 Jenny U. Tran
Jenny U. Tran Breann L. Brown
Breann L. Brown- 1Department of Biochemistry, Vanderbilt University School of Medicine, Nashville, TN, United States
- 2Center for Structural Biology, Vanderbilt University School of Medicine, Nashville, TN, United States
Pyridoxal 5′-phosphate (PLP)-dependent enzymes are found ubiquitously in nature and are involved in a variety of biological pathways, from natural product synthesis to amino acid and glucose metabolism. The first structure of a PLP-dependent enzyme was reported over 40 years ago, and since that time, there is a steady wealth of structural and functional information revealed for a wide array of these enzymes. A functional mechanism that is gaining more appreciation due to its relevance in drug design is that of protein allostery, where binding of a protein or ligand at a distal site influences the structure, organization, and function at the active site. Here, we present a review of current structure-based mechanisms of allostery for select members of each PLP-dependent enzyme family. Knowledge of these mechanisms may have a larger potential for identifying key similarities and differences among enzyme families that can eventually be exploited for therapeutic development.
1 Introduction
Pyridoxal 5′-phosphate (PLP), the active form of vitamin B6, is a coenzyme that is required for the activity of many proteins from bacteria to humans. These enzymes carry out a number of processes, including amino acid metabolism and biosynthesis of antibiotic compounds (Schneider et al., 2000). PLP-dependent enzymes typically bind the cofactor covalently via a conserved catalytic lysine residue, forming an internal aldimine. After binding the amino acid substrate, the internal aldimine is exchanged for an external aldimine Schiff base with the amino group of the substrate and the PLP aldehyde, regenerating the free lysine (Schneider et al., 2000). The reaction then proceeds through a quinonoid intermediate with the PLP cofactor providing an electron sink to stabilize the transient reaction intermediates (Eliot and Kirsch 2004). After these initial steps, there is a diverse number of reactions PLP-dependent enzymes mediate, including decarboxylation, transamination, and racemization (Liang et al., 2019). In fact, PLP-dependent enzymes are known to mediate more than 140 distinct activities (Percudani and Peracchi 2003).
To date, there are seven PLP-dependent protein families that are classified based on their 3-dimensional folds (Figure 1). Although proteins within each family have little overall sequence homology, they exhibit characteristic structures. Notably, a particular fold does not necessarily dictate a particular reaction, as each fold type can mediate multiple types of reactions [for review see (Eliot and Kirsch 2004)]. Fold Type I is the largest family typified by the enzyme aspartate aminotransferase. These enzymes are homodimers with each protomer containing a large and small subdomain; however, the PLP-binding sites are comprised of residues from both subunits. Although structurally similar to Type I, Fold Type II, or the tryptophan synthase β family, typically contains enzymes that catalyze β-elimination, β-replacement, and racemization reactions. They differ from Fold Type I in that the active sites are usually made up of residues from only one protomer, and there may be additional regulatory domains present. Fold Type III (alanine racemase) enzymes, characterized by an α/β-barrel attached to a β-strand domain, also function as dimers and include several amino-acid decarboxylases. Fold Type IV enzymes include D-amino acid aminotransferase and a few other enzymes that also function as homodimers, but the PLP-binding mode differs from Folds I and II. The Fold Type V group includes glycogen phosphorylase and is atypical from other families in that this clade uses the PLP phosphate group for catalysis (Eliot and Kirsch 2004). More recently, two new PLP-dependent families were identified with structural divergence from the other families, and they are denoted Fold Type VI (lysine 5,6-aminomutase) and Fold Type VII (lysine 2,3-aminomutase) (Percudani and Peracchi 2009).
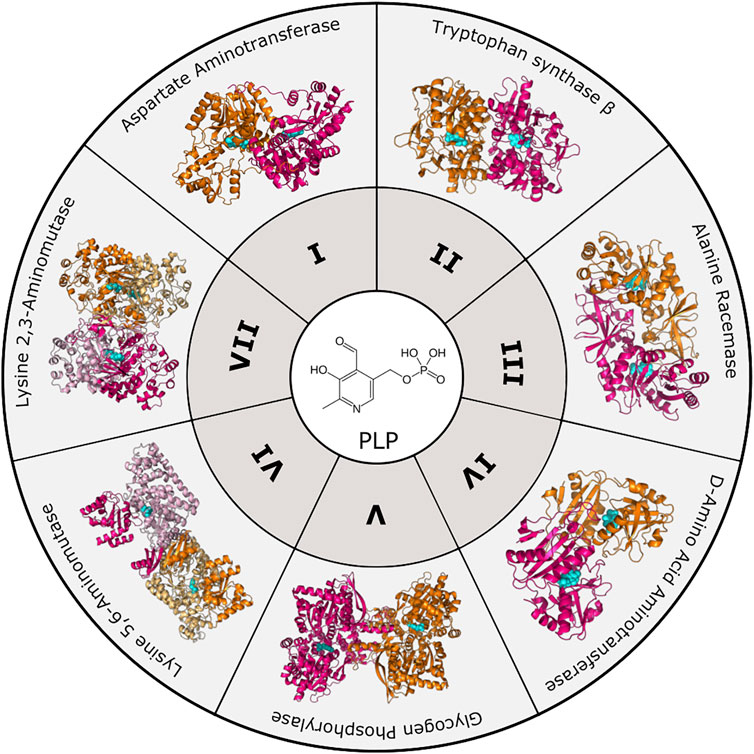
FIGURE 1. PLP-dependent enzyme families. To date, PLP-dependent enzymes are categorized into seven different families based on structural homology to an archetypal enzyme. These include Aspartate Aminotransferase (Fold Type I, PDB 8AAT), Tryptophan Synthase β-subunit (Fold Type II, PDB 1BKS), Alanine Racemase (Fold Type III, PDB 1SFT), D-Amino Acid Aminotransferase (Fold Type IV, PDB 1DAA), Glycogen Phosphorylase (Fold Type V, PDB 1GPB), Lysine 5,6-Aminomutase (Fold Type VI, PDB 1XRS), and Lysine 2,3-Aminomutase (Fold Type VII, PDB 2A5H). Protomers are colored orange and magenta with the PLP cofactor shown in cyan.
In addition to structural diversity, PLP-dependent enzymes also display a diverse repertoire of allosteric mechanisms. Protein allostery is key for the modulation of enzyme activity given a particular cellular context or binding partner. Protein allostery, a term first coined 60 years ago (Monod and Jacob 1961), is defined as the ability for ligand or protein binding at one site to affect the binding or activity of another distal site (Monod et al., 1965). A seminal example of allostery is the Bohr effect, where factors affecting blood pH cause a change in the binding affinity of hemoglobin for oxygen. Since these early studies, allosteric control has been identified for many proteins mediating nearly all functions in the cell. Along with the expansion of our understanding of allostery, the methods to determine and study allostery have also grown. Regardless of the mechanism, allostery presents a powerful tool for proteins to regulate their own functions and can even be harnessed for drug design (Nussinov and Tsai 2013). This review will focus on the various allosteric mechanisms and their structural bases invoked by members of each PLP-dependent enzyme family. Although we focus on a set of key proteins, PLP-dependent enzymes exhibit a broad range of allosteric mechanisms in addition to these specific examples (Supplementary Table S1). Future work in therapeutic development will benefit from a thorough understanding of these burgeoning principles.
2 Diverse PLP-dependent Proteins and Their Allosteric Mechanisms
2.1 Fold Type I: 5-Aminolevulinic Acid Synthase (ALAS)
PLP-dependent enzymes belonging to Fold Type I exhibit a conserved structure typical of many aminotransferases, decarboxylases, and enzymes that catalyze α-, β- or γ-eliminations (Percudani and Peracchi 2003). 5-aminolevulinic acid synthase (ALAS) is the first and rate-limiting enzyme for heme biosynthesis in α-proteobacteria and the mitochondria of non-plant eukaryotes. ALAS catalyzes the condensation of glycine and succinyl-CoA to yield aminolevulinic acid (Gibson et al., 1958; Kikuchi et al., 1958; Laver et al., 1958). ALAS is a member of the α-oxoamine family of Fold Type I PLP-dependent enzymes (Schneider et al., 2000; Eliot and Kirsch 2004) and exists as a homodimer with the two PLP cofactor binding pockets buried at the subunit interface (Figure 2A) (Astner et al., 2005). Currently, there are several published structures of ALAS enzymes from multiple organisms either bound covalently (internal aldimine) or non-covalently to PLP. The crystal structures of ALAS from Rhodobacter capsulatus were captured with both the internal aldimine (PDB 2BWN) and the glycine-bound external aldimine (PDB 2BWP) (Astner et al., 2005). Subsequently, ALAS structures from Saccharomyces cerevisiae and the erythroid-specific Homo sapiens isoforms were also crystallized in the presence of PLP, either bound covalently or non-covalently (Brown et al., 2018; Bailey et al., 2020). Importantly, these structures revealed the position and conformation of the eukaryote-specific ALAS C-terminal extension—a region absent from bacterial ALAS enzymes.
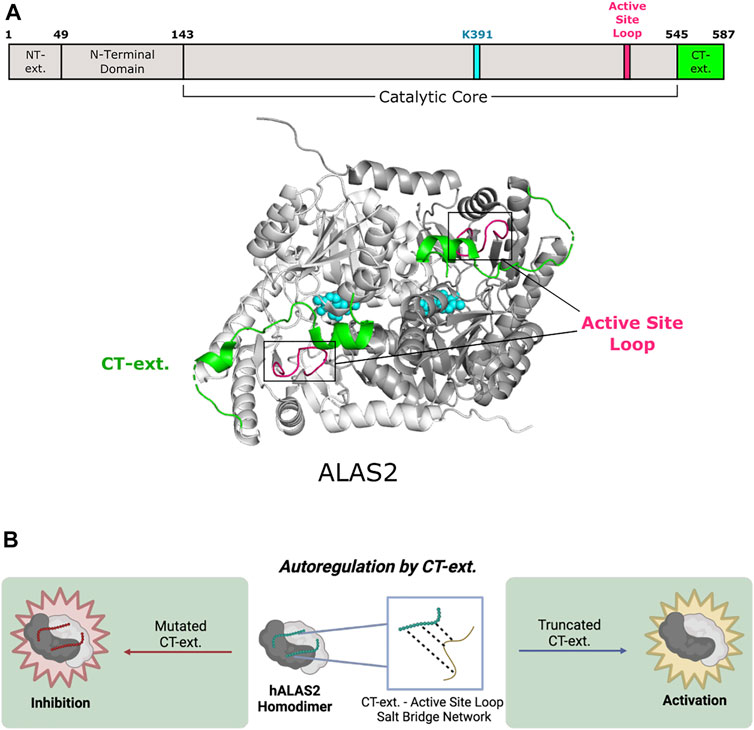
FIGURE 2. Human 5-aminolevulinic acid synthase 2 of Fold Type I undergoes autoregulation via its C-terminal extension. (A) Human ALAS2 (PDB 6HRH) has a C-terminal extension (CT-ext.; aa 545–587; green) that interacts with the active site loop (aa 500–517; pink) from the catalytic core to block access to the active site. ALAS2 protomers are shown in white and gray. PLP and the active site lysine (K391) are shown in cyan. (B) The CT-ext. of ALAS2 exhibits an autoregulatory mechanism by occluding the active site. Truncation of the CT-ext. disrupts the salt bridge network between the CT-ext. and active site loop, leading to gain of function. Mutations of the CT-ext. may also cause loss of function by changing the active site conformation (figure created with BioRender.com).
2.1.1 C-Terminal Extension
The eukaryotic ALAS C-terminal extension allosterically communicates to the enzyme active site by controlling the position of the ALAS active site loop (Fratz et al., 2015; Bailey et al., 2020). The conformation and dynamics of this loop are reported to control the overall rate of ALA product release, which is the rate-limiting step of the ALAS reaction (Hunter and Ferreira 1999; Astner et al., 2005; Hunter et al., 2007; Lendrihas et al., 2010). In yeast ALAS, the extreme portion of the C-terminus makes trans interactions with the neighboring subunit via an interaction with a conserved arginine residue located in the ALAS catalytic glycine-rich motif. Importantly, mutation of this arginine in yeast or eukaryotic ALAS leads to a decrease in enzyme activity (Gong and Ferreira 1995; Katsurada et al., 2016; Brown et al., 2018). Thus, for yeast ALAS, the C-terminus has two points of allosteric control—first by regulating the position of the active site loop and second by interacting with the ALAS glycine-rich loop adjacent to the enzyme’s active site (Brown et al., 2018). The mammalian C-terminal extension also allosterically regulates enzyme activity of ALAS2, the erythroid-specific isoform (Figure 2B). Notably, deletion or modification of this region in humans underlies toxic hyperactivity leading to porphyrin accumulation and the disease X-linked protoporphyria (XLP) (Whatley et al., 2008; Bishop et al., 2013; Ducamp et al., 2013). The recent crystal structure of human ALAS2 identified key interactions between the C-terminal extension and other regulatory regions of the enzyme (Bailey et al., 2020). A short helix in this extension (helix α15, Ser568-Phe575) forms a lid over the active site but does not directly contact the non-covalently bound PLP cofactor. Nonetheless, in vitro biochemical experiments showed that disruption of the human C-terminal extension alters the PLP microenvironment and changes the tautomeric equilibrium of the cofactor (Fratz et al., 2015). Additionally, it was determined that the orientation of the internal aldimine in the XLP variants is different from wild-type ALAS2 in the presence of bound succinyl-CoA substrate. The ALAS2 crystal structure reveals a direct interaction between an arginine in the active site loop and the C-terminal extension, leading to the hypothesis that the conformation and flexibility of the active site loop are coupled to changes in the C-terminus, thus controlling overall ALAS2 activity (Bailey et al., 2020). Finally, the ALAS2 C-terminal extension may auto regulate enzyme activity by acting as a signal for degradation (Kadirvel et al., 2012). Thus, the eukaryote-specific ALAS C-terminal extension acts as a homo-allosteric regulator of enzyme activity at multiple nodes.
2.2 Fold Type II: Cystathionine β-Synthase (CBS)
Fold Type II (also known as the tryptophan synthase β family) encompasses numerous allosteric enzymes (Liang et al., 2019), including the tryptophan synthase α2β2 complex (Hyde et al., 1988), threonine deaminase (Eisenstein 1995), threonine synthase (Curien et al., 1998), and O-acetylserine sulfhydrylase (Burkhard et al., 2000). Although enzymes in this fold have active sites composed of residues from one subunit, they are active in various oligomeric states (usually dimers or tetramers) that also accommodate allosteric regulation (Gallagher et al., 1998; Garrido-Franco et al., 2002; Fatmi and Chang 2010). For example, fungal threonine synthase functions as a monomer and is not subject to allosteric regulation, whereas plant threonine synthase is found as a homodimer and is activated by S-adenosyl-L-methionine (AdoMet) (Garrido-Franco et al., 2002). Cystathionine β-synthase (CBS) is involved in the initial step of sulfur-containing amino acid biosynthesis (Gerritsen and Waisman 1964) where it catalyzes the condensation of serine and potentially toxic homocysteine to yield cystathionine. It assembles as a tetramer (a dimer of dimers), with each subunit consisting of a catalytic N-terminal domain that binds PLP and heme and a regulatory C-terminal domain (Figure 3A) (Taoka et al., 2002). In addition to PLP, CBS uses heme as a cofactor and is further activated by AdoMet. CBS distinguishes itself from other family members in how both the N-terminal and C-terminal domains participate in allostery (Meier et al., 2001).
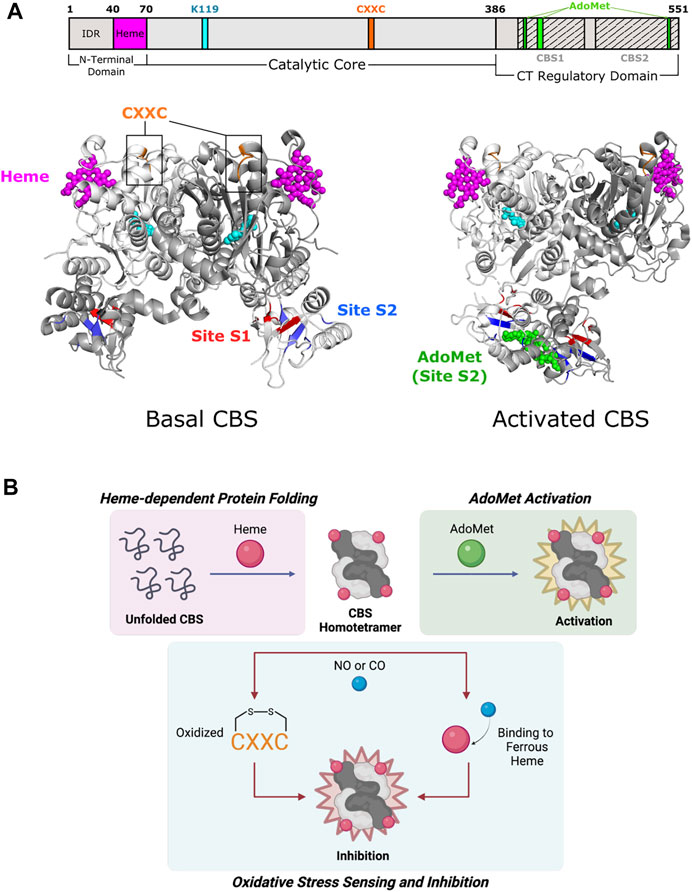
FIGURE 3. Cystathionine β-synthase of Fold Type II involves each of its domains in allostery. (A) Human CBS contains three domains that are all implicated in allostery. Heme binding occurs in a shallow pocket (aa 40–70; magenta) at the N-terminal domain. The catalytic core contains a CXXC oxidoreductase motif (aa 272–275; orange). The C-terminal regulatory domain has a Bateman module consisting of two tandem CBS motifs, CBS1 (aa 412–471) and CBS2 (aa 477–551), which fold to form Site S1 (M458, V459, Y484, F487, F508, and A509; red) and Site S2 (P422, L423, F443, A446, P447, V448, V533, and V534; blue) for AdoMet binding. Basal CBS (PDB 4L0D) has little activity due to the regulatory domain of one subunit blocking access to the active site of a neighboring subunit. Activated CBS (PDB 4PCU) binds AdoMet (green) in Site S2, which moves the regulatory domain away from the active sites to allow access. CBS protomers are shown in white and gray. PLP and the active site lysine (K119) are shown in cyan. (B) Formation of the CBS homotetramer is mediated by heme binding, in which the heme porphyrin scaffold facilitates protein folding. AdoMet binds and activates the CBS homotetramer. CBS inhibition takes place via oxidation of the CXXC motif and/or gaseous signaling molecule (i.e., NO, CO) binding to ferrous heme.
2.2.1 C-Terminal Regulatory Domain
Alignment of Fold Type II enzymes show a highly conserved catalytic core and minimally conserved N- and C-terminal extensions (Miles and Kraus 2004). A key feature of allosteric enzymes in this fold is the C-terminal regulatory domain, which is usually involved in effector binding (Gallagher et al., 1998; Garrido-Franco et al., 2002). In CBS, truncation of this domain yields the “active core” that is not activated by AdoMet, has twice the enzymatic activity of full-length CBS, and forms dimers instead of tetramers (Meier et al., 2001). Available CBS crystal structures only represent mutant CBS dimers, whereas native wildtype CBS exists as tetramers (Ereno-Orbea et al., 2013).
The C-terminal regulatory domain of human CBS has two tandem β-α-β-β-α secondary structure motifs known as “CBS domains” that can be found in other proteins (Bateman 1997; Ignoul and Eggermont 2005). These motifs interact to form intramolecular structures known as Bateman modules or CBS pairs. PLP is deeply buried in a cleft between the N-terminal and C-terminal domains of a subunit (Meier et al., 2001). The CBS1 (amino acids 415–468) and CBS2 (aa 511–531) domains from one subunit associate to block the narrow active site channel of a neighboring subunit and form AdoMet binding clefts called Site S1 and Site S2 (Figure 3A). Site S1 is blocked by hydrophobic residues, leaving Site S2 as the effective AdoMet binding site ∼10 Å from the active site. Site S2 is therefore known as an autoinhibitory region, where AdoMet binding displaces the regulatory domain of one subunit from the catalytic cavity of another subunit. Rotation of Site S2 occurs and weakens interactions with the loops of the catalytic cavity. The active site thus becomes less sterically hindered and is kinetically stabilized (Ereno-Orbea et al., 2013). Because of its effect on active site accessibility, AdoMet is known as a V-type allosteric activator (which increases Vmax) and can bind to each subunit to cause a 2- to 3- fold increase in activity (Taoka et al., 1999a).
2.2.2 Heme Binding Domain
CBS is one of few PLP-dependent enzymes that binds heme for regulatory purposes. Heme is not essential for catalysis in yeast and parasitic CBS as they are active but do not bind heme (Jhee et al., 2000; Nozaki et al., 2001). However, alterations in heme binding modulate human CBS activity and stability (Badawy 2021). The N-terminal domain of CBS comprises two distinct regions. The first few residues contain an intrinsically disordered region (IDR) that may play a part in heme binding. The IDR has a canonical cysteine-proline (CP) motif that is found in other heme-binding proteins such as human ALAS enzymes, but the function of this site in CBS is unknown (Kumar et al., 2018). The remaining residues in the N-terminal domain fold into a shallow, hydrophobic pocket known to non-covalently bind heme ∼20 Å from the active site (Figure 3A) (Meier et al., 2001).
The mechanism of heme regulation is not fully understood, but the components of heme may play different roles. The porphyrin moiety of heme acts as a scaffold to facilitate protein folding and maintain stability (Majtan et al., 2008). Alternatively, the heme iron may be involved in oxido-reducing reactions that allow for proper enzyme function (Figure 3B) (Taoka et al., 1998). Redox sensitivity is an important property in determining CBS activity since the metabolism of homocysteine is directly related to cellular redox homeostasis (Weiss et al., 2002; Prudova et al., 2006). The heme iron can exist in two redox states, Fe(III)-CBS (ferric heme) and the reduced Fe(II)-CBS (ferrous heme) (Carballal et al., 2013). Ferrous heme binds available gaseous signaling molecules (e.g., NO or CO) with relatively high affinity (Taoka et al., 1999b; Vicente et al., 2014), which inhibits CBS activity by up to 2-fold compared to ferric CBS (Taoka et al., 1998). A possible pathway for allosteric regulation by heme is through the formation of salt bridges, a mechanism that has been shown to be important in conformational changes of other heme proteins. In CBS, Arg266 is located at the distal end of a helix that makes hydrogen bonds with the phosphate moiety of PLP. This same arginine forms a salt bridge with Cys52, a heme-binding residue, which allows for conformational changes to propagate to the active site (Taoka et al., 2002).
Certain data questions the existence of the ferrous form of CBS in vivo because of the low redox potential of heme in wild-type CBS (Singh et al., 2009). However, it was reported that methionine synthase reductase can indeed reduce the CBS heme in an NADPH-dependent manner, supporting the biological relevance of ferrous CBS (Kabil et al., 2011). Another potential site for redox sensing is an oxidoreductase motif, CXXC, located ∼20 Å away from the active site. Mutagenesis of either cysteine within the motif affects CBS activity, but overall structural changes have not yet been elucidated due to the lack of a structure for full-length, oxidized CBS (Niu et al., 2018).
2.3 Fold Type III: Ornithine Decarboxylase (ODC)
Fold Type III, or the alanine racemase family, is characterized by a mixed α/β barrel structure found in certain amino-acid decarboxylases (Percudani and Peracchi 2003). L-Ornithine decarboxylase (ODC), a member of the Group IV family of decarboxylases, catalyzes the first and rate-limiting step of polyamine biosynthesis, which is the formation of putrescine from ornithine (Sandmeier et al., 1994). Putrescine is ultimately converted into the polyamines spermidine and spermine (Gale 1940; Tabor and Tabor 1976; Cohen 1998; Pegg 2006). There are structures of ODC enzymes from multiple organisms, including bacteria and mammals. Each ODC subunit contains an N-terminal PLP-binding domain with a TIM-like α/β-barrel fold and a C-terminal β-sheet domain (Figure 4A) (Kern et al., 1999; Almrud et al., 2000). The active sites, which accommodate PLP and L-ornithine, are formed at the dimer interface between the N-terminal domain of one subunit and the C-terminal domain of the partner subunit (Almrud et al., 2000; Jackson et al., 2004). Wild-type ODC dimers are weakly-interacting and exist in equilibrium with the monomeric form (Pegg 2006).
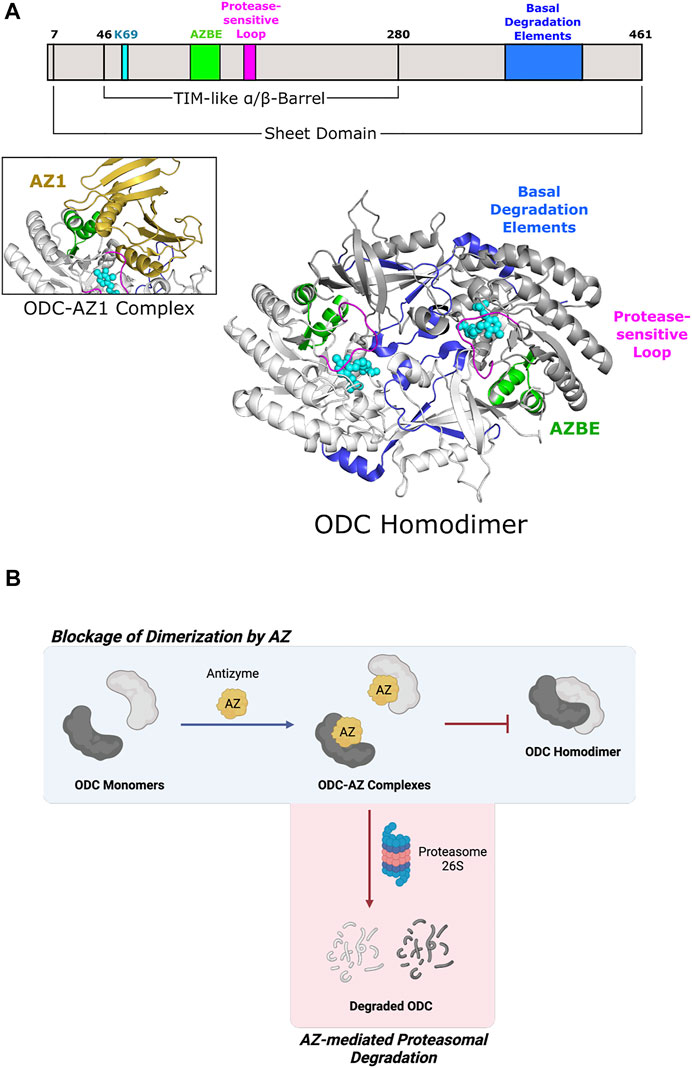
FIGURE 4. Eukaryotic ornithine decarboxylase of Fold Type III is targeted for proteasomal degradation by interaction with Antizyme 1. (A) Human ODC (PDB 1D7K) binds AZ1 at the antizyme binding element (AZBE; aa 117–140; green) to form an ODC-AZ1 heterodimer (AZ1 in yellow; PDB 4ZGY). Two basal degradation elements (aa 376–427; blue) are subsequently exposed to allow ubiquitin-independent, proteasomal degradation of ODC. A protease-sensitive loop (aa 158–168; magenta) positions active site residues for catalysis. ODC protomers are shown in white and gray. PLP and the active site lysine (K69) are shown in cyan. (B) ODC dimerization is blocked by Antizyme 1 binding, leading to ODC-AZ1 heterodimer formation. This inactive form of ODC is then targeted by the 26S proteasome for degradation.
2.3.1 Antizyme Binding
Antizyme is a non-competitive protein inhibitor of ODC that is produced in response to an increase in cellular polyamine levels (Heller et al., 1976). Antizyme binds to the free ODC monomer, forming an inactive heterodimer (Fujita et al., 1984) that sterically blocks the ODC homodimerization interface (Figure 4B) (Wu et al., 2015). Antizyme also abrogates ODC function by increasing the interaction of ODC with the proteasome in a ubiquitin-independent manner (Murakami et al., 1992). The crystal structure of human ODC in complex with a portion of antizyme isoform 1 indicates a conformational rearrangement in ODC which may reveal a structural feature that is then recognized by the proteasome (Wu et al., 2015) (Figure 4A). The C-terminal extension of ODC, which is absent from Trypanosoma brucei ODC (Persson et al., 2003), represents another point of allosteric control via serving as a signal for degradation by the 26S proteasome. Experiments using murine ODC showed deletion of the C-terminal 37 amino acids prevents proteasomal degradation (Ghoda et al., 1989). Additional work showed this peptide serves as a protein degron as appending it to T. brucei ODC leads to degradation (Zhang et al., 2003). Although not required for proteasome binding, the ODC C-terminus is necessary for degradation as a truncated version was stable even in the presence of antizyme (Wu et al., 2015). Unfortunately, there is no structural information pertaining to this region since it remains disordered in known mammalian ODC crystal structures (Almrud et al., 2000; Wu et al., 2015). How this region is recognized by the proteasome remains an outstanding question in the field.
2.3.2 Allosteric Inhibitors
In addition to homo-allostery, much work is focused on developing direct and allosteric ODC inhibitors since it promotes cell transformation and is overexpressed in many cancers (Pegg 1988; Auvinen et al., 1992; O'Brien et al., 1997). Additionally, T. brucei ODC is a validated drug target to treat African Sleeping Sickness (trypanosomiasis) (Barrett et al., 2007; Smithson et al., 2010). Difluoromethylornithine (DFMO) is a structural analog of L-ornithine that works as an irreversible suicide ODC inhibitor by binding at the active site and forming a covalent adduct between the PLP cofactor and the conserved Cys360 (Metcalf et al., 1978; Poulin et al., 1992; Grishin et al., 1999); this same residue is also affected by S-nitrosylation via nitric oxide treatment (Bauer et al., 2001). Unfortunately, DFMO exhibits a low affinity for ODC and high doses of DFMO can result in permanent hearing loss (Lao et al., 2004), so ongoing work is targeted toward finding inhibitors with lower toxicity. To this end, multiple groups have located allosteric inhibitory sites on both T. brucei and human ODC. For example, Geneticin is a weak, non-competitive inhibitor that binds at the interface between the ODC N- and C-terminal domains, inducing an order-to-disorder transition in a key catalytic loop located at the dimer interface (Jackson et al., 2003). Herbacetin is a natural product that was shown through computational modeling and in vitro studies to bind at an allosteric site on ODC (comprised of residues Asp44, Asp243, and Glu384) to inhibit ODC activity (Kim et al., 2016). Another natural product, allicin, reversibly S-thioallylates accessible ODC cysteines, causing reduced polyamine levels and cell proliferation (Schultz et al., 2020) It is unclear whether the allicin-induced deactivation of ODC is due to thioallylation of the conserved Cys360, disruption of ODC dimerization, or some other unknown mechanism. More recent work is directed toward characterizing multipurpose inhibitors that might inhibit ODC’s activity, target the ODC-Antizyme1 interaction, and enhance non-functional ODC dimerization (Chai et al., 2020)
2.4 Fold Type IV: Branched-chain L-amino Acid Aminotransferase (BCAT)
Unlike other fold types, the D-amino acid aminotransferase family is not yet reported to contain enzymes that display clear allostery. Fold Type IV consists of four broad categories: (S)-selective branched-chain L-amino acid aminotransferases (BCATs) (Taylor and Jenkins 1966; Grishin et al., 1995), (R)-selective D-amino acid aminotransferases (DAATs) (Peisach et al., 1998), (R)-amine:pyruvate transaminases (R-ATAs) (Iwasaki et al., 2012), and 4-amino-4-deoxychroismate lyases (ADCLs) (Nakai et al., 2000). Out of these four protein types, the first three are transaminases. Although Fold Type I also contains transaminases that use similar enzymatic mechanisms, those of Fold Type IV have strict (R)- or (S)-stereospecificity and orient PLP differently in the active site (Okada et al., 1997; Bezsudnova et al., 2020).
Generally, most Fold Type IV enzymes form homodimers for catalysis (Sugio et al., 1995; Bezsudnova et al., 2020), but some BCATs and R-ATAs form tetramers or hexamers (Inoue et al., 1988; Iwasaki et al., 2012; Isupov et al., 2019). It is not known if these different oligomeric states are important for function. In an active dimer, one subunit comprises two domains connected by an interdomain loop (Figure 5A). The small N-terminal domain has an α/β structure. The large C-terminal domain has a pseudo β-barrel structure. The active site exists at the bottom of the subunit interface cleft formed by residues from both domains of one subunit and the small domain of a neighboring subunit (Okada et al., 2001). Although the active site is geometrically similar within proteins of this family, strict stereospecificity is based on different amino acid compositions (Bezsudnova et al., 2020). Another important note is that most enzymes belonging to this family are bacterial, archaeal, or plant proteins, and only BCATs have mammalian homologs (Ichihara and Koyama 1966; Taylor and Jenkins 1966). There has been a focus on industrial, antibiotic, and herbicidal applications because of the unique stereospecificity that can be exploited with Fold Type IV enzymes (Nakai et al., 2000; Pavkov-Keller et al., 2016). Therefore, much industrial research has focused on using the differences in active sites to obtain stereoselective products.
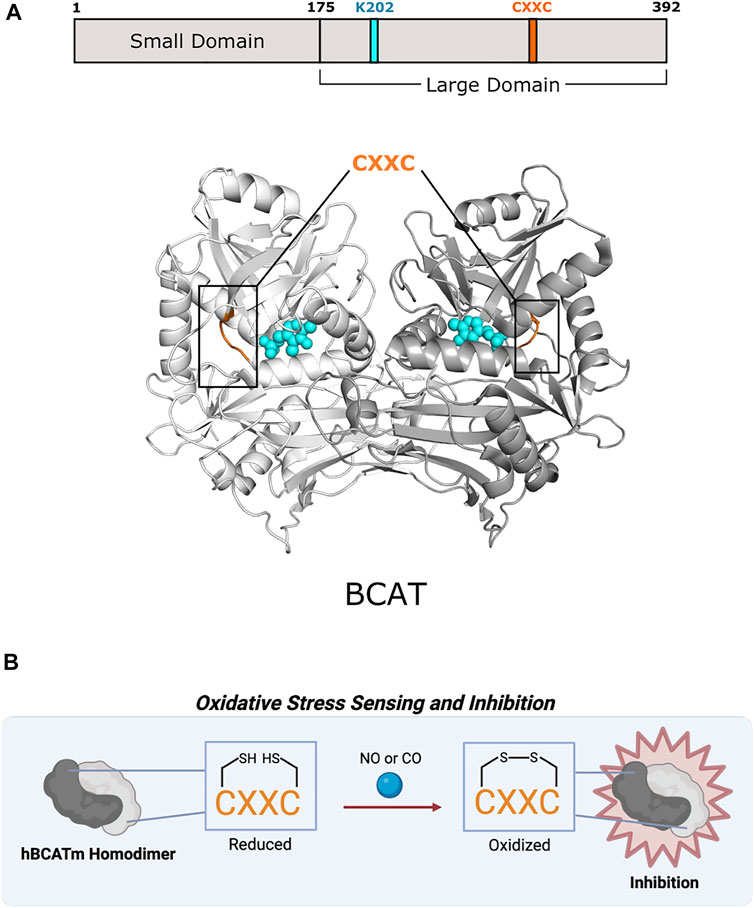
FIGURE 5. Human mitochondrial branched-chain amino acid aminotransferase of Fold Type IV uses a CXXC motif for preventing overoxidation. (A) Human mitochondrial BCAT (PDB 1EKF) has a CXXC motif (aa 315–318; orange) in which one cysteine (C315) acts as an oxidative sensor and the other (C318) is a “resolving cysteine” to prevent overoxidation of hBCATm. BCAT protomers are shown in white and gray. PLP and the active site lysine (K202) are shown in cyan. (B) BCAT is inhibited via oxidation of the CXXC motif.
Excluding antibiotic development, therapeutic efforts have only extended toward BCATs. In humans, branched-chain amino acids are nutrient signals, so BCATs are important facets in cancer and metabolic diseases (Lynch and Adams 2014; Hattori et al., 2017). BCAT inhibitor design has concentrated on obstructing reaction mechanisms, leading mainly toward irreversible, competitive inhibitors (Deng et al., 2015). Studies have shown that mammalian BCATs have an oxidoreductase CXXC motif that could be further investigated as a potential allosteric site as found in CBS in Fold Type II (Figure 5B). Humans have two BCAT isozymes, a mitochondrial form (hBCATm) and a cytosolic form (hBCATc). In hBCATm, the two cysteines of 315CXXC318 participate in thiol-thiolate interaction and are responsible for redox sensitivity. Cys315 acts as a sensor for redox regulation and helps in substrate orientation (Yennawar et al., 2006). Cys318 is a “resolving cysteine” that forms a reversible disulfide bond to prevent overoxidation or irreversible oxidation to sulfinic or sulfonic acid (Conway et al., 2004). Oxidation of the CXXC motif located ∼10 Å from the active site disrupts the hydrogen-bonding network necessary for PLP coordination and substrate channeling, thus inhibiting hBCATm (Yennawar et al., 2006). This motif also allows hBCAT to play a potential role as a redox chaperone in protein misfolding of neurodegenerative diseases like Alzheimer’s disease (El Hindy et al., 2014). There are currently no therapeutics that target the CXXC motif of mammalian BCATs, but further exploration into its function and role may provide new avenues for drug discovery.
2.5 Fold Type V: Glycogen Phosphorylase (GP)
This fold type contains only glycogen phosphorylase (GP), which is the first PLP-dependent enzyme to be structurally determined (Weber et al., 1978). Fold Type V enzymes use the phosphate moiety of PLP for proton transfer, whereas Fold Types I through IV use PLP as an electrophilic sink. As a result, the active site is completely divergent from other folds and binds PLP in a unique way (Schneider et al., 2000). GP is a well-known allosteric enzyme that, with the help of inorganic phosphate, catalyzes the phosphorolytic cleavage of α-1,4-glycosidic bonds to liberate the terminal glucose (glucose 1-phosphate; G1P) of a glycogen molecule in glycogenolysis (Hestrin 1949; Sprang et al., 1991). In mammals, glycogen is the main carbohydrate source and is found throughout the body, however, its function is tissue-dependent. GP has three isozymes, liver GP (lGP), muscle GP (mGP), and brain GP (bGP) (David and Crerar 1986; Newgard et al., 1988). Although they are encoded by separate genes, they are highly similar in sequence and differ mostly in expression and regulation.
2.5.1 Ser14 Phosphorylation
One of the main allosteric mechanisms of glycogen phosphorylase is phosphorylation at Ser14 located ∼45 Å from the active site (Figure 6B) (Krebs and Fischer 1956; Sprang et al., 1991). Unphosphorylated GP is dependent on adenosine monophosphate (AMP) for activity and inhibited by glucose-6-phosphate (G6P) and adenosine triphosphate (ATP). However, phosphorylation causes the 22 amino-terminal residues of GP to become ordered, which induces the rotation of subunits within the functional dimer to activate the enzyme and further enhance AMP activation (Barford and Johnson 1989; Newgard et al., 1989). Since phosphorylation occurs because of physiological changes communicated by hormonal or neuronal signals, the three tissue-specific isozymes respond differently to this mode of regulation (Agius 2015). In lGP, phosphorylation is the main regulatory mechanism because of the function of lGP in maintaining plasma glucose levels (Wolf et al., 1970). In mGP, phosphorylation helps activate the enzyme with the onset of exercise, but modulator binding (e.g., AMP, ATP, glucose, glycogen, and caffeine) provides another level of control based on cellular energy levels (Howlett et al., 1998). Lastly, since brain glycogen is an emergency glucose store, bGP is tightly regulated solely by modulator binding to respond to hypoxic stress and support high cognitive processes (Mathieu et al., 2016).
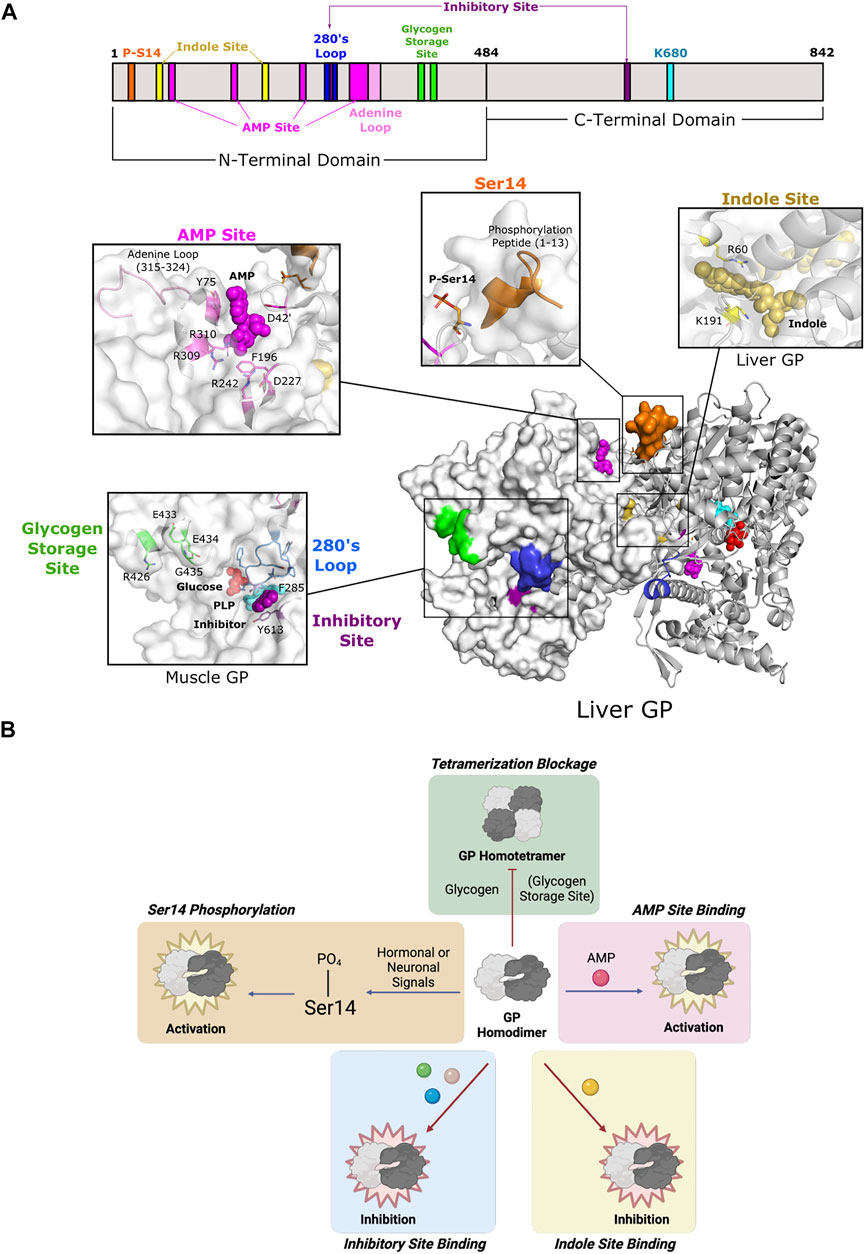
FIGURE 6. Glycogen phosphorylase of Fold Type V utilizes diverse modes of allostery. (A) One protomer of human liver GP homodimer (PDB 1FA9) is shown as a surface representation (white) and the second as a cartoon representation (gray). GP is phosphorylated at Ser14 (orange), which causes the phosphorylation peptide (aa 1–13; orange) to stabilize the active conformation. Ligand binding may occur at three sites: the AMP site, the indole site, and the inhibitory site. AMP binding at the AMP site (D42 of one subunit; Y75, F195, D227, R242, R309, and R310 of the other subunit; dark pink) is coordinated by the adenine loop (aa 315–324; light pink). The indole site (human liver GP, PDB 1XOI; R60, and K191; yellow) is located at the dimer interface. The inhibitory site (human muscle GP, PDB 1Z8D; F285, Y613; purple) is a shallow pocket at the entrance of the active site. The inhibitory site is also part of the 280s loop (aa 280–289; blue), which forms a gate for the active site. The glycogen storage site (R426, E433, E434, and G435; green) is part of the tetrameric interface (not shown). PLP and the active site lysine (K202) are shown in cyan. (B) GP is active as a homodimer and can be further activated via phosphorylation at Ser14 and/or AMP binding. Phosphorylation and AMP binding triggers GP tetramerization, which inactivates GP, but tetramerization can be blocked by glycogen binding. GP can also be inhibited by ligand binding at the indole and inhibitory sites.
2.5.2 AMP-Binding Site
All three isozymes have a well-conserved AMP-binding site that is formed by a bundle of helices located at the interface between dimer subunits (Figure 6A) (Sprang et al., 1991; Rath et al., 2000a). However, isozyme-specific amino acid substitutions within the site lead to differential binding affinity, thus affecting allosteric control (Hudson et al., 1993; Mathieu et al., 2016). This site is also known as the allosteric site because it contains three subsites (sugar, nucleotide base, and phosphate) that allows for promiscuous effector binding (e.g., AMP, ATP, and G6P). Because of the low level of specificity, the nature of the bound effector determines whether it exerts inhibition or activation of enzyme function (Wang et al., 1970). Since the position of the AMP site allows for effectors to bind between subunits, they are often associated with quaternary structural changes (Sprang et al., 1991).
The function of the AMP site corresponds with intracellular energy demands. As energy needs increase, the intracellular concentration of AMP increases because of ATP hydrolysis. AMP binds in this site to stabilize the active relaxed state of GP (R state) that allows for access to the catalytic site (Barford and Johnson 1989). ATP and G6P can then displace AMP and destabilize quaternary interactions to switch the enzyme conformation back to the less active tense state (T state) (Kasvinsky 1982; Sprang et al., 1987; Gaboriaud-Kolar and Skaltsounis 2013). Importantly, Ser14 phosphorylation complements AMP site binding by inducing conformational changes that bury the AMP site and increase effector binding through additional intermolecular forces (Barford et al., 1991).
2.5.3 Glycogen-Binding Site
The GP oligomeric state is an important facet in mediating GP activity. Homodimeric GP is the active form of the enzyme, but activation by AMP or Ser14 phosphorylation causes a change in the tertiary and quaternary structures of the enzyme to promote pairs of dimers to form tetramers. Tetramerization partially blocks access to the active site, thus decreasing enzyme activity to 12–33% of the fully active dimers (Huang and Graves 1970). Furthermore, formation of the tetrameric interface leads to global structural changes that affect the propagation of other allosteric effects (Barford and Johnson 1992). The glycogen storage site is located ∼30 Å from the catalytic site and is situated at the entrance of the catalytic tunnel (Figure 6A). This site forms a contact for the tetrameric interface, so binding of glycogen discourages further oligomerization and leaves the enzyme as an active homodimer (Figure 6B).
2.5.4 Inhibitory Site and Indole-Binding Site
Two additional effector binding sites have been described, the inhibitory site (also known as the nucleoside site or the purine site) and the indole site. Both sites inhibit glycogen phosphorylase by stabilizing the T state and working synergistically with other GP inhibitors (Kasvinsky et al., 1978a; Martin et al., 1998). The inhibitory site is a hydrophobic binding pocket ∼10 Å from the active site (Figure 6A). Its low specificity allows for binding to a diverse set of ligands, including purines, nucleosides, nucleotides, and other related heterocyclic compounds (Oikonomakos et al., 2000a). Inhibitory site ligands interact with a loop called the 280s loop that forms a gate to block substrate access to the active site. Communication with the AMP-binding site decreases AMP binding to hinder GP activation (Kasvinsky et al., 1978b; Buchbinder and Fletterick 1996; Ekstrom et al., 2002). The indole site was first discovered in human lGP during a screening of antidiabetic agents targeting GP (Rath et al., 2000b). The natural ligand is unknown, but synthetic effector binding forms a bridge between dimeric subunits, thus stabilizing the less active conformation and inhibiting GP. Without ligand binding at the indole site, the cavity is instead solvent-filled, which allows for the necessary rotation of subunits during activation (Ercan-Fang et al., 2005).
2.6 Fold Types VI and VII
Homology searches identified many PLP-dependent enzymes belonging to the above five fold types. However, other PLP-dependent enzymes that do not resemble the archetypal enzymes were also discovered and subsequently categorized into new fold types (Percudani and Peracchi 2003). Fold Types VI and VII are sparsely populated but contain examples of allostery. Lysine fermentation in anaerobic bacteria uses two analogous PLP-dependent enzymes, lysine 5,6-aminomutase (5,6-LAM) and lysine 2,3-aminomutase (2,3-LAM), to catalyze non-classical, free radical reactions (Ballinger et al., 1992; Chang and Frey 2000). Although both aminomutases have similar reaction mechanisms and analogous intermediates, their structures differ enough to organize into Fold Types VI and VII.
2.6.1 Fold Type VI: Lysine 5,6-Aminomutase (5,6-LAM)
Lysine 5,6-aminomutase (5,6-LAM) catalyzes the reversible 1,2 rearrangement of the terminal amino group of DL-lysine and L-β-lysine. Unlike other PLP-dependent enzymes, 5,6-LAM requires radical propagation from another cofactor, adenosylcobalamin (AdoCbl; vitamin B12), to the external aldimine for its reaction mechanism to occur. This protein forms an α2β2 tetramer (a dimer of αβ units) (Berkovitch et al., 2004), and the complete holoenzyme is composed of the tetramer in addition to an auxiliary activating protein (Figure 7A). The 5,6-LAM tetramer is known as the active core enzyme E1 that uses the coenzymes PLP and AdoCbl. The other component, a sulfhydryl protein E2, is responsible for the reactivation and ATP-dependent allosteric regulation of E1 (Baker et al., 1973; Chang and Frey 2000). The function of E2 may be tied to the exchange of free AdoCbl with bound cobalamins to reactivate the holoenzyme (Toraya and Mori 1999). The PLP- and AdoCbl-dependent D-ornithine 4,5-aminomutase (4,5-OAM) is structurally similar and has a similar reaction mechanism to 5,6-LAM (Barker 1981). 4,5-OAM is an α2β2 heterotetramer made of the catalytic β subunit and the α subunit necessary for folding (Chen et al., 2001; Wolthers et al., 2008). The α subunit can form a complex with the 5,6-LAM heterotetramer to restore ATP regulation and may work together with E2 to reactivate E1 (Tseng et al., 2007; Wolthers et al., 2008).
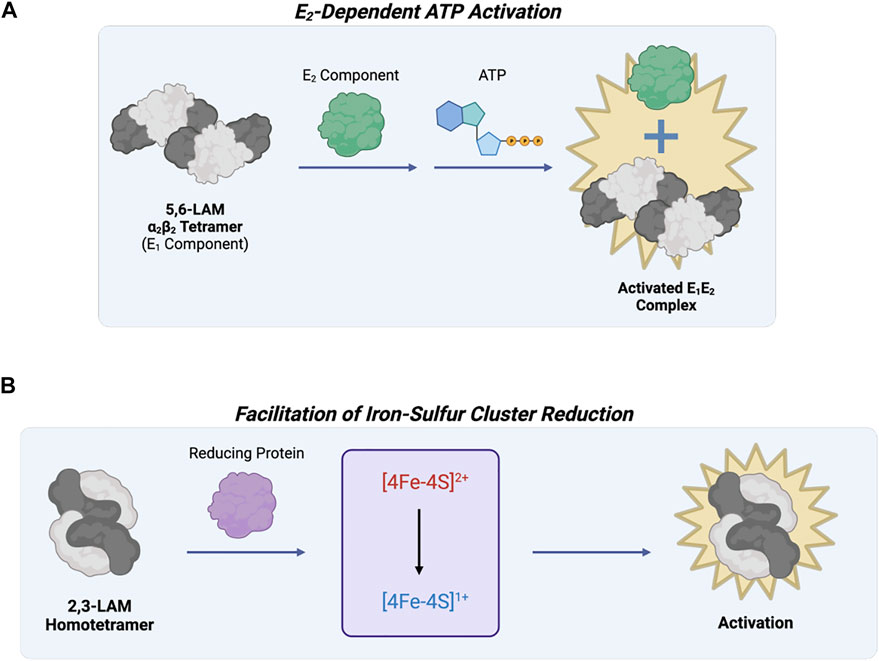
FIGURE 7. Lysine 5,6-aminomutase of Fold Type VI and Lysine 2,3-aminomutase of Fold Type VII are involved in the same metabolic pathway but are differentially regulated. (A) 5,6-LAM (Fold Type VI) is active as an α2β2 tetramer (known as the E1 component), but reactivation and ATP regulation are possible through interaction with another protein referred to as the E2 component. (B) 2,3-LAM (Fold Type VII) uses three cofactors, PLP, AdoMet, and [4Fe-4S]+, for catalysis. AdoMet and [4Fe-4S]+ work together for adenosyl radical formation, but this step is limited by 4Fe-4S cluster formation and reduction to the +1 state. Interaction between 2,3-LAM and a reducing protein (e.g., flavodoxin NADP+ reductase, flavodoxin, or ferredoxin) activates 2,3-LAM.
2.6.2 Fold Type VII: Lysine 2,3-Aminomutase (2,3-LAM)
Lysine 2,3-aminomutase (2,3-LAM) was the first aminomutase to be discovered (Chirpich et al., 1970). It is a homotetramer (a dimer of dimers) that catalyzes the interconversion of L-α-lysine and L-β-lysine using PLP, AdoMet, and a [4Fe-4S]+ cluster as coenzymes (Lepore et al., 2005). Most isomerization reactions require the use of AdoCbl as a coenzyme, but 2,3-LAM uses AdoMet and a [4Fe-4S]+ cluster to mediate hydrogen transfer by radical propagation. Since AdoMet is not as easily cleaved as AdoCbl, the iron-sulfur cluster is needed as an electron source to convert AdoMet into methionine and an Ado radical (Baraniak et al., 1989; Ballinger et al., 1992). Because of the essential nature of the iron-sulfur cluster, 2,3-LAM is catalytically limited by cluster formation and reduction to the +1 state. Previous work showed reducing proteins (e.g., flavodoxin NADP+ reductase, flavodoxin, and ferredoxin) can help activate 2,3-LAM by facilitating cluster reduction (Figure 7B) (Brazeau et al., 2006).
3 Discussion
Pyridoxal 5′-phosphate is a coenzyme involved in a number of essential cellular processes within nearly all organisms. The diversity of reactions catalyzed by PLP-dependent enzymes is not fully realized solely by examination of their 3-D structures and active site architectures. Rather, this review provides an overview of the diverse mechanisms of protein allostery utilized by members of PLP-dependent enzyme families. It is through allostery that PLP-dependent activities are tuned to meet a particular cellular need. Although there are some reoccurring themes, such as oxidation of a CXXC motif or regulation via an N-terminal or C-terminal extension, there are also many divergent mechanisms. These mechanisms include binding of diverse protein or small molecule effectors at distal sites which induce conformational changes that alter oligomerization or active site accessibility and architecture. Here, we present allosteric mechanisms from various members of each fold type. However, as structural and biochemical investigations continue, it is likely that more allosteric routes will be discovered.
A common approach in drug development is the design of competitive inhibitors that directly disrupt catalysis by active site interference. This concept proves difficult with PLP-dependent enzymes because of the common reaction mechanisms within fold types. How can we target a particular enzyme without affecting a litany of other biological processes? Nature surmounts this problem by the evolution of allosteric regulation, where structural changes distal to the active site induce conformational changes to fine-tune catalysis. Allostery does not center around a certain mechanism but rather encompasses a multitude of means. By exploiting the natural phenomenon of allostery, the issue of active site similarity can be circumvented, and PLP-dependent enzymes that have been previously structurally characterized can be reexamined for novel therapeutic development. Not only will this expand drug discovery opportunities, but it will also aid in exploring novel structure-based means of allosteric regulation.
Author Contributions
JT and BB conceptualized, wrote, and edited the manuscript.
Funding
JT is supported by the Vanderbilt Chemical-Biology Interface Training Grant (5T32GM065086). BB is supported by the United Mitochondrial Disease Foundation (ESPI-2020-0003) and by the National Institute of General Medical Sciences of the National Institutes of Health (DP2GM146255).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.884281/full#supplementary-material
References
Agius, L. (2015). Role of Glycogen Phosphorylase in Liver Glycogen Metabolism. Mol. Aspects Med. 46, 34–45. doi:10.1016/j.mam.2015.09.002
Almrud, J. J., Oliveira, M. A., Kern, A. D., Grishin, N. V., Phillips, M. A., and Hackert, M. L. (2000). Crystal Structure of Human Ornithine Decarboxylase at 2.1 Å Resolution: Structural Insights to Antizyme Binding. J. Mol. Biol. 295, 7–16. doi:10.1006/jmbi.1999.3331
Andréll, J., Hicks, M. G., Palmer, T., Carpenter, E. P., Iwata, S., and Maher, M. J. (2009). Crystal Structure of the Acid-Induced Arginine Decarboxylase from Escherichia coli: Reversible Decamer Assembly Controls Enzyme Activity. Biochemistry 48, 3915–3927. doi:10.1021/bi900075d
Astner, I., Schulze, J. O., van den Heuvel, J., Jahn, D., Schubert, W.-D., and Heinz, D. W. (2005). Crystal Structure of 5-aminolevulinate Synthase, the First Enzyme of Heme Biosynthesis, and its Link to XLSA in Humans. EMBO J. 24, 3166–3177. doi:10.1038/sj.emboj.7600792
Auvinen, M., Paasinen, A., Andersson, L. C., and Hölttä, E. (1992). Ornithine Decarboxylase Activity Is Critical for Cell Transformation. Nature 360, 355–358. doi:10.1038/360355a0
Badawy, A. A. (2021). Multiple Roles of Haem in Cystathionine β-synthase Activity: Implications for Hemin and Other Therapies of Acute Hepatic Porphyria. Biosci. Rep. 41, BSR20210935. doi:10.1042/BSR20210935
Bailey, H. J., Bezerra, G. A., Marcero, J. R., Padhi, S., Foster, W. R., Rembeza, E., et al. (2020). Human Aminolevulinate Synthase Structure Reveals a Eukaryotic-specific Autoinhibitory Loop Regulating Substrate Binding and Product Release. Nat. Commun. 11, 2813. doi:10.1038/s41467-020-16586-x
Baker, J. J., van der Drift, C., and Stadtman, T. C. (1973). Purification and Properties of β-lysine Mutase, a Pyridoxal Phosphate and B12 Coenzyme Dependent Enzyme. Biochemistry 12, 1054–1063. doi:10.1021/bi00730a006
Ballinger, M. D., Frey, P. A., and Reed, G. H. (1992). Structure of a Substrate Radical Intermediate in the Reaction of Lysine 2,3-aminomutase. Biochemistry 31, 10782–10789. doi:10.1021/bi00159a020
Baraniak, J., Moss, M. L., and Frey, P. A. (1989). Lysine 2,3-Aminomutase. J. Biol. Chem. 264, 1357–1360. doi:10.1016/s0021-9258(18)94194-3
Barford, D., Hu, S.-H., and Johnson, L. N. (1991). Structural Mechanism for Glycogen Phosphorylase Control by Phosphorylation and AMP. J. Mol. Biol. 218, 233–260. doi:10.1016/0022-2836(91)90887-c
Barford, D., and Johnson, L. N. (1989). The Allosteric Transition of Glycogen Phosphorylase. Nature 340, 609–616. doi:10.1038/340609a0
Barford, D., and Johnson, L. N. (1992). The Molecular Mechanism for the Tetrameric Association of Glycogen Phosphorylase Promoted by Protein Phosphorylation. Protein Sci. 1, 472–493. doi:10.1002/pro.5560010403
Barker, H. A. (1981). Amino Acid Degradation by Anaerobic Bacteria. Annu. Rev. Biochem. 50, 23–40. doi:10.1146/annurev.bi.50.070181.000323
Barrett, M. P., Boykin, D. W., Brun, R., and Tidwell, R. R. (2007). Human African Trypanosomiasis: Pharmacological Re-engagement with a Neglected Disease. Br. J. Pharmacol. 152, 1155–1171. doi:10.1038/sj.bjp.0707354
Bateman, A. (1997). The Structure of a Domain Common to Archaebacteria and the Homocystinuria Disease Protein. Trends Biochem. Sci. 22, 12–13. doi:10.1016/s0968-0004(96)30046-7
Bauer, P. M., Buga, G. M., Fukuto, J. M., Pegg, A. E., and Ignarro, L. J. (2001). Nitric Oxide Inhibits Ornithine Decarboxylase viaS-Nitrosylation of Cysteine 360 in the Active Site of the Enzyme. J. Biol. Chem. 276, 34458–34464. doi:10.1074/jbc.M105219200
Berkovitch, F., Behshad, E., Tang, K.-H., Enns, E. A., Frey, P. A., and Drennan, C. L. (2004). A Locking Mechanism Preventing Radical Damage in the Absence of Substrate, as Revealed by the X-ray Structure of Lysine 5,6-aminomutase. Proc. Natl. Acad. Sci. U.S.A. 101, 15870–15875. doi:10.1073/pnas.0407074101
Bertoldi, M., Gonsalvi, M., Contestabile, R., and Voltattorni, C. B. (2002). Mutation of Tyrosine 332 to Phenylalanine Converts Dopa Decarboxylase into a Decarboxylation-dependent Oxidative Deaminase. J. Biol. Chem. 277, 36357–36362. doi:10.1074/jbc.M204867200
Bertoldi, M. (2014). Mammalian Dopa Decarboxylase: Structure, Catalytic Activity and Inhibition. Arch. Biochem. Biophys. 546, 1–7. doi:10.1016/j.abb.2013.12.020
Bezsudnova, E. Y., Popov, V. O., and Boyko, K. M. (2020). Structural Insight into the Substrate Specificity of PLP Fold Type IV Transaminases. Appl. Microbiol. Biotechnol. 104, 2343–2357. doi:10.1007/s00253-020-10369-6
Bishop, D. F., Tchaikovskii, V., Nazarenko, I., and Desnick, R. J. (2013). Molecular Expression and Characterization of Erythroid-specific 5-aminolevulinate Synthase Gain-Of-Function Mutations Causing X-Linked Protoporphyria. Mol. Med. 19, 18–25. doi:10.2119/molmed.2013.00003
Bosken, Y. K., Ai, R., Hilario, E., Ghosh, R. K., Dunn, M. F., Kan, S. H., et al. (2022). Discovery of Antimicrobial Agent Targeting Tryptophan Synthase. Protein Sci. 31, 432–442. doi:10.1002/pro.4236
Brazeau, B. J., Gort, S. J., Jessen, H. J., Andrew, A. J., and Liao, H. H. (2006). Enzymatic Activation of Lysine 2,3-aminomutase from Porphyromonas Gingivalis. Appl. Environ. Microbiol. 72, 6402–6404. doi:10.1128/AEM.01143-06
Brown, B. L., Kardon, J. R., Sauer, R. T., and Baker, T. A. (2018). Structure of the Mitochondrial Aminolevulinic Acid Synthase, a Key Heme Biosynthetic Enzyme. Structure 26, 580–589. doi:10.1016/j.str.2018.02.012
Bruno, S., Margiotta, M., Marchesani, F., Paredi, G., Orlandi, V., Faggiano, S., et al. (2017). Magnesium and Calcium Ions Differentially Affect Human Serine Racemase Activity and Modulate its Quaternary Equilibrium toward a Tetrameric Form. Biochim. Biophys. Acta (Bba) - Proteins Proteomics 1865, 381–387. doi:10.1016/j.bbapap.2017.01.001
Buchbinder, J. L., and Fletterick, R. J. (1996). Role of the Active Site Gate of Glycogen Phosphorylase in Allosteric Inhibition and Substrate Binding. J. Biol. Chem. 271, 22305–22309. doi:10.1074/jbc.271.37.22305
Buller, A. R., Brinkmann-Chen, S., Romney, D. K., Herger, M., Murciano-Calles, J., and Arnold, F. H. (2015). Directed Evolution of the Tryptophan Synthase β-subunit for Stand-Alone Function Recapitulates Allosteric Activation. Proc. Natl. Acad. Sci. U.S.A. 112, 14599–14604. doi:10.1073/pnas.1516401112
Burkhard, P., Tai, C.-H., Jansonius, J. N., and Cook, P. F. (2000). Identification of an Allosteric Anion-Binding Site on O-Acetylserine Sulfhydrylase: Structure of the Enzyme with Chloride Bound. J. Mol. Biol. 303, 279–286. doi:10.1006/jmbi.2000.4109
Campanini, B., Speroni, F., Salsi, E., Cook, P. F., Roderick, S. L., Huang, B., et al. (2005). Interaction of Serine Acetyltransferase withO-Acetylserine Sulfhydrylase Active Site: Evidence from Fluorescence Spectroscopy. Protein Sci. 14, 2115–2124. doi:10.1110/ps.051492805
Canosa, A. V., Faggiano, S., Marchetti, M., Armao, S., Bettati, S., Bruno, S., et al. (2018). Glutamine 89 Is a Key Residue in the Allosteric Modulation of Human Serine Racemase Activity by ATP. Sci. Rep. 8, 9016. doi:10.1038/s41598-018-27227-1
Carballal, S., Cuevasanta, E., Marmisolle, I., Kabil, O., Gherasim, C., Ballou, D. P., et al. (2013). Kinetics of Reversible Reductive Carbonylation of Heme in Human Cystathionine β-Synthase. Biochemistry 52, 4553–4562. doi:10.1021/bi4004556
Chai, X., Zhan, J., Pan, J., He, M., Li, B., Wang, J., et al. (2020). The Rational Discovery of Multipurpose Inhibitors of the Ornithine Decarboxylase. FASEB j. 34, 10907–12921. doi:10.1096/fj.202001222R
Chang, C. H., and Frey, P. A. (2000). Cloning, Sequencing, Heterologous Expression, Purification, and Characterization of Adenosylcobalamin-Dependentd-Lysine 5,6-Aminomutase from Clostridium Sticklandii. J. Biol. Chem. 275, 106–114. doi:10.1074/jbc.275.1.106
Changeux, J.-P. (1963). Allosteric Interactions on Biosynthetic L-Threonine Deaminase from E. coli K12. Cold Spring Harbor Symposia Quantitative Biol. 28, 497–504. doi:10.1101/SQB.1963.028.01.066
Chant, E. L., and Summers, D. K. (2007). Indole Signalling Contributes to the Stable Maintenance of Escherichia coli Multicopy Plasmids. Mol. Microbiol. 63, 35–43. doi:10.1111/j.1365-2958.2006.05481.x
Chen, H.-P., Wu, S.-H., Lin, Y.-L., Chen, C.-M., and Tsay, S.-S. (2001). Cloning, Sequencing, Heterologous Expression, Purification, and Characterization of Adenosylcobalamin-Dependentd-Ornithine Aminomutase from Clostridium Sticklandii. J. Biol. Chem. 276, 44744–44750. doi:10.1074/jbc.M108365200
Chen, L., Chen, Z., Zheng, P., Sun, J., and Zeng, A.-P. (2013). Study and Reengineering of the Binding Sites and Allosteric Regulation of Biosynthetic Threonine Deaminase by Isoleucine and Valine in Escherichia coli. Appl. Microbiol. Biotechnol. 97, 2939–2949. doi:10.1007/s00253-012-4176-z
Chirpich, T. P., Zappia, V., Costilow, R. N., and Barker, H. A. (1970). Lysine 2,3-Aminomutase. J. Biol. Chem. 245, 1778–1789. doi:10.1016/s0021-9258(19)77160-9
Chou, C.-C., Modi, J. P., Wang, C.-Y., Hsu, P.-C., Lee, Y.-H., Huang, K.-F., et al. (2017). Activation of Brain L-Glutamate Decarboxylase 65 Isoform (GAD65) by Phosphorylation at Threonine 95 (T95). Mol. Neurobiol. 54, 866–873. doi:10.1007/s12035-015-9633-0
Conway, M. E., Poole, L. B., and Hutson, S. M. (2004). Roles for Cysteine Residues in the Regulatory CXXC Motif of Human Mitochondrial Branched Chain Aminotransferase Enzyme. Biochemistry 43, 7356–7364. doi:10.1021/bi0498050
Curien, G., Job, D., Douce, R., and Dumas, R. (1998). Allosteric Activation of Arabidopsis Threonine Synthase by S-Adenosylmethionine. Biochemistry 37, 13212–13221. doi:10.1021/bi980068f
D’Amico, R. N., Bosken, Y. K., O’Rourke, K. F., Murray, A. M., Admasu, W., Chang, C.-e. A., et al. (2021). Substitution of a Surface-Exposed Residue Involved in an Allosteric Network Enhances Tryptophan Synthase Function in Cells. Front. Mol. Biosci. 8, 679915. doi:10.3389/fmolb.2021.679915
David, E., and Crerar, M. (1986). Quantitation of Muscle Glycogen Phosphorylase mRNA and Enzyme Amounts in Adult Rat Tissues. Biochim. Biophys. Acta (Bba) - Gen. Subjects 880, 78–90. doi:10.1016/0304-4165(86)90122-4
Deng, H., Zhou, J., Sundersingh, F. S., Summerfield, J., Somers, D., Messer, J. A., et al. (2015). Discovery, SAR, and X-ray Binding Mode Study of BCATm Inhibitors from a Novel DNA-Encoded Library. ACS Med. Chem. Lett. 6, 919–924. doi:10.1021/acsmedchemlett.5b00179
Ducamp, S., Schneider-Yin, X., de Rooij, F., Clayton, J., Fratz, E. J., Rudd, A., et al. (2013). Molecular and Functional Analysis of the C-Terminal Region of Human Erythroid-specific 5-aminolevulinic Synthase Associated with X-Linked Dominant Protoporphyria (XLDPP). Hum. Mol. Genet. 22, 1280–1288. doi:10.1093/hmg/dds531
Dunn, M. F. (2012). Allosteric Regulation of Substrate Channeling and Catalysis in the Tryptophan Synthase Bienzyme Complex. Arch. Biochem. Biophys. 519, 154–166. doi:10.1016/j.abb.2012.01.016
Eisenstein, E. (1995). Allosteric Regulation of Biosynthetic Threonine Deaminase from Escherichia coli: Effects of Isoleucine and Valine on Active-Site Ligand Binding and Catalysis. Arch. Biochem. Biophys. 316, 311–318. doi:10.1006/abbi.1995.1042
Ekstrom, J. L., Pauly, T. A., Carty, M. D., Soeller, W. C., Culp, J., Danley, D. E., et al. (2002). Structure-activity Analysis of the Purine Binding Site of Human Liver Glycogen Phosphorylase. Chem. Biol. 9, 915–924. doi:10.1016/s1074-5521(02)00186-2
El Hindy, M., Hezwani, M., Corry, D., Hull, J., El Amraoui, F., Harris, M., et al. (2014). The Branched-Chain Aminotransferase Proteins: Novel Redox Chaperones for Protein Disulfide Isomerase-Implications in Alzheimer's Disease. Antioxid. Redox Signaling 20, 2497–2513. doi:10.1089/ars.2012.4869
Eliot, A. C., and Kirsch, J. F. (2004). Pyridoxal Phosphate Enzymes: Mechanistic, Structural, and Evolutionary Considerations. Annu. Rev. Biochem. 73, 383–415. doi:10.1146/annurev.biochem.73.011303.074021
Ercan-Fang, N., Taylor, M. R., Treadway, J. L., Levy, C. B., Genereux, P. E., Gibbs, E. M., et al. (2005). Endogenous Effectors of Human Liver Glycogen Phosphorylase Modulate Effects of Indole-Site Inhibitors. Am. J. Physiology-Endocrinology Metab. 289, E366–E372. doi:10.1152/ajpendo.00264.2004
Ereño-Orbea, J., Majtan, T., Oyenarte, I., Kraus, J. P., and Martínez-Cruz, L. A. (2013). Structural Basis of Regulation and Oligomerization of Human Cystathionine β-synthase, the central Enzyme of Transsulfuration. Proc. Natl. Acad. Sci. U.S.A. 110, E3790–E3799. doi:10.1073/pnas.1313683110
Fatmi, M. Q., and Chang, C.-e. A. (2010). The Role of Oligomerization and Cooperative Regulation in Protein Function: the Case of Tryptophan Synthase. Plos Comput. Biol. 6, e1000994. doi:10.1371/journal.pcbi.1000994
Fenalti, G., Law, R. H. P., Buckle, A. M., Langendorf, C., Tuck, K., Rosado, C. J., et al. (2007). GABA Production by Glutamic Acid Decarboxylase Is Regulated by a Dynamic Catalytic Loop. Nat. Struct. Mol. Biol. 14, 280–286. doi:10.1038/nsmb1228
Fratz, E. J., Clayton, J., Hunter, G. A., Ducamp, S., Breydo, L., Uversky, V. N., et al. (2015). Human Erythroid 5-Aminolevulinate Synthase Mutations Associated with X-Linked Protoporphyria Disrupt the Conformational Equilibrium and Enhance Product Release. Biochemistry 54, 5617–5631. doi:10.1021/acs.biochem.5b00407
Fujii, K., Maeda, K., Hikida, T., Mustafa, A. K., Balkissoon, R., Xia, J., et al. (2006). Serine Racemase Binds to PICK1: Potential Relevance to Schizophrenia. Mol. Psychiatry 11, 150–157. doi:10.1038/sj.mp.4001776
Fujita, K., Matsufuji, S., Murakami, Y., and Hayashi, S. (1984). Antizyme to Ornithine Decarboxylase Is Present in the Liver of Starved Rats. Biochem. J. 218, 557–562. doi:10.1042/bj2180557
Gaboriaud-Kolar, N., and Skaltsounis, A.-L. (2013). Glycogen Phosphorylase Inhibitors: a Patent Review (2008 - 2012). Expert Opin. Ther. Patents 23, 1017–1032. doi:10.1517/13543776.2013.794790
Gale, E. F. (1940). The Production of Amines by Bacteria. Biochem. J. 34, 392–413. doi:10.1042/bj0340392
Gallagher, D. T., Gilliland, G. L., Xiao, G., Zondlo, J., Fisher, K. E., Chinchilla, D., et al. (1998). Structure and Control of Pyridoxal Phosphate Dependent Allosteric Threonine Deaminase. Structure 6, 465–475. doi:10.1016/s0969-2126(98)00048-3
Garrido-Franco, M., Ehlert, S., Messerschmidt, A., Marinković, S., Huber, R., Laber, B., et al. (2002). Structure and Function of Threonine Synthase from Yeast. J. Biol. Chem. 277, 12396–12405. doi:10.1074/jbc.M108734200
Gerritsen, T., and Waisman, H. A. (1964). Homocystinuria: Absence of Cystathionine in the Brain. Science 145, 588. doi:10.1126/science.145.3632.58810.1126/science.145.3632.588.a
Ghoda, L., van Daalen Wetters, T., Macrae, M., Ascherman, D., and Coffino, P. (1989). Prevention of Rapid Intracellular Degradation of ODC by a Carboxyl-Terminal Truncation. Science 243, 1493–1495. doi:10.1126/science.2928784
Ghosh, R. K., Hilario, E., Liu, V., Wang, Y., Niks, D., Holmes, J. B., et al. (2021). Mutation of βGln114 to Ala Alters the Stabilities of Allosteric States in Tryptophan Synthase Catalysis. Biochemistry 60, 3173–3186. doi:10.1021/acs.biochem.1c00383
Giardina, G., Brunotti, P., Fiascarelli, A., Cicalini, A., Costa, M. G. S., Buckle, A. M., et al. (2015). How Pyridoxal 5′-phosphate Differentially Regulates Human Cytosolic and Mitochondrial Serine Hydroxymethyltransferase Oligomeric State. FEBS J. 282, 1225–1241. doi:10.1111/febs.13211
Giardina, G., Montioli, R., Gianni, S., Cellini, B., Paiardini, A., Voltattorni, C. B., et al. (2011). Open Conformation of Human DOPA Decarboxylase Reveals the Mechanism of PLP Addition to Group II Decarboxylases. Proc. Natl. Acad. Sci. U.S.A. 108, 20514–20519. doi:10.1073/pnas.1111456108
Gibson, K. D., Laver, W. G., and Neuberger, A. (1958). Initial Stages in the Biosynthesis of Porphyrins. 2. The Formation of δ-aminolaevulic Acid from glycine and Succinyl-Coenzyme A by Particles from Chicken Erythrocytes. Biochem. J. 70, 71–81. doi:10.1042/bj0700071
Gong, J., and Ferreira, G. C. (1995). Aminolevulinate Synthase: Functionally Important Residues at a glycine Loop, a Putative Pyridoxal Phosphate Cofactor-Binding Site. Biochemistry 34, 1678–1685. doi:10.1021/bi00005a024
Grishin, N. V., Osterman, A. L., Brooks, H. B., Phillips, M. A., and Goldsmith, E. J. (1999). X-ray Structure of Ornithine Decarboxylase from Trypanosoma Brucei: The Native Structure and the Structure in Complex with α-Difluoromethylornithine,. Biochemistry 38, 15174–15184. doi:10.1021/bi9915115
Grishin, N. V., Phillips, M. A., and Goldsmith, E. J. (1995). Modeling of the Spatial Structure of Eukaryotic Ornithine Decarboxylases. Protein Sci. 4, 1291–1304. doi: DOI doi:10.1002/pro.5560040705
Harmon, J. M., Bacikova, D., Gable, K., Gupta, S. D., Han, G., Sengupta, N., et al. (2013). Topological and Functional Characterization of the ssSPTs, Small Activating Subunits of Serine Palmitoyltransferase. J. Biol. Chem. 288, 10144–10153. doi:10.1074/jbc.M113.451526
Hattori, A., Tsunoda, M., Konuma, T., Kobayashi, M., Nagy, T., Glushka, J., et al. (2017). Cancer Progression by Reprogrammed BCAA Metabolism in Myeloid Leukaemia. Nature 545, 500–504. doi:10.1038/nature22314
Heller, J. S., Fong, W. F., and Canellakis, E. S. (1976). Induction of a Protein Inhibitor to Ornithine Decarboxylase by the End Products of its Reaction. Proc. Natl. Acad. Sci. U.S.A. 73, 1858–1862. doi:10.1073/pnas.73.6.1858
Hennig, M., Grimm, B., Contestabile, R., John, R. A., and Jansonius, J. N. (1997). Crystal Structure of Glutamate-1-Semialdehyde Aminomutase: An α 2 -dimeric Vitamin B 6 -dependent Enzyme with Asymmetry in Structure and Active Site Reactivity. Proc. Natl. Acad. Sci. U.S.A. 94, 4866–4871. doi:10.1073/pnas.94.10.4866
Hestrin, S. (1949). Action Pattern of Crystalline Muscle Phosphorylase. J. Biol. Chem. 179, 943–955. doi:10.1016/s0021-9258(19)51288-1
Hohenester, E., Keller, J. W., and Jansonius, J. N. (1994). An Alkali Metal Ion Size-dependent Switch in the Active Site Structure of Dialkylglycine Decarboxylase. Biochemistry 33, 13561–13570. doi:10.1021/bi00250a008
Howlett, R. A., Parolin, M. L., Dyck, D. J., Hultman, E., Jones, N. L., Heigenhauser, G. J. F., et al. (1998). Regulation of Skeletal Muscle Glycogen Phosphorylase and PDH at Varying Exercise Power Outputs. Am. J. Physiology-Regulatory, Integr. Comp. Physiol. 275, R418–R425. doi:10.1152/ajpregu.1998.275.2.R418
Huang, C. Y., and Graves, D. J. (1970). Correlation between Subunit Interactions and Enzymic Activity of Phosphorylase a. Method for Determining Equilibrium Constants from Initial Rate Measurements. Biochemistry 9, 660–671. doi:10.1021/bi00805a028
Hudson, J. W., Golding, G. B., and Crerar, M. M. (1993). Evolution of Allosteric Control in Glycogen Phosphorylase. J. Mol. Biol. 234, 700–721. doi:10.1006/jmbi.1993.1621
Hunter, G. A., and Ferreira, G. C. (1999). Pre-steady-state Reaction of 5-Aminolevulinate Synthase. J. Biol. Chem. 274, 12222–12228. doi:10.1074/jbc.274.18.12222
Hunter, G. A., Zhang, J., and Ferreira, G. C. (2007). Transient Kinetic Studies Support Refinements to the Chemical and Kinetic Mechanisms of Aminolevulinate Synthase. J. Biol. Chem. 282, 23025–23035. doi:10.1074/jbc.M609330200
Hyde, C. C., Ahmed, S. A., Padlan, E. A., Miles, E. W., and Davies, D. R. (1988). Three-dimensional Structure of the Tryptophan Synthase Alpha 2 Beta 2 Multienzyme Complex from Salmonella typhimurium. J. Biol. Chem. 263, 17857–17871. doi:10.1016/s0021-9258(19)77913-7
Ichihara, A., and Koyama, E. (1966). Transaminase of Branched Chain Amino Acids. J. Biochem. 59, 160–169. doi:10.1093/oxfordjournals.jbchem.a128277
Ignoul, S., and Eggermont, J. (2005). CBS Domains: Structure, Function, and Pathology in Human Proteins. Am. J. Physiology-Cell Physiol. 289, C1369–C1378. doi:10.1152/ajpcell.00282.2005
Inoue, K., Kuramitsu, S., Aki, K., Watanabe, Y., Takagi, T., Nishigai, M., et al. (1988). Branched-Chain Amino Acid Aminotransferase of Escherichia coli: Overproduction and Properties1. J. Biochem. 104, 777–784. doi:10.1093/oxfordjournals.jbchem.a122549
Isupov, M. N., Boyko, K. M., Sutter, J. M., James, P., Sayer, C., Schmidt, M., et al. (2019). Thermostable Branched-Chain Amino Acid Transaminases from the Archaea Geoglobus Acetivorans and Archaeoglobus Fulgidus: Biochemical and Structural Characterization. Front. Bioeng. Biotechnol. 7, 7. doi:10.3389/fbioe.2019.00007
Isupov, M. N., Antson, A. A., Dodson, E. J., Dodson, G. G., Dementieva, I. S., Zakomirdina, L. N., et al. (1998). Crystal Structure of Tryptophanase. J. Mol. Biol. 276, 603–623. doi:10.1006/jmbi.1997.1561
Iwasaki, A., Matsumoto, K., Hasegawa, J., and Yasohara, Y. (2012). A Novel Transaminase, (R)-amine:pyruvate Aminotransferase, from Arthrobacter Sp. KNK168 (FERM BP-5228): Purification, Characterization, and Gene Cloning. Appl. Microbiol. Biotechnol. 93, 1563–1573. doi:10.1007/s00253-011-3580-0
Jackson, L. K., Baldwin, J., Akella, R., Goldsmith, E. J., and Phillips, M. A. (2004). Multiple Active Site Conformations Revealed by Distant Site Mutation in Ornithine Decarboxylase,. Biochemistry 43, 12990–12999. doi:10.1021/bi048933l
Jackson, L. K., Goldsmith, E. J., and Phillips, M. A. (2003). X-ray Structure Determination of Trypanosoma Brucei Ornithine Decarboxylase Bound to D-Ornithine and to G418. J. Biol. Chem. 278, 22037–22043. doi:10.1074/jbc.M300188200
Janošík, M., Kery, V., Gaustadnes, M., Maclean, K. N., and Kraus, J. P. (2001). Regulation of Human Cystathionine β-Synthase by S-Adenosyl-L-Methionine: Evidence for Two Catalytically Active Conformations Involving an Autoinhibitory Domain in the C-Terminal Region. Biochemistry 40, 10625–10633. doi:10.1021/bi010711p
Jhee, K.-H., McPhie, P., and Miles, E. W. (2000). Yeast Cystathionine β-Synthase Is a Pyridoxal Phosphate Enzyme but, unlike the Human Enzyme, Is Not a Heme Protein. J. Biol. Chem. 275, 11541–11544. doi:10.1074/jbc.c000056200
Jyothikumar, J., Chandani, S., and Ramakrishna, T. (2018). Computational Evidence of a New Allosteric Communication Pathway between Active Sites and Putative Regulatory Sites in the Alanine Racemase ofMycobacterium Tuberculosis. bioRxiv, 346130. doi:10.1101/346130
Kabil, O., Weeks, C. L., Carballal, S., Gherasim, C., Alvarez, B., Spiro, T. G., et al. (2011). Reversible Heme-dependent Regulation of Human Cystathionine β-Synthase by a Flavoprotein Oxidoreductase. Biochemistry 50, 8261–8263. doi:10.1021/bi201270q
Kadirvel, S., Furuyama, K., Harigae, H., Kaneko, K., Tamai, Y., Ishida, Y., et al. (2012). The Carboxyl-Terminal Region of Erythroid-specific 5-aminolevulinate Synthase Acts as an Intrinsic Modifier for its Catalytic Activity and Protein Stability. Exp. Hematol. 40, 477–486. doi:10.1016/j.exphem.2012.01.013
Kasvinsky, P. J., Madsen, N. B., Sygusch, J., and Fletterick, R. J. (1978a). The Regulation of Glycogen Phosphorylase Alpha by Nucleotide Derivatives. Kinetic and X-ray Crystallographic Studies. J. Biol. Chem. 253, 3343–3351. doi:10.1016/s0021-9258(17)40842-8
Kasvinsky, P. J., Shechosky, S., and Fletterick, R. J. (1978b). Synergistic Regulation of Phosphorylase a by Glucose and Caffeine. J. Biol. Chem. 253, 9102–9106. doi:10.1016/s0021-9258(17)34291-6
Kasvinsky, P. J. (1982). The Effect of AMP on Inhibition of Muscle Phosphorylase a by Glucose Derivatives. J. Biol. Chem. 257, 10805–10810. doi:10.1016/s0021-9258(18)33896-1
Katsurada, T., Kawabata, H., Kawabata, D., Kawahara, M., Nakabo, Y., Takaori-Kondo, A., et al. (2016). A Japanese Family with X-Linked Sideroblastic Anemia Affecting Females and Manifesting as Macrocytic Anemia. Int. J. Hematol. 103, 713–717. doi:10.1007/s12185-016-1949-7
Kaushik, A., Rahisuddin, R., Saini, N., Singh, R. P., Kaur, R., Koul, S., et al. (2021). Molecular Mechanism of Selective Substrate Engagement and Inhibitor Disengagement of Cysteine Synthase. J. Biol. Chem. 296, 100041. doi:10.1074/jbc.RA120.014490
Kern, A. D., Oliveira, M. A., Coffino, P., and Hackert, M. L. (1999). Structure of Mammalian Ornithine Decarboxylase at 1.6 Å Resolution: Stereochemical Implications of PLP-dependent Amino Acid Decarboxylases. Structure 7, 567–581. doi:10.1016/S0969-2126(99)80073-2
Kikuchi, G., Kumar, A., Talmage, P., and Shemin, D. (1958). The Enzymatic Synthesis of δ-Aminolevulinic Acid. J. Biol. Chem. 233, 1214–1219. doi:10.1016/s0021-9258(19)77371-2
Kim, D. J., Roh, E., Lee, M.-H., Oi, N., Lim, D. Y., Kim, M. O., et al. (2016). Herbacetin Is a Novel Allosteric Inhibitor of Ornithine Decarboxylase with Antitumor Activity. Cancer Res. 76, 1146–1157. doi:10.1158/0008-5472.Can-15-0442
Kim, P. M., Aizawa, H., Kim, P. S., Huang, A. S., Wickramasinghe, S. R., Kashani, A. H., et al. (2005). Serine Racemase: Activation by Glutamate Neurotransmission via Glutamate Receptor Interacting Protein and Mediation of Neuronal Migration. Proc. Natl. Acad. Sci. U.S.A. 102, 2105–2110. doi:10.1073/pnas.0409723102
Krebs, E. G., and Fischer, E. H. (1956). The Phosphorylase B to a Converting Enzyme of Rabbit Skeletal Muscle. Biochim. Biophys. Acta 20, 150–157. doi:10.1016/0006-3002(56)90273-6
Kumar, A., Wissbrock, A., Goradia, N., Bellstedt, P., Ramachandran, R., Imhof, D., et al. (2018). Heme Interaction of the Intrinsically Disordered N-Terminal Peptide Segment of Human Cystathionine-β-Synthase. Sci. Rep. 8, 2474. doi:10.1038/s41598-018-20841-z
Langendorf, C. G., Tuck, K. L., Key, T. L. G., Fenalti, G., Pike, R. N., Rosado, C. J., et al. (2013). Structural Characterization of the Mechanism through Which Human Glutamic Acid Decarboxylase Auto-Activates. Biosci. Rep. 33, 137–144. doi:10.1042/BSR20120111
Lao, C. D., Backoff, P., Shotland, L. I., McCarty, D., Eaton, T., Ondrey, F. G., et al. (2004). Irreversible Ototoxicity Associated with Difluoromethylornithine. Cancer Epidemiol. Biomarkers Prev. 13, 1250–1252.
Laver, W. G., Neuberger, A., and Udenfriend, S. (1958). Initial Stages in the Biosynthesis of Porphyrins. 1. The Formation of δ-aminolaevulic Acid by Particles Obtained from Chicken Erythrocytes. Biochem. J. 70, 4–14. doi:10.1042/bj0700004
Lendrihas, T., Hunter, G. A., and Ferreira, G. C. (2010). Targeting the Active Site Gate to Yield Hyperactive Variants of 5-aminolevulinate Synthase. J. Biol. Chem. 285, 13704–13711. doi:10.1074/jbc.M109.074237
Lepore, B. W., Ruzicka, F. J., Frey, P. A., and Ringe, D. (2005). The X-ray crystal Structure of Lysine-2,3-Aminomutase from Clostridium Subterminale. Proc. Natl. Acad. Sci. U.S.A. 102, 13819–13824. doi:10.1073/pnas.0505726102
Liang, J., Han, Q., Tan, Y., Ding, H., and Li, J. (2019). Current Advances on Structure-Function Relationships of Pyridoxal 5'-phosphate-dependent Enzymes. Front. Mol. Biosci. 6, 4. doi:10.3389/fmolb.2019.00004
Lynch, C. J., and Adams, S. H. (2014). Branched-chain Amino Acids in Metabolic Signalling and Insulin Resistance. Nat. Rev. Endocrinol. 10, 723–736. doi:10.1038/nrendo.2014.171
Majtan, T., Singh, L. R., Wang, L., Kruger, W. D., and Kraus, J. P. (2008). Active Cystathionine β-Synthase Can Be Expressed in Heme-free Systems in the Presence of Metal-Substituted Porphyrins or a Chemical Chaperone. J. Biol. Chem. 283, 34588–34595. doi:10.1074/jbc.M805928200
Mangold, U. (2005). The Antizyme Family: Polyamines and beyond. IUBMB Life (International Union Biochem. Mol. Biol. Life) 57, 671–676. doi:10.1080/15216540500307031
Marchetti, M., Bruno, S., Campanini, B., Bettati, S., Peracchi, A., and Mozzarelli, A. (2015). Regulation of Human Serine Racemase Activity and Dynamics by Halides, ATP and Malonate. Amino Acids 47, 163–173. doi:10.1007/s00726-014-1856-2
Maria-Solano, M. A., Iglesias-Fernández, J., and Osuna, S. (2019). Deciphering the Allosterically Driven Conformational Ensemble in Tryptophan Synthase Evolution. J. Am. Chem. Soc. 141, 13049–13056. doi:10.1021/jacs.9b03646
Martin, W. H., Hoover, D. J., Armento, S. J., Stock, I. A., McPherson, R. K., Danley, D. E., et al. (1998). Discovery of a Human Liver Glycogen Phosphorylase Inhibitor that Lowers Blood Glucose In Vivo. Proc. Natl. Acad. Sci. U.S.A. 95, 1776–1781. doi:10.1073/pnas.95.4.1776
Mas-Droux, C., Biou, V., and Dumas, R. (2006). Allosteric Threonine Synthase. J. Biol. Chem. 281, 5188–5196. doi:10.1074/jbc.M509798200
Mathieu, C., de la Sierra-Gallay, I. L., Duval, R., Xu, X., Cocaign, A., Léger, T., et al. (2016). Insights into Brain Glycogen Metabolism. J. Biol. Chem. 291, 18072–18083. doi:10.1074/jbc.M116.738898
Meier, M., Janosik, M., Kery, V., Kraus, J. P., and Burkhard, P. (2001). Structure of Human Cystathionine Beta-Synthase: a Unique Pyridoxal 5'-phosphate-dependent Heme Protein. Embo J. 20, 3910–3916. doi:10.1093/emboj/20.15.3910
Metcalf, B. W., Bey, P., Danzin, C., Jung, M. J., Casara, P., and Vevert, J. P. (1978). Catalytic Irreversible Inhibition of Mammalian Ornithine Decarboxylase (E.C.4.1.1.17) by Substrate and Product Analogs. J. Am. Chem. Soc. 100, 2551–2553. doi:10.1021/ja00476a050
Michalska, K., Wellington, S., Maltseva, N., Jedrzejczak, R., Selem‐Mojica, N., Rosas‐Becerra, L. R., et al. (2021). Catalytically Impaired TrpA Subunit of Tryptophan Synthase from Chlamydia trachomatis Is an Allosteric Regulator of TrpB. Protein Sci. 30, 1904–1918. doi:10.1002/pro.4143
Miles, E. W., and Kraus, J. P. (2004). Cystathionine β-Synthase: Structure, Function, Regulation, and Location of Homocystinuria-Causing Mutations. J. Biol. Chem. 279, 29871–29874. doi:10.1074/jbc.R400005200
Miles, E. W., Rhee, S., and Davies, D. R. (1999). The Molecular Basis of Substrate Channeling. J. Biol. Chem. 274, 12193–12196. doi:10.1074/jbc.274.18.12193
Miles, E. W. (2013). The Tryptophan Synthase α2β2 Complex: A Model for Substrate Channeling, Allosteric Communication, and Pyridoxal Phosphate Catalysis. J. Biol. Chem. 288, 10084–10091. doi:10.1074/jbc.X113.463331
Monod, J., and Jacob, F. (1961). General Conclusions: Teleonomic Mechanisms in Cellular Metabolism, Growth, and Differentiation. Cold Spring Harbor Symposia Quantitative Biol. 26, 389–401. doi:10.1101/sqb.1961.026.01.048
Monod, J., Wyman, J., and Changeux, J.-P. (1965). On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 12, 88–118. doi:10.1016/s0022-2836(65)80285-6
Murakami, Y., Matsufuji, S., Kameji, T., Hayashi, S.-i., Igarashi, K., Tamura, T., et al. (1992). Ornithine Decarboxylase Is Degraded by the 26S Proteasome without Ubiquitination. Nature 360, 597–599. doi:10.1038/360597a0
Nakai, T., Mizutani, H., Miyahara, I., Hirotsu, K., Takeda, S., Jhee, K.-H., et al. (2000). Three-dimensional Structure of 4-Amino-4-Deoxychorismate Lyase from Escherichia coli. J. Biochem. 128, 29–38. doi: DOI doi:10.1093/oxfordjournals.jbchem.a022727
Nardella, C., Boi, D., di Salvo, M. L., Barile, A., Stetefeld, J., Tramonti, A., et al. (2019). Isolation of a Complex Formed between Acinetobacter Baumannii HemA and HemL, Key Enzymes of Tetrapyrroles Biosynthesis. Front. Mol. Biosci. 6, 6. doi:10.3389/fmolb.2019.00006
Newgard, C. B., Hwang, P. K., and Fletterick, R. J. (1989). The Family of Glycogen Phosphorylases: Structure and Functio. Crit. Rev. Biochem. Mol. Biol. 24, 69–99. doi:10.3109/10409238909082552
Newgard, C. B., Littman, D. R., van Genderen, C., Smith, M., and Fletterick, R. J. (1988). Human Brain Glycogen Phosphorylase. Cloning, Sequence Analysis, Chromosomal Mapping, Tissue Expression, and Comparison with the Human Liver and Muscle Isozymes. J. Biol. Chem. 263, 3850–3857. doi:10.1016/s0021-9258(18)69003-9
Niu, W., Wang, J., Qian, J., Wang, M., Wu, P., Chen, F., et al. (2018). Allosteric Control of Human Cystathionine β-synthase Activity by a Redox Active Disulfide Bond. J. Biol. Chem. 293, 2523–2533. doi:10.1074/jbc.RA117.000103
Nozaki, T., Shigeta, Y., Saito-Nakano, Y., Imada, M., and Kruger, W. D. (2001). Characterization of Transsulfuration and Cysteine Biosynthetic Pathways in the Protozoan Hemoflagellate, Trypanosoma Cruzi. J. Biol. Chem. 276, 6516–6523. doi:10.1074/jbc.M009774200
Nussinov, R., and Tsai, C.-J. (2013). Allostery in Disease and in Drug Discovery. Cell 153, 293–305. doi:10.1016/j.cell.2013.03.034
O'Brien, T. G., Megosh, L. C., Gilliard, G., and Soler, A. P. (1997). Ornithine Decarboxylase Overexpression Is a Sufficient Condition for Tumor Promotion in Mouse Skin. Cancer Res. 57, 2630–2637.
O'Rourke, K. F., D'Amico, R. N., Sahu, D., and Boehr, D. D. (2021). Distinct Conformational Dynamics and Allosteric Networks in Alpha Tryptophan Synthase during Active Catalysis. Protein Sci. 30, 543–557. doi:10.1002/pro.4011
Oikonomakos, N. G., Schnier, J. B., Zographos, S. E., Skamnaki, V. T., Tsitsanou, K. E., and Johnson, L. N. (2000a). Flavopiridol Inhibits Glycogen Phosphorylase by Binding at the Inhibitor Site. J. Biol. Chem. 275, 34566–34573. doi:10.1074/jbc.M004485200
Oikonomakos, N. G., Skamnaki, V. T., Tsitsanou, K. E., G Gavalas, N., and Johnson, L. N. (2000b). A New Allosteric Site in Glycogen Phosphorylase B as a Target for Drug Interactions. Structure 8, 575–584. doi:10.1016/s0969-2126(00)00144-1
Okada, K., Hirotsu, K., Hayashi, H., and Kagamiyama, H. (2001). Structures of Escherichia coli Branched-Chain Amino Acid Aminotransferase and its Complexes with 4-Methylvalerate and 2-Methylleucine: Induced Fit and Substrate Recognition of the Enzyme,. Biochemistry 40, 7453–7463. doi:10.1021/bi010384l
Okada, K., Hirotsu, K., Sato, M., Hayashi, H., and Kagamiyama, H. (1997). Three-Dimensional Structure of Escherichia coli Branched-Chain Amino Acid Aminotransferase at 2.5 Resolution. J. Biochem. 121, 637–641. doi:10.1093/oxfordjournals.jbchem.a021633
Pavkov-Keller, T., Strohmeier, G. A., Diepold, M., Peeters, W., Smeets, N., Schürmann, M., et al. (2016). Discovery and Structural Characterisation of New Fold Type IV-Transaminases Exemplify the Diversity of This Enzyme Fold. Sci. Rep. 6, 38183. doi:10.1038/srep38183
Pegg, A. E. (1988). Polyamine Metabolism and its Importance in Neoplastic Growth and a Target for Chemotherapy. Cancer Res. 48, 759–774.
Pegg, A. E. (2006). Regulation of Ornithine Decarboxylase. J. Biol. Chem. 281, 14529–14532. doi:10.1074/jbc.R500031200
Peisach, D., Chipman, D. M., Van Ophem, P. W., Manning, J. M., and Ringe, D. (1998). Crystallographic Study of Steps along the Reaction Pathway of D-Amino Acid Aminotransferase,. Biochemistry 37, 4958–4967. doi:10.1021/bi972884d
Percudani, R., and Peracchi, A. (2003). A Genomic Overview of Pyridoxal‐phosphate‐dependent Enzymes. EMBO Rep. 4, 850–854. doi:10.1038/sj.embor.embor914
Percudani, R., and Peracchi, A. (2009). The B6 Database: a Tool for the Description and Classification of Vitamin B6-dependent Enzymatic Activities and of the Corresponding Protein Families. BMC Bioinformatics 10, 273. doi:10.1186/1471-2105-10-273
Persson, L., Jeppsson, A., and Nasizadeh, S. (2003). Turnover of Trypanosomal Ornithine Decarboxylases. Biochem. Soc. Trans. 31, 411–414. doi:10.1042/bst0310411
Phillips, R. S., and Harris, A. P. (2021). Structural Basis of the Stereochemistry of Inhibition of Tryptophan Synthase by Tryptophan and Derivatives. Biochemistry 60, 231–244. doi:10.1021/acs.biochem.0c00635
Poulin, R., Lu, L., Ackermann, B., Bey, P., and Pegg, A. E. (1992). Mechanism of the Irreversible Inactivation of Mouse Ornithine Decarboxylase by Alpha-Difluoromethylornithine. Characterization of Sequences at the Inhibitor and Coenzyme Binding Sites. J. Biol. Chem. 267, 150–158. doi:10.1016/s0021-9258(18)48472-4
Prudova, A., Bauman, Z., Braun, A., Vitvitsky, V., Lu, S. C., and Banerjee, R. (2006). S -adenosylmethionine Stabilizes Cystathionine β-synthase and Modulates Redox Capacity. Proc. Natl. Acad. Sci. U.S.A. 103, 6489–6494. doi:10.1073/pnas.0509531103
Raboni, S., Marchetti, M., Faggiano, S., Campanini, B., Bruno, S., Marchesani, F., et al. (2018). The Energy Landscape of Human Serine Racemase. Front. Mol. Biosci. 5, 112. doi:10.3389/fmolb.2018.00112
Rath, V. L., Ammirati, M., Danley, D. E., Ekstrom, J. L., Gibbs, E. M., Hynes, T. R., et al. (2000a). Human Liver Glycogen Phosphorylase Inhibitors Bind at a New Allosteric Site. Chem. Biol. 7, 677–682. doi: Doi doi:10.1016/S1074-5521(00)00004-1
Rath, V. L., Ammirati, M., LeMotte, P. K., Fennell, K. F., Mansour, M. N., Danley, D. E., et al. (2000b). Activation of Human Liver Glycogen Phosphorylase by Alteration of the Secondary Structure and Packing of the Catalytic Core. Mol. Cel 6, 139–148. doi:10.1016/s1097-2765(05)00006-7
Rhee, S., Parris, K. D., Ahmed, S. A., Miles, E. W., and Davies, D. R. (1996). Exchange of K+ or Cs+ for Na+ Induces Local and Long-Range Changes in the Three-Dimensional Structure of the Tryptophan Synthase α2β2 Complex. Biochemistry 35, 4211–4221. doi:10.1021/bi952506d
Sandmeier, E., Hale, T. I., and Christen, P. (1994). Multiple Evolutionary Origin of Pyridoxal-5'-phosphate-dependent Amino Acid Decarboxylases. Eur. J. Biochem. 221, 997–1002. doi:10.1111/j.1432-1033.1994.tb18816.x
Schneider, G., Käck, H., and Lindqvist, Y. (2000). The Manifold of Vitamin B6 Dependent Enzymes. Structure 8, R1–R6. doi:10.1016/s0969-2126(00)00085-x
Schneider, T. R., Gerhardt, E., Lee, M., Liang, P.-H., Anderson, K. S., and Schlichting, I. (1998). Loop Closure and Intersubunit Communication in Tryptophan Synthase,. Biochemistry 37, 5394–5406. doi:10.1021/bi9728957
Schultz, C. R., Gruhlke, M. C. H., Slusarenko, A. J., and Bachmann, A. S. (2020). Allicin, a Potent New Ornithine Decarboxylase Inhibitor in Neuroblastoma Cells. J. Nat. Prod. 83, 2518–2527. doi:10.1021/acs.jnatprod.0c00613
Sekula, B., Ruszkowski, M., and Dauter, Z. (2018). Structural Analysis of Phosphoserine Aminotransferase (Isoform 1) from Arabidopsis Thaliana- the Enzyme Involved in the Phosphorylated Pathway of Serine Biosynthesis. Front. Plant Sci. 9, 876. doi:10.3389/fpls.2018.00876
Shulman, A., Zalyapin, E., Vyazmensky, M., Yifrach, O., Barak, Z. e., and Chipman, D. M. (2008). Allosteric Regulation of Bacillus Subtilis Threonine Deaminase, a Biosynthetic Threonine Deaminase with a Single Regulatory Domain. Biochemistry 47, 11783–11792. doi:10.1021/bi800901n
Singh, A. K., Ekka, M. K., Kaushik, A., Pandya, V., Singh, R. P., Banerjee, S., et al. (2017). Substrate-Induced Facilitated Dissociation of the Competitive Inhibitor from the Active Site of O-Acetyl Serine Sulfhydrylase Reveals a Competitive-Allostery Mechanism. Biochemistry 56, 5011–5025. doi:10.1021/acs.biochem.7b00500
Singh, S., Madzelan, P., Stasser, J., Weeks, C. L., Becker, D., Spiro, T. G., et al. (2009). Modulation of the Heme Electronic Structure and Cystathionine β-synthase Activity by Second Coordination Sphere Ligands: The Role of Heme Ligand Switching in Redox Regulation. J. Inorg. Biochem. 103, 689–697. doi:10.1016/j.jinorgbio.2009.01.009
Siow, D. L., and Wattenberg, B. W. (2012). Mammalian ORMDL Proteins Mediate the Feedback Response in Ceramide Biosynthesis. J. Biol. Chem. 287, 40198–40204. doi:10.1074/jbc.C112.404012
Smithson, D. C., Lee, J., Shelat, A. A., Phillips, M. A., and Guy, R. K. (2010). Discovery of Potent and Selective Inhibitors of Trypanosoma Brucei Ornithine Decarboxylase. J. Biol. Chem. 285, 16771–16781. doi:10.1074/jbc.M109.081588
Snell, E. E. (1975). Tryptophanase: Structure, Catalytic Activities, and Mechanism of Action. Adv. Enzymol. Relat. Areas Mol. Biol. 42, 287–333. doi:10.1002/9780470122877.ch6
Sprang, S., Goldsmith, E., and Fletterick, R. (1987). Structure of the Nucleotide Activation Switch in Glycogen Phosphorylase a. Science 237, 1012–1019. doi:10.1126/science.3616621
Sprang, S. R., Withers, S. G., Goldsmith, E. J., Fletterick, R. J., and Madsen, N. B. (1991). Structural Basis for the Activation of Glycogen Phosphorylase B by Adenosine Monophosphate. Science 254, 1367–1371. doi:10.1126/science.1962195
Stetefeld, J., Jenny, M., and Burkhard, P. (2006). Intersubunit Signaling in Glutamate-1-Semialdehyde-Aminomutase. Proc. Natl. Acad. Sci. U.S.A. 103, 13688–13693. doi:10.1073/pnas.0600306103
Sugio, S., Petsko, G. A., Manning, J. M., Soda, K., and Ringe, D. (1995). Crystal Structure of a D-Amino Acid Aminotransferase: How the Protein Controls Stereoselectivity. Biochemistry 34, 9661–9669. doi: DOI doi:10.1021/bi00030a002
Tabor, C. W., and Tabor, H. (1976). 1,4-Diaminobutane (Putrescine), Spermidine, and Spermine. Annu. Rev. Biochem. 45, 285–306. doi:10.1146/annurev.bi.45.070176.001441
Tai, C.-H., Burkhard, P., Gani, D., Jenn, T., Johnson, C., and Cook, P. F. (2001). Characterization of the Allosteric Anion-Binding Site of O-Acetylserine Sulfhydrylase. Biochemistry 40, 7446–7452. doi:10.1021/bi015511s
Taoka, S., Lepore, B. W., Kabil, Ö., Ojha, S., Ringe, D., and Banerjee, R. (2002). Human Cystathionine β-Synthase Is a Heme Sensor Protein. Evidence that the Redox Sensor Is Heme and Not the Vicinal Cysteines in the CXXC Motif Seen in the Crystal Structure of the Truncated Enzyme,. Biochemistry 41, 10454–10461. doi:10.1021/bi026052d
Taoka, S., Ohja, S., Shan, X., Kruger, W. D., and Banerjee, R. (1998). Evidence for Heme-Mediated Redox Regulation of Human Cystathionine β-Synthase Activity. J. Biol. Chem. 273, 25179–25184. doi:10.1074/jbc.273.39.25179
Taoka, S., West, M., and Banerjee, R. (1999a). Characterization of the Heme and Pyridoxal Phosphate Cofactors of Human Cystathionine β-Synthase Reveals Nonequivalent Active Sites. Biochemistry 38, 2738–2744. doi:10.1021/bi9826052
Taoka, S., Widjaja, L., and Banerjee, R. (1999b). Assignment of Enzymatic Functions to Specific Regions of the PLP-dependent Heme Protein Cystathionine β-Synthase. Biochemistry 38, 13155–13161. doi:10.1021/bi990865t
Taylor, R. T., and Jenkins, W. T. (1966). Leucine Aminotransferase. J. Biol. Chem. 241, 4396–4405. doi:10.1016/s0021-9258(18)99734-6
Toney, M. D., Hohenester, E., Cowan, S. W., and Jansonius, J. N. (1993). Dialkylglycine Decarboxylase Structure: Bifunctional Active Site and Alkali Metal Sites. Science 261, 756–759. doi:10.1126/science.8342040
Toney, M. D., Hohenester, E., Keller, J. W., and Jansonius, J. N. (1995). Structural and Mechanistic Analysis of Two Refined crystal Structures of the Pyridoxal Phosphate-dependent Enzyme Dialkylglycine Decarboxylase. J. Mol. Biol. 245, 151–179. doi:10.1006/jmbi.1994.0014
Toraya, T., and Mori, K. (1999). A Reactivating Factor for Coenzyme B12-dependent Diol Dehydratase. J. Biol. Chem. 274, 3372–3377. doi:10.1074/jbc.274.6.3372
Tseng, C.-H., Yang, C.-H., Lin, H.-J., Wu, C., and Chen, H.-P. (2007). The S Subunit of D-Ornithine Aminomutase fromClostridium Sticklandiiis Responsible for the Allosteric Regulation in D-α-Lysine Aminomutase. Fems Microbiol. Lett. 274, 148–153. doi:10.1111/j.1574-6968.2007.00820.x
Tsesin, N., Kogan, A., Gdalevsky, G. Y., Himanen, J.-P., Cohen-Luria, R., Parola, A. H., et al. (2007). The Structure of Apo Tryptophanase fromEscherichia Colireveals a Wide-Open Conformation. Acta Crystallogr. D Biol. Cryst. 63, 969–974. doi:10.1107/S0907444907036396
Ubonprasert, S., Jaroensuk, J., Pornthanakasem, W., Kamonsutthipaijit, N., Wongpituk, P., Mee-Udorn, P., et al. (2019). A Flap Motif in Human Serine Hydroxymethyltransferase Is Important for Structural Stabilization, Ligand Binding, and Control of Product Release. J. Biol. Chem. 294, 10490–10502. doi:10.1074/jbc.RA119.007454
Umbarger, H. E., and Brown, B. (1958). Isoleucine and Valine Metabolism in Escherichia coli. J. Biol. Chem. 233, 415–420. doi:10.1016/s0021-9258(18)64775-1
Vicente, J. B., Colaço, H. G., Mendes, M. I. S., Sarti, P., Leandro, P., and Giuffrè, A. (2014). NO Binds Human Cystathionine β-Synthase Quickly and Tightly. J. Biol. Chem. 289, 8579–8587. doi:10.1074/jbc.M113.507533
Wang, J. H., Tu, J.-I., and Lo, F. M. (1970). Effect of Glucose 6-Phosphate on the Nucleotide Site of Glycogen Phosphorylase B. J. Biol. Chem. 245, 3115–3121. doi:10.1016/s0021-9258(18)63030-3
Wang, Y., Niu, Y., Zhang, Z., Gable, K., Gupta, S. D., Somashekarappa, N., et al. (2021). Structural Insights into the Regulation of Human Serine Palmitoyltransferase Complexes. Nat. Struct. Mol. Biol. 28, 240–248. doi:10.1038/s41594-020-00551-9
Weber, I. T., Johnson, L. N., Wilson, K. S., Yeates, D. G. R., Wild, D. L., and Jenkins, J. A. (1978). Crystallographic Studies on the Activity of Glycogen Phosphorylase B. Nature 274, 433–437. doi:10.1038/274433a0
Weiss, N., Heydrick, S., Zhang, Y.-Y., Bierl, C., Cap, A., and Loscalzo, J. (2002). Cellular Redox State and Endothelial Dysfunction in Mildly Hyperhomocysteinemic Cystathionine β-Synthase-Deficient Mice. Atvb 22, 34–41. doi:10.1161/hq1201.100456
Whatley, S. D., Ducamp, S., Gouya, L., Grandchamp, B., Beaumont, C., Badminton, M. N., et al. (2008). C-terminal Deletions in the ALAS2 Gene lead to Gain of Function and Cause X-Linked Dominant Protoporphyria without Anemia or Iron Overload. Am. J. Hum. Genet. 83, 408–414. doi:10.1016/j.ajhg.2008.08.003
Wolf, D. P., Fischer, E. H., and Krebs, E. G. (1970). Amino Acid Sequence of the Phosphorylated Site in Rabbit Liver Glycogen Phosphorylase. Biochemistry 9, 1923–1929. doi:10.1021/bi00811a010
Wolthers, K. R., Rigby, S. E. J., and Scrutton, N. S. (2008). Mechanism of Radical-Based Catalysis in the Reaction Catalyzed by Adenosylcobalamin-dependent Ornithine 4,5-aminomutase. J. Biol. Chem. 283, 34615–34625. doi:10.1074/jbc.M807911200
Wu, H.-Y., Chen, S.-F., Hsieh, J.-Y., Chou, F., Wang, Y.-H., Lin, W.-T., et al. (2015). Structural Basis of Antizyme-Mediated Regulation of Polyamine Homeostasis. Proc. Natl. Acad. Sci. U.S.A. 112, 11229–11234. doi:10.1073/pnas.1508187112
Yennawar, N. H., Islam, M. M., Conway, M., Wallin, R., and Hutson, S. M. (2006). Human Mitochondrial Branched Chain Aminotransferase Isozyme. J. Biol. Chem. 281, 39660–39671. doi:10.1074/jbc.M607552200
Keywords: pyridoxal 5-phosphate, allostery, enzyme structure, protein structural & functional analysis, coenzyme, protein-protein interaction
Citation: Tran JU and Brown BL (2022) Structural Basis for Allostery in PLP-dependent Enzymes. Front. Mol. Biosci. 9:884281. doi: 10.3389/fmolb.2022.884281
Received: 26 February 2022; Accepted: 25 March 2022;
Published: 25 April 2022.
Edited by:
Tayana Victorovna Demidkina, Engelhardt Institute of Molecular Biology (RAS), RussiaReviewed by:
Andrea Mozzarelli, University of Parma, ItalyEkaterina Bezsudnova, A N Bach Institute of Biochemistry (RAS), Russia
Copyright © 2022 Tran and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Breann L. Brown, YnJlYW5uLmJyb3duQHZhbmRlcmJpbHQuZWR1