 Julia Hernandez-Rapp1,2,3
Julia Hernandez-Rapp1,2,3 Séverine Martin-Lannerée1,2
Séverine Martin-Lannerée1,2 Théo Z. Hirsch1,2
Théo Z. Hirsch1,2 Jean-Marie Launay4,5
Jean-Marie Launay4,5 Sophie Mouillet-Richard1,2*
Sophie Mouillet-Richard1,2*- 1INSERM UMR-S1124, Paris, France
- 2Sorbonne Paris Cité, UMR-S1124, Université Paris Descartes, Paris, France
- 3Université Paris Sud 11, ED419 Biosigne, Orsay, France
- 4AP-HP Service de Biochimie, Fondation FondaMental, INSERM U942 Hôpital Lariboisière, Paris, France
- 5Pharma Research Department, F. Hoffmann-La-Roche Ltd., Basel, Switzerland
The cellular prion protein PrPc is the normal counterpart of the scrapie prion protein PrPSc, the main component of the infectious agent of transmissible spongiform encephalopathies. The recent discovery that PrPc can serve as a receptor for the amyloid beta (Aβ) peptide and relay its neurotoxicity is sparking renewed interest on this protein and its involvement in signal transduction processes. Disease-associated PrPSc shares with Aβ the ability to hijack PrPc-dependent signaling cascades, and thereby instigate pathogenic events. Among these is an impairment of Aβ clearance, uncovered in prion-infected neuronal cells. These findings add another facet to the intricate interplay between PrPc and Aβ. Here, we summarize the connection between PrP-mediated signaling and Aβ clearance and discuss its pathological implications.
Introduction
Up to recent years, commonalities between the amyloid precursor protein (APP) and the cellular prion protein PrPc were considered to mainly reside in their capacity to give rise to aggregation-prone proteins, amyloid beta (Aβ) and PrPSc (standing for scrapie isoform of the prion protein), both involved in neurodegenerative disorders, Alzheimer’s disease (AD) and transmissible spongiform encephalopathies (TSEs), respectively (Haass and Selkoe, 2007; Aguzzi and Calella, 2009). These two diseases share neuropathological features, including synaptic damage, neuronal loss and astrogliosis (Reiniger et al., 2011). As for AD, human prion diseases may have a genetic origin, while most cases are sporadic (Aguzzi and Calella, 2009). Sporadic human prion diseases are thought to arise from the spontaneous conformational conversion of PrPc into its pathogenic PrPSc counterpart (Aguzzi and Calella, 2009). PrPSc has a propensity to aggregate, form amyloid-like structures and can act as a seed to transmit its aberrant conformation to native PrPc molecules (Aguzzi and Calella, 2009). Such template-directed misfolding is also now established in the case of Aβ species [(Jucker and Walker, 2013) for review]. Like Aβ (Langer et al., 2011), much evidence suggests that PrPSc-associated toxicity is imparted by small oligomers (Silveira et al., 2005). This toxicity is assumed to be driven by the subversion of the normal function of PrPc (Harris and True, 2006), which now also emerges as a key event in Aβ-induced neuronal damage (Lauren et al., 2009). The discovery that PrPc may serve as a receptor for Aβ is calling for a better understanding of the role played by PrPc in neurons. The relationship between Aβ and PrPc actually extends beyond that of a ligand-receptor connection, since the prion-induced subversion of PrPc-dependent signaling causes impaired Aβ clearance (Pradines et al., 2013). In this review, we summarize the recent advances focusing on the Aβ-PrP duo and discuss the ensuing challenges.
The Basics of PrPc
Understanding the interplay between Aβ and PrPc necessitates some focus on the latter protein. PrPc is encoded by a unique gene, Prnp, whose open reading frame is contained within a single exon (Aguzzi and Calella, 2009). While ubiquitous, this protein is most abundantly expressed in neurons (Linden et al., 2008). It is located at the outer leaflet of the plasma membrane, to which it is attached through a glycosyl-phosphatidylinositol (GPI) moiety. PrPc contains two potentially glycosylated aspargine residues, at the origin of a great diversity of isoforms (Ermonval et al., 2003). It can also undergo three types of proteolytical cleavages. The best studied are a so-called alpha-cleavage at position 111/112, yielding a N-terminal N1 fragment and a C-terminal C1 fragment, and a beta-cleavage in its N-terminal region giving rise to N2 and C2 fragments [reviewed in (Checler and Vincent, 2002), Figure 1]. The third cleavage leads to the release of a full-length anchorless isoform of PrPc through the action of the metalloprotease ADAM10 (Altmeppen et al., 2011; Figure 1). Of note, similarly to APP processing, the alpha-cleavage of PrPc occurs within a region bearing toxicity (residues 106 to 126), and may involve the TNF-alpha converting enzyme (TACE) metalloprotease (Checler and Vincent, 2002), although the nature of the proteases driving this alpha-cleavage is subject to controversy (Altmeppen et al., 2011; Beland et al., 2012). PrPc thus exists under a plethora of isoforms, whose distribution may vary in distinct brain regions, cell types or even subcellular compartments and whose respective functions remain to be thoroughly resolved (Linden et al., 2008). Clues as to PrPc normal function were greatly anticipated from the development of PrP-null mice. However, these mice are viable, develop normally and do not suffer from major abnormalities (Steele et al., 2007). Subtle behavioral and cognitive deficits were subsequently reported in PrP-deficient mice (Linden et al., 2008), in addition to an increased vulnerability to various types of stresses (Resenberger et al., 2011b). Noteworthy, these animals have been instrumental in substantiating the central role exerted by PrPc in the development of prion diseases, since the knockout of the Prnp gene confers resistance to prion infection (Bueler et al., 1993). Further, post-infection knockdown of PrPc in neurons is sufficient to counteract the progression of prion neuropathogenesis (Mallucci et al., 2003). Finally, transgenic mice expressing an anchorless variant of PrPc (ΔGPI-PrP) were found to efficiently replicate prions upon infection, while not showing any sign of neurodegeneration (Chesebro et al., 2005). Overall, it is now quite well established that neuronal, GPI-anchored PrPc serves as a relay of PrPSc-induced neuropathogenesis. On this basis, understanding the normal function held by PrPc in neurons appears as a necessary step to grasp how it is corrupted by its pathogenic counterpart, PrPSc. Actually, this notion now also applies to Aβ-related pathogenesis, as will be emphasized below.
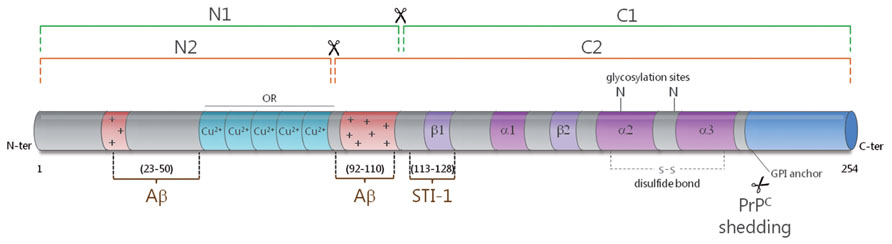
FIGURE 1. Schematic representation of the secondary structure of PrPc. The alpha-cleavage (green) occurs at position 111/112 and generates the N1 and C1 fragments. The beta-cleavage (orange) occurs in the vicinity of octapeptide repeats (ORs), which bind copper ions, and generates the N2 and C2 fragments. PrPc can also be shed from the plasma membrane through the action of ADAM10. The protective factor STI-1 binds to PrPc at amino acids 113–128. Two main binding sites have been mapped for Aβ to the very N-terminus (AA 23–50) and near the alpha-cleavage site at amino acids 92–110. The alpha helices (α) and beta sheets (β) are shown in purple and the polybasic regions (+) are shown in red.
PrPc-Dependent Signaling in Neurons: Findings and Implications
The involvement of PrPc in signal transduction was initially suspected from its cell surface location, as well as its enrichment in lipid rafts (Lewis and Hooper, 2011), which allow the spatial segregation of cell signaling components (Parton and del Pozo, 2013). Using a neuronal cell line (1C11) endowed with the capacity to differentiate into either serotonergic (1C115-HT) or noradrenergic (1C11NE) neurons, we demonstrated that PrPc has the capacity to trigger the activation of the Fyn kinase in neuronal cells (Mouillet-Richard et al., 2000). This signaling cascade is exclusively imparted by PrPc molecules located on the neurites of differentiated cells and is relayed by the scaffold protein caveolin-1 (Mouillet-Richard et al., 2000). Downstream from Fyn, PrPc can mobilize the reactive oxygen species (ROS) generating enzyme NADPH oxidase, and the transcription factors ERK1/2, cAMP response element-binding protein (CREB), Egr-1 and c-Fos (Schneider et al., 2003; Pradines et al., 2008). From a functional point of view, the PrPc-Fyn coupling was shown to mediate neural cell adhesion molecule (NCAM)-dependent neurite outgrowth (Santuccione et al., 2005) and to control calcium influxes in hippocampal neurons (Krebs et al., 2007). As for NADPH oxidase-derived ROS, they can promote the catalytic activation of TACE, subsequent TNFα shedding and bioamine catabolism, thus exerting a neuromodulatory function (Pradines et al., 2009). The demonstration that PrPc could serve as a receptor for the chaperone protein STI-1 (Zanata et al., 2002) further set the stage for the identification of various cell signaling pathways involved in neuroprotection (Chiarini et al., 2002; Lopes et al., 2005), neurite outgrowth (Lopes et al., 2005), and/or memory consolidation (Coitinho et al., 2007). Interestingly, the contribution of PrPc to some of these processes may involve additional partners, such as laminin (Coitinho et al., 2006; Beraldo et al., 2011; Santos et al., 2013). The PrPc-STI-1 interaction was also reported to sustain neuronal protein synthesis via the mTOR pathway (Roffe et al., 2010). Finally, multiple connections have been depicted between PrPc and neurotransmitter signaling including crosstalk with serotonergic (Mouillet-Richard et al., 2005), nicotinic (Beraldo et al., 2010) and mostly glutamatergic receptors, whether metabotropic (Beraldo et al., 2011) or ionotropic (Stys et al., 2012). Collectively, there is now compelling evidence for the involvement of PrPc in multiple signal transduction cascades, whose deviation may drastically impact on neuronal function and activity.
Pathogenic Prions Deviate PrPc Signaling
While it is now acknowledged that PrPc is mandatory for the neurotoxic action of PrPSc, our current understanding of how prions corrupt the physiological function of PrPc is still far from complete (Harris and True, 2006). Achieving this goal is all the more challenging since PrPc encompasses a great variety of isoforms, which may be associated with distinct partners and sustain promiscuous functions. In addition, the repertoire of PrPc species eligible for de novo conversion into PrPSc may vary according to the prion strain and the cell type considered (Ermonval et al., 2003). Despite these hurdles, the development of in vitro modelsof prion infection has allowed to shed some light on this issue. Taking advantage of thecapacity of the 1C11 cell line to replicate various prion strains (Mouillet-Richard et al., 2008), we recently reported on the constitutive activation of signaling targets normally coupled with PrPc in chronically infected cells (Pradines et al., 2013). In these cells as well as in prion-infected neurospheres, prion accumulation is associated with increased activities of Fyn, ERK1/2 and CREB (Figure 2). Another consequence of prion infection is the recruitment of the stress-associated kinases p38 and JNK, as a result of unbalanced ROS production. Downstream from CREB, we documented that prion-infected cells exhibit a reduced activity of the matrix metalloprotease MMP-9. This observation led us to delineate an impact on the clearance of the Aβ peptide, as will be discussed below. Collectively, these findings support the view that PrPSc exerts a toxic gain of PrPc function. In line with this “gain of function” scheme, a central role of the NADPH oxidase NOX2 subunit in prion-induced neuronal damage was established using cerebellar organotypic culture slices (Falsig et al., 2012). Notwithstanding, some PrPc functions may conversely be disrupted within an infected context. One such example is the loss of TACE activity, which renders cells highly sensitive to TNFα-induced cell death (Pietri et al., 2013). The “loss of function” hypothesis accommodates well with the neuroprotective activity ascribed to PrPc (Resenberger et al., 2011b). Notably, the loss of PrPc protective function within an infected context may relate to the inability of PrPSc to undergo proteolytic processing at position 111/112 (McMahon, 2012), and thereby generate the N1 fragment, endowed with neuroprotective activity (Guillot-Sestier and Checler, 2012). Most likely, prion-associated neuropathogenesis involves both gain and loss of function events, that may altogether promote synaptic dysfunction, oxidative stress, loss of neuronal homeostasis and ultimately neurodegeneration (Kovacs and Budka, 2008).
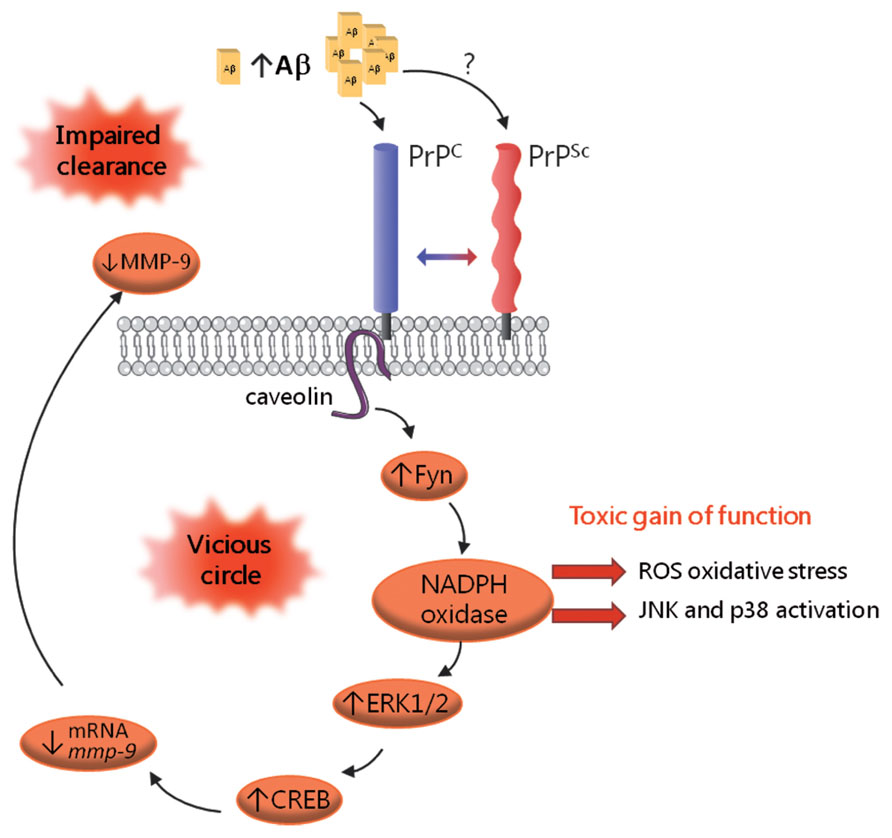
FIGURE 2. Schematic representation of the corruption of PrPc-mediated signaling in prion-infected cells. PrPSc accumulation is associated with the constitutive recruitment of various effectors of PrPc, including the Fyn kinase, the MAP kinases ERK1/2 and the CREB transcription factor. Prion-infected cells also exhibit recruitment of the stress-associated kinases p38 and JNK, which mirrors oxidative stress conditions. A downstream event in this cascade is the drastic reduction in MMP-9 mRNAs and activity, which, in turn, causes an impairment in Aβ clearance, leading to Aβ accumulation. Aβ oligomers would bind PrPc and possibly PrPSc and further fuel the activation of this signaling cascade, thereby sustaining a vicious circle.
PrPc Transduces Aβ Toxicity
The first demonstration that PrPc can bind Aβ came from the Strittmatter group in 2009, via an unbiased screening for receptors that could bind Aβ42 oligomers in the form of Aβ-derived diffusible ligands (ADDLs; Lauren et al., 2009). Lauren et al. (2009) mapped the interaction site to residues 92-110 in PrPc, i.e., the extremity of the N1 fragment (Figure 1), and showed that PrPc supports about 50% of Aβ binding, in line with the multiplicity of Aβ receptors (Benilova and De Strooper, 2013). In that study, the impairment of synaptic plasticity fostered by ADDL was shown to be PrPc-dependent. While the involvement of PrPc in Aβ-induced neurotoxicity was initially subject to controversy (Balducci et al., 2010; Calella et al., 2010; Kessels et al., 2010; Cisse et al., 2011), possibly as a result of distinct experimental paradigms, notably in the preparation of Aβ mixtures (Nicoll et al., 2013), all groups agreed that PrPc has very good affinity for Aβ (within the nanomolar range). Over the past three years, the contribution of PrPc as a relay of Aβ-dependent pathogenic effects has been corroborated and refined by several studies. For instance, targeting PrPc using neutralizing antibodies was shown to counteract the Aβ-mediated impairment in synaptic plasticity (Barry et al., 2011; Freir et al., 2011; Kudo et al., 2012). Molecular analyses have further revealed that Aβ oligomers crosslink PrPc to induce synaptic damage (Bate and Williams, 2011). As for PrPSc, Aβ-induced toxic signaling via PrPc requires its GPI-anchor, as well as lipid raft integrity (Resenberger et al., 2011a; Um et al., 2012; Rushworth et al., 2013). Besides, Aβ was shown to enhance the clustering of PrPc at the cell surface of neuronal cells (Caetano et al., 2011). Taken together, these studies argue that Aβ oligomers amplify the duration or strength of PrPc signaling. This scenario is also supported by several reports that have emphasized the corruption of the PrPc-Fyn signaling cascade by Aβ (Larson et al., 2012; Um et al., 2012, 2013). Um et al. (2013) were able to relate the Aβ-dependent subversion of the PrPc coupling to Fyn to overactivation of the mGluR5 or NMDA receptor (Um et al., 2012), the latter observation being recapitulated in various other studies (Resenberger et al., 2011a; You et al., 2012). Taking a step further, Larson et al. (2012) demonstrated that the PrPc-Fyn complex promotes tau hyperphosphorylation in response to oligomeric Aβ, thus providing some molecular explanation for the well-established role of Fyn as a mediator of Aβ/tau-associated toxicity (Ittner and Gotz, 2011; Roberson et al., 2011). Whether the recruitment of the PrPc-Fyn complex by Aβ impacts on additional signaling effectors such as NADPH oxidase, similarly to PrPSc (Pradines et al., 2013), deserves further investigation. Finally, the recent demonstration that the binding of Aβ to PrPc and the ensuing toxic events can be efficiently inhibited by the PrPc alternate ligand STI-1 is providing a tangible avenue to disrupt the deleterious Aβ-PrPc interaction (Ostapchenko et al., 2013).
Multifaceted Control of PrPc on Aβ Production and Availability
Aβ is generated from APP through the amyloidogenic processing pathway, which involves the beta-secretase BACE1 and occurs in a mutually exclusive fashion with the non-amyloidogenic pathway largely controlled by the alpha-secretase TACE (Checler and Vincent, 2002). In 2007, the Hooper group reported that PrPc decreases the amyloidogenic processing of APP, thereby decreasing Aβ levels, a property that is lost in the context of prion infection (Parkin et al., 2007). Griffiths et al. (2011) went on to show that PrPc interacts with BACE1 and controls its subcellular compartmentation. Of note, while this spatial control is protective toward the cleavage of wild-type APP, it does not prevent BACE1 from processing the Swedish mutant form of APP (Griffiths et al., 2011). Adding yet another layer of complexity to the picture, PrPc also regulates the activity of TACE, a function that is deviated in an Alzheimer’s context and causes an imbalance of APP cleavage toward the amyloidogenic pathway (Pietri et al., 2013). PrPc also regulates the availability of Aβ at several levels. For instance, PrPc could favor the accumulation of Aβ in the brain by contributing to its transcytosis across the blood brain barrier (Pflanzner et al., 2012). Finally, in accordance with the binding of Aβ to various sites located within the N-terminal region of PrPc (Chen et al., 2010; see Figure 1) and with the physiological processing of PrPc at position 111/112, the resulting PrPc-derived N1 fragment was reported to trap Aβ and thereby exert a protective action against Aβ-induced cell death (Beland et al., 2012; Guillot-Sestier et al., 2012).
Hijacking PrPc Signaling by Pathogenic Prions Causes Impaired Aβ Clearance
As mentioned above, we documented that the subversion of PrPc signaling by PrPSc in prion-infected cells leads to decreased activity of the MMP-9 metalloprotease (Pradines et al., 2013; Figure 2). MMP-9 exhibits alpha-secretase activity (Fragkouli et al., 2012) and also features among the Aβ degrading enzymes [(De Strooper, 2010) for review]. On this basis, we examined the potential outcome of the prion-dependent reduction in MMP-9 activity on Aβ metabolism. By assessing the production and elimination rates of Aβ peptides in prion-infected 1C11 cells, we substantiated an imbalance in the clearance of Aβ under prion infection, which was canceled after knockdown of the Fyn kinase (Pradines et al., 2013; Figure 2). We further observed a similarly impaired Aβ clearance in uninfected cells upon inhibition of MMP-9, highlighting the involvement of MMP-9 in this process. In agreement, we found increased levels of Aβ in the supernatants of prion-infected 1C11 cells or MMP-9 inhibitor-treated non-infected cells. Besides, we monitored an increase in the cerebrospinal fluid (CSF) levels of Aβ in mice treated with a MMP-9 inhibitor as well as in mice inoculated with prion-infected 1C11 cells. Altogether, these results delineate a reciprocal connection between PrP and Aβ that may sustain a pathogenic loop. Indeed, by promoting an accumulation of Aβ in the extracellular space, prion infection would fuel the recruitment of Fyn and its downstream targets through the interaction of Aβ with PrP, whether in its cellular or aggregated form (Chen et al., 2010; Zou et al., 2011). Whether the subversion of PrPc dependent signal transduction by Aβ in Alzheimer’s disease also negatively impacts on MMP-9 activity and thereby impinges on Aβ clearance remains to be investigated. Should it be confirmed, disrupting this vicious circle would appear as a promising avenue to combat both prion and Alzheimer’s neuropathogenesis.
Concluding Remarks
As outlined above, tremendous progress has been achieved in our understanding of the Aβ-PrPc interplay in very recent years. The bidirectional relationship between these two proteins in signal transduction cascades may account for some -once puzzling- observations that deposits of Aβ and PrP may co-occur in patients with mixed AD and CJD clinical manifestation (Muramoto et al., 1992; Hainfellner et al., 1998; Debatin et al., 2008). Beyond the various PrPc-Aβ connections summarized above, the possibility that the interaction between the two proteins may favor their aggregation in a “cross-seeding” fashion is beginning to be explored (Morales et al., 2010). Overall, the more we learn in the field, the more we appraise the complexity of the PrPc-Aβ relationship. The latest advance has further extended the points of convergence in the molecular pathogenic pathways at play in the two diseases, including loss of TACE activity (Pietri et al., 2013) and impairment of the unfolded protein response (Moreno et al., 2012; Ma et al., 2013). Striking differences however remain between the two disorders, most notably the infectious properties specific to prions (Jucker and Walker, 2013). The only partial overlap between the two pathologies may originate from the extremely diverse array of PrPc and Aβ species as well as their promiscuous partners (Linden et al., 2008; Benilova et al., 2012). Notwithstanding, the central role ascribed to PrPc in defined Aβ-related toxic pathways opens new avenues for therapeutic intervention in AD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to colleagues in the field whose work we were unable to cite owing to space limitations. We acknowledge financial support from the ANR, INSERM, as well as the Region Ile de France (DIM-Stem Pôle).
References
Aguzzi, A., and Calella, A. M. (2009). Prions: protein aggregation and infectious diseases. Physiol. Rev. 89, 1105–1152. doi: 10.1152/physrev.00006.2009
Altmeppen, H. C., Prox, J., Puig, B., Kluth, M. A., Bernreuther, C., Thurm, D., et al. (2011). Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol. Neurodegener. 6, 36. doi: 10.1186/1750-1326-6-36
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 2295–2300. doi: 10.1073/pnas.0911829107
Barry, A. E., Klyubin, I., Mc Donald, J. M., Mably, A. J., Farrell, M. A., Scott, M., et al. (2011). Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 31, 7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011
Bate, C., and Williams, A. (2011). Amyloid-beta-induced synapse damage is mediated via cross-linkage of cellular prion proteins. J. Biol. Chem. 286, 37955–37963. doi: 10.1074/jbc.M111.248724
Beland, M., Motard, J., Barbarin, A., and Roucou, X. (2012). PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 32, 13255–13263. doi: 10.1523/JNEUROSCI.2236-12.2012
Benilova, I., and De Strooper, B. (2013). Neuroscience. Promiscuous Alzheimer’s amyloid: yet another partner. Science 341, 1354–1355. doi: 10.1126/science.1244166
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Beraldo, F. H., Arantes, C. P., Santos, T. G., Machado, C. F., Roffe, M., Hajj, G. N., et al. (2011). Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 25, 265–279. doi: 10.1096/fj.10-161653
Beraldo, F. H., Arantes, C. P., Santos, T. G., Queiroz, N. G., Young, K., Rylett, R. J., et al. (2010). Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 285, 36542–36550. doi: 10.1074/jbc.M110.157263
Bueler, H., Aguzzi, A., Sailer, A., Greiner, R. A., Autenried, P., Aguet, M., et al. (1993). Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347. doi: 10.1016/0092-8674(93)90360-3
Caetano, F. A., Beraldo, F. H., Hajj, G. N., Guimaraes, A. L., Jurgensen, S., Wasilewska-Sampaio, A. P., et al. (2011). Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 117, 538–553. doi: 10.1111/j.1471-4159.2011.07225.x
Calella, A. M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., et al. (2010). Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314. doi: 10.1002/emmm.201000082
Checler, F., and Vincent, B. (2002). Alzheimer’s and prion diseases: distinct pathologies, common proteolytic denominators. Trends Neurosci. 25, 616–620. doi: 10.1016/S0166-2236(02)02263-4
Chen, S., Yadav, S. P., and Surewicz, W. K. (2010). Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role of N-terminal residues. J. Biol. Chem. 285, 26377–26383. doi: 10.1074/jbc.M110.145516
Chesebro, B., Trifilo, M., Race, R., Meade-White, K., Teng, C., Lacasse, R., et al. (2005). Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1435–1439. doi: 10.1126/science.1110837
Chiarini, L. B., Freitas, A. R., Zanata, S. M., Brentani, R. R., Martins, V. R., and Linden, R. (2002). Cellular prion protein transduces neuroprotective signals. EMBO J. 21, 3317–3326. doi: 10.1093/emboj/cdf324
Cisse, M., Halabisky, B., Harris, J., Devidze, N., Dubal, D. B., Sun, B., et al. (2011). Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature 469, 47–52. doi: 10.1038/nature09635
Coitinho, A. S., Freitas, A. R., Lopes, M. H., Hajj, G. N., Roesler, R., Walz, R., et al. (2006). The interaction between prion protein and laminin modulates memory consolidation. Eur. J. Neurosci. 24, 3255–3264. doi: 10.1111/j.1460-9568.2006.05156.x
Coitinho, A. S., Lopes, M. H., Hajj, G. N., Rossato, J. I., Freitas, A. R., Castro, C. C., et al. (2007). Short-term memory formation and long-term memory consolidation are enhanced by cellular prion association to stress-inducible protein 1. Neurobiol. Dis. 26, 282–290. doi: 10.1016/j.nbd.2007.01.005
Debatin, L., Streffer, J., Geissen, M., Matschke, J., Aguzzi, A., and Glatzel, M. (2008). Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener. Dis. 5, 347–354. doi: 10.1159/000121389
De Strooper, B. (2010). Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90, 465–494. doi: 10.1152/physrev.00023.2009
Ermonval, M., Mouillet-Richard, S., Codogno, P., Kellermann, O., and Botti, J. (2003). Evolving views in prion glycosylation: functional and pathological implications. Biochimie 85, 33–45. doi: 10.1016/S0300-9084(03)00040-3
Falsig, J., Sonati, T., Herrmann, U. S., Saban, D., Li, B., Arroyo, K., et al. (2012). Prion pathogenesis is faithfully reproduced in cerebellar organotypic slice cultures. PLoS Pathog. 8:e1002985. doi: 10.1371/journal.ppat.1002985
Fragkouli, A., Papatheodoropoulos, C., Georgopoulos, S., Stamatakis, A., Stylianopoulou, F., Tsilibary, E. C., et al. (2012). Enhanced neuronal plasticity and elevated endogenous sAPPalpha levels in mice over-expressing MMP9. J. Neurochem. 121, 239–251. doi: 10.1111/j.1471-4159.2011.07637.x
Freir, D. B., Nicoll, A. J., Klyubin, I., Panico, S., Mc Donald, J. M., Risse, E., et al. (2011). Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat. Commun. 2, 336. doi: 10.1038/ncomms1341
Griffiths, H. H., Whitehouse, I. J., Baybutt, H., Brown, D., Kellett, K. A., Jackson, C. D., et al. (2011). Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 286, 33489–33500. doi: 10.1074/jbc.M111.278556
Guillot-Sestier, M. V., and Checler, F. (2012). α-Secretase-derived cleavage of cellular prion yields biologically active catabolites with distinct functions. Neurodegener. Dis. 10, 294–297. doi: 10.1159/000333804
Guillot-Sestier, M. V., Sunyach, C., Ferreira, S. T., Marzolo, M. P., Bauer, C., Thevenet, A., et al. (2012). α-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J. Biol. Chem. 287, 5021–5032. doi: 10.1074/jbc.M111.323626
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Hainfellner, J. A., Wanschitz, J., Jellinger, K., Liberski, P. P., Gullotta, F., and Budka, H. (1998). Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 96, 116–122. doi: 10.1007/s004010050870
Harris, D. A., and True, H. L. (2006). New insights into prion structure and toxicity. Neuron 50, 353–357. doi: 10.1016/j.neuron.2006.04.020
Ittner, L. M., and Gotz, J. (2011). Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 12, 65–72. doi: 10.1038/nrn2967
Jucker, M., and Walker, L. C. (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. doi: 10.1038/nature12481
Kessels, H. W., Nguyen, L. N., Nabavi, S., and Malinow, R. (2010). The prion protein as a receptor for amyloid-beta. Nature 466, E3–E4; discussion E4–E5. doi: 10.1038/nature09217
Kovacs, G. G., and Budka, H. (2008). Prion diseases: from protein to cell pathology. Am. J. Pathol. 172, 555–565. doi: 10.2353/ajpath.2008.070442
Krebs, B., Wiebelitz, A., Balitzki-Korte, B., Vassallo, N., Paluch, S., Mitteregger, G., et al. (2007). Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J. Neurochem. 100, 358–367. doi: 10.1111/j.1471-4159.2006.04256.x
Kudo, W., Lee, H. P., Zou, W. Q., Wang, X., Perry, G., Zhu, X., et al. (2012). Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genet. 21, 1138–1144. doi: 10.1093/hmg/ddr542
Langer, F., Eisele, Y. S., Fritschi, S. K., Staufenbiel, M., Walker, L. C., and Jucker, M. (2011). Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J. Neurosci. 31, 14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011
Larson, M., Sherman, M. A., Amar, F., Nuvolone, M., Schneider, J. A., Bennett, D. A., et al. (2012). The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 32, 16857–16871a. doi: 10.1523/JNEUROSCI.1858-12.2012
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Lewis, V., and Hooper, N. M. (2011). The role of lipid rafts in prion protein biology. Front. Biosci. 16:151–168. doi: 10.2741/3681
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Lopes, M. H., Hajj, G. N., Muras, A. G., Mancini, G. L., Castro, R. M., Ribeiro, K. C., et al. (2005). Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 25, 11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Mallucci, G., Dickinson, A., Linehan, J., Klohn, P. C., Brandner, S., and Collinge, J. (2003). Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871–874. doi: 10.1126/science.1090187
McMahon, H. E. (2012). Prion processing: a double-edged sword? Biochem. Soc. Trans. 40, 735–738. doi: 10.1042/BST20120031
Morales, R., Estrada, L. D., Diaz-Espinoza, R., Morales-Scheihing, D., Jara, M. C., Castilla, J., et al. (2010). Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J. Neurosci. 30, 4528–4535. doi: 10.1523/JNEUROSCI.5924-09.2010
Moreno, J. A., Radford, H., Peretti, D., Steinert, J. R., Verity, N., Martin, M. G., et al. (2012). Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485, 507–511. doi: 10.1038/nature11058
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M., et al. (2000). Signal transduction through prion protein. Science 289, 1925–1928. doi: 10.1126/science.289.5486.1925
Mouillet-Richard, S., Nishida, N., Pradines, E., Laude, H., Schneider, B., Feraudet, C., et al. (2008). Prions impair bioaminergic functions through serotonin- or catecholamine-derived neurotoxins in neuronal cells. J. Biol. Chem. 283, 23782–23790.. doi: 10.1074/jbc.M802433200
Mouillet-Richard, S., Pietri, M., Schneider, B., Vidal, C., Mutel, V., Launay, J. M., et al. et al. (2005). Modulation of serotonergic receptor signaling and cross-talk by prion protein. J. Biol. Chem. 280, 4592–4601. doi: 10.1074/jbc.M406199200
Muramoto, T., Kitamoto, T., Koga, H., and Tateishi, J. (1992). The coexistence of Alzheimer’s disease and Creutzfeldt-Jakob disease in a patient with dementia of long duration. Acta Neuropathol. 84, 686–689. doi: 10.1007/BF00227747
Nicoll, A. J., Panico, S., Freir, D. B., Wright, D., Terry, C., Risse, E., et al. (2013). Amyloid-beta nanotubes are associated with prion protein-dependent synaptotoxicity. Nat. Commun. 4, 2416. doi: 10.1038/ncomms3416
Ostapchenko, V. G., Beraldo, F. H., Mohammad, A. H., Xie, Y. F., Hirata, P. H., Magalhaes, A. C., et al. (2013). The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 33, 16552–16564. doi: 10.1523/JNEUROSCI.3214-13.2013
Parkin, E. T., Watt, N. T., Hussain, I., Eckman, E. A., Eckman, C. B., Manson, J. C., et al. (2007). Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 104, 11062–11067. doi: 10.1073/pnas.0609621104
Parton, R. G., and del Pozo, M. A. (2013). Caveolae as plasma membrane sensors, protectors and organizers. Nat. Rev. Mol. Cell Biol. 14, 98–112. doi: 10.1038/nrm3512
Pflanzner, T., Petsch, B., Andre-Dohmen, B., Muller-Schiffmann, A., Tschickardt, S., Weggen, S., et al. (2012). Cellular prion protein participates in amyloid-beta transcytosis across the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 628–632. doi: 10.1038/jcbfm.2012.7
Pietri, M., Dakowski, C., Hannaoui, S., Alleaume-Butaux, A., Hernandez-Rapp, J., Ragagnin, A., et al. (2013). PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat. Med. 19, 1124–1131. doi: 10.1038/nm.3302
Pradines, E., Hernandez-Rapp, J., Villa-Diaz, A., Dakowski, C., Ardila-Osorio, H., Haik, S., et al. (2013). Pathogenic prions deviate PrP(C) signaling in neuronal cells and impair A-beta clearance. Cell Death Dis. 4:e456. doi: 10.1038/cddis.2012.195
Pradines, E., Loubet, D., Mouillet-Richard, S., Manivet, P., Launay, J. M., Kellermann, O., et al. (2009). Cellular prion protein coupling to TACE-dependent TNF-alpha shedding controls neurotransmitter catabolism in neuronal cells. J. Neurochem. 110, 912–923. doi: 10.1111/j.1471-4159.2009.06176.x
Pradines, E., Loubet, D., Schneider, B., Launay, J. M., Kellermann, O., and Mouillet-Richard, S. (2008). CREB-dependent gene regulation by prion protein: impact on MMP-9 and beta-dystroglycan. Cell. Signal. 20, 2050–2058. doi: 10.1016/j.cellsig.2008.07.016
Reiniger, L., Lukic, A., Linehan, J., Rudge, P., Collinge, J., Mead, S., et al. (2011). Tau, prions and Abeta: the triad of neurodegeneration. Acta Neuropathol. 121, 5–20. doi: 10.1007/s00401-010-0691-0
Resenberger, U. K., Harmeier, A., Woerner, A. C., Goodman, J. L., Muller, V., Krishnan, R., et al. (2011a). The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070. doi: 10.1038/emboj.2011.86
Resenberger, U. K., Winklhofer, K. F., and Tatzelt, J. (2011b). Neuroprotective and neurotoxic signaling by the prion protein. Top. Curr. Chem. 305, 101–119. doi: 10.1007/128_2011_160
Roberson, E. D., Halabisky, B., Yoo, J. W., Yao, J., Chin, J., Yan, F., et al. (2011). Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 31, 700–711. doi: 10.1523/JNEUROSCI.4152-10.2011
Roffe, M., Beraldo, F. H., Bester, R., Nunziante, M., Bach, C., Mancini, G., et al. (2010). Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proc. Natl. Acad. Sci. U.S.A. 107, 13147–13152. doi: 10.1073/pnas.1000784107
Rushworth, J. V., Griffiths, H. H., Watt, N. T., and Hooper, N. M. (2013). Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 288, 8935–8951. doi: 10.1074/jbc.M112.400358
Santos, T. G., Beraldo, F. H., Hajj, G. N., Lopes, M. H., Roffe, M., Lupinacci, F. C., et al. (2013). Laminin-gamma1 chain and stress inducible protein 1 synergistically mediate PrPC-dependent axonal growth via Ca2+ mobilization in dorsal root ganglia neurons. J. Neurochem. 124, 210–223. doi: 10.1111/jnc.12091
Santuccione, A., Sytnyk, V., Leshchyns’ka, I., and Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169, 341–354. doi: 10.1083/jcb.200409127
Schneider, B., Mutel, V., Pietri, M., Ermonval, M., Mouillet-Richard, S., and Kellermann, O. (2003). NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. U.S.A. 100, 13326–13331. doi: 10.1073/pnas.2235648100
Silveira, J. R., Raymond, G. J., Hughson, A. G., Race, R. E., Sim, V. L., Hayes, S. F., et al. (2005). The most infectious prion protein particles. Nature 437, 257–261. doi: 10.1038/nature03989
Steele, A. D., Lindquist, S., and Aguzzi, A. (2007). The prion protein knockout mouse: a phenotype under challenge. Prion 1, 83–93.. doi: 10.4161/pri.1.2.4346
Stys, P. K., You, H., and Zamponi, G. W. (2012). Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. J. Physiol. 590, 1357–1368. doi: 10.1113/jphysiol.2011.225276
Um, J. W., Kaufman, A. C., Kostylev, M., Heiss, J. K., Stagi, M., Takahashi, H., et al. (2013). Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. doi: 10.1016/j.neuron.2013.06.036
Um, J. W., Nygaard, H. B., Heiss, J. K., Kostylev, M. A., Stagi, M., Vortmeyer, A., et al. (2012). Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 15, 1227–1235. doi: 10.1038/nn.3178
You, H., Tsutsui, S., Hameed, S., Kannanayakal, T. J., Chen, L., Xia, P., et al. (2012). Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 1737–1742. doi: 10.1073/pnas.1110789109
Zanata, S. M., Lopes, M. H., Mercadante, A. F., Hajj, G. N., Chiarini, L. B., Nomizo, R., et al. (2002). Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 21, 3307–3316. doi: 10.1093/emboj/cdf325
Keywords: cellular prion protein, prion infection, Aβ clearance, signal transduction, Alzheimer’s disease
Citation: Hernandez-Rapp J, Martin-Lannerée S, Hirsch TZ, Launay J-M and Mouillet-Richard S (2014) Hijacking PrPC-dependent signal transduction: when prions impair Aβ clearance. Front. Aging Neurosci. 6:25. doi: 10.3389/fnagi.2014.00025
Received: 28 January 2014; Paper pending published: 06 February 2014;
Accepted: Published online: 28 February 2014.
Edited by:
Robert Marr, Rosalind Franklin University of Medicine and Science, USACopyright © 2014 Hernandez-Rapp, Martin-Lannerée, Hirsch, Launay and Mouillet-Richard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie Mouillet-Richard, INSERM UMR-S1124, Sorbonne Paris Cité, UMR-S1124, Université Paris Descartes, 45, rue des Saints Pères, 75006 Paris, France email:c29waGllLm1vdWlsbGV0LXJpY2hhcmRAcGFyaXNkZXNjYXJ0ZXMuZnI=