 Natalya Gertsik
Natalya Gertsik Danica Chiu
Danica Chiu Yue-Ming Li
Yue-Ming Li- 1Molecular Pharmacology and Chemistry Program, Memorial Sloan-Kettering Cancer Center, New York, NY, USA
- 2Biochemistry and Molecular Biology Program, Weill Graduate School of Medical Sciences of Cornell University, New York, NY, USA
- 3Program of Pharmacology, Weill Graduate School of Medical Sciences of Cornell University, New York, NY, USA
γ-Secretase is a four subunit, 19-pass transmembrane enzyme that cleaves amyloid precursor protein (APP), catalyzing the formation of amyloid beta (Aβ) peptides that form amyloid plaques, which contribute to Alzheimer’s disease (AD) pathogenesis. γ-Secretase also cleaves Notch, among many other type I transmembrane substrates. Despite its seemingly promiscuous enzymatic capacity, γ-secretase activity is tightly regulated. This regulation is a function of many cellular entities, including but not limited to the essential γ-secretase subunits, nonessential (modulatory) subunits, and γ-secretase substrates. Regulation is also accomplished by an array of cellular events, such as presenilin (active subunit of γ-secretase) endoproteolysis and hypoxia. In this review we discuss how γ-secretase is regulated with the hope that an advanced understanding of these mechanisms will aid in the development of effective therapeutics for γ-secretase-associated diseases like AD and Notch-addicted cancer.
Introduction
γ-Secretase is an intramembrane aspartyl protease that cleaves an array of type 1 transmembrane substrates, of which amyloid precursor protein (APP) and Notch are the most widely studied. APP undergoes sequential proteolytic processing by β-secretase (BACE1) and γ-secretase to generate amyloid beta (Aβ) peptides, which are 37–43 amino acids long. Notch, a protein that resides on the surface of signal-receiving cells as a heterodimeric receptor, is also subject to a series of proteolytic cleavages (Figure 1). The scientific scrutiny sustained by both APP and Notch results from their role in disease: aberrant γ-secretase cleavage of APP and Notch can lead to Alzheimer’s disease (AD) and cancer, respectively. γ-Secretase is an important potential drug target for both diseases and γ-secretase inhibitors (GSIs) and modulators (GSMs) are currently in clinical trials.
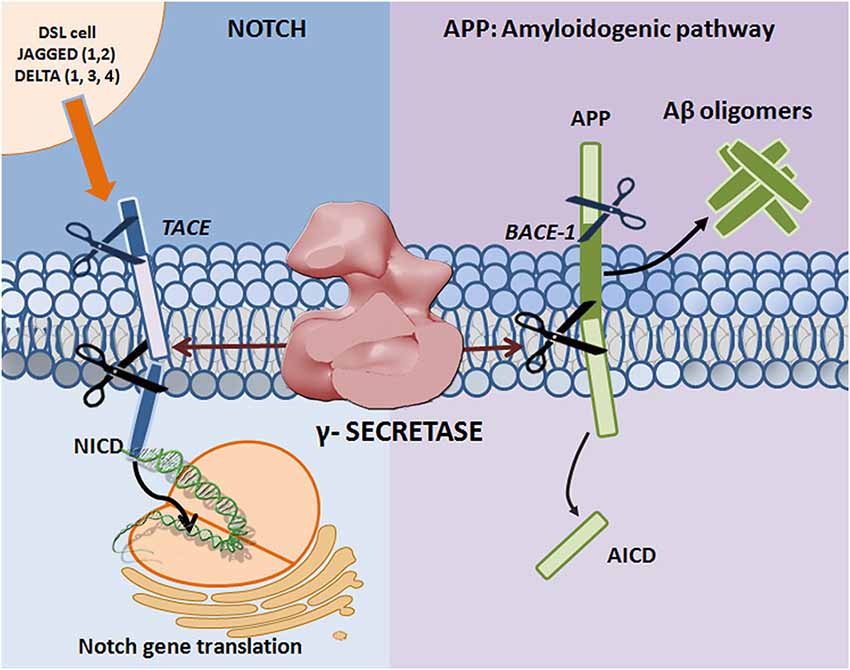
Figure 1. Proteolytic processing of APP and Notch. Mature Notch receptors are activated by binding to ligands (Jagged-1, -2 and Delta-like -1, -3, and -4) located on adjacent signal-presenting cells. An induced conformational change exposes a cleavage site (S2) for ADAM family metalloproteases that cleave Notch at an extracellular, membrane-proximal region. The membrane-bound Notch segment that results from this cleavage, known as Notch Intracellular Truncation domain (NEXT), is a γ-secretase substrate (Kopan and Ilagan, 2009). γ-Secretase performs the subsequent cleavage at S3 (De Strooper et al., 1999), releasing Notch intracellular domain (NICD) from the membrane and allowing for signal transduction through binding with the CBL-1, Su(H), Lag-1 (CSL; Schroeter et al., 1998; Struhl and Adachi, 1998) family of DNA binding proteins. APP undergoes sequential proteolytic processing first by β-secretase (BACE1, aspartyl protease) and then by γ-secretase, in the amyloidogenic pathway. The first cleavage results in ectodomain shedding in which the amino-terminal of APP is removed, yielding a soluble APP derivative (sAPPβ) and a carboxy-terminal membrane stub known as βCTF (C99). βCTF is a substrate for γ-secretase, and is cleaved in its transmembrane domain to form AICD and the potentially toxic Aβ. Mutations in presenilin (the catalytic subunit of γ-secretase) and APP can lead to increases in the Aβ42 to Aβ40 ratio, resulting in Aβ deposition and plaque formation.
Biochemical studies indicated that γ-secretase activity is catalyzed by the presenilin (PS)-containing macromolecular complex (Li et al., 2000a). The search for other components of the complex revealed three additional proteins: nicastrin (Nct), anterior pharynx-defective-1 (Aph-1), and presenilin enhancer-2 (Pen-2) (Yu et al., 2000; Francis et al., 2002; Goutte et al., 2002). It has since been established that these four proteins constitute the mature γ-secretase complex (De Strooper, 2003; Selkoe and Wolfe, 2007), and their stepwise assembly, followed by endoproteolysis of PS into amino-terminal (PS-NTF)and carboxy-terminal fragments (PS-CTF), is necessary for active complex formation (Takasugi et al., 2003) (Figure 2). The recent report of a 4.5Å cryo-electron microscopy structure of intact human γ-secretase and identification of novel γ-secretase modulating mechanisms have provided insight into the flexibility and complexity of this enzyme (Lu et al., 2014).
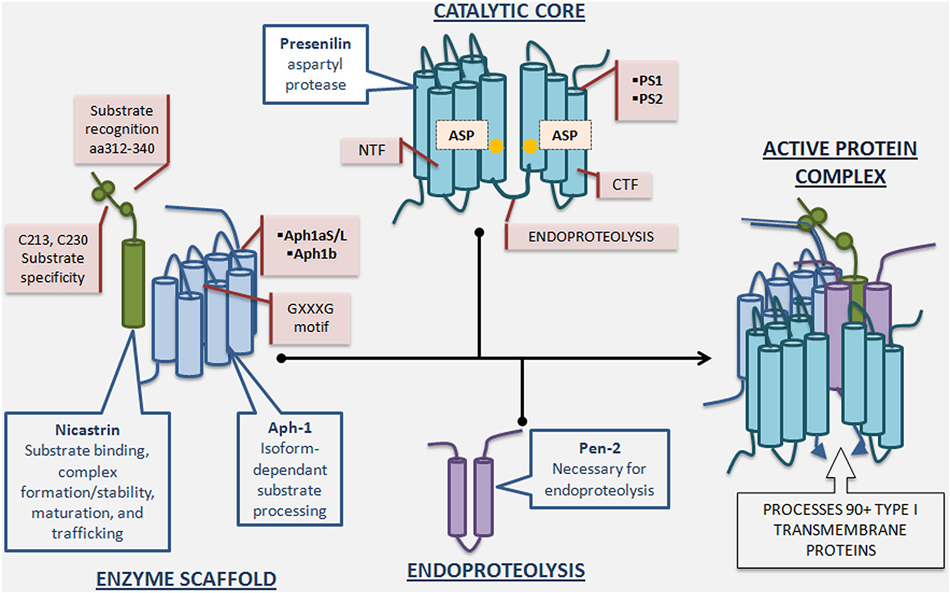
Figure 2. γ-Secretase complex formation and regulatory roles of individual essential subunits. The γ-Secretase complex is formed by the sequential assembly of Aph1, nicastrin, presenilin, and Pen-2. First, Aph-1 and nicastrin come together to form the scaffold. Next, full length presenilin is incorporated. Last, Pen-2 is recruited and full length presenilin is endoproteolysed into presenilin-NTF/CTF, activating the enzyme. Nicastrin, a heavily glycosylated single-pass transmembrane protein, plays a role in scaffolding, enzyme stabilization, substrate recognition, and trafficking. Nicastrin’s amino acids 312–340 are important for substrate recognition and deletion of these residues reduces γ-secretase activity and nicastrin’s interaction with APP and Notch. Furthermore, mutation of nicastrin’s C213 and C230 leads to different impact on processing of APP and Notch, underscoring nicastrin’s role in substrate selectivity. Aph-1, a 7-pass transmembrane protein with 3 human isoforms, is crucial for scaffolding and stability, and may have an additional role in determining length of Aβ species produced depending on which isoform is incorporated into the γ-secretase complex. The GXXXG motif in Aph-1 is critical for γ-secretase complex assembly. Presenilin, a 9-pass transmembrane protein with 2 isoforms, is the catalytic subunit of γ-secretase, and full length presenilin is a zymogen that must be endoproteolysed into NTF/CTF to be enzymatically active. Mutations in presenilin1 and presenilin2-encoding genes account for the majority of genetic mutations leading to Familial Alzheimer’s disease. Pen-2, a 2-pass transmembrane protein, is required for presenilin endoproteolysis and γ-secretase activation, but also may play an endoproteolysis-independent role in γ-secretase regulation. Active γ-secretase constitutes a small percentage of total γ-secretase and resides primarily in the plasma membrane.
γ-Secretase processing of its substrates produces distinct amino and carboxy termini with variable functions; some products of γ-secretase cleavage function as transcriptional regulators while others are thought to play roles in signaling, cell adhesion, and cytoskeletal dynamics. As the list of putative γ-secretase substrates continues to grow, now reaching over 90 reported proteins (Haapasalo and Kovacs, 2011), the lack of homology between these substrates becomes increasingly apparent: other than the fact that they are all type I transmembrane proteins that have undergone ectodomain shedding, γ-secretase substrates are surprisingly dissimilar (Beel and Sanders, 2008; Lleó, 2008) (of note, how many of these substrates are actually processed by γ-secretase in vivo remains to be investigated). Not only are the substrates themselves widely variable, but cleavage of some substrates (i.e., CD44 and Notch-1) (Lammich et al., 2002; Okochi et al., 2002) leads to release of peptides with variable carboxy-terminal ends, further confirming γ-secretase’s astounding promiscuity. In short, γ-secretase not only cleaves many substrates, but it cleaves the same substrate in many places. The permutation is daunting, and may be evolutionary evidence for γ-secretase’s initial function in regulated degradation of transmembrane proteins (Kopan and Ilagan, 2004). However, even if γ-secretase was ever simply a “proteasome of the membrane,” its function now is certainly much more complex. As a result, γ-secretase regulation must be at least as intricate and diverse as its function.
γ-Secretase activity is regulated by the assembly of its four essential subunits as well as at the level of the entire complex. Extensive investigation of the former revealed that each of the four essential γ-secretase subunits is tightly and independently controlled. More recently, the importance of regulation of the entire complex has emerged, suggesting additional levels of modulation in γ-secretase function. CD147, phospholipase D1, TMP21, GPR3, γ-secretase activating protein (GSAP), syntaxin-1, Arc, voltage-dependent anion channel 1 (VDAC1), contactin-associated protein 1 (CNTNAP1), TPPP, NDUFS7, Erlin-2, β-arrestin-1, β-arrestin-2, Hif-1α and Nexin 27 have all been implicated as nonessential γ-secretase interacting partners that modulate γ-secretase activity (Zhou et al., 2005; Cai et al., 2006; Chen et al., 2006; Thathiah et al., 2009; He et al., 2010; Teranishi et al., 2010; Wu et al., 2011; Frykman et al., 2012; Hur et al., 2012; Teranishi et al., 2012; Liu et al., 2013; Thathiah et al., 2013; Villa et al., 2014; Wang et al., 2014). However, some of this work is controversial and it remains to be seen whether many of these proteins play a specific and functionally significant role in γ-secretase regulation (Vetrivel et al., 2007, 2008; Hussain et al., 2013). Despite the uncertainty, much can be learned from the way in which a promiscuous enzyme is spatially and temporally modulated by its nonessential cofactors.
We begin by discussing the four essential subunits of γ-secretase, their physiological roles, and, where known, the way in which they regulate γ-secretase activity. Next we move to γ-secretase modulation by nonessential γ-secretase interacting partners, in particular GSAP and Hif-1α, which have been convincingly shown to specifically engage with the active γ-secretase complex. Last we comment on γ-secretase’s regulation by its substrate, APP.
γ-Secretase is Regulated by its Four Essential Subunits
γ-Secretase’s enzymatic promiscuity may on first glance suggest a kind of rampant cleavage activity that indiscriminately chops up anything in its way. Actually, the opposite is true. γ-Secretase may be flexible in its choice of substrate and cleavage site, but its activity is controlled in the cell by a variety of mechanisms, not the least of which is regulation of active complex formation. Perhaps the most basic evidence for this is the finding that γ-secretase activity cannot be increased through the overexpression of PS alone (Levitan et al., 2001), and can be reconstituted only when all four γ-secretase subunits are present (Edbauer et al., 2003). Put another way, the selective ablation of any one of the essential subunits leads to a loss of active complex and enzymatic activity (De Strooper, 2003). This implies that each subunit must be in the proper place and time if there is to be any hope for γ-secretase formation. However, the presence of all four essential subunits does not guarantee active complex formation. This is evidenced by the fact that only a small fraction of steady-state γ-secretase in the cell is actually catalytically active (Beher et al., 2003; Lai et al., 2003; Gu et al., 2004). Furthermore, while overexpression of wild type PS1 in mice is sufficient to increase the amount of γ-secretase complex and protease activity in brains, this is not the case in cellular studies (Li et al., 2011). The implication is that even when all four subunits are in complex with one another, additional events may be necessary to render that complex catalytically active. Some of these events are discussed in the “γ-secretase is regulated by modulatory proteins” section of this review.
The issue is further complicated by the fact that active complexes fall into a wide range of activities with respect to both catalytic efficiency and substrate specificity. Despite the deceptive language used here to describe γ-secretase as “active” and “inactive,” γ-secretase activity is far from a simple on/off switch. Let’s look at the statistics: assuming γ-secretase complexes have a 1:1:1:1 ratio of all essential subunits (Sato et al., 2007), at least four different complexes can be theoretically constructed, keeping in mind the existence of PS and Aph-1 (PS1/PS2 and Aph-1a/Aph-1b) isoforms and the finding that these isoforms do not co-exist in the same complexes (Lai et al., 2003; Shirotani et al., 2004). Identification of the Aph-1a splice variants (Aph-1aS and Aph-1aL) increased the permutation further, to a total of six (Shirotani et al., 2004). Experimentally, different γ-secretase complexes have indeed been identified and shown to vary in catalytic activity (Lai et al., 2003). Some tissue specificity has been observed in the expression of Aph-1 and PS variants, but this alone cannot account for determining which complex gets formed and which does not, especially since different variants of the γ-secretase complex exist dynamically in the same tissue, and even in the same cell line (Placanica et al., 2009a,b). More complicated mechanisms of regulating complex formation pervade, such as the ability of one isoform/mutant to outcompete the other for limiting factors (Placanica et al., 2009a).
The inherent complexity of γ-secretase can be put into perspective by comparing it to signal peptide peptidase (SPP), the only other family of intramembrane aspartyl proteases. Unlike γ-secretase, SPP appears to function alone without the participation of other protein co-factors (Weihofen et al., 2002), although it does form higher order oligomers (Nyborg et al., 2004, 2006; Miyashita et al., 2011). SPP’s simpler structure fits its function as a processor of signal peptides in the membrane, which may not require the same extent of regulation as γ-secretase. Below we discuss each essential γ-secretase subunit individually, paying particular attention to its role in regulating activity.
Presenilin
PS1 and and its less abundant isoform PS2, are ~50 kDa multipass transmembrane proteins that contain the catalytic core of the γ-secretase complex. These proteins were implicated in γ-secretase function when knock-out of PS1 resulted in severely reduced γ-secretase activity (De Strooper et al., 1998). For a long time it was unclear whether PS contains the active site of γ-secretase or is a chaperone involved in γ-secretase activity or colocalization to substrate. Several critical studies indicated that PS is indeed the catalytic subunit of γ-secretase. First, mutation of the two conserved aspartates in both PS1 (Wolfe et al., 1999) and PS2 (Steiner et al., 1999) significantly reduced Aβ production, suggesting that the aspartates are catalytic or essential residues for γ-secretase activity. Second, aspartyl protease transition-state analogs were shown to directly label and inhibit γ-secretase activity through covalent binding to PS, providing compelling evidence for PS as the catalytic core of γ-secretase (Esler et al., 2000; Li et al., 2000b). Finally, recombinant PS reconstituted into proteoliposomes was shown to be catalytically active even in the absence of other γ-secretase subunits (in contrast to cellular activity which requires all four subunits), providing conclusive evidence for PS’s role as the catalytic subunit of γ-secretase (Ahn et al., 2010).
PS is synthesized as a full length (FL) protein but PS-FL is unstable and is quickly either endoproteolysed or degraded (Ratovitski et al., 1997; Thinakaran et al., 1997; Zhang et al., 1998). Endoproteolysis, a process required for γ-secretase activation, results in the formation of PS1 amino-terminal and carboxy-terminal fragments (PS1-NTF/CTF), which remain associated as a stable heterodimer (Podlisny et al., 1997). In fact, the requirement for endoproteolysis is an onerous one to meet, as evidenced by the fact that in cells it is dependent on the assembly of all four subunits. Furthermore, the ability of FAD mutant PS1ΔE9 (Thinakaran et al., 1996) to be constitutively active despite its inability to be endoproteolysed suggests that evolution has found a way to increase enzymatic activity by circumnavigating an important requirement. A parallel can again be drawn to the PS-like enzyme SPP, which is not endoproteolysed and is active in its FL form, suggesting a simpler form of regulation. The exact molecular mechanism of γ-secretase endoproteolysis remains unclear, but one approach to study it is through the use of active-site directed probes (Gertsik et al., 2014).
As mentioned previously, the formation of an active complex is only the first hurdle in regulating γ-secretase activity in the cell. An added layer of regulation resides in the choice to form PS1 vs. PS2-containing complexes. Different PS variants play different, albeit overlapping, roles as evidenced by genetic knock out studies (De Strooper et al., 1998; Herreman et al., 1999). Later biochemical studies showed that PS1 complexes display a much higher activity than PS2 complexes for a truncated APP substrate (Lai et al., 2003).
PS1 is not only the more active of the two isoforms, but also may be the more amyloidogenic. A reconstitution study in which four γ-secretase isoforms (PS1–aph-1a, PS1–aph-1b, PS2–aph-1a, PS2–aph-1b) were analyzed showed that PS1 complexes form more aggregation-prone Aβ42 (relative to Aβ40) compared to PS2 complexes (Lee et al., 2011). This provides further evidence that the 67% homologous PS isoforms may have different cleavage-site preferences. It would be interesting to investigate whether the PS isoforms also have different substrate preferences. For example, do PS1/PS2 complexes differentially cleave APP/Notch? The decision to form PS1 vs. PS2 complexes probably determines not only “how much” but also “what” is cleaved.
Pen-2
Pen-2, a ~10 kDa protein with two TMDs (Crystal et al., 2003), was discovered as a gene product that can interfere with PS activity in a genetic study involving C. elegans (Francis et al., 2002). Pen-2 is required for endoproteolysis of PS-FL into PS-NTF/CTF and for γ-secretase activity. The following evidence supports that Pen-2 is indispensable for endoproteolysis of PS: first, knock-down of Pen-2 by RNAi resulted in a decrease of PS1-NTF/CTF and a stabilization of the PS1 holoprotein in the Nct-Aph-1 complex, while transient overexpression of Pen-2 in Pen-2 deficient cells led to the recovery of PS fragments (Takasugi et al., 2003). Second, coincorporation of recombinant PS1 and Pen-2 in liposomes showed Pen-2 to be necessary and sufficient for endoproteolysis of PS1 (Ahn et al., 2010). Pen-2 is also required for γ-secretase activity: Pen-2 knockdown in mammalian cells resulted not only in an accumulation of the PS1 holoprotein, but also in a drastic decrease in γ-secretase activity (Takasugi et al., 2003). Pen-2−/– mouse embryos exhibited a Notch-deficiency phenotype and Pen-2−/– MEFs displayed no γ-secretase activity toward APP processing (Bammens et al., 2011). Furthermore, overexpression of human Pen-2 in Pen-2 deficient mice recapitulated AD-like symptoms such as increase in Aβ42, behavioral dysfunctions, and feeding defects, underscoring the importance of Pen-2 in γ-secretase activity and AD pathogenesis (Nam et al., 2011).
The roles of Pen-2 in PS endoproteolysis and γ-secretase activity raised the question—is Pen-2 necessary for γ-secretase activity per se, or is Pen-2-inspired endoproteolysis of PS the only requirement for activity? To answer this question the catalytically active PS1ΔE9 endoproteolysis deficient mutant was expressed in Pen-2−/– MEFs and found to have no activity, suggesting that Pen-2 is required for γ-secretase activity per se, and not just for endoproteolysis of PS (Bammens et al., 2011). This implies that Pen-2 regulates γ-secretase on multiple levels. We already discussed that Pen-2 dictates activity, as PS-FL is a zymogen that relies on Pen-2-dependent endoproteolysis. This type of regulation is close to being an “on” switch, since lack of WT PS endoproteolysis precludes γ-secretase activity. However, Pen-2 is also capable of more subtle regulation in which it modulates the composition of the γ-secretase complex: overexpression of Pen-2 shifted the equilibrium from PS1 containing complexes to PS2 containing complexes and increased the Aβ42:Aβ40 ratio (Placanica et al., 2009a). Clearly, Pen-2 regulates γ-secretase through a variety of mechanisms, not the least of which are endoproteolysis of PS and complex assembly.
Nicastrin
In the search for cofactors required for γ-secretase activity, Nct, a type I transmembrane glycoprotein, was the first to be discovered through coimmunoprecipitation with PS1-directed antibody (Yu et al., 2000). A 1.95 Å-resolution crystal structure of Nct from an amoeboid eukaryote Dictyostelium purpureum has been solved (Xie et al., 2014). Four hydrophilic residues in the proximal one third of the N-terminal portion of the Nct TMD are critical for interaction between Nct and the rest of the γ-secretase complex (Capell et al., 2003). Nct interacts initially with Aph-1, followed by the incorporation of PS and Pen-2 (LaVoie et al., 2003). Not only does Nct, together with Aph-1, provide a scaffold for the γ-secretase complex, but it also may recognize γ-secretase substrates by binding to their amino termini. Particularly, Nct’s ectodomain has been shown to bind the extracellular regions of APP and Notch after they undergo ectodomain shedding, with Nct’s residues 312–340, and especially Glu333, being most important for substrate recognition (Shah et al., 2005). This finding branded Nct as the substrate-recruiting subunit of γ-secretase. Further confirmation came from work with anti-Nct antibodies: the monoclonal antibody A5226A binds the extracellular domain of Nct causing both a disruption in Nct binding to Notch-based substrate (N100) and a decrease in γ-secretase activity (Hayashi et al., 2011). Additionally, the use of synthetic antibodies showed that a certain structured region in Nct, homologous to the TPR domain involved in peptide recognition, is critical for substrate binding (Zhang et al., 2012). However, several studies called the “substrate-binding” capacity of Nct into question: mutation of mouse Nct-Glu332 (equivalent to human Glu333) to alanine or glutamine was reported to hinder assembly of the γ-secretase complex but not its specific activity, suggesting that substrate recognition/binding was not affected (Chávez-Gutiérrez et al., 2008). Furthermore, Nct-independent, L-685,458 specific, γ-secretase activity has since been detected in two separate MEF lines, suggesting that Nct is in fact not required for substrate recognition (Zhao et al., 2010). These results support Nct’s role in complex assembly and maturation, but not substrate recognition. A subsequent study reexamined the role of Nct and Glu333 in substrate recognition, overturning the previous finding and suggesting once again that Nct is indeed involved in substrate binding (Dries et al., 2009).
While Nct’s role as the substrate-binding subunit of γ-secretase may be controversial, its importance in γ-secretase regulation is uncontested: mutation of two conserved cysteine residues to serine (C213S and C230S) in Nct’s ectodomain resulted in differential γ-secretase processing of APP and Notch in MEF cells lacking endogenous Nct. In particular, APP processing was reduced compared to Notch, suggesting that Nct plays a role in substrate selectivity, although the exact mechanism was not identified (Pamrén et al., 2011). Furthermore, synthetic anti-Nct antibodies were shown to impact substrate selectivity by changing γ-secretase sub-cellular localization (Zhang et al., 2014). Quite possibly Nct regulates γ-secretase activity, and particularly substrate selectivity, through a variety of mechanisms including direct substrate binding, complex formation/stabilization, maturation, and trafficking.
Aph-1
Aph-1, a ~29 kDa protein with seven TMDs, was discovered in the same genetic screen as Pen-2 (Francis et al., 2002). The GXXXG motif of Aph-1’s TMD4 is crucial for assembly into the γ-secretase complex as it plays a major role in intramembrane helix-helix interactions. Mutation of Gly123 and Gly122 to aspartic acid in C. elegans and humans, respectively, results in a loss-of-function phenotype (LOF). The C. elegans LOF phenotype gave Aph-1 its name: anterior-pharynx-defective (Goutte et al., 2002). In mammals, mutation of Gly122 to aspartic acid renders Aph-1 incapable of associating with the γ-secretase complex, thereby leading to deficiency in Notch cleavage (Lee et al., 2004).
Humans have two Aph-1 genes that give rise to three versions of the Aph-1 protein (Aph-1aS, Aph-1aL, Aph-1b) due to alternative splicing of the Aph-1a gene (Shirotani et al., 2004). Rodents have an additional isoform, Aph1c, which is a duplication of the Aph-1b gene. Aph-1 isoforms have been reported to differ in their production of longer and shorter Aβ peptides. Particularly, when Aph-1a, Aph-1b, and Aph-1c were individually reintroduced into an Aph1-a−/–b−/–c−/– mouse, Aph1a rescue of γ-secretase activity resulted in production of shorter Aβ peptides while Aph-1b and Aph-1c rescue led to formation of longer Aβ species. A potential mechanism for this variability may stem from the structural changes evident in PS upon binding to one or the other Aph isoform: fluorescence lifetime imaging microscopy showed that PS may adopt a more closed conformation upon binding to Aph-1b compared to its more open conformation when in complex with Aph-1a (Serneels et al., 2009). This data implies that the choice to incorporate one Aph1 isoform over another can have a profound impact on Aβ production and plaque formation.
γ-Secretase is Regulated by Modulatory Proteins
γ-Secretase is regulated not only by its four essential subunits, but also by other pathways and proteins, many of which have been identified through LCMS analysis (Teranishi et al., 2010, 2012; Frykman et al., 2012). However, it is unclear whether most of these alleged γ-secretase-interacting partners actually bind the active complex and impact γ-secretase activity. Two γ-secretase-interacting partners, GSAP and Hif-1α, have been reproducibly shown to bind active γ-secretase and modulate γ-secretase activity, rendering them both biologically interesting and potentially clinically relevant. Here we describe what is known about the roles these “nonessential” γ-secretase subunits play in regulation.
GSAP
GSAP is a ~98 kDa holoprotein whose ~16 kDa processed fragment was identified as a γ-secretase-interacting partner. The story began when GSAP was implicated in the mechanism by which imatinib (Gleevec) selectively lowers Aβ without affecting Notch cleavage: a photoactivatable form of imatinib specifically labeled GSAP and not any of the γ-secretase subunits. Further investigation revealed that GSAP may form a complex with γ-secretase and APP-CTF, thereby imparting γ-secretase with substrate specificity for APP over Notch. GSAP RNAi mice crossed with double transgenic APPswe and PS1ΔE9 AD model mice presented a reduced Aβ burden (He et al., 2010). However, a subsequent study questioned GSAP’s role in γ-secretase regulation, reporting that GSAP does not interact with APP-CTF and does not enhance Aβ production in cells or in vitro (Hussain et al., 2013). The recent finding that imatinib does in fact regulate GSAP levels and γ-secretase activity supports GSAP’s role as a γ-secretase modulator (Chu et al., 2014). Furthermore, a SNP in GSAP has been found to associate with AD, revealing a new potential risk factor for the disease (Zhu et al., 2014). Taken together, the data provide strong evidence for GSAP’s role in modulating Aβ production, but the precise mechanism by which this occurs needs to be further investigated. GSAP’s function in Aβ formation is the topic of an ongoing debate (Alzforum, 2014).
Hif-1α
Hif-1α, a ~93 kDa protein, is a master regulator of cellular response to hypoxia. While Hif-1α’s properties as a transcription factor and role in Notch signaling are well documented (Gustafsson et al., 2005; Mukherjee et al., 2011; Wang et al., 2011), its role in γ-secretase regulation was recently discovered: Hif-1α directly binds to γ-secretase, stimulating its activity for Notch cleavage. Data show that hypoxic conditions enhance γ-secretase cleavage of Notch in a Hif-1α-dependent manner. Furthermore, it appears that the mechanism for γ-secretase activation is through a shift in equilibrium from inactive to active complexes upon Hif-1α binding, suggesting that Hif-1α has the capacity to turn an inactive γ-secretase into an active one (Villa et al., 2014). This is a common form of enzymatic regulation in which the enzyme is abundant in the cell in its “off” state and can be quickly mobilized should an “on” signal appear. Hif-1α studies presented the first evidence for the cell’s ability to activate γ-secretase complexes that were previously inactive, and for γ-secretase’s subjugation to temporal regulation (Figure 3).
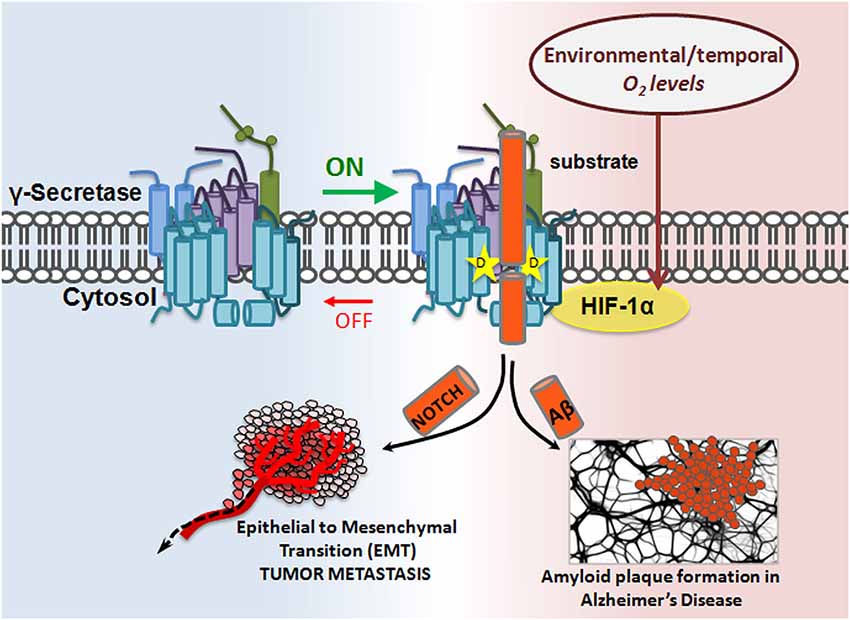
Figure 3. Modulatory-protein-based regulation of γ-secretase. Hif-1α, a master regulator of cellular response to hypoxia, is a γ-secretase interacting partner. In the presence of sufficient oxygen, no Hif-1α is produced. However, in oxygen-deficient conditions, Hif-1α is upregulated. Hif-1α binding to γ-secretase enhances γ-secretase activity by increasing the ratio of active:inactive γ-secretase complexes in the cell, thereby increasing γ-secretase cleavage of its substrates, such as APP and Notch. The implication is twofold: first, inactive γ-secretase complexes are physiologically important, and second, γ-secretase can be temporally regulated.
γ-Secretase is Regulated Via its Substrate, APP
APP proteolysis falls into one of two pathways: either APP is cleaved sequentially by α and γ-secretase in the nonamyloidogenic pathway, or it is cleaved by β and γ-secretase in the amyloidogenic pathway. α-Secretase cleavage of APP occurs in the Aβ region, which precludes Aβ production, so the choice to preferentially undergo more α cleavage can mean the difference between healthy and disease states. What factors determine the decision to participate in one pathway over the other? It has been suggested that α-secretase competes with β-secretase for the APP substrate, thereby lowering Aβ formation (Lammich et al., 1999; Skovronsky et al., 2000; Postina et al., 2004). However, compelling evidence exists for an alternate mechanism in which α-secretase cleavage results in formation of a substrate inhibitory domain (ASID) within αCTF that binds to an allosteric site in γ-secretase, thereby inhibiting γ-secretase processing of βCTF and ultimate Aβ production. In this model, α-secretase plays a dual anti-amyloidogenic role: first, it cleaves APP in the Aβ region, thereby directly precluding Aβ formation, and second, it initiates a feedback loop in which αCTF binds γ-secretase and acts as a γ-secretase modulator which specifically lowers Aβ production (Tian et al., 2010b). ASID is also present in βCTF, suggesting that the product of β-secretase cleavage is imbued with γ-secretase-regulating capacity as well. The Flemish FAD mutation, located in the ASID domain, interferes with βCTF’s inhibitory potency, leading to increased Aβ (Tian et al., 2010a). ASID is the first example of a substrate’s inherent ability to regulate γ-secretase, but it is probably not the last (reviewed in Zhang and Xu, 2010) (Figure 4).
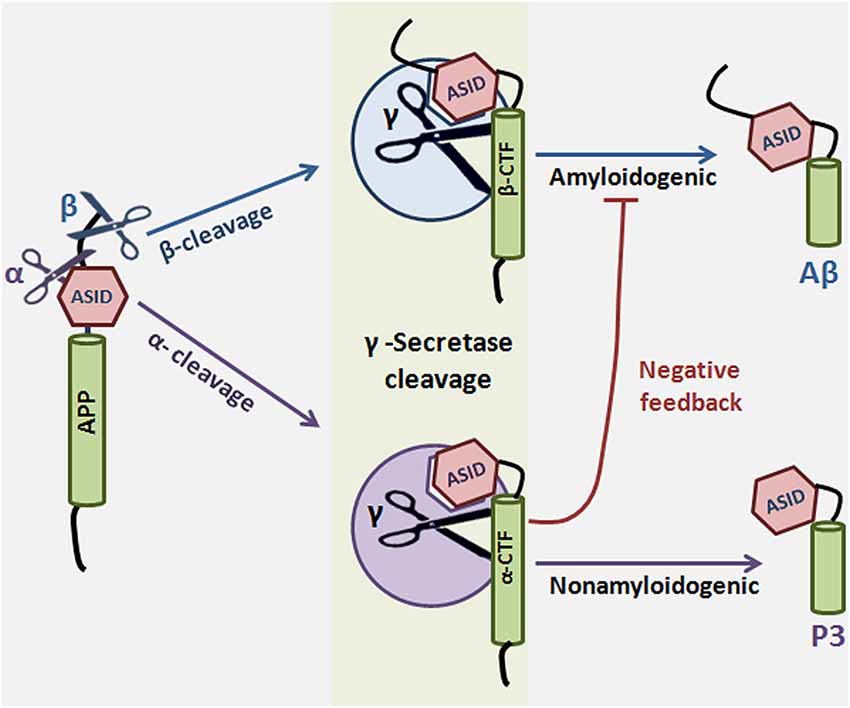
Figure 4. Substrate-based regulation of γ-Secretase. α or β-secretase cleave amyloid precursor protein to generate αCTF or βCTF, respectively. αCTF and βCTF, which are γ-secretase substrates, can then interact with γ-secretase to form a substrate-enzyme complex. The substrate inhibitory domain (ASID) in αCTF and βCTF interacts with γ-secretase in a location distinct from both the active site and the substrate binding site of the enzyme. Binding of αCTF to γ-secretase results in a low-productivity complex: the ASID in αCTF inhibits γ-secretase activity for Aβ production. ASID in βCTF is a less potent inhibitor of γ-secretase. The implication is that α-secretase cleavage potentiates γ-secretase inhibition by at least two mechanisms: first, α-secretase cleaves APP in the middle of the Aβ region, precluding Aβ formation, and second, α-secretase cleavage results in formation of αCTF, which inhibits γ-secretase activity through its ASID. γ-Secretase cleavage of αCTF produces p3 and cleavage of βCTF produces the amyloidogenic Aβ.
Conclusion
γ-Secretase is regulated at many levels, including but not limited to regulation by “essential” subunits, complex formation, “nonessential” subunits, substrates, and lipid composition (Holmes et al., 2012; Walter and Van Echten-Deckert, 2013). Although the importance of PS in γ-secretase activity is well established, it appears that the other essential subunits also notably contribute to regulating activity and substrate specificity beyond just their roles in complex assembly. Furthermore, nonessential subunits like GSAP and Hif-1α fine-tune modulation of the already stringently-regulated enzyme, thereby presenting potential therapeutic opportunities for modulating γ-secretase activity without the mechanism-based toxicities that result from γ-secretase inhibition (i.e., GSIs).
Currently, GSMs are being developed as a promising treatment for AD (Crump et al., 2013) and animal studies indicate that they could provide advantages over GSIs (Mitani et al., 2012; Rogers et al., 2012). Other Aβ-based therapies, such as immunotherapy and autophagic restoration, are being explored for AD as well (Lemere, 2013; Li et al., 2013). In addition to mice (Eimer and Vassar, 2013; Veeraraghavalu et al., 2013), other models have been investigated for AD studies (Mccoll et al., 2012; Do Carmo and Cuello, 2013), which help to elucidate the role of γ-secretase in pathogenesis. Recently, it has been suggested that a set of γ-secretase product peptides in the cerebrospinal fluid may serve as biomarkers for AD (Hata et al., 2012; Rosén et al., 2013). No doubt, developing a better understanding of γ-secretase in AD pathogenesis will facilitate the discovery of effective biomarkers and safe treatments. The complex levels of γ-secretase regulation that are now emerging may improve our ability to develop targeted therapies for AD and cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David B. Iaea for critically reading the manuscript. This work is supported by NIH grant R01AG026660 (Yue-Ming Li), R01NS076117 (Yue-Ming Li), Alzheimer Association IIRG-12-242137 (Yue-Ming Li), the MetLife Foundation (Yue-Ming Li), the JPB Foundation (Yue-Ming Li), training grant NIH T32 CA062948 (Danica Chiu), Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, the Experimental Therapeutics Center of MSKCC, and the William Randolph Hearst Fund in Experimental Therapeutics.
References
Ahn, K., Shelton, C. C., Tian, Y., Zhang, X., Gilchrist, M. L., Sisodia, S. S., et al. (2010). Activation and intrinsic γ-secretase activity of presenilin 1. Proc. Natl. Acad. Sci. U S A 107, 21435–21440. doi: 10.1073/pnas.1013246107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alzforum. (2014). GSAP revisited: does it really play a role in processing Aβ? http://www.alzforum.org/news/research-news/gsap-revisited-does-it-really-play-role-processing-av
Bammens, L., Chávez-Gutiérrez, L., Tolia, A., Zwijsen, A., and De Strooper, B. (2011). Functional and topological analysis of Pen-2, the fourth subunit of the gamma-secretase complex. J. Biol. Chem. 286, 12271–12282. doi: 10.1074/jbc.M110.216978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beel, A. J., and Sanders, C. R. (2008). Substrate specificity of gamma-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 65, 1311–1334. doi: 10.1007/s00018-008-7462-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beher, D., Fricker, M., Nadin, A., Clarke, E. E., Wrigley, J. D., Li, Y. M., et al. (2003). In vitro characterization of the presenilin-dependent gamma-secretase complex using a novel affinity ligand. Biochemistry 42, 8133–8142. doi: 10.1021/bi034045z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cai, D., Netzer, W. J., Zhong, M., Lin, Y., Du, G., Frohman, M., et al. (2006). Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc. Natl. Acad. Sci. U S A 103, 1941–1946. doi: 10.1073/pnas.0510708103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Capell, A., Kaether, C., Edbauer, D., Shirotani, K., Merkl, S., Steiner, H., et al. (2003). Nicastrin interacts with gamma-secretase complex components via the N-terminal part of its transmembrane domain. J. Biol. Chem. 278, 52519–52523. doi: 10.1074/jbc.c300435200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chávez-Gutiérrez, L., Tolia, A., Maes, E., Li, T., Wong, P. C., and De Strooper, B. (2008). Glu(332) in the Nicastrin ectodomain is essential for gamma-secretase complex maturation but not for its activity. J. Biol. Chem. 283, 20096–20105. doi: 10.1074/jbc.m803040200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, F., Hasegawa, H., Schmitt-Ulms, G., Kawarai, T., Bohm, C., Katayama, T., et al. (2006). TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature 440, 1208–1212. doi: 10.1038/nature04667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chu, J., Lauretti, E., Craige, C. P., and Pratico, D. (2014). Pharmacological modulation of GSAP reduces amyloid-beta levels and tau phosphorylation in a mouse model of Alzheimer’s disease with plaques and tangles. J. Alzheimers Dis. 41, 729–737. doi: 10.3233/JAD-140105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crump, C. J., Johnson, D. S., and Li, Y.-M. (2013). Development and mechanism of γ-secretase modulators for Alzheimer’s disease. Biochemistry 52, 3197–3216. doi: 10.1021/bi400377p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Crystal, A. S., Morais, V. A., Pierson, T. C., Pijak, D. S., Carlin, D., Lee, V. M., et al. (2003). Membrane topology of gamma-secretase component PEN-2. J. Biol. Chem. 278, 20117–20123. doi: 10.1074/jbc.m213107200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Strooper, B. (2003). Aph-1, Pen-2 and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 38, 9–12. doi: 10.1016/s0896-6273(03)00205-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Strooper, B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., et al. (1999). A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522.
De Strooper, B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., et al. (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390.
Do Carmo, S., and Cuello, A. (2013). Modeling Alzheimer’s disease in transgenic rats. Mol. Neurodegener. 8:37. doi: 10.1186/1750-1326-8-37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dries, D. R., Shah, S., Han, Y. H., Yu, C., Yu, S., Shearman, M. S., et al. (2009). Glu-333 of nicastrin directly participates in gamma-secretase activity. J. Biol. Chem. 284, 29714–29724. doi: 10.1074/jbc.m109.038737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edbauer, D., Winkler, E., Regula, J. T., Pesold, B., Steiner, H., and Haass, C. (2003). Reconstitution of gamma-secretase activity. Nat. Cell Biol. 5, 486–488. doi: 10.1038/ncb960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eimer, W., and Vassar, R. (2013). Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener. 8:2. doi: 10.1186/1750-1326-8-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Esler, W. P., Kimberly, W. T., Ostaszewski, B. L., Diehl, T. S., Moore, C. L., Tsai, J. Y., et al. (2000). Transition-state analogue inhibitors of gamma-secretase bind directly to presenilin-1. Nat. Cell Biol. 2, 428–434. doi: 10.1038/35017062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Francis, R., Mcgrath, G., Zhang, J., Ruddy, D. A., Sym, M., Apfeld, J., et al. (2002). aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP and presenilin protein accumulation. Dev. Cell 3, 85–97. doi: 10.1016/s1534-5807(02)00189-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frykman, S., Teranishi, Y., Hur, J. Y., Sandebring, A., Yamamoto, N. G., Ancarcrona, M., et al. (2012). Identification of two novel synaptic gamma-secretase associated proteins that affect amyloid beta-peptide levels without altering Notch processing. Neurochem. Int. 61, 108–118. doi: 10.1016/j.neuint.2012.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gertsik, N., Ballard, T. E., Am Ende, C. W., Johnson, D. S., and Li, Y. M. (2014). Development of CBAP-BPyne, a probe for gamma-secretase and presenilinase. Medchemcomm 5, 338–341. doi: 10.1039/c3md00281k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goutte, C., Tsunozaki, M., Hale, V. A., and Priess, J. R. (2002). APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U S A 99, 775–779. doi: 10.1073/pnas.022523499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gu, Y., Sanjo, N., Chen, F., Hasegawa, H., Petit, A., Ruan, X., et al. (2004). The presenilin proteins are components of multiple membrane-bound complexes that have different biological activities. J. Biol. Chem. 279, 31329–31336. doi: 10.1074/jbc.m401548200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gustafsson, M. V., Zheng, X., Pereira, T., Gradin, K., Jin, S., Lundkvist, J., et al. (2005). Hypoxia requires Notch signaling to maintain the undifferentiated cell state. Dev. Cell 9, 617–628. doi: 10.1016/j.devcel.2005.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haapasalo, A., and Kovacs, D. M. (2011). The many substrates of presenilin/gamma-secretase. J. Alzheimers Dis. 25, 3–28. doi: 10.3233/JAD-2011-101065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hata, S., Taniguchi, M., Piao, Y., Ikeuchi, T., Fagan, A., Holtzman, D., et al. (2012). Multiple gamma-secretase product peptides are coordinately increased in concentration in the cerebrospinal fluid of a subpopulation of sporadic Alzheimer’s disease subjects. Mol. Neurodegener. 7:16. doi: 10.1186/1750-1326-7-16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hayashi, I., Takatori, S., Urano, Y., Miyake, Y., Takagi, J., Sakata-Yanagimoto, M., et al. (2011). Neutralization of the gamma-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene 31, 787–798. doi: 10.1038/onc.2011.265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
He, G., Luo, W., Li, P., Remmers, C., Netzer, W. J., Hendrick, J., et al. (2010). Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. doi: 10.1038/nature09325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Herreman, A., Hartmann, D., Annaert, W., Saftig, P., Craessaerts, K., Serneels, L., et al. (1999). Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. U S A 96, 11872–11877. doi: 10.1073/pnas.96.21.11872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Holmes, O., Paturi, S., Ye, W., Wolfe, M. S., and Selkoe, D. J. (2012). Effects of membrane lipids on the activity and processivity of purified gamma-secretase. Biochemistry 51, 3565–3575. doi: 10.1021/bi300303g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hur, J. Y., Teranishi, Y., Kihara, T., Yamamoto, N. G., Inoue, M., Hosia, W., et al. (2012). Identification of novel gamma-secretase-associated proteins in detergent-resistant membranes from brain. J. Biol. Chem. 287, 11991–12005. doi: 10.1074/jbc.M111.246074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hussain, I., Fabrègue, J., Anderes, L., Ousson, S., Borlat, F., Eligert, V., et al. (2013). The role of gamma-secretase activating protein (GSAP) and imatinib in the regulation of gamma-secretase activity and amyloid-beta generation. J. Biol. Chem. 288, 2521–2531. doi: 10.1074/jbc.m112.370924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kopan, R., and Ilagan, M. X. (2004). Gamma-secretase: proteasome of the membrane? Nat. Rev. Mol. Cell Biol. 5, 499–504. doi: 10.1038/nrm1406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kopan, R., and Ilagan, M. X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. doi: 10.1016/j.cell.2009.03.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lai, M. T., Chen, E., Crouthamel, M. C., Dimuzio-Mower, J., Xu, M., Huang, Q., et al. (2003). Presenilin-1 and presenilin-2 exhibit distinct yet overlapping gamma-secretase activities. J. Biol. Chem. 278, 22475–22481. doi: 10.1074/jbc.m300974200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lammich, S., Kojro, E., Postina, R., Gilbert, S., Pfeiffer, R., Jasionowski, M., et al. (1999). Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. U S A 96, 3922–3927.
Lammich, S., Okochi, M., Takeda, M., Kaether, C., Capell, A., Zimmer, A. K., et al. (2002). Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Abeta-like peptide. J. Biol. Chem. 277, 44754–44759. doi: 10.1074/jbc.m206872200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
LaVoie, M. J., Fraering, P. C., Ostaszewski, B. L., Ye, W., Kimberly, W. T., Wolfe, M. S., et al. (2003). Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J. Biol. Chem. 278, 37213–37222. doi: 10.1074/jbc.m303941200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, J., Song, L., Terracina, G., Bara, T., Josien, H., Asberom, T., et al. (2011). Identification of presenilin 1-selective gamma-secretase inhibitors with reconstituted gamma-secretase complexes. Biochemistry 50, 4973–4980. doi: 10.1021/bi200026m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S. F., Shah, S., Yu, C., Wigley, W. C., Li, H., Lim, M., et al. (2004). A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. J. Biol. Chem. 279, 4144–4152. doi: 10.1074/jbc.m309745200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lemere, C. (2013). Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol. Neurodegener. 8:36. doi: 10.1186/1750-1326-8-36
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levitan, D., Lee, J., Song, L., Manning, R., Wong, G., Parker, E., et al. (2001). PS1 N- and C-terminal fragments form a complex that functions in APP processing and Notch signaling. Proc. Natl. Acad. Sci. U S A 98, 12186–12190. doi: 10.1073/pnas.211321898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y. M., Lai, M. T., Xu, M., Huang, Q., Dimuzio-Mower, J., Sardana, M. K., et al. (2000a). Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc. Natl. Acad. Sci. U S A 97, 6138–6143. doi: 10.1073/pnas.110126897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, T., Li, Y. M., Ahn, K., Price, D. L., Sisodia, S. S., and Wong, P. C. (2011). Increased expression of PS1 is sufficient to elevate the level and activity of gamma-secretase in vivo. PLoS One 6:e28179. doi: 10.1371/journal.pone.0028179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, W., Tang, Y., Fan, Z., Meng, Y., Yang, G., Luo, J., et al. (2013). Autophagy is involved in oligodendroglial precursor-mediated clearance of amyloid peptide. Mol. Neurodegener. 8:27. doi: 10.1186/1750-1326-8-27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y. M., Xu, M., Lai, M. T., Huang, Q., Castro, J. L., Dimuzio-Mower, J., et al. (2000b). Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. doi: 10.1038/35015085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, X., Zhao, X., Zeng, X., Bossers, K., Swaab, D. F., Zhao, J., et al. (2013). beta-Arrestin1 regulates gamma-secretase complex assembly and modulates amyloid-beta pathology. Cell Res. 23, 351–365. doi: 10.1038/cr.2012.167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lleó, A. (2008). Activity of gamma-secretase on substrates other than APP. Curr. Top. Med. Chem. 8, 9–16. doi: 10.2174/156802608783334060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, P., Bai, X. C., Ma, D., Xie, T., Yan, C., Sun, L., et al. (2014). Three-dimensional structure of human gamma-secretase. Nature 512, 166–170. doi: 10.1038/nature13567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mccoll, G., Roberts, B., Pukala, T., Kenche, V., Roberts, C., Link, C., et al. (2012). Utility of an improved model of amyloid-beta (Abeta1–42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 7:57. doi: 10.1186/1750-1326-7-57
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mitani, Y., Yarimizu, J., Saita, K., Uchino, H., Akashiba, H., Shitaka, Y., et al. (2012). Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J. Neurosci. 32, 2037–2050. doi: 10.1523/jneurosci.4264-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyashita, H., Maruyama, Y., Isshiki, H., Osawa, S., Ogura, T., Mio, K., et al. (2011). Three-dimensional structure of the signal peptide peptidase. J. Biol. Chem. 286, 26188–26197. doi: 10.1074/jbc.M111.260273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mukherjee, T., Kim, W. S., Mandal, L., and Banerjee, U. (2011). Interaction between Notch and Hif-alpha in development and survival of Drosophila blood cells. Science 332, 1210–1213. doi: 10.1126/science.1199643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nam, S. H., Seo, S. J., Goo, J. S., Kim, J. E., Choi, S. I., Lee, H. R., et al. (2011). Pen-2 overexpression induces Abeta-42 production, memory defect, motor activity enhancement and feeding behavior dysfunction in NSE/Pen-2 transgenic mice. Int. J. Mol. Med. 28, 961–971. doi: 10.3892/ijmm.2011.767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nyborg, A. C., Herl, L., Berezovska, O., Thomas, A. V., Ladd, T. B., Jansen, K., et al. (2006). Signal peptide peptidase (SPP) dimer formation as assessed by fluorescence lifetime imaging microscopy (FLIM) in intact cells. Mol. Neurodegener. 1:16. doi: 10.1186/1750-1326-1-16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nyborg, A. C., Kornilova, A. Y., Jansen, K., Ladd, T. B., Wolfe, M. S., and Golde, T. E. (2004). Signal peptide peptidase forms a homodimer that is labeled by an active site-directed gamma-secretase inhibitor. J. Biol. Chem. 279, 15153–15160. doi: 10.1074/jbc.m309305200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Okochi, M., Steiner, H., Fukumori, A., Tanii, H., Tomita, T., Tanaka, T., et al. (2002). Presenilins mediate a dual intramembranous gamma-secretase cleavage of Notch-1. EMBO J. 21, 5408–5416. doi: 10.1093/emboj/cdf541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pamrén, A., Wanngren, J., Tjernberg, L. O., Winblad, B., Bhat, R., Näslund, J., et al. (2011). Mutations in nicastrin protein differentially affect amyloid beta-peptide production and notch protein processing. J. Biol. Chem. 286, 31153–31158. doi: 10.1074/jbc.c111.235267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Placanica, L., Tarassishin, L., Yang, G., Peethumnongsin, E., Kim, S. H., Zheng, H., et al. (2009a). Pen2 and presenilin-1 modulate the dynamic equilibrium of presenilin-1 and presenilin-2 gamma-secretase complexes. J. Biol. Chem. 284, 2967–2977. doi: 10.1074/jbc.m807269200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Placanica, L., Zhu, L., and Li, Y. M. (2009b). Gender- and age-dependent gamma-secretase activity in mouse brain and its implication in sporadic Alzheimer disease. PLoS One 4:e5088. doi: 10.1371/journal.pone.0005088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Podlisny, M. B., Citron, M., Amarante, P., Sherrington, R., Xia, W., Zhang, J., et al. (1997). Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol. Dis. 3, 325–337. doi: 10.1006/nbdi.1997.0129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Postina, R., Schroeder, A., Dewachter, I., Bohl, J., Schmitt, U., Kojro, E., et al. (2004). A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 113, 1456–1464. doi: 10.1172/jci200420864e1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ratovitski, T., Slunt, H. H., Thinakaran, G., Price, D. L., Sisodia, S. S., and Borchelt, D. R. (1997). Endoproteolytic processing and stabilization of wild-type and mutant presenilin. J. Biol. Chem. 272, 24536–24541. doi: 10.1074/jbc.272.39.24536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rogers, K., Felsenstein, K., Hrdlicka, L., Tu, Z., Albayya, F., Lee, W., et al. (2012). Modulation of gamma-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Mol. Neurodegener. 7:61. doi: 10.1186/1750-1326-7-61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rosén, C., Hansson, O., Blennow, K., and Zetterberg, H. (2013). Fluid biomarkers in Alzheimer’s disease - current concepts. Mol. Neurodegener. 8:20. doi: 10.1186/1750-1326-8-20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, T., Diehl, T. S., Narayanan, S., Funamoto, S., Ihara, Y., De Strooper, B., et al. (2007). Active gamma-secretase complexes contain only one of each component. J. Biol. Chem. 282, 33985–33993. doi: 10.1074/jbc.m705248200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schroeter, E. H., Kisslinger, J. A., and Kopan, R. (1998). Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386.
Selkoe, D. J., and Wolfe, M. S. (2007). Presenilin: running with scissors in the membrane. Cell 131, 215–221. doi: 10.1016/j.cell.2007.10.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Serneels, L., Van Biervliet, J., Craessaerts, K., Dejaegere, T., Horré, K., Van Houtvin, T., et al. (2009). gamma-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer’s disease. Science 324, 639–642. doi: 10.1126/science.1171176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shah, S., Lee, S. F., Tabuchi, K., Hao, Y. H., Yu, C., Laplant, Q., et al. (2005). Nicastrin functions as a gamma-secretase-substrate receptor. Cell 122, 435–447. doi: 10.1016/j.cell.2005.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shirotani, K., Edbauer, D., Prokop, S., Haass, C., and Steiner, H. (2004). Identification of distinct gamma-secretase complexes with different APH-1 variants. J. Biol. Chem. 279, 41340–41345. doi: 10.1074/jbc.m405768200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Skovronsky, D. M., Moore, D. B., Milla, M. E., Doms, R. W., and Lee, V. M. (2000). Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J. Biol. Chem. 275, 2568–2575. doi: 10.1074/jbc.275.4.2568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steiner, H., Duff, K., Capell, A., Romig, H., Grim, M. G., Lincoln, S., et al. (1999). A loss of function mutation of presenilin-2 interferes with amyloid beta-peptide production and notch signaling. J. Biol. Chem. 274, 28669–28673. doi: 10.1074/jbc.274.40.28669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Struhl, G., and Adachi, A. (1998). Nuclear access and action of notch in vivo. Cell 93, 649–660. doi: 10.1016/s0092-8674(00)81193-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takasugi, N., Tomita, T., Hayashi, I., Tsuruoka, M., Niimura, M., Takahashi, Y., et al. (2003). The role of presenilin cofactors in the gamma-secretase complex. Nature 422, 438–441. doi: 10.1038/nature01506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Teranishi, Y., Hur, J. Y., Gu, G. J., Kihara, T., Ishikawa, T., Nishimura, T., et al. (2012). Erlin-2 is associated with active gamma-secretase in brain and affects amyloid beta-peptide production. Biochem. Biophys. Res. Commun. 424, 476–481. doi: 10.1016/j.bbrc.2012.06.137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Teranishi, Y., Hur, J. Y., Welander, H., Frånberg, J., Aoki, M., Winblad, B., et al. (2010). Affinity pulldown of gamma-secretase and associated proteins from human and rat brain. J. Cell. Mol. Med. 14, 2675–2686. doi: 10.1111/j.1582-4934.2009.00907.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thathiah, A., Horré, K., Snellinx, A., Vandewyer, E., Huang, Y., Ciesielska, M., et al. (2013). beta-arrestin 2 regulates Abeta generation and gamma-secretase activity in Alzheimer’s disease. Nat. Med. 19, 43–49. doi: 10.1038/nm.3023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thathiah, A., Spittaels, K., Hoffmann, M., Staes, M., Cohen, A., Horré, K., et al. (2009). The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science 323, 946–951. doi: 10.1126/science.1160649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thinakaran, G., Borchelt, D. R., Lee, M. K., Slunt, H. H., Spitzer, L., Kim, G., et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. doi: 10.1016/s0896-6273(00)80291-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thinakaran, G., Harris, C. L., Ratovitski, T., Davenport, F., Slunt, H. H., Price, D. L., et al. (1997). Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J. Biol. Chem. 272, 28415–28422. doi: 10.1074/jbc.272.45.28415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tian, Y., Bassit, B., Chau, D., and Li, Y. M. (2010a). An APP inhibitory domain containing the Flemish mutation residue modulates gamma-secretase activity for Abeta production. Nat. Struct. Mol. Biol. 17, 151–158. doi: 10.1038/nsmb.1743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tian, Y., Crump, C. J., and Li, Y. M. (2010b). Dual role of alpha-Secretase Cleavage in the regulation of gamma-secretase activity for amyloid production. J. Biol. Chem. 285, 32549–32556. doi: 10.1074/jbc.M110.128439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Veeraraghavalu, K., Choi, S., Zhang, X., and Sisodia, S. (2013). Endogenous expression of FAD-linked PS1 impairs proliferation, neuronal differentiation and survival of adult hippocampal progenitors. Mol. Neurodegener. 8:41. doi: 10.1186/1750-1326-8-41
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vetrivel, K. S., Gong, P., Bowen, J. W., Cheng, H., Chen, Y., Carter, M., et al. (2007). Dual roles of the transmembrane protein p23/TMP21 in the modulation of amyloid precursor protein metabolism. Mol. Neurodegener. 2:4. doi: 10.1186/1750-1326-2-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vetrivel, K. S., Zhang, X., Meckler, X., Cheng, H., Lee, S., Gong, P., et al. (2008). Evidence that CD147 modulation of beta-amyloid (Abeta) levels is mediated by extracellular degradation of secreted Abeta. J. Biol. Chem. 283, 19489–19498. doi: 10.1074/jbc.m801037200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villa, J. C., Chiu, D., Brandes, A. H., Escorcia, F. E., Villa, C. H., Maguire, W. F., et al. (2014). Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell Rep. 8, 1077–1092. doi: 10.1016/j.celrep.2014.07.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walter, J., and Van Echten-Deckert, G. (2013). Cross-talk of membrane lipids and Alzheimer-related proteins. Mol. Neurodegener. 8:34. doi: 10.1186/1750-1326-8-34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, X., Huang, T., Zhao, Y., Zheng, Q., Thompson, R. C., Bu, G., et al. (2014). Sorting Nexin 27 regulates abeta production through modulating gamma-secretase activity. Cell Rep. 9, 1023–1033. doi: 10.1016/j.celrep.2014.09.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Liu, Y., Malek, S. N., and Zheng, P. (2011). Targeting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8, 399–411. doi: 10.1016/j.stem.2011.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weihofen, A., Binns, K., Lemberg, M. K., Ashman, K., and Martoglio, B. (2002). Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218. doi: 10.1126/science.1070925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398, 513–517.
Wu, J., Petralia, R. S., Kurushima, H., Patel, H., Jung, M. Y., Volk, L., et al. (2011). Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell 147, 615–628. doi: 10.1016/j.cell.2011.09.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xie, T., Yan, C., Zhou, R., Zhao, Y., Sun, L., Yang, G., et al. (2014). Crystal structure of the gamma-secretase component nicastrin. Proc. Natl. Acad. Sci. U S A 111, 13349–13354. doi: 10.1073/pnas.1414837111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., et al. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407, 48–54. doi: 10.1038/35024009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Hoey, R., Koide, A., Dolios, G., Paduch, M., Nguyen, P., et al. (2014). Synthetic antibody fragment targeting nicastrin affects assembly and trafficking of gamma-secretase. J. Biol. Chem. doi: 10.1074/jbc.m114.609636. [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, X., Hoey, R. J., Lin, G., Koide, A., Leung, B., Ahn, K., et al. (2012). Identification of a tetratricopeptide repeat-like domain in the nicastrin subunit of gamma-secretase using synthetic antibodies. Proc. Natl. Acad. Sci. U S A 109, 8534–8539. doi: 10.1073/pnas.1202691109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, J., Kang, D. E., Xia, W., Okochi, M., Mori, H., Selkoe, D. J., et al. (1998). Subcellular distribution and turnover of presenilins in transfected cells. J. Biol. Chem. 273, 12436–12442. doi: 10.1074/jbc.273.20.12436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, Y. W., and Xu, H. (2010). Substrate check of gamma-secretase. Nat. Struct. Mol. Biol. 17, 140–141. doi: 10.1038/nsmb0210-140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, G., Liu, Z., Ilagan, M. X., and Kopan, R. (2010). Gamma-secretase composed of PS1/Pen2/Aph1a can cleave notch and amyloid precursor protein in the absence of nicastrin. J. Neurosci. 30, 1648–1656. doi: 10.1523/JNEUROSCI.3826-09.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, S., Zhou, H., Walian, P. J., and Jap, B. K. (2005). CD147 is a regulatory subunit of the gamma-secretase complex in Alzheimer’s disease amyloid beta-peptide production. Proc. Natl. Acad. Sci. U S A 102, 7499–7504. doi: 10.1073/pnas.0502768102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhu, M., Tao, Y., He, Q., Gao, H., Song, F., Sun, Y. M., et al. (2014). A common GSAP promoter variant contributes to Alzheimer’s disease liability. Neurobiol. Aging 35, 2656.e1–2656.e7. doi: 10.1016/j.neurobiolaging.2014.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: γ-Secretase, Alzheimer’s disease, presenilin, β-amyloid, Notch, APP, Hif-1α
Citation: Gertsik N, Chiu D and Li Y-M (2014) Complex regulation of γ-secretase: from obligatory to modulatory subunits. Front. Aging Neurosci. 6:342. doi: 10.3389/fnagi.2014.00342
Received: 25 November 2014; Paper pending published: 03 December 2014;
Accepted: 09 December 2014; Published online: 06 January 2015.
Edited by:
Xiongwei Zhu, Case Western Reserve University, USACopyright © 2014 Gertsik, Chiu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yue-Ming Li, Molecular Pharmacology and Chemistry Program, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, 10065 NY, USA e-mail:bGl5MkBtc2tjYy5vcmc=