 Jesús Avila
Jesús Avila Alberto Gómez-Ramos
Alberto Gómez-Ramos Marta Bolós
Marta Bolós- 1Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas, Madrid, Spain
- 2Centro de Biología Molecular “Severo Ochoa” CSIC-UAM, Madrid, Spain
Development of tau pathology is associated with progressive neuronal loss and cognitive decline. In the brains of Alzheimer's disease (AD) patients, tau pathology propagates according to an anatomically defined pattern with relatively uniform distribution, and contributes to cognitive decline in age-associated tauopathy (Braak and Braak, 1991; Saito et al., 2004). Recently, it has been revealed that tau, which is an intracellular protein, can appear in the extracellular space, likely due to an exocytosis mechanism. Such extracellular tau could then be internalized into neighboring cells in at least two different ways depending on its aggregation state. In the case of soluble monomeric or small oligomeric tau protein, the endocytosis appears to be clathrin dependent (reviewed in Rubinsztein, 2006). In contrast, larger aggregates of tau could bind heparin in the extracellular matrix and be internalized through macropinocytosis (Holmes et al., 2014). As a result of exocytosis and endocytosis, the spreading of tau can occur in various neurodegenerative diseases (tauopathies) including AD. In this opinion article we have focused on the endocytosis mechanism.
Several genetic risk factors have been associated with a higher probability of developing sporadic Alzheimer's disease (SAD). The Alzheimer Association (http://www.alzforum.org/) has ranked the top six risk genes, shown in Table 1, based on genome-wide association studies (GWAS).
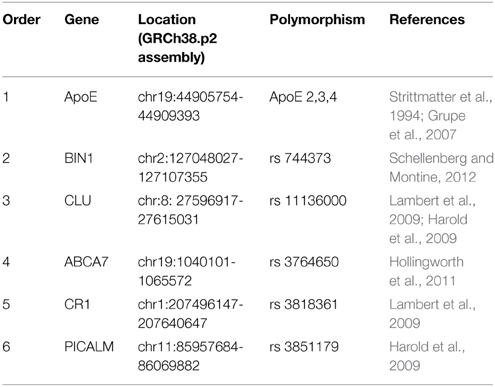
Table 1. The top six AD risk genes that interact with tau.
Binding of SAD Genetic Risk Factors to Tau Protein
The myc box-dependent-interacting protein 1, also known as bridging integrator 1 (BIN1), clusterin (clu), phosphatidylinositol binding clathrin assembly protein (PICALM), and apolipoprotein E (ApoE) are encoded by the BIN1, CLU, PICALM, and ApoE genes, respectively. These proteins are involved in the endocytosis of tau in either a direct (BIN1, CLU, and PICALM) or an indirect (ApoE) way, and all of them can also interact directly with tau (Chapuis et al., 2013; Tan et al., 2013; Holler et al., 2014; Moreau et al., 2014; Zhou et al., 2014). ApoE - mainly isoform ApoE3 - can bind efficiently to tau protein (Strittmatter et al., 1994). For BIN1, a novel brain-specific allele containing a 3 bp insertion has been reported that may be responsible for the interaction of BIN1 with tau (Tan et al., 2013). Intracellular clusterin interacts with brain isoforms of BIN1 and with tau (Zhou et al., 2014). Less is known about the interaction of PICALM and tau. However, Moreau et al. (2014) carried out groundbreaking work describing the relationship between PICALM and tau. They showed how PICALM-dependent autophagy can modulate tau accumulation in cells. Impaired autophagy could result in neurotoxicity and, consequently, might also be related to the spreading of tau pathology.
ApoE and clu (ApoJ) are related proteins. They are involved in cholesterol and lipid transport and can regulate Aβ endocytosis and Aβ clearance (Bertrand et al., 1995; Nuutinen et al., 2009). They also share some cellular receptors (Leeb et al., 2014). For example, both ApoE and clusterin bind to heparin (Cardin et al., 1986; Pankhurst et al., 1998), which in turn may affect endocytic processes such as macropinocytosis. Thus, it can be hypothesized that ApoE (and ApoJ) may, in this indirect way, regulate tau endocytosis.
BIN1 has been ranked as the second most important susceptibility locus for developing SAD. It is expressed from a single locus located on human chromosome 2 (Ren et al., 2006). The gene is transcribed into nuclear RNA that can produce different proteins by alternative splicing (Pineda-Lucena et al., 2005). Some of the BIN1 isoforms, such as isoforms 1-6, are specifically located in the brain (Butler et al., 1997; Tsutsui et al., 1997; Wechsler-Reya et al., 1997). Furthermore, BIN1 is mainly expressed in neurons and some brain isoforms are mainly expressed in the axon initial segment (Holler et al., 2014).
Extracellular Tau Endocytosis
Extracellular soluble tau (monomers, small oligomers) or larger aggregates of tau can be endocytosed by neurons in several ways. It was shown that neurons have cell receptors for extracellular tau, for example M1 and M3 muscarinic receptors (Gomez-Ramos et al., 2006, 2008). Once extracellular tau is bound to muscarinic receptors, it can be endocytosed in a clathrin-dependent process. This uptake mechanism could facilitate the spreading of tau from neuron to neuron, perhaps through synaptic transmission (De Calignon et al., 2012; Liu et al., 2012; Pooler et al., 2013). For aggregated tau, endocytosis is mediated by macropinocytosis (Holmes et al., 2014), in which components of the extracellular matrix, such as heparin sulfate, seem to play a role. Thus, two clearly different endocytic pathways have been proposed for the neuronal uptake of tau: receptor-clathrin dependent uptake of soluble tau species versus cell matrix-dependent endocytosis of tau aggregates.
In SAD, an increase in the expression of BIN1 and PICALM has been described, which could induce an increase in tau clathrin-mediated endocytosis. This could lead to the accumulation of soluble tau in tau recipient cells and may result in a toxic effect. On the other hand, a decrease in the level of clusterin, which like ApoE, is a heparin-binding protein, could enhance the binding of aggregated tau to the extracellular matrix and hence also enhance its endocytosis, resulting in a toxic effect in the recipient cell.
In summary, it is remarkable that four of the main genetic risk factors for Alzheimer's disease are tau-binding partners. Identifying how these risk factors affect tau propagation may unveil new therapeutic targets to stop or delay progression of pathology. For example, one possible way to reduce tau endocytosis and subsequent spreading of tau pathology may be to decrease neuronal levels of BIN1 and PICALM (which are increased in Alzheimer's disease) in early stages of the disease. Future research is therefore needed to better understand the interactions of tau with these proteins.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants of MINECO (SAF2014-5040-P), Spain and CIBERNED.
References
Bertrand, P., Poirier, J., Oda, T., Finch, C. E., and Pasinetti, G. M. (1995). Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Brain Res. Mol. Brain Res. 33, 174–178. doi: 10.1016/0169-328X(95)00097-C
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Butler, M. H., David, C., Ochoa, G. C., Freyberg, Z., Daniell, L., Grabs, D., et al. (1997). Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of ranvier in brain and around T tubules in skeletal muscle. J. Cell Biol. 137, 1355–1367. doi: 10.1083/jcb.137.6.1355
Cardin, A. D., Hirose, N., Blankenship, D. T., Jackson, R. L., Harmony, J. A., Sparrow, D. A., et al. (1986). Binding of a high reactive heparin to human apolipoprotein E: identification of two heparin-binding domains. Biochem. Biophys. Res. Commun. 29, 783–789. doi: 10.1016/S0006-291X(86)80489-2
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
De Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
Gomez-Ramos, A., Diaz-Hernandez, M., Cuadros, R., Hernandez, F., and Avila, J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850. doi: 10.1016/j.febslet.2006.07.078
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell. Neurosci. 37, 673–681. doi: 10.1016/j.mcn.2007.12.010
Grupe, A., Abraham, R., Li, Y., Rowland, C., Hollingworth, P., Morgan, A., et al. (2007). Evidence for novel susceptibility genes for late-onset Alzheimer's disease from a genome-wide association study of putative functional variants. Hum. Mol. Genet. 16, 865–873. doi: 10.1093/hmg/ddm031
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
Holler, C. J., Davis, P. R., Beckett, T. L., Platt, T. L., Webb, R. L., Head, E., et al. (2014). Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers. Dis. 42, 1221–1227. doi: 10.3233/JAD-132450
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J.-C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Holmes, B. B., Furman, J. L., Mahan, T. E., Yamasaki, T. R., Mirbaha, H., Eades, W. C., et al. (2014). Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E4376–E4385. doi: 10.1073/pnas.1411649111
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Leeb, C., Eresheim, C., and Nimpf, J. (2014). Clusterin is a ligand for apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and signals via the Reelin-signaling pathway. J. Biol. Chem. 289, 4161–4172. doi: 10.1074/jbc.M113.529271
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS ONE 7:e31302. doi: 10.1371/journal.pone.0031302
Moreau, K., Fleming, A., Imarisio, S., Lopez Ramirez, A., Mercer, J. L., Jimenez-Sanchez, M., et al. (2014). PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 5, 4998. doi: 10.1038/ncomms5998
Nuutinen, T., Suuronen, T., Kauppinen, A., and Salminen, A. (2009). Clusterin: a forgotten player in Alzheimer's disease. Brain Res. Rev. 61, 89–104. doi: 10.1016/j.brainresrev.2009.05.007
Pankhurst, G. J., Bennett, C. A., and Easterbrook-Smith, S. B. (1998). Characterization of the heparin-binding properties of human clusterin. Biochemistry 37, 4823–4830. doi: 10.1021/bi972367v
Pineda-Lucena, A., Ho, C. S., Mao, D. Y., Sheng, Y., Laister, R. C., Muhandiram, R., et al. (2005). A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J. Mol. Biol. 351, 182–194. doi: 10.1016/j.jmb.2005.05.046
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Ren, G., Vajjhala, P., Lee, J. S., Winsor, B., and Munn, A. L. (2006). The BAR domain proteins: molding membranes in fission, fusion, and phagy. Microbiol. Mol. Biol. Rev. 70, 37–120. doi: 10.1128/MMBR.70.1.37-120.2006
Rubinsztein, D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 doi: 10.1038/nature05291
Saito, Y., Ruberu, N. N., Sawabe, M., Arai, T., Tanaka, N., Kakuta, Y., et al. (2004). Staging of argyrophilic grains: an age-associated tauopathy. J. Neuropathol. Exp. Neurol. 63, 911–918.
Schellenberg, G. D., and Montine, T. J. (2012). The genetics and neuropathology of Alzheimer's disease. Acta Neuropathol. (Berl.) 124, 305–323. doi: 10.1007/s00401-012-0996-2
Strittmatter, W. J., Saunders, A. M., Goedert, M., Weisgraber, K. H., Dong, L. M., Jakes, R., et al. (1994). Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 11183–11186. doi: 10.1073/pnas.91.23.11183
Tan, M. S., Yu, J. T., and Tan, L. (2013). Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol. Med. 19, 594–603. doi: 10.1016/j.molmed.2013.06.004
Tsutsui, K., Maeda, Y., Seki, S., and Tokunaga, A. (1997). cDNA cloning of a novel amphiphysin isoform and tissue-specific expression of its multiple splice variants. Biochem. Biophys. Res. Commun. 236, 178–183. doi: 10.1006/bbrc.1997.6927
Wechsler-Reya, R., Sakamuro, D., Zhang, J., Duhadaway, J., and Prendergast, G. C. (1997). Structural analysis of the human BIN1 gene. Evidence for tissue-specific transcriptional regulation and alternate RNA splicing. J. Biol. Chem. 272, 31453–31458. doi: 10.1074/jbc.272.50.31453
Keywords: endocytosis, tau proteins, genetic factors, PICALM, BIN1 bridging Integrator 1/amphiphysin-2 gene
Citation: Avila J, Gómez-Ramos A and Bolós M (2015) AD genetic risk factors and tau spreading. Front. Aging Neurosci. 7:99. doi: 10.3389/fnagi.2015.00099
Received: 06 April 2015; Accepted: 08 May 2015;
Published: 21 May 2015.
Edited by:
Elena Galea, Universitat Autònoma de Barcelona, SpainReviewed by:
Amy Pooler, Nestlé Institute of Health Sciences, SwitzerlandSusanne Wegmann, Massachusetts General Hospital, USA
Copyright © 2015 Avila, Gómez-Ramos and Bolós. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Avila,amF2aWxhQGNibS5jc2ljLmVz