 Barbara B. Bendlin1,2*
Barbara B. Bendlin1,2* Lisa M. Newman1,2
Lisa M. Newman1,2 Michele L. Ries1,2
Michele L. Ries1,2 Luigi Puglielli1,2
Luigi Puglielli1,2 Cynthia M. Carlsson1,2
Cynthia M. Carlsson1,2 Mark A. Sager1,2
Mark A. Sager1,2 Howard A. Rowley2
Howard A. Rowley2 Catherine L. Gallagher1,2
Catherine L. Gallagher1,2 Auriel A. Willette2,3
Auriel A. Willette2,3 Andrew L. Alexander4,5,6
Andrew L. Alexander4,5,6 Sanjay Asthana1,2
Sanjay Asthana1,2 Sterling C. Johnson1,2
Sterling C. Johnson1,2
- 1 William S. Middleton Memorial Veterans Hospital, Geriatric Research Education and Clinical Center, Madison, WI, USA
- 2 Department of Medicine, Wisconsin Alzheimer’s Disease Research Center, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
- 3 Harlow Primate Laboratory, Department of Psychology, University of Wisconsin-Madison, Madison, WI, USA
- 4 Department of Medical Physics, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
- 5 Department of Psychiatry, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
- 6 Waisman Laboratory for Brain Imaging and Behavior, Madison, WI, USA
The use of non-steroidal anti-inflammatory drugs (NSAIDs) in humans is associated with brain differences including decreased number of activated microglia. In animals, NSAIDs are associated with reduced microglia, decreased amyloid burden, and neuronal preservation. Several studies suggest NSAIDs protect brain regions affected in the earliest stages of AD, including hippocampal and parahippocampal regions. In this cross-sectional study, we examined the protective effect of NSAID use on gray matter volume in a group of middle-aged and older NSAID users (n = 25) compared to non-user controls (n = 50). All participants underwent neuropsychological testing and T1-weighted magnetic resonance imaging. Non-user controls showed smaller volume in portions of the left hippocampus compared to NSAID users. Age-related loss of volume differed between groups, with controls showing greater medial temporal lobe volume loss with age compared to NSAID users. These results should be considered preliminary, but support previous reports that NSAIDs may modulate age-related loss of brain volume.
Introduction
McGeer et al. (1990) published the remarkable finding that of 923 arthritis patients older than 64 years, only four had clinical signs of Alzheimer’s disease (AD), a prevalence of only 0.4% compared to a prevalence of 5.1% for the general population at that time Canadian Study of Health and Aging (1994). Since then, additional studies have indicated a protective effect on cognition (Hee Kang and Grodstein, 2003; Jonker et al., 2003; Hayden et al., 2007) and decreased risk for AD with anti-inflammatory drug use (McGeer et al., 2006), chiefly non-steroidal anti-inflammatory drugs (NSAIDs). Epidemiological evidence for a beneficial effect of NSAIDs has been bolstered by the results of animal studies. NSAIDs reduce the number of activated microglia and amyloid plaque burden (Lim et al., 2000; Yan et al., 2003; van Groen and Kadish, 2005) in amyloid precursor protein (APP) transgenic mouse models of AD. Additionally, NSAIDs attenuate inflammation and subsequent loss of neurons in models that mimic AD associated inflammation via lipopolysaccharide infusion (Willard et al., 2000).
Only a few studies have examined the effect of anti-inflammatory drugs on the human brain. NSAIDS appear to decrease microglial activation; a comparison of brains from arthritis patients to control brains at post mortem indicated that NSAID users with senile plaques had only one-third of the activated microglia found in control brains with senile plaques (Mackenzie and Munoz, 1998). A follow-up study by the same investigator found similar results and confirmed that the effect was specific to NSAIDs and not steroidal anti-inflammatory medications (Mackenzie, 2000). Microglia under normal conditions scavenge the extracellular milieu and tissue to remove debris and endogenous toxic molecules, as well as phagocytize pathogens. However, even slight perturbations in the CNS can induce activation and endocytic damage (Banati et al., 1993). A post mortem study of a subset of brains donated by participants in the Religious Orders Study did not show brain differences between NSAID-users and non-users (Arvanitakis et al., 2008); yet only plaque and tangle burdens were compared, but not activated microglia nor other indices of neuroinflammation (Mackenzie and Munoz, 1998; Mackenzie, 2000). The results of a recent brain imaging study suggest a neuroprotective effect of anti-inflammatory drugs on brain volume (Walther et al., 2009). Walther et al. (2009) examined gray and white matter volume in a group of participants ranging in age from 65 to 93 years who were taking NSAIDs or other anti-inflammatory medications. Anti-inflammatory drugs were associated with widespread attenuated age-related volume decline.
In the present study, we extended upon the findings of Walther et al. (2009) by examining participants ranging in age from 42 to 75 years of age, who were NSAID users or non-user controls. Since NSAIDs protect against AD and are involved in preservation of memory function, we hypothesized that NSAID use would be associated with greater volume in brain regions typically affected by AD pathology. Analyses were restricted to the hippocampus and parahippocampal gray matter, as these regions are known to be profoundly affected in AD (Hyman et al., 1984; de Leon et al., 1989) and are also known to be affected by risk factors for AD such as APOE ε4 genotype (Trivedi et al., 2006; Burggren et al., 2008; Mueller and Weiner, 2009) and parental family history of AD (Johnson et al., 2006; Mosconi et al., 2007). Based on previous findings indicating that NSAIDs modify cognitive and brain aging trajectories (Rozzini et al., 1996; Hayden et al., 2007; Walther et al., 2009), we tested for an interaction between NSAID use and age on gray matter volume. We hypothesized that non-users would exhibit more age-related brain volume loss compared to the NSAID user group.
Materials and Methods
Participants
Cognitively normal participants from the community recruited as part of ongoing studies of brain function and aging were included in this study. Prior to participation in these studies, participants were screened for eligibility including medical history and MRI scanner compatibility. Exclusion criteria were: a diagnosis of dementia, mild cognitive impairment, or other cognitive disorder; significant cognitive complaints; prior or current neurological disease or neurosurgery; current diagnosis of major Axis I psychiatric disorder; chronic major medical conditions (e.g., poorly controlled diabetes or hypertension, cardiac disease, cancer treated with radiation or chemotherapy within the past 5 years); and a modified Hachinski score (Rosen et al., 1980) greater than four. Health history was obtained through a comprehensive health and medical history questionnaire as well as screening interviews. Cognitive function was tested using a battery of neuropsychological tests administered by experienced technicians. The cognitive data were scored independently by the testing technician and a second technician. The double-scored results were reviewed by a neuropsychologist who resolved any non-clerical discrepancies. Participants who scored two standard deviations below the mean on any test were excluded from the analysis. Current mood was assessed with the Center for Epidemiologic Study-Depression Scale (CES-D) and the State-Trait Anxiety Inventory (STAI). Participants showing evidence of significant affective dysfunction (CES-D > 16; STAI trait scale in > 95th percentile of age- and gender-corrected normative data) were excluded. Most participants were administered both the STAI and the CES-D, however in a few cases, participants completed only one of these tests. The number of participants that underwent each test is indicated in Table 1. MRI scans were read by a neuroradiologist and were required to be read as normal for inclusion in statistical analyses.
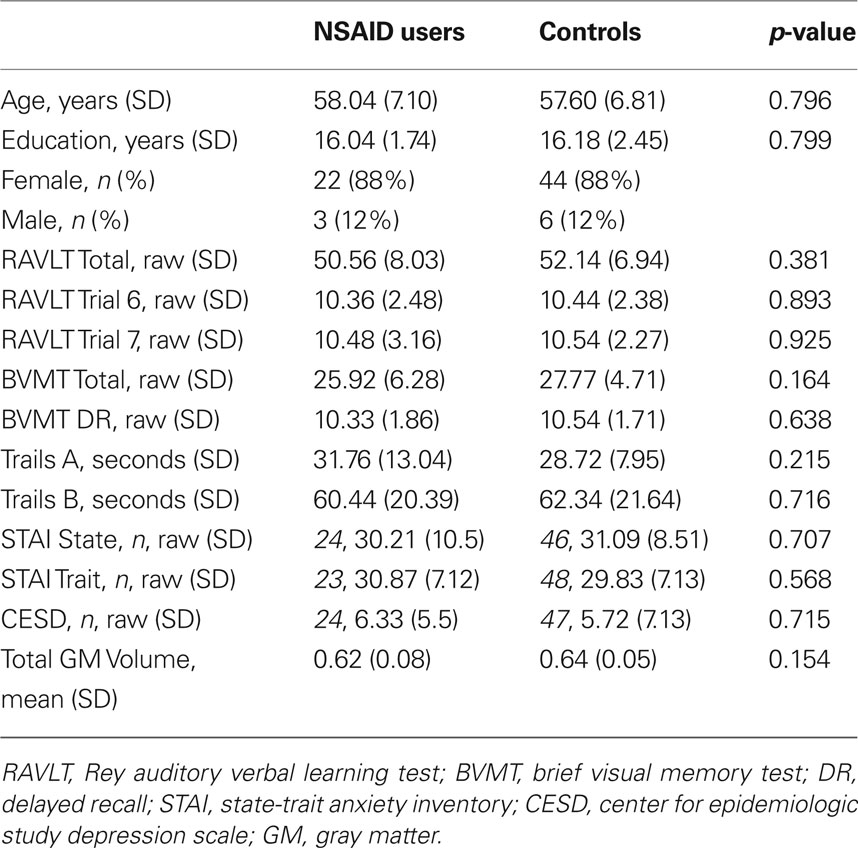
Table 1. Demographic, neuropsychological, and gray matter volume data.
Information regarding medication usage was obtained via the health and medical history questionnaire. Using this information, a total of 25 NSAID users were identified from the original pool of subjects (mean age 58.04 ± 7.10 years; mean education 16.04 ± 1.74 years; 22 women, 3 men). From the remaining pool of participants, two non-user controls were identified for every one NSAID user, matched for age within 1 year, educational achievement within 1 year, and sex (mean age 57.60 ± 6.81 years; mean education 16.18 ± 2.45 years; 44 women, 6 men). NSAID users were defined as individuals who had self-reportedly been taking NSAIDs at least once per week for a minimum of 6 months prior to their visit. Sixteen of the 25 NSAID users took ibuprofen; six of the 25 users took one of the following: diclofenac, etodolac, meloxicam, nabumetone, piroxicam, rofecoxib; and the remaining three users took a combination of NSAIDS: one of the three took naproxen and ibuprofen, a second user took rofecoxib and ibuprofen, and the third user took rofecoxib and nabumetone. The duration of NSAID use ranged from a minimum of 6 months to 16 years. The most frequently occurring duration of NSAID was 6 years (modal score) prior to MRI scan, with an average of approximately 3 years of use prior to scan. Controls were defined as cognitively normal individuals who had never reported taking NSAIDS for conditions other than occasional headache or minor transient (not chronic) pain. All participants gave written informed consent approved by the University of Wisconsin Health Sciences Institutional Review Board prior to their brain scan and neuropsychological assessment. The demographic characteristics of the participants and descriptive statistics of the neuropsychological testing are reported in Table 1.
Neuropsychological Testing
Participants underwent neuropsychological evaluation to characterize cognitive function and ensure normal functioning. Test results included in this report were common across all participants and were: the Rey Auditory Verbal Learning Test (RAVLT), Brief Visual Memory Test (BVMT), the Trail Making Test parts A and B, CES-D, and STAI.
Magnetic Resonance Imaging
Magnetic resonance imaging (MRI) scans were obtained using a General Electric 3.0 Tesla scanner (Waukesha, WI, USA). A 3D T1-weighted image was acquired with an inversion recovery prepared fast gradient echo pulse sequence. Parameters were: inversion time = 600 ms, fast gradient echo read-out with TR/TE/flip angle = 9 ms/1.8 ms/20°; acquisition matrix = 256 × 192 × 124, interpolated to 256 × 256 × 124; field of view = 240 mm; slice thickness = 1.2 mm (124 slices), no gap; receiver bandwidth = ± 16 kHz; acquisition time ∼7.5 min. Other scans were collected but are not included here. An experienced neuroradiologist reviewed all images prior to the current analysis for clinical evidence of any neurovascular disease or structural abnormality that would exclude the subjects from the analysis.
Voxel-Based Morphometry (VBM)
T1-weighted images were processed using Statistical Parametric Mapping (SPM5) software (http://www.fil.ion.ucl.ac.uk/spm), which employs a unified approach for segmentation, combining segmentation of the original anatomical images into gray matter (GM), white matter, and cerebrospinal fluid probability images; spatial normalization (12-parameter affine transformation and non-linear deformation with a warp frequency cutoff of 25) of the segmented images to the Montreal Neurological Institute (MNI) template; and bias correction, in a single iterative process. The resulting segmented images were modulated using the Jacobian values obtained from spatial normalization in order to preserve GM volume by scaling the final images by the amount of contraction required to warp the images to the template. The results from this step were GM volume maps for each participant, where the total amount of GM remained the same as in the original images. Finally, the normalized maps were smoothed using an 8-mm isotropic Gaussian kernel to optimize signal to noise and facilitate comparison across participants. Analysis of GM volume employed an absolute threshold masking of 0.1 to minimize the inclusion of white matter voxels in the analysis.
Statistical Analysis
We hypothesized that non-users would show lower GM volume compared to NSAID users. Groups were compared using a voxel-wise two-sample t-test in SPM5. Due to the well-matched sample, we did not include age, education, or gender as covariates. Total GM volume (in mm3) was derived by summing the voxels in the modulated, spatially normalized images, and multiplying by the voxel volume. GM volume was entered into the model as a proportional scaling factor to control for global differences. Additionally, we examined the effect of age across all participants, and in order to test the hypothesis that NSAID use would be protective against age-related volume changes, we tested for an age by group (NSAID, non-user) interaction using a regression model implemented in SPM5. All analyses were restricted to hippocampi and parahippocampal gyri bilaterally using the Wake Forest University (WFU) Pickatlas and Anatomic Automatic labeling atlas (AAL) atlas. An ROI was generated by combining hippocampi and parahippocampal gyri and dilating by two which expanded the ROI by two voxels in each direction. Additionally, we masked the effects of group and age (main effects) by the interaction effect to exclude regions were the group × age interaction results were significant (thresholded at p < 0.05). Main effects and the interaction effect were considered significant at p < 0.01. A cluster size threshold of 20 contiguous voxels was employed in order to exclude clusters of a very small size. The Statistical Package for the Social Sciences SPSS version 16.0 (SPSS, Inc., Chicago, IL, USA) was used for the analysis of behavioral data.
Results
Neuropsychological Characterization
Performance on the RAVLT, BVMT, and Trail Making Test A did not differ significantly between groups. Group means, and results of t-tests, are reported in Table 1.
MRI Results
Group × age interaction
As shown in Figure 1, there was a significant interaction between age and group in bilateral hippocampi (p < 0.01). Increasing age in controls was associated with lower volume compared to NSAID users. The greater age-related slope in controls compared to NSAID users is plotted in Figure 2. Cluster sizes, MNI coordinates, and t values for significant regions are listed in Table 2.
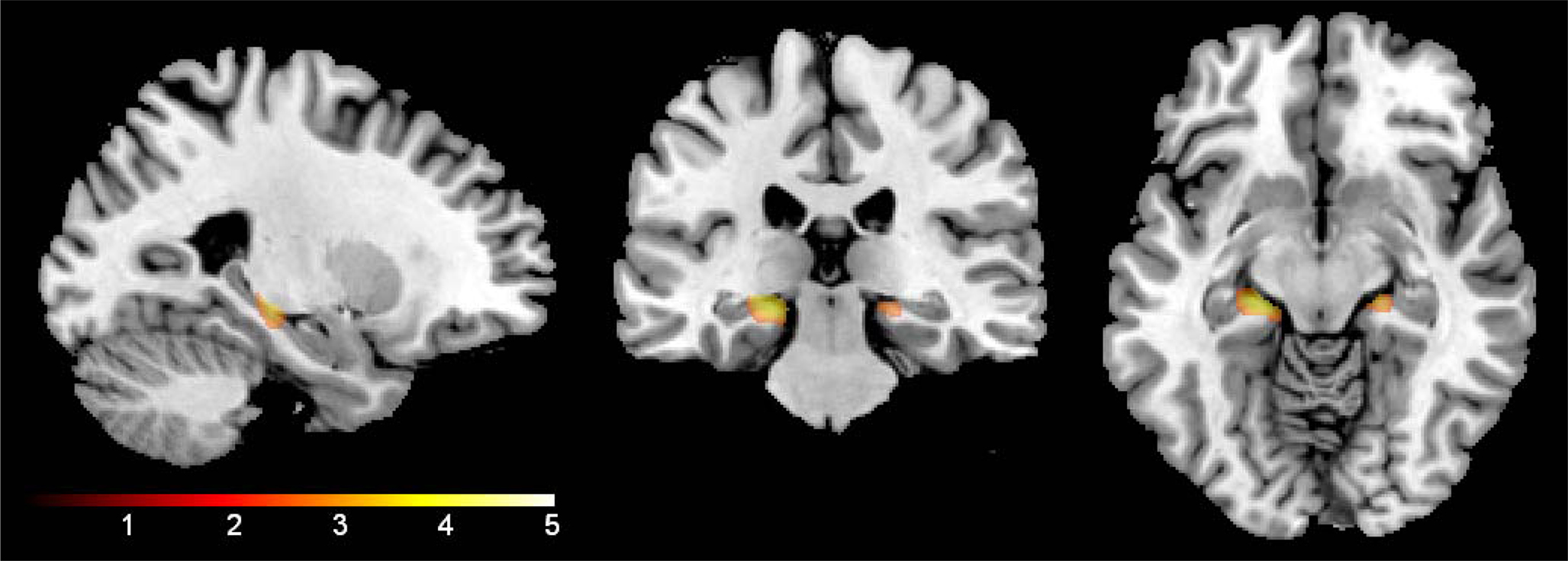
Figure 1. Age by group interaction. NSAID users showed an attenuated age slope compared to non-user controls in bilateral hippocampi (shown below) and right parahippocampal gyrus (not pictured). The results are confined to the study’s a priori regions of interest (hippocampi and parahippocampal gyri). The color bar represents the height of the t-statistic.
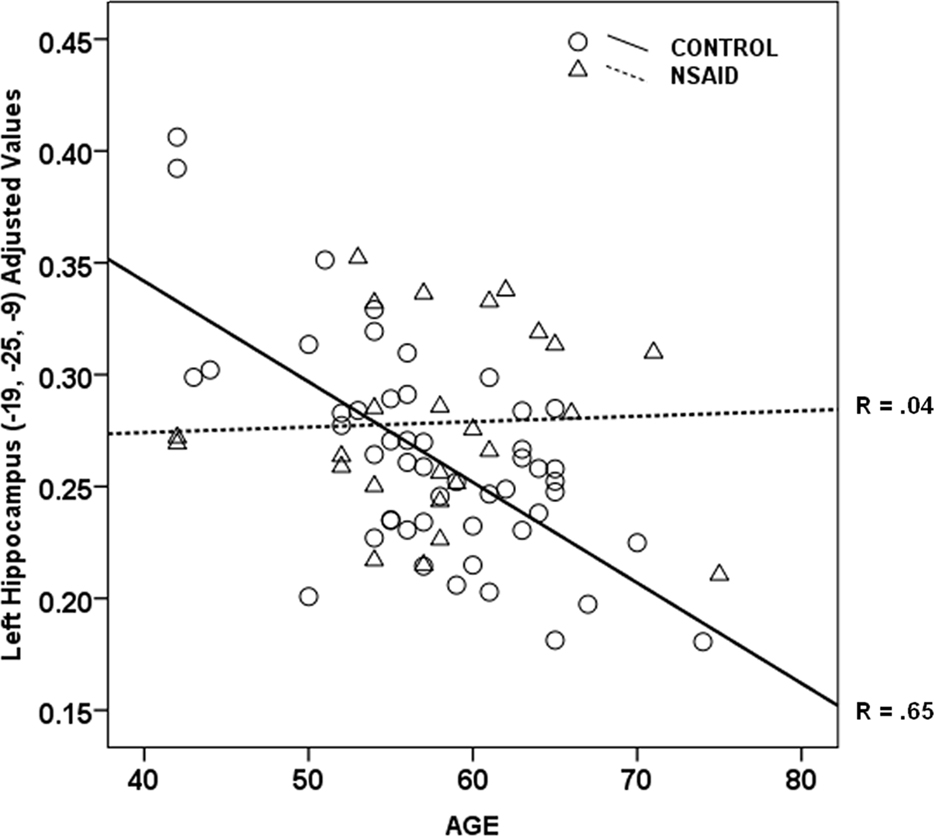
Figure 2. Age by group interaction plot. Non-users controls showed a strong relationship between older age and lower hippocampal volume (r = 0.65) compared to the NSAID user group (r = 0.04). Values plotted below are from left hippocampus (Y axis) were adjusted for total gray volume.
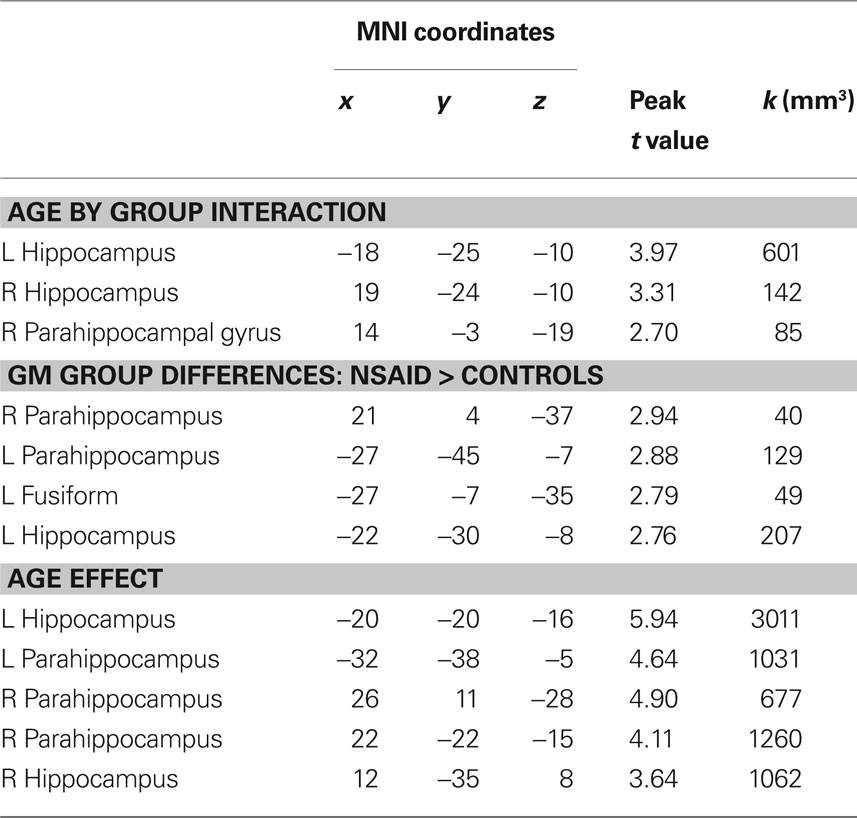
Table 2. MNI coordinates, t-values, and cluster sizes.
Main effects
Group. Total gray matter volume (in mm3) compared between groups using a t-test did not differ significantly between NSAID users and non-users, t(73) = 1.44, p = 1.54. A voxel-wise comparison of gray matter probability maps between NSAID users and non-users revealed a main effect of group in small portions of left bilateral parahippocampal gray matter, these regions are shown in Figure 3. Cluster sizes, MNI coordinates, and t values for regions where non-users showed less volume compared to NSAID users are listed in Table 2. A contrast in the opposite direction (non-users showing greater volume than NSAID users) did not show any significant differences.
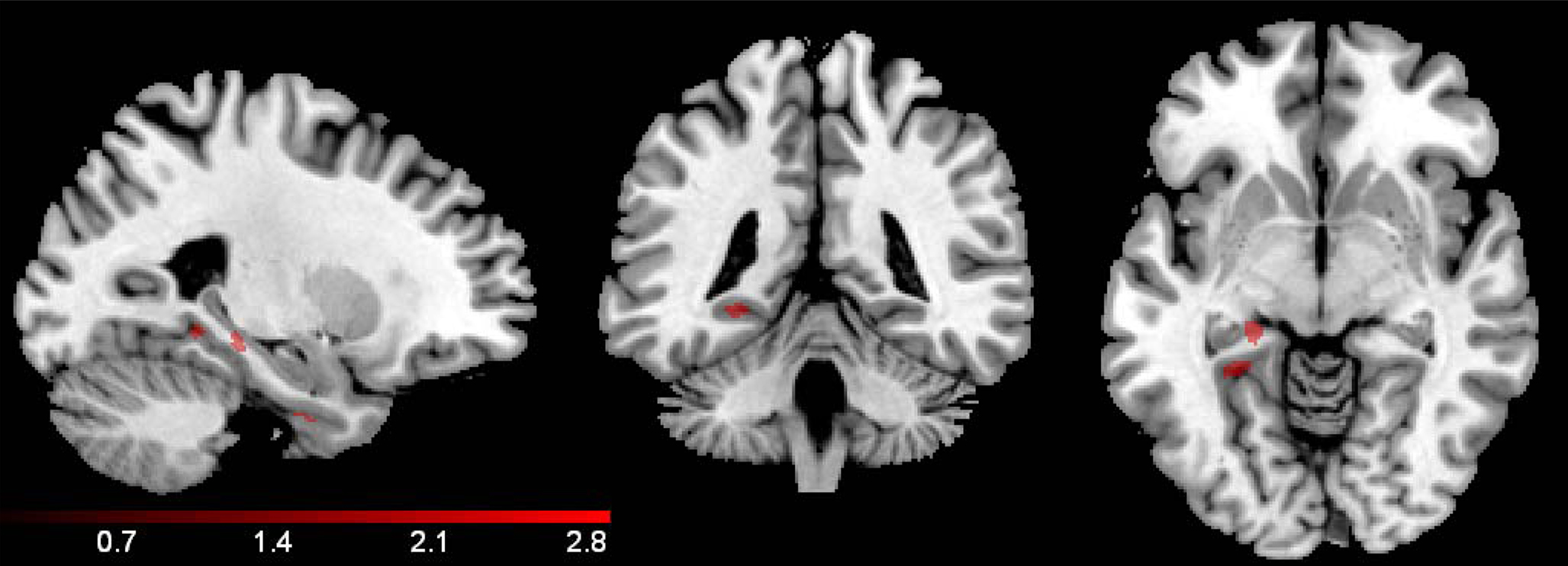
Figure 3. Group effect. A voxel-wise comparison of gray matter volume between NSAID users and non-user controls showed small regions of medial temporal lobe difference where non-users demonstrated smaller volume, including left hippocampus, and parahippocampal gyrus. The results below are confined by the inclusive region of interest mask, and a mask that excludes the significant interaction effect shown in Figure 1. The color bar represents the height of the t-statistic.
Age. The main effect of age was examined using voxel-wise regression which indicated a significant correlation between age and gray matter volume. Regions where volume declined with age are shown in Figure 4. Cluster sizes, MNI coordinates, and t values for regions where older age was associated with smaller volume are shown in Table 2.
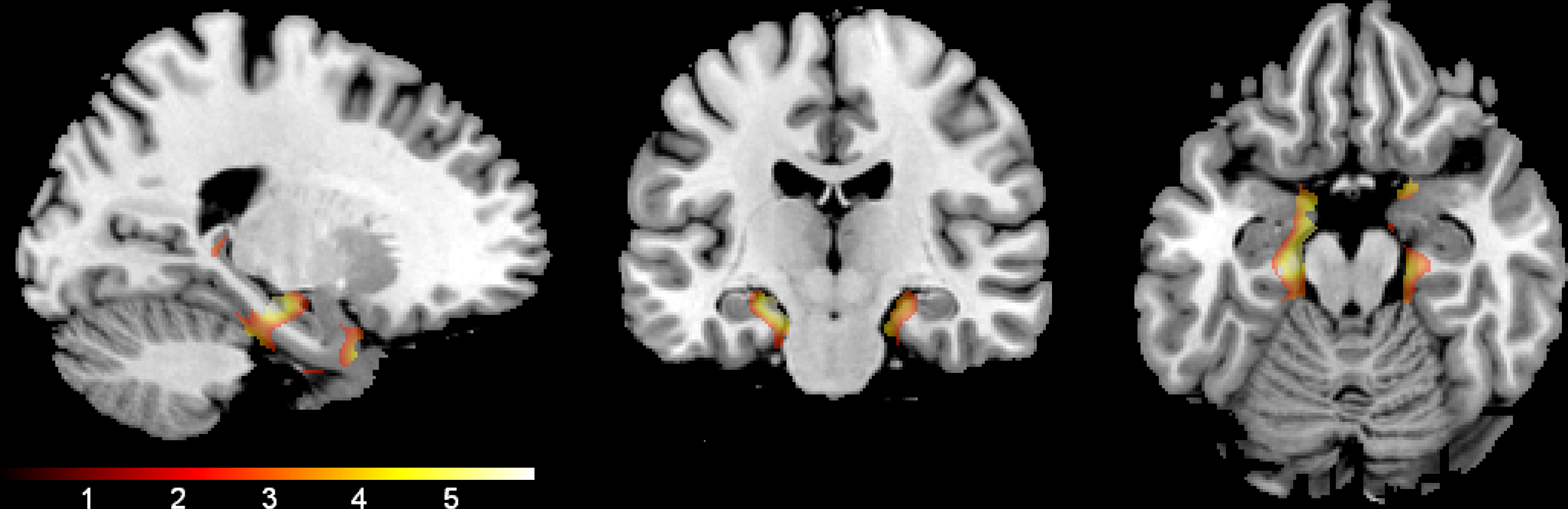
Figure 4. Age effect. The regions where older age was associated with lower gray matter volume are shown. The results are confined to the study’s a priori regions of interest (hippocampi and parahippocampal gyri) and a mask that excludes the significant interaction effect shown in Figure 1. The color bar represents the height of the t-statistic.
Discussion
We performed a voxel-wise analysis of gray matter maps obtained in healthy middle to older-age adults and found significant group differences in medial temporal lobe. NSAID users showed greater volume in bilateral temporal lobe in addition to showing attenuated age-related volume decline compared to non-user controls. These results support previous literature indicating a neuroprotective effect of NSAIDs on the brain (Mackenzie and Munoz, 1998; Mackenzie, 2000; Walther et al., 2009).
At present, the mechanisms by which NSAIDs offer neural protection remain unclear. Recent studies have examined the role of NSAIDs in reducing amyloid burden; NSAIDs that appear to have an effect on amyloid include ibuprofen, sulindac sulfide, flurbiprofen, and indomethacin. There may be several mechanisms by which this occurs, either via modulation of APP cleavage by gamma secretase to favor production of shorter Aβ peptides that are less toxic, thus reducing Aβ42, action on BACE via a peroxisome proliferator-activated receptor gamma (PPARγ) mediated pathway, by direct action on Aβ peptide, or possibly increasing amyloid removal by microglia. NSAIDs limit amyloid accumulation in vitro (Blasko et al., 2001; Thomas et al., 2001; Weggen et al., 2001), and in transgenic mice (Lim et al., 2000; Heneka et al., 2005; McKee et al., 2008). Studies in humans, however, have not supported a decrease in AD pathology in NSAID users, namely decreased burden of β-amyloid plaques or neurofibrillary tangles (Mackenzie and Munoz, 1998; Arvanitakis et al., 2008). Surprisingly, no human post mortem studies have examined the relationship between neuronal loss and NSAID use. Of the characteristics which define AD, it is neuronal loss and synaptic pathology – not β-amyloid plaques or neurofibrillary tangles – that show the stronger relationship to dementia severity and cognitive deficits in AD (DeKosky and Scheff, 1990; Lassmann et al., 1993; Gomez-Isla et al., 1997). Although our study only used an indirect measure of neuronal loss, namely gray matter volume, our results together with a previous report (Walther et al., 2009) support the possibility that beneficial effects of NSAIDs are realized through neuronal preservation.
The alternative and originally proposed mechanism for beneficial actions of NSAIDs is via reduction in neuroinflammation. NSAIDs inhibit cyclooxygenase (COX), which in turn decreases production of prostaglandins, hence decreasing the downstream inflammatory cascade. It is well established that inflammation plays a role in AD related neurodegeneration (McGeer and McGeer, 1995). In addition to COX-1 and COX-2, a full complement of inflammatory mediators have been found in the AD brain, including c-reactive protein; amyloid P; complement proteins; the proinflammatory cytokines interleukin-1, interleukin-6, and tumor necrosis factor-alpha (McGeer and McGeer, 2001). Animal models of neuroinflammation indicate that lipopolysaccharide (LPS)-induced inflammation results in a pattern that has many similarities to the pattern of disease found in AD. For example, rats infused with LPS show an increase in activated microglia in medial temporal lobe especially hippocampus, an increase in β-amyloid precursor protein production, degeneration of hippocampal CA3 pyramidal neurons (Hauss-Wegrzyniak et al., 1998), a loss of pyramidal cells in entorhinal cortex (Hauss-Wegrzyniak et al., 2002), loss of basal forebrain cholinergic neurons (Willard et al., 2000), decreased spatial memory performance (Hauss-Wegrzyniak et al., 2000a), and no recovery of memory function over time (Hauss-Wegrzyniak et al., 2000b).
Although inflammation in AD is likely secondary to other primary pathology (Rogers and Shen, 2000), it is probable that neuroinflammation plays a role in neuronal and synaptic damage, with several studies indicating that accumulation of inflammatory mediators are neurotoxic (see Glass et al., 2010 for review). Cumulative loss of neurons is measurable as atrophy on MRI and recently, the increase in a marker of inflammation, interleukin-6, was found to correspond with lower regional brain volume in rhesus macaque monkeys (Willette et al., 2010) and in middle-aged humans (Marsland et al., 2008). Conversely, treatment with NSAIDs appears to protect against neuronal damage. In an LPS model of neuroinflammation, mice pretreated with sulindac sulfide 3 weeks prior to LPS treatment were protected against the neuronal loss found in the LPS-only group (Lee et al., 2008). Similarly, rats treated with the COX-2 inhibiting NSAID rofecoxib prior to treatment with the excitotoxin N-methyl-d-aspartate, were protected against hippocampal neurodegeneration (Hewett et al., 2006).
The protective effect of NSAIDs found in the present study may have been mediated via either anti-amyloidogenic or COX-inhibiting properties; although several observations suggest that anti-amyloid effects are less likely. Medial temporal gray matter, where a beneficial effect of NSAIDs was found, is not known to show high levels of amyloid plaque burden in either normal aging or in mild to moderate AD (Arriagada et al., 1992; Rowe et al., 2007), and post mortem evaluation of amyloid burden has not been found to relate strongly to volume loss (Josephs et al., 2008). Additionally, the sample of adults in the present study was cognitively normal and the maximum age was 75 years, further decreasing the likelihood that amyloid build-up in the non-user control group mediated volume loss. Inhibition of COX with a decrease in downstream inflammation is probable, but requires further study using either peripheral or central markers of inflammation that can be evaluated in conjunction with the imaging data Furthermore, it is important to note that even though anti-inflammatory effects are likely, it is quite possible that the beneficial effects of NSAIDs are realized through multiple mechanisms, including anti-amyloid effects.
Our results confirm previous reports that NSAID users follow a different aging trajectory than non-users. The next quandary, then, concerns the age boundaries of the critical period where NSAIDs exert beneficial effects; when do individuals predisposed to decline need intervention? Failed trials in moderate AD (Aisen et al., 2003; Reines et al., 2004; de Jong et al., 2008), mild AD (Green et al., 2009), and mild cognitive impairment (Thal et al., 2005) indicate that once neuropathology has advanced sufficiently to affect cognition, it is too late. In fact, it is possible that once pathological processes are underway, limiting immune response via NSAIDs may be harmful, since microglia perform a role in clearing β-amyloid (Jantzen et al., 2002; Chakrabarty et al., 2009).
Post mortem data combined with in vivo imaging studies using PET and MRI suggest that pathological processes in AD begin several years in advance of cognitive decline (Reiman et al., 1996; Braak and Braak, 1997; Small et al., 2000; Johnson et al., 2006; Reiman et al., 2009; Xu et al., 2009; Bendlin et al., in press). In transgenic mouse models of AD, the association of NSAIDs with decreased amyloid pathology (Lim et al., 2000) is especially effective when initiated at the age of onset of amyloid plaque formation, and less so when initiated once pathological processes have progressed (Lim et al., 2001). In the Cache county study, epidemiological results indicated that NSAIDs need to be commenced before the age 65, and that beneficial effects of NSAIDs may be driven by their effect on people who are APOE4 positive, a genetic profile which places them at greater risk for developing AD (Hayden et al., 2007). Based on data showing that brain alterations can be detected in people at risk for AD in middle age, it is possible that intervention would be prudent even earlier.
There are several limitations to the present study that bear mention. The study included only a small number of NSAID users, and the results here will need replication in larger studies. Additionally, data on NSAID use was obtained via self-report, and a wide variety of NSAIDs were included in the analysis, limiting our interpretation of the potential mechanisms that underlie the observed effects. Studies that combine brain-imaging analysis with medical record review of NSAID data usage will be needed to fill this gap. Although we found brain differences between the groups, cognitive differences were not evident. This is perhaps unsurprising, as normal cognition was a criterion for inclusion. Future studies including a larger range of cognitive ability may shed more light on the relationship between NSAIDs, brain structure, and cognitive function.
In conclusion, this study, together with previous findings, provides preliminary support for the notion that NSAIDs exert a beneficial effect on the brain. Beneficial effects manifested as preserved volume, and although the study was cross-sectional, the significant interaction between group and age point toward attenuated volume decline with age, suggesting neuronal protection. Medial temporal lobe gray matter volume of NSAID users and controls showed increasing difference with greater age, lending support to the supposition that intervention should occur in earlier stages, perhaps middle age, in order to advance a favorable aging trajectory. At present however, despite the promising epidemiological literature, there is no definitive evidence that NSAIDs will protect against future cognitive decline and brain disease. Non-selective NSAIDs carry a risk of gastrointestinal bleeding (Graham et al., 2005) and COX-2 inhibitors are now known to carry their own health risks. These attributes make NSAIDs less than ideal for a long-term prevention trial. Options include short-term prevention trials in middle-aged adults that evaluate markers of inflammation and CNS pathology via cerebrospinal fluid (CSF) or other mechanisms, rather than waiting for final outcome measures such as conversion to disease. Longitudinal studies may be used to monitor CSF markers together with brain imaging markers that may prove to be sensitive to neuroprotection over time. Additionally, further work will be needed to evaluate anti-inflammatory therapies or regimes that carry less risk than prolonged and intense exposure to NSAIDs.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Institutes of Health (AG021155 to Sterling C. Johnson, MH62015 to Andrew L. Alexander, and R01 AG27161 to Mark A. Sager, and P50 AG033514); a Merit Review Grant from the Department of Veterans Affairs (to Sterling C. Johnson), and by the facilities and resources at the William S. Middleton Memorial Veterans Hospital (GRECC manuscript number 2010-15). The authors gratefully acknowledge the assistance of Brent Thiel, Britta Jabbar, Michele Fitzgerald, Erik Kastman, Amy Hawley, and Sandra Harding at the University of Wisconsin-Madison for their roles in data collection and technical assistance. In addition, we would like to acknowledge the kind support of researchers and staff at the Waisman Center, University of Wisconsin-Madison, where MR imaging took place. Finally, we extend thanks to our volunteers who participated in this research.
References
Aisen, P. S., Schafer, K. A., Grundman, M., Pfeiffer, E., Sano, M., Davis, K. L., Farlow, M. R., Jin, S., Thomas, R. G., and Thal, L. J. (2003). Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826.
Arriagada, P. V., Marzloff, K., and Hyman, B. T. (1992). Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology 42, 1681–1688.
Arvanitakis, Z., Grodstein, F., Bienias, J. L., Schneider, J. A., Wilson, R. S., Kelly, J. F., Evans, D. A., and Bennett, D. A. (2008). Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 70, 2219–2225.
Banati, R. B., Gehrmann, J., Schubert, P., and Kreutzberg, G. W. (1993). Cytotoxicity of microglia. Glia 7, 111–118.
Bendlin, B. B., Ries, M. L., Canu, E., Sodhi, A., Lazar, M., Alexander, A. L., Carlsson, C. M., Sager, M. A., Asthana, S., and Johnson, S. C. (In press). White matter is altered with parental family history of Alzheimer’s disease. Alzheimer’s Dementia, doi:10.1016/j.jalz.2009.11.003.
Blasko, I., Apochal, A., Boeck, G., Hartmann, T., Grubeck-Loebenstein, B., and Ransmayr, G. (2001). Ibuprofen decreases cytokine-induced amyloid beta production in neuronal cells. Neurobiol. Dis. 8, 1094–1101.
Braak, H., and Braak, E. (1997). Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357.
Burggren, A. C., Zeineh, M. M., Ekstrom, A. D., Braskie, M. N., Thompson, P. M., Small, G. W., and Bookheimer, S. Y. (2008). Reduced cortical thickness in hippocampal subregions among cognitively normal apolipoprotein E e4 carriers. Neuroimage 41, 1177–1183.
Canadian study of health and aging: study methods and prevalence of dementia (1994). CMAJ 150, 899–913.
Chakrabarty, P., Jansen-West, K., Beccard, A., Ceballos-Diaz, C., Levites, Y., Verbeeck, C., Zubair, A. C., Dickson, D., Golde, T. E., and Das, P. (2009). Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 24, 548–559.
de Jong, D., Jansen, R., Hoefnagels, W., Jellesma-Eggenkamp, M., Verbeek, M., Borm, G., and Kremer, B. (2008). No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS One 3, e1475. doi:10.1371/journal.pone.0001475.
de Leon, M. J., George, A. E., Stylopoulos, L. A., Smith, G., and Miller, D. C. (1989). Early marker for Alzheimer’s disease: the atrophic hippocampus. Lancet 2, 672–673.
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464.
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934.
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., Parisi, J. E., and Hyman, B. T. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24.
Graham, D. Y., Opekun, A. R., Willingham, F. F., and Qureshi, W. A. (2005). Visible small-intestinal mucosal injury in chronic NSAID users. Clin. Gastroenterol. Hepatol. 3, 55–59.
Green, R. C., Schneider, L. S., Amato, D. A., Beelen, A. P., Wilcock, G., Swabb, E. A., and Zavitz, K. H. (2009). Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564.
Hauss-Wegrzyniak, B., Dobrzanski, P., Stoehr, J. D., and Wenk, G. L. (1998). Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res. 780, 294–303.
Hauss-Wegrzyniak, B., Lynch, M. A., Vraniak, P. D., and Wenk, G. L. (2002). Chronic brain inflammation results in cell loss in the entorhinal cortex and impaired LTP in perforant path-granule cell synapses. Exp. Neurol. 176, 336–341.
Hauss-Wegrzyniak, B., Vannucchi, M. G., and Wenk, G. L. (2000a). Behavioral and ultrastructural changes induced by chronic neuroinflammation in young rats. Brain Res. 859, 157–166.
Hauss-Wegrzyniak, B., Vraniak, P. D., and Wenk, G. L. (2000b). LPS-induced neuroinflammatory effects do not recover with time. Neuroreport 11, 1759–1763.
Hayden, K. M., Zandi, P. P., Khachaturian, A. S., Szekely, C. A., Fotuhi, M., Norton, M. C., Tschanz, J. T., Pieper, C. F., Corcoran, C., Lyketsos, C. G., Breitner, J. C., and Welsh-Bohmer, K. A. (2007). Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology 69, 275–282.
Hee Kang, J., and Grodstein, F. (2003). Regular use of nonsteroidal anti-inflammatory drugs and cognitive function in aging women. Neurology 60, 1591–1597.
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Hanke, A., Dewachter, I., Kuiperi, C., O’Banion, K., Klockgether, T., Van Leuven, F., and Landreth, G. E. (2005). Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 128, 1442–1453.
Hewett, S. J., Silakova, J. M., and Hewett, J. A. (2006). Oral treatment with rofecoxib reduces hippocampal excitotoxic neurodegeneration. J. Pharmacol. Exp. Ther. 319, 1219–1224.
Hyman, B. T., Van Hoesen, G. W., Damasio, A. R., and Barnes, C. L. (1984). Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science 225, 1168–1170.
Jantzen, P. T., Connor, K. E., DiCarlo, G., Wenk, G. L., Wallace, J. L., Rojiani, A. M., Coppola, D., Morgan, D., and Gordon, M. N. (2002). Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J. Neurosci. 22, 2246–2254.
Johnson, S. C., Schmitz, T. W., Trivedi, M. A., Ries, M. L., Torgerson, B. M., Carlsson, C. M., Asthana, S., Hermann, B. P., and Sager, M. A. (2006). The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J. Neurosci. 26, 6069–6076.
Jonker, C., Comijs, H. C., and Smit, J. H. (2003). Does aspirin or other NSAIDs reduce the risk of cognitive decline in elderly persons? Results from a population-based study. Neurobiol. Aging 24, 583–588.
Josephs, K. A., Whitwell, J. L., Ahmed, Z., Shiung, M. M., Weigand, S. D., Knopman, D. S., Boeve, B. F., Parisi, J. E., Petersen, R. C., Dickson, D. W., and Jack, C. R. Jr. (2008). Beta-amyloid burden is not associated with rates of brain atrophy. Ann. Neurol. 63, 204–212.
Lassmann, H., Fischer, P., and Jellinger, K. (1993). Synaptic pathology of Alzheimer’s disease. Ann. N.Y. Acad. Sci. 695, 59–64.
Lee, J. W., Lee, Y. K., Yuk, D. Y., Choi, D. Y., Ban, S. B., Oh, K. W., and Hong, J. T. (2008). Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflammation 5, 37.
Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., Tran, T., Ubeda, O., Ashe, K. H., Frautschy, S. A., and Cole, G. M. (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 20, 5709–5714.
Lim, G. P., Yang, F., Chu, T., Gahtan, E., Ubeda, O., Beech, W., Overmier, J. B., Hsiao-Ashec, K., Frautschy, S. A., and Cole, G. M. (2001). Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol. Aging 22, 983–991.
Mackenzie, I. R. (2000). Anti-inflammatory drugs and Alzheimer-type pathology in aging. Neurology 54, 732–734.
Mackenzie, I. R., and Munoz, D. G. (1998). Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 50, 986–990.
Marsland, A. L., Gianaros, P. J., Abramowitch, S. M., Manuck, S. B., and Hariri, A. R. (2008). Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol. Psychiatry 64, 484–490.
McGeer, P. L., McGeer, E., Rogers, J., and Sibley, J. (1990). Anti-inflammatory drugs and Alzheimer disease. Lancet 335, 1037.
McGeer, P. L., and McGeer, E. G. (1995). The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Brain Res. Rev. 21, 195–218.
McGeer, P. L., and McGeer, E. G. (2001). Inflammation, autotoxicity and Alzheimer disease. Neurobiol. Aging 22, 799–809.
McGeer, P. L., Rogers, J., and McGeer, E. G. (2006). Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J. Alzheimer’s Dis. 9, 271–276.
McKee, A. C., Carreras, I., Hossain, L., Ryu, H., Klein, W. L., Oddo, S., LaFerla, F. M., Jenkins, B. G., Kowall, N. W., and Dedeoglu, A. (2008). Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 1207, 225–236.
Mosconi, L., Brys, M., Switalski, R., Mistur, R., Glodzik, L., Pirraglia, E., Tsui, W., De Santi, S., and de Leon, M. J. (2007). Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc. Natl. Acad. Sci. U.S.A. 104, 19067–19072.
Mueller, S. G., and Weiner, M. W. (2009). Selective effect of age, Apo e4, and Alzheimer’s disease on hippocampal subfields. Hippocampus 19, 558–564.
Reiman, E. M., Caselli, R. J., Yun, L. S., Chen, K., Bandy, D., Minoshima, S., Thibodeau, S. N., and Osborne, D. (1996). Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein. E. N. Engl. J. Med. 334, 752–758.
Reiman, E. M., Chen, K., Liu, X., Bandy, D., Yu, M., Lee, W., Ayutyanont, N., Keppler, J., Reeder, S. A., Langbaum, J. B., Alexander, G. E., Klunk, W. E., Mathis, C. A., Price, J. C., Aizenstein, H. J., DeKosky, S. T., and Caselli, R. J. (2009). Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 6820–6825.
Reines, S. A., Block, G. A., Morris, J. C., Liu, G., Nessly, M. L., Lines, C. R., Norman, B. A., and Baranak, C. C. (2004). Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62, 66–71.
Rogers, J., and Shen, Y. (2000). A perspective on inflammation in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 924, 132–135.
Rosen, W. G., Terry, R. D., Fuld, P. A., Katzman, R., and Peck, A. (1980). Pathological verification of ischemic score in differentiation of dementias. Ann. Neurol. 7, 486–488.
Rowe, C. C., Ng, S., Ackermann, U., Gong, S. J., Pike, K., Savage, G., Cowie, T. F., Dickinson, K. L., Maruff, P., Darby, D., Smith, C., Woodward, M., Merory, J., Tochon-Danguy, H., O’Keefe, G., Klunk, W. E., Mathis, C. A., Price, J. C., Masters, C. L., and Villemagne, V. L. (2007). Imaging beta-amyloid burden in aging and dementia. Neurology 68, 1718–1725.
Rozzini, R., Ferrucci, L., Losonczy, K., Havlik, R. J., and Guralnik, J. M. (1996). Protective effect of chronic NSAID use on cognitive decline in older persons. J. Am. Geriatr. Soc. 44, 1025–1029.
Small, G. W., Ercoli, L. M., Silverman, D. H., Huang, S. C., Komo, S., Bookheimer, S. Y., Lavretsky, H., Miller, K., Siddarth, P., Rasgon, N. L., Mazziotta, J. C., Saxena, S., Wu, H. M., Mega, M. S., Cummings, J. L., Saunders, A. M., Pericak-Vance, M. A., Roses, A. D., Barrio, J. R., and Phelps, M. E. (2000). Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 6037–6042.
Thal, L. J., Ferris, S. H., Kirby, L., Block, G. A., Lines, C. R., Yuen, E., Assaid, C., Nessly, M. L., Norman, B. A., Baranak, C. C., and Reines, S. A. (2005). A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30, 1204–1215.
Thomas, T., Nadackal, T. G., and Thomas, K. (2001). Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-beta aggregation. Neuroreport 12, 3263–3267.
Trivedi, M. A., Schmitz, T. W., Ries, M. L., Torgerson, B. M., Sager, M. A., Hermann, B. P., Asthana, S., and Johnson, S. C. (2006). Reduced hippocampal activation during episodic encoding in middle-aged individuals at genetic risk of Alzheimer’s disease: a cross-sectional study. BMC Med. 4, 1.
van Groen, T., and Kadish, I. (2005). Transgenic AD model mice, effects of potential anti-AD treatments on inflammation and pathology. Brain Res. Brain Res. Rev. 48, 370–378.
Walther, K., Bendlin, B. B., Glisky, E. L., Trouard, T. P., Lisse, J. R., Posever, J. O., and Ryan, L. (2009). Anti-inflammatory drugs reduce age-related decreases in brain volume in cognitively normal older adults. Neurobiol. Aging. doi:10.1016/j.neurobiolaging.2009.03.006.
Weggen, S., Eriksen, J. L., Das, P., Sagi, S. A., Wang, R., Pietrzik, C. U., Findlay, K. A., Smith, T. E., Murphy, M. P., Bulter, T., Kang, D. E., Marquez-Sterling, N., Golde, T. E., and Koo, E. H. (2001). A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414, 212–216.
Willard, L. B., Hauss-Wegrzyniak, B., Danysz, W., and Wenk, G. L. (2000). The cytotoxicity of chronic neuroinflammation upon basal forebrain cholinergic neurons of rats can be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibition. Exp. Brain Res. 134, 58–65.
Willette, A. A., Bendlin, B. B., McLaren, D. G., Canu, E., Kastman, E. K., Kosmatka, K. J., Xu, G., Field, A. S., Alexander, A. L., Colman, R. J., Weindruch, R. H., Coe, C. L., and Johnson, S. C. (2010). Age-related changes in neural volume and microstructure associated with interleukin-6 are ameliorated by a calorie-restricted diet in old rhesus monkeys. Neuroimage 51, 987–994.
Xu, G., McLaren, D. G., Ries, M. L., Fitzgerald, M. E., Bendlin, B. B., Rowley, H. A., Sager, M. A., Atwood, C., Asthana, S., and Johnson, S. C. (2009). The influence of parental history of Alzheimer’s disease and apolipoprotein E epsilon4 on the BOLD signal during recognition memory. Brain 132, 383–391.
Keywords: Alzheimer’s disease, aging, risk factors, volumetric MRI, NSAIDs
Citation: Bendlin BB, Newman LM, Ries ML, Puglielli L, Carlsson CM, Sager MA, Rowley HA, Gallagher CL, Willette AA, Alexander AL, Asthana S and Johnson SC (2010) NSAIDs may protect against age-related brain atrophy. Front. Ag. Neurosci. 2:35. doi: 10.3389/fnagi.2010.00035
Received: 30 April 2010;
Paper pending published: 03 June 2010;
Accepted: 20 July 2010;
Published online: 03 September 2010
Edited by:
Elena Galea, Universitat Autònoma de Barcelona, SpainReviewed by:
Bruno P. Imbimbo, Chiesi Farmaceutici S. P. A., ItalyWolfgang J. Streit, University of Florida, USA
Copyright: © 2010 Bendlin, Newman, Ries, Puglielli, Carlsson, Sager, Rowley, Gallagher, Willette, Alexander, Asthana and Johnson. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Barbara B. Bendlin, Geriatric Research Education and Clinical Center, Veterans Administration Hospital, 2500 Overlook Terrace, Madison, WI 53705, USA. e-mail:YmJiQG1lZGljaW5lLndpc2MuZWR1