao, Q., Zhang, F., Nacev, B. A., Liu, J. O., and Pei, D. (2010). Protein N-Terminal Processing: Substrate Specificity of Escherichia coli and Human Methionine Aminopeptidases. Biochemistry 49 (26), 5588–5599. doi:10.1021/bi1005464
Yang, C.-I., Hsieh, H.-H., and Shan, S.-o. (2019). Timing and Specificity of Cotranslational Nascent Protein Modification in Bacteria. Proc. Natl. Acad. Sci. U.S.A. 116 (46), 23050–23060. doi:10.1073/pnas.1912264116
Yi, L., Celebi, N., Chen, M., and Dalbey, R. E. (2004). Sec/SRP Requirements and Energetics of Membrane Insertion of Subunits a, B, and C of the Escherichia coli F1F0 ATP Synthase. J. Biol. Chem. 279 (38), 39260–39267. doi:10.1074/jbc.M405490200
Zhang, B., and Miller, T. F. (2012). Long-timescale Dynamics and Regulation of Sec-Facilitated Protein Translocation. Cel Rep. 2 (4), 927–937. doi:10.1016/j.celrep.2012.08.039
Zhang, X., Rashid, R., Wang, K., and Shan, S. o. (2010). Sequential Checkpoints Govern Substrate Selection during Cotranslational Protein Targeting. Science 328 (5979), 757–760. doi:10.1126/science.1186743
Zhu, L., Klenner, C., Kuhn, A., and Dalbey, R. E. (2012). Both YidC and SecYEG Are Required for Translocation of the Periplasmic Loops 1 and 2 of the Multispanning Membrane Protein TatC. J. Mol. Biol. 424 (5), 354–367. doi:10.1016/j.jmb.2012.09.026
Keywords: N-terminal processing, membrane targeting, membrane insertion, membrane protein topology, translocon, cotranslational folding, YidC
Citation: Mercier E, Wang X, Bögeholz LAK, Wintermeyer W and Rodnina MV (2022) Cotranslational Biogenesis of Membrane Proteins in Bacteria. Front. Mol. Biosci. 9:871121. doi: 10.3389/fmolb.2022.871121
Received: 07 February 2022; Accepted: 12 April 2022;
Published: 29 April 2022.
Edited by:
Stephen D. Fried, Johns Hopkins University, United StatesReviewed by:
Huan Bao, The Scripps Research Institute, United StatesChristian Kaiser, Johns Hopkins University, United States
Copyright © 2022 Mercier, Wang, Bögeholz, Wintermeyer and Rodnina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina V. Rodnina, cm9kbmluYUBtcGluYXQubXBnLmRl