 Jan Klohs
Jan Klohs Markus Rudin
Markus Rudin Derya R. Shimshek
Derya R. Shimshek Nicolau Beckmann
Nicolau Beckmann- 1Institute for Biomedical Engineering, University of Zurich and ETH Zurich, Zurich, Switzerland
- 2Neuroscience Center Zurich, University of Zurich and ETH Zurich, Zurich, Switzerland
- 3Institute of Pharmacology and Toxicology, University of Zurich, Zurich, Switzerland
- 4Autoimmunity, Transplantation and Inflammation/Neuroinflammation Department, Novartis Institutes for BioMedical Research, Basel, Switzerland
- 5Analytical Sciences and Imaging, Novartis Institutes for BioMedical Research, Basel, Switzerland
In Alzheimer's disease (AD), vascular pathology may interact with neurodegeneration and thus aggravate cognitive decline. As the relationship between these two processes is poorly understood, research has been increasingly focused on understanding the link between cerebrovascular alterations and AD. This has at last been spurred by the engineering of transgenic animals, which display pathological features of AD and develop cerebral amyloid angiopathy to various degrees. Transgenic models are versatile for investigating the role of amyloid deposition and vascular dysfunction, and for evaluating novel therapeutic concepts. In addition, research has benefited from the development of novel imaging techniques, which are capable of characterizing vascular pathology in vivo. They provide vascular structural read-outs and have the ability to assess the functional consequences of vascular dysfunction as well as to visualize and monitor the molecular processes underlying these pathological alterations. This article focusses on recent in vivo small animal imaging studies addressing vascular aspects related to AD. With the technical advances of imaging modalities such as magnetic resonance, nuclear and microscopic imaging, molecular, functional and structural information related to vascular pathology can now be visualized in vivo in small rodents. Imaging vascular and parenchymal amyloid-β (Aβ) deposition as well as Aβ transport pathways have been shown to be useful to characterize their dynamics and to elucidate their role in the development of cerebral amyloid angiopathy and AD. Structural and functional imaging read-outs have been employed to describe the deleterious affects of Aβ on vessel morphology, hemodynamics and vascular integrity. More recent imaging studies have also addressed how inflammatory processes partake in the pathogenesis of the disease. Moreover, imaging can be pivotal in the search for novel therapies targeting the vasculature.
Introduction to Cerebrovascular Pathology in Alzheimer's Disease
Alzheimer's disease (AD) is the most common form of dementia in elderly individuals. The disease has been classically viewed as the accumulation of amyloid-β (Aβ), generated by proteolytic cleavage of the amyloid precursor protein (APP), in the brain parenchyma (Aβ plaques), leading to Aβ-related neuropathology and loss of cognitive function (Hardy and Selkoe, 2002). Increasing evidence has implicated cerebrovascular dysfunction in the etiology of the disease (for reviews see Thal et al., 2008a,b; Bell and Zlokovic, 2009; Weller et al., 2009; Biffi and Greenberg, 2011). Epidemiological studies indicate a strong overlap between AD pathology and cardiovascular disease, suggesting that they might share common mechanisms and risk factors. Among all cerebrovascular comorbidities in AD, cerebral amyloid angiopathy (CAA) is the most common pathological finding, present in up to 90% of AD patients (Vinters, 1987; Jellinger, 2002). CAA results from the failure to eliminate Aβ from the cerebral vasculature (Weller et al., 2009). Both AD and CAA can lead to pronounced cerebrovascular dysfunction, characterized by impaired neurovascular and metabolic regulation of cerebral blood flow (CBF) and aberrations in vascular morphology and density. In addition, changes in the proteolytic microenvironment and inflammation lead to impairment of blood-brain barrier (BBB) integrity and the occurrence of cerebral microbleeds (CMBs) and intracerebral hemorrhages (Snowdon, 2003; Cordonnier and van der Flier, 2011). It has been suggested that the vascular pathology may mutually interact with neurodegeneration in AD, and thus aggravate cognitive decline, though the relationship between these two processes is poorly understood. Hence, recent research has been increasingly focused on understanding the link between cerebrovascular alterations and AD. This has on one hand been spurred by the engineering of transgenic animals, which display pathological features of AD and develop CAA to various degrees. These models have been proven versatile for elucidating the role of amyloid deposition, vascular dysfunction and for evaluating novel therapeutic concepts. On the other hand, research has benefited from the development of novel imaging techniques, which are capable of characterizing vascular pathology in vivo. They provide vascular structural read-outs and have the ability to assess the functional consequences of vascular dysfunction as well as to visualize and monitor the molecular processes underlying these pathological alterations.
This article focusses on recent in vivo small animal imaging studies addressing vascular aspects related to AD. We first introduce transgenic mouse models of AD displaying CAA and their main characteristics, followed by a summary of the current imaging techniques and discuss their advantages and limitations. The potential of imaging vascular pathology will be illustrated by discussing applications on the visualization of vascular amyloid deposition and amyloid clearance pathways, the assessment of the cerebrovascular architecture to elucidate the dynamics and mechanism of CAA and to understand how amyloid deposition induces vascular remodeling. The use of functional imaging read-outs to monitor the deleterious consequences of amyloid deposition, namely chronic hypoperfusion and reduced hemodynamic response are presented. The role of neurovascular inflammation, loss of BBB as well as CMBs in advanced stages of the disease are then addressed. The fact that CAA may be halted or even reversed is evaluated by a glance on therapeutic studies involving the animal models. Finally, we evaluated how biological lessons learned from these models may be translated into the clinic.
Transgenic Models of Alzheimer's Disease Displaying Cerebral Amyloid Angiopathy
To date, several mouse and rat lines have been genetically engineered to serve as preclinical models for AD. Most of these models have been generated by transgenic overexpression of the gene encoding for the human APP, which leads to progressive accumulation of Aβ and amyloidosis in the brain. Some of these strains develop CAA to various degrees, which allow studying the effect of Aβ accumulation on vascular function. Further strains have been established addressing the other pathological hallmark of AD, neurofibrillary tangles, by expressing different forms of tau (Gotz et al., 1995; Duff et al., 2000; Allen et al., 2002; SantaCruz et al., 2005). Furthermore, mouse models with multiple mutations have also been engineered, e.g., APP/PS1 and 3xTg-AD, to study either the enhancement of amyloid pathology by presenilin mutations or the interaction between amyloid and tau (Blanchard et al., 2003; Oddo et al., 2003a,b). However, most studies utilize strains with a single mutation, which have the advantage of assessing one pathological process at a time without being confounded by the complex pathophysiology of sporadic AD. Transgenic mouse models are very valuable for drug discovery as the pathology usually develops within months (as compared to years or decades in AD patients) and disease pathological stages are well characterized.
Mice overexpressing APP have been utilized as valid models for CAA (summarized in Table 1). These transgenic models use various promoters to drive transgene expression in different genetic backgrounds. Interestingly, it has been shown that neuronal Aβ is the driver for CAA (Calhoun et al., 1999) and an impaired Aβ clearance seems to enhance CAA (Herzig et al., 2004). Early onset CAA is observed in models with multiple autosomal dominant mutations like Thy1-APP751, Tg-SwDI, TgCRND8, whereas late-onset CAA (>9 months of age) is usually detected in mice with expression of mutated APP restricted to one familiar mutation as in Tg2576, PDAPP, APPDutch, APP/London, APP23, or TgAPPArc animals. CAA is observed earlier in mice that additionally carry a presenilin mutation like APPswe/PS1dE9 or Thy1-APP751SLxHMG-PS1M146L.
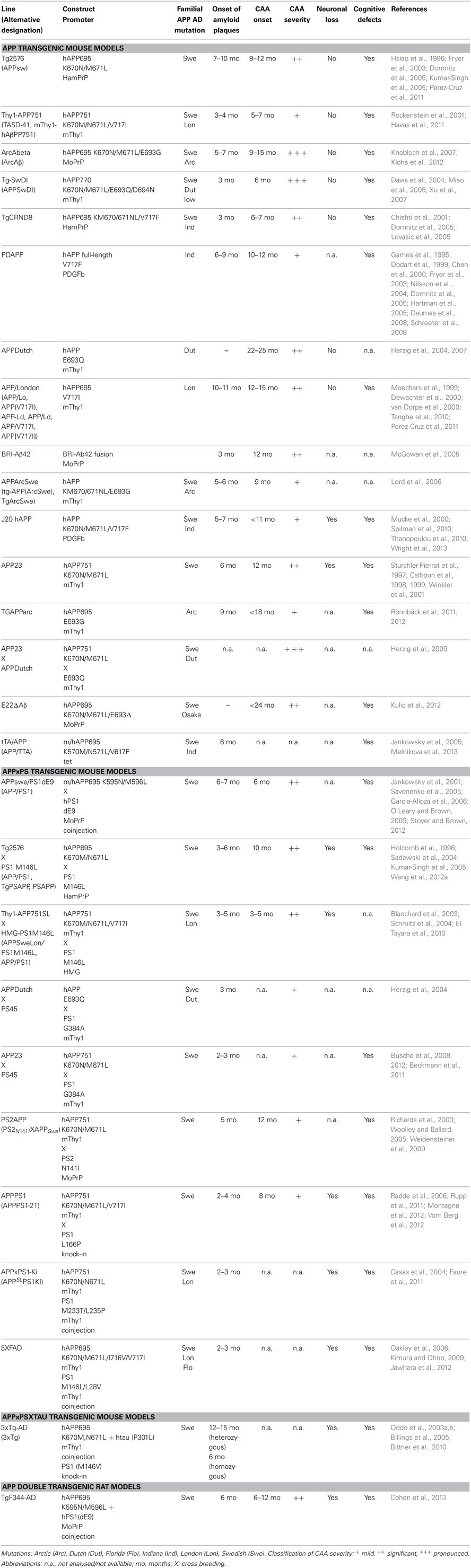
Table 1. Summary of transgenic models used in AD research and their relation to CAA development.
Experimental evidence suggests that total levels of Aβ as well as the ratio of the Aβ40 and Aβ42 peptides (Aβ40/Aβ42), generated by proteolytic cleavage of β- and γ-secretase, are factors determining both onset and the severity of CAA (Herzig et al., 2004, 2007; for reviews see Herzig et al., 2006; Kumar-Singh, 2009). APPDutch animals displaying a very high ratio of Aβ40/Aβ42 develop severe CAA (Herzig et al., 2004), indicating that the majority of Aβ deposited in vascular deposits is Aβ40. Furthermore, APP23xAPPDutch double transgenic animals with an overall increase of Aβ load and a high ratio Aβ40/Aβ42 show enhanced CAA compared to APP23 single transgenic mice. In contrast, APPDutchxPS45 with a lower Aβ40/Aβ42 ratio develop more pronounced parenchymal than vascular deposits (Herzig et al., 2004). Also, autosomal dominant mutations with reduced Aβ40/Aβ42 ratio such as the Indiana mutation in PDAPP and APP/London mice display less pronounced CAA. These observations are in line with findings in hereditary cerebral hemorrhage patients with amyloidosis of the Dutch type. Individuals with this rare autosomal dominant disorder, caused by an APP 693 mutation that leads to recurrent hemorrhagic strokes and dementia, have decreased Aβ42 levels in the brain (Bornebroek et al., 2003). However, contrasting data have been reported for the BRI-Aβ42 animals, where overexpression solely of Aβ42 led to CAA, while overexpression of Aβ40 did not (McGowan et al., 2005). Other reports suggested that Aβ40 can inhibit fibril formation and even inhibit amyloid deposition (Jarrett et al., 1993; Kim et al., 2007). Taken together, most studies indicate that Aβ42 might be essential for the initial amyloid deposition in vessels and that an increase of overall total Aβ as well as of the Aβ40/Aβ42 ratio favors subsequent vascular Aβ load.
In this review, we focus on effects of Aβ on the cerebral vasculature and the consequences thereof. Since the transgenic AD models exhibit quite different levels of CAA vs. neuritic Aβ deposits, they enable studying the effect of these processes on AD pathology.
Imaging Modalities for the Characterization of Cerebral Vasculature
Multimodal imaging offers an impressive number of approaches for characterizing cerebral vasculature from the cellular to the whole organ scale (Table 2). The dimensions of cerebral vessels span a range of 2–3 orders of magnitude with large arteries and veins of dimensions of approximately 1 mm to capillaries with typical diameters of 5–10 μm. Correspondingly, the phenotypic characterization of cerebral vasculature under normal and pathological conditions requires information at multiple length and time scales addressing various aspects of vascular anatomy and function/physiology (Figure 1).
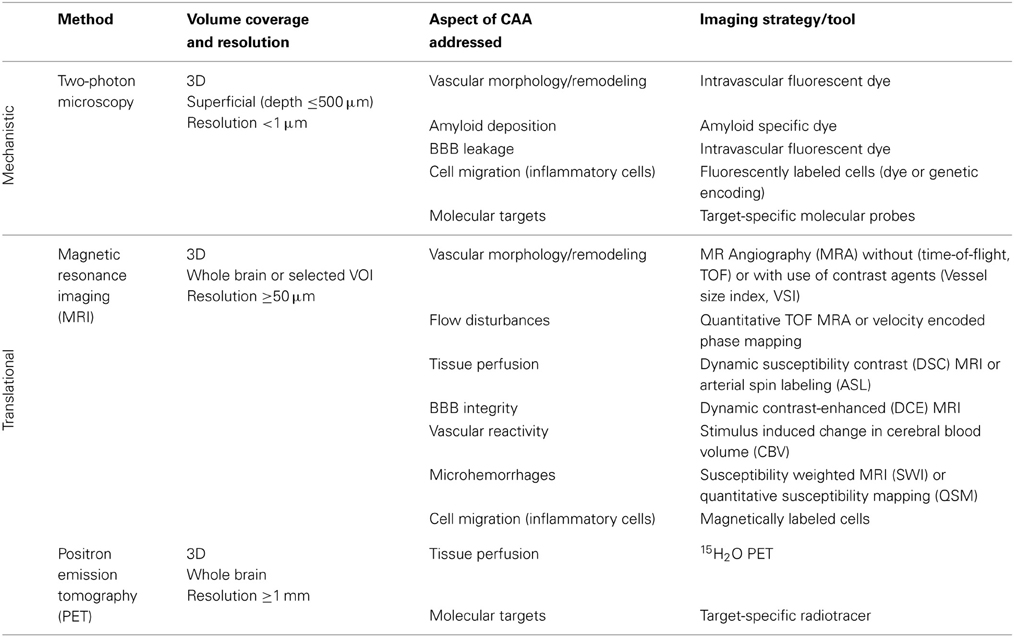
Table 2. In vivo imaging techniques applied to AD models.
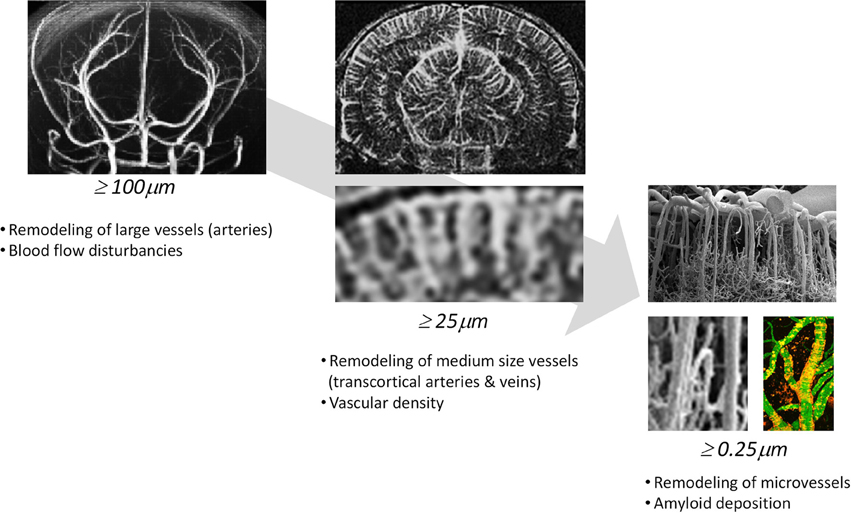
Figure 1. Phenotypic characterization of cerebrovascular structures at various length scales. Time-of-flight magnetic resonance angiography can depict large vessels (≥100μm), contrast-enhanced magnetic resonance angiography can depict medium sized vessels (≥50μm) and two-photon microscopy can visualize microvessels (≥0.25μm).
Light-based microscopy methods such as two-photon microscopy or optical coherence tomography provide exquisite information at submicrometer resolution though they are limited to superficial structures due to light scattering by turbid tissue. The cortex is ideally suited for in vivo microscopy of adult mice. Technically, it involves the preparation of an optical window in anesthetized animals, comprising either a thinned skull region or a sealed craniotomy. For short- and long-term imaging experiments the thinned skull preparation is the preferred method because it is the least invasive to parenchymal tissue (Helm et al., 2009). Recording duration ranges from minutes to months depending on the biological process investigated. Some applications, for instance high-resolution two-photon imaging of extensive cortical areas or micrometerscale structures deep inside the cortex (~250 μm), require the use of an open skull window providing direct access to the brain parenchyma (Holtmaat et al., 2009). To target deeper brain structures like the hippocampus, removal of parts of the cortex have been proven to be feasible (Busche et al., 2012). It should be kept in mind that skull removal may lead to mechanical injuries to the cortical surface or immediate disturbances in local blood perfusion, BBB permeability, and brain homeostasis, while removal of whole brain structures might even lead to damage of brain structures.
Modalities such as computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography (PET), single-photon emission computed tomography (SPECT) and near-infrared fluorescence (NIRF) imaging allow for non-invasive three-dimensional (3D) coverage of large volumes at the expense of spatial resolution. Imaging solutions based on these technologies are potentially translatable. Due to its versatility, MRI has been extensively used for characterizing cerebral vasculature: gross vascular architecture, tissue perfusion, integrity of the BBB, occurrence of hemorrhages and immune cell infiltration, all have been studied both in patients and animal models of the disease. These structural and functional read-outs can be complemented by molecular information derived from the use of target specific probes (Klohs and Rudin, 2011).
A brief summary of imaging activities and techniques related to murine AD models is provided in Table 3. A glance at Tables 2, 3 reveals that both macroscopic techniques, like MRI and PET, as well as in vivo microscopy are at the center of attention. The method of choice depends on the required resolution of the method, and thus on the specific research questions addressed.
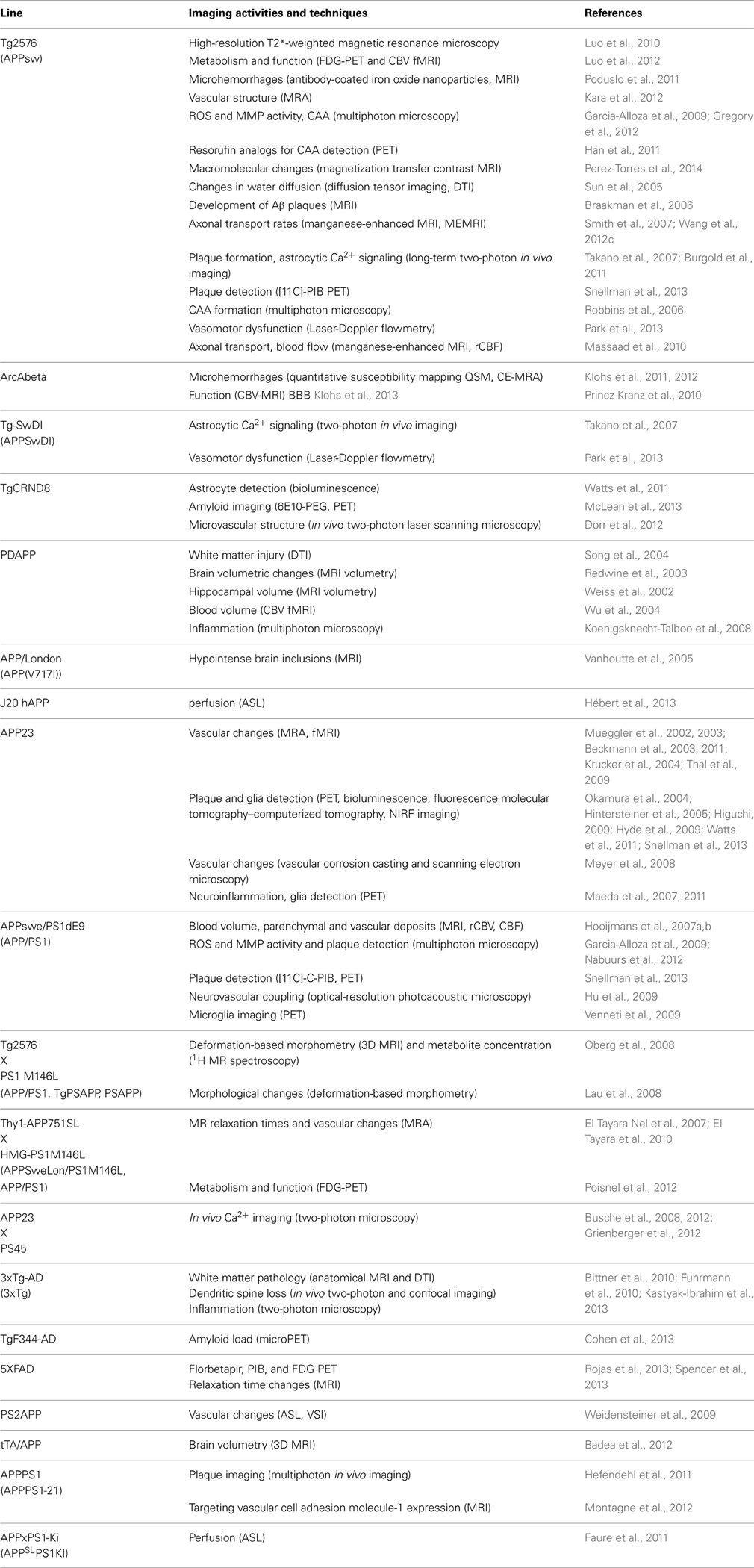
Table 3. In vivo imaging activities and techniques related to AD models.
Visualizing Vascular vs. Parenchymal Amyloid Depositions
The amyloid hypothesis proposes that AD is caused by an imbalance between Aβ production and clearance which leads to parenchymal and vascular Aβ deposits (Hardy and Selkoe, 2002). Visualizing Aβ deposition in general is desirable to characterize the dynamics of this process and for testing of Aβ-directed therapeutics. To investigate the role of CAA in AD requires discriminating vascular from parenchymal Aβ deposits. Differentiation of the two compartments would enable monitoring the effects of Aβ removal strategies, which have been shown in some instances to increase CAA (Wilcock et al., 2004a, 2011). To date, assessment of Aβ load and CAA requires time-consuming postmortem neuropathological analysis. Imaging approaches enabling to assess Aβ deposition at the microscopic and macroscopic scale in situ are therefore welcome. The sub-micrometer spatial resolution of in vivo microscopic techniques allows differentiating CAA from neuritic amyloid deposits in a straightforward manner based on their spatial distribution within tissue. In contrast, monitoring vascular amyloid deposition using non-invasive macroscopic imaging with voxel dimensions of ≥50 μm is hampered by the fact that spatial resolution does not allow discriminating between the parenchymal and vascular compartments. Instead plaque subtype specific labeling is required, which remains a major challenge, given the chemical and structural similarity of the amyloid deposits. As addressed next, in vivo microscopy has evolved as an indispensable tool for studying the dynamics of CAA under experimental conditions and also for the development of amyloid subtype specific probes, which can then be appropriately labeled for macroscopic imaging investigations with e.g., optical techniques or PET.
The dynamics of CAA has been studied in real time in Tg2576 mice using multiphoton microscopy through cranial windows (Robbins et al., 2006). Affected vessels were labeled by methoxy-X04. Earliest appearance of CAA was observed as multifocal deposits of band-like Aβ in leptomeningeal arteries at approximately 9 months of age. Serial imaging sessions enabled monitoring growth of these deposits as well as appearance of new bands. The CAA progression in Tg2576 mice was found to be linear in the range of 9–16 months of age (Robbins et al., 2006). In contrast, APPswe/PS1dE9 mice showed CAA deposition in leptomeningeal arteries by 6 months of age (Garcia-Alloza et al., 2006). However, compared to Tg2576 animals, CAA progressed at a lower rate in these mice, which may be accounted for an increase of the Aβ42/Aβ40 ratio in APPswe/PS1dE9 mice.
Amyloid specific dyes such as Thioflavin, Congo red or curcumin have been used for the histopathological assessment of cerebral amyloidosis and CAA. Chemical modifications of these dyes have led to the development of specific imaging probes which can be employed to detect amyloid deposition in vivo. Alternative approaches explore the use of antibodies or antibody fragments for targeting vascular Aβ deposition. For this purpose, Aβ targeted compounds can be labeled with radionuclides such as 11C and 18F, fluorescent dyes or paramagnetic nanoparticles. However, the delivery of intravenously injected compounds can be affected by the status of the BBB as an impairment of the BBB function may lead to unspecific leakage of the probe. Moreover, species specific differences in the affinity sites of Aβ exist (Klunk et al., 2005), which in some cases does not allow for simple translation of approaches targeting Aβ across different species.
Established PET tracers such as the [11C]-Pittsburgh compound B or [18F]-florbetapir enable cerebral Aβ detection (Johnson et al., 2007; Wong et al., 2010), but do not allow the differentiation of neuritic plaques from CAA. Thus, a new series of [18F]-styrylpyridine derivatives has been developed which showed labeling of vascular Aβ in in vitro autoradiography of brain sections of patients with CAA or AD (Zha et al., 2011). Fluorescent dyes with specificity for vascular Aβ have also been synthesized. For example, Han et al. (2011) found that the phenoxazine derivative resorufin binds preferentially to vascular amyloid deposits as compared to neuritic plaques in aged Tg2576 transgenic mice, in contrast to methoxy-X04 which binds to both (Figure 2). Along the same lines, McLean et al. (2013) developed a method to translate a panel of anti-Aβ antibodies, which show excellent histological properties, into live animal imaging contrast agents. The antibodies M116 and M64 targeting neuritic plaques and M31 binding to vascular Aβ were labeled with 64Cu and injected into TgCRND8 mice. M31 and M116 were found to be significantly retained in the brains of transgenic mice after intravenous injection, while M64 was not (Figure 3). Immunohistological examination confirmed the specificity of the antibodies for either vascular or parenchymal Aβ deposits. Similarly, Nabuurs et al. (2012) investigated the properties of two heavy chain antibody fragments, ni3A and pa2H (Harmsen and De Haard, 2007; Rutgers et al., 2009), which in APP/PS1 mice showed affinity for neuritic plaques and CAA, in contrast to observations in human tissue, where ni3A was found to specifically target vascular Aβ. An antibody-based approach for MRI detection was developed by Poduslo et al. (2011). The monoclonal antibody, IgG4.1, was labeled with monocrystalline iron oxide nanoparticles. These conjugated nanoparticles bound to vascular amyloid deposits in arterioles of Tg2576 mice after infusion into the external carotid artery. The selectivity of the nanoparticle approach was fostered by the fact that the nanoparticles cannot cross the BBB and thus remained in the vascular compartment.
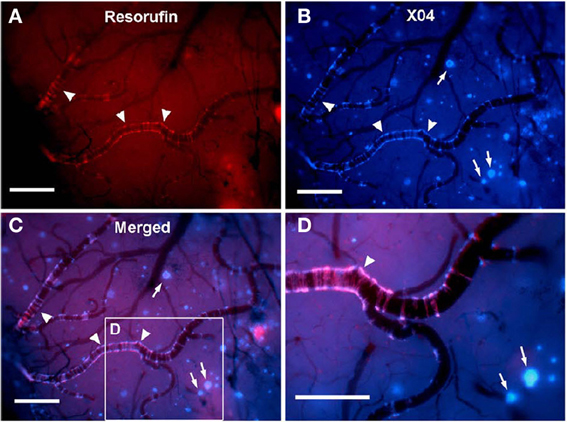
Figure 2. In vivo live imaging of CAA amyloid deposits through cranial window. Closed cranial windows were prepared on the right parietal bone of 16-month-old Tg2576 mice and the congophilic amyloid binding dye, methoxy-X04 (X04), was administered (6 mg/kg i.p.). On the next day, 2 μM resorufin (dissolved in artificial CSF) was superfused over the brain through a closed cranial window for 5 min. After washing with artificial CSF for 10 min, live fluorescent images of resorufin (red) and X04 (blue) were taken. (A) Intense fluorescent labeling detected within the walls of the leptomeningeal arteries (arrowheads) but not in neuritic plaques after topical application of resorufin. (B) In contrast, topical application of methoxy-X04 labeled Aβ aggregates in both cerebral arteries (arrowheads) and parenchymal neuritic plaques (arrows). (C) Resorufin- and X04-images merged. (D) Magnified detail of (C). Scale bars: 100 μm. Reproduced with permission from Han et al. (2011), © 2011 Han et al.
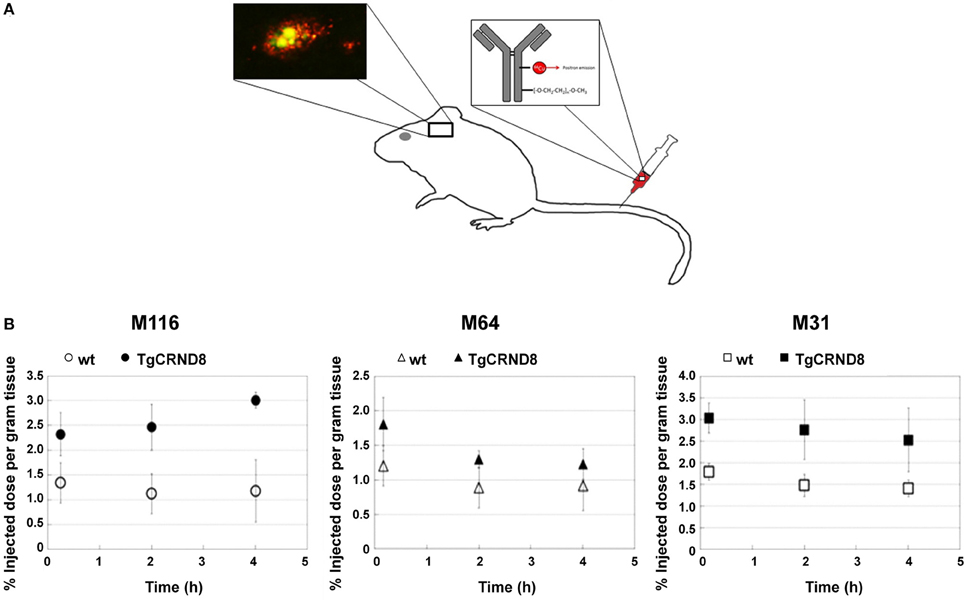
Figure 3. Targeting specific of Aβ with PET compatible radiolabelled antibodies in the brains of living mice. (A) Antibodies offer an opportunity to image specific types of Aβ pathology because of their excellent specificity. In the TgCRND8 mouse model of AD, two antibodies, M64 and M116, that target parenchyma aggregated Aβ plaques and one antibody, M31, that targets vascular Aβ were tested. All three antibodies were administered i.v. after labeling with both poly(ethylene glycol) (PEG) to enhance circulation and 64Cu to allow PET detection. (B) Quantitation of PET images (% of injected dose per gram tissue) in the brain 5 min, 2 h, and 4 h after i.v. of the probes: M116 showed progressive accumulation of M116 in the TgCRND8 brain vs. a lower, constant amount in the wild-type brain; M64 showed no difference in accumulation between TgCRND8 and wild-type mice at any time point; M31 showed greater accumulation in TgCRND8 mice than wild-type, but at a constant amount. Modified with permission from McLean et al. (2013), © 2013 American Chemical Society.
In summary, several studies have successfully demonstrated that CAA can be visualized in transgenic mice in vivo using different targeting strategies. While microscopic techniques are invasive and therefore confined to yield mechanistic information in animals, they constitute an important complement to macroscopic imaging approaches like PET and MRI which can also be used in humans. These imaging assays could be used in the future to address how vasculopathy is temporally linked to vascular Aβ deposition, but also how risk factors of AD, for example hypertension, affects this process. Moreover, the tools might be useful to evaluate Aβ removal strategies.
Imaging Cerebral Amyloid Clearance
It has been implicated that Aβ accumulation in the brain is not only the result of faulty Aβ production but also of an impaired Aβ clearance (Bell and Zlokovic, 2009). Mechanisms of cerebral Aβ clearing include degradation by proteases, interstitial fluid drainage, and transport of Aβ across the BBB (Weller, 1998; Deane et al., 2004, 2008). As discussed in the present section, imaging approaches have revealed aberrant vascular clearance mechanisms in transgenic models of AD.
Arbel-Ornath et al. (2013) have used multi-photon microscopy to visualize interstitial fluid drainage along perivascular spaces in APPswe/PS1dE9 in real time. The kinetics of dye clearance was studied after parenchymal dye injections in transgenic mice and wildtype controls 2.5–3 and 6–8 months of age. A significant impairment of the interstitial fluid drainage was observed in the old transgenic mice compared to young transgenic mice and age-matched wildtype mice.
Moreover, it has been shown that Aβ is a substrate for efflux transporters, enabling to traffick Aβ across the BBB (Kuhnke et al., 2007). Imaging strategies have been developed to visualize efflux transporter function by quantifying the uptake of substrates of these transporters. For example, (R)-[11C]-verapamil has been developed as a PET tracer to study P-glycoprotein function (van Assema et al., 2012). Higher (R)-[11C]-verapamil binding potential values were observed in AD patients compared to healthy controls, indicative of a decreased P-glycoprotein function.
In a different approach the role of the drug efflux transporter ABCG2 was studied in a transgenic mouse model. ABCG2 is a 72 kDa transmembrane protein that forms functional homodimers and operates as BBB drug efflux transporter (Doyle and Ross, 2003). It has been shown that this transporter is significantly upregulated in AD/CAA brains at both the mRNA and protein levels (Zhang et al., 2003). It has been shown that ABCG2 is also increased in Tg-SwDI and 3XTg-AD mouse models (Xiong et al., 2009). The role of ABCG2 in Aβ transport at the BBB was investigated by Xiong et al. (2009) in Abcg2-null and wildtype mice after intravenous injection of Cy5.5-labeled Aβ1–40 or scrambled Aβ1–40. NIRF imaging of live animals showed that Abcg2-null mice accumulated significantly more Aβ in their brains than wildtype mice (Figure 4), a finding that was confirmed by immunohistochemistry. These results suggest that ABCG2 may act as a gatekeeper at the BBB to prevent blood Aβ from entering into the brain.
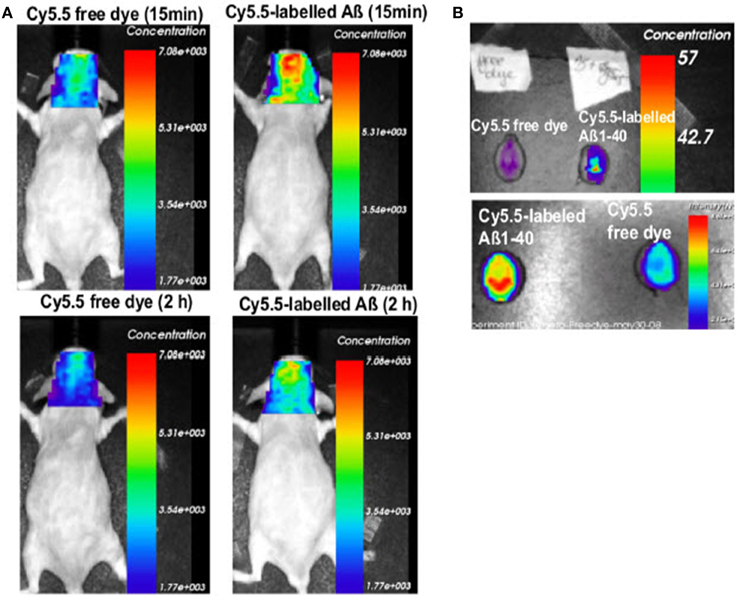
Figure 4. Absence of Abcg2 allows more A β peptides to be transported into the brain. (A) Two pairs of Abcg2 knockout mice were injected i.v. Cy5.5-free dye or Cy5.5-labeled Aβ1 − 40 peptides in equal fluorescence intensity. Animals were scanned alive using a NIRF imager at 15 min and 2 h. (B) NIRF scans of ex vivo brains collected at the end of the experiment. Signal intensity was significantly higher in the brains of Abcg knockout mice injected with Cy5.5-labeled Aβ peptides compared with Cy5.5 free dye (t-test, p < 0.001). This demonstrates that Cy5.5 was brought into the brain as a form of Cy5.5-labeled Aβ1 − 40 peptide, indicating that Abcg2 is required at the BBB to prevent the entry into the brain of circulating Aβ peptides. Modified with permission from Xiong et al. (2009), © 2009 Society for Neuroscience.
Taken together, these imaging studies provide a mechanistic link between cerebrovascular disease and AD where an impaired Aβ clearance promotes further amyloid deposition. If a defective clearance might constitute an initiating event for Aβ deposition needs to be investigated, but should be considered a new target for therapy in AD and CAA.
Detection of Chronic Cerebral Hypoperfusion
This section is devoted to studies comprising the use of microscopic or macroscopic imaging techniques to assess alterations in hemodynamic function due to deposition of Aβ in and around vessels as well as to changes in vasoactive mediators. For example, Dorr et al. (2012) observed a prolonged transit time of a fluorescent dye bolus in TgCRND8 mice compared to wildtype littermates using two-photon microscopy. Assessment of hemodynamic parameters covering the whole brain can be made with MRI. For dynamic susceptibility contrast MRI (DSC-MRI), T2- or T*2-weighted images are acquired serially. Regional changes in MRI signal intensity are measured as the contrast agent traverses the cerebral vasculature during its first-pass following intravenous bolus injection (Villringer et al., 1998). This information is then converted into contrast-time curves. The intravascular indicator dilution theory has been used to derive the hemodynamic parameters mean transient time, cerebral blood volume (CBV) and flow (CBF). Determination of absolute hemodynamic parameters requires calibration of the perfusion maps by the arterial input function (Rausch et al., 2000). Moreover, the theory assumes that the contrast agent remains intravascular during its passage. This is often not the case under pathological conditions where the BBB function may be compromised, thus leading to leakage of the injected tracer. Modeling of the leakage contribution to the image signal intensity changes has been used to obtain information on the vascular transfer constant, i.e., BBB permeability (Johnson et al., 2004). Instead of introducing an exogenous label, moving blood can also be labeled magnetically. These MRI methods are based on arterial water as a freely diffusible tracer (Williams et al., 1992). For arterial spin labeling (ASL) a non-equilibrium state (typically spin inversion) is generated to tag inflowing spins at a level proximal to the imaging slab. Images are recorded following a transit delay to allow these tagged spins to enter the imaging plane and exchange with tissue. Control images are required to compensate for direct saturation effects (Williams et al., 1992). Quantitative CBF values can be obtained from ASL images.
Cerebral hypoperfusion has been observed in AD patients using MRI (Johnson et al., 2005; Chen et al., 2011a), and has been suggested to be an early biomarker for the disease (Alsop et al., 2010; Chao et al., 2010). However, the mechanism underlying the perfusion deficits are poorly understood (Chen et al., 2011a). A decreased metabolic demand (Chen et al., 2011b) and decreased microvascular density (Buee et al., 1994) have been suggested as plausible causes, however, studies linking directly perfusion with pathological and molecular postmortem read-outs have not been attempted in humans and might be difficult to achieve. Studies assessing impairment of hemodynamic function in mice overexpressing APP can be pivotal in this regard, as a correlation of imaging studies with postmortem analysis of brain tissue can be conveniently performed. In several studies ASL was applied to APP mouse strains which have only sporadic CAA. A significant reduction in CBF has been observed in the occipital cortex of 10- to 17-month-old PS2APP (Weidensteiner et al., 2009), in 6-month-old APPxPS1-Ki (Faure et al., 2011), in 12-month old APP/PS1 (Poisnel et al., 2012), in 3-, 12- and 18-month-old J20 hAPP (Hébert et al., 2013) (Figure 5), and in 12- to 16-month-old Tg2576 mice (Massaad et al., 2010) compared to the respective age-matched controls. Perfusion was normal in subcortical (thalamic) areas in the transgenic mice (Faure et al., 2011; Poisnel et al., 2012). Reduced CBV levels at rest were also observed in the cerebral cortex, hippocampus, and thalamus of PDAPP mice compared to wildtype controls, while values were similar in other brain regions (Wu et al., 2004). In contrast, Hooijmans et al. (2007a) reported that CBF was not significantly reduced in 18-month-old APP/PS1 mice when performing bolus tracking of D2O using deuterium MRS.
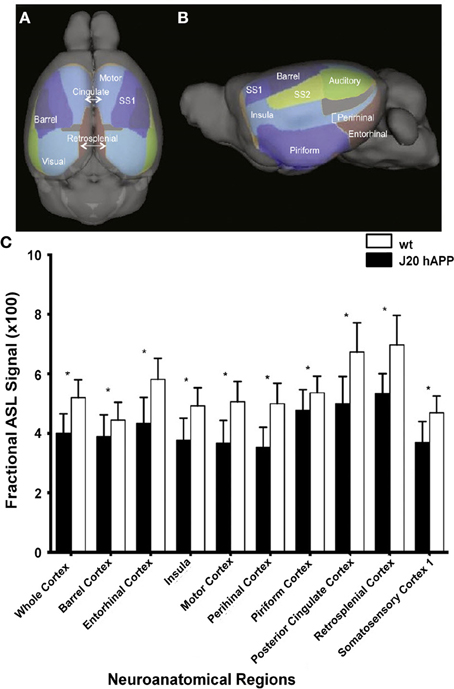
Figure 5. Hypoperfusion in 3-month-old J20 hAPP mice modeling AD. Superior (A) and lateral (B) views of the cortical surface atlas with 14 regions-of-interest labels derived from high resolution 3D MRI data sets. (C) ASL perfusion MRI measurements from representative regions-of-interest in young transgenic and age-matched wild-type mice. Note that the whole cortex and most regions demonstrated significantly lower perfusion (*p < 0.05) in J20 hAPP compared with wild-type animals. Modified with permission from Hébert et al. (2013), © 2013 Elsevier Inc.
The question which needs to be addressed is why these mice show reduced cerebral perfusion. A histopathological study has shown a decreased capillary density around senile Aβ plaques (Koutnetsova et al., 2006) which might explain the perfusion deficit, but areas of decreased perfusion did not correlate with plaque load (Weidensteiner et al., 2009). Another possibility is that reduced CBF may be due to lower cerebral metabolic demand of the brain tissue in APP mice. However, a study comparing cerebral glucose metabolism as assessed with [18F]-fluoro-2-deoxy-D-glucose PET and ASL-derived perfusion showed no correlation between the two read-outs (Poisnel et al., 2012). Cerebral glucose uptake decreased in the hippocampus, cortex and striatum of 3-month-old APP/PS1 mice, but increased in these brain regions in 12-month-old mice at an age when CBF is compromised, thus suggesting alternative mechanisms. Several studies using transgenic APP mice demonstrated alterations in vasoactive signaling (Niwa et al., 2001) and in the renin-angiotensin system (Takeda et al., 2009), as well as the generation of reactive oxygen species (Iadecola et al., 1999; Tong et al., 2005) in the brains of APP mice, all of which can directly affect vascular tone. Impairment of vascular function is observed in APP overexpressing mice prior the onset of plaque deposition and appears to be mediated by soluble Aβ (Han et al., 2008; Park et al., 2013). Indeed hypoperfusion was observed in mouse strains at this young age (Faure et al., 2011; Hébert et al., 2013).
Vascular deposition of Aβ is not a prerequisite for vascular dysfunction in AD, but CAA aggravates the functional deficit (Park et al., 2013). Aβ can exert direct vascular effects by attenuating the endothelium-dependent vasodilation (Paris et al., 2000, 2003; Luo et al., 2008), triggering the production of reactive oxygen species (Tong et al., 2005; Park et al., 2008; Massaad et al., 2010) and inducing remodeling of the vessel wall (Merlini et al., 2011). In addition, vascular accumulation of Aβ has been associated with the deposition of fibrin, which can lead to vessel stenosis and occlusion (Paul et al., 2007; Cortes-Canteli et al., 2010; Klohs et al., 2012). Cerebral hypoperfusion accelerates CAA (Okamoto et al., 2012), induces oxidative stress and alterations of the renin-angiotensin system (Washida et al., 2010), and may thus initiate a vicious cycle. Moreover, it has been shown that transgenic mice overexpressing APP have an impaired cerebral autoregulation (Niwa et al., 2002). The disability of the cerebral vasculature in the presence of Aβ to respond to changes in perfusion pressure constitutes another mechanism of vascular pathology in AD.
Hypoperfusion seems to be a critical process in the pathogenesis of AD and further investigation into its mechanism is warranted for developing therapeutic interventions that can abrogate the functional deficits. MRI has been demonstrated to be a robust technique to assess perfusion in large areas of the human and small animal brain, with or without administration of contrast agent. To obtain information at a higher spatial resolution, laser Doppler flowmetry or two-photon microscopy may be applied. But for this, cranial windows are necessary, and the information is obviously limited to upper cortical regions.
Alteration of Stimulus Evoked Response in Functional Imaging—Changed Neurovascular Coupling or Impaired Neuronal Function?
Functional imaging read-outs may constitute early sensitive markers of underlying pathology, since alterations in neuronal function and vascular reactivity are expected to precede any gross changes in anatomy as detected with structural imaging techniques. One caveat for functional imaging studies are that they are routinely performed in anesthetized animals. As the anesthetic may affect neuronal activation and/or neurovascular coupling and thus have an effect on hemodynamic read-outs (Masamoto and Kanno, 2012), the protocol needs to be carefully controlled. It is discussed next how imaging based on CBF and CBV read-outs are suitable to conduct functional imaging studies in order to investigate changes in these parameters in response to neuronal activation in transgenic animals modeling AD.
Two-photon imaging has been shown to be a unique approach to studying vascular dysfunction in mouse models of AD, by evaluating neurovascular function e.g., through analyses of functional hyperemia evoked by sensory stimulation. Using this technique, Takano et al. (2007) demonstrated in vivo that reactive changes of astrocytes and abnormalities of the microcirculation occur in early stages of the disease preceding amyloid deposition and neuronal loss. In contrast to the low Ca2+ signaling activity in non-stimulated control animals, astrocytes in 2–4-month-old Tg2576 mice exhibited a higher frequency of spontaneous Ca2+ oscillations. Animals with abnormal astrocytic activity also displayed instability of the vascular tone with oscillatory cycles of relaxation/constriction of small arteries. Aβ administration increased the frequency of spontaneous astrocytic Ca2+ increases. Because astrocytes control local microcirculation and contribute to functional hyperemia (Anderson and Nedergaard, 2003; Takano et al., 2006), abnormal astrocytic activity may contribute to vascular instability in AD and thereby to compromised neuronal function.
Dorr et al. (2012) observed a prolongation of bolus transit times of a fluorescent dye in TgCRND8 mice during hypercapnia using two-photon microscopy. While in wildtype mice, due to CO2-induced vessel dilatation, the hypercapnic challenge led to a reduction of transit time as compared to animals breathing air, the opposite effect has been observed in transgenic animals. Also in the transgenic group there was an increase in transit time with age, i.e., with more severe Aβ pathology. It was concluded that this paradoxical response to hypercapnia resulted from compromised CO2-induced dilatation of the feeding arteries/arterioles in the presence of preserved venous dilatation and reflected a profoundly impaired vascular function in TgCRND8 mice.
Functional MRI (fMRI) comprises a number of techniques to non-invasively study brain function in humans and animals. In addition to CBV and CBF read-outs, a blood oxygenation level dependent (BOLD) contrast can be used for fMRI (Ogawa et al., 1992). fMRI can be performed at rest (Jonckers et al., 2011) or with different types of physiological stimuli like sensory, thermal, or electrical stimulation (Mueggler et al., 2003; Bosshard et al., 2010). Moreover, pharmacological fMRI can measure the hemodynamic responses induced by central nervous system active drugs or vasoactive compounds and can thus be used as surrogate reflecting the effects of these drugs on neural transmission and/or vessel function.
APP23 mice of various ages have been analyzed using fMRI (Mueggler et al., 2002, 2003). CBV changes were detected in 6-, 13–15- and 25-month-old mutant mice in response to pharmacological stimulation using the GABAA receptor antagonist, bicuculline, physiological stimulation by inducing hypercapnia using the carbonic anhydrase inhibitor, acetazolamide, and peripheral sensory activation using electrical stimulation of the hind paws. In 13–15- and 25-month-old APP23 mice, all three stimulation paradigms evoked CBV responses that were significantly smaller when compared to age-matched, control littermates (Mueggler et al., 2002, 2003). In young animals of 6 months of age, there was no difference between the transgenic and wildtype group. Princz-Kranz et al. (2010) demonstrated a diminuished CBV response upon stimulation with acetazolamide in the cortex of 16- and 23-month-old arcAβ mice compared to age-matched wildtype littermates, while there was no difference between 3-month-old ArcAβ mice and controls (Figure 6). Both the rate of vascular adaptation (vascular reactivity) and the extent of the dilatation (as a measure for the reserve capacity) were found to be impaired in aged ArcAβ mice.
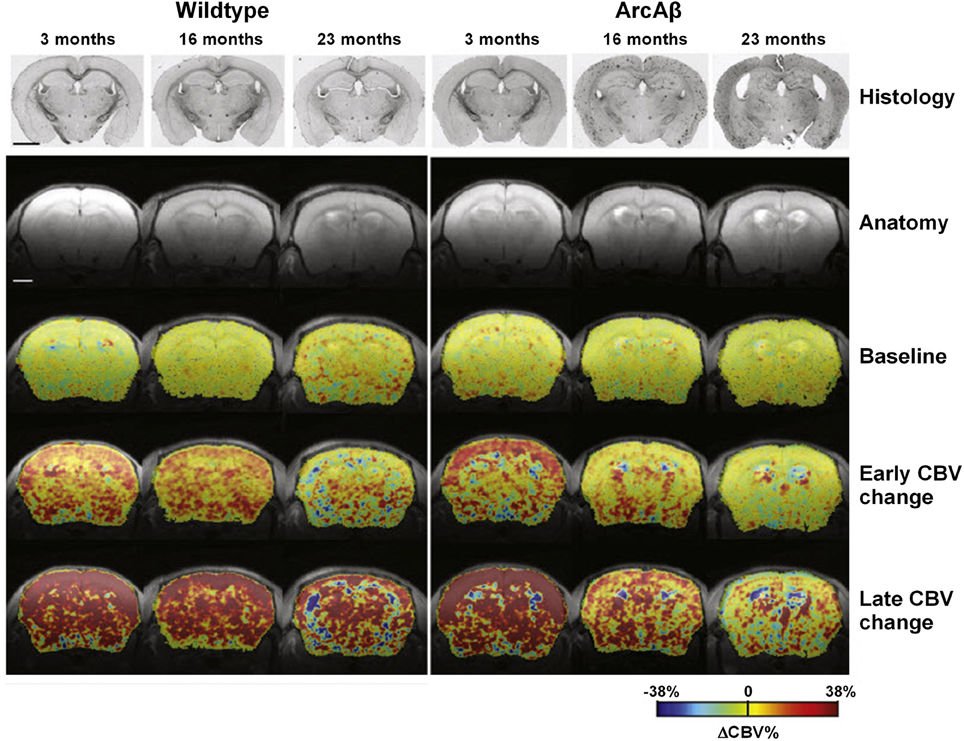
Figure 6. Vascular response to acetazolamide decreased as a function of age in the arcA β mouse model of cerebral amyloidosis, exemplified in color-coded MRI-derived CBV maps. Images for a representative age-matched wild-type control littermate and an arcAβ mouse of each age group. Histological sections stained for Aβ amyloid deposition as well as anatomical MR reference images are displayed in the two top rows. Histology reveals Aβ deposition in 16- and 23-month-old but not 3-month-old arcAβ mice, while none of the wild-type animals displayed any amyloid pathology. The color-coded CBV maps superimposed on the anatomical scans represent baseline ΔCBV% values, early changes in ΔCBV% and maximum ΔCBV% values (ΔCBV%, max). The early ΔCBV% response in arcAβ mice decreased significantly as a function of age as compared to age-matched wild-type mice. Similarly ΔCBV%, max significantly decreased in arcAβ mice as a function of age. In 3-month-old animals no difference between wild-type and arcAβ mice has been found in either parameter. The scale bar represents 2 mm. Reproduced with permission from Princz-Kranz et al. (2010), © 2010 Elsevier Inc.
A challenge in fMRI is the interpretation in animal models of AD. Under physiological conditions neurovascular coupling is rather tight (Logothetis et al., 2001; Schulz et al., 2012), but under pathological conditions a reduced fMRI response may indicate either a decrease in neuronal activity, an impaired neurovascular coupling or both. Sanganahalli et al. (2013) have shown in a non-transgenic rat model without CAA that the cortical BOLD response and neuronal activity upon sensory stimulation are reduced in rats with inducible amyloid pathology while the neurovascular coupling remains unaffected. But neurovascular coupling may be impacted in the presence of CAA as Aβ exerts direct vascular effects. Luo et al. (2008) intravenously injected Aβ1–40 in anesthetized C57BL/6 mice.Injection of the peptide led to a significant reduction in CBV in a dose-dependent and region-specific manner while the injection of phosphate buffered solution or of the reversed peptide, Aβ40–1, did not induce any significant change in vascular response. This vasconstrictive effect might also explain the impaired vascular reactivity in mice with CAA upon acetazolamide and hypercapnia challenge (Mueggler et al., 2003; Princz-Kranz et al., 2010; Dorr et al., 2012). Given the attractiveness of performing fMRI also in AD patients, further studies are warranted to examine how changes in neurovascular mediators impact fMRI read-outs.
Visualizing Amyloid-Induced Vascular Remodeling
Changes in hemodynamics of the vasculature will inevitably lead to vascular remodeling. In patients, Aβ deposits are seen in leptomeningeal and cortical arteries, and less frequently in veins and capillaries (Buee et al., 1994; Thal et al., 2008a,b). Transgenic mice show a larger heterogeneity of phenotypes with capillaries and large arteries affected by Aβ deposition. Imaging approaches targeting the vasculature at a phenotypic level are attractive tools to study remodeling of the vascular architecture as a consequence of CAA. The use of MRI to detect vascular remodeling in transgenic models is discussed in this section.
Magnetic resonance angiography (MRA) comprises a number of MRI techniques capable of visualizing the vascular architecture non-invasively. The method has limited spatial resolution, but is translational and routinely used in the clinics. Time-of-flight MRA (TOF-MRA) generates contrast between signals arising from stationary tissue and flowing blood. Maximum intensity projection or volume-rendered visualization delivers 3D representations of the cerebral vasculature in humans (Talagala et al., 1995) and rodents (Beckmann et al., 1999; Reese et al., 1999; Beckmann, 2000). However, the technique is inherently dependent on the orientation of the blood vessels with respect to the imaging plane and the actual flow velocity of the blood (Lin et al., 1997; Reese et al., 1999). While TOF-MRA can depict major brain arteries, parts of the vasculature such as small intracortical arteries, which branch off the larger cerebral vessels, and veins displaying slower blood flow velocities than arteries, are difficult to be visualized. The quality of TOF-MR angiograms is governed by the vascular anatomy and the blood flow characteristics. Signal voids in TOF-MRA may indicate absence of flow, low flow velocity, or turbulent flow. Microturbulences for instance translate into MRA signal voids due to the loss of signal coherence despite the fact that the vessel is still fully perfused (Krucker et al., 2004). Nevertheless, the degree of vasculopathy may be graded based on number and extent of signal voids detected on the angiograms in a semiquantitative manner (El Tayara et al., 2010; Kara et al., 2012). In contrast-enhanced MRA (CE-MRA) a paramagnetic contrast agent is intravenously administered, which causes a signal loss due to increased signal dephasing (El Tayara et al., 2010; Klohs et al., 2012). The CE-MRA data image can be used like in TOF-MRA data to visualize the 3D vessel architecture. However, in CE-MRA flow and motion artifacts are smaller compared to TOF-MRA (Mellin et al., 1994; Lin et al., 1997).
TOF-MRA has been applied to probe vascular remodeling in APP23 mice in vivo (Beckmann et al., 2003). Flow voids were detected at the internal carotid artery of 11-month-old APP23 mice. At the age of 20 months, additional flow disturbances were observed in the circle of Willis. Vascular corrosion casts (Krucker et al., 2004; Meyer et al., 2008) obtained from the same mice revealed that vessel elimination, deformation, or both had taken place at the sites where flow voids were detected by TOF-MRA. The detailed 3D architecture of the vasculature visible in the casts assisted the identification of smaller vessels most likely formed as substitution or anastomosis within the Circle of Willis. Thal et al. (2009) observed blood flow disturbances in TOF-MR angiograms in 25- to 26-month-old APP23 mice which corresponded to CAA-related capillary occlusion in the branches of the thalamoperforating arteries as seen with histology. El Tayara et al. (2010) evaluated vascular alterations in APP/PS1 and in PS1 mice. The double transgenic model is relatively aggressive as extracellular amyloid deposition starts at the age of 2.5 months (Blanchard et al., 2003). However, unlike plaque deposition, severity of cerebrovascular alterations is stabilized in older animals. Alterations of the middle cerebral artery were detected in old APP/PS1 mice by evaluating the severity of signal voids and the reduction of patent length of the vessel using TOF-MRA and CE-MRA. MRA obtained at very high magnetic fields (17.6 T) improved the capability to visualize smaller vessels (Kara et al., 2012). Visual and quantitative analysis of angiograms revealed severe blood flow defects in large and medium sized arteries in Tg2576 mice (Figure 7). In particular blood flow defects were observed in the middle cerebral and in the anterior communicating artery in Tg2576 mice. Histological data show that Aβ deposits in the vessel wall may be responsible for impaired CBF.
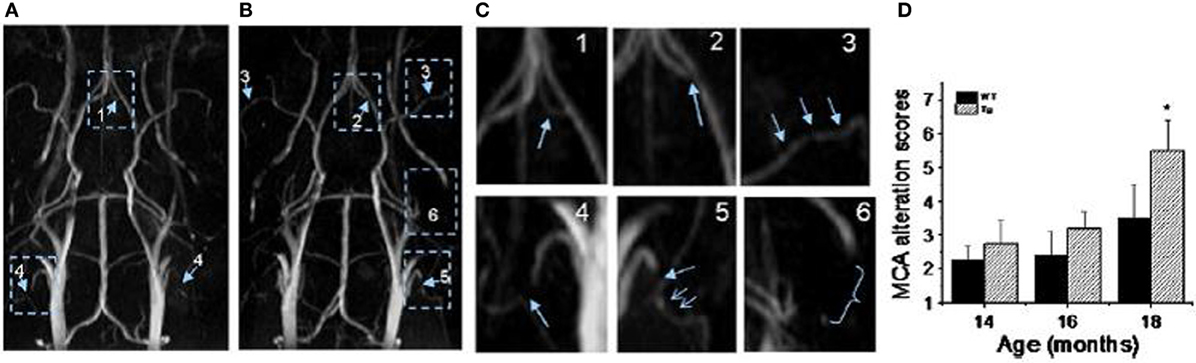
Figure 7. MR angiography of transgenic mice modeling AD. (A,B) MR angiograms of 18-month-old Tg2576 mice collected at 17.6 T showing various levels of severities of morphological changes appointed in 3D maximum intensity projection. The number indicates the appointed score to the level of severity of alterations. For example: 1, a flow disturbance (as seen in anterior communicating artery in image A); 2, a small signal void (as observed at the origin of anterior communicating artery in image B); 3, more than two small voids in same artery (as observed on the middle cerebral artery (MCA) on both sides in image B); 4, an extended void (as observed in the external carotid artery on both sides in image A); 5, a combination of an extended void and several small signal voids (as observed in the external carotid artery on both sides in image B); 6, the signal is no longer visible (as shown at the pterygo portion of the pterygopalatine artery in image A,B). The enlarge view of alterations is shown in (C). (D) MCA alteration mean score in control and Tg2576 mice with age. Values are mean ± SE (error bars); one-tail student t-test; *P < 0.05; n = 4. Reproduced with permission from Kara et al. (2012), © 2011 Elsevier Inc.
The use of cryogenic radiofrequency probes improves the quality of mouse brain angiograms at lower magnetic fields (Baltes et al., 2009). Klohs et al. (2012) employed this technology to quantitatively assess age-dependent changes of the cortical vasculature in the ArcAβ model of cerebral amyloidosis. To estimate the density of the cortical microvasculature in vivo, CE-MRA was used, based on the acquisition of data before and after administration of superparamagnetic iron oxide (SPIO) nanoparticles allowing the visualization of intracortical microvessels with high-resolution. A significant reduction in the number of functional vessels (radii of 20–80 μm) has been observed in 24-month-old ArcAβ mice compared with age-matched wildtype mice, whereas there was no difference between transgenic and wildtype mice at 4 months of age. Immunohistochemistry demonstrated strong fibrinogen and Aβ deposition in small- and medium-sized vessels, but not in large cerebral arteries, of 24-month-old ArcAβ mice. The reduced density of transcortical functional vessels may thus be attributed to vascular occlusion caused by deposition of Aβ and fibrin, which translated into impaired perfusion. Fibrin deposition has been observed previously in TgCRND8 mice (Paul et al., 2007; Cortes-Canteli et al., 2010) and since fibrin-binding probes are currently under development (Starmans et al., 2013) it may become possible to visualize cerebral fibrin deposition in these transgenic models in vivo in the near future.
The microvasculature which includes capillaries cannot be visualized directly with current MRA techniques. For this purpose, methods have been developed based on measuring the changes in the relaxation rates R*2 and R2 after administration of a paramagnetic contrast agent with long blood half-life. Relaxation has been exploited in vessel size imaging, where maps can provide insight into the composition of vessel sizes in the brain in vivo (Tropres et al., 2001). A method closely related is to measure the relaxation rate shift index Q (Jensen and Chandra, 2000), where the index is sensitive to the density but not the size of microvessels. Weidensteiner et al. (2009) determined vessel size and density in different brain regions in PS2APP mice but observed no significant differences to wildtype littermates. However, in this strain CAA is sparse and affects only large arteries.
MRI has rendered itself the most versatile methodology to visualize vascular networks of large regions or even of the whole brain while retaining a sufficient high resolution to assess smaller vessels and to provide an estimation of microvascular density. Despite the fact that in transgenic mouse models it has been shown that CAA affects the cerebral vasculature at different hierarchical levels, what causes such structural alterations in cerebral vessels is not yet known. Chronic changes in levels of vasoactive mediators like soluble Aβ, vascular endothelial growth factor, transforming growth factor-1, and altered signaling or density of vascular receptors might be implicated and future imaging studies might address this by visualizing vascular remodeling in models where these medidators are modified. Moreover, imaging studies might be useful to elucidate the role of risk factors of AD like diabetes and hypertension on the vasculature of the AD brain. Indeed, hypertension, atherosclerosis, diabetes, dyslipidemia and adiposity may impact on vascular structure and function to promote neurodegenerative processes and instigate AD (see Kalaria et al., 2012 for a recent review). The presence of vascular pathology involving arterial stiffness, arteriolosclerosis, endothelial degeneration and BBB dysfunction leads to chronic cerebral hypoperfusion, which in turn induces several features of AD pathology including selective brain atrophy, white matter changes and accumulation of abnormal proteins such Aβ. To our knowledge, no imaging studies addressing specific questions related to atherosclerosis in AD mouse models have been reported so far. Nevertheless, it is worth stressing the fact that very important developments have been achieved in molecular imaging of atherosclerosis (the interested reader is referred to reviews by Lobatto et al., 2011; Owen et al., 2011). Linking plaque anatomy and function to inflammation may help considerably to elucidate the mechanisms and complications related to atherosclerosis in AD.
Targeting Neurovascular Inflammation
A hallmark of AD is neuroinflammation which has been implicated to drive and even trigger neurodegeneration (Krstic and Knuesel, 2013). Inflammation involves also the cerebral vasculature, though the role of inflammation in the vasculopathy is not well understood. Macrophages and microglia surround amyloid affected vessels (Maat-Schieman et al., 1997; Vinters et al., 1998) and circulating macrophages have been shown to migrate from the lumen into the vessel wall (Vinters et al., 1998). Intercellular adhesion molecule-1 is upregulated at the endothelium in the AD brain (Frohman et al., 1991). Moreover, the inflammatory response of the vasculature is increased in the presence of Aβ (Vromman et al., 2013). A few imaging studies suggesting that inflammation might have deleterious consequences on vascular function are discussed next.
Different strategies to image vascular inflammation have been pursued comprising the labeling of inflammatory cells, the use of fluorogenic substrates for enzymes and fluorescent or PET probes targeted against inflammatory receptors (Wunder and Klohs, 2008; Wunder et al., 2009; Aalto et al., 2011; Li et al., 2013). Garcia-Alloza et al. (2009) have observed a strong association between CAA, matrix metalloproteinases and oxidative stress in leptomeningeal vessels of APPswe/PS1dE9 and Tg2576 with multiphoton microscopy and fluorogenic probes. The matrix metalloproteinases activity was found to be associated with matrix degradation and loss of vascular integrity.
In a different approach, mice of different transgenic lines have been examined with MRI following the intravenous administration of SPIO nanoparticles (Beckmann et al., 2011), which were hypothesized to having been taken up by circulating monocytes through absorptive endocytosis (Weissleder et al., 1990; Beckmann et al., 2009). Foci of signal attenuation were detected in cortical and thalamic brain regions of aged APP23 mice (Figure 8). Histology confirmed the presence of iron-containing macrophages in the vicinity of CAA-affected blood vessels, suggesting that the foci of signal attenuation detected in vivo might be associated with CAA in the transgenic model. A fraction of the sites additionally showed thickened vessel walls and vasculitis. Consistent with the visualization of CAA-associated lesions, MRI detected a much smaller number of attenuated signal sites in APP23xPS45, APP24, and APP51 mice, which develop significantly less CAA and microvascular pathology than APP23. These results are consistent with monocytes and microglia being involved in amyloid deposition in the wall of capillaries and in perivascular plaques (Wegiel et al., 2004). Montagne et al. (2012) have used an antibody targeting the vascular adhesion molecule-1 (VCAM-1) coupled to microparticles of iron oxide (MPIO). After injection of the probe MRI showed a significantly higher number of signal voids in the brains of 20-month-old APP/PS1 compared to age-matched wildtype controls. Immunohistochemistry revealed that VCAM-1 was overexpressed in APP/PS1 mice in all the brain regions studied (cortex, hippocampus and cerebellum). Despite APP/PS1 mice develop only minimal amyloid angiopathy (Radde et al., 2006) a significant cerebrovascular inflammation was detected in the cerebellum of these animals, which was associated with intravascular Aβ deposition (Montagne et al., 2012). Interestingly, the expression of VCAM-1 was significantly higher in the cerebellum compared to the cortex in transgenic mice. Accordingly, signal voids induced by MPIOs-αVCAM-1 and detected by MRI were significantly increased in APP/PS1 mice in all structures compared to age-matched wildtype mice.
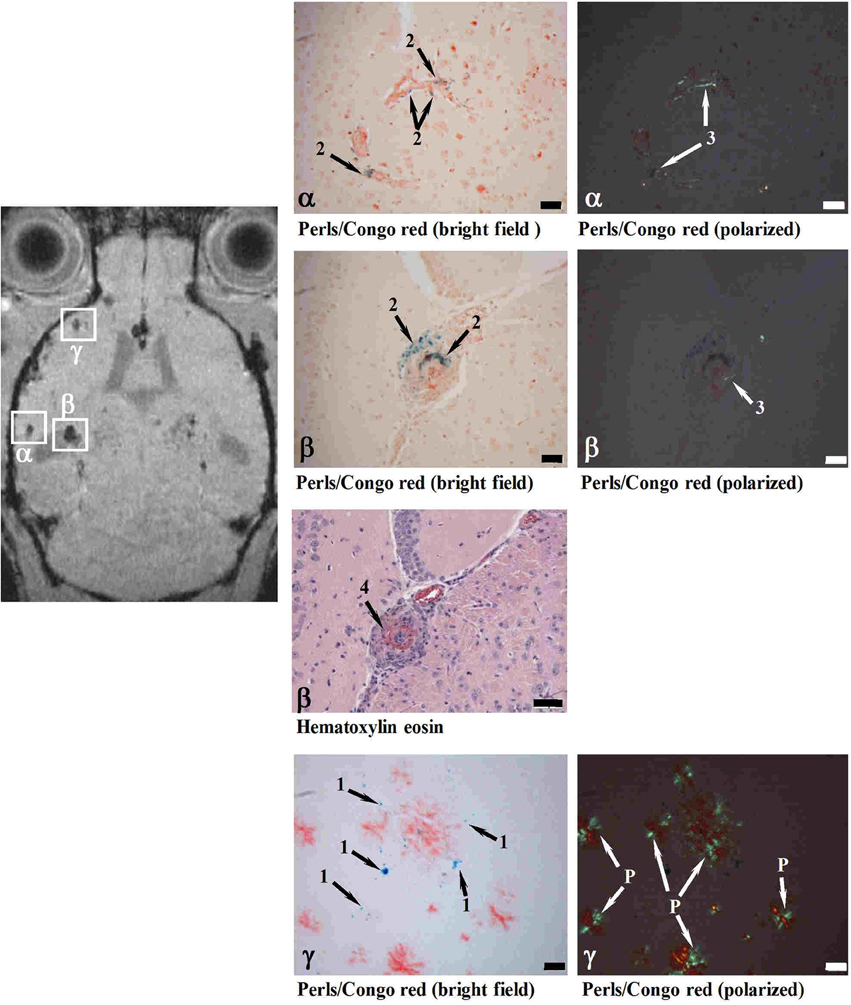
Figure 8. MRI detection of CAA-related microvascular alterations utilizing superparamagnetic iron oxide (SPIO) particles. Histological examination of cerebral cortex sites with foci of attenuated MRI signal (α, β, γ). At 24 h following SPIO administration, a male 28-month-old APP23 mouse was analyzed in vivo by MRI and processed for histology immediately thereafter. Perls/Prussian blue staining showed iron-loaded macrophages in CAA-laden vessels (Congo red positive) at both sites (α and β). While the vessel walls were thickened at both α and β locations, only site β showed in addition vasculitis characterized by lymphocyte infiltration (Hematoxylin eosin). At site γ, isolated iron-loaded macrophages were present close to amyloid vessels. 1, Iron in isolated macrophages; 2, iron in macrophages at the vessel wall; 3, amyloid deposit in vessel wall; 4, vasculitis; P, amyloid plaque. Scale bars, 50 μm. Congo red-stained sections were observed under bright field or polarized light. Reproduced from Beckmann et al. (2011), © 2011 the authors.
Microglial-vascular interactions may play a critical role in the amplification and perpetuation of inflammatory reactivity in AD brain. Indeed, post-mortem examination of medial temporal cortical tissue from humans revealed that microgliosis was progressively increased from non-demented controls to mild AD to severe AD with the latter demonstrating areas of clustered microglia (Jantaratnotai et al., 2010). Microglial clusters in severe AD brain were in close proximity with extravascular laminin and also plasma protein, fibrinogen, implicating vascular perturbation as a component of inflammatory reactivity. Microscopy studies of microglial function in murine AD models may help to better understand microglial-vascular interactions.
So far high resolution in vivo studies of microglial function were conducted in mice with genetically labeled microglia. However, because of the low expression levels of green fluorescent protein, some mouse lines are less suitable for studying the role of microglia under pathological conditions. The availability of a non-genetically encoded, easy to use marker, enabling high quality staining of microglia in any mouse strain at any experimental age would obviously be very attractive. Schwendele et al. (2012) utilized tomato lectin from Lycopersicon esculentum (Acarin et al., 1994; Boucsein et al., 2000) for high resolution in vivo imaging of microglia. A brief pressure injection of tomato lectin conjugated with a fluorescent dye (DyLight® 594) into the mouse cortex resulted in robust staining of microglial cells and blood vessels. The latter were easily distinguished from microglia based on their morphological appearance. The reliability of the in vivo staining protocol was tested in different mouse lines.
Since vascular inflammation has been implicated to partake in the deleterious consequences of CAA like degeneration of vascular smooth muscle cells and hemorrhage (Maat-Schieman et al., 1997), but still very little is known between the interaction of inflammation and vascular pathology. Further studies are warranted to investigate when and how inflammation is involved.
Detection of Blood-Brain Barrier Integrity Loss and of Cerebral Microbleeds
Severe CAA is characterized by the degeneration of the vessel wall, leading to a double-barreled appearance of the vessels with an intact adventitia, a thickened basement membrane that contains Aβ-deposits, and a widely degenerated smooth muscle cell layer (Thal et al., 2008a). Areas of fibrinoid necrosis can be frequently observed in these vessels. Degeneration of vascular smooth muscle cells lead to a loss of BBB function and eventually to vessel rupture with the occurrence of CMBs and hemorrhage. In this section, we address the detection of BBB leakage and of CMBs using MRI.
Imaging of the BBB with MRI has been widely applied to pathologies such as brain tumors and metastases, stroke and head trauma (Giesel et al., 2010). In dynamic contrast-enhanced MRI (DCE-MRI), a series of images is acquired during intravenous bolus injection of Gd-based contrast agents. Kinetic modeling of the contrast agent can provide information on vascular leakage (Tofts and Kernode, 1991). The method has for example been used to predict the occurrence of hemorrhage after ischemic stroke (Kassner et al., 2005). Klohs et al. (2013) have performed a longitudinal MRI study where DCE-MRI was applied in ArcAβ and wildtype mice. While vascular leakage of the contrast agent was significantly associated with age, there was no effect of genotype. This finding was surprising as compromised BBB function has been described for the ArcAβ strain (Merlini et al., 2011). When aged ArcAβ mice were injected intravenously with Trypan blue, leakage of the dye was observed around Aβ-affected vessels. Moreover, aged ArcAβ mice showed CMBs indicative of severe vascular pathology (Klohs et al., 2011, 2013).
The observation made in the transgenic animals is in line with studies in AD and MCI patients, where no differences in contrast agent kinetics have been detected with respect to healthy controls (Caserta et al., 1998; Starr et al., 2009). The discrepancy of the DCE-MRI findings might be explained by the fact that BBB dysfunction in AD is subtle and diffuse when compared to diseases such as brain tumors, multiple sclerosis and stroke, for which the impairment is relatively large and focal (Giesel et al., 2010). Hence, DCE-MRI may not be sensitive enough for detecting BBB impairment in mouse models of AD in vivo.
CMBs and hemorrhages can be detected with CT and MRI techniques with the latter being more often used for diagnosis (Sperling et al., 2011). T*2-weighted gradient-echo MRI protocols, which are sensitive to paramagnetic iron compounds such as hemosiderin found in blood degradation products, reveal CMBs in patients as focal hypointensities typically occurring as round or ovoid areas (Pettersen et al., 2008; Ayaz et al., 2010; Sperling et al., 2011). The occurrence of CMBs has also been reported for transgenic APP mice with CAA. For example, Beckmann et al. (2011) described the presence of foci of low signal intensity in cortical and thalamic brain regions of aged APP23 mice. Klohs et al. (2011) demonstrated in ArcAβ mice that quantitative susceptibility mapping provides increased detection sensitivity of CMBs and improved contrast when compared with conventional T*2-weighted gradient-echo magnitude imaging (Figure 9). Quantitative susceptibility maps were generated from phase data acquired with a high-resolution T*2-weighted gradient-echo sequence depicting both the localization and spatial extent of CMBs with high accuracy.
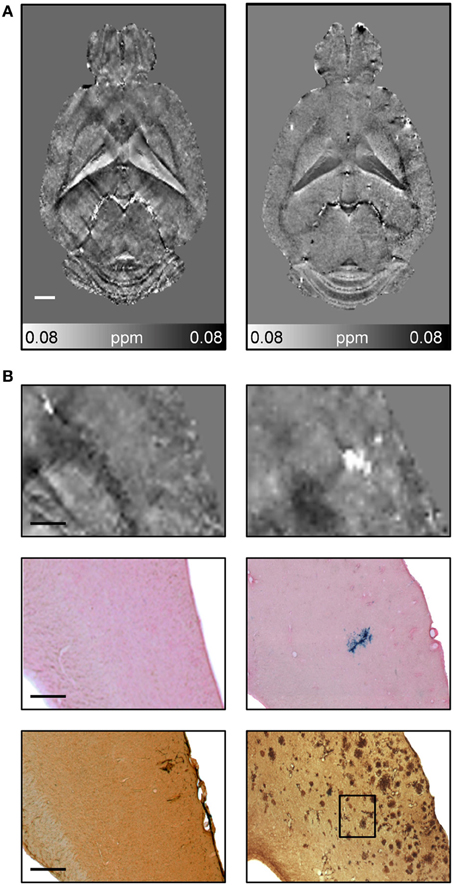
Figure 9. Detection of CMBs using quantitative susceptibility mapping MRI. (A) Horizontal quantitative susceptibility maps of an 18-month-old wild type animal and of an age-matched transgenic arcAβ mouse. (B) Quantitative susceptibility maps with corresponding tissue section after Prussian blue/eosin staining and anti-Aβ immunohistochemistry. Focal areas of high susceptibility in the cortex of 18-month old arcAβ mice correspond to areas of focal iron accumulation, indicating the occurrence of cerebral microbleeds in this mouse strain. Modified from Klohs et al. (2011), © 2011 ISCBFM.
Taken together, assessment of BBB with current DCE-MRI methods does not seem to be sensitive enough to detect vascular leakage. Advances in imaging technology enable the improved diagnostic detection of CMBs in patients and animal models. The assessment of CMB load can be used in studies to estimate the severity of CAA and to monitor the effect of therapy.
Assessing the Effects of Therapies Targeting Vascular Pathology in AD
Clinical therapeutic trials in AD patients performed so far were disappointing. Based on activities in animal models suggesting that prevention or early intervention may be a viable strategy for AD treatment, there is a trend toward treating patients at very early stages of disease or even preventatively (Bateman et al., 2012; Fleisher et al., 2012). Obviously, biomarker development, including imaging, is an essential part of this endeavor, in order to select the right patients to be treated early (Reiman et al., 2012). In this section, we briefly discuss a few therapy-intervention studies in animals addressing vascular pathology related to AD.
Therapeutic strategies have targeted APP processing, as well as the trafficking of soluble Aβ and strategies to remove aggregated Aβ. Gregory et al. (2012) analyzed in Tg2576 mice Aβ deposition in vessels and clearance from vascular walls and their relationship to the concentration of Aβ in the brain. Levels of Aβ in the brain were modulated either by peripheral clearance through administration of gelsolin which binds with high affinity to plasma Aβ (Matsuoka et al., 2003), or by directly inhibiting its formation via administration of LY-411575, a small-molecule γ-secretase inhibitor. Both gelsolin and LY-411575 reduced the rate of CAA progression in Tg2576 mice in the absence of an immune response. The progression of CAA was also halted when gelsolin was combined with LY-411575. These data suggest that CAA progression can be prevented with non-immune therapy approaches that may reduce the availability of soluble Aβ. Yet there was no evidence for substantial clearance of Aβ already deposited at vessels.
Yang et al. (2011) assessed the therapeutic potential of blocking apolipoprotein E (ApoE)/Aβ interactions, by administering an Aβ fragment (Aβ12–28P) to young TgSwDI mice (from 3 to 9 months of age). Increased cognitive function, decreased cortical, hippocampal and thalamic fibrillar vascular amyloid burden, and decreased extent of cerebral hemorrhages was found in treated compared with untreated TgSwDI mice. While this therapeutic strategy holds promise in young mice, it would be of interest to verify whether it would be effective in older animals.
Two studies demonstrated the stereoisomer of inositol, scyllo-inositol, to have potential therapeutic properties to treat CAA. When given orally to TgCRND8 mice, scyllo-inositol inhibited Aβ aggregation into high-molecular-weight oligomers in the brain and ameliorated several AD-like phenotypes in these mice, including impaired cognition, altered synaptic physiology, and cerebral Aβ pathology (McLaurin et al., 2006). These therapeutic effects, which occurred regardless of whether the compound was given before or well after the onset of the AD-like phenotypes, support the idea that the accumulation of Aβ oligomers plays a central role in the disease pathogenesis. Multiphoton laser scanning microscopy examinations of TgCRND8 mice in vivo revealed that structural changes of cortical arterioles (increase in tortuosity and decrease in caliber) with amyloid-β peptide accumulation were accompanied by progressive functional compromise, reflected in higher dispersion of microvascular network transit times, elongation of the transit times, and impaired microvascular reactivity to hypercapnia in the transgenic mice (Dorr et al., 2012). However, administration of scyllo-inositol rescued both structural and functional impairment of the cortical microvasculature (Figure 10). Overall, these results suggest microvascular impairment to be directly correlated with Aβ accumulation, highlighting the importance of targeting CAA clearance for effective diagnosis, monitoring of disease progression and treatment of AD.
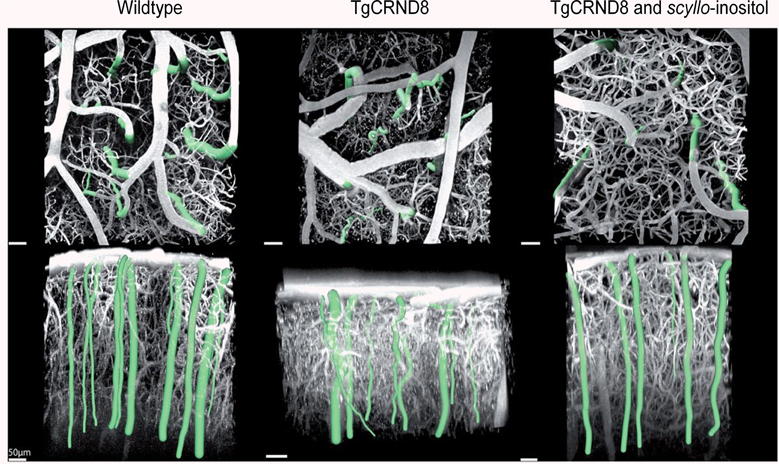
Figure 10. Segmentation of cortical penetrating vessels overlaid on maximum intensity projections of cortical microvasculature obtained from in vivo two-photon fluorescence microscopy images of the cortical microcirculation of 6.5–12-month-old mice: parallel to cortical surface (top row) and perpendicular to cortical surface (bottom row). Penetrating vessels for each individual mouse are highlighted. Average tortuosity of penetrating vessels for each sample subject (mean ± standard error): wild-type mice 1.03 ± 0.003, transgenic TgCRND8 mice 1.10 ± 0.006, scyllo-inositol-treated TgCRND8 mice 1.03 ± 0.005. Reproduced with permission from Dorr et al. (2012), © 2012 the authors.
Epidemiological studies have provided evidence that statins, cholesterol-lowering drugs broadly used in the treatment of cardiovascular diseases, have therapeutic potential in AD (Jick et al., 2000; Wolozin et al., 2000) and to a slower cognitive decline in mild-to-moderate AD patients (Sparks et al., 2006). Studies in animals have revealed that the effects of statin treatment are not due to their vascular and anti-inflammatory effects rather than their cholesterol-lowering effect. Tong et al. (2012) reported that a 3–6 months treatment with simvastatin completely rescued cerebrovascular reactivity, basal endothelial nitric oxide synthesis, and activity-induced neurometabolic and neurovascular coupling in adult (6 months) and aged (12 months) J20 hAPP transgenic mice. Remarkably, simvastatin fully restored short- and long-term memory in adult mice, but not in aged AD mice. These beneficial effects occurred without any decreasing effect of simvastatin on brain Aβ levels or plaque load. However, in AD mice with recovered memory, protein levels of the learning- and memory-related immediate early genes c-Fos and Egr-1 were normalized or upregulated in hippocampal CA1 neurons, indicative of restored neuronal function. Simvastatin also restored the CBF response in the somatosenory cortex to whisker simulation in 6- and 12-month-old J20 hAPP mice, and restored whisker-stimulated cerebral glucose uptake in the somatosensory cortex of 12-month-old APP mice as assessed by FDG-PET. These findings disclose new sites of action for statins against Aβ-induced neuronal and cerebrovascular deficits that could be predictive of therapeutic benefit in AD patients. They further indicate that simvastatin and, possibly, other brain penetrant statins bear high therapeutic promise in early AD and in patients with vascular diseases who are at risk of developing AD.
Treatment with angiotensin receptor blockers has been associated to reduce AD-related pathology (Hajjar et al., 2012) or with a lower risk and slower disease progression compared to other antihypertensive agents (Li et al., 2010). These data indicate that angiotensin receptor blockers not only decrease blood pressure but also decrease vascular inflammation may effectively reduce the risk of developing AD (Hajjar et al., 2012). Wang et al. (2007) screened 55 clinically prescribed antihypertensive medications for AD-modifying activity using primary cortico-hippocampal neuron cultures generated from Tg2576 mice. Despite 7 antihypertensive agents reduced Aβ accumulation, only valsartan was capable of attenuating oligomerization of Aβ peptides into high-molecular-weight oligomeric peptides, known to be involved in cognitive deterioration. Preventive treatment of Tg2576 mice with valsartan significantly reduced AD-type neuropathology and the content of soluble extracellular oligomeric Aβ peptides in the brain. Most importantly, valsartan administration also attenuated the development of Aβ-mediated cognitive deterioration. These preclinical studies suggest that certain antihypertensive drugs may have AD-modifying activity and may protect against progressive Aβ-related memory deficits in subjects with AD or in those at high risk of developing AD.
Aβ removing therapies are currently tested in clinical trials and have also been studied in transgenic animals, yielding conflicting results. Using multiphoton microscopy, Prada et al. (2007) showed that anti-Aβ passive immunotherapy can remove cerebral Aβ in Tg2576 mice, depending on the duration of treatment. Clearance of CAA and neuritic deposits was detected within 1 week after a single administration of 10D5, an antibody against the N-terminal of Aβ, directly to the brain, but the effect on CAA was only transient. Moreover, the progression rate of CAA became greater in the antibody treated group, suggesting that vascular Aβ deposition may accelerate after short-lived clearance. Chronic administration of the antibody over 2 weeks led to a more robust clearance of CAA. Other studies have shown that Aβ immunotherapy may also at least transiently worsen CAA, with increased incidence of cerebral microhemorrhages in aged transgenic mice (Bard et al., 2000; Pfeifer et al., 2002; Wilcock et al., 2006; Schroeter et al., 2008; Thakker et al., 2009). This is in line with a study where SPIO-enhanced MRI revealed a higher number of sites with signal attenuation in APP23 mice following a chronic treatment with the Aβ antibody β 1 (Beckmann et al., 2011). Histological analyses demonstrated an increased number of CAA vessels and of iron loaded macrophages in the vicinity of CAA vessels, for mice receiving the β 1 antibody. In addition, a study using T*2-weighted MRI for the detection of CMBs in Tg2576 mice treated with either a non-selective antibody (6G1) targeting soluble and insoluble Aβ or a more selective antibody (8F5) targeting primarily soluble Aβ (Luo et al., 2010). Both antibodies increased CMB incidence in aged APP transgenic mice compared with baseline or vehicle treatment.
It has been hypothesized that the antibodies exert their beneficial as well as their deleterious effects via an antibody Fc domain-mediated microglial activation and Aβ phagocytosis (Bard et al., 2000; Wilcock et al., 2004b). Koenigsknecht-Talboo et al. (2008) demonstrated that the effects of antibodies that recognize aggregated Aβ are rapid and involve microglia. The anti-Aβ antibody, m3D6, that binds to aggregated Aβ (Cirrito et al., 2003), was administered to PDAPP mice, an AD mouse model that was bred to contain fluorescent microglia. Three days after systemic administration of m3D6, there was a marked increase in both the number of microglial cells and processes per cell visualized in vivo by multiphoton microscopy (Figure 11). These changes required the Fc domain of m3D6 and were not observed with mHJ5.1, an antibody specific to soluble Aβ.
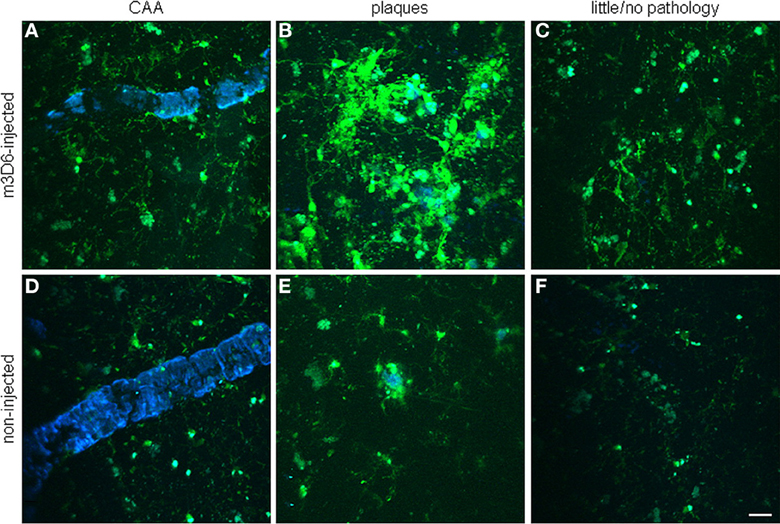
Figure 11. Rapid microglial response around amyloid pathology after systemic anti-Aβ antibody administration in PDAPP mice. Peripheral m3D6 administration results in marked morphological changes in microglia. Three-dimensional reconstructed z-series stack two-photon microscopy images taken of 22-month-old PDAPP±;CX3CR1/green fluorescent protein± mice injected with 500 μ g of m3D6 (A–C), an anti-Aβ antibody, or not injected (D–F). Green fluorescent protein-labeled microglia are green. Fibrillar amyloid was labeled with methoxy-XO4 (blue). Scale bar, 20 μm. Reproduced with permission from Koenigsknecht-Talboo et al. (2008), © 2008 Society for Neuroscience.
Taken together, these studies demonstrate the potential of animal studies in therapy studies. Imaging studies are expected to play a pivotal role in this regard; their application ranging from safety testing of putative drugs e.g., detection of microbleeds, to elucidating mechanism of action, to monitoring of treatment efficacy.
Translating Imaging Findings from Animal Models of AD
Translational research from animal models to clinical studies and from human studies back to animal models is relevant for the development and validation of imaging biomarkers of AD. In particular, MRI as well as PET assays are well suited for translational applications. While many imaging findings presented here, for example chronic cerebral hypoperfusion or the occurrence of CMBs, have been observed both in transgenic mice overexpressing APP and in AD patients, some studies have reported a lack of concordance between imaging findings in mice and men. For example, the uptake of an amyloid PET tracer reflected the amount of Aβ in AD patients but not in transgenic mice due to species-specific differences in the affinity sites of Aβ between mice and humans (Klunk et al., 2005). In another example, Luo et al. (2012) observed a glucose hypermetabolism in the brain of 7-month-old Tg2576 mice, despite the fact that hemodynamic read-outs were not different to wildtype mice. This is in contrast to AD patients, where glucose hypometabolism and hypoperfusion were concomitantly observed (Nagata et al., 2000). The increase in glucose metabolism in Tg2576 has been attributed to a neuronal compensatory mechanism due to the APP overexpression and might thus constitute an artifact of the model.
In general, there are limitations in employing models for developing biomarkers of AD. Transgenic strains overexpressing APP have been widely used because they reproduce essential histopathological features and molecular mechanisms of AD. But these models might resemble more the familial forms of AD (<1% of AD cases) or Down syndrome rather than the sporadic, late-onset form of AD. Some double and triple transgenic lines have been generated to overlay amyloid with tau and presenilin pathology (Blanchard et al., 2003; Oddo et al., 2003a,b). But even these strains do not recapitulate the complex pathophysiology of sporadic AD, which affects most AD patients. Moreover, while it is advantageous from an experimental perspective that the disease pathology in animals develops quickly, it could be an important feature of human AD that converging mechanisms which contribute to the impairment of brain function occur over a very long span and might be modulated by life-style choices and comorbidies.
Additional challenges occur when developing new therapies. Main potential reasons for the lack of concordance between preclinical models and human clinical trials could be wrong targets, incomplete models, lack of individual variability in the animal models, patients enrolled too late and comorbidities. As patients participating in clinical trials are heterogenous whereas most models utilize in-bred mouse strains, evaluating novel treatments in multiple lines may help to address this point. Also, the lack of substantial cell loss in the majority of rodent models may indicate that they better represent some aspects of the prodromal phase of the disease.
Emerging studies foster the relationship between vascular disease and tauopathy in AD. Recently, several post mortem studies on the brains of AD patients provided evidence that cardiovascular pathology like atherosclerosis is highly correlated with neuritic plaques, tau neurofibrillary tangles, and CAA (Roher et al., 2003; Beach et al., 2007; Yarchoan et al., 2012). However, imaging approaches addressing vascular aspects have not been applied to transgenic tau models so far. Similarly to the use of probes for targeting amyloid, such studies may profit from the recent development of PET tracer to image tau (Okamura et al., 2012; Maruyama et al., 2013).
Finally, animal models displaying chronic hypoperfusion (Shibata et al., 2004) or multiple microinfarcts (Wang et al., 2012b) and spontaneous hypertensive rats (Calcinaghi et al., 2013) are being used to investigate the role of vascular risk factors and pathological mechanisms in the etiology of dementia.
Conclusion
The potential of imaging techniques to study cerebrovascular pathology in animal models of AD has just been started to be fully exploited. The presented examples using genetically engineered mice demonstrate the extreme versatility of the application of imaging tools, which can provide information on gross vascular morphology and probe hemodynamic function down to address the basic mechanisms underlying CAA and neurovascular dysfunction. While the microscopic techniques are inherently invasive and thus not translatable to AD patients, the non-invasive techniques like PET and MRI can be translated and might thus provide biomarkers of the disease. Imaging of animal models is ideally suited for developing such diagnostic assays. But challenges remain in the selection of a suitable animal model to address a specific research question related to the vascular aspects of AD.
Conflict of Interest Statement
Nicolau Beckmann and Derya R. Shimshek are employees of Novartis Pharma AG, Basel, Switzerland. The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aalto, K., Autio, A., Kiss, E. A., Elima, K., Nymalm, Y., Veres, T. Z., et al. (2011). Siglec-9 is a novel leukocyte ligand for vascular adhesion protein-1 and can be used in PET imaging of inflammation and cancer. Blood 118, 3725–3733. doi: 10.1182/blood-2010-09-311076
Acarin, L., Vela, J. M., González, B., and Castellano, B. (1994). Demonstration of poly-N-acetyl lactosamine residues in ameboid and ramified microglial cells in rat brain by tomato lectin binding. J. Histochem. Cytochem. 42, 1033–1041. doi: 10.1177/42.8.8027523
Allen, B., Ingram, E., Takao, M., Smith, M. J., Jakes, R., Virdee, K., et al. (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci. 22, 9340–9351.
Alsop, D. C., Dai, W., Grossman, M., and Detre, J. A. (2010). Arterial spin labeling blood flow MRI: its role in the early characterization of Alzheimer's disease. J. Alzheimer Dis. 20, 871–880. doi: 10.3233/JAD-2010-091699
Anderson, C. M., and Nedergaard, M. (2003). Astrocyte-mediated control of cerebral microcirculation. Trends Neurosci. 26, 340–344. doi: 10.1016/S0166-2236(03)00141-3
Arbel-Ornath, M., Hudry, E., Eikermann-Haerter, K., Hou, S., Gregory, J. L., Zhao, L., et al. (2013). Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer's disease mouse models. Acta Neuropathol. 126, 353–364. doi: 10.1007/s00401-013-1145-2
Ayaz, M., Boikov, A. S., Haacke, E. M., Kido, D. K., and Kirsch, W. M. (2010). Imaging cerebral microbleeds using susceptibility weighted imaging: one step toward detecting vascular dementia. J. Magn. Reson. Imaging 31, 142–148. doi: 10.1002/jmri.22001
Badea, A., Gewalt, S., Avants, B. B., Cook, J. J., and Johnson, G. A. (2012). Quantitative mouse brain phenotyping based on single and multispectral MR protocols. Neuroimage 63, 1633–1645. doi: 10.1016/j.neuroimage.2012.07.021
Baltes, C., Radzwill, N., Bosshard, S., Marek, D., and Rudin, M. (2009). Micro MRI of the mouse brain using a novel 400 MHz cryogenic quadrature RF probe. NMR Biomed. 22, 834–842. doi: 10.1002/nbm.1396
Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., et al. (2000). Peripherally administered antibodies against amyloid Aβ-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 6, 916–919. doi: 10.1038/78682
Bateman, R. J., Xiong, C., Benzinger, T. L., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Beach, T. G., Wilson, J. R., Sue, L. I., Newell, A., Poston, M., Cisneros, R., et al. (2007). Circle of Willis atherosclerosis: association with Alzheimer's disease, neuritic plaques and neurofibrillary tangles. Acta Neuropathol. 113, 13–21. doi: 10.1007/s00401-006-0136-y
Beckmann, N. (2000). High resolution magnetic resonance angiography non-invasively reveals mouse strain differences in the cerebrovascular anatomy in vivo. Magn. Reson. Med. 44, 252–258. doi: 10.1002/1522-2594(200008)44:2<252::AID-MRM12>3.0.CO;2-G
Beckmann, N. (2011). Probing cerebrovascular alterations in Alzheimer's disease using MRI: from transgenic models to patients. Curr. Med. Imag. Rev. 7, 51–61. doi: 10.2174/157340511794653531
Beckmann, N., Cannet, C., Babin, A. L., Blé, F. X., Zurbruegg, S., Kneuer, R., et al. (2009). In vivo visualization of macrophage infiltration and activity in inflammation using MRI. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 1, 272–298. doi: 10.1002/wnan.16
Beckmann, N., Gérard, C., Abramowski, D., Cannet, C., and Staufenbiel, M. (2011). Non-invasive MRI detection of cerebral amyloid angiopathy related microvascular alterations utilizing superparamagnetic iron oxide particles in APP transgenic mouse models of Alzheimer's disease: application to passive Aβ immunotherapy. J. Neurosci. 31, 1023–1031. doi: 10.1523/JNEUROSCI.4936-10.2011
Beckmann, N., Schuler, A., Mueggler, T., Meyer, E. P., Wiederhold, K. H., Staufenbiel, M., et al. (2003). Age-dependent cerebrovascular abnormalities and blood flow disturbances in APP23 mice modeling Alzheimer's disease. J. Neurosci. 23, 8453–8459.
Beckmann, N., Stirnimann, R., and Bochelen, D. (1999). High resolution magnetic resonance angiography of the mouse brain: application to murine focal cerebral ischemia models. J. Magn. Reson. 140, 442–450. doi: 10.1006/jmre.1999.1864
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 118, 103–113. doi: 10.1007/s00401-009-0522-3
Biffi, A., and Greenberg, S. M. (2011). Cerebral amyloid angiopathy: a systematic review. J. Clin. Neurol. 7, 1–9. doi: 10.3988/jcn.2011.7.1.1
Billings, L. M., Oddo, S., Green, K. N., McGaugh, J. L., and LaFerla, F. M. (2005). Intraneuronal abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. doi: 10.1016/j.neuron.2005.01.040
Bittner, T., Fuhrmann, M., Burgold, S., Ochs, S. M., Hoffmann, N., Mitteregger, G., et al. (2010). Multiple events lead to dendritic spine loss in triple transgenic Alzheimer's disease mice. PLoS ONE 5:e15477. doi: 10.1371/journal.pone.0015477
Blanchard, V., Moussaoui, S., Czech, C., Touchet, N., Bonici, B., and Planche, M. (2003). Time sequence of maturation of dystrophic neurites associated with Ab deposits in APP/PS1 transgenic mice. Exp. Neurol. 184, 247–263. doi: 10.1016/S0014-4886(03)00252-8
Boche, D., Zotova, E., Weller, R. O., Love, S., Neal, J. W., and Pickering, R. M. (2008). Consequence of A{beta} immunization on the vasculature of human Alzheimer's disease brain. Brain 131, 3299–3310. doi: 10.1093/brain/awn261
Bornebroek, M., De Jonghe, C., Haan, J., Kumar-Singh, S., Younkin, S., Roos, R., et al. (2003). Hereditary cerebral hemorrhage with amyloidosis Dutch type (AbetaPP 693): decreased plasma amyloid-beta 42 concentration. Neurobiol. Dis. 14, 619–623. doi: 10.1016/j.nbd.2003.08.019
Bosshard, S. C., Baltes, C., Wyss, M. T., Mueggler, T., Weber, B., and Rudin, M. (2010). Assessment of brain responses to innocuous and noxious electrical forepaw stimulation in mice using BOLD fMRI. Pain 151, 655–663. doi: 10.1016/j.pain.2010.08.025
Boucsein, C., Kettenmann, H., and Nolte, C. (2000). Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur. J. Neurosci. 12, 2049–2058. doi: 10.1046/j.1460-9568.2000.00100.x
Braakman, N., Matysik, J., van Duinen, S. G., Verbeek, F., Schliebs, R., de Groot, H. J., et al. (2006). Longitudinal assessment of Alzheimer's beta-amyloid plaque development in transgenic mice monitored by in vivo magnetic resonance microimaging. J. Magn. Reson. Imaging 24, 530–536. doi: 10.1002/jmri.20675
Buee, L., Hof, P. R., Bouras, C., Delacourte, A., Perl, D. P., and Morrison, J. H. (1994). Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol. 87, 469–480. doi: 10.1007/BF00294173
Burgold, S., Bittner, T., Dorostkar, M. M., Kieser, D., Fuhrmann, M., Mitteregger, G., et al. (2011). In vivo multiphoton imaging reveals gradual growth of newborn amyloid plaques over weeks. Acta Neuropathol. 121, 327–335. doi: 10.1007/s00401-010-0787-6
Busche, M. A., Chen, X., Henning, H. A., Reichwald, J., Staufenbiel, M., Sakmann, B., et al. (2012). Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 109, 8740–8745. doi: 10.1073/pnas.1206171109
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Wiederhold, K. H., and Haass, C. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Calcinaghi, N., Wyss, M. T., Jolivet, R., Singh, A., Keller, A. L., Winnik, S., et al. (2013). Multimodal imaging in rats reveals impaired neurovascular coupling in sustained hypertension. Stroke 44, 1957–1964. doi: 10.1161/STROKEAHA.111.000185
Calhoun, M. E., Wiederhold, K. H., Abramowski, D., Phinney, A. L., Probst, A., Sturchler-Pierrat, C., et al. (1998). Neuron loss in APP transgenic mice. Nature 395, 755–756. doi: 10.1038/27351
Calhoun, M. E., Burgermeister, P., Phinney, A. L., Stalder, M., Tolnay, M., Wiederhold, K. H., et al. (1999). Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc. Natl. Acad. Sci. U.S.A. 96, 14088–14093. doi: 10.1073/pnas.96.24.14088
Casas, C., Sergeant, N., Itier, J. M., Blanchard, V., Wirths, O., van der Kolk, N., et al. (2004). Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model. Am. J. Pathol. 165, 1289–1300. doi: 10.1016/S0002-9440(10)63388-3
Caserta, M. T., Caccioppo, D., Lapin, G. D., Ragin, A., and Groothuis, D. R. (1998). Blood-brain barrier integrity in Alzheimer's disease patients and elderly control subjects. J. Neuropsychiatry Clin. Neurosci. 10, 78–84.
Chao, L. L., Buckleys, S. T., Kornak, J., Schuff, N., Madison, C., Yaffe, K., et al. (2010). ASL, perfusion MRI predicts cognitive decline and conversion from MCI to dementia. Alzheimer Dis. Assoc. Disord. 24, 19–27. doi: 10.1097/WAD.0b013e3181b4f736
Chen, G., Chen, K. S., Knox, J., Inglis, J., Bernard, A., Martin, S. J., et al. (2000). A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature 408, 975–979. doi: 10.1038/35046031
Chen, J. J., Rosas, H. D., and Salat, D. H. (2011a). Age-associated reductions in cerebral blood flow are independent from regional atrophy. Neuroimage 55, 468–478. doi: 10.1016/j.neuroimage.2010.12.032
Chen, Y., Wolk, D. A., Reddin, J. S., Korczykowski, M., Martinez, P. M., and Musiek, E. S. (2011b). Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology 77, 1977–1185. doi: 10.1212/WNL.0b013e31823a0ef7
Chishti, M. A., Yang, D. S., Janus, C., Phinney, A. L., Horne, P., Pearson, J., et al. (2001). Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. doi: 10.1074/jbc.M100710200
Cirrito, J. R., May, P. C., O'Dell, M. A., Taylor, J. W., Parsadanian, M., Cramer, J. W., et al. (2003). In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J. Neurosci. 23, 8844–8853.
Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., et al. (2013). A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci. 33, 6245–6256. doi: 10.1523/JNEUROSCI.3672-12.2013
Cordonnier, C., and van der Flier, W. M. (2011). Brain microbleeds and Alzheimer's disease: innocent observation or key player? Brain 134, 335–344. doi: 10.1093/brain/awq321
Cortes-Canteli, M., Paul, J., Norris, E. H., Bronstein, R., Ahn, H. J., Zamolodchikov, D., et al. (2010). Fibrinogen and betaamyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron 66, 695–709. doi: 10.1016/j.neuron.2010.05.014
Daumas, S., Sandin, J., Chen, K. S., Kobayashi, D., Tulloch, J., Martin, S. J., et al. (2008). Faster forgetting contributes to impaired spatial memory in the PDAPP mouse: deficit in memory retrieval associated with increased sensitivity to interference? Learn. Mem. 15, 625–632. doi: 10.1101/lm.990208
Davis, J., Xu, F., Deane, R., Romanov, G., Previti, M. L., Zeigler, K., et al. (2004). Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 279, 20296–20306. doi: 10.1074/jbc.M312946200
Deane, R., Sagare, A., and Zlokovic, B. V. (2008). The role of the cell surface LRP and soluble LRP in blood– brain barrier Abeta clearance in Alzheimer's disease. Curr. Pharm. Des. 14, 1601–1605. doi: 10.2174/138161208784705487
Deane, R., Wu, Z., and Zlokovic, B. V. (2004). RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood– brain barrier. Stroke 35 (11 Suppl. 1), 2628–2631. doi: 10.1161/01.STR.0000143452.85382.d1
Dewachter, I., Van Dorpe, J., Smeijers, L., Gilis, M., Kuipéri, C., Laenen, I., et al. (2000). Aging increased amyloid peptide and caused amyloid plaques in brain of old APP/V717I transgenic mice by a different mechanism than mutant presenilin1. J. Neurosci. 20, 6452–6458.
Dodart, J. C., Meziane, H., Mathis, C., Bales, K. R., Paul, S. M., and Ungerer, A. (1999). Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav. Neurosci. 113, 982–990. doi: 10.1037/0735-7044.113.5.982
Domnitz, S. B., Robbins, E. M., Hoang, A. W., Garcia-Alloza, M., Hyman, B. T., Rebeck, G. W., et al. (2005). Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J. Neuropathol. Exp. Neurol. 64, 588–594. doi: 10.1097/01.jnen.0000171644.00180.fc
Dorr, A., Sahota, B., Chinta, L. V., Brown, M. E., Lai, A. Y., Ma, K., et al. (2012). Amyloid-β-dependent compromise of microvascular structure and function in a model of Alzheimer's disease. Brain 135, 3039–3050. doi: 10.1093/brain/aws243
Doyle, L. A., and Ross, D. D. (2003). Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22, 7340–7358. doi: 10.1038/sj.onc.1206938
Duff, K., Knight, H., Refolo, L. M., Sanders, S., Yu, X., Picciano, M., et al. (2000).Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis. 7, 87–98. doi: 10.1006/nbdi.1999.0279
El Tannir El Tayara, N., Delatour, B., Volk, A., and Dhenain, M. (2010). Detection of vascular alterations by in vivo magnetic resonance angiography and histology in APP/PS1 mouse model of Alzheimer's disease. MAGMA 23, 53–64. doi: 10.1007/s10334-009-0194-y
El Tayara Nel, T., Volk, A., Dhenain, M., and Delatour, B. (2007). Transverse relaxation time reflects brain amyloidosis in young APP/PS1 transgenic mice. Magn. Reson. Med. 58, 179–184. doi: 10.1002/mrm.21266
Faure, A., Verret, L., Bozon, B., El Tannir El Tayara, N., Ly, M., Kober, F., et al. (2011). Impaired neurogenesis, neuronal loss, and brain functional deficits in the APPxPS1-Ki mouse model of Alzheimer's disease. Neurobiol. Aging 32, 407–418. doi: 10.1016/j.neurobiolaging.2009.03.009
Fleisher, A. S., Chen, K., Quiroz, Y. T., Jakimovich, L. J., Gutierrez-Gomez, M., Langoi, C. M., et al. (2012). Florbetapir PET analysis of amyloid-b deposition in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross sectional study. Lancet Neurol. 11, 1057–1065. doi: 10.1016/S1474-4422(12)70227-2
Frohman, E. M., Frohman, T. C., Gupta, S., de Fougerolles, A., and van den Noort, S. (1991). Expression of intercellular adhesion molecule 1 (ICAM-1) in Alzheimer's disease. J. Neurol. Sci. 106, 105–111. doi: 10.1016/0022-510X(91)90202-I
Fryer, J. D., Taylor, J. W., Demattos, R. B., Bales, K. R., Paul, S. M., Parsadanian, M., et al. (2003). Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J. Neurosci. 23, 7889–7896.
Fuhrmann, M., Bittner, T., Jung, C. K., Burgold, S., Page, R. M., Mitteregger, G., et al. (2010). Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat. Neurosci. 13, 411–413. doi: 10.1038/nn.2511
Games, D., Adams, D., Alessandrini, R., Barbour, R., Berthelette, P., Blackwell, C., et al. (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373, 523–527. doi: 10.1038/373523a0
Garcia-Alloza, M., Prada, C., Lattarulo, C., Fine, S., Borrelli, L. A., Betensky, R., et al. (2009). Matrix metalloproteinase inhibition reduces oxidative stress associated with cerebral amyloid angiopathy in vivo in transgenic mice. J. Neurochem. 109, 1636–1647. doi: 10.1111/j.1471-4159.2009.06096.x
Garcia-Alloza, M., Robbins, E. M., Zhang-Nunes, S. X., Purcell, S. M., Betensky, R. A., Raju, S., et al. (2006). Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524. doi: 10.1016/j.nbd.2006.08.017
Giesel, F. L., Mehndiratta, A., and Essig, M. (2010). High-relaxivity contrast-enhanced magnetic resonance neuroimaging: a review. Eur. Radiol. 20, 2461–2474. doi: 10.1007/s00330-010-1805-8
Gotz, J., Probst, A., Spillantini, M. G., Schafer, T., Jakes, R., Burki, K., et al. (1995). Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 14, 1304–1313.
Gregory, J. L., Prada, C. M., Fine, S. J., Garcia-Alloza, M., Betensky, R. A., Arbel-Ornath, M., et al., 2012. Reducing available soluble β-amyloid prevents progression of cerebral amyloid angiopathy in transgenic mice. J. Neuropathol. Exp. Neurol. 71, 1009–1017. doi: 10.1097/NEN.0b013e3182729845
Grienberger, C., Rochefort, N. L., Adelsberger, H., Henning, H. A., Hill, D. N., Reichwald, J., et al. (2012). Staged decline of neuronal function in vivo in an animal model of Alzheimer's disease. Nat. Commun. 3, 774. doi: 10.1038/ncomms1783
Hajjar, I., Brown, L., Mack, W. J., and Chui, H. (2012). Impact of angiotensin receptor blockers on Alzheimer's disease pathology in a large brain autopsy series. Arch. Neurol. 69, 1632–1638. doi: 10.1001/archneurol.2012.1010
Han, B. H., Zhou, M. L., Abousaleh, F., Brendza, R. P., Dietrich, H. H., Koenigsknecht-Talboo, J., et al. (2008). Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: contribution of soluble and insoluble amyloid-β peptide, partial restoration via γ-secretase inhibition. J. Neurosci. 28, 13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008
Han, B. H., Zhou, M. L., Vellimana, A. K., Milner, E., Kim, D. H., Greenberg, J. K., et al. (2011). Resorufin analogs preferentially bind cerebrovascular amyloid: potential use as imaging ligands for cerebral amyloid angiopathy. Mol. Neurodegener. 6, 86. doi: 10.1186/1750-1326-6-86
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Harmsen, M. M., and De Haard, H. J. (2007). Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22. doi: 10.1007/s00253-007-1142-2
Hartman, R. E., Izumi, Y., Bales, K. R., Paul, S. M., Wozniak, D. F., and Holtzman, D. M. (2005). Treatment with an amyloid-beta antibody ameliorates plaque load, learning deficits, and hippocampal long-term potentiation in a mouse model of Alzheimer's disease. J. Neurosci. 25, 6213–6220. doi: 10.1523/JNEUROSCI.0664-05.2005
Havas, D., Hutter-Paier, B., Ubhi, K., Rockenstein, E., Crailsheim, K., Masliah, E., et al. (2011). A longitudinal study of behavioral deficits in an Aβ PP transgenic mouse model of Alzheimer's disease. J. Alzheimers Dis. 25, 231–243. doi: 10.3233/JAD-2011-101866
Hébert, F., Grand'maison, M., Ho, M. K., Lerch, J. P., Hamel, E., and Bedell, B. J. (2013). Cortical atrophy and hypoperfusion in a transgenic mouse model of Alzheimer's disease. Neurobiol. Aging 34, 1644–1652. doi: 10.1016/j.neurobiolaging.2012.11.022
Hefendehl, J. K., Wegenast-Braun, B. M., Liebig, C., Eicke, D., Milford, D., and Calhoun, M. E. (2011). Long-term in vivo imaging of beta-amyloid plaque appearance and growth in a mouse model of cerebral beta-amyloidosis. J. Neurosci. 31, 624–629. doi: 10.1523/JNEUROSCI.5147-10.2011
Helm, P. J., Ottersen, O. P., and Nase, G. (2009). Analysis of optical properties of the mouse cranium - implications for in vivo multiphoton laser scanning microscopy. J. Neurosci. Methods 178, 316–322. doi: 10.1016/j.jneumeth.2008.12.032
Herzig, M. C., Paganetti, P., Staufenbiel, M., and Jucker, M. (2007). BACE1 and mutated presenilin-1 differently modulate Aβ40 and Aβ42 levels and cerebral amyloidosis in APP dutch transgenic mice. Neurodegener. Dis. 4, 127–135. doi: 10.1159/000101837
Herzig, M. C., Eisele, Y. S., Staufenbiel, M., and Jucker, M. (2009). E22Q-mutant Abeta peptide (AbetaDutch) increases vascular but reduces parenchymal Abeta deposition. Am. J. Pathol. 174, 722–726. doi: 10.2353/ajpath.2009.080790
Herzig, M. C., van Nostrand, W. E., and Jucker, M. (2006). Mechanism of cerebral β-amyloid angiopathy: murine and cellular models. Brain Pathol. 16, 40–54. doi: 10.1111/j.1750-3639.2006.tb00560.x
Herzig, M. C., Winkler, D. T., Burgermeister, P., Pfeifer, M., Kohler, E., Schmidt, S. D., et al. (2004). Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 7, 954–960. doi: 10.1038/nn1302
Higuchi, M. (2009). Visualization of brain amyloid and microglial activation in mouse models of Alzheimer's disease. Curr. Alzheimer Res. 6, 137–143. doi: 10.2174/156720509787602906
Hintersteiner, M., Enz, A., Frey, P., Jaton, A. L., Kinzy, W., Kneuer, R., et al. (2005). In vivo detection of amyloid-ß deposits by near-infrared imaging using an oxazine-derivative probe. Nat. Biotechnol. 23, 577–583. doi: 10.1038/nbt1085
Holcomb, L., Gordon, M. N., McGowan, E., Yu, X., Benkovic, S., Jantzen, P., et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100. doi: 10.1038/nm0198-097
Holtmaat, A., Bonhoeffer, T., Chow, D. K., Chuckowree, J., De Paola, V., Hofer, S. B., et al. (2009). Long-term, high resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 4, 1128–1144. doi: 10.1038/nprot.2009.89
Hooijmans, C. R., Graven, C., Dederen, P. J., Tanila, H., van Groen, T., and Kiliaan, A. J. (2007b). Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 1181, 93–103. doi: 10.1016/j.brainres.2007.08.063
Hooijmans, C. R., Rutters, F., Dederen, P. J., Gambarota, G., Veltien, A., and van Groen, T. (2007a). Changes in cerebral blood volume and amyloid pathology in aged Alzheimer's APP/PS1 mice on a docosahexaenoic acid (DHA) diet or cholesterol enriched Typical Western Diet (TWD). Neurobiol. Dis. 28, 16–29. doi: 10.1016/j.nbd.2007.06.007
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. doi: 10.1126/science.274.5284.99
Hu, S., Yan, P., Maslov, K., Lee, J. M., and Wang, L. V. (2009). Intravital imaging of amyloid plaques in a transgenic mouse model using optical-resolution photoacoustic microscopy. Opt. Lett. 34, 3899–3901. doi: 10.1364/OL.34.003899
Hyde, D., de Kleine, R., MacLaurin, S. A., Miller, E., Brooks, D. H., Krucker, T., et al. (2009). Hybrid FMT-CT imaging of amyloid-beta plaques in a murine Alzheimer's disease model. Neuroimage 44, 1304–1311. doi: 10.1016/j.neuroimage.2008.10.038
Iadecola, C., Zhang, F., Niwa, K., Eckman, C., Turner, S. K., Fischer, E., et al. (1999). SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat. Neurosci. 2, 157–161. doi: 10.1038/5715
Jankowsky, J. L., Slunt, H. H., Ratovitski, T., Jenkins, N. A., Copeland, N. G., and Borchelt, D. R. (2001). Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Eng. 17, 157–165. doi: 10.1016/S1389-0344(01)00067-3
Jankowsky, J. L., Slunt, H. H., Gonzales, V., Savonenko, A. V., Wen, J. C., and Jenkins, N. A. (2005). Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med 2:e355. doi 10.1371/journal.pmed.0020355
Jantaratnotai, N., Schwab, C., Ryu, J. K., McGeer, P. L., and McLarnon, J. G. (2010). Converging perturbed microvasculature and microglial clusters characterize Alzheimer disease brain. Curr. Alzheimer Res. 7, 625–636. doi: 10.2174/156720510793499039
Jarrett, J. T., Berger, E. P., and Lansbury, P. T. Jr. (1993). The carboxy terminus of the beta amyloid protein is critical for the seeding of the amyloid formation: implications for the pathogenesis of Alzheimer's diseases. Biochemistry 32, 4693–4697. doi: 10.1021/bi00069a001
Jawhara, S., Trawickaa, A., Jenneckensa, C., Bayera, T. A., and Wirths, O. (2012). Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol. Aging 33, 196.e29–196.e40. doi: 10.1016/j.neurobiolaging.2010.05.027
Jellinger, K. A. (2002). Alzheimer disease and cerebrovascular pathology: an update. J. Neural Transm. 109, 813–836. doi: 10.1007/s007020200068
Jensen, J. H., and Chandra, R. (2000). MR imaging of microvasculature. Magn. Reson. Med. 44, 224–230. doi: 10.1002/1522-2594(200008)44:2<224::AID-MRM9>3.0.CO;2-M
Jick, H., Zornberg, G. L., Jick, S. S., Seshadri, S., and Drachman, D. A. (2000). Statins and the risk of dementia. Lancet 356, 1627–1631. doi: 10.1016/S0140-6736(00)03155-X
Johnson, G., Wetzel, S. G., Cha, S., Babb, J., and Tofts, P. S. (2004). Measuring blood volume and vascular transfer constant from dynamic, T(2)*-weighted contrast-enhanced MRI. Magn. Res. Med. 51, 961–968. doi: 10.1002/mrm.20049
Johnson, K. A., Gregas, M., Becker, J. A., Kinnecom, C., Salat, D. H., Moran, E. K., et al. (2007). Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol. 62, 229–234. doi: 10.1002/ana.21164
Johnson, N. A., Jahng, G. H., Weiner, M. W., Miller, B. L., Chui, H. C., Jagust, W. J., et al. (2005). Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234, 851–859. doi: 10.1148/radiol.2343040197
Jonckers, E., Van Audekerke, J., De Visscher, G., Van der Linden, A., and Verhoye, M. (2011). Functional connectivity fMRI of the rodent brain: comparison of functional connectivity networks in rat and mouse PLoS ONE 6:e18876. doi: 10.1371/journal.pone.0018876
Kalaria, R. N., Akinyemi, R., and Ihara, M. (2012). Does vascular pathology contribute to Alzheimer changes? J. Neurol. Sci. 322, 141–147. doi: 10.1016/j.jns.2012.07.032
Kara, F., Dongen, E. S., Schliebs, R., Buchem, M. A., Groot, H. J., and Alia, A. (2012). Monitoring blood flow alterations in the Tg2576 mouse model of Alzheimer's disease by in vivo magnetic resonance angiography at 17.6 T. Neuroimage 60, 958–966. doi: 10.1016/j.neuroimage.2011.12.055
Kassner, A., Roberts, T., Taylor, K., Silver, F., and Mikulis, D. (2005). Prediction of hemorrhage in acute ischemic stroke using permeability MR imaging. AJNR Am. J. Neuroradiol. 26, 2213–2217.
Kastyak-Ibrahim, M. Z., Di Curzio, D. L., Buist, R., Herrera, S. L., Albensi, B. C., Del Bigio, M. R., et al. (2013). Neurofibrillary tangles and plaques are not accompanied by white matter pathology in aged triple transgenic-Alzheimer disease mice. Magn. Reson. Imaging 31, 1515–1521. doi: 10.1016/j.mri.2013.06.013
Kim, J., Onstead, L., Randle, S., Price, R., Smithson, L., Zwizinski, C., et al. (2007). Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 27, 627–633. doi: 10.1523/JNEUROSCI.4849-06.2007
Kimura, R., and Ohno, M. (2009). Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 33, 229–235. doi: 10.1016/j.nbd.2008.10.006
Klohs, J., Baltes, C., Princz-Kranz, F., Ratering, D., Nitsch, R. M., Knuesel, I., et al. (2012). Contrast-enhanced magnetic resonance microangiography reveals remodeling of the cerebral microvasculature in transgenic ArcAβ mice. J. Neurosci. 32, 1705–1713. doi: 10.1523/JNEUROSCI.5626-11.2012
Klohs, J., Deistung, A., Schweser, F., Grandjean, J., Dominietto, M., Waschkies, C., et al. (2011). Detection of cerebral microbleeds with quantitative susceptibility mapping in the ArcAbeta mouse model of cerebral amyloidosis. J. Cereb. Blood Flow. Metab. 31, 2282–2292. doi: 10.1038/jcbfm.2011.118
Klohs, J., Politano, I. W., Deistung, A., Grandjean, J., Drewek, A., Dominietto, M., et al. (2013). Longitudinal assessment of amyloid pathology in transgenic ArcAβ mice using multi-parametric magnetic resonance imaging. PLoS ONE 8:e66097. doi: 10.1371/journal.pone.0066097
Klohs, J., and Rudin, M. (2011). Unveiling molecular events in the brain by noninvasive imaging. Neuroscientist 17, 539–559. doi: 10.1177/1073858410383433
Klunk, W. E., Lopresti, B. J., Ikonomovic, M. D., Lefterov, I. M., Koldamova, R. P., Abrahamson, E. E., et al. (2005). Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-beta in Alzheimer's disease brain but not in transgenic mouse brain. J. Neurosci. 25, 10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005
Knobloch, M., Konietzko, U., Krebs, D. C., and Nitsch, R. M. (2007). Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol. Aging 28, 1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019
Koenigsknecht-Talboo, J., Meyer-Luehmann, M., Parsadanian, M., Garcia-Alloza, M., Finn, M. B., Hyman, B. T., et al. (2008). Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J. Neurosci. 28, 14156–14164. doi: 10.1523/JNEUROSCI.4147-08.2008
Koutnetsova, E., Klinger, M., Sorger, D., Sabri, O., Grossmann, U., Steinbach, J., et al. (2006). Developmental and amyloid plaque-related changes in cerebral cortical capillaries in transgenic Tg2576 Alzheimer mice. Int. J. Dev. Neurosci. 24, 187–193. doi: 10.1016/j.ijdevneu.2005.11.011
Krstic, D., and Knuesel, I. (2013). Deciphering the mechanism underlying late-onset Alzheimer disease. Nat. Rev. Neurol. 9, 25–34. doi: 10.1038/nrneurol.2012.236
Krucker, T., Schuler, A., Meyer, E. P., Staufenbiel, M., and Beckmann, N. (2004). Magnetic resonance angiography and vascular corrosion casting as tools in biomedical research: application to transgenic mice modeling Alzheimer's disease. Neurol. Res. 26, 507–516. doi: 10.1179/016164104225016281
Kuhnke, D., Jedlitschky, G., Grube, M., Krohn, M., Jucker, M., Mosyagin, I., et al. (2007). MDR1-P-glycoprotein (ABCB1) mediates transport of Alzheimer's amyloid-beta peptides–implications for the mechanisms of Abeta clearance at the blood-brain barrier. Brain Pathol. 17, 347–353. doi: 10.1111/j.1750-3639.2007.00075.x
Kulic, L., McAfoose, J., Welt, T., Tackenberg, C., Spani, C., Wirth, F., et al. (2012). Early accumulation of intracellular fibrillar oligomers and late congophilic amyloid angiopathy in mice expressing the Osaka intra-Abeta APP mutation. Transl. Psychiatry 2, e183. doi: 10.1038/tp.2012.109
Kumar-Singh, S. (2009). Hereditary and sporadic forms of Aβ-cerebrovascular amyloidosis and relevant transgenic mouse models. Int. J. Mol. Sci. 10, 1872–1895. doi: 10.3390/ijms10041872
Kumar-Singh, S., Pirici, D., McGowan, E., Serneels, S., Ceuterick, C., Hardy, J., et al. (2005). Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer's disease are centered on vessel walls. Am. J. Pathol. 167, 527–543. doi: 10.1016/S0002-9440(10)62995-1
Lau, J. C., Lerch, J. P., Sled, J. G., Henkelman, R. M., Evans, A. C., and Bedell, B. J. (2008). Longitudinal neuroanatomical changes determined by deformation-based morphometry in a mouse model of Alzheimer's disease. Neuroimage 42, 19–27. doi: 10.1016/j.neuroimage.2008.04.252
Li, N. C., Lee, A., Whitmer, R. A., Kivipelto, M., Lawler, E., Kazis, L. E., et al. (2010). Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 340, b5465. doi: 10.1136/bmj.b5465
Li, X. G., Autio, A., Ahtinen, H., Helariutta, K., Liljenbäck, H., and Jalkanen, S. (2013). Translating the concept of peptide labeling with 5-deoxy-5-[18F]fluororibose into preclinical practice: 18F-labeling of Siglec-9 peptide for PET imaging of inflammation. Chem. Commun. (Camb.) 49, 3682–3684. doi: 10.1039/c3cc40738a
Lin, W., Abendschein, D. R., Celik, A., Dolan, R. P., Lauffer, R. B., Walovitch, R. C., et al. (1997). Intravascular contrast agent improves magnetic resonance angiography of carotid areries in minipigs. J. Magn. Reson. Imaging 7, 963–971. doi: 10.1002/jmri.1880070605
Lobatto, M. E., Fuster, V., Fayad, Z. A., and Mulder, W. J. (2011). Perspectives and opportunities for nanomedicine in the management of atherosclerosis. Nat. Rev. Drug. Discov. 10, 835–852. doi: 10.1038/nrd3578
Logothetis, N. K., Pauls, J., Augath, M., Trinath, T., and Oeltermann, A. (2001). Neurophysiological investigation of the basis of the fMRI signal. Nature 412, 150–157. doi: 10.1038/35084005
Lord, A., Kalimo, H., Eckman, C., Zhang, X. Q., Lannfelt, L., and Nilsson, L. N. (2006). The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol. Aging 27, 67–77. doi: 10.1016/j.neurobiolaging.2004.12.007
Lovasic, L., Bauschke, H., and Janus, C. (2005). Working memory impairment in a transgenic amyloid precursor protein TgCRND8 mouse model of Alzheimer's disease. Genes Brain Behav. 4, 197–208. doi: 10.1111/j.1601-183X.2004.00104.x
Luo, F., Rustay, N. R., Ebert, U., Hradil, V. P., Cole, T. B., Llano, D. A., et al. (2012). Characterization of 7- and 19-month-old Tg2576 mice using multimodal in vivo imaging: limitations as a translatable model of Alzheimer's disease. Neurobiol. Aging 33, 933–944. doi: 10.1016/j.neurobiolaging.2010.08.005
Luo, F., Rustay, N. R., Seifert, T., Roesner, B., Hradil, V., Hillen, H., et al. (2010). Magnetic resonance imaging detection and time course of cerebral microhemorrhages during passive immunotherapy in living amyloid precursor protein transgenic ice. J. Pharmacol. Exp. Ther. 335, 580–588. doi: 10.1124/jpet.110.172932
Luo, F., Seifert, T. R., Edalji, R., Loebbert, R. W., Hradil, V. P., Harlan, J., et al. (2008). Non-invasive characterization of beta-amyloid(1-40) vasoactivity by functional magnetic resonance imaging in mice. Neuroscience 155, 263–269. doi: 10.1016/j.neuroscience.2008.04.021
Maat-Schieman, M. L., van Duinen, S. G., Rozemuller, A. J., Haan, J., and Roos, R. A. (1997). Association of vascular amyloid beta and cells of the mononuclear phagocyte system in hereditary cerebral hemorrhage with amyloidosis (Dutch) and Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 273–284. doi: 10.1097/00005072-199703000-00006
Maeda, J., Ji, B., Irie, T., Tomiyama, T., Maruyama, M., Okauchi, T., et al. (2007). Longitudinal, quantitative assessment of amyloid, neuroinflammation, and anti-amyloid treatment in a living mouse model of Alzheimer's disease enabled by positron emission tomography. J. Neurosci. 27, 10957–10968. doi: 10.1523/JNEUROSCI.0673-07.2007
Maeda, J., Zhang, M. R., Okauchi, T., Ji, B., Ono, M., Hattori, S., et al. (2011). In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer's disease and related disorders. J. Neurosci. 31, 4720–4730. doi: 10.1523/JNEUROSCI.3076-10.2011
Maruyama, M., Shimada, H., Suhara, T., Shinotoh, H., Ji, B., Maeda, J., et al. (2013). Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 79, 1094–1108. doi: 10.1016/j.neuron.2013.07.037
Masamoto, K., and Kanno, I. (2012). Anesthesia and the quantitative evaluation of neurovascular coupling. J. Cereb. Blood Flow. Metab. 32, 1233–1247. doi: 10.1038/jcbfm.2012.50
Massaad, C. A., Amin, S. K., Hu, L., Mei, Y., Klann, E., and Pautler, R. G. (2010). Mitochondrial superoxide contributes to blood flow and axonal transport deficits in the Tg2576 mouse model of Alzheimer's disease. PLoS ONE 5:e10561. doi: 10.1371/journal.pone.0010561
Matsuoka, Y., Saito, M., LaFrancois, J., Saito, M., Gaynor, K., Olm, V., et al. (2003). Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J. Neurosci. 23, 29–33.
McGowan, E., Pickford, F., Kim, J., Onstead, L., Eriksen, J., Yu, C., et al. (2005). Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47, 191–199. doi: 10.1016/j.neuron.2005.06.030
McLaurin, J., Kierstead, M. E., Brown, M. E., Hawkes, C. A., Lambermon, M. H., Phinney, A. L., et al. (2006). Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nat. Med. 12, 801–808. doi: 10.1038/nm1423
McLean, D., Cooke, M. J., Albay, R. 3rd., Glabe, C., and Shoichet, M. S. (2013). Positron emission tomography imaging of fibrillar parenchymal and vascular amyloid-β in TgCRND8 mice. ACS Chem. Neurosci. 4, 613–623. doi: 10.1021/cn300226q
Mellin, A. F., Cofer, G. P., Smith, B. R., Suddarth, S. A., Hedlund, L. W., and Johnson, G. A. (1994). Three dimensional magnetic resonance microangiography of rat neurovasculature. Mag. Res. Med. 32, 199–205. doi: 10.1002/mrm.1910320208
Melnikova, T., Fromholt, S., Kim, H., Lee, D., Xu, G., Price, A., et al. (2013). Reversible pathologic and cognitive phenotypes in an inducible model of Alzheimer-amyloidosis. J. Neurosci. 33, 3765–3779. doi: 10.1523/JNEUROSCI.4251-12.2013
Merlini, M., Meyer, E. P., Ulmann-Schuler, A., and Nitsch, R. M. (2011). Vascular β-amyloid and early astroctye alterations impair cerebrovascular function and cerebral metabolism in transgenic arcAβ mice. Acta Neuropathol. 122, 293–311. doi: 10.1007/s00401-011-0834-y
Meyer, E. P., Ulmann-Schuler, A., Staufenbiel, M., and Krucker, T. (2008). Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 105, 3587–3592. doi: 10.1073/pnas.0709788105
Miao, J., Xu, F., Davis, J., Otte-Holler, I., Verbeek, M. M., and van Nostrand, W. E. (2005). Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am. J. Pathol. 167, 505–515. doi: 10.1016/S0002-9440(10)62993-8
Moechars, D., Dewachter, I., Lorent, K., Reversé, D., Baekelandt, V., Naidu, A., et al. (1999). Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J. Biol. Chem. 274, 6483–6492. doi: 10.1074/jbc.274.10.6483
Montagne, A., Gauberti, M., Macrez, R., Jullienne, A., Briens, A., Raynaud, J. S., et al. (2012). Ultra-sensitive molecular MRI of cerebrovascular cell activation enables early detection of chronic central nervous system disorders. Neuroimage 63, 760–770. doi: 10.1016/j.neuroimage.2012.07.018
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058.
Mueggler, T., Baumann, D., Rausch, M., Staufenbiel, M., and Rudin, M. (2003). Age-dependent impairment of somatosensory response in the amyloid precursor protein 23 transgenic mouse model of Alzheimer's disease. J. Neurosci. 23, 8231–8236.
Mueggler, T., Sturchler-Pierrat, C., Baumann, D., Rausch, M., Staufenbiel, M., and Rudin, M. (2002). Compromized hemodynamic response in amyloid precursor protein transgenic mice. J. Neurosci. 22, 7218–7224.
Nabuurs, R. J., Rutgers, K. S., Welling, M. M., Metaxas, A., de Backer, M. E., Rotman, M., et al. (2012). In vivo detection of amyloid-b deposits using heavy chain antibody fragments in a transgenic mouse model for Alzheimer's disease. PLoS ONE 7:e38284. doi: 10.1371/journal.pone.0038284
Nagata, K., Maruya, H., Yuya, H., Terashi, H., Mito, Y., Kato, H., et al. (2000). Can PET data differentiate Alzheimer's disease from vascular dementia? Ann. N.Y. Acad. Sci. 903, 252–261. doi: 10.1111/j.1749-6632.2000.tb06375.x
Nilsson, L. N., Arendash, G. W., Leighty, R. E., Costa, D. A., Low, M. A., Garcia, M. F., et al. (2004). Cognitive impairment in PDAPP mice depends on ApoE and ACT-catalyzed amyloid formation. Neurobiol. Aging 25, 1153–1167. doi: 10.1016/j.neurobiolaging.2003.12.011
Niwa, K., Kazama, K., Younkin, L., Younkin, S. G., Carlson, G. A., and Iadecola, C. (2002). Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am. J. Physiol. Heart Circ. Physiol. 283, H315–H323. doi: 10.1152/ajpheart.00022.2002
Niwa, K., Porter, V. A., Kazama, K., Cornfield, D., Carlson, G. A., and Iadecola, C. (2001). Abeta-peptides enhance vasoconstriction in cerebral circulation. Am. J. Physiol. Heart Circ. Physiol. 281, H2417–H2424.
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
Oberg, J., Spenger, C., Wang, F. H., Andersson, A., Westman, E., Skoglund, P., et al. (2008). Age related changes in brain metabolites observed by H-1 MRS in APP/PS1 mice. Neurobiol. Aging 29, 1423–1433. doi: 10.1016/j.neurobiolaging.2007.03.002
Oddo, S., Caccamo, A., Kitazawa, M., Tseng, B. P., and LaFerla, F. M. (2003a). Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol. Aging 24, 1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., and Kayed, R. (2003b). Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/S0896-6273(03)00434-3
Ogawa, S., Tank, D. W., Menon, R., Ellermann, J. M., Kim, S. G., and Merkle, H. (1992). Intrinsic signal changes accompanying sensory stimulation: functional brain mapping with magnetic resonance imaging. Proc. Nat. Acad. Sci. U.S.A. 89, 5951–5955. doi: 10.1073/pnas.89.13.5951
Okamoto, Y., Yamamoto, T., Kalaria, R. N., Senzaki, H., Maki, T., Hase, Y., et al. (2012). Cerebral hypoperfusion accelaretes cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 123, 381–394. doi: 10.1007/s00401-011-0925-9
Okamura, N., Furumoto, S., Harada, R., Tago, T., Yoshikawa, T., Fodero-Tavoletti, M., et al. (2012). Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J. Nucl. Med. 54, 1420–1427. doi: 10.2967/jnumed.112.117341
Okamura, N., Suemoto, T., Shiomitsu, T., Suzuki, M., Shimadzu, H., Akatsu, H., et al. (2004). A novel imaging probe for in vivo detection of neuritic and diffuse amyloid plaques in the brain. J. Mol. Neurosci. 24, 247–255. doi: 10.1385/JMN:24:2:247
O'Leary, T. P., and Brown, R. E. (2009). Visuo-spatial learning and memory deficits on the Barnes maze in the 16-month-old APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav. Brain Res. 201, 120–127. doi: 10.1016/j.bbr.2009.01.039
Owen, D. R., Lindsay, A. C., Choudhury, R. P., and Fayad, Z. A. (2011). Imaging of atherosclerosis. Annu. Rev. Med. 62, 25–40. doi: 10.1146/annurev-med-041709-133809
Paris, D., Humphrey, J., Quadros, A., Patel, N., Crescentini, R., Crawford, F., et al. (2003). Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer's disease: role of inflammation. Neurol. Res. 25, 642–651. doi: 10.1179/016164103101201940
Paris, D., Town, T., Mori, T., Parker, T. A., Humphrey, J., and Mullan, M. (2000). Soluble beta-amyloid peptides mediate vasoactivity via activation of a pro-inflammatory pathway. Neurobiol. Aging 21, 183–197. doi: 10.1016/S0197-4580(00)82162-4
Park, L., Zhou, P., Koizumi, K., El Jamal, S. E., Previti, M. L., van Nostrand, W. E., et al. (2013). Brain and circulating levels of Aβ 1-40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke 44, 198–204. doi: 10.1161/STROKEAHA.112.670976
Park, L., Zhou, P., Pistick, R., Capone, C., Anrather, J., Norris, E. H., et al. (2008). Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 105, 1347–1352. doi: 10.1073/pnas.0711568105
Paul, J., Strickland, S., and Melchor, J. P. (2007). Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J. Exp. Med. 204, 1999–2008. doi: 10.1084/jem.20070304
Perez-Cruz, C., Nolte, M. W., van Gaalen, M. M., Rustay, N. R., Termont, A., Tanghe, A., et al. (2011). Reduced spine density in specific regions of CA1 pyramidal neurons in two transgenic mouse models of Alzheimer's disease. J. Neurosci. 31, 3926–3934. doi: 10.1523/JNEUROSCI.6142-10.2011
Perez-Torres, C. J., Reynolds, J. O., and Pautler, R. G. (2014). Use of magnetization transfer contrast MRI to detect early molecular pathology in Alzheimer's disease. Magn. Reson. Med. 71, 333–338. doi: 10.1002/mrm.24665
Pettersen, J. A., Sathiyamoorthy, G., Gao, F. Q., Szilagyi, G., Nadkarni, N. K., St. George-Hyslop, P., et al. (2008). Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook Dementia study. Arch. Neurol. 65, 790–795. doi: 10.1001/archneur.65.6.790
Pfeifer, M., Boncristiano, S., Bondolfi, L., Stalder, A., Deller, T., Staufenbiel, M., et al. (2002). Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 298, 1379. doi: 10.1126/science.1078259
Poduslo, J. F., Hultman, K. L., Curran, G. L., Preboske, G. M., Chamberlain, R., Marjañska, M., et al. (2011). Targeting vascular amyloid in arterioles of Alzheimer disease transgenic mice with amyloid β protein antibody-coated nanoparticles. J. Neuropathol. Exp. Neurol. 70, 653–661. doi: 10.1097/NEN.0b013e318225038c
Poisnel, G., Herard, A. S., El Tannir El Tayara, N., Bourrin, E., Vol, A., Kober, F., et al. (2012). Increased regional cerebral glucose uptake in an APP/PS1 model of Alzheimer's disease. Neurobiol. Aging 33, 1995–2005. doi: 10.1016/j.neurobiolaging.2011.09.026
Prada, C. M., Garcia-Alloza, M., Betensky, R. A., Zhang-Nunes, S. X., Greenberg, S. M., Bacskai, B. J., et al. (2007). Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J. Neurosci. 27, 1973–1980. doi: 10.1523/JNEUROSCI.5426-06.2007
Princz-Kranz, F. L., Mueggler, T., Knobloch, M., Nitsch, R. M., and Rudin, M. (2010). Vascular response to acetazolamide decreases as a function of age in the arcA beta mouse model of cerebral amyloidosis. Neurobiol. Dis. 40, 284–292. doi: 10.1016/j.nbd.2010.06.002
Radde, R., Bolmont, T., Kaeser, S. A., Coomaraswamy, J., Lindau, D., Stoltze, L., et al. (2006). Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946. doi: 10.1038/sj.embor.7400784
Rausch, M., Scheffler, K., Rudin, M., and Radü, E. W. (2000). Analysis of input functions from different arterial branches with gamma variate functions and cluster analysis for quantitative blood volume measurements. Magn. Reson. Imaging 18, 1235–1243. doi: 10.1016/S0730-725X(00)00219-8
Redwine, J. M., Kosofsky, B., Jacobs, R. E., Games, D., Reilly, J. F., Morrison, J. H., et al. (2003). Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc. Natl. Acad. Sci. U.S.A. 100, 1381–1386. doi: 10.1073/pnas.242746599
Reese, T., Bochelen, D., Sauter, A., Beckmann, N., and Rudin, M. (1999). Magnetic resonance angiography of the rat cerebrovascular system without the use of contrast agents. NMR Biomed. 12, 189–196.
Reiman, E. M., Quiroz, Y. T., Fleisher, A. S., Chen, K., Velez-Pardo, C., Jimenez-Del-Rio, M., et al. (2012). Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 11, 1048–1056. doi: 10.1016/S1474-4422(12)70228-4
Richards, J. G., Higgins, G. A., Ouagazzal, A. M., Ozmen, L., Kew, J. N., Bohrmann, B., et al. (2003). PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 23, 8989–9003.
Robbins, E. M., Betensky, R. A., Domnitz, S. B., Purcell, S. M., Garcia-Alloza, M., Greenberg, C., et al. (2006). Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J. Neurosci. 26, 365–371. doi: 10.1523/JNEUROSCI.3854-05.2006
Rockenstein, E., Mallory, M., Mante, M., Sisk, A., and Masliaha, E. (2001). Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42). J. Neurosci. Res. 66, 573–582. doi: 10.1002/jnr.1247
Roher, A. E., Esh, C., Kokjohn, T. A., Kalback, W., Luehrs, D. C., Seward, J. D., et al. (2003). Circle of willis atherosclerosis is a risk factor for sporadic Alzheimer's disease. Arterioscler. Thromb. Vasc. Biol. 23, 2055–2062. doi: 10.1161/01.ATV.0000095973.42032.44
Rojas, S., Herance, J. R., Gispert, J. D., Abad, S., Torrent, E., Jiménez, X., et al. (2013). In vivo evaluation of amyloid deposition and brain glucose metabolism of 5xFAD mice using positron emission tomography. Neurobiol. Aging 34, 1790–1798. doi: 10.1016/j.neurobiolaging.2012.12.027
Rönnbäck, A., Sagelius, H., Bergstedt, K. D., Näslund, J., Westermark, G. T., Winblad, B., et al. (2012). Amyloid neuropathology in the single Arctic APP transgenic model affects interconnected brain regions. Neurobiol. Aging 33, 831.e11–831.e19. doi: 10.1016/j.neurobiolaging.2011.07.012
Rönnbäck, A., Zhu, S., Dillner, K., Aoki, M., Lilius, L., Näslund, J., et al. (2011). Progressive neuropathology and cognitive decline in a single Arctic APP transgenic mouse model. Neurobiol. Aging 32, 280–292. doi: 10.1016/j.neurobiolaging.2009.02.021
Rupp, N. J., Wegenast-Braun, B. M., Radde, R., Calhoun, M. E., and Jucker, M. (2011). Early onset amyloid lesions lead to severe neuritic abnormalities and local, but not global neuron loss in APPPS1 transgenic mice. Neurobiol. Aging 32, 2324.e2321–2324.e2326. doi: 10.1016/j.neurobiolaging.2010.08.014
Rutgers, K. S., van Remoortere, A., van Buchem, M. A., Verrips, C. T., Greenberg, S. M., and Bacskai, B. J. (2009). Differential recognition of vascular and parenchymal beta amyloid deposition. Neurobiol. Aging 32, 1774–1783. doi: 10.1016/j.neurobiolaging.2009.11.012
Sadowski, M., Pankiewicz, J., Scholtzova, H., Yong, J., Quartermain, D., Jensen, C. H., et al. (2004). Amyloid-beta deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 63, 418–428.
Sanganahalli, B. G., Herman, P., Behar, K. L., Blumenfeld, H., Rothman, D. L., and Hyder, F. (2013). Functional MRI and neural responses in a rat model of Alzheimer's disease. Neuroimage 79, 404–411. doi: 10.1016/j.neuroimage.2013.04.099
SantaCruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Savonenko, A., Xu, G. M., Melnikova, T., Morton, J. L., Gonzales, V., Wong, M. P., et al. (2005). Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer's disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 18, 602–617. doi: 10.1016/j.nbd.2004.10.022
Schmitz, C., Rutten, B. P., Pielen, A., Schäfer, S., Wirths, O., Tremp, G., et al. (2004). Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer's disease. Am. J. Pathol. 164, 1495–1502. doi: 10.1016/S0002-9440(10)63235-X
Schroeter, S., Khan, K., Barbour, R., Doan, M., Chen, M., Guido, T., et al. (2008). Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J. Neurosci. 28, 6787–6793. doi: 10.1523/JNEUROSCI.2377-07.2008
Schulz, K., Sydekum, E., Krueppel, R., Engelbrecht, C. J., Schlegel, F., Schröter, A., et al. (2012). Simultaneous BOLD fMRI and fiber-optic calcium recording in rat neocortex. Nat. Methods 9, 597–602. doi: 10.1038/nmeth.2013
Schwendele, B., Brawek, B., Hermes, M., and Garaschuk, O. (2012). High-resolution in vivo imaging of microglia using a versatile nongenetically encoded marker. Eur. J. Immunol. 42, 2187–2196. doi: 10.1002/eji.201242436
Shibata, M., Ohtani, R., Ihara, M., and Tomimoto, H. (2004). White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35, 2598–2603. doi: 10.1161/01.STR.0000143725.19053.60
Smith, K. D., Kallhoff, V., Zheng, H., and Pautler, R. G. (2007). In vivo axonal transport rates decrease in a mouse model of Alzheimer's disease. Neuroimage 35, 1401–1408. doi: 10.1016/j.neuroimage.2007.01.046
Snellman, A., Lopez-Picon, F. R., Rokka, J., Salmona, M., Forloni, G., Scheinin, M., et al. (2013). Longitudinal amyloid imaging in mouse brain with 11C-PIB: comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of Alzheimer disease. J. Nucl. Med. 54, 1434–1441. doi: 10.2967/jnumed.112.110163
Snowdon, D. A. (2003). Nun study, healthy aging and dementia: findings from the Nun study. Ann. Intern. Med. 139(5 Pt 2), 450–454. doi: 10.7326/0003-4819-139-5_Part_2-200309021-00014
Song, S. K., Kim, J. H., Lin, S. J., Brendza, R. P., and Holtzman, D. M. (2004). Diffusion tensor imaging detects age-dependent white matter changes in a transgenic mouse model with amyloid deposition. Neurobiol. Dis. 15, 640–647. doi: 10.1016/j.nbd.2003.12.003
Sparks, D. L., Sabbagh, M., Connor, D., Soares, H., Lopez, J., Stankovic, G., et al. (2006). Statin therapy in Alzheimer's disease. Acta Neurol. Scand. Suppl. 185, 78–86. doi: 10.1111/j.1600-0404.2006.00689.x
Spencer, N. G., Bridges, L. R., Elderfield, K., Amir, K., Austen, B., and Howe, F. A. (2013). Quantitative evaluation of MRI and histological characteristics of the 5xFAD Alzheimer mouse brain. Neuroimage 76, 108–115. doi: 10.1016/j.neuroimage.2013.02.071
Sperling, R. A., Jack, C. R. Jr., Black, S. E., Frosch, M. P., Greenberg, S. M., Hyman, B. T., et al. (2011). Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 7, 367–385. doi: 10.1016/j.jalz.2011.05.2351
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE 5:e9979. doi: 10.1371/journal.pone.0009979
Starmans, L. W., van Duijnhoven, S. M., Rossin, R., Aime, S., Daemen, M. J., Nicolay, K., et al. (2013). SPECT imaging of fibrin using fibrin-binding peptides. Contrast Media Mol. Imaging 8, 229–237. doi: 10.1002/cmmi.1521
Starr, J. M., Farrall, A. J., Armitage, P., McGurn, B., and Wardlaw, J. (2009). Blood-brain barrier permeability in Alzheimer's disease: a case-control MRI study. Psychiatry Res. 171, 232–241. doi: 10.1016/j.pscychresns.2008.04.003
Stover, K. R., and Brown, R. E. (2012). Age-related changes in visual acuity, learning and memory in the APPswe/PS1dE9 mouse model of Alzheimer's disease. Behav. Brain Res. 231, 75–85. doi: 10.1016/j.bbr.2012.02.044
Sturchler-Pierrat, C., Abramowski, D., Duke, M., Wiederhold, K. H., Mistl, C., Rothacher, S., et al. (1997). Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. U.S.A. 94, 13287–13292. doi: 10.1073/pnas.94.24.13287
Sun, S. W., Song, S. K., Harms, M. P., Lin, S. J., Holtzman, D. M., Merchant, K. M., et al. (2005). Detection of age-dependent brain injury in a mouse model of brain amyloidosis associated with Alzheimer's disease using magnetic resonance diffusion tensor imaging. Exp. Neurol. 191, 77–85. doi: 10.1016/j.expneurol.2004.09.006
Takano, T., Han, X., Deane, R., Zlokovic, B., and Nedergaard, M. (2007). Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer's disease. Ann. N.Y. Acad. Sci. 1097, 40–50. doi: 10.1196/annals.1379.004
Takano, T., Tian, G. F., Peng, W., Lou, N., Libionka, W., and Han, X. (2006). Astrocyte-mediated control of cerebral blood flow. Nat. Neurosci. 9, 260–267. doi: 10.1038/nn1623
Takeda, S., Sato, N., Takeuchi, D., Kurinami, H., Shinohara, M., Niisato, K., et al. (2009). Angiotensin receptor blocker prevented beta-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension 54, 1345–1352. doi: 10.1161/HYPERTENSIONAHA.109.138586
Talagala, S. L., Jungreis, C. A., Kanal, E., Meyers, S. P., Foo, T. K. F., Rubin, R. A., et al. (1995). Fast three-dimensional time-of-flight MR angiography of the intracranial vasculature. J. Magn. Reson. Imaging 5, 317–323. doi: 10.1002/jmri.1880050316
Tanghe, A., Termont, A., Merchiers, P., Schilling, S., Demuth, H. U., Scrocchi, L., et al. (2010). Pathological hallmarks, clinical parallels, and value for drug testing in Alzheimer's disease of the APP[V717I] London transgenic mouse model. Int. J. Alzheimers Dis. 2010:pii: 417314. doi: 10.4061/2010/417314
Thakker, D. R., Weatherspoon, M. R., Harrison, J., Keene, T. E., Lane, D. S., Kaemmerer, W. F., et al. (2009). Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc. Natl. Acad. Sci. U.S.A. 106, 4501–4506. doi: 10.1073/pnas.0813404106
Thal, D. R., Capetillo-Zarate, E., Larionov, S., Staufenbiel, M., Zurbruegg, S., and Beckmann, N. (2009). Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol. Aging 30, 1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017
Thal, D. R., Griffin, W. S., and Braak, H. (2008b). Parenchymal and vascular Abeta-deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J. Cell. Mol. Med. 12, 1848–1862. doi: 10.1111/j.1582-4934.2008.00411.x
Thal, D. R., Griffin, W. S., de Vos, R. A., and Ghebremedhin, E. (2008a). Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol. 115, 599–609. doi: 10.1007/s00401-008-0366-2
Thanopoulou, K., Fragkouli, A., Stylianopoulou, F., and Georgopoulos, S. (2010). Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 20816–20821. doi: 10.1073/pnas.1005888107
Tofts, P. S., and Kernode, A. G. (1991). Measurement of the blood-brain barrier permeability and leakage space using dynamic MR imaging: 1. Fundamental concepts. Magn. Reson. Med. 17, 357–367. doi: 10.1002/mrm.1910170208
Tong, X. K., Lecrux, C., and Hamel, E. (2012). Age-dependent rescue by simvastatin of Alzheimer's disease cerebrovascular and memory deficits. J. Neurosci. 32, 4705–4715. doi: 10.1523/JNEUROSCI.0169-12.2012
Tong, X. K., Nicolakakis, N., KocharyanA., and Hamel, E. (2005). Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J. Neurosci. 25, 11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005
Tropres, I., Grimault, S., Vaeth, A., Grillon, E., Julien, C., Payen, J. F., et al. (2001). Vessel size imaging. Magn. Reson. Med. 45, 397–408. doi: 10.1002/1522-2594(200103)45:3<397::AID-MRM1052>3.0.CO;2-3
van Assema, D. M., Lubberink, M., Bauer, M., van der Flier, W. M., Schuit, R. C., Windhorst, A. D., et al. (2012). Blood-brain barrier P-glycoprotein function in Alzheimer's disease. Brain 135(Pt 1), 181–189. doi: 10.1093/brain/awr298
van Dorpe, J., Smeijers, L., Dewachter, I., Nuyens, D., Spittaels, K., Van Den Haute, C., et al. (2000). Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am. J. Pathol. 157, 1283–1298. doi: 10.1016/S0002-9440(10)64644-5
Vanhoutte, G., Dewachter, I., Borghgraef, P., Van Leuven, F., and Van der Linden, A. (2005). Noninvasive in vivo MRI detection of neuritic plaques associated with iron in APP[V717I] transgenic mice, a model for Alzheimer's disease. Magn. Reson. Med. 53, 607–613. doi: 10.1002/mrm.20385
Venneti, S., Lopresti, B. J., Wang, G., Hamilton, R. L., Mathis, C. A., and Klunk, W. E. (2009). PK11195 labels activated microglia in Alzheimer's disease and in vivo in a mouse model using PET. Neurobiol. Aging 30, 1217–1226. doi: 10.1016/j.neurobiolaging.2007.11.005
Villringer, A., Rosen, B. R., Bellieau, J. W., Ackerman, J. L., Lauffer, R. B., Buxton, R. B., et al. (1998). Dynamic imaging with lanthanide chelates in normal brain contrast due to magnetic susceptibility effects. Magn. Res. Med. 6, 164–174. doi: 10.1002/mrm.1910060205
Vinters, H. V. (1987). Cerebral amyloid angiopathy. A critical review. Stroke 18, 311–324. doi: 10.1161/01.STR.18.2.311
Vinters, H. V., Natté, R., Maat-Schieman, M. L., van Duinen, S. G., Hegeman-Kleinn, I., Welling-Graafland, C., et al. (1998). Secondary microvascular degeneration in amyloid angiopathy of patients with hereditary cerebral hemorrhage with amyloidosis, Dutch type (HCHWA-D). Acta Neuropathol. (Berl.) 95, 235–244. doi: 10.1007/s004010050793
Vom Berg, J., Prokop, S., Miller, K. R., Obst, J., Kalin, R. E., Lopategui-Cabezas, I., et al. (2012). Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat. Med. 18, 1812–1819. doi: 10.1038/nm.2965
Vromman, A., Trabelsi, N., Rouxel, C., Béréziat, G., Limon, I., and Blaise, R. (2013). β-Amyloid context intensifies vascular smooth muscle cells induced inflammatory response and de-differentiation. Aging Cell 12, 358–369. doi: 10.1111/acel.12056
Wang, F. H., Appelkvist, P., Klason, T., Gissberg, O., Bogstedt, A., Eliason, K., et al. (2012c). Decreased axonal transport rates in the Tg2576 APP transgenic mouse: improvement with the gamma-secretase inhibitor MRK-560 as detected by manganese-enhanced MRI. Eur. J. Neurosci. 36, 3165–3172. doi: 10.1111/j.1460-9568.2012.08258.x
Wang, H., He, J., Zhang, R., Zhu, S., Wang, J., Kong, L., et al. (2012a). Sensorimotor gating and memory deficits in an APP/PS1 double transgenic mouse model of Alzheimer's disease. Behav. Brain Res. 233, 237–243. doi: 10.1016/j.bbr.2012.05.007
Wang, J., Ho, L., Chen, L., Zhao, Z., Zhao, W., Qian, X., et al. (2007). Valsartan lowers brain b-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J. Clin. Invest. 117, 3393–3402. doi: 10.1172/JCI31547
Wang, M., Iliff, J. J., Liao, Y., Chen, M. J., Shinseki, M. S., Venkataraman, A., et al. (2012b). Cognitive deficits and delayed neuronal loss in a mouse model of multiple microinfarcts. J. Neurosci. 32, 17948–17960. doi: 10.1523/JNEUROSCI.1860-12.2012
Washida, K., Ihara, M., Nishio, K., Fujita, Y., Maki, T., Yamada, M., et al. (2010). Nonhypotensive dose of telmisartan attenuates cognitive impairment partially due to peroxisome proliferator-activated receptor-gamma activation in mice with chronic cerebral hypoperfusion. Stroke 41, 1798–1806. doi: 10.1161/STROKEAHA.110.583948
Watts, J. C., Giles, K., Grillo, S. K., Lemus, A., DeArmond, S. J., and Prusiner, S. B. (2011). Bioluminescence imaging of Abeta deposition in bigenic mouse models of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 108, 2528–2533. doi: 10.1073/pnas.1019034108
Wegiel, J., Imaki, H., Wang, K. C., Wegiel, J., and Rubenstein, R. (2004). Cells of monocyte/microglial lineage are involved in both microvessel amyloidosis and fibrillar plaque formation in APPsw tg mice. Brain Res. 1022, 19–29. doi: 10.1016/j.brainres.2004.06.058
Weidensteiner, C., Metzger, F., Bruns, A., Bohrmann, B., Kuennecke, B., and von Kienlin, M. (2009). Cortical hypoperfusion in the B6.PS2APP mouse model for Alzheimer's disease: comprehensive phenotyping of vascular and tissular parameters by MRI. Magn. Reson. Med. 62, 35–45. doi: 10.1002/mrm.21985
Weiss, C., Venkatasubramanian, P. N., Aguado, A. S., Power, J. M., Tom, B. C., Li, L., et al. (2002). Impaired eyeblink conditioning and decreased hippocampal volume in PDAPP V717F mice. Neurobiol. Dis. 11, 425–433. doi: 10.1006/nbdi.2002.0555
Weissleder, R., Elizondo, G., Wittenberg, J., Rabito, C., Bengele, H., and Josephson, L. (1990). Ultrasmall superparamagnetic iron oxide: characterization of a new class of contrast agents for MR imaging. Radiology 175, 489–493.
Weller, R. O. (1998). Cerebral amyloid angiopathy: amyloid β accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am. J. Pathol. 153, 725–733. doi: 10.1016/S0002-9440(10)65616-7
Weller, R. O., Boche, D., and Nicoll, J. A. (2009). Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol. 118, 87–102. doi: 10.1007/s00401-009-0498-z
Wilcock, D. M., Alamed, J., Gottschall, P. E., Grimm, J., Rosenthal, A., Pons, J., et al. (2006). Deglycosylated anti-amyloid-β antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J. Neurosci. 26, 5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006
Wilcock, D. M., Morgan, D., Gordon, M. N., Taylor, T. L., Ridnour, L. A., Wink, D. A., et al. (2011). Activation of matrix metalloproteinases following anti-Aβ immunotherapy; implications for microhemorrhage occurrence. J. Neuroinflammation 8, 115. doi: 10.1186/1742-2094-8-115
Wilcock, D. M., Rojiani, A., Rosenthal, A., Levkowitz, G., Subbarao, S., and Alamed, J. (2004b). Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J. Neurosci. 24, 6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004
Wilcock, D. M., Rojiani, A., Rosenthal, A., Subbarao, S., Freeman, M. J., Gordon, M. N., et al. (2004a). Passive immunotherapy against Aβ in aged APP transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J. Neuroinflammation 1, 24. doi: 10.1186/1742-2094-1-24
Williams, D. S., Detre, J. A., Leigh, J. S., and Koretsky, A. P. (1992). Magnetic resonance imaging of perfusion using spin inversion of arterial water. Proc. Natl. Acad. Sci. U.S.A. 89, 212–216. doi: 10.1073/pnas.89.1.212
Winkler, D. T., Bondolfi, L., Herzig, M. C., Jann, L., Calhoun, M. E., Wiederhold, K. H., et al. (2001). Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci. 21, 1619–1627.
Wolozin, B., Kellman, W., Ruosseau, P., Celesia, G. G., and Siegel, G. (2000). Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57, 1439–1443. doi: 10.1001/archneur.57.10.1439
Wong, D. F., Rosenberg, P. B., Zhou, Y., Kumar, A., Raymont, V., Ravert, H. T., et al. (2010). In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J. Nucl. Med. 51, 913–920. doi: 10.2967/jnumed.109.069088
Woolley, M. L., and Ballard, T. M. (2005). Age-related impairments in operant DMTP performance in the PS2APP mouse, a transgenic mouse model of Alzheimer's disease. Behav. Brain Res. 161, 220–228. doi: 10.1016/j.bbr.2005.02.007
Wright, A. L., Zinn, R., Hohensinn, B., Konen, L. M., Beynon, S. B., Tan, R. P., et al. (2013). Neuroinflammation and neuronal loss precede Aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease. PLoS ONE 8:e59586. doi: 10.1371/journal.pone.0059586
Wu, E. X., Tang, H., Asai, T., and Yan, S. D. (2004). Regional cerebral blood volume reduction in transgenic mutant APP (V717F, K670N/M671L) mice. Neurosci. Lett. 365, 223–227. doi: 10.1016/j.neulet.2004.05.004
Wunder, A., and Klohs, J. (2008). Optical imaging of vascular pathophysiology. Basic Res. Cardiol. 103, 182–190. doi: 10.1007/s00395-008-0712-5
Wunder, A., Klohs, J., and Dirnagl, U. (2009). Non-invasive visualization of CNS inflammation with nuclear and optical imaging. Neuroscience 158, 1161–1173. doi: 10.1016/j.neuroscience.2008.10.005
Xiong, H., Callaghan, D., Jones, A., Bai, J., Rasquinha, I., Smith, C., et al. (2009). ABCG2 is upregulated in Alzheimer's brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J. Neurosci. 29, 5463–5475. doi: 10.1523/JNEUROSCI.5103-08.2009
Xu, F., Grande, A. M., Robinson, J. K., Previti, M. L., Vasek, M., Davis, J., et al. (2007). Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid beta-protein precursor transgenic mice. Neuroscience 146, 98–107. doi: 10.1016/j.neuroscience.2007.01.043
Yang, J., Ji, Y., Mehta, P., Bates, K. A., Sun, Y., and Wisniewski, T. (2011). Blocking the ApolipoproteinE/Amyloid-b interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J. Alzheimers Dis. 24, 269–285. doi: 10.3233/JAD-2011-101401
Yarchoan, M., Xie, S. X., Kling, M. A., Toledo, J. B., Wolk, D. A., Lee, E. B., et al. (2012). Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 135, 3749–3756. doi: 10.1093/brain/aws271
Zha, Z., Choi, S. R., Ploessl, K., Lieberman, B. P., Qu, W., Hefti, F., et al. (2011). Multidentate (18)F-polypegylated styrylpyridines as imaging agents for Aβ plaques in cerebral amyloid angiopathy (CAA). J. Med. Chem. 54, 8085–8098. doi: 10.1021/jm2009106
Keywords: Alzheimer's disease (AD), amyloid-beta (Aβ), angiography, cerebral amyloid angiopathy (CAA), cerebral blood flow (CBF), magnetic resonance imaging (MRI), microscopy, transgenic mice
Citation: Klohs J, Rudin M, Shimshek DR and Beckmann N (2014) Imaging of cerebrovascular pathology in animal models of Alzheimer's disease. Front. Aging Neurosci. 6:32. doi: 10.3389/fnagi.2014.00032
Received: 25 October 2013; Accepted: 19 February 2014;
Published online: 13 March 2014.
Edited by:
Paula I. Moreira, University of Coimbra, PortugalReviewed by:
Marc Dhenain, Commissariat à l'Énergie Atomique, FranceSylvia Villeneuve, University of California, Berkeley, USA
Copyright © 2014 Klohs, Rudin, Shimshek and Beckmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicolau Beckmann, Analytical Sciences and Imaging, Novartis Institutes for BioMedical Research, Forum 1, Novartis Campus, WSJ-386.2.09, CH-4056 Basel, Switzerland e-mail:bmljb2xhdS5iZWNrbWFubkBub3ZhcnRpcy5jb20=