 Sónia C. Correia1,2
Sónia C. Correia1,2 Cristina Carvalho1,2
Cristina Carvalho1,2 Susana Cardoso1,2
Susana Cardoso1,2 Renato X. Santos1,2
Renato X. Santos1,2 Maria S. Santos1,2
Maria S. Santos1,2 Catarina R. Oliveira1,3
Catarina R. Oliveira1,3 George Perry4,5
George Perry4,5 Xiongwei Zhu5
Xiongwei Zhu5 Mark A. Smith5
Mark A. Smith5 Paula I. Moreira1,6*
Paula I. Moreira1,6*
- 1 Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
- 2 Department of Life Sciences – Faculty of Sciences and Technology, University of Coimbra, Coimbra, Portugal
- 3 Institute of Biochemistry – Faculty of Medicine, University of Coimbra, Coimbra, Portugal
- 4 UTSA Neurosciences Institute and Department of Biology, University of Texas at San Antonio, San Antonio, TX, USA
- 5 Department of Pathology, Case Western Reserve University, Cleveland, OH, USA
- 6 Institute of Physiology – Faculty of Medicine, University of Coimbra, Coimbra, Portugal
Mitochondria have long been known as the powerhouse of the cell. However, these organelles are also pivotal players in neuronal cell death. Mitochondrial dysfunction is a prominent feature of chronic brain disorders, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), and cerebral ischemic stroke. Data derived from morphologic, biochemical, and molecular genetic studies indicate that mitochondria constitute a convergence point for neurodegeneration. Conversely, mitochondria have also been implicated in the neuroprotective signaling processes of preconditioning. Despite the precise molecular mechanisms underlying preconditioning-induced brain tolerance are still unclear, mitochondrial reactive oxygen species generation and mitochondrial ATP-sensitive potassium channels activation have been shown to be involved in the preconditioning phenomenon. This review intends to discuss how mitochondrial malfunction contributes to the onset and progression of cerebral ischemic stroke and AD and PD, two major neurodegenerative disorders. The role of mitochondrial mechanisms involved in the preconditioning-mediated neuroprotective events will be also discussed. Mitochondrial targeted preconditioning may represent a promising therapeutic weapon to fight neurodegeneration.
Introduction
Neurodegenerative diseases represent one of the major health problems. In fact, the prevalence of neurodegenerative diseases is rising dramatically due to the increase in life expectancy and demographic changes in the population. The etiology of most neurodegenerative disorders is complex and multifactorial, involving genetic predisposition, environmental and endogenous factors (Correia and Moreira, 2010; Przedborski et al., 2003; Migliore and Coppedè, 2009). Nevertheless, mitochondria have emerged as a pivotal “convergence point” for neurodegeneration (Lin and Beal, 2006; Moreira et al., 2010).
Mitochondria play a critical role in the regulation of both cell survival and death (Green and Kroemer, 2004; Beal, 2005). These organelles are essential for the production of ATP through oxidative phosphorylation and regulation of intracellular calcium (Ca2+) homeostasis. Thus, dysfunction of mitochondrial energy metabolism culminates in ATP production and Ca2+ buffering impairment and exacerbated generation of reactive oxygen species (ROS; Beal, 2005). High levels of ROS cause, among other things, damage of cell membranes through lipid peroxidation and accelerate the high mutation rate of mitochondrial DNA (mtDNA; Petrozzi et al., 2007). Accumulation of mtDNA mutations enhances oxidative damage, causes energy depletion and increases ROS production, in a vicious cycle (Petrozzi et al., 2007). Moreover, the brain is especially prone to oxidative stress-induced damage due to its high levels of polyunsaturated fatty acids, high oxygen consumption, high content in transition metals, and poor antioxidant defenses (Nunomura et al., 2006).
Although exaggerated mitochondrial ROS production has been associated with mitochondrial dysfunction and neuronal cell degeneration and death, it has also been shown that a slight rise of mitochondrial ROS levels can trigger preconditioning- mediated brain tolerance, suggesting that mitochondria might be gateways on endogenous neuroprotection (Ravati et al., 2000, 2001; Dirnagl and Meisel, 2008; Jou, 2008; Dirnagl et al., 2009). Further, mitochondrial ATP-sensitive potassium (mitoKATP) channels activation has also been involved in the preconditioning phenomenon (Busija et al., 2008). It was demonstrated that antioxidants and mitoKATP blockers abolish preconditioning-induced protection (Vanden Hoek et al., 1998; Oldenburg et al., 2002). Jou (2008) proposed that this “minor” mitochondrial ROS generation induces fission and fusion of mitochondria and relocates mitochondrial network to form a mitochondria free gap, which may play a crucial role in mitochondrial ROS-mediated protective “preconditioning” by preventing propagation of ROS during oxidative insult.
In light of these evidences, we may say that mitochondria take a center stage in both neurodegeneration and neuroprotection. Herein, we review the current knowledge pertaining to mitochondrial dysfunction involvement on the onset and progression of chronic neurodegenerative disorders, namely Alzheimer’s disease (AD) and Parkinson’s disease (PD), and cerebral ischemic stroke. We also intend to explore the crucial role of mitochondria in preconditioning-induced neuroprotection, putting focus on the role of mitochondrial ROS and mitoKATP channels.
Mitochondrial Dysfunction as a Critical Event Implicated in Neurodegeneration
It is well established that mitochondria have a prominent role in neuronal physiology. Neurons are cells with extremely high energy demands, since mitochondrial oxidative phosphorylation is essential for neurons to meet their high ATP requirements. Therefore, neurons are very vulnerable to bioenergetic crisis and dysfunction of mitochondrial machinery (Murphy et al., 1999; Moreira et al., 2009; Simpkins et al., 2009). Data from postmortem brain tissue and genetic analysis in humans and biochemical and pathological studies in in vitro and in vivo models of neurodegeneration suggest that mitochondrial dysfunction is a common and critical event involved in neurodegenerative processes. The following subsections are devoted to highlight the involvement of mitochondrial malfunction in AD, PD, and cerebral ischemic stroke.
Alzheimer’s Disease and Mitochondrial Dysfunction
Alzheimer’s disease is the most common neurodegenerative disease, being characterized by the progressive neuronal impairment and loss and cognitive decline. The pathological hallmarks of AD are the formation of extracellular senile plaques, mainly composed of amyloid-β (Aβ) peptide, and intracellular neurofibrillary tangles (NFT) containing hyperphosphorylated tau protein (Selkoe, 2001; Moreira et al., 2006, 2007). Aβ peptides are generated by successive proteolysis of amyloid-β precursor protein (AβPP), a large transmembrane glycoprotein that is initially cleaved by the β-site AβPP-cleaving enzyme 1 (BACE1) and subsequently by γ-secretase in the transmembrane domain (Greenfield et al., 2000; Findeis, 2007).
Mitochondrial dysfunction and bioenergetics failure have been demonstrated to be early events implicated in the pathogenesis of AD (Figure 1). In fact, it was observed an impairment in the activities of the three key tricarboxylic acid cycle (TCA) enzyme complexes, pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase in postmortem AD brain and fibroblasts from AD patients (Huang et al., 2003; Bubber et al., 2005). In addition, a decline in respiratory chain complexes I, III, and IV activities is seen in platelets and lymphocytes from AD patients and postmortem AD brain tissue (Kish et al., 1992; Parker et al., 1994; Bosetti et al., 2002; Valla et al., 2006). Several lines of evidence also reported increased free radical production, lipid peroxidation, oxidative DNA damage, oxidative protein damage, and decreased ATP production and cell viability in AD brains compared to age-matched control subjects (Smith et al., 1996; Gibson et al., 1998; Maurer et al., 2000; Wang et al., 2005; Devi et al., 2006; Sultana et al., 2006).
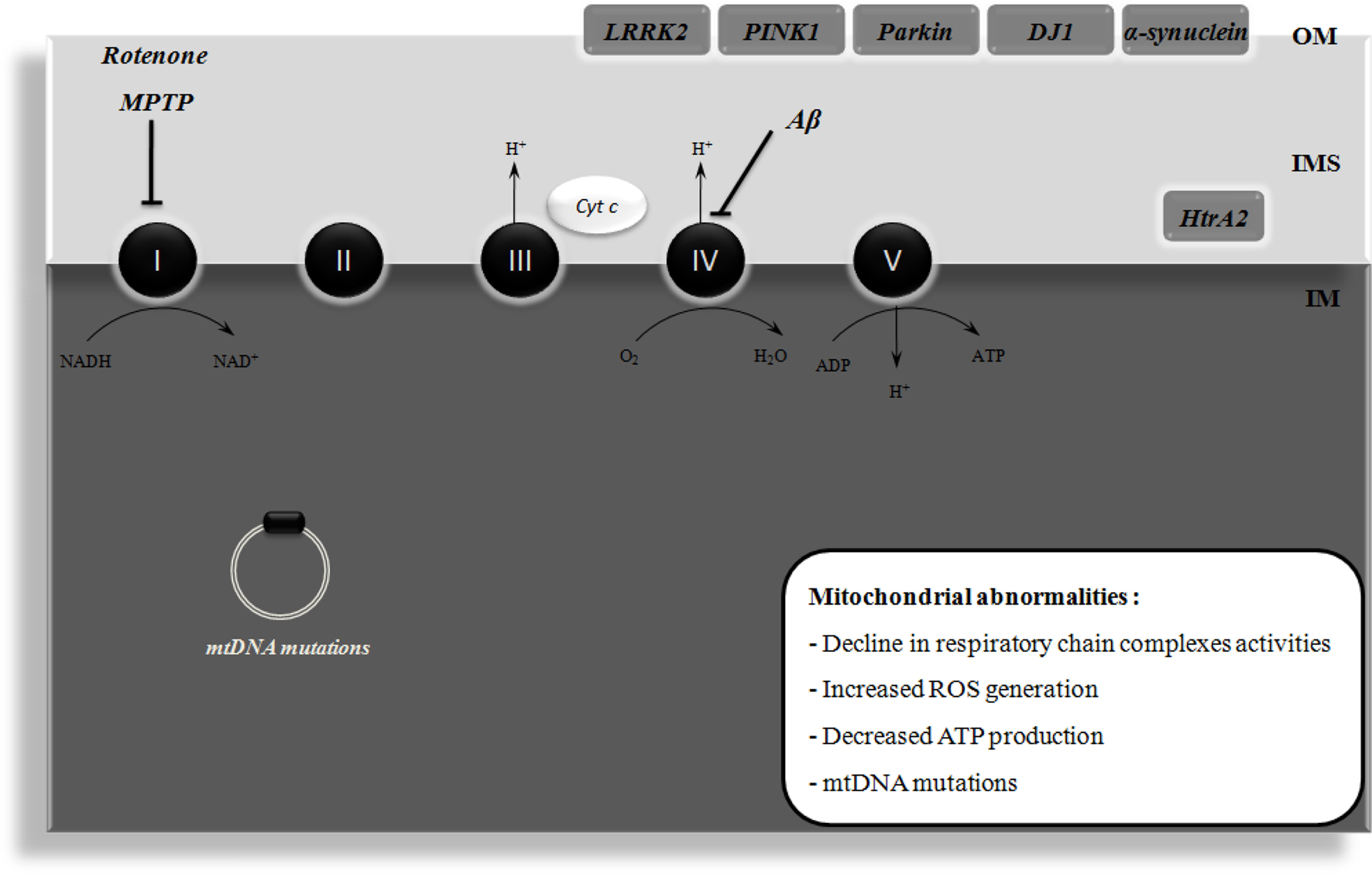
Figure 1. Mitochondrial dysfunction in Alzheimer’s disease (AD) and Parkinson’s disease (PD). Mitochondrial dysfunction has been implicated in the etiology of AD and PD. In AD, it has been shown that amyloid β peptide (Aβ) impairs the activity of respiratory chain complex IV, leading to increased reactive oxygen species (ROS) levels and ATP depletion. Moreover, mitochondrial DNA (mtDNA) mutations have also been implicated in mitochondrial dysfunction that occurs in AD. PD is associated with an impairment of mitochondrial complex I activity. It has been demonstrated that the pharmacological inhibition of this complex with rotenone or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) causes degeneration of the nigral dopaminergic neurons and PD symptoms in in vivo models. The familial forms of PD are associated with mutations in leucine-rich repeat kinase 2 (LRRK2), α-synuclein, parkin, DJ1, and PTEN-induced putative kinase 1 (PINK1), these proteins being associated with the mitochondrial outer membrane (OM) and involved in ROS production or defense. High temperature requirement A2 (HtrA2) is another protein that is mutated in familial PD and is localized in the intermembrane space (IMS) of mitochondria. ADP, adenosine diphosphate; Cyt c, cytochrome c; IM, inner membrane; NAD+, oxidized nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; H+, proton.
Mitochondrial DNA mutations have also been implicated in mitochondrial dysfunction in the pathogenesis of AD. It was detected 20 point mutations in the mitochondrial-encoded cytochrome c oxidase subunits I, II, and III genes in AD patients (Hamblet et al., 2006). Similarly, Qiu et al. (2001) identified two missense mutations in the mtDNA of cytochrome c oxidase in a patient with AD. Further, a high aggregate burden of somatic mtDNA mutations was observed in postmortem brain tissue from AD patients (Lin et al., 2002; Coskun et al., 2004).
Accumulating evidence also indicate that Aβ and AβPP could directly target mitochondria. Mitochondrial dysfunction was linked to the accumulation of full-length and carboxy- terminally truncated AβPP across mitochondrial import channels in brain tissue from AD patients (Devi et al., 2006). The authors also observed that this accumulation of AβPP inhibited the entrance of nuclear-encoded cytochrome c oxidase subunits IV and Vb proteins, which was associated with a decrease in cytochrome c oxidase activity and increased hydrogen peroxide (H2O2) production (Devi et al., 2006). Anandatheerthavarada et al. (2003) reported an accumulation of full-length AβPP in the mitochondrial compartment in a transmembrane-arrested form that impaired mitochondrial functionality and energy metabolism. Also detected was a progressive accumulation of Aβ monomers and oligomers within the mitochondria of both transgenic mice overexpressing mutant AβPP and postmortem brain from AD patients (Caspersen et al., 2005; Crouch et al., 2005; Devi et al., 2006; Manczak et al., 2006). Indeed, Aβ can disrupt mitochondrial cytochrome c oxidase activity (Crouch et al., 2005; Takuma et al., 2005) in a sequence- and conformation-dependent manner (Crouch et al., 2005). A direct link between Aβ-induced toxicity and mitochondrial dysfunction in AD pathology has been suggested by the interaction between mitochondrial Aβ and the mitochondrial protein, Aβ-binding alcohol dehydrogenase (ABAD; Lustbader et al., 2004; Yan and Stern, 2005). It was observed that this interaction culminates in mitochondrial failure via changes in mitochondrial membrane permeability and a reduction in the activities of enzymes involved in mitochondrial respiration (Lustbader et al., 2004). Hansson Petersen et al. (2008) have also shown that Aβ peptide is imported into mitochondria via the translocase of the outer membrane (TOM) import machinery and localized to mitochondrial cristae. Thus, it has been suggested that the transport of Aβ species to mitochondria cause mitochondrial dysfunction and oxidative damage and, consequently, damage neurons both structurally and functionally (Caspersen et al., 2005; Crouch et al., 2005; Devi et al., 2006; Manczak et al., 2006; Hansson Petersen et al., 2008). Previous in vitro studies from our laboratory also show an increased susceptibility to mitochondrial permeability transition pore (MPTP) induction promoted by Aβ peptides (Moreira et al., 2001, 2002). Accordingly, Du et al. (2008) provide a plausible mechanism underlying Aβ-induced mitochondrial dysfunction, in which Aβ interacts with cyclophilin D, a critical molecule involved in MPTP formation and cell death. The authors showed that the interaction of cyclophilin D with mitochondrial Aβ potentiates mitochondrial, neuronal and synaptic stress (Du et al., 2008). In contrast, cyclophilin D ablation protects neurons from Aβ-induced MPTP opening and the resultant mitochondrial and cellular stress (Du et al., 2008). Additionally, cyclophilin D deficiency substantially improves learning and memory and synaptic function in an AD mouse model and alleviates Aβ-mediated reduction of long-term potentiation (LTP; Du et al., 2008). Another study reported that the presequence protease (PreP) is responsible for the degradation of the accumulated Aβ in mitochondria, supporting the association of Aβ with mitochondria and mitochondrial dysfunction in AD (Falkevall et al., 2006).
Recent studies also demonstrated that Aβ induce mitochondrial dynamic changes, including mitochondrial fission/fusion perturbations (Wang et al., 2008a). Wang et al. (2008a) reported abnormal mitochondrial fission and fusion in AD fibroblasts. It was found that fibroblasts from sporadic AD patients express lower levels of dynamin-related protein 1 (Drp1), a key regulator of mitochondrial fission and display elongated mitochondria, which form collapsed perinuclear networks (Wang et al., 2008a, 2009a). It was also observed that AβPP overexpression in M17 neuroblastoma cells resulted in predominantly fragmented mitochondria, decreased Drp1 and optic atrophy protein 1 (OPA1) levels, and a defect in neuronal differentiation (Wang et al., 2008b). Moreover, reduced levels of Drp1, OPA1, mitofusin (Mfn)1 and 2 and increased mitochondria fission protein 1 (Fis1) levels were found in hippocampal tissues from AD patients compared with age-matched controls (Wang et al., 2009b). These studies suggest that Aβ potentiates mitochondrial fission/fusion imbalance and, consequently, mitochondrial fragmentation and abnormal distribution contributing to mitochondrial and neuronal dysfunction in AD.
Parkinson’s Disease and Mitochondrial Dysfunction
Parkinson’s disease is the second most prevalent neurodegenerative disease, affecting approximately 2% of individuals over 65 years of age. PD is characterized by a progressive and massive loss of midbrain dopaminergic neurons of the substantia nigra pars compacta (de Rijk et al., 1997; de Lau et al., 2004; Cardoso et al., 2005). Consequently, substantia nigra pars compacta dopaminergic neuronal loss leads to the degeneration of neurons terminals and dopamine (DA) depletion in the striatum, which is required for normal motor function. One of the pathological hallmarks of PD and related synucleinopathies is the presence of intracellular inclusions called Lewy bodies (LBs), which are constituted by aggregates of the presynaptic soluble protein called α-synuclein (Cardoso et al., 2005).
Over the last decades, evidence indicates that mitochondrial dysfunction plays a critical role in the etiology of PD (Figure 1) (Schapira, 2008; Banerjee et al., 2009). Indeed, an impairment of mitochondrial complex I activity has been observed in the substantia nigra (Schapira et al., 1989; Mann et al., 1994), platelets (Parker et al., 1989; Krige et al., 1992; Haas et al., 1995; Blandini et al., 1998), lymphocytes (Yoshino et al., 1992; Barroso et al., 1993), and skeletal muscle tissue (Taylor et al., 1994; Penn et al., 1995) from PD patients. Consistently, cybrids containing mtDNA from PD patients present a significant impairment in complex I activity associated with increased oxidative stress levels (Swerdlow et al., 1996), suggesting mtDNA encoded defects in PD. Additionally, it was observed that LBs within these cybrids also react positively with cytochrome c antibodies, which imply a mitochondrial origin (Trimmer et al., 2004). Mitochondrial involvement in the etiology of PD is also provided by the use of specific complex I inhibitors, such as 1-methyl-4-phenyl- 1,2,3,6-tetrahydropyridine, rotenone and 6-hydroxydopamine (6-OHDA) which cause degeneration of the nigral dopaminergic neurons and PD symptoms in in vivo models (Betarbet et al., 2000; Gash et al., 2007; Sherer et al., 2007). Furthermore, a proteomic analysis of mitochondria-enriched fractions from postmortem PD substantia nigra revealed differential expression of multiple mitochondrial proteins in PD brain as compared to control, including complex I subunits and mitochondrial creatine kinase (Jin et al., 2006). It was also reported that mortalin, a mitochondrial stress protein, is substantially decreased in PD brains and cellular models of PD (Jin et al., 2006). Manipulations of mortalin levels in dopaminergic neurons resulted in significant changes in sensitivity to PD phenotypes via pathways involving mitochondrial and proteasomal function as well as oxidative stress (Jin et al., 2006). Decreased immunostaining for mitochondrial α-ketoglutarate was also observed in postmortem brain from PD patients (Mizuno et al., 1994).
Genetic evidence reveals that mutations in genes encoding both mitochondrially targeted proteins and proteins involved in mitochondrial function and/or oxidative stress responses also play a role in the pathogenesis of PD (Thomas and Beal, 2007). Ekstrand et al. (2007) created conditional knockout “MitoPark” mice, which have a disrupted mitochondrial transcription factor A (Tfam) gene in dopaminergic neurons. The authors observed that these knockout mice have reduced mtDNA expression and respiratory chain deficiency in midbrain dopaminergic neurons, which lead to a Parkinsonism phenotype with adult onset and characterized by slowly progressive impairment of motor function accompanied by the formation of intraneuronal inclusions and dopamine nerve cell death (Ekstrand et al., 2007). Moreover, familial forms of PD are associated with mutations in leucine-rich repeat kinase 2 (LRRK2), α-synuclein, parkin, DJ1, and PTEN-induced putative kinase 1 (PINK1), these proteins being associated with the mitochondrial outer membrane and involved in ROS production or defense (Knott et al., 2008). High temperature requirement A2 (HtrA2) is another protein that is mutated in familial PD and localizes in the intermembrane space of mitochondria (Knott et al., 2008). In vitro cell culture studies showed that mutant PINK1 or PINK1 knock-down induce mitochondrial respiration, ATP synthesis and proteasome function impairment and increased α-synuclein aggregation (Liu et al., 2009). Additionally, it was reported that HtrA2 loss results in transcriptional upregulation of nuclear genes characteristic of the integrated stress response, including the transcription factor C/EBP homologous protein (CHOP), selectively in the brain (Moisoi et al., 2009). HtrA2 loss also induces accumulation of unfolded proteins in the mitochondria, defective mitochondrial respiration and enhanced ROS production, which contribute to the induction of CHOP expression and neuronal cell death (Moisoi et al., 2009). Previous studies also showed that the overexpression of α-synuclein in cell culture and in transgenic mice impairs mitochondrial function and increases the susceptibility to MPTP induction (Hsu et al., 2000; Song et al., 2004). In contrast, α-synuclein-null mice are resistant to respiratory chain inhibitors implicating an involvement of mitochondria in α-synuclein-mediated toxicity (Dauer et al., 2002; Klivenyi et al., 2006).
Cerebral Ischemic Stroke and Mitochondrial Dysfunction
Stroke is one of the major causes of death and severe long-term disability in adults. According to the World Health Organization, 15 million people suffer stroke worldwide each year (http://www.strokecenter.org/patients/stats.htm). Due to a dramatic increase in life expectancy and demographic changes in the population, the burden will increase greatly during the next 20 years (Donnan et al., 2008). Of all strokes, 87% are ischemic, 10% are intracerebral hemorrhage, and 3% are subarachnoid hemorrhage strokes (Bolaños et al., 2009). In cerebral ischemic stroke, the reduction in blood supply to brain tissue results in a decrease in the availability of glucose and oxygen, which are necessary for normal brain function (Lipton, 1999; Lo et al., 2003; Rami et al., 2008). The mechanisms underlying neuronal cell death after cerebral ischemia are very complex. However, mitochondrial dysfunction is known to occur after cerebral ischemia and play an important role in mediating ischemic neuronal cell death via either necrosis or apoptosis (Figure 2) (Green and Reed, 1998; Nicotera and Lipton, 1999; Fiskum, 2000; Chan, 2004). Apoptotic cell damage, concomitant with necrotic cell loss, was found in postmortem brain tissue from a cohort of human ischemic stroke patients (Sairanen et al., 2006). A recent postmortem analysis of brain tissue from stroke patients also revealed an involvement of mitochondrial signaling in cerebral ischemia pathobiology (Mitsios et al., 2007). One of the key events intimately involved with mitochondrial dysfunction is the abnormal increase in intracellular Ca2+ levels (Puka-Sundvall et al., 2000; Schild et al., 2003). Transient ischemia is accompanied by a gradual rise in intracellular Ca2+ levels, by Ca2+ sequestration in mitochondria and by mitochondrial bioenergetic failure (Dux et al., 1987; Sims and Pulsinelli, 1987; Silver and Erecinska, 1992; Zaidan and Sims, 1994). Indeed, evidence from the literature demonstrated that during brain ischemia the activities of the complexes I, II, and III of the mitochondrial respiratory chain are decreased (Sims, 1991; Allen et al., 1995). Despite the fact that ischemia does not affect complex IV activity, it has been shown that its activity is remarkably inhibited by a long period of reperfusion, which suggests an irreversible damage of this mitochondrial complex probably due to the production of free radicals (Nelson and Silverstein, 1994; Allen et al., 1995; Almeida et al., 1995). Consequently, the maintenance of ATP levels and other high-energy metabolites after ischemia is affected, leading to an energy crisis (Siesjö et al., 1995; Kuroda et al., 1996). Excessive mitochondrial Ca2+ accumulation following the raise in intracellular Ca2+ is also responsible for ischemia-induced exacerbated ROS production (Murphy et al., 1999). It has been shown that the extensive Ca2+ accumulation by mitochondria induces MPTP opening, which can mediate the release of cytochrome c, favoring superoxide anion (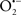 ) formation at complex I and leading to mitochondrial membrane potential (ΔΨm) collapse, mitochondrial swelling, and rupture of the mitochondrial outer membrane (Nicholls, 2009). The release of cytochrome c and apoptosis inducing factor (AIF) from the mitochondria together with caspase-9 and caspase-3 activation following ischemia suggest that the induction of MPTP is an integral apoptotic mechanism involved in ischemia-triggered mitochondrial dysfunction and neuronal cell death (Dirnagl et al., 1999; Lipton, 1999; Fiskum, 2000; Liou et al., 2003; Lo et al., 2003). It has been shown that cyclosporine A (CsA), which inhibits MPTP, has neuroprotective effects against ischemia-induced brain injury (Soane et al., 2007; Tsujimoto and Shimizu, 2007). Cyclophilin D-knockout mice display a dramatic reduction in the size of brain infarcts (Schinzel et al., 2005).
) formation at complex I and leading to mitochondrial membrane potential (ΔΨm) collapse, mitochondrial swelling, and rupture of the mitochondrial outer membrane (Nicholls, 2009). The release of cytochrome c and apoptosis inducing factor (AIF) from the mitochondria together with caspase-9 and caspase-3 activation following ischemia suggest that the induction of MPTP is an integral apoptotic mechanism involved in ischemia-triggered mitochondrial dysfunction and neuronal cell death (Dirnagl et al., 1999; Lipton, 1999; Fiskum, 2000; Liou et al., 2003; Lo et al., 2003). It has been shown that cyclosporine A (CsA), which inhibits MPTP, has neuroprotective effects against ischemia-induced brain injury (Soane et al., 2007; Tsujimoto and Shimizu, 2007). Cyclophilin D-knockout mice display a dramatic reduction in the size of brain infarcts (Schinzel et al., 2005).
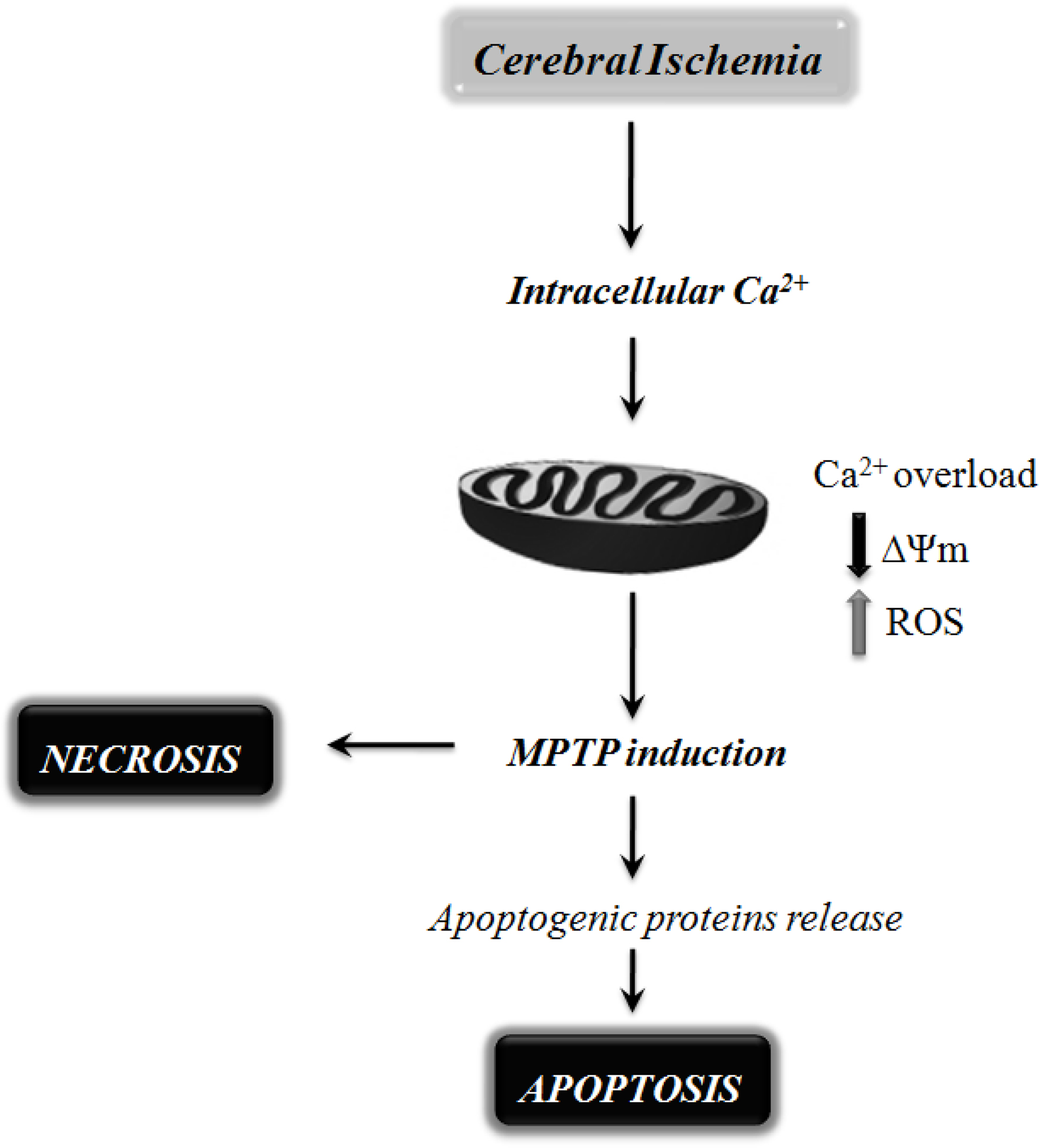
Figure 2. Schematic representation of the mitochondrial mechanisms involved in neuronal cell death following cerebral ischemia. Mitochondrial dysfunction has been shown to be a critical player in mediating ischemic neuronal cell death via either necrosis or apoptosis. During cerebral ischemia, the absence of glucose and oxygen causes exacerbated intracellular and mitochondrial calcium (Ca2+) uptake, leading to mitochondrial dysfunction and bioenergetic failure. The extensive Ca2+ accumulation by mitochondria has been shown to increase the susceptibility to mitochondrial permeability transition pore (MPTP) opening, favoring reactive oxygen species (ROS) formation, mitochondrial membrane potential (ΔΨm) collapse, mitochondrial swelling, and rupture of the mitochondrial outer membrane. MPTP opening can also trigger the release of pro-apoptotic proteins, leading to neuronal cell death by apoptosis.
These studies indicate that during cerebral ischemia the absence of glucose and oxygen causes exacerbated intracellular and mitochondrial Ca2+ uptake, which ultimately causes mitochondrial dysfunction and bioenergetic failure compromising neuronal function and survival in ischemic brain (Figure 2).
Mitochondrial Preconditioning-Mediated Neuroprotection: Triggers and Mediators
Preconditioning is a phenomenon in which the brain protects itself against future injury by adapting to low doses of noxious insults. It has been shown that certain stimuli such as ischemia, low doses of endotoxin and hypoxia can induce preconditioning-dependent protective responses (Cadet and Krasnova, 2009). Despite the molecular mechanisms underlying preconditioning remain unclear, it has been postulated that mitochondria actively participate in the preconditioning signaling pathway by the generation of ROS (Kowaltowski, 2000; Suleiman et al., 2001; Ravagnan et al., 2002). It has also been shown that preconditioning can be prevented with the pharmacological inhibition of mitoKATP channels and activated by mitoKATP agonists, which emphasize the crucial involvement of mitochondria in the preconditioning phenomenon (Auchampach et al., 1992; Yao and Gross, 1993; Garlid et al., 1997; Jaburek et al., 1998). Interestingly, oxidant signals contribute to the activation of the mitoKATP (Ardehali and O’Rourke, 2005). ROS, particularly 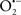 , may directly activate mitoKATP channels (Zhang et al., 2001). It was shown in a transient focal cerebral ischemia model that mitoKATP opening was not followed by ROS generation suggesting that ROS signaling acts upstream of mitoKATP (Mayanagi et al., 2007a). On the other hand, there is also evidence that ROS act downstream of mitoKATP activation in preconditioning (Patel and Gross, 2001). The next subsections intend to discuss the role of mitochondria as major integrators of preconditioning-induced endogenous neuroprotection.
, may directly activate mitoKATP channels (Zhang et al., 2001). It was shown in a transient focal cerebral ischemia model that mitoKATP opening was not followed by ROS generation suggesting that ROS signaling acts upstream of mitoKATP (Mayanagi et al., 2007a). On the other hand, there is also evidence that ROS act downstream of mitoKATP activation in preconditioning (Patel and Gross, 2001). The next subsections intend to discuss the role of mitochondria as major integrators of preconditioning-induced endogenous neuroprotection.
Mitochondrial ROS Production as a Signaling Mechanism Underlying Preconditioning-Induced Neuroprotection
Mitochondria are one of the major sources of ROS. Within the four protein complexes associated with the respiratory chain, the primary sites of ROS production and release are complexes I and III (Zhang and Gutterman, 2007). As mentioned above, ROS have been implicated in the preconditioning phenomenon (Ohtsuki et al., 1992; Peters et al., 1998; Ravati et al., 2000). Mitochondria regulate several signaling pathways through the generation of moderate levels of ROS (Allen and Tresini, 2000; Droge, 2002; Zhang and Gutterman, 2007). Ravati et al. (2000) demonstrated that preconditioning stimulated by moderate ROS levels protect cultured neurons against different damaging agents and prevent against the subsequent massive oxygen radical formation. Moreover, an immediate and constant radical scavenger abolishes this ROS-induced neuronal preconditioning (Ravati et al., 2000). It was also shown that preconditioning with xanthine/xanthine oxidase (X/XO) and ferrous sulfate (FeSO4) protected cultured neurons against staurosporine-induced damage by an increase in ROS followed by the activation of transcription factor nuclear factor-κB (NF-κB) and the subsequent increase in NF-κB-regulated gene expression, namely manganese superoxide dismutase (MnSOD; Ravati et al., 2001). Preconditioning with low concentrations of H2O2 was also shown to protect PC12 cells against apoptosis induced by subsequent lethal oxidative stress (Tang et al., 2005a). Moreover, the mechanisms behind H2O2-induced neuroprotective effects include blockage of ΔΨm loss, increase in ROS generation and overexpression of Bcl-2 (Tang et al., 2005b). Accordingly, Furuichi et al. (2005) demonstrated that the generation of H2O2 during brief oxygen–glucose deprivation (OGD) is the main trigger involved in the mechanism of preconditioning-induced neuronal protection. Recently, it was reported a preconditioning effect of an in situ administration of H2O2 inside the brain cortex which suggests a direct implication of ROS during the triggering phase of cerebral preconditioning (Simerabet et al., 2008). Also found was a relationship between mitoKATP channels and ROS since the protection induced by H2O2 against cerebral ischemia-reperfusion injury was blocked by mitoKATP channels antagonist and the antioxidant N-acetyl-cysteine (NAC) blocked protection induced by diazoxide, a mitoKATP channels opener (Simerabet et al., 2008). The strong relationship observed between ROS and mitoKATP suggests a central position of mitochondria in the neuroprotection induced by delayed cerebral preconditioning (Simerabet et al., 2008). Additionally, it has been shown that the inhibition of succinate dehydrogenase (SDH) with 3-nitropropionic acid (3-NPA), an agent known to increase the production of ROS probably at mitochondrial complex I, induces tolerance to focal cerebral ischemia (Wiegand et al., 1999) implicating mitochondrial ROS in cerebral preconditioning. Horiguchi et al. (2003) also demonstrated that 3-NPA is able to induce delayed preconditioning in rats when administered 3 days after transient middle cerebral artery occlusion (MCAO) by reducing infarct volume by about 20%. More recently, it was observed that NS1619, which inhibits complex I of mitochondrial respiratory chain, induces neuronal preconditioning by increasing ROS production and mitochondrial depolarization (Gáspár et al., 2008a, 2009). The same authors observed that ROS scavenging during the preconditioning phase significantly reduces the neuroprotective effect of NS1619 (Gáspár et al., 2009), which emphasizes the role of mitochondria and mitochondrial ROS in the preconditioning phenomenon. Moreover, it was demonstrated that immediate NS1619 preconditioning include decreased Ca2+ influx through glutamate receptors, increased SOD activity, reduced ROS response during glutamate stimulation, and a better preservation of ATP levels (Gáspár et al., 2009).
One key transcription factor involved in preconditioning-induced brain tolerance is the hypoxia-inducible factor 1 (HIF-1). HIF-1 is a heterodimeric protein composed of a constitutively expressed HIF-1β subunit and an inducible HIF-1α subunit. Under normoxia, HIF-1α is hydroxylated by prolyl hydroxylase enzymes (PHDs) and rapidly degraded by the ubiquitin-proteasome system. On the other hand, under hypoxic conditions the PHDs enzymatic inhibition abrogates HIF-1α proteasomal degradation, resulting in HIF-1α stabilization and translocation to the nucleus. In the nucleus, HIF-1α recruits HIF-1β and modulates the expression of several target genes involved in angiogenesis, metabolism, apoptosis and cell survival (Correia and Moreira, 2010). Accumulating evidence demonstrated that HIF-1 induction has neuroprotective effects in cerebral ischemic stroke and chronic neurodegenerative disorders, such as AD and PD (Correia and Moreira, 2010). Mitochondria and mitochondrial ROS seems to be critical players involved in HIF-1α stabilization. Indeed, Klimova and Chandel (2008) suggested that mitochondrial ROS are essential to HIF-1α protein stabilization and activation. There is a crucial role of ROS generated by the Q0 site of complex III in the hypoxia-mediated survival signaling (Bell et al., 2007). Additionally, mitochondrial ROS generation was shown to be able to prevent the hydroxylation of HIF-1α, thereby stabilizing HIF-1α and allowing its translocation to the nucleus and dimerization with HIF-1β, initiating the transcription of target genes (Figure 3) (Brunelle et al., 2005; Guzy et al., 2005; Bell et al., 2007). Several studies show that the exogenous application of H2O2 can induce HIF-1α under normoxic conditions and ROS scavengers can block hypoxic induction of HIF (Guzy et al., 2005; Mansfield et al., 2005). Moreover, it was demonstrated that mitochondrial DNA-depleted (ρ0) cells, without a functional mitochondrial respiratory chain, failed to increase ROS generation and HIF-1α accumulation under hypoxic conditions (Chandel et al., 1998, 2000). Chandel et al. (2000) also found that low levels of H2O2 stabilize HIF-1α protein during normoxia and increase hypoxia responsive element (HRE)-luciferase expression in ρ0 cells (Chandel et al., 2000).
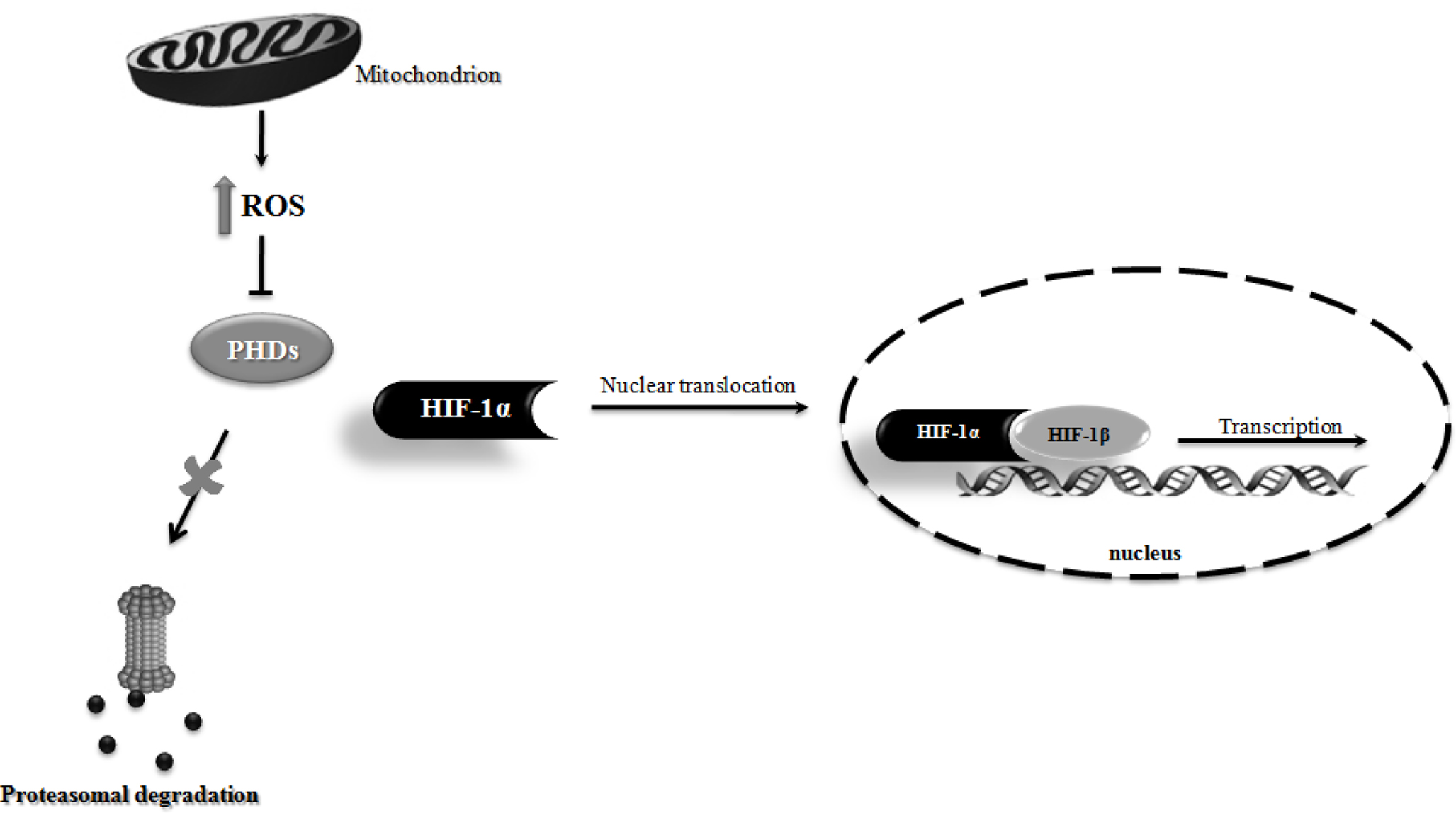
Figure 3. Schematic illustration of the involvement of mitochondrial reactive oxygen species (ROS) in hypoxia-inducible factor 1 (HIF-1) stabilization. Mitochondrial ROS production has been shown to inhibit prolyl hydroxylase enzymes (PHDs) activity, thus preventing HIF-1α proteasomal degradation. Consequently, HIF-1α is stabilized and translocated to the nucleus, where it dimerizes with HIF-1β, initiating the transcription of HIF-1 responsiveness genes.
Collectively these findings reinforce the critical involvement of mitochondria and mitochondrial ROS on neuroprotective mechanisms triggered by preconditioning.
Mechanisms of Neuroprotection Mediated by MitoKATP Channels Opening
The mitoKATP channels are heteromultimers consisting of a 55-kDa inwardly rectifying potassium (K+) channel, mitoKIR, and a 63-kDa sulfonylurea receptor, mitoSUR and are localized in the inner mitochondrial membrane, regulating mitochondrial function in several tissues, including the brain (Figure 4)(Bajgar et al., 2001; Debska et al., 2001; Kulawiak and Bednarczyk, 2005). The physiological role mitoKATP channels has been proposed to buffer potential perturbations of matrix volume and the intermembrane space, consequently ATP synthesis and transport fulfill cellular demands. For instance, during high rates of ATP production, increased current flow through the electron transport system will cause ΔΨm to fall, thus inhibiting K+ diffusion into the matrix and osmotically contracting the matrix (Garlid et al., 2003). Brain mitochondria contain seven times more mitoKATP channels than liver or heart mitochondria, which reflect the importance of these channels in neuronal functionality and integrity (Bajgar et al., 2001). Accumulating evidence suggests a key role for the mitoKATP channels as both triggers and end effectors of acute and delayed neuroprotection of preconditioning (Busija et al., 2008; Dirnagl and Meisel, 2008). Indeed, activation of mitoKATP channels with pharmacological agents mimics the protective effects mediated by preconditioning (Garlid et al., 1997; Szewczyk and Wojtczak, 2002). On the other hand, it has been shown that physiological or chemical preconditioning is abrogated by mitoKATP channels blockers, such as glibenclamide and 5-hydroxydecanoate (5-HD; Szewczyk and Wojtczak, 2002). It has been hypothesized that mitoKATP channels opening may decrease ΔΨm, promoting an increase in the electron transport chain rate, and, consequently, increasing ATP production (Inoue et al., 1991). Wu et al. (2006) demonstrated that the activation of mitoKATP channels protects the brain against injury through the attenuation of mitochondrial Ca2+ overload and, thus, preventing MPTP induction. More recently, it was also observed that the signal transduction pathways initiated by epsilon protein kinase C (εPKC) mediate preconditioning-induced neuroprotection through the activation of mitoKATP channels (Raval et al., 2007). The authors observed that the inhibition of both mitoKATP channels or εPKC abolishes the beneficial effects of preconditioning (Raval et al., 2007).
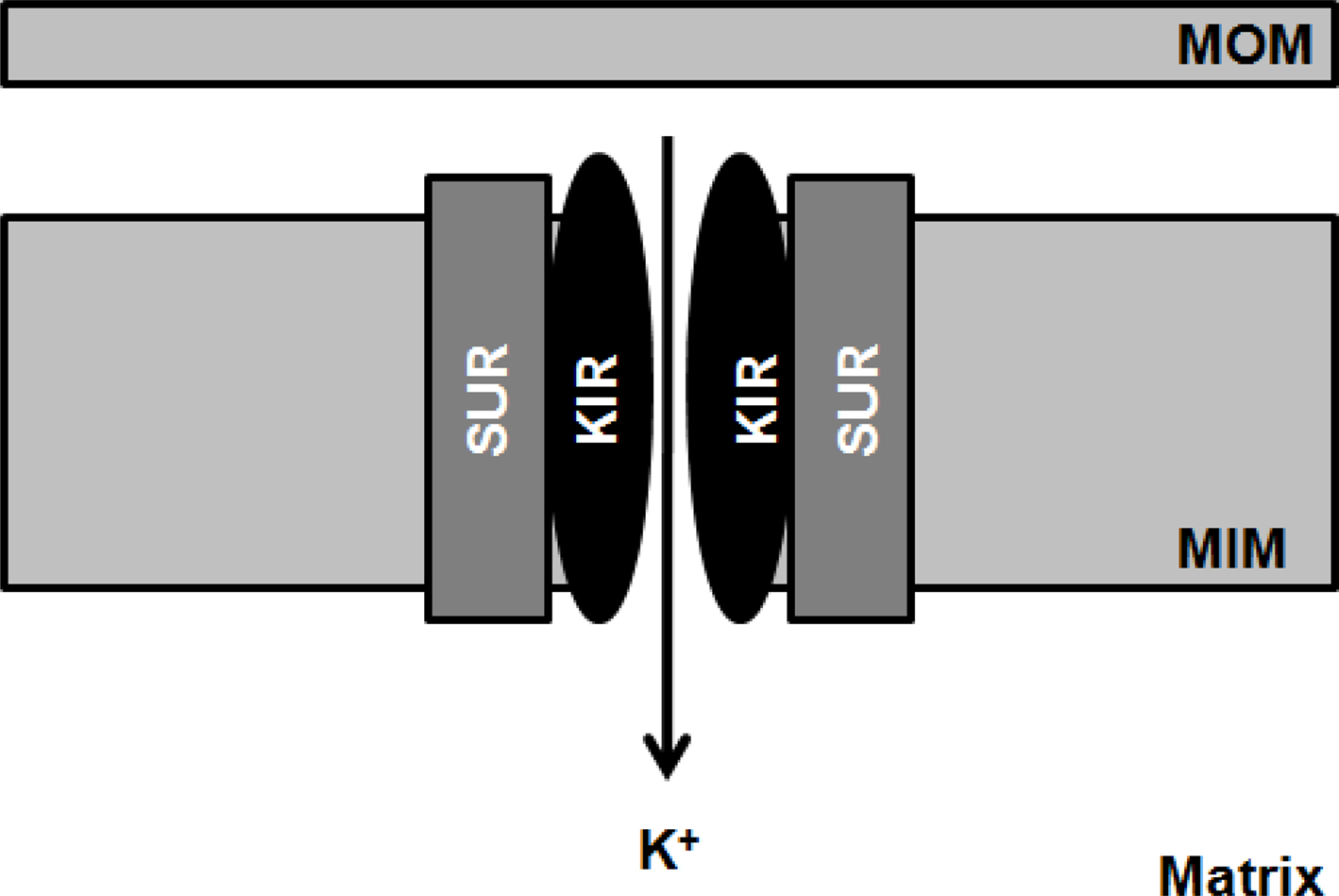
Figure 4. Schematic illustration of the mitochondrial ATP-sensitive K+-channel. The mitochondrial ATP-sensitive K+-channel (mitoKATP) channels are heteromultimers consisting of a 55-kDa inwardly rectifying potassium (K+) channel, mitoKIR, and a 63-kDa sulfonylurea receptor, mitoSUR and are localized in the inner mitochondrial membrane (MIM).
Diazoxide has been suggested to induce mild oxidative stress and preconditioning-like neuroprotection (Samavati et al., 2002). Diazoxide is a selective mitoKATP channels opener (Kis et al., 2003; Lacza et al., 2003; Nagy et al., 2004; Busija et al., 2005). However, it has been shown that high doses of diazoxide also inhibit SDH, the complex II of the mitochondrial respiratory chain, leading to the release of ROS in a mitoKATP channel-independent manner (Kis et al., 2003). Recently, Busija et al. (2008) proposed that diazoxide is the most potent inducer of preconditioning-mediated protection due to the combined effects: mitochondrial membrane depolarization and enhanced ROS production through SDH inhibition. It was also demonstrated that the immediate preconditioning induced by low doses of diazoxide preserves neuronal and vascular function after cerebral ischemia (Domoki et al., 1999). Moreover, it was observed that this immediate preconditioning with diazoxide protects against ischemia-reperfusion injury by preventing mitochondrial swelling and Ca2+ accumulation in brain cells (Domoki et al., 2004). Diazoxide also induces delayed preconditioning in neurons via acute generation of 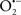 and activation of protein kinases protecting against the oxidative stress induced by OGD, which is a well-established in vitro model of cerebral ischemia-reperfusion (Kis et al., 2003). It was also demonstrated that diazoxide protects neurons against ischemia-induced death by increasing mitochondrial Bcl-2 levels and suppressing Bax translocation to mitochondria and subsequent cytochrome c release, suggesting that mitoKATP channels activation may stabilize mitochondrial function by differentially modulating pro- apoptotic and anti-apoptotic proteins (Liu et al., 2002). Some in vivo studies also revealed that the acute and delayed preconditioning with diazoxide had a neuroprotective effect against transient focal cerebral ischemia (Shimizu et al., 2002; Mayanagi et al., 2007b). Moreover, Goodman and Mattson (1996) demonstrated that diazoxide is effective in protecting hippocampal neurons against oxidative injury induced by exposure to FeSO4 and Aβ, leading to a suppression of intracellular peroxide formation. Accordingly, a protective effect of diazoxide against Aβ-induced cytotoxicity in endothelial cells was observed (Chi et al., 2000). Ma and Chen (2004) reported that diazoxide counteracts the effects of Aβ1-42, protecting neurons against the increase of ΔΨm and intracellular ROS levels induced by this amyloidogenic peptide. A recent study reported that exposure to Aβ1-42 enhances the expression of KATP channel subunits in cholinergic neurons, being suggested that the change in the composition of KATP channels may contribute to the dysfunction of KATP channels and membrane excitability disturbance (Ma et al., 2009). Moreover, it was demonstrated that the pretreatment with diazoxide reverses the Aβ1-42-induced enhancement in the expression of KATP channels subunits (Ma et al., 2009). In an in vitro model of PD, it was also observed that diazoxide protects neurons against MPP+-induced cytotoxicity via inhibition of ROS overproduction, which improve mitochondrial function (Xie et al., 2009). Similarly, Yang et al. (2006) found that this mitoKATP opener improves both parkinsonian symptoms and neurochemistry alterations found in rats treated with rotenone, a model of PD. These results suggest that mitoKATP activation could provide a new therapeutic strategy for the treatment of early PD. The authors also observed that 5-HD abolishes all neurorestorative effects of diazoxide (Yang et al., 2006). This is consistent with previous studies showing that activation of mitoKATP channels with diazoxide in PC12 cells induces protection against the neurotoxic effects of rotenone, this protection being attenuated by 5-HD (Tai and Truong, 2002; Tai et al., 2003). An in vitro study also revealed that diazoxide prevents rotenone-induced microglial activation and the subsequent production of pro-inflammatory factors, such as tumor necrosis factor alpha (TNF-α) and inducible isoform of nitric oxide synthase (iNOS; Liu et al., 2006).
and activation of protein kinases protecting against the oxidative stress induced by OGD, which is a well-established in vitro model of cerebral ischemia-reperfusion (Kis et al., 2003). It was also demonstrated that diazoxide protects neurons against ischemia-induced death by increasing mitochondrial Bcl-2 levels and suppressing Bax translocation to mitochondria and subsequent cytochrome c release, suggesting that mitoKATP channels activation may stabilize mitochondrial function by differentially modulating pro- apoptotic and anti-apoptotic proteins (Liu et al., 2002). Some in vivo studies also revealed that the acute and delayed preconditioning with diazoxide had a neuroprotective effect against transient focal cerebral ischemia (Shimizu et al., 2002; Mayanagi et al., 2007b). Moreover, Goodman and Mattson (1996) demonstrated that diazoxide is effective in protecting hippocampal neurons against oxidative injury induced by exposure to FeSO4 and Aβ, leading to a suppression of intracellular peroxide formation. Accordingly, a protective effect of diazoxide against Aβ-induced cytotoxicity in endothelial cells was observed (Chi et al., 2000). Ma and Chen (2004) reported that diazoxide counteracts the effects of Aβ1-42, protecting neurons against the increase of ΔΨm and intracellular ROS levels induced by this amyloidogenic peptide. A recent study reported that exposure to Aβ1-42 enhances the expression of KATP channel subunits in cholinergic neurons, being suggested that the change in the composition of KATP channels may contribute to the dysfunction of KATP channels and membrane excitability disturbance (Ma et al., 2009). Moreover, it was demonstrated that the pretreatment with diazoxide reverses the Aβ1-42-induced enhancement in the expression of KATP channels subunits (Ma et al., 2009). In an in vitro model of PD, it was also observed that diazoxide protects neurons against MPP+-induced cytotoxicity via inhibition of ROS overproduction, which improve mitochondrial function (Xie et al., 2009). Similarly, Yang et al. (2006) found that this mitoKATP opener improves both parkinsonian symptoms and neurochemistry alterations found in rats treated with rotenone, a model of PD. These results suggest that mitoKATP activation could provide a new therapeutic strategy for the treatment of early PD. The authors also observed that 5-HD abolishes all neurorestorative effects of diazoxide (Yang et al., 2006). This is consistent with previous studies showing that activation of mitoKATP channels with diazoxide in PC12 cells induces protection against the neurotoxic effects of rotenone, this protection being attenuated by 5-HD (Tai and Truong, 2002; Tai et al., 2003). An in vitro study also revealed that diazoxide prevents rotenone-induced microglial activation and the subsequent production of pro-inflammatory factors, such as tumor necrosis factor alpha (TNF-α) and inducible isoform of nitric oxide synthase (iNOS; Liu et al., 2006).
BMS-191095, a selective mitoKATP channels opener, has been shown to induce both immediate and delayed preconditioning in neurons. Kis et al. (2004) demonstrated that the selective opening of mitoKATP channels with BMS-191095 leads to a remarkable neuroprotection via mechanisms that involve mitochondrial depolarization, PKC activation, and attenuated free radical production during neuronal stress. In addition, it was found that BMS-191095 depolarizes mitochondria without ROS generation, activates the phosphoinositide 3-kinase (PI3-K) signaling pathway, increases ATP content and catalase expression; these mechanisms playing important roles in the neuroprotective effect afforded by this mitoKATP channels opener (Gáspár et al., 2008b). Similarly, Mayanagi et al. (2007a) examined the effects of BMS-191095 pretreatment on transient ischemia induced by MCAO in rats and concluded that BMS-191095 afforded protection against cerebral ischemia by delayed preconditioning via selective opening of mitoKATP channels without ROS generation.
These evidences indicate that mitoKATP channels activation is a key mechanism involved in the neuroprotective events elicited by the preconditioning phenomenon, which suggest that these channels could represent promising therapeutic targets to counteract brain cells degeneration and death.
Concluding Remarks
Mitochondria are dynamic organelles critically involved in cell survival and death, since they are main sources of cellular ATP and ROS, buffers of intracellular Ca2+ and integrate several survival and death signaling pathways. Therefore, disturbances in the mitochondrial machinery, dynamics and turnover are important events underlying neurodegeneration. On the other hand, strong evidence demonstrate the involvement of mitochondrial ROS and mitoKATP channels on the neuroprotective mechanisms triggered by preconditioning establishing a central role of mitochondria in the preconditioning phenomenon. Understanding the mechanism underlying mitochondrial preconditioning-induced brain protection may provide important information that can be used for the development of new and more effective therapeutic strategies to prevent and/or mitigate neurodegeneration.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work is supported by the European Foundation for the Study of Diabetes/Servier Programme 2007. Sónia C. Correia has a PhD fellowship from the Fundação para a Ciência e a Tecnologia (SFRH/BD/40702/2007).
References
Allen, K. L., Almeida, A., Bates, T. E., and Clark J. B. (1995). Changes of respiratory chain activity in mitochondrial and synaptosomal fractions isolated from the gerbil brain after graded ischemia. J. Neurochem. 64, 2222–2229.
Allen, R. G., and Tresini, M. (2000). Oxidative stress and gene regulation. Free Radic. Biol. Med. 28, 463–499.
Almeida, A., Allen, K. L., Bates, T. E., and Clark, J. B. (1995). Effect of reperfusion following cerebral ischemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J. Neurochem. 65, 1698–1703.
Anandatheerthavarada, H. K., Biswas, G., Robin, M. A., and Avadhani, N. G. (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 161, 41–54.
Ardehali, H., and O’Rourke, B. (2005). Mitochondrial K(ATP) channels in cell survival and death. J. Mol. Cell. Cardiol. 39, 7–16.
Auchampach, J. A., Grover, G. J., and Gross, G. J. (1992). Blockade of ischaemic preconditioning in dogs by the novel ATP dependent potassium channel antagonist sodium 5-hydroxydecanoate. Cardiovasc. Res. 26, 1054–1062.
Bajgar, R., Seetharaman, S., Kowaltowski, A. J., Garlid, K. D., and Paucek, P. (2001). Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. J. Biol. Chem. 276, 33369–33374.
Banerjee, R., Starkov, A. A., Beal, M. F., and Thomas, B. (2009). Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta 1792, 651–663.
Barroso, N., Campos, Y., Huertas, R., Esteban, J., Molina, J. A., Alonso, A., Gutierrez-Rivas, E., and Arenas, J. (1993). Respiratory chain enzyme activities in lymphocytes from untreated patients with Parkinson disease. Clin. Chem. 39, 667–669.
Beal, M. F. (2005). Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 58, 495–505.
Bell, E. L., Klimova, T. A., Eisenbart, J., Moraes, C. T., Murphy, M. P., Budinger, G. R., and Chandel, N. S. (2007). The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 177, 1029–1036.
Betarbet, R., Sherer, T. B., MacKenzie, G., Garcia-Osuna, M., Panov, A. V., and Greenamyre, J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 3, 1301–1306.
Blandini, F., Nappi, G., and Greenamyre, J. T. (1998). Quantitative study of mitochondrial complex I in platelets of parkinsonian patients. Mov. Disord. 13, 11–15.
Bolaños, J. P., Moro, M. A., Lizasoain, I., and Almeida, A. (2009). Mitochondria and reactive oxygen and nitrogen species in neurological disorders and stroke: therapeutic implications. Adv. Drug Deliv. Rev. 61, 1299–1315.
Bosetti, F., Brizzi, F., Barogi, S., Mancuso, M., Siciliano, G., Tendi, E. A., Murri, L., Rapoport, S. I., and Solaini, G. (2002). Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol. Aging 23, 371–376.
Brunelle, J. K., Bell, E. L., Quesada, N. M., Vercauteren, K., Tiranti, V., Zeviani, M., Scarpulla, R. C., and Chandel, N. S. (2005). Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 1, 409–414.
Bubber, P., Haroutunian, V., Fisch, G., Blass, J. P., and Gibson, G. E. (2005). Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 57, 695–703.
Busija, D. W., Gaspar, T., Domoki, F., Katakam, P. V., and Bari, F. (2008). Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv. Drug Deliv. Rev. 60, 1471–1477.
Busija, D. W., Katakam, P., Rajapakse, N. C., Kis, B., Grover, G., Domoki, F., and Bari, F. (2005). Effects of ATP-sensitive potassium channel activators diazoxide and BMS-191095 on membrane potential and reactive oxygen species production in isolated piglet mitochondria. Brain Res. Bull. 66, 85–90.
Cadet, J. L., and Krasnova, I. N. (2009). Cellular and molecular neurobiology of brain preconditioning. Mol. Neurobiol. 39, 50–61.
Cardoso, S. M., Moreira, P. I., Agostinho, P., Pereira, C., and Oliveira, C. R. (2005). Neurodegenerative pathways in Parkinson’s disease: therapeutic strategies. Curr. Drug Targets CNS Neurol. Disord. 4, 405–419.
Caspersen, C., Wang, N., Yao, J., Sosunov, A., Chen, X., Lustbader, J. W., Xu, H. W., Stern, D., McKhann, G., and Yan, S. D. (2005). Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 19, 2040–2041.
Chan, P. H. (2004). Future targets and cascades for neuroprotective strategies. Stroke 35, 2748–2750.
Chandel, N. S., Maltepe, E., Goldwasser, E., Mathieu, C. E., Simon, M. C., and Schumacker, P. T. (1998). Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA. 95, 11715–11720.
Chandel, N. S., McClintock, D. S., Feliciano, C. E., Wood, T. M., Melendez, J. A., Rodriguez, A. M., and Schumacker, P. T. (2000). Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138.
Chi, X., Sutton, E. T., Hellermann, G., and Price, J. M. (2000). Potassium channel openers prevent beta-amyloid toxicity in bovine vascular endothelial cells. Neurosci. Lett. 290, 9–12.
Correia, S. C., and Moreira, P. I. (2010). Hypoxia-inducible factor 1: a new hope to counteract neurodegeneration? J. Neurochem. 112, 1–12.
Coskun, P. E., Beal, M. F., and Wallace, D. C. (2004). Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. U.S.A. 101, 10726–10731.
Crouch, P. J., Blake, R., Duce, J. A., Ciccotosto, G. D., Li, Q. X., Barnham, K. J., Curtain, C. C., Cherny, R. A., Cappai, R., Dyrks, T., Masters, C. L., and Trounce, I. A. (2005). Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid- beta1-42. J. Neurosci. 25, 672–679.
Dauer, W., Kholodilov, N., Vila, M., Trillat, A. C., Goodchild, R., Larsen, K. E., Staal, R., Tieu, K., Schmitz, Y., Yuan, C. A., Rocha, M., Jackson-Lewis, V., Hersch, S., Sulzer, D., Przedborski, S., Burke, R., and Hen, R. (2002). Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U.S.A. 99, 14524–14529.
Debska, G., May, R., Kicinska, A., Szewczyk, A., Elger, C. E., and Kunz, W. S. (2001). Potassium channel openers depolarize hippocampal mitochondria. Brain Res. 892, 42–50.
de Lau, L. M., Giesbergen, P. C., de Rijk, M. C., Hofman, A., Koudstaal, P. J., and Breteler, M. M. (2004). Incidence of parkinsonism and Parkinson disease in a general population: the Rotterdam Study. Neurology 63, 1240–1244.
de Rijk, M. C., Rocca, W. A., Anderson, D. W., Melcon, M. O., Breteler, M. M., and Maraganore, D. M. (1997). A population perspective on diagnostic criteria for Parkinson’s disease. Neurology 48, 1277–1281.
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068.
Dirnagl, U., Becker, K., and Meisel, A. (2009). Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 8, 398–412.
Dirnagl, U., Iadecola, C., and Moskowitz, M. A. (1999). Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397.
Dirnagl, U., and Meisel, A. (2008). Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology 55, 334–344.
Domoki, F., Bari, F., Nagy, K., Busija, D. W., and Siklos, L. (2004). Diazoxide prevents mitochondrial swelling and Ca (2+) accumulation in CAI pyramidal cells after cerebral ischemia in newborn pigs. Brain Res. 1019, 97–104.
Domoki, F., Perciaccante, J. V., Veltkamp, R., Bari, F., and Busija, D. W. (1999). Mitochondrial potassium channel opener diazoxide preserves neuronal-vascular function after cerebral ischemia in newborn pigs. Stroke 30, 2713–2718.
Droge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95.
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., Yan, Y., Wang, C., Zhang, H., Molkentin, J. D., Gunn-Moore, F. J., Vonsattel, J. P., Arancio, O., Chen, J. X., and Yan, S. D. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105.
Dux, E., Mies, G., Hossmann, K. A., and Siklos, L. (1987). Calcium in the mitochondria following brief ischemia of gerbil brain. Neurosci. Lett. 78, 295–300.
Ekstrand, M. I., Terzioglu, M., Galter, D., Zhu, S., Hofstetter, C., Lindqvist, E., Thams, S., Bergstrand, A., Hansson, F. S., Trifunovic, A., Hoffer, B., Cullheim, S., Mohammed, A. H., Olson, L., and Larsson, N. G. (2007). Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA. 104, 1325–1330.
Falkevall, A., Alikhani, N., Bhushan, S., Pavlov, P. F., Busch, K., Johnson, K. A., Eneqvist, T., Tjernberg, L., Ankarcrona, M., and Glaser, E. (2006). Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J. Biol. Chem. 281, 29096–29104.
Findeis, M. A. (2007). The role of amyloid beta peptide 42 in Alzheimer’s disease. Pharmacol. Ther. 116, 266–286.
Fiskum, G. (2000). Mitochondrial participation in ischemic and traumatic neuronal cell death. J. Neurotrauma 17, 843–855.
Furuichi, T., Liu, W., Shi, H., Miyake, M., and Liu, K. J. (2005). Generation of hydrogen peroxide during brief oxygen–glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. J. Neurosci. Res. 79, 816–824.
Garlid, K. D., Dos Santos, P., Xie, Z. J., Costa, A. D., and Paucek P. (2003). Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim. Biophys. Acta 1606, 1–21.
Garlid, K. D., Paucek, P., Yarov-Yarovoy, V., Murray, H. N., Darbenzio, R. B., D’Alonzo, A. J., Lodge, N. J., Smith, M. A., and Grover, G. J. (1997). Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 81, 1072–1082.
Gash, D. M., Rutland, K., Hudson, N. L., Sullivan, P. G., Bing, G., Cass, W. A., Pandya, J. D., Liu, M., Choi, D. Y., Hunter, R. L., Gerhardt, G. A., Smith, C. D., Slevin, J. T., and Prince, T. S. (2007). Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann. Neurol. 63, 184–192.
Gáspár, T., Domoki, F., Lenti, L., Katakam, P. V., Snipes, J. A., Bari, F., and Busija, D. W. (2009). Immediate neuronal preconditioning by NS1619. Brain Res. 1285, 196–207.
Gáspár, T., Katakam, P., Snipes, J. A., Kis, B., Domoki, F., Bari, F., and Busija, D. W. (2008a). Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J. Neurochem. 105, 1115–1128.
Gáspár, T., Snipes, J. A., Busija, A. R., Kis, B., Domoki, F., Bari, F., and Busija, D. W. (2008b). ROS-independent preconditioning in neurons via activation of mitoKATP channels by BMS-191095. J. Cereb. Blood Flow Metab. 28, 1090–1103.
Gibson, G. E., Sheu, K. F., and Blass, J. P. (1998). Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 105, 855–870.
Goodman, Y., and Mattson, M. P. (1996). K+ channel openers protect hippocampal neurons against oxidative injury and amyloid beta-peptide toxicity. Brain Res. 706, 328–332.
Green, D. R., and Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629.
Greenfield, J. P., Gross, R. S., Gouras, G. K., and Xu, H. (2000). Cellular and molecular basis of beta-amyloid precursor protein metabolism. Front. Biosci. 5, 72–83.
Guzy, R. D., Hoyos, B., Robin, E., Chen, H., Liu, L., Mansfield, K. D., Simon, M. C., Hammerling, U., and Schumacker, P. T. (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408.
Haas, R. H., Nasirian, F., Nakano, K., Ward, D., Pay, M., Hill, R., and Shults, C. W. (1995). Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 37, 714–722.
Hamblet, N. S., Ragland. B., Ali, M., Conyers, B., and Castora, F. J. (2006). Mutations in mitochondrial-encoded cytochrome c oxidase subunits I, II, and III genes detected in Alzheimer’s disease using single-strand conformation polymorphism. Electrophoresis 27, 398–408.
Hansson Petersen, C. A., Alikhani, N., Behbahani, H., Wiehager, B., Pavlov, P. F., Alafuzoff, I., Leinonen, V., Ito, A., Winblad, B., Glaser, E., and Ankarcrona, M. (2008). The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 105, 13145–13150.
Horiguchi, T., Kis, B., Rajapakse, N., Shimizu, K., and Busija, D. W. (2003). Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke 34, 1015–1020.
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., Wong, J., Takenouchi, T., Hashimoto, M., and Masliah, E. (2000). Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157, 401–410.
Huang, H. M., Ou, H. C., Xu, H., Chen, H. L., Fowler, C., and Gibson, G. E. (2003). Inhibition of alpha-ketoglutarate dehydrogenase complex promotes cytochrome c release from mitochondria, caspase-3 activation, and necrotic cell death. J. Neurosci. Res. 74, 309–317.
Inoue, I., Nagase, H., Kishi, K., and Higuti, T. (1991). ATP-sensitive K channel mitochondrial inner membrane. Nature 352, 244–247.
Jaburek, M., Yarov-Yarovoy, V., Paucek, P., and Garlid, K. D. (1998). State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J. Biol. Chem. 273, 13578–13582.
Jin, J., Hulette, C., Wang, Y., Zhang, T., Pan, C., Wadhwa, R., and Zhang, J. (2006). Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol. Cell. Proteomics 5, 1193–1204.
Jou, M. J. (2008). Pathophysiological and pharmacological implications of mitochondria-targeted reactive oxygen species generation in astrocytes. Adv. Drug Deliv. Rev. 60, 1512–1526.
Kis, B., Nagy, K., Snipes, J. A., Rajapakse, N., Horiguchi, T., Grover, G. J., and Busija, D. W. (2004). The mitochondrial KATP channel opener BMS191095 induces neuronal preconditioning. Neuroreport 15, 345–349.
Kis, B., Rajapakse, N., Snipes, J. A., Nagy, K., Horiguchi, T., and Busija, D. W. (2003). Diazoxide induces delayed preconditioning in cultured rat cortical neurons. J. Neurochem. 87, 969–980.
Kish, S. J., Bergeron, C., Rajput, A., Dozic, S., Mastrogiacomo, F., Chang, L. J., Wilson, J. M., DiStefano, L. M., and Nobrega, J. N. (1992). Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 59, 776–779.
Klimova, T., and Chandel, N. S. (2008). Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 15, 660–666.
Klivenyi, P., Siwek, D., Gardian, G., Yang, L., Starkov, A., Cleren, C., Ferrante, R. J., Kowall, N. W., Abeliovich, A., and Beal, M. F. (2006). Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 21, 541–548.
Knott, A. B., Perkins, G., Schwarzenbacher, R., and Bossy-Wetzel, E. (2008). Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518.
Kowaltowski, A. J. (2000). Alternative mitochondrial functions in cell physiopathology: beyond ATP production. Braz. J. Med. Biol. Res. 33, 241–250.
Krige, D., Carroll, M. T., Cooper, J. M., Marsden, C. D., and Schapira, A. H. (1992). Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann. Neurol. 32, 782–788.
Kulawiak, B., and Bednarczyk, P. (2005). Reconstitution of brain mitochondria inner membrane into planar lipid bilayer. Acta Neurobiol. Exp. 65, 271–276.
Kuroda, S., Katsura, K. I., Tsuchidate, R., and Siesjo, B. K. (1996). Secondary bioenergetic failure after transient focal ischaemia is due to mitochondrial injury. Acta Physiol. Scand. 156, 149–150.
Lacza, Z., Snipes, J. A., Kis, B., Szabo, C., Grover, G., and Busija, D. W. (2003). Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Res. 19, 27–36.
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795.
Lin, M. T., Simon, D. K., Ahn, C. H., Kim, L. M., and Beal, M. F. (2002). High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum. Mol. Genet. 11, 133–145.
Liou, A. K., Clark, R. S., Henshall, D. C., Yin, X. M., and Chen, J. (2003). To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog. Neurobiol. 69, 103–142.
Liu, D., Lu, C., Wan, R., Auyeung, W. W., and Mattson, M. P. (2002). Activation of mitochondrial ATP-dependent potassium channels protects neurons against ischemia-induced death by a mechanism involving suppression of Bax translocation and cytochrome c release. J. Cereb. Blood Flow Metab. 22, 431–443.
Liu, W., Vives-Bauza, C., Acín-Peréz-, R., Yamamoto, A., Tan, Y., Li, Y., Magrané, J., Stavarache, M. A., Shaffer, S., Chang, S., Kaplitt, M. G., Huang, X. Y., Beal, M. F., Manfredi, G., and Li, C. (2009). PINK1 defect causes mitochondrial dysfunction, proteasomal deficit and alpha-synuclein aggregation in cell culture models of Parkinson’s disease. PLoS ONE 4, 4597. doi:10.1371/journal.pone.0004597.
Liu, X., Wu, J. Y., Zhou, F., Sun, X. L., Yao, H. H., Yang, Y., Ding, J. H., and Hu, G. (2006). The regulation of rotenone-induced inflammatory factor production by ATP-sensitive potassium channel expressed in BV-2 cells. Neurosci. Lett. 394, 131–135.
Lo, E. H., Dalkara, T., and Moskowitz, M. A. (2003). Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 4, 399–415.
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., Caspersen, C., Chen, X., Pollak, S., Chaney, M., Trinchese, F., Liu, S., Gunn-Moore, F., Lue, L. F., Walker, D. G., Kuppusamy, P., Zewier, Z. L., Arancio, O., Stern, D., Yan, S. S., and Wu, H. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452.
Ma, G., and Chen, S. (2004). Diazoxide and N omega-nitro-l-arginine counteracted A beta 1-42-induced cytotoxicity. Neuroreport 15, 1813–1817.
Ma , G., Gao, J., Fu, Q., Jiang, L., Wang, R., Zhang, Y., and Liu, K. (2009). Diazoxide reverses the enhanced expression of K(ATP) subunits in cholinergic neurons caused by exposure to Abeta(1-42). Neurochem. Res. (in press).
Manczak, M., Anekonda, T. S., Henson, E., Park, B. S., Quinn, J., and Reddy, P. H. (2006). Mitochondria are a direct site of Abeta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449.
Mann, V. M., Cooper, J. M., Daniel, S. E., Srai, K., Jenner, P., Marsden, C. D., and Schapira, A. H. (1994). Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 36, 876–881.
Mansfield, K. D., Guzy, R. D., Pan, Y., Young, R. M., Cash, T. P., Schumacker, P. T., and Simon, M. C. (2005). Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 1, 393–399.
Maurer, I., Zierz, S., and Möller, H. J. (2000). A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol. Aging 21, 455–462.
Mayanagi, K., Gaspar, T., Katakam, P. V., Kis, B., and Busija, D. W. (2007a). The mitochondrial K(ATP) channel opener BMS-191095 reduces neuronal damage after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 27, 348–355.
Mayanagi, K., Gaspar, T., Katakam, P. V., and Busija, D. W. (2007b). Systemic administration of diazoxide induces delayed preconditioning against transient focal cerebral ischemia in rats. Brain Res. 1168, 106–111.
Migliore, L., and Coppedè, F. (2009). Environmental-induced oxidative stress in neurodegenerative disorders and aging. Mutat. Res. 674, 73–84.
Mitsios, N., Gaffney, J., Krupinski, J., Mathias, R., Wang, Q., Hayward, S., Rubio, F., Kumar, P., Kumar, S., and Slevin, M. (2007). Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem. Biophys. 47, 73–86.
Mizuno, Y., Matuda, S., Yoshino, H., Mori, H., Hattori, N., and Ikebe, S. (1994). An immunohistochemical study on alpha-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 35, 204–210.
Moisoi, N., Klupsch, K., Fedele, V., East, P., Sharma, S., Renton, A., Plun-Favreau, H., Edwards, R. E., Teismann, P., Esposti, M. D., Morrison, A. D., Wood, N. W., Downward, J., and Martins, L. M. (2009). Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 16, 449–464.
Moreira, P. I., Duarte, A. I., Santos, M. S., Rego, A. C., and Oliveira, C. R. (2009). An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J. Alzheimers Dis. 16, 741–761.
Moreira, P. I., Honda, K., Zhu, X., Nunomura, A., Casadesus, G., Smith, M. A., and Perry, G. (2006). Brain and brawn: parallels in oxidative strength. Neurology 66, 97–101.
Moreira, P. I., Santos, M. S., Moreno, A., and Oliveira, C. (2001). Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci. Rep. 21, 789–800.
Moreira, P. I., Santos, M. S., Moreno, A., Rego, A. C., and Oliveira, C. (2002). Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J. Neurosci. Res. 69, 257–267.
Moreira, P. I., Santos, M. S., and Oliveira, C. R. (2007). Alzheimer’s disease: a lesson from mitochondrial dysfunction. Antioxid. Redox Signal. 9, 1621–1630.
Moreira, P. I., Zhu, X., Wang, X., Lee, H. G., Nunomura, A., Petersen, R. B., Perry, G., and Smith, M. A. (2010). Mitochondria: a therapeutic target in neurodegeneration. Biochim. Biophys. Acta 1802, 212–220.
Murphy, A. N., Fiskum, G., and Beal, M. F. (1999). Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab. 19, 231–245.
Nagy, K., Kis, B., Rajapakse, N. C., Bari, F., and Busija, D. W. (2004). Diazoxide preconditioning protects against neuronal cell death by attenuation of oxidative stress upon glutamate stimulation. J. Neurosci. Res. 76, 697–704.
Nelson, C., and Silverstein, F. S. (1994). Acute disruption of cytochrome oxidase activity in brain in a perinatal rat stroke model. Pediatr. Res. 36, 12–19.
Nicholls, D. G. (2009). Mitochondrial calcium function and dysfunction in the central nervous system. Biochim. Biophys. Acta 1787, 1416–1424.
Nicotera, P., and Lipton, S. A. (1999). Excitotoxins in neuronal apoptosis and necrosis. J. Cereb. Blood Flow Metab. 19, 583–591.
Nunomura, A., Honda, K., Takeda, A., Hirai, K., Zhu, X., Smith, M.A., and Perry, G. (2006). Oxidative damage to RNA in neurodegenerative diseases. J. Biomed. Biotechnol. 2006, 82323.
Ohtsuki, T., Matsumoto, M., Kuwabara, K., Kitagawa, K., Suzuki, K., Taniguchi, N., and Kamada, T. (1992). Influence of oxidative stress on induced tolerance to ischemia in gerbil hippocampal neurons. Brain Res. 599, 246–252.
Oldenburg, O., Cohen, M. V., Yellon, D. M., and Downey, J. M. (2002). Mitochondrial KATP channels: role in cardioprotection. Cardiovasc. Res. 55, 429–437.
Parker, W. D. Jr., Boyson, S. J., and Parks, J. K. (1989). Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 26, 719–723.
Parker, W. D. Jr., Mahr, N. J., Filley, C. M., Parks, J. K., Hughes, D., Young, D. A., and Cullum, C. M. (1994). Reduced platelet cytochrome c oxidase activity in Alzheimer’s disease. Neurology 44, 1086–1090.
Patel, H. H., and Gross, G. J. (2001). Diazoxide induced cardioprotection: what comes first, KATP channels or reactive oxygen species? Cardiovasc. Res. 51, 633–636.
Penn, A. M., Roberts, T., Hodder, J., Allen, P. S., Zhu, G., and Martin, W. R. (1995). Generalized mitochondrial dysfunction in Parkinson’s disease detected by magnetic resonance spectroscopy of muscle. Neurology 45, 2097–2099.
Peters, O., Back, T., Lindauer, U., Busch, C., Megow, D., Dreier, J., and Dirnagl, U. (1998). Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J. Cereb. Blood Flow Metab. 18, 196–205.
Petrozzi, L., Ricci, G., Giglioli, N. J., Siciliano, G., and Mancuso, M. (2007). Mitochondria and neurodegeneration. Biosci. Rep. 27, 87–104.
Przedborski, S., Vial, M., and Jackson-Lewis, V. (2003). Neurodegeneration: what is it and where are we? J. Clin. Invest. 111, 3–10.
Puka-Sundvall, M., Gajkowska, B., Cholewinski, M., Blomgren, K., Lazarewicz, J. W., and Hagberg, H. (2000). Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Brain Res. Dev. Brain Res. 125, 31–41.
Qiu, X., Chen, Y., and Zhou, M. (2001). Two point mutations in mitochondrial DNA of cytochrome c oxidase coexist with normal mtDNA in a patient with Alzheimer’s disease. Brain Res. 893, 261–263.
Rami, A., Bechmann, I., and Stehle, J. H. (2008). Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog. Neurobiol. 85, 273–296.
Ravagnan, L., Roumier, T., and Kroemer, G. (2002). Mitochondria, the killer organelles and their weapons. J. Cell. Physiol. 192, 131–137.
Raval, A. P., Dave, K. R., Defazio, R. A., and Pérez-Pinzón, M. A. (2007). EpsilonPKC phosphorylates the mitochondrial K(+) (ATP) channel during induction of ischemic preconditioning in the rat hippocampus. Brain Res. 1184, 345–353.
Ravati, A., Ahlemeyer, B., Becker, A., Klumpp, S., and Krieglstein, J. (2001). Preconditioning-induced neuroprotection is mediated by reactive oxygen species and activation of the transcription factor nuclear factor-kappaB. J. Neurochem. 78, 909–919.
Ravati, A., Ahlemeyer, B., Becker, A., and Krieglstein, J. (2000). Preconditioning-induced neuroprotection is mediated by reactive oxygen species. Brain Res. 866, 23–32.
Sairanen, T., Karjalainen-Lindsberg, M. L., Paetau, A., Ijas, P., and Lindsberg, P. J. (2006). Apoptosis dominant in the periinfarct area of human ischaemic stroke–a possible target of antiapoptotic treatments. Brain 129, 189–199.
Samavati, L., Monick, M. M., Sanlioglu, S., Buettner, G. R., Oberley, L. W., and Hunninghake, G. W. (2002). Mitochondrial K(ATP) channel openers activate the ERK kinase by an oxidant-dependent mechanism. Am. J. Physiol. Cell Physiol. 283, 273–281.
Schapira, A. H. (2008). Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 7, 97–109.
Schapira, A. H., Cooper, J. M., Dexter, D., Jenner, P., Clark, J. B., and Marsden, C. D. (1989). Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269.
Schild, L., Huppelsberg, J., Kahlert, S., Keilhoff, G., and Reiser, G. (2003). Brain mitochondria are primed by moderate Ca2+rise upon hypoxia/reoxygenation for functional breakdown and morphological disintegration. J. Biol. Chem. 278, 25454–25460.
Schinzel, A. C., Takeuchi, O., Huang, Z., Fisher, J. K., Zhou, Z., Rubens, J., Hetz, C., Danial, N. N., Moskowitz, M. A., and Korsmeyer, S. J. (2005). Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 102, 12005–12010.
Selkoe, D. J. (2001). Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta- protein. J. Alzheimers Dis. 3, 75–80.
Sherer, T. B., Richardson, J. R., Testa, C. M., Seo, B. B., Panov, A. V., Yagi, T., Matsuno-Yagi, A., Miller, G. W., and Greenamyre, J. T. (2007). Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J. Neurochem. 100, 1469–1479.
Shimizu, K., Lacza, Z., Rajapakse, N., Horiguchi, T., Snipes, J., and Busija, D. W. (2002). MitoKATP opener, diazoxide, reduces neuronal damage after middle cerebral artery occlusion in the rat. Am. J. Physiol. 283, 1005–1011.
Siesjö, B. K., Katsura, K. I., Zhao, Q., Folbergrova, J., Pahlmark, K., Siesjö, P., and Smith, M. L. (1995). Mechanisms of secondary brain damage in global and focal ischemia: a speculative synthesis. J. Neurotrauma 12, 943–956.
Silver, I. A., and Erecinska, M. (1992). Ion homeostasis in rat brain in vivo: intra- and extracellular [Ca2+] and [H+] in the hippocampus during recovery from short-term, transient ischemia. J. Cereb. Blood Flow Metab. 12, 759–772.
Simerabet, M., Robin, E., Aristi, I., Adamczyk, S., Tavernier, B., Vallet, B., Bordet, R., and Lebuffe, G. (2008). Preconditioning by an in situ administration of hydrogen peroxide: involvement of reactive oxygen species and mitochondrial ATP-dependent potassium channel in a cerebral ischemia-reperfusion model. Brain Res. 1240, 177–184.
Simpkins, J. W., Yi, K. D., Yang, S., and Dykens, J. A. (2009). Mitochondrial mechanisms of estrogen neuroprotection. Biochim. Biophys. Acta (in press).
Sims, N. R. (1991). Selective impairment of respiration in mitochondria isolated from brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 56, 1835–1844.
Sims, N. R., and Pulsinelli, W. A. (1987). Altered mitochondrial respiration in selectively vulnerable brain subregions following transient forebrain ischemia in the rat. J. Neurochem. 49, 1367–1374.
Smith, M. A., Perry, G., Richey, P. L., Sayre, L. M., Anderson, V. E., Beal, M. F., and Kowall, N. (1996). Oxidative damage in Alzheimer’s. Nature 382, 120–121.
Soane, L., Kahraman, S., Kristian, T., and Fiskum, G. (2007). Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. J. Neurosci. Res. 85, 3407–3415.
Song, D. D., Shults, C. W., Sisk, A., Rockenstein, E., and Masliah, E. (2004). Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp. Neurol. 186, 158–172.
Suleiman, M. S., Halestrap, A. P., and Griffiths, E. J. (2001). Mitochondria: a target for myocardial protection. Pharmacol. Ther. 89, 29–46.
Sultana, R., Perluigi, M., and Butterfield, D. A. (2006). Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid. Redox Signal. 8, 2021–2037.
Swerdlow, R. H., Parks, J. K., Miller, S. W., Tuttle, J. B., Trimmer, P. A., Sheehan, J. P., Bennett, J.P. Jr., Davis, R. E., and Parker, W. D. Jr. (1996). Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann. Neurol. 40, 663–671.
Szewczyk, A., and Wojtczak, L. (2002). Mitochondria as a pharmacological target. Pharmacol. Rev. 54, 101–127.
Tai, K. K., McCrossan, Z. A., and Abbott, G. W. (2003). Activation of mitochondrial ATP-sensitive potassium channels increases cell viability against rotenone-induced cell death. J. Neurochem. 84, 1193–1200.
Tai, K. K., and Truong DD. (2002). Activation of adenosine triphosphate-sensitive potassium channels confers protection against rotenone-induced cell death: therapeutic implications for Parkinson’s disease. J. Neurosci. Res. 69, 559–566.
Takuma, K., Yao, J., Huang, J., Xu, H., Chen, X., Luddy, J., Trillat, A. C., Stern, D. M., Arancio, O., and Yan, S. S. (2005). ABAD enhances Abeta-induced cell stress via mitochondrial dysfunction. FASEB J. 19, 597–598.
Tang, X. Q., Chen, J., Tang, E. H., Feng, J. Q., and Chen, P. X. (2005a). Hydrogen peroxide preconditioning protects PC12 cells against apoptosis induced by oxidative stress. Sheng Li Xue Bao 57, 211–216.
Tang, X. Q., Feng, J. Q., Chen, J., Chen, P. X., Zhi, J. L., Cui, Y., Guo, R. X., and Yu, H. M. (2005b). Protection of oxidative preconditioning against apoptosis induced by H2O2 in PC12 cells: mechanisms via MMP, ROS, and Bcl-2. Brain Res. 1057, 57–64.
Taylor, D. J., Krige, D., Barnes, P. R., Kemp, G. J., Carroll, M. T., Mann, V. M., Cooper, J. M., Marsden, C. D., and Schapira, A. H. (1994). A 31P magnetic resonance spectroscopy study of mitochondrial function in skeletal muscle of patients with Parkinson’s disease. J. Neurol. Sci. 125, 77–81.
Trimmer, P. A., Borland, M. K., Keeney, P. M., Bennett, J.P. Jr., and Parker, W. D. Jr. (2004). Parkinson’s disease transgenic mitochondrial cybrids generate Lewy inclusion bodies. J. Neurochem. 88, 800–812.
Tsujimoto, Y., and Shimizu, S. (2007). Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12, 835–840.
Valla, J., Schneider, L., Niedzielko, T., Coon, K. D., Caselli, R., Sabbagh, M. N., Ahern, G. L., Baxter, L., Alexander, G., Walker, D. G., and Reiman, E. M. (2006). Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion 6, 323–330.
Vanden Hoek, T. L., Becker, L. B., Shao, Z., Li, C., and Schumacker, P. T. (1998). Reactive oxygen species released from mitochondria during brief hypoxia induce preconditioning in cardiomyocytes. J. Biol. Chem. 273, 18092–18098.
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962.
Wang, X., Su, B., Fujioka, H., and Zhu, X. (2008a). Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 173, 470–482.
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., Casadesus, G., and Zhu, X. (2008b). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 19318–19323.
Wang, X., Su, B., Zheng, L., Perry, G., Smith, M. A., and Zhu, X. (2009a). The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J. Neurochem. 109, 153–159.
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., and Zhu, X. (2009b). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103.
Wiegand, F., Liao, W., Busch, C., Castell, S., Knapp, F., Lindauer, U., Megow, D., Meisel, A., Redetzky, A., Ruscher, K., Trendelenburg, G., Victorov, I., Riepe, M., Diener, H. C., and Dirnagl, U. (1999). Respiratory chain inhibition induces tolerance to focal cerebral ischemia. J. Cereb. Blood Flow Metab. 19, 1229–1237.
Wu, L., Shen, F., Lin, L., Zhang, X., Bruce, I. C., and Xia, Q. (2006). The neuroprotection conferred by activating the mitochondrial ATP-sensitive K+ channel is mediated by inhibiting the mitochondrial permeability transition pore. Neurosci. Lett. 402, 184–189.
Xie, J., Duan, L., Qian, X., Huang, X., Ding, J., and Hu, G. (2009). K(ATP) channel openers protect mesencephalic neurons against MPP(+)-induced cytotoxicity via inhibition of ROS production. J. Neurosci. Res. 88, 428–437.
Yan, S. D., and Stern, D. M. (2005). Mitochondrial dysfunction and Alzheimer’s disease: role of amyloid-beta peptide alcohol dehydrogenase (ABAD). Int. J. Exp. Pathol. 86, 161–171.
Yang, Y., Liu, X., Long, Y., Wang, F., Ding, J. H., Liu, S. Y., Sun, Y. H., Yao, H. H., Wang, H., Wu, J., and Hu, G. (2006). Activation of mitochondrial ATP-sensitive potassium channels improves rotenone-related motor and neurochemical alterations in rats. Int. J. Neuropsychopharmacol. 9, 51–61.
Yao, Z., and Gross, G. J. (1993). Role of nitric oxide, muscarinic receptors, and the ATP-sensitive K+ channel in mediating the effects of acetylcholine to mimic preconditioning in dogs. Circ. Res. 73, 1193–1201.
Yoshino, H., Nakagawa-Hattori, Y., Kondo, T., and Mizuno, Y. (1992). Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect. 4, 27–34.
Zaidan, E., and Sims, N. R. (1994). The calcium content of mitochondria from brain subregions following short-term forebrain ischemia and recirculation in the rat. J. Neurochem. 63, 1812–1819.
Zhang, D. X., and Gutterman, D. D. (2007). Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 292, 2023–2031.
Keywords: mitochondria, mitoKATP channels, neurodegeneration, neuroprotection, preconditioning, reactive oxygen species
Citation: Correia SC, Carvalho C, Cardoso S, Santos RX, Santos MS, Oliveira CR, Perry G, Zhu X, Smith MA and Moreira PI (2010) Mitochondrial preconditioning: a potential neuroprotective strategy. Front. Ag. Neurosci. 2:138. doi: 10.3389/fnagi.2010.00138
Received: 29 December 2009;
Paper pending published: 14 July 2010;
Accepted: 11 August 2010;
Published online: 26 August 2010
Edited by:
Massimo Tabaton, University of Genoa, ItalyReviewed by:
Russell H. Swerdlow, University of Kansas Medical Center, USAMassimo Tabaton, University of Genoa, Italy
Copyright: © 2010 Correia, Carvalho, Cardoso, Santos, Santos, Oliveira, Perry, Zhu, Smith and Moreira. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Paula I. Moreira, Center for Neuroscience and Cell Biology, Institute of Physiology – Faculty of Medicine, University of Coimbra, 3000-354 Coimbra, Portugal. e-mail:dmVudGFAY2kudWMucHQ=;cGlzbW9yZWlyYUBnbWFpbC5jb20=