 Pearl Morosky1
Pearl Morosky1 Cody Comyns1
Cody Comyns1 Lance G. A. Nunes2Christina Z. Chung1
Lance G. A. Nunes2Christina Z. Chung1 Peter R. Hoffmann2
Peter R. Hoffmann2 Dieter Söll1,3
Dieter Söll1,3 Oscar Vargas-Rodriguez1
Oscar Vargas-Rodriguez1 Natalie Krahn1*
Natalie Krahn1*- 1Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT, United States
- 2Department of Cell and Molecular Biology, John A. Burns School of Medicine, University of Hawaii, Honolulu, HI, United States
- 3Department of Chemistry, Yale University, New Haven, CT, United States
Post-translational modifications (PTMs) can occur on almost all amino acids in eukaryotes as a key mechanism for regulating protein function. The ability to study the role of these modifications in various biological processes requires techniques to modify proteins site-specifically. One strategy for this is genetic code expansion (GCE) in bacteria. The low frequency of post-translational modifications in bacteria makes it a preferred host to study whether the presence of a post-translational modification influences a protein’s function. Genetic code expansion employs orthogonal translation systems engineered to incorporate a modified amino acid at a designated protein position. Selenoproteins, proteins containing selenocysteine, are also known to be post-translationally modified. Selenoproteins have essential roles in oxidative stress, immune response, cell maintenance, and skeletal muscle regeneration. Their complicated biosynthesis mechanism has been a hurdle in our understanding of selenoprotein functions. As technologies for selenocysteine insertion have recently improved, we wanted to create a genetic system that would allow the study of post-translational modifications in selenoproteins. By combining genetic code expansion techniques and selenocysteine insertion technologies, we were able to recode stop codons for insertion of Nε-acetyl-l-lysine and selenocysteine, respectively, into multiple proteins. The specificity of these amino acids for their assigned position and the simplicity of reverting the modified amino acid via mutagenesis of the codon sequence demonstrates the capacity of this method to study selenoproteins and the role of their post-translational modifications. Moreover, the evidence that Sec insertion technology can be combined with genetic code expansion tools further expands the chemical biology applications.
1 Introduction
Selenium is an essential micronutrient required for the biosynthesis of selenocysteine (Sec), which is exclusively found in selenoproteins. Selenoproteins are involved in many key cellular functions, including calcium and redox homeostasis, cell maintenance, and immune and inflammatory responses (Ye et al., 2022). Twenty-five selenoproteins have been identified in humans, but their functions remain poorly characterized. This is partly due to the complicated and highly regulated Sec biosynthesis and its insertion into proteins. In eukaryotes, this process depends on cis and trans-acting factors in the mRNA and transportation of the tRNA to the ribosome, respectively (Pinkerton and Copeland, 2016). Similar to bacterial systems, the Sec-charged tRNA (Sec-tRNASec) recognizes a dedicated UGA non-sense codon with the assistance of the Sec-specific elongation factor (eEFSec). Redefinition of the UGA codon as Sec is mediated by a hairpin element known as the Sec insertion sequence (SECIS) found up to 1,600 nucleotides away from the UGA codon in the 3′-untranslated region (UTR) of eukaryotic selenoprotein mRNA. eEFSec brings Sec-tRNASec to the ribosome in response to the SECIS element in a process that involves an additional factor, the SECIS binding protein 2 (SBP2) (Copeland et al., 2000). SBP2 is predicted to bind to the SECIS element, bending the 3′-UTR to interact with eEFSec, though the detailed mechanism is still not fully understood (Kossinova et al., 2014; Hilal et al., 2022).
The complicated translation path of eukaryotic selenoproteins poses a challenge to overexpress and purify these proteins for functional characterization studies. Some strategies [e.g., pSelExpress1, orthogonal aminoacyl-tRNA synthetase (aaRS):tRNA pairs] have been developed to address this challenge in eukaryotes [reviewed in (Novoselov et al., 2007; Chung and Krahn, 2022)]. However, the bacterial Sec insertion system is simpler, only requiring a specialized elongation factor (SelB) to recognize Sec-tRNASec and the SECIS element for insertion of Sec. As a result, more methodologies have been developed for recombinant Sec insertion in bacteria [reviewed in (Chung and Krahn, 2022)]. We have specifically focused on engineering SECIS-independent translation by removing the requirement for SelB and instead using the canonical elongation factor (EF-Tu) (Aldag et al., 2013). This method depends on allo-tRNAs, a tRNA species with an unusual cloverleaf structure (Mukai et al., 2017). We have since verified that some of the allo-tRNAs are recognized by EF-Tu (Mukai et al., 2018) with tertiary structures that facilitates accommodation in the E. coli ribosome (Prabhakar et al., 2022). Through engineering strategies, they have been altered for efficient Sec incorporation (Mukai et al., 2018).
As with standard protein expression, eukaryotic selenoproteins can also be expressed in bacteria but with minimal post-translational modifications (PTMs). To install PTMs into E. coli expressed proteins, genetic code expansion (GCE) utilizes orthogonal aaRS:tRNA synthetase pairs and non-canonical amino acids (ncAAs) (Chen et al., 2018; Porter and Mehl, 2018; Yang et al., 2018). The aaRS is engineered to accept diverse ncAAs typically for insertion at a non-sense or stop codon. Some commonly used orthogonal translation systems include the Methanocaldococcus jannaschii tyrosyl-tRNA synthetase (TyrRS) and archaeal pyrrolysyl-tRNA synthetase (PylRS) systems. The tolerance of PylRS to anticodon changes in its cognate tRNAPyl makes it an attractive tool to recode any codon, while the codon choice for TyrRS can be more limiting due to its anticodon recognition (Crnković et al., 2016). These systems have been used to insert common PTMs such as acetylation (Lacoursiere et al., 2020), phosphorylation (Balasuriya et al., 2020), or both (Venkat et al., 2018).
The presence of three non-sense codons in the genetic code offers an opportunity to insert multiple ncAAs into a single protein by combining existing orthogonal systems. To this end, several studies have demonstrated the synthesis of proteins containing two and even three ncAAs (Neumann et al., 2010; Chatterjee et al., 2013; Wang et al., 2014; Dunkelmann et al., 2020; Tharp et al., 2021). Since tRNASec is acylated with Ser by endogenous seryl-tRNA synthetase (SerRS) before conversion to Sec via dedicated enzymes, it may be compatible with many of these orthogonal translation systems. This was tested with the natural Sec machinery in E. coli. Using a natural eukaryotic selenoprotein with a penultimate Sec amino acid, mRNA engineering facilitated a SECIS element to be inserted into the 3′-UTR of the mRNA while still promoting recognition by endogenous SelB for Sec insertion. Combining this with AcKRS (an aaRS evolved from Methanosarcinae PylRS) for incorporation of Nε-acetyl-l-lysine (AcK) showed the compatibility of these two systems and their capability of introducing AcK into a natural selenoprotein (Wright et al., 2018). However, the requirement of a SECIS element immediately downstream of the UGA codon limits this strategy to natural bacterial selenoproteins or eukaryotic selenoproteins with a Sec residue at the C-terminal end (penultimate or ultimate state).
Here we present a new strategy for the simultaneous insertion of Sec and AcK into super-folder green fluorescent protein (sfGFP) and human glutathione peroxidase 1 (GPx1). We achieved this by combining a previously engineered Sec-incorporation system to site-specifically insert Sec anywhere in a polypeptide chain (Mukai et al., 2018) with a M. alvus PylRS engineered for insertion of AcK (Figure 1) (Seki et al., 2020). With the correct choice of anticodon for each of these translation systems, we facilitated increased codon orthogonality and suppression to yield post-translationally modified selenoproteins. This further expands the genetic code expansion toolbox, enabling the capability to design novel proteins and study eukaryotic selenoproteins in E. coli.
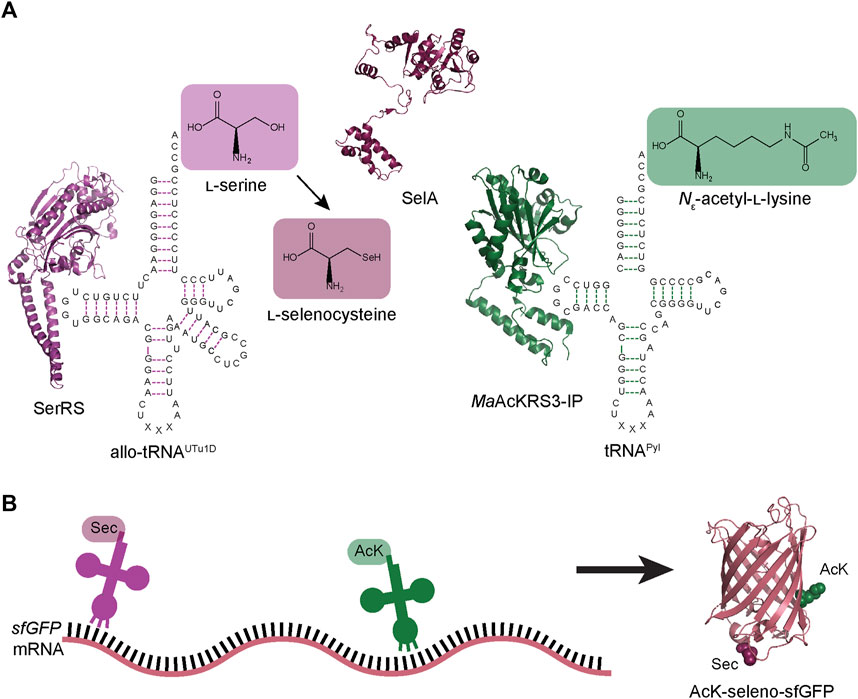
FIGURE 1. Schematic of dual ncAA incorporation to produce post-translationally modified seleno-sfGFP. (A) Endogenous E. coli seryl-tRNA synthetase (SerRS represented by PDB:1SER) aminoacylates allo-tRNAUTu1D with l-serine, which is then converted to l-Sec by A. salmonicida selenocysteine synthase (SelA represented by PDB:3W1K). M. alvus PylRS containing mutations for recognition of Nε-acetyl-l-lysine (AcK) (MaAcKRS3-IP represented by PDB:4Q6G) aminoacylates M. alvus tRNAPyl. (B) Aminoacylated allo-tRNAUTu1D and tRNAPyl insert Sec and AcK, respectively, at specific positions in the mRNA. These tRNAs are encoded to suppress stop codons (UAG or UGA), leading to the biosynthesis of AcK-seleno-sfGFP (represented by PDB:1EMB).
2 Materials and methods
2.1 General
Enzymes for molecular cloning were purchased from New England Biolabs and Takara Bio. Gene fragments were purchased from Integrated DNA Technologies and Twist Bioscience. W.M. Keck Foundation at Yale University provided oligonucleotide synthesis (Supplementary Table S1) and DNA plasmid (Supplementary Table S2) sequencing. Antibiotics and media additives were used at the following final concentrations: ampicillin (amp), 100 μg/mL; spectinomycin (spec), 50 μg/mL; glucose, 1% (w/v); arabinose, 0.1% (w/v); sodium selenite, 10 μM; Nε-acetyl-l-lysine (AcK), 10 mM; nicotinamide, 20 mM.
2.2 Plasmid construction
2.2.1 pB_sfGFP_2TGA (pB_PM02) and pB_sfGFP_151TAG (pB_PM03)
The stop codon of the sfGFP gene in the previously reported pB_sfGFP plasmid (Chung et al., 2022) was mutated to TAA using the primer pair PM01/02 (pB_PM01). PM03/04 and PM05/06 primers were used separately to introduce a TGA at position 2 (2TGA) or a TAG at position 151 (151TAG) of sfGFP in the pB_PM01 plasmid. This provided the plasmids (pB_PM02 and pB_PM03) to test each suppression system individually.
2.2.2 pB_sfGFP_2TGA-MaAcLysRS3-tRNAPylUCA (pB_PM06) and pB_sfGFP_151TAG-MaAcLysRS3- tRNAPylUCA (pB_PM07)
To insert the PylRS and tRNAPyl genes for AcK insertion into the previously designed plasmids, the primer pair PM07/08 was used to amplify the chemically synthesized MaAcLysRS3-IP gene (Seki et al., 2020) and insert a proK promoter. Primers PM09/10 opened the pB_PM01, pB_PM02 or pB_PM03 plasmids downstream of the origin of replication with overhangs complementary to the amplified MaAcLysRS3-IP fragment to insert in the reverse direction. The opened pB_PM01, pB_PM02 or pB_PM03 and MaAcLysRS3-IP fragment were assembled with NEBuilder HiFi to generate pB_PM04, pB_PM05, and pB_PM06, respectively. These plasmids were then opened upstream of MaAcLysRS3-IP with PM11/12 to insert the M. alvus tRNAPylUCA fragment containing an Lpp promoter and rrcn terminator. The tRNAPylUCA fragment was amplified with PM13/14 from a pBAD-sfGFP-tRNA plasmid (Tharp et al., 2020) containing the sequence for tRNAPylUCA. The opened pB_PM04, pB_PM05, and pB_PM06 plasmids and tRNAPylUCA fragment were assembled with NEBuilder HiFi to generate pB_PM07, pB_PM08, and pB_PM09, respectively.
2.2.3 pB_sfGFP_2TGA_151TAG-MaAcLysRS3- tRNAPylUCA (pB_PM10)
Starting with the pB_PM08 plasmid, PM05/06 primers were used to add the second stop codon within the coding region of sfGFP for dual suppression studies.
2.2.4 pB_04_2TGA-MaAcLysRS3- tRNAPylUCA (pB_PM13)
The pB_04 plasmid containing sfGFP disrupted by the M86 DnaB mini-intein at position 204 was previously used to detect Sec insertion (Chung et al., 2022). The primer pair PM01/02 was used first to mutate the stop codon to TAA (pB_PM11). Due to the presence of a TAG already in the intein-sfGFP fusion, the second in-frame stop codon, TGA, was inserted at position 2 using the PM03/04 primers (pB_PM12). Insertion of the AcK-translation system (MaAcLysRS3-mA17) was achieved as described in Section 2.2.2 (pB_PM13).
2.2.5 pB_sfGFP-MaAcLysRS3-tRNAPylCUA plasmids (pB_PM14, pB_PM15, pB_PM16, and pB_PM17)
The anticodon sequence of the tRNAPylUCA gene in plasmids pB_PM07, pB_PM08, pB_PM09, and pB_PM10 plasmids was mutated to CUA to suppress the UAG codon using the primer pair PM15/16. This generated plasmids pB_PM14, pB_PM15, pB_PM16, and pB_PM17, respectively.
2.2.6 pB_04TGA_2TAG-MaAcLysRS3-tRNAPylCUA (pB_PM18)
The anticodon sequence of tRNAPylUCA from pB_PM13 was also converted to CUA following the strategy described in 2.2.5. However, since the intein is specific for Sec insertion, the codons at position 2 of sfGFP and position 204 (position 1 of the intein) had to also be switched. Primer pairs PM17/18 and PM19/20 were used to convert 2TGA to 2TAG and pB_04 to pB_04TGA, respectively.
2.2.7 pSecUGA
To recode UGA with Sec, the anticodon of allo-tRNAUTu1D in the pSecUAG plasmid (Mukai et al., 2018; Chung et al., 2021) was changed to UCA. This was done using primer pair PM21/22 on the pSecUAG plasmid to generate pSecUGA.
2.2.8 pB_GPx1_49TAG-MaAcLysRS3-tRNAPylCUA (pB_PM19)
For GPx1 expression, the human GPx1 gene was inserted into the pB-MaAcLysRS3-tRNAPylCUA vector containing genes for AcK insertion at UAG. Primer pair PM23/24 amplified GPx1_49TAG from pET-GPx1 (Mukai et al., 2018), while primer pair PM25/26 was used to open pB_PM17. Digestion with DpnI removed any parental plasmid from the PCR product before NEBuilder HiFi assembled the two PCR purified samples (Gpx1_49TAG and pB-MaAcLysRS3-tRNAPylCUA). This produced plasmid pB_PM19.
2.2.9 pB_GPx1_49TGA_114TAG-MaAcLysRS3-tRNAPylCUA (pB_PM21) and pB_GPx1_49TGA_148TAG-MaAcLysRS3-tRNAPylCUA plasmids (pB_PM22)
Mutagenesis of position 49 to encode a UGA was accomplished with primer pair PM27/28 on pB_PM19. This resulting plasmid (pB_PM20) was then subject to further mutagenesis to install UAG codons at position 114 or 148 (using primer pairs PM29/30 or PM31/32, respectively) for insertion of AcK. Final plasmids (pB_PM21 and pB_PM22) were then ready for dual insertion of Sec and AcK.
2.3 sfGFP fluorescence assay
Plasmids were transformed into electrocompetent E. coli cells (B-95.ΔA.ΔfabRΔselABC) (Mukai et al., 2015) and plated on Luria Broth (LB) agar containing appropriate antibiotics and incubated overnight at 37°C. Single colonies were grown in 150 μL media at 37°C for 6 h in a 96-well black plate with clear bottoms. Unless otherwise noted, the media containing appropriate antibiotics included glucose, arabinose, sodium selenite, AcK, and nicotinamide was used. After 6 h, 75 μL from each well was transferred to a clean well in the same plate. The original wells were replenished with 75 μL of fresh media, while protein expression was induced in the new wells by adding 75 μL of media containing 2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Fluorescent measurements and analysis were performed as described previously (Chung et al., 2022) using a minimum of four biological replicates.
2.4 Post-translationally modified selenoprotein production
For protein expression, pB_PM17, pB_PM21, or pB_PM22 plasmids were co-transformed with pSecUGA into electrocompetent E. coli cells (B-95.ΔA.ΔfabRΔselABC) (Mukai et al., 2015) before plated on LB agar containing appropriate antibiotics and incubated overnight at 37°C. Single colonies were grown overnight at 37°C in 25 mL LB containing the appropriate antibiotics. Precultures were transferred to 1 L of LB containing antibiotics, arabinose, and sodium selenite and grown at 37°C. At an OD600 = 0.6, protein expression was induced with 0.5 mM IPTG, following the addition of nicotinamide, AcK, and extra sodium selenite. Proteins were expressed at 20°C for 16 h before pelleting the cells for purification.
All purification steps were performed under anaerobic conditions (90% N2, 5% H2, 5% CO2) in an anaerobic tent (Coy Laboratories). Each cell pellet was resuspended in 18 mL lysis buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 30 mM imidazole, 10% (v/v) glycerol, 120 μg/mL lysozyme, 30 μg/mL Dnase, 0.5 mM phenylmethylsulfonyl fluoride [PMSF]) and 2 mL BugBuster® 10 X Extraction Reagent (EMD Millipore). The lysed cells were incubated at room temperature for 20 min before the lysate was ultracentrifuged at 45,000 rpm (150,000 x g) for 45 min at 4°C. The supernatant was 0.45 μm filtered before being loaded onto a 1 mL Ni-NTA column pre-equilibrated with wash buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 30 mM imidazole, 10% (v/v) glycerol). The beads were washed with 50 mL of wash buffer and then eluted in 1 mL fractions with elution buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 250 mM imidazole, 10% (v/v) glycerol). Protein fractions were combined, concentrated, and buffer exchanged into storage buffer (50 mM sodium phosphate [pH 8.0], 300 mM NaCl, 10% (v/v) glycerol) before being stored at −80°C.
2.5 Mass spectrometry analysis
LC-MS analysis of dithiothreitol reduced samples were performed on a ThermoFisher Scientific Orbitrap Exploris 240 mass spectrometer, equipped with a heated electrospray ionization source (H-ESI) in positive ion mode with a ThermoFisher Ultimate 3000 RSLCnano HPLC System. On H-ESI source, sheath gas was set to 2 arbitrary units (arb), and auxiliary gas was set to 6 arb. The ion transfer tube was set at 275°C and the vaporizer temp at 200°C. The sample was analyzed on a MAbPac RP, 4 μM, 3.0 mm × 50 mm analytical column (ThermoFisher Scientific), held at 60°C. The protein was eluted at a rate of 500 μL/min for a 10-min gradient, where 0–7 min: 10%–70% acetonitrile + 0.1% formic acid; 7–8.2 min: 95% acetonitrile + 0.1% formic acid, 8.2–10 min: 20% acetonitrile + 0.1% formic acid. MS spectra were acquired using full scans at 15,000 resolution in the orbitrap within a range of 700–2,200 m/z. The maximum injection time was set at auto with a standard AGC target. Ten micro scans were employed, and the RF lens was set to 100%. 15 V of insource CID was applied. Thermo BioPharma Finder 5.1 was used for intact mass deconvolution and peak identification.
2.6 Western blot to identify acetylation
Approximately 5 μg of GPx1 protein samples were loaded onto a 4%–20% Mini-PROTEAN TGX stain-free gel (Bio-Rad) in reducing conditions. Samples were transferred to nitrocellulose and acetylation was detected with a primary mouse anti-acetylated lysine mAb (Novus Biologicals) and secondary anti-mouse IgG HRP-linked antibody (Cell Signaling Technology).
3 Results
3.1 Determination of suppression codon choice
Using our established pSecUAG plasmid system for insertion of Sec at UAG codons, we first engineered the anticodon of M. alvus tRNAPyl to recode UGA codons (tRNAPylUCA). Before testing the orthogonality of each tRNA for its respective codon, we confirmed the specificity of MaAcLysRS3-IP for AcK (Seki et al., 2020). E. coli cells expressing only pB_PM08 showed significantly higher fluorescence (p < 0.0001) in the presence of AcK compared to cells without the amino acid (Figure 2A). This suggests that AcK promotes readthrough of the UGA codon in sfGFP with the MaAcLysRS3:tRNAPylUCA pair.
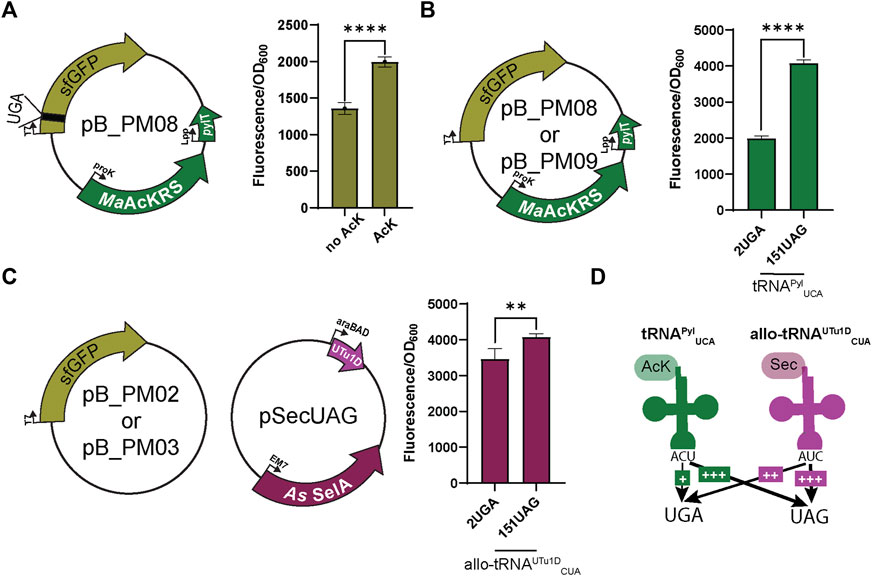
FIGURE 2. Fluorescence assay observes bias for UAG suppression. (A) To first test the specificity of the MaAcKRS system for the presence of Nε-acetyl-l-lysine (AcK), pB_PM06 was used. This plasmid contains sfGFP under the control of a T7 promoter encoding a UGA at position 2 (2UGA), MaAcKRS3-IP under a proK promoter, and tRNAPylUCA (pylT) under an Lpp promoter. The presence of AcK promoted sfGFP fluorescence (p < 0.0001, n = 4). (B) pB_PM06 and pB_PM07 plasmids encoding 2UGA or 151UAG, respectively, were tested for the specificity of tRNAPylUCA for its cognate codon (UGA). Significant increase in readthrough fluorescence (p < 0.0001, n = 4) was observed for 151UAG, the non-cognate codon. (C) Combining plasmids pB_PM02 or pB_PM03 encoding 2UGA or 151UAG, respectively, and pSecUAG allowed a test for UAG suppression by allo-tRNAUTu1DCUA. This plasmid contains the machinery for insertion of Sec, namely allo-tRNAUTu1DCUA (UTu1D) under the araBAD promoter and A. salmonicida (As) SelA under EM7. Similar readthrough fluorescence was observed for both codons with a preference for its cognate codon, UAG (p < 0.01, n = 4). (D) Combining this information, tRNAPylUCA (green) poorly suppresses its cognate codon UGA but is much better at suppressing UAG, while allo-tRNAUTu1DCUA (magenta) also suppresses both codons with a bias towards UAG. Increase in suppression efficiency is denoted by an increase in + and bolder arrow.
When using multiple translation systems to suppress more than one stop codon, confirming the codon specificity and orthogonality of engineered suppressor tRNAs is imperative. Therefore, we tested the ability of each system to readthrough UAG or UGA independently. To first investigate the ability of tRNAPylUCA to specifically suppress UGA, we expressed the plasmid pB_PM08 or pB_PM09 in the presence of AcK. Notably, a significant increase (p < 0.0001) in GFP fluorescence for readthrough of 151UAG relative to 2UGA was observed (Figure 2B), suggesting that tRNAPylUCA has a propensity to decode UAG codons better than its cognate UGA codon. We also tested this for allo-tRNAUTu1DCUA by expressing plasmids pB_PM02 or pB_PM03 together with pSecUAG in the presence of sodium selenite. Interestingly, allo-tRNAUTu1DCUA translated both 2UGA and 151UAG, with a significant increase in fluorescence (p < 0.01) observed for the intended UAG codon (Figure 2C). Together, these results show that tRNAPylUCA and allo-tRNAUTu1DCUA can suppress both UGA and UAG codons in isolation, displaying a higher efficiency to recode UAG (Figure 2D). The propensity to recode UAG was also observed when testing the suppression efficiency of each codon individually (pB_PM08 or pB_PM09) and together (pB_PM10) compared to wild-type (pB_PM07) in the presence of AcK and sodium selenite (Supplementary Figure S1A). Thus, these two tRNA variants are not entirely orthogonal.
To test whether codon specificity can be attained, we swapped the anticodons of both tRNAs, resulting in tRNAPylCUA and allo-tRNAUTu1DUCA. This led to the pB series (pB_PM14, pB_PM15, pB_PM16, and pB_PM17) and pSecUGA to decode UAG and UGA, respectively. We again confirmed that the MaAcLysRS3:tRNAPylCUA pair relies on AcK for efficient readthrough (p < 0.01) (Figure 3A). Importantly, we found that tRNAPylCUA almost exclusively translates the UAG codon, with minimal decoding of UGA. Expressing only pB_PM15 or pB_PM16 we observed fluorescence when 151UAG is present but not in the presence of 2UGA (p < 0.0001) (Figure 3B). Similarly, allo-tRNAUTu1DUCA displayed a higher specificity for UGA than UAG. Expressing pSecUGA with pB_PM02 or pB_PM03, we observed the opposite, fluorescence with 2UGA and not 151UAG (p < 0.0001) (Figure 3C). Furthermore, the suppression efficiencies of each codon individually (pB_PM15 or pB_PM16) and together (pB_PM17) were comparable and roughly 60%–70% of wild-type sfGFP (pB_PM14) in the presence of AcK and sodium selenite (Supplementary Figure S1B). These data suggest that allo-tRNAUTu1DUCA and tRNAPylCUA can simultaneously insert AcK at UAG and Sec at UGA into proteins efficiently (Figure 3D).
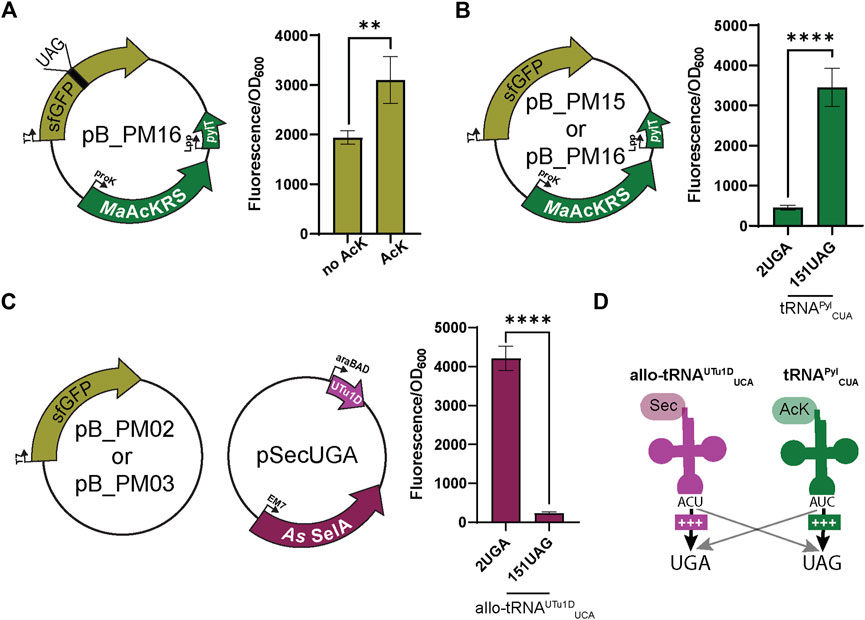
FIGURE 3. Anticodon swap creates codon orthogonality. (A) Nε-acetyl-l-lysine (AcK) was still found to be required for significant fluorescence (p < 0.0001, n = 4) of sfGFP with plasmid pB_PM13. This plasmid contains sfGFP under the control of a T7 promoter encoding a UAG at position 151 (151UAG), MaAcKRS3-IP under a proK promoter, and tRNAPylCUA (pylT) under an Lpp promoter. (B) pB_PM12 and pB_PM13 encoding 2UGA or 151UAG, respectively, were tested for the specificity of tRNAPylCUA for its cognate codon (UAG). Significant increase in readthrough fluorescence (p < 0.0001, n = 4) was observed for 151UAG, the cognate codon, compared to 2UGA. (C) Combining plasmids pB_PM02 or pB_PM03 encoding 2UGA or 151UAG, respectively, and pSecUGA allowed a test for UGA suppression by allo-tRNAUTu1DUCA. This plasmid contains the machinery for insertion of Sec, namely allo-tRNAUTu1DUCA (UTu1D) under the araBAD promoter and Aeromonas salmonicida (As) SelA under EM7. Significant increase in readthrough fluorescence (p < 0.0001, n = 4) was observed for 2UGA, the cognate codon, compared to 151UAG. (D) Combining this information, allo-tRNAUTu1DUCA (magenta) is efficient at UGA suppression, while tRNAPylCUA (green) is efficient at UAG suppression, creating an orthogonal system for dual suppression. Increase in suppression efficiency is denoted by an increase in + and bolder arrow. Grey arrows denote that no suppression was observed.
3.2 Dual insertion of AcK and Sec in a fluorescent reporter
Next, we combined the AcK and Sec incorporation systems to produce a synthetic post-translationally modified selenoprotein (AcK-seleno-sfGFP). We first tested the activities of the MaAcLysRS3:tRNAPylCUA and the Sec-translation systems (Figure 4A) in the presence and absence of AcK and arabinose (for allo-tRNAUTu1DUCA expression). Significant sfGFP fluorescence (p < 0.001) was only observed in the presence of both AcK and arabinose but not if one or both is missing (Figure 4B), confirming the feasibility and compatibility of these two systems. Due to the possibility of Ser misincorporation by the pSecUGA system, we confirmed Sec incorporation using pB_PM18 (Figure 5A) which was engineered based on our previously developed Sec-specific sfGFP reporter (pB_04) (Chung et al., 2022). The sfGFP reporter is only functional (fluorescent) when Sec is inserted at amino acid 204 (position 1 of the M86 mini intein) to facilitate cleavage and splicing of the two sfGFP fragments (Appleby-Tagoe et al., 2011). Moreover, mutagenesis of codon 2 to UAG and the stop codon of the sfGFP to UAA allows for simultaneously monitoring the insertion of two ncAAs. By monitoring the fluorescence of the intein-sfGFP reporter, we only observed significant activity when both suppression systems and sodium selenite (Figure 5B) were present. These results corroborate that the fluorescence observed with the pB_PM17 plasmid (Figure 4B) is due to the presence of Sec. Moreover, the lack of fluorescence in the absence of AcK verifies its requirement and insertion at UAG (Figures 3A, 4B).
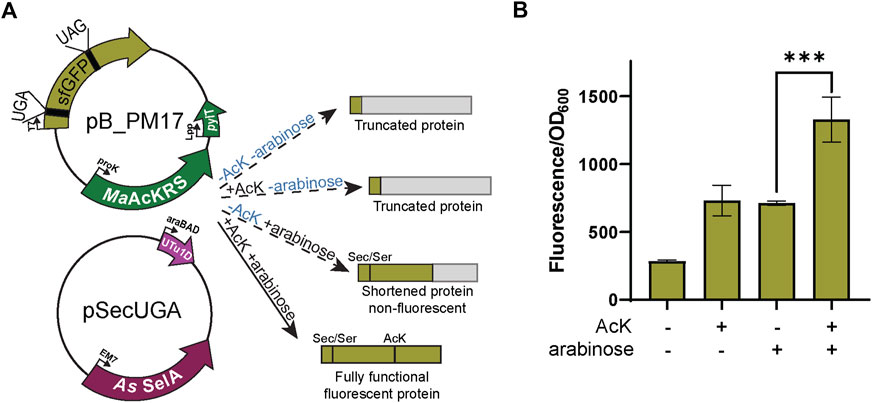
FIGURE 4. Fluorescence from dual suppression in sfGFP. (A) The assay was performed with a two-plasmid system: pB_PM14 and pSecUGA. The first plasmid contains the genes for MaAcKRS3-IP and tRNAPylCUA (pylT) for insertion of Nε-acetyl-l-lysine (AcK) at a UAG and pSecUGA contains the machinery for insertion of Sec at a UGA. In the presence of AcK, arabinose, and sodium selenite, the entire sfGFP gene should be expressed to produce fluorescence. In the absence of AcK or arabinose (or both), translation stops resulting in truncated non-fluorescent protein. Without arabinose, only the first amino acid would be translated. Without AcK, translation would continue until the UAG at position 151 to produce a truncated protein. (B) sfGFP assay shows successful readthrough of both codons in the presence of AcK and Sec with a significant increase in fluorescence (p < 0.001) compared to when only one or none of the components were added. Data are shown as the average of at least four biological replicates with the corresponding standard deviation shown with error bars.
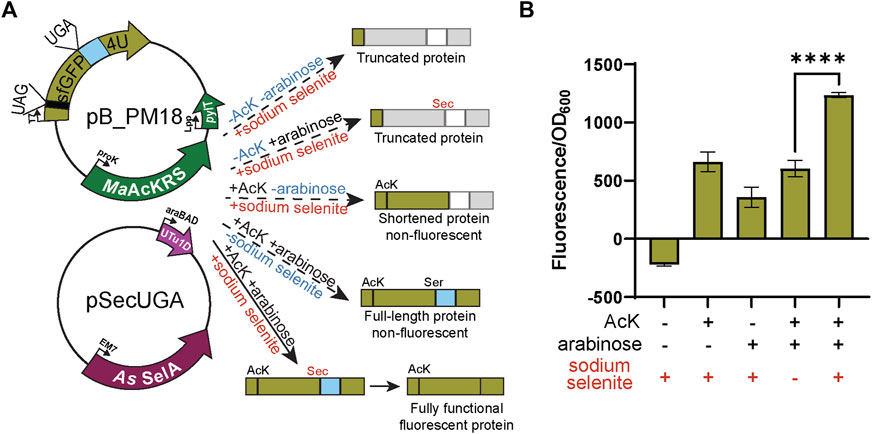
FIGURE 5. Confirmation of Sec insertion using intein reporter. (A) The assay was performed with a two-plasmid system: pB_PM15 and pSecUGA. The first plasmid contains the genes for MaAcKRS3-IP and tRNAPylCUA (pylT) for insertion of Nε-acetyl-l-lysine (AcK) at a UAG and a modified pB_04 sfGFP intein reporter gene. This gene has been modified from the original (Chung et al., 2022) to encode a UAG at position 2 and a UGA at position 204, the first position of the M86 mini-intein. The second plasmid, pSecUGA, contains machinery for Sec insertion at UGA. In the presence of AcK, arabinose, and sodium selenite, the entire sfGFP gene should be expressed. When Sec is inserted at position 204, the intein will splice out to produce a fluorescent functional reporter. In the absence of sodium selenite alone, the entire sfGFP gene should be expressed except serine (Ser) should be inserted at position 204, which does not induce splicing. This results in a non-functional, non-fluorescent protein. In the absence of AcK or arabinose (or both), translation stops resulting in truncated non-fluorescent protein. Only the first amino acid would be translated without AcK, while without arabinose, translation would stop at 204UGA. (B) The sfGFP_intein assay shows successful readthrough of position 2 and splicing of the intein in the presence of AcK, arabinose, and sodium selenite, with a significant increase in fluorescence (p < 0.0001) compared to when any one of the components is missing or in the absence of them all. Data are shown as the average of at least four biological replicates with the corresponding standard deviation shown with error bars.
3.3 Expression of post-translationally modified seleno-sfGFP in E. coli
The sfGFP readthrough and intein assays demonstrated that AcK and Sec could be inserted into a single protein using two different stop codons. Therefore, we applied our integrated plasmid setup and growth conditions to express and purify acetylated seleno-sfGFP from E. coli. The location of the His6-tag on the C-terminus allowed the separation of full-length protein from truncated versions due to early termination (Figure 5A). This strategy produced a robust band at roughly 25 kDa correlating to our expected protein size (28 kDa) (Figure 6). We also compared our optimized system, which involves recoding UGA with Sec and UAG with AcK, to our original system (recoding UGA with AcK and UAG with Sec) (Figure 6A). In both systems, we obtained pure protein; however, our yields were increased roughly 5-fold with our optimized codon set compared to the original (Figure 6B). Mass spectrometry analysis of the optimized codon set confirmed the presence of acetylated seleno-sfGFP. Protein mass was also detected that corresponded to Gln instead of AcK at a proportion of 67%, suggesting that near-cognate suppression by tRNAGln can impact the final protein purity (Figure 6C). This was further verified with MS/MS data to show that indeed AcK or Gln was inserted at position 151 while Sec was always observed at position 2 (Supplementary Figure S2).
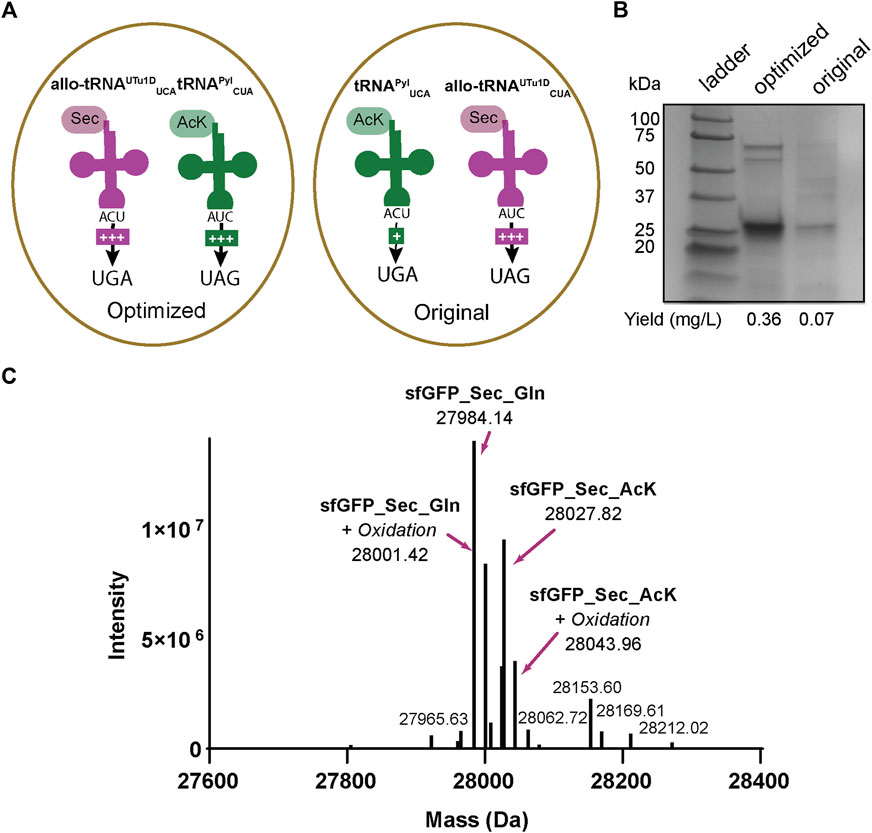
FIGURE 6. Production of AcK-seleno-sfGFP. (A) Schematic of tRNAs used and the respective codons they are expected to recode. Optimized conditions involved Sec insertion at position 2UGA with allo-tRNAUTu1DUCA and Nε-acetyl-l-lysine (AcK) at 151UAG with tRNAPylCUA. Original conditions involved AcK insertion at position 2UGA with tRNAPylUCA and Sec insertion at 151UAG with allo-tRNAUTu1DCUA. An increase in the amount of + signs correspond to the increased suppression observed. (B) SDS-PAGE shows a strong band around the 25 kDa marker, corresponding to the AcK-seleno-sfGFP size of 28 kDa. An increase in protein yield was found for the anticodon-optimized conditions compared to the original. (C) Intact MS data has multiple peaks which can be assigned to AcK-seleno-sfGFP (37%) and Gln-seleno-sfGFP (63%) with or without an oxidation state.
3.4 Recombinant purification of acetylated human selenoprotein GPx1
To prove the versatility and utility of our system, we produced an authentic selenoprotein, human GPx1. GPx1 is predicted to be acetylated at different Lys residues based on murine GPx1 MS studies (Rardin et al., 2013). We individually targeted K114 and K148 together with the Sec at position 49. The protein was expressed under the same conditions with the same plasmid backbone as described for sfGFP (Figure 7A). Three bands were visible on the polyacrylamide gel, with the lower band corresponding to the expected size of monomeric GPx1 (25 kDa). In terms of protein yield, we found that GPx1_148AcK had roughly twice as much protein as GPx1_114AcK. This may be a result of codon context or the closer proximity of position 114 to 49Sec (Figure 7B). To confirm incorporation of AcK, a western blot with an anti-AcK antibody was used (Figure 7B). The western blot revealed the presence of acetylation in the higher molecular weight band which is approximately 30%–35% of the total protein (Supplementary Figure S3). We surmise that the lower molecular weight band corresponds to a GPx1 variant containing Gln (65%–70%) instead of AcK as suggested by the sfGFP MS data. This provides evidence that imply the potential role of AcK in GPx1 oligomerization, and emphasizes the necessity for a system which can study post-translationally modified selenoproteins.
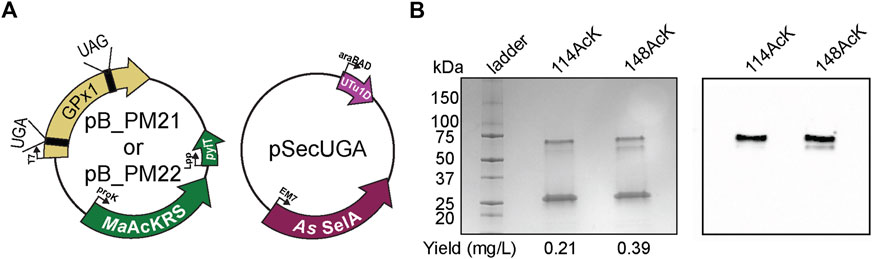
FIGURE 7. Production of post-translationally modified human GPx1. (A) Plasmids used for insertion of selenocysteine and Nε-acetyl-l-lysine (AcK) into GPx1 at UGA and UAG, respectively. Two variants were made with AcK at either position 114 or 148. (B) 4%–20% PAGE shows three bands on the gel for each variant, with the strongest one at the 25-kDa marker corresponding to monomeric GPx1 (24 kDa). An increase in protein yield was observed for 148AcK. Western blot with anti-AcK antibody targeted the two higher molecular weight bands.
4 Conclusion
Here we established a method for the simultaneous incorporation of selenocysteine and the ncAA Nε-acetyl-l-lysine into a protein at two targeted positions in E. coli. This method integrates pSec_Evol and MaPylRS:tRNAPyl non-sense translation systems for the first time (Figure 1). This was accomplished by characterizing the decoding efficiency, specificity, and compatibility of these systems. We found that allo-tRNAUTu1D and MaAcLysRS3:tRNAPyl are compatible when UGA and UAG are assigned to Sec and AcK, respectively (Figures 3B, C). In contrast, allo-tRNAUTu1DCUA and tRNAPylUCA lacked decoding fidelity as they translated both UGA and UAG codons, albeit with different efficiencies (Figures 2B, C). Thus, our results indicate that the codon specificity of these two translation systems should be an essential consideration when developing GCE applications, particularly when combining them with other systems. Another important consideration is the position of the non-sense codons within each protein as mRNA context can influence the suppressor tRNAs as well as the proximity of the two ncAAs. The off-target activity of these tRNAs with non-specific anticodons may hinder the accurate incorporation of the desired amino acid and drastically decrease protein yields by impacting the fitness of the host organism. Using this knowledge, we demonstrated that pSecUGA and MaAcLysRS3:tRNAPylCUA systems could be combined in E. coli to create a platform for synthesizing proteins containing these two important amino acids. This provides the groundwork to produce acetylated natural or synthetic selenoproteins. The utility of our method can be expanded by combining the pSecUGA system with other existing GCE platforms to facilitate the synthesis of selenoproteins containing other important PTMs or ncAAs with reactive side chains to expand protein functionalization (Young and Schultz, 2018; Shandell et al., 2021). Ultimately, this technology can enable the investigation of PTMs in naturally occurring selenoproteins and the development of artificial proteins endowed with unique chemical properties.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author contributions
PM and NK performed the cloning and fluorescence assays. CC, LN, and NK performed protein expression and purification. PM, NK, and OV-R wrote the manuscript. OV-R, CZC, and NK conceptualized the experiments and edited the manuscript. DS and PH edited the manuscript and supported the research.
Funding
This work was supported by grants from the National Institute of General Medical Sciences (R35GM122560-05S1 to DS) and the National Institute of Allergy and Infectious Diseases (5R01AI147496 to PH). Genetic experiments were supported by the Department of Energy Office of Basic Energy Sciences (DE-FG0298ER2031 to DS). CZC holds a Postdoctoral Fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC).
Acknowledgments
We are grateful to Kyle Hoffman (Bioinformatics Solutions Inc.) for intellectual and technical assistance with the mass spectrometry analyses. We also thank the Keck Oligo Synthesis Resource at Yale for assisting with nucleotide synthesis services and Kexin Meng for experimental advice and support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1096261/full#supplementary-material
References
Aldag, C., Bröcker, M. J., Hohn, M. J., Prat, L., Hammond, G., Plummer, A., et al. (2013). Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew. Chem. Int. Ed. Engl. 52 (5), 1441–1445. doi:10.1002/anie.201207567
Appleby-Tagoe, J. H., Thiel, I. V., Wang, Y., Wang, Y., Mootz, H. D., and Liu, X. Q. (2011). Highly efficient and more general cis- and trans-splicing inteins through sequential directed evolution. J. Biol. Chem. 286 (39), 34440–34447. doi:10.1074/jbc.M111.277350
Balasuriya, N., Davey, N. E., Johnson, J. L., Liu, H., Biggar, K. K., Cantley, L. C., et al. (2020). Phosphorylation-dependent substrate selectivity of protein kinase B (AKT1). J. Biol. Chem. 295 (24), 8120–8134. doi:10.1074/jbc.RA119.012425
Chatterjee, A., Sun, S. B., Furman, J. L., Xiao, H., and Schultz, P. G. (2013). A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52 (10), 1828–1837. doi:10.1021/bi4000244
Chen, H., Venkat, S., McGuire, P., Gan, Q., and Fan, C. (2018). Recent development of genetic code expansion for posttranslational modification studies. Molecules 23 (7), 1662. doi:10.3390/molecules23071662
Chung, C. Z., and Krahn, N. (2022). The selenocysteine toolbox: A guide to studying the 21st amino acid. Arch. Biochem. Biophys. 730, 109421. doi:10.1016/j.abb.2022.109421
Chung, C. Z., Miller, C., Söll, D., and Krahn, N. (2021). Introducing selenocysteine into recombinant proteins in Escherichia coli. Curr. Protoc. 1 (2), e54. doi:10.1002/cpz1.54
Chung, C. Z., Krahn, N., Crnković, A., and Söll, D. (2022). Intein-based design expands diversity of selenocysteine reporters. J. Mol. Biol. 434 (8), 167199. doi:10.1016/j.jmb.2021.167199
Copeland, P. R., Fletcher, J. E., Carlson, B. A., Hatfield, D. L., and Driscoll, D. M. (2000). A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 19 (2), 306–314. doi:10.1093/emboj/19.2.306
Crnković, A., Suzuki, T., Söll, D., and Reynolds, N. M. (2016). Pyrrolysyl-tRNA synthetase, an aminoacyl-tRNA synthetase for genetic code expansion. Croat. Chem. Acta 89 (2), 163–174. doi:10.5562/cca2825
Dunkelmann, D. L., Willis, J. C. W., Beattie, A. T., and Chin, J. W. (2020). Engineered triply orthogonal pyrrolysyl-tRNA synthetase/tRNA pairs enable the genetic encoding of three distinct non-canonical amino acids. Nat. Chem. 12 (6), 535–544. doi:10.1038/s41557-020-0472-x
Hilal, T., Killam, B. Y., Grozdanović, M., Dobosz-Bartoszek, M., Loerke, J., Burger, J., et al. (2022). Structure of the mammalian ribosome as it decodes the selenocysteine UGA codon. Science 376 (6599), 1338–1343. doi:10.1126/science.abg3875
Kossinova, O., Malygin, A., Krol, A., and Karpova, G. (2014). The SBP2 protein central to selenoprotein synthesis contacts the human ribosome at expansion segment 7L of the 28S rRNA. RNA 20 (7), 1046–1056. doi:10.1261/rna.044917.114
Lacoursiere, R. E., O'Donoghue, P., and Shaw, G. S. (2020). Programmed ubiquitin acetylation using genetic code expansion reveals altered ubiquitination patterns. FEBS Lett. 594 (7), 1226–1234. doi:10.1002/1873-3468.13702
Mukai, T., Hoshi, H., Ohtake, K., Takahashi, M., Yamaguchi, A., Hayashi, A., et al. (2015). Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep. 5, 9699. doi:10.1038/srep09699
Mukai, T., Vargas-Rodriguez, O., Englert, M., Tripp, H. J., Ivanova, N. N., Rubin, E. M., et al. (2017). Transfer RNAs with novel cloverleaf structures. Nucleic Acids Res. 45 (5), 2776–2785. doi:10.1093/nar/gkw898
Mukai, T., Sevostyanova, A., Suzuki, T., Fu, X., and Söll, D. (2018). A facile method for producing selenocysteine-containing proteins. Angew. Chem. Int. Ed. Engl. 57 (24), 7215–7219. doi:10.1002/anie.201713215
Neumann, H., Slusarczyk, A. L., and Chin, J. W. (2010). De novo generation of mutually orthogonal aminoacyl-tRNA synthetase/tRNA pairs. J. Am. Chem. Soc. 132 (7), 2142–2144. doi:10.1021/ja9068722
Novoselov, S. V., Lobanov, A. V., Hua, D., Kasaikina, M. V., Hatfield, D. L., and Gladyshev, V. N. (2007). A highly efficient form of the selenocysteine insertion sequence element in protozoan parasites and its use in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 104 (19), 7857–7862. doi:10.1073/pnas.0610683104
Pinkerton, M. H., and Copeland, P. R. (2016). “Eukaryotic mechanisms of selenocysteine incorporation and its reconstitution in vitro,” in Selenium: its molecular biology and role in human health. Fourth ed (Springer International Publishing), 13–24.
Porter, J. J., and Mehl, R. A. (2018). Genetic code expansion: A powerful tool for understanding the physiological consequences of oxidative stress protein modifications. Oxid. Med. Cell Longev. 2018, 7607463. doi:10.1155/2018/7607463
Prabhakar, A., Krahn, N., Zhang, J., Vargas-Rodriguez, O., Krupkin, M., Fu, Z., et al. (2022). Uncovering translation roadblocks during the development of a synthetic tRNA. Nucleic Acids Res. 50 (18), 10201–10211. doi:10.1093/nar/gkac576
Rardin, M. J., Newman, J. C., Held, J. M., Cusack, M. P., Sorensen, D. J., Li, B., et al. (2013). Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. U. S. A. 110 (16), 6601–6606. doi:10.1073/pnas.1302961110
Seki, E., Yanagisawa, T., Kuratani, M., Sakamoto, K., and Yokoyama, S. (2020). Fully productive cell-free genetic code expansion by structure-based engineering of Methanomethylophilus alvus pyrrolysyl-tRNA synthetase. ACS Synth. Biol. 9 (4), 718–732. doi:10.1021/acssynbio.9b00288
Shandell, M. A., Tan, Z., and Cornish, V. W. (2021). Genetic code expansion: A brief history and perspective. Biochemistry 60 (46), 3455–3469. doi:10.1021/acs.biochem.1c00286
Tharp, J. M., Ad, O., Amikura, K., Ward, F. R., Garcia, E. M., Cate, J. H. D., et al. (2020). Initiation of protein synthesis with non-canonical amino acids in vivo. Angew. Chem. Int. Ed. Engl. 59 (8), 3122–3126. doi:10.1002/anie.201914671
Tharp, J. M., Vargas-Rodriguez, O., Schepartz, A., and Söll, D. (2021). Genetic encoding of three distinct noncanonical amino acids using reprogrammed initiator and nonsense codons. ACS Chem. Biol. 16 (4), 766–774. doi:10.1021/acschembio.1c00120
Venkat, S., Sturges, J., Stahman, A., Gregory, C., Gan, Q., and Fan, C. (2018). Genetically incorporating two distinct post-translational modifications into one protein simultaneously. ACS Synth. Biol. 7 (2), 689–695. doi:10.1021/acssynbio.7b00408
Wang, K., Sachdeva, A., Cox, D. J., Wilf, N. M., Lang, K., Wallace, S., et al. (2014). Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat. Chem. 6 (5), 393–403. doi:10.1038/nchem.1919
Wright, D. E., Altaany, Z., Bi, Y., Alperstein, Z., and O'Donoghue, P. (2018). Acetylation regulates thioredoxin reductase oligomerization and activity. Antioxid. Redox Signal 29 (4), 377–388. doi:10.1089/ars.2017.7082
Yang, A., Cho, K., and Park, H. S. (2018). Chemical biology approaches for studying posttranslational modifications. RNA Biol. 15 (4-5), 427–440. doi:10.1080/15476286.2017.1360468
Ye, R., Huang, J., Wang, Z., Chen, Y., and Dong, Y. (2022). The role and mechanism of essential selenoproteins for homeostasis. Antioxidants (Basel) 11 (5), 973. doi:10.3390/antiox11050973
Keywords: selenoproteins, acetyl-lysine, post-translational modifications, genetic code expansion, selenocysteine, synthetic biology
Citation: Morosky P, Comyns C, Nunes LGA, Chung CZ, Hoffmann PR, Söll D, Vargas-Rodriguez O and Krahn N (2023) Dual incorporation of non-canonical amino acids enables production of post-translationally modified selenoproteins. Front. Mol. Biosci. 10:1096261. doi: 10.3389/fmolb.2023.1096261
Received: 11 November 2022; Accepted: 11 January 2023;
Published: 24 January 2023.
Edited by:
Ned Budisa, University of Manitoba, CanadaReviewed by:
Weimin Xuan, Tianjin University, ChinaXian Fu, Beijing Genomics Institute (BGI), China
Chenguang Fan, University of Arkansas, United States
Copyright © 2023 Morosky, Comyns, Nunes, Chung, Hoffmann, Söll, Vargas-Rodriguez and Krahn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natalie Krahn, bmF0YWxpZS5rcmFobkB5YWxlLmVkdQ==