 Bing Bai
Bing Bai- Department of Laboratory Medicine, Nanjing Drum Tower Hospital, Nanjing University Medical School, Nanjing, China
The aberrancy of U1 small nuclear ribonucleoprotein (snRNP) complex and RNA splicing has been demonstrated in Alzheimer’s disease (AD). Importantly, the U1 proteopathy is AD-specific, widespread and early-occurring, thus providing a very unique clue to the AD pathogenesis. The prominent feature of U1 histopathology is its nuclear depletion and redistribution in the neuronal cytoplasm. According to the preliminary data, the initial U1 cytoplasmic distribution pattern is similar to the subcellular translocation of the spliceosome in cells undergoing mitosis. This implies that the U1 mislocalization might reflect the neuronal cell cycle-reentry (CCR) which has been extensively evidenced in AD brains. The CCR phenomenon explains the major molecular and cellular events in AD brains, such as Tau and amyloid precursor protein (APP) phosphorylation, and the possible neuronal death through mitotic catastrophe (MC). Furthermore, the CCR might be mechanistically linked to inflammation, a critical factor in the AD etiology according to the genetic evidence. Therefore, the discovery of U1 aberrancy might strengthen the involvement of CCR in the AD neuronal degeneration.
Introduction
Alzheimer’s disease (AD) is the most common dementia that is caused by the aging-related irreversible progression of neurodegeneration in the brain (Goedert and Spillantini, 2006; Roberson and Mucke, 2006). Besides the obvious brain atrophy with massive neuronal loss, AD brains are hallmarked by the deposition of extracellular amyloid plaques and intracellular neurofibrillary tangles, whose major components are Aβ peptides and the hyperphosphorylated microtubule-associated protein Tau (MAPT), respectively.
Currently, all three established familial AD genes (APP, PSEN1 and PSEN2) are directly involved in the Aβ generation (Guerreiro et al., 2013). Mutations that lead to enhanced Aβ production or fibrillization accelerate the onset of AD (Citron et al., 1992; Nilsberth et al., 2001), and those inhibiting Aβ generation reduce the likelihood of AD development (Jonsson et al., 2012). In combination with other lines of evidence from pathology (Serrano-Pozo et al., 2011; Selkoe, 2013), biochemistry (Cleary et al., 2005; Masters and Selkoe, 2012), cell biology (Shankar et al., 2008; Kuperstein et al., 2010) and animal models (Lesne et al., 2006; Oakley et al., 2006), it is well accepted that the amyloid cascade is the initiating event in the AD pathogenesis (Selkoe et al., 2012; Bloom, 2014; Selkoe and Hardy, 2016). However, the brain Aβ load has a fair correlation with the severity of dementia in patients (Giannakopoulos et al., 2003; Nelson et al., 2012); and the Aβ overexpression in mouse brains fails to elicit the dementia and neurofibrillary tangles to the extent manifested in AD patients (Oakley et al., 2006).
Although the neurofibrillary tangles have a better correlation with the severity of dementia, they are not specific for AD and appear in almost any kinds of brain diseases (Nelson et al., 2012). In addition, the neuronal stress has already been observed prior to tangle formation in the neuron and the number of dying neurons is often greater than the tangle-bearing neurons (Gómez-Isla et al., 1997; Hoozemans et al., 2009). Moreover, almost all people will eventually develop tangles during aging even their cognition is intact (Nelson et al., 2012). Collectively, the tangle formation might possibly be considered as a universal event that appears at the later stage of the neuronal dyshomeostasis. Therefore, the critical pathway besides the amyloid cascade that mediates the final neuronal degeneration in the AD pathogenesis still remains unclear.
U1 snRNP Proteopathy and RNA Splicing Deficiency in AD
It is known that some neurodegenerative disorders are caused by the aberrancy of RNA processing proteins (Neumann et al., 2006; Sreedharan et al., 2008; Mackenzie et al., 2010), but whether the pathogenesis of AD possibly involves a similar mechanism had remained unclear, until we have found the U1 small nuclear ribonucleoprotein (snRNP) complex pathology and the RNA splicing deficiency in AD patents (Bai et al., 2013).
The major human spliceosome is composed of five subunits, each comprising snRNPs in conjunction with a specific small nuclear RNA (snRNA) named as U1, U2, U4, U5 and U6 respectively (Wahl et al., 2009). The U1 snRNP contains the U1 snRNA in complex with U1-70K, U1A, SmD and other protein components, which are normally located in the neuronal nucleus. However, these proteins are aggregated and form tangle-like structures in the neuronal cytoplasm in AD brains (Bai et al., 2013; Hales et al., 2014a).
Notably, this U1 proteopathy, unlike Tau, is almost exclusively present in AD and Down’s syndrome (Bai et al., 2013; Hales et al., 2014b), but not in any other types of dementia or neurodegenerative diseases that involve no amyloid and Tau tangle pathologies, providing a unique clue to the mechanism of AD. Importantly, in these reports, the U1 aggregation occurs in the mild cognitive impairment, an early stage of dementia and often evolves into AD eventually.
Besides, we have also found that the RNA splicing is impaired in the AD brains, probably as a functional consequence of the U1 proteopathy. The transcriptomic analysis by RNA deep sequencing shows that the overall intronic reads are increased in AD as compared to those in the non-demented controls. Further examination reveals that the insufficient RNA splicing occurs extensively in individual genes. Nevertheless, the strongest evidence of U1 dysfunction might come from the phenomenon of PCPA (premature cleavage and polyadenylation) observed in the RNA transcriptome data. Besides RNA splicing, the U1 SnRNP complex has a unique function. It prevents the PCPA of the pre-RNA during transcription through binding to the putative polyadenylation sites (Kaida et al., 2010). This is also found the AD brains in our study in addition to the evidence of RNA splicing deficiency (Bai et al., 2013), confirming the dysfunction of U1 proteopathy. The following up studies by other groups demonstrate more possible roles of U1 in the autophagy-lysosome system and the presenilin protein (Cheng et al., 2017a,b, 2018), both of which are key players in the AD etiology. Indeed, the U1 deregulation in AD on particular genes has been reported previously (Manabe et al., 2007; Ohe and Mayeda, 2010).
Spliceosome Alterations Cause Neurodegeneration
It is well known that alterations in the RNA processing machineries can cause neurodegeneration. The first example is the spinal muscular atrophy (SMA), arising from mutations in the gene SMN that encodes the survival motor neuron protein (Lefebvre et al., 1995, 1997; Lorson et al., 1999). The SMA disease is characterized by the loss of motor neurons and the progressive muscle atrophy. The SMN protein, together with other proteins, forms the heptameric protein ring that commonly exists as a core in the U1, U2, U4, U5 and U6 snRNP complexes to catalyze their assembly (Matera and Wang, 2014; Wahl and Lührmann, 2015). The homozygous disruption of SMN causes this inheritable neuromuscular disorder and often leads to death in patients (Lunn and Wang, 2008). Consistently, experimental reduction in the SMN level in zebra fish or mice leads to motor neuron degeneration and its restoration rescues this deleterious effect (Winkler et al., 2005; Hua et al., 2011).
The next strong evidence is about U2 snRNA, the specific component of U2 snRNP subunit. Mutations in one of the copies of U2 snRNA genes lead to ataxia in mice, with extensive neurodegeneration and RNA splicing aberrancy in the cerebellum where the U2 snRNA is highly expressed (Jia et al., 2012). Another example is from hnRNPA2B1 and hnRNPA1, both of which are involved in the RNA processing and splicing regulation (Gabut et al., 2008). Their mutations have been found in the familial multisystem proteinopathy and ALS, two devastating diseases resulting from the progressive degeneration of the neural system; and expression of the mutant hnRNPA2B1 and hnRNPA1 in transgenic Drosophila recapitulates the phenotype and pathology to some extent demonstrated in human (Kim et al., 2013). These lines of evidence support the notion that disruption in the RNA splicing system is sufficient to cause the degeneration of the neural system.
The protein TDP-43 binds DNA and RNA to regulate the transcription and splicing processes. Although it is not a typical member of the spliceosomal proteins based on the currently available knowledge (Hegele et al., 2012; Korneta et al., 2012), proteopathy of TDP-43 was discovered in the central nervous system that includes hippocampus, neocortex, and spinal cord in patients with frontotemporal lobar degeneration (FTLD-U) or amyotrophic lateral sclerosis (ALS; Neumann et al., 2006; Maekawa et al., 2009). Further genetic evidence establishes the causative role of TDP-43 in these two neurodegenerative diseases by its mutations in the familial cases (Sreedharan et al., 2008), strengthened by evidence from other biological studies (Gitcho et al., 2008; Wegorzewska et al., 2009; Wils et al., 2010; Alami et al., 2014).
Despite the direct evidence of mutations in U1 snRNP components is currently rare, probably because their knockouts are embryonically lethal (Hilleren et al., 1995; Salz et al., 2004); and the direct association between the U1 proteopathy and the cellular stress within the same neuron in the brain is currently under investigation, the U1 dysfunction in AD brains is presumably a disaster in neurons. However, what causes it? Insights might be gained from the characteristics of its histopathology in the brain.
Cytplasmic Distribution of U1 snRNP in AD Brain Neurons
The prominent feature of the neuronal U1 pathology in AD brains is its depletion from the nucleus and redistribution in the cytoplasm where it largely overlays with the neurofibrillary phospho-Tau (Bai et al., 2013). Besides, when the phospho-Tau is not obvious yet in the neuron, we often find U1-70K has already redistributed into the cytoplasm surrounding the nucleus without forming the tangle-like structure (Data to be published). This not only indicates that the U1 alteration and the Tau proteopathy are two independent events, but also suggests that the original characteristic of the U1 pathology is its nuclear exclusion redistribution into the cytoplasm.
Actually, the cytoplasmic redistribution of nuclear proteins is quite common in neurodegenerative disorders. TDP-43 and FUS, the nuclear proteins that bind RNA/DNA, are normally located in the neuronal nucleus, but relocate into the cytoplasm and colocalize with the ubiquitin-positive inclusion body in brains or spines of FTLD-U and ALS patients (Arai et al., 2006; Neumann et al., 2006). SFPQ, another nuclear RNA/DNA binding protein that is mainly involved in RNA splicing, relocates from the nucleus to the perinuclear region of the cytoplasm in the hippocampal neurons in AD and Pick’s Disease (Ke et al., 2012). In addition, similar nuclear exclusion phenomenon is also seen with the proteins hnRNPA2/B1 which form aberrant sarcoplasmic inclusions in multisystem proteinopathy and ALS patients, and also in the animal models (Kim et al., 2013). However, unlike TDP-43 and FUS whose cytoplasmic accumulations appear focal and might be related to stress granules, the U1 snRNPs show a more diffusive distribution pattern and display filamentous structure (Hales et al., 2014b), indicating a distinct underlying mechanism.
U1 snRNP Cytoplasmic Redistribution Indicates Neuronal Cell Cycle Reentry (CCR)
The first possible biological event that causes the U1 subcellular location change is the apoptosis (Dieker et al., 2008). During this cellular process, U1-70K is phosphorylated, fragmented, and largely excluded from the DNA-staining region and become surrounding the chromatins in clusters. However, it is notable that this cluster-like appearance is largely different from that observed in the AD brain, in which the distribution of U1 is more diffusive (Bai et al., 2013). This is consistent with the fact that apoptosis is not likely the major way of neuronal death in AD (Stadelmann et al., 1998; Yuan and Yankner, 2000; Zhu et al., 2006). Indeed, the U1-70K fragment in the AD brain is not generated through cleavage by caspase-3 (Bai et al., 2014). Therefore, the U1 subcellular mislocation is not likely a reflection of apoptosis.
The next event that elicits the U1 subcellular distribution in a pattern similarly observed in AD is mitosis. In this cellular process, the U snRNPs initially remain in the nucleus in the interphase, then move to the cytoplasm after the nuclear envelope is broken down during the metaphase and anaphase, and finally return into the nuclei of the two daughter cells in the telophase (Verheijen et al., 1986). The nucleus-to-cytoplasm redistribution of spliceosome during mitosis is extensively demonstrated in many studies (Goldstein et al., 1977; Reuter et al., 1985; Spector and Smith, 1986; Leser et al., 1989; Carmo-Fonseca et al., 1993; Azum-Gélade et al., 1994; Ferreira et al., 1994; Blencowe, 2003). It is worth reiterating that the cytoplasmic distribution of U1 snRNPs during the mitosis is very similar to the U1 pathology when Tau tangles are not present in AD brains (Supplementary Figure S1).
Indeed, the neuronal cell cycle activation has already been widely evidenced. Neurons are postmitotic and usually not dividable, in which the cell cycle is arrested in the G1 phase. However, a significant number of hippocampal pyramidal and basal forebrain neurons in AD brains display duplicated genetic loci on different chromosomes (Yang et al., 2001), indicating a progression from G1 phase into the S phase. In addition, several critical cell proliferation and cycle-related proteins, such as PCNA (proliferating cell nuclear antigen), cyclin D and cyclin B, are evidenced to increase in hippocampus, basal nucleus of Meynert, and entorhinal cortex in AD brain sections (Yang et al., 2003). Similar cell cycle-reentry (CCR events have been extensively demonstrated and studied by several major research groups (Andorfer et al., 2005; Bauer and Patterson, 2005; Webber et al., 2005; Neve and Mcphie, 2006; Herrup and Yang, 2007; Varvel et al., 2008). The major CCR related molecules and events in AD human and animals are summarized (Table 1).
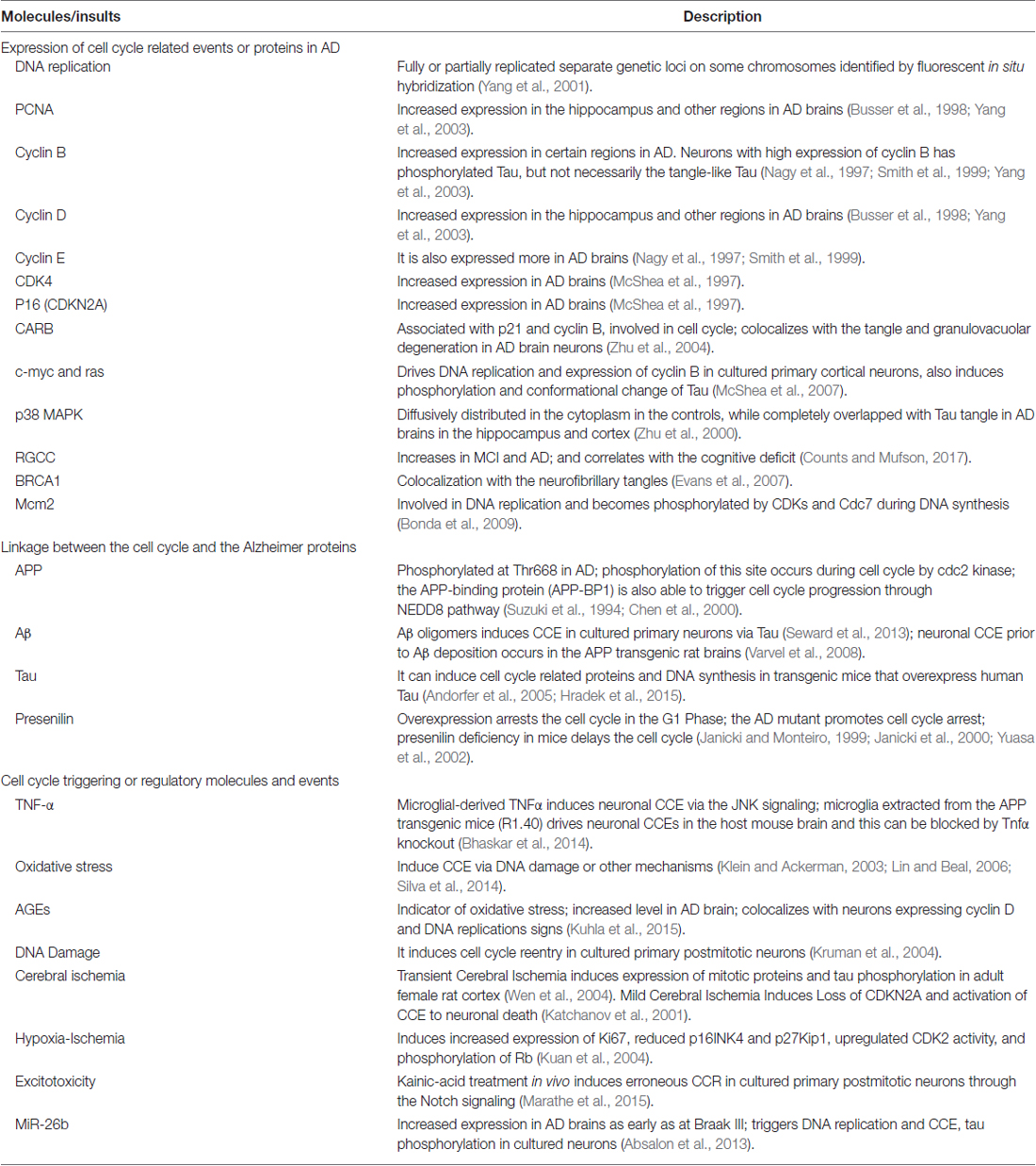
Table 1. Cell cycle related molecules and events in Alzheimer’s disease (AD).
Neuronal CCR Explains Major Cellular and Molecular Alterations in AD
The neuronal CCR theory gains more research attention because it explains several critical events during the AD pathogenesis. The first is the Tau phosphorylation, one of the hallmarks in AD pathology. As a member of the Ser/Thr cyclin-dependent kinases, CDK5 phosphorylates Tau at the sites that are most frequently hyperphosphorylated in AD brains (Kimura et al., 2014). CDK5 is active mainly in the postmitotic neurons where its regulatory protein subunits p35/p39 are predominantly expressed and activated in the AD brain (Patrick et al., 1999). Actually, the fact of Tau phosphorylation during mitosis is evidenced by the in vitro study (Illenberger et al., 1998). Therefore, it is reasonable to speculate that the Tau hyperphosphorylation possibly involves the CCR attempt in AD brain neurons.
The neuronal CCR might also account for the APP phosphorylation in AD brains. The phosphorylation of T668 on APP695, the major form in neurons (Kang and Müller-Hill, 1990), is known to be significantly increased in AD and leads to accelerated Aβ generation. Further study has demonstrated that the phosphorylation of APP on T668 can be achieved during cell cycle by CDK5 and CDC2 kinases (Suzuki et al., 1994; Iijima et al., 2000; Liu et al., 2003). As a critical evidence, such phosphorylated APP is largely accumulated in neurons that bear phosphorylated Tau (Lee et al., 2003), strongly suggestive of CCR as their common upstream inducer.
Besides, the neuronal CCR might provide insights to the mechanism of AD neuronal death. As mentioned earlier, the AD neuronal loss is not likely due to apoptosis. If CCR is widely activated, then the mitotic catastrophe (MC) might be a mechanism of neuronal death in AD. The MC is a type of cell death that results from failed completion of mitosis (Kroemer et al., 2009). In AD hippocampus, the phosphorylated histone H3 appears in the neuronal cytoplasm instead of its normal localization in the nucleus during mitosis in actively dividing cells, indicating an aberrant mitosis undergoing in neurons which might proceed into necrosis by MC (Ogawa et al., 2003). Okadaic acid (OA), a potent phosphatase inhibitor, induces the expression of G2/M phase cyclins B1 and D1 in neuroblastoma cells to activate cell cycle, making neurons become dying with signs of MC (Chen et al., 2006). Interestingly, these OA treatments can induce the paired helical filament-like phosphorylation of Tau in rats (Arendt et al., 1995), possibly through the activation of CDK5. Collectively, these lines of evidence suggest CCR can be activated in neurons and lead to MC eventually, providing a potential mechanism of neuronal death in AD.
Indeed, the aging process is associated with the activation of cell cycle and this is well demonstrated in the mouse model. The senescence-accelerated mice 8 (SAMP8) is a model of aging and displays typical AD pathological characteristics (Pallas et al., 2008), including Aβ amyloid accumulation and Tau phosphorylation and other events (Del Valle et al., 2010). The SAMP8 mice not only have elevated CDK5 and GSK3β, but also demonstrate a significant increase of cell cycle progression markers, including cyclin A, cyclin D1, cyclin E, Cdk2, cyclin B, pRb, and E2F1 (Casadesüs et al., 2012). Taken together, lessons from the SAMP8 mice might highlight the CCR during the aging process as a major driver in the AD pathogenesis.
Mechanistic Link Between Neuronal CCR and Inflammation in AD
Study has shown the soluble Aβ oligomers induce neuronal CCR through the phosphorylation of Tau (Seward et al., 2013). The CCR events are also observed in APP transgenic mice at about ~6 months of age at which a substantial amount of soluble Aβ peptides are expressed (Varvel et al., 2008). However, the CCR events are usually only sparsely seen in these animal models. The more extensive CCRs in AD brains likely involve other factors.
Inflammation is probably the most important causative insult besides the amyloid cascade in the AD pathogenesis (Wyss-Coray and Rogers, 2012; Heppner et al., 2015); and it is able to trigger the cell cycle process. Because the activation of cell cycle is closely related to the cellular proliferation which is a hallmark in tumorigenesis, insights about the role of inflammation in AD neuronal CCR might be gained from its role as a driver in the cancer development (Crusz and Balkwill, 2015).
The cancer and AD are apparently two opposite diseases with a common molecular basis: loss of control on cellular growth due to chronic accumulation of biological alterations (López-Otín et al., 2013). Therefore, the neuronal CCR might be considered as a result of an aborted tumorigenesis. In fact, both diseases have age as their strongest risk factor; and presents a similar incidence trend during aging: from ~4% under 65 years old and up to 40% after 75 years old (Crusz and Balkwill, 2015; Siegel et al., 2018). Because these two diseases are probably the opposite manifestations of the same disorder, they tend to be exclusive in a particular individual and therefore have a negative association in a general population as expected (Roe et al., 2010; Musicco et al., 2013).
The inflammation drives the cancer development through factors including IL-1, IL-6, IL-13, IL-22, TNFα, TGFβ, ROS and other possible agents, with a converge on two major signaling pathways: STAT3 and NF-κB (Elinav et al., 2013). The IL-1 and TNFα activate the NF-κB pathway to increase the expression of IL-6 which, in turn, stimulates through STAT3 the upregulation of cyclins D1, D2 and B to initiate the cell cycle for cellular proliferation. This pathway is widely demonstrated in cancer tissues where it associates with the inflammation (He and Karin, 2011; Dmitrieva et al., 2016; Taniguchi and Karin, 2018), suggesting a causative relationship between the inflammation and the cell cycle activation. Among these factors, IL-1, IL-6 and TGF-β are evidenced to be increased in AD brains (Blum-Degen et al., 1995; Alvarez et al., 1996; Ye and Johnson, 1999; Luterman et al., 2000; Quintanilla et al., 2004; Patel et al., 2005; Rota et al., 2006; Ghosh et al., 2013; Zheng et al., 2016), in which STAT3 and NF-κB pathways are also activated (Tarkowski et al., 2002; Tesseur et al., 2006; Town et al., 2008; Chiba et al., 2009; Wan et al., 2010; Ben Haim et al., 2015). Besides, the complements of the innate immune system that are upregulated and activated in AD, can also modulate the cell cycle (Rus et al., 1996, 2001; Fosbrink et al., 2005). Therefore, the inflammation in AD brains, probably initiated from amyloid plaques but exacerbated by other possible factors (e.g., chronic accumulation of other aberrant proteins, the blood-brain barrier leakage, the reactivation of latent microorganisms, etc.; Glass et al., 2010), might be the major inducer for the fatal CCR in neurons.
Conclusion
The U1 snRNP pathology provides a very unique mechanism in the AD pathogenesis. Although other mechanisms might exist, the most possible cellular alteration that mechanistically links U1 alteration is the neuronal cell cycle reentry, based on the preliminary data that we have obtained. It is possible that AD is caused by the continued excessive accumulation of Aβ that elicits the immune response which is exacerbated by other inflammatory insults, initiating the neuronal cell cycle activation that eventually causes neuronal death by MC (Figure 1). Nevertheless, thorough studies are required to evaluate this bold assumption, which will facilitate understanding of the fundamental mechanism of the AD etiology.
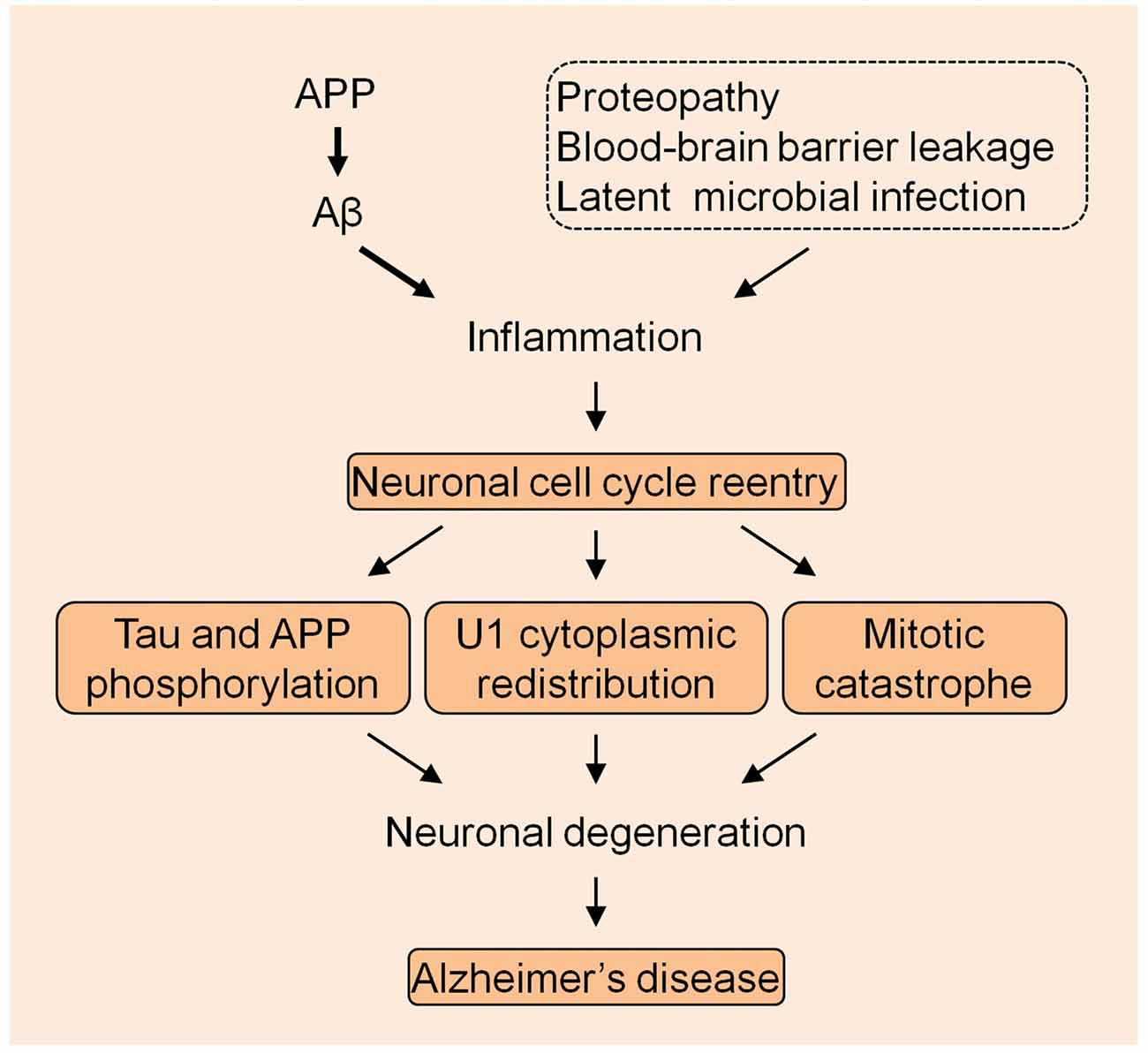
Figure 1. The summarized model of Alzheimer’s disease (AD) pathogenesis. The amyloid precursor protein (APP) derived Aβ species initiates the inflammation with the exacerbation by other insults to drive the neuronal cell cycle events.
Author Contributions
BB conceived the idea and wrote the manuscript.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author appreciates the research experience with the principle investigators who have supervised the U1 discovery (Bai et al., 2013). This article only represents my personal perspective. This work was partially supported by the Chinese Fundamental Research Funds for the Central Universities (021414380299, to BB) and the Nanjing Drum Tower Hospital Research Initiation (RE445, to BB).
Abbreviations
AD, Alzheimer’s Disease; APP, amyloid precursor protein; CCR, cell cycle reentry; MC, mitotic catastrophe; snRNP, small nuclear ribonucleoprotein.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2018.00075/full#supplementary-material
FIGURE S1 | Immunofluorescent staining to demonstrate the cytoplasmic distribution of U1-70K in a Alzheimer’s disease (AD) brain and HEK293T cells. (A) Coimmunostaining of U1-70K and phospho-Tau (AT8) on an AD brain cortical tissue slide. The dotted circles point to the neuron with nuclear depletion and cytoplasmic distribution of U1-70K, but no obvious phospho-Tau staining. (B) U1-70K in HEK293T cells. The dotted circles point the cytoplasmic distribution of U1-70K in cells that seem in the mitotic process. DAPI: 4′,6-Diamidine-2′-phenylindole dihydrochloride, a DNA staining dye.
References
Absalon, S., Kochanek, D. M., Raghavan, V., and Krichevsky, A. M. (2013). MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 33, 14645–14659. doi: 10.1523/JNEUROSCI.1327-13.2013
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S. W., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Alvarez, X. A., Franco, A., Fernández-Novoa, L., and Cacabelos, R. (1996). Blood levels of histamine, IL-1 β, and TNF-α in patients with mild to moderate Alzheimer disease. Mol. Chem. Neuropathol. 29, 237–252. doi: 10.1007/bf02815005
Andorfer, C., Acker, C. M., Kress, Y., Hof, P. R., Duff, K., and Davies, P. (2005). Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J. Neurosci. 25, 5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Arendt, T., Holzer, M., Fruth, R., Brückner, M. K., and Gärtner, U. (1995). Paired helical filament-like phosphorylation of tau, deposition of β/A4-amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience 69, 691–698. doi: 10.1016/0306-4522(95)00347-l
Azum-Gélade, M. C., Noaillac-Depeyre, J., Caizergues-Ferrer, M., and Gas, N. (1994). Cell cycle redistribution of U3 snRNA and fibrillarin. Presence in the cytoplasmic nucleolus remnant and in the prenucleolar bodies at telophase. J. Cell Sci. 107, 463–475.
Bai, B., Chen, P. C., Hales, C. M., Wu, Z., Pagala, V., High, A. A., et al. (2014). Integrated approaches for analyzing U1b–70K cleavage in Alzheimer’s disease. J. Proteome Res. 13, 4526–4534. doi: 10.1021/pr5003593
Bai, B., Hales, C. M., Chen, P. C., Gozal, Y., Dammer, E. B., Fritz, J. J., et al. (2013). U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 110, 16562–16567. doi: 10.1073/pnas.1310249110
Bauer, S., and Patterson, P. H. (2005). The cell cycle-apoptosis connection revisited in the adult brain. J. Cell Biol. 171, 641–650. doi: 10.1083/jcb.200505072
Ben Haim, L., Ceyzériat, K., Carrillo-de Sauvage, M. A., Aubry, F., Auregan, G., Guillermier, M., et al. (2015). The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 35, 2817–2829. doi: 10.1523/JNEUROSCI.3516-14.2015
Bhaskar, K., Maphis, N., Xu, G., Varvel, N. H., Kokiko-Cochran, O. N., Weick, J. P., et al. (2014). Microglial derived tumor necrosis factor-α drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol. Dis. 62, 273–285. doi: 10.1016/j.nbd.2013.10.007
Blencowe, B. J. (2003). Splicing regulation: the cell cycle connection. Curr. Biol. 13, R149–R151. doi: 10.1016/s0960-9822(03)00079-4
Bloom, G. S. (2014). Amyloid-β and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Blum-Degen, D., Müller, T., Kuhn, W., Gerlach, M., Przuntek, H., and Riederer, P. (1995). Interleukin-1 β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 202, 17–20. doi: 10.1016/0304-3940(95)12192-7
Bonda, D. J., Evans, T. A., Santocanale, C., Llosá, J. C., Vina, J., Bajic, V., et al. (2009). Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging 1, 382–388. doi: 10.18632/aging.100044
Busser, J., Geldmacher, D. S., and Herrup, K. (1998). Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci. 18, 2801–2807.
Carmo-Fonseca, M., Ferreira, J., and Lamond, A. I. (1993). Assembly of snRNP-containing coiled bodies is regulated in interphase and mitosis—evidence that the coiled body is a kinetic nuclear structure. J. Cell Biol. 120, 841–852. doi: 10.1083/jcb.120.4.841
Casadesüs, G., Gutierrez-Cuesta, J., Lee, H. G., Jiménez, A., Tajes, M., Ortuno-Sahagun, D., et al. (2012). Neuronal cell cycle re-entry markers are altered in the senescence accelerated mouse P8 (SAMP8). J. Alzheimers Dis. 30, 573–583. doi: 10.3233/JAD-2012-120112
Chen, B., Cheng, M., Hong, D. J., Sun, F. Y., and Zhu, C. Q. (2006). Okadaic acid induced cyclin B1 expression and mitotic catastrophe in rat cortex. Neurosci. Lett. 406, 178–182. doi: 10.1016/j.neulet.2006.06.074
Chen, Y., McPhie, D. L., Hirschberg, J., and Neve, R. L. (2000). The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J. Biol. Chem. 275, 8929–8935. doi: 10.1074/jbc.275.12.8929
Cheng, Z., Du, Z., Shang, Y., Zhang, Y., and Zhang, T. (2017a). A preliminary study: PS1 increases U1 snRNA expression associated with AD. J. Mol. Neurosci. 62, 269–275. doi: 10.1007/s12031-017-0932-y
Cheng, Z., Shang, Y., Gao, S., and Zhang, T. (2017b). Overexpression of U1 snRNA induces decrease of U1 spliceosome function associated with Alzheimer’s disease. J. Neurogenet. 31, 337–343. doi: 10.1080/01677063.2017.1395425
Cheng, Z., Du, Z., Zhai, B., Yang, Z., and Zhang, T. (2018). U1 small nuclear RNA overexpression implicates autophagic-lysosomal system associated with AD. Neurosci. Res. doi: 10.1016/j.neures.2018.01.006 [Epub ahead of print].
Chiba, T., Yamada, M., Sasabe, J., Terashita, K., Shimoda, M., Matsuoka, M., et al. (2009). Amyloid-β causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol. Psychiatry 14, 206–222. doi: 10.1038/mp.2008.105
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature 360, 672–674. doi: 10.1038/360672a0
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Counts, S. E., and Mufson, E. J. (2017). Regulator of cell cycle (RGCC) expression during the progression of Alzheimer’s disease. Cell Transplant. 26, 693–702. doi: 10.3727/096368916x694184
Crusz, S. M., and Balkwill, F. R. (2015). Inflammation and cancer: advances and new agents. Nat. Rev. Clin. Oncol. 12, 584–596. doi: 10.1038/nrclinonc.2015.105
Del Valle, J., Duran-Vilaregut, J., Manich, G., Casadesús, G., Smith, M. A., Camins, A., et al. (2010). Early amyloid accumulation in the hippocampus of SAMP8 mice. J. Alzheimers Dis. 19, 1303–1315. doi: 10.3233/JAD-2010-1321
Dieker, J., Cisterna, B., Monneaux, F., Decossas, M., van der Vlag, J., Biggiogera, M., et al. (2008). Apoptosis-linked changes in the phosphorylation status and subcellular localization of the spliceosomal autoantigen U1–70K. Cell Death Differ. 15, 793–804. doi: 10.1038/sj.cdd.4402312
Dmitrieva, O. S., Shilovskiy, I. P., Khaitov, M. R., and Grivennikov, S. I. (2016). Interleukins 1 and 6 as main mediators of inflammation and cancer. Biochemistry Mosc. 81, 80–90. doi: 10.1134/s0006297916020024
Elinav, E., Nowarski, R., Thaiss, C. A., Hu, B., Jin, C., and Flavell, R. A. (2013). Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771. doi: 10.1038/nrc3611
Evans, T. A., Raina, A. K., Delacourte, A., Aprelikova, O., Lee, H. G., Zhu, X., et al. (2007). BRCA1 may modulate neuronal cell cycle re-entry in Alzheimer disease. Int. J. Med. Sci. 4, 140–145. doi: 10.7150/ijms.4.140
Ferreira, J. A., Carmo-Fonseca, M., and Lamond, A. I. (1994). Differential interaction of splicing snRNPs with coiled bodies and interchromatin granules during mitosis and assembly of daughter cell nuclei. J. Cell Biol. 126, 11–23. doi: 10.1083/jcb.126.1.11
Fosbrink, M., Niculescu, F., and Rus, H. (2005). The role of c5b-9 terminal complement complex in activation of the cell cycle and transcription. Immunol. Res. 31, 37–46. doi: 10.1385/ir:31:1:37
Gabut, M., Chaudhry, S., and Blencowe, B. J. (2008). SnapShot: the splicing regulatory machinery. Cell 133:192.e1. doi: 10.1016/j.cell.2008.03.010
Ghosh, S., Wu, M. D., Shaftel, S. S., Kyrkanides, S., LaFerla, F. M., Olschowka, J. A., et al. (2013). Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J. Neurosci. 33, 5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013
Giannakopoulos, P., Herrmann, F. R., Bussiere, T., Bouras, C., Kovari, E., Perl, D. P., et al. (2003). Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60, 1495–1500. doi: 10.1212/01.WNL.0000063311.58879.01
Gitcho, M. A., Baloh, R. H., Chakraverty, S., Mayo, K., Norton, J. B., Levitch, D., et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538. doi: 10.1002/ana.21344
Glass, C. K., Saijo, K., Winner, B., Marchetto, M. C., and Gage, F. H. (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. doi: 10.1016/j.cell.2010.02.016
Goedert, M., and Spillantini, M. G. (2006). A century of Alzheimer’s disease. Science 314, 777–781. doi: 10.1126/science.1132814
Goldstein, L., Wise, G. E., and Ko, C. (1977). Small nuclear RNA localization during mitosis. An electron microscope study. J. Cell Biol. 73, 322–331. doi: 10.1083/jcb.73.2.322
Gómez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Guerreiro, R., Brás, J., and Hardy, J. (2013). SnapShot: genetics of Alzheimer’s disease. Cell 155, 968.e1–968.e1. doi: 10.1016/j.cell.2013.10.037
Hales, C. M., Dammer, E. B., Diner, I., Yi, H., Seyfried, N. T., Gearing, M., et al. (2014a). Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 24, 344–351. doi: 10.1111/bpa.12133
Hales, C. M., Seyfried, N. T., Dammer, E. B., Duong, D., Yi, H., Gearing, M., et al. (2014b). U1 small nuclear ribonucleoproteins (snRNPs) aggregate in Alzheimer’s disease due to autosomal dominant genetic mutations and trisomy 21. Mol. Neurodegener. 9:15. doi: 10.1186/1750-1326-9-15
He, G., and Karin, M. (2011). NF-κB and STAT3—key players in liver inflammation and cancer. Cell Res. 21, 159–168. doi: 10.1038/cr.2010.183
Hegele, A., Kamburov, A., Grossmann, A., Sourlis, C., Wowro, S., Weimann, M., et al. (2012). Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580. doi: 10.1016/j.molcel.2011.12.034
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Herrup, K., and Yang, Y. (2007). Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378. doi: 10.1038/nrn2124
Hilleren, P. J., Kao, H. Y., and Siliciano, P. G. (1995). The amino-terminal domain of yeast U1–70K is necessary and sufficient for function. Mol. Cell. Biol. 15, 6341–6350. doi: 10.1128/mcb.15.11.6341
Hoozemans, J. J., van Haastert, E. S., Nijholt, D. A., Rozemuller, A. J., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Hradek, A. C., Lee, H. P., Siedlak, S. L., Torres, S. L., Jung, W., Han, A. H., et al. (2015). Distinct chronology of neuronal cell cycle re-entry and tau pathology in the 3xTg-AD mouse model and Alzheimer’s disease patients. J. Alzheimers Dis. 43, 57–65. doi: 10.3233/JAD-141083
Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C. F., et al. (2011). Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478, 123–126. doi: 10.1038/nature10485
Iijima, K., Ando, K., Takeda, S., Satoh, Y., Seki, T., Itohara, S., et al. (2000). Neuron-specific phosphorylation of Alzheimer’s β-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 75, 1085–1091. doi: 10.1046/j.1471-4159.2000.0751085.x
Illenberger, S., Zheng-Fischhofer, Q., Preuss, U., Stamer, K., Baumann, K., Trinczek, B., et al. (1998). The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer’s disease. Mol. Biol. Cell 9, 1495–1512. doi: 10.1091/mbc.9.6.1495
Janicki, S. M., and Monteiro, M. J. (1999). Presenilin overexpression arrests cells in the G1 phase of the cell cycle. Arrest potentiated by the Alzheimer’s disease PS2(N141I)mutant. Am. J. Pathol. 155, 135–144. doi: 10.1016/s0002-9440(10)65108-5
Janicki, S. M., Stabler, S. M., and Monteiro, M. J. (2000). Familial Alzheimer’s disease presenilin-1 mutants potentiate cell cycle arrest. Neurobiol. Aging 21, 829–836. doi: 10.1016/s0197-4580(00)00222-0
Jia, Y., Mu, J. C., and Ackerman, S. L. (2012). Mutation of a U2 snRNA gene causes global disruption of alternative splicing and neurodegeneration. Cell 148, 296–308. doi: 10.1016/j.cell.2011.11.057
Jonsson, T., Atwal, J. K., Steinberg, S., Snaedal, J., Jonsson, P. V., Bjornsson, S., et al. (2012). A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488, 96–99. doi: 10.1038/nature11283
Kaida, D., Berg, M. G., Younis, I., Kasim, M., Singh, L. N., Wan, L., et al. (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. doi: 10.1038/nature09479
Kang, J., and Müller-Hill, B. (1990). Differential splicing of Alzheimer’s disease amyloid A4 precursor RNA in rat tissues: PreA4(695) mRNA is predominantly produced in rat and human brain. Biochem. Biophys. Res. Commun. 166, 1192–1200. doi: 10.1016/0006-291x(90)90992-v
Katchanov, J., Harms, C., Gertz, K., Hauck, L., Waeber, C., Hirt, L., et al. (2001). Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J. Neurosci. 21, 5045–5053.
Ke, Y. D., Dramiga, J., Schütz, U., Kril, J. J., Ittner, L. M., Schroder, H., et al. (2012). Tau-mediated nuclear depletion and cytoplasmic accumulation of SFPQ in Alzheimer’s and Pick’s disease. PLoS One 7:e35678. doi: 10.1371/journal.pone.0035678
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Kimura, T., Ishiguro, K., and Hisanaga, S. (2014). Physiological and pathological phosphorylation of tau by Cdk5. Front. Mol. Neurosci. 7:65. doi: 10.3389/fnmol.2014.00065
Klein, J. A., and Ackerman, S. L. (2003). Oxidative stress, cell cycle, and neurodegeneration. J. Clin. Invest. 111, 785–793. doi: 10.1172/jci18182
Korneta, I., Magnus, M., and Bujnicki, J. M. (2012). Structural bioinformatics of the human spliceosomal proteome. Nucleic Acids Res. 40, 7046–7065. doi: 10.1093/nar/gks347
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 16, 3–11. doi: 10.1038/cdd.2008.150
Kruman, I. I., Wersto, R. P., Cardozo-Pelaez, F., Smilenov, L., Chan, S. L., Chrest, F. J., et al. (2004). Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41, 549–561. doi: 10.1016/s0896-6273(04)00017-0
Kuan, C. Y., Schloemer, A. J., Lu, A., Burns, K. A., Weng, W. L., Williams, M. T., et al. (2004). Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J. Neurosci. 24, 10763–10772. doi: 10.1523/JNEUROSCI.3883-04.2004
Kuhla, A., Ludwig, S. C., Kuhla, B., Münch, G., and Vollmar, B. (2015). Advanced glycation end products are mitogenic signals and trigger cell cycle reentry of neurons in Alzheimer’s disease brain. Neurobiol. Aging 36, 753–761. doi: 10.1016/j.neurobiolaging.2014.09.025
Kuperstein, I., Broersen, K., Benilova, I., Rozenski, J., Jonckheere, W., Debulpaep, M., et al. (2010). Neurotoxicity of Alzheimer’s disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420. doi: 10.1038/emboj.2010.211
Lee, M. S., Kao, S. C., Lemere, C. A., Xia, W., Tseng, H. C., Zhou, Y., et al. (2003). APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 163, 83–95. doi: 10.1083/jcb.200301115
Lefebvre, S., Burglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., et al. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet. 16, 265–269. doi: 10.1038/ng0797-265
Leser, G. P., Fakan, S., and Martin, T. E. (1989). Ultrastructural distribution of ribonucleoprotein complexes during mitosis. snRNP antigens are contained in mitotic granule clusters. Eur. J. Cell Biol. 50, 376–389.
Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-β protein assembly in the brain impairs memory. Nature 440, 352–357. doi: 10.1038/nature04533
Lin, M. T., and Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. doi: 10.1038/nature05292
Liu, F., Su, Y., Li, B., Zhou, Y., Ryder, J., Gonzalez-Dewhitt, P., et al. (2003). Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 547, 193–196. doi: 10.1016/s0014-5793(03)00714-2
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Lorson, C. L., Hahnen, E., Androphy, E. J., and Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U S A 96, 6307–6311. doi: 10.1073/pnas.96.11.6307
Lunn, M. R., and Wang, C. H. (2008). Spinal muscular atrophy. Lancet 371, 2120–2133. doi: 10.1016/S0140-6736(08)60921-6
Luterman, J. D., Haroutunian, V., Yemul, S., Ho, L., Purohit, D., Aisen, P. S., et al. (2000). Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch. Neurol. 57, 1153–1160. doi: 10.1001/archneur.57.8.1153
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/s1474-4422(10)70195-2
Maekawa, S., Leigh, P. N., King, A., Jones, E., Steele, J. C., Bodi, I., et al. (2009). TDP-43 is consistently co-localized with ubiquitinated inclusions in sporadic and Guam amyotrophic lateral sclerosis but not in familial amyotrophic lateral sclerosis with and without SOD1 mutations. Neuropathology 29, 672–683. doi: 10.1111/j.1440-1789.2009.01029.x
Manabe, T., Ohe, K., Katayama, T., Matsuzaki, S., Yanagita, T., Okuda, H., et al. (2007). HMGA1a: sequence-specific RNA-binding factor causing sporadic Alzheimer’s disease-linked exon skipping of presenilin-2 pre-mRNA. Genes Cells 12, 1179–1191. doi: 10.1111/j.1365-2443.2007.01123.x
Marathe, S., Liu, S., Brai, E., Kaczarowski, M., and Alberi, L. (2015). Notch signaling in response to excitotoxicity induces neurodegeneration via erroneous cell cycle reentry. Cell Death Differ. 22, 1775–1784. doi: 10.1038/cdd.2015.23
Masters, C. L., and Selkoe, D. J. (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262. doi: 10.1101/cshperspect.a006262
Matera, A. G., and Wang, Z. (2014). A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 15, 108–121. doi: 10.1038/nrm3742
McShea, A., Harris, P. L., Webster, K. R., Wahl, A. F., and Smith, M. A. (1997). Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am. J. Pathol. 150, 1933–1939.
McShea, A., Lee, H. G., Petersen, R. B., Casadesus, G., Vincent, I., Linford, N. J., et al. (2007). Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim. Biophys. Acta 1772, 467–472. doi: 10.1016/j.bbadis.2006.09.010
Musicco, M., Adorni, F., Di Santo, S., Prinelli, F., Pettenati, C., Caltagirone, C., et al. (2013). Inverse occurrence of cancer and Alzheimer disease: a population-based incidence study. Neurology 81, 322–328. doi: 10.1212/WNL.0b013e31829c5ec1
Nagy, Z., Esiri, M. M., Cato, A. M., and Smith, A. D. (1997). Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol. 94, 6–15. doi: 10.1007/s004010050665
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Neve, R. L., and Mcphie, D. L. (2006). The cell cycle as a therapeutic target for Alzheimer’s disease. Pharmacol. Ther. 111, 99–113. doi: 10.1016/j.pharmthera.2005.09.005
Nilsberth, C., Westlind-Danielsson, A., Eckman, C. B., Condron, M. M., Axelman, K., Forsell, C., et al. (2001). The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 4, 887–893. doi: 10.1038/nn0901-887
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal β-amyloid aggregates, neurodegeneration and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. doi: 10.1523/jneurosci.1202-06.2006
Ogawa, O., Zhu, X., Lee, H. G., Raina, A., Obrenovich, M. E., Bowser, R., et al. (2003). Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathol. 105, 524–528. doi: 10.1007/s00401-003-0684-3
Ohe, K., and Mayeda, A. (2010). HMGA1a trapping of U1 snRNP at an authentic 5′ splice site induces aberrant exon skipping in sporadic Alzheimer’s disease. Mol. Cell. Biol. 30, 2220–2228. doi: 10.1128/mcb.00114-10
Pallas, M., Camins, A., Smith, M. A., Perry, G., Lee, H. G., and Casadesus, G. (2008). From aging to Alzheimer’s disease: unveiling “the switch” with the senescence-accelerated mouse model (SAMP8). J. Alzheimers Dis. 15, 615–624. doi: 10.3233/jad-2008-15408
Patel, N. S., Paris, D., Mathura, V., Quadros, A. N., Crawford, F. C., and Mullan, M. J. (2005). Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflammation 2:9. doi: 10.1186/1742-2094-2-9
Patrick, G. N., Zukerberg, L., Nikolic, M., De La Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. doi: 10.1038/45159
Quintanilla, R. A., Orellana, D. I., Gonzalez-Billault, C., and Maccioni, R. B. (2004). Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 295, 245–257. doi: 10.1016/j.yexcr.2004.01.002
Reuter, R., Appel, B., Rinke, J., and Luhrmann, R. (1985). Localization and structure of snRNPs during mitosis. Immunofluorescent and biochemical studies. Exp. Cell Res. 159, 63–79. doi: 10.1016/s0014-4827(85)80038-0
Roberson, E. D., and Mucke, L. (2006). 100 years and counting: prospects for defeating Alzheimer’s disease. Science 314, 781–784. doi: 10.1126/science.1132813
Roe, C. M., Fitzpatrick, A. L., Xiong, C., Sieh, W., Kuller, L., Miller, J. P., et al. (2010). Cancer linked to Alzheimer disease but not vascular dementia. Neurology 74, 106–112. doi: 10.1212/WNL.0b013e3181c91873
Rota, E., Bellone, G., Rocca, P., Bergamasco, B., Emanuelli, G., and Ferrero, P. (2006). Increased intrathecal TGF-β1, but not IL-12, IFN-γ and IL-10 levels in Alzheimer’s disease patients. Neurol. Sci. 27, 33–39. doi: 10.1007/s10072-006-0562-6
Rus, H. G., Niculescu, F. I., and Shin, M. L. (1996). Sublytic complement attack induces cell cycle in oligodendrocytes. J. Immunol. 156, 4892–4900.
Rus, H. G., Niculescu, F. I., and Shin, M. L. (2001). Role of the C5b-9 complement complex in cell cycle and apoptosis. Immunol. Rev. 180, 49–55. doi: 10.1034/j.1600-065x.2001.1800104.x
Salz, H. K., Mancebo, R. S., Nagengast, A. A., Speck, O., Psotka, M., and Mount, S. M. (2004). The Drosophila U1–70K protein is required for viability, but its arginine-rich domain is dispensable. Genetics 168, 2059–2065. doi: 10.1534/genetics.104.032532
Selkoe, D. J. (2013). SnapShot: pathobiology of Alzheimer’s disease. Cell 154:468.e1. doi: 10.1016/j.cell.2013.07.003
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Selkoe, D., Mandelkow, E., and Holtzman, D. (2012). Deciphering Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a011460. doi: 10.1101/cshperspect.a011460
Serrano-Pozo, A., Frosch, M. P., Masliah, E., and Hyman, B. T. (2011). Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1:a006189. doi: 10.1101/cshperspect.a006189
Seward, M. E., Swanson, E., Norambuena, A., Reimann, A., Cochran, J. N., Li, R., et al. (2013). Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 126, 1278–1286. doi: 10.1242/jcs.1125880
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Siegel, R. L., Miller, K. D., and Jemal, A. (2018). Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30. doi: 10.3322/caac.21442
Silva, A. R., Santos, A. C., Farfel, J. M., Grinberg, L. T., Ferretti, R. E., Campos, A. H., et al. (2014). Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PLoS One 9:e99897. doi: 10.1371/journal.pone.0099897
Smith, M. Z., Nagy, Z., and Esiri, M. M. (1999). Cell cycle-related protein expression in vascular dementia and Alzheimer’s disease. Neurosci. Lett. 271, 45–48. doi: 10.1016/s0304-3940(99)00509-1
Spector, D. L., and Smith, H. C. (1986). Redistribution of U-snRNPs during mitosis. Exp. Cell Res. 163, 87–94. doi: 10.1016/0014-4827(86)90560-4
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672. doi: 10.1126/science.1154584
Stadelmann, C., Brück, W., Bancher, C., Jellinger, K., and Lassmann, H. (1998). Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J. Neuropathol. Exp. Neurol. 57, 456–464. doi: 10.1097/00005072-199805000-00009
Suzuki, T., Oishi, M., Marshak, D. R., Czernik, A. J., Nairn, A. C., and Greengard, P. (1994). Cell cycle-dependent regulation of the phosphorylation and metabolism of the Alzheimer amyloid precursor protein. EMBO J. 13, 1114–1122.
Taniguchi, K., and Karin, M. (2018). NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. doi: 10.1038/nri.2017.142 [Epub ahead of print].
Tarkowski, E., Issa, R., Sjögren, M., Wallin, A., Blennow, K., Tarkowski, A., et al. (2002). Increased intrathecal levels of the angiogenic factors VEGF and TGF-β in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 23, 237–243. doi: 10.1016/s0197-4580(01)00285-8
Tesseur, I., Zou, K., Esposito, L., Bard, F., Berber, E., Can, J. V., et al. (2006). Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer’s pathology. J. Clin. Invest. 116, 3060–3069. doi: 10.1172/jci27341
Town, T., Laouar, Y., Pittenger, C., Mori, T., Szekely, C. A., Tan, J., et al. (2008). Blocking TGF-β-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat. Med. 14, 681–687. doi: 10.1038/nm1781
Varvel, N. H., Bhaskar, K., Patil, A. R., Pimplikar, S. W., Herrup, K., and Lamb, B. T. (2008). Aβ oligomers induce neuronal cell cycle events in Alzheimer’s disease. J. Neurosci. 28, 10786–10793. doi: 10.1523/JNEUROSCI.2441-08.2008
Verheijen, R., Kuijpers, H., Vooijs, P., Van Venrooij, W., and Ramaekers, F. (1986). Distribution of the 70K U1 RNA-associated protein during interphase and mitosis. Correlation with other U RNP particles and proteins of the nuclear matrix. J. Cell Sci. 86, 173–190.
Wahl, M. C., and Lührmann, R. (2015). SnapShot: spliceosome dynamics I. Cell 161:1474.e1. doi: 10.1016/j.cell.2015.05.050
Wahl, M. C., Will, C. L., and Luhrmann, R. (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. doi: 10.1016/j.cell.2009.02.009
Wan, J., Fu, A. K., Ip, F. C., Ng, H. K., Hugon, J., Page, G., et al. (2010). Tyk2/STAT3 signaling mediates β-amyloid-induced neuronal cell death: implications in Alzheimer’s disease. J. Neurosci. 30, 6873–6881. doi: 10.1523/jneurosci.0519-10.2010
Webber, K. M., Raina, A. K., Marlatt, M. W., Zhu, X., Prat, M. I., Morelli, L., et al. (2005). The cell cycle in Alzheimer disease: a unique target for neuropharmacology. Mech. Ageing Dev. 126, 1019–1025. doi: 10.1016/j.mad.2005.03.024
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U S A 106, 18809–18814. doi: 10.1073/pnas.0908767106
Wen, Y., Yang, S., Liu, R., Brun-Zinkernagel, A. M., Koulen, P., and Simpkins, J. W. (2004). Transient cerebral ischemia induces aberrant neuronal cell cycle re-entry and Alzheimer’s disease-like tauopathy in female rats. J. Biol. Chem. 279, 22684–22692. doi: 10.1074/jbc.m311768200
Wils, H., Kleinberger, G., Janssens, J., Pereson, S., Joris, G., Cuijt, I., et al. (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U S A 107, 3858–3863. doi: 10.1073/pnas.0912417107
Winkler, C., Eggert, C., Gradl, D., Meister, G., Giegerich, M., Wedlich, D., et al. (2005). Reduced U snRNP assembly causes motor axon degeneration in an animal model for spinal muscular atrophy. Genes Dev. 19, 2320–2330. doi: 10.1101/gad.342005
Wyss-Coray, T., and Rogers, J. (2012). Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2:a006346. doi: 10.1101/cshperspect.a006346
Yang, Y., Geldmacher, D. S., and Herrup, K. (2001). DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 21, 2661–2668.
Yang, Y., Mufson, E. J., and Herrup, K. (2003). Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J. Neurosci. 23, 2557–2563.
Ye, S. M., and Johnson, R. W. (1999). Increased interleukin-6 expression by microglia from brain of aged mice. J. Neuroimmunol. 93, 139–148. doi: 10.1016/s0165-5728(98)00217-3
Yuan, J., and Yankner, B. A. (2000). Apoptosis in the nervous system. Nature 407, 802–809. doi: 10.1038/35037739
Yuasa, S., Nakajima, M., Aizawa, H., Sahara, N., Koizumi, K., Sakai, T., et al. (2002). Impaired cell cycle control of neuronal precursor cells in the neocortical primordium of presenilin-1-deficient mice. J. Neurosci. Res. 70, 501–513. doi: 10.1002/jnr.10430
Zheng, C., Zhou, X. W., and Wang, J. Z. (2016). The dual roles of cytokines in Alzheimer’s disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 5:7. doi: 10.1186/s40035-016-0054-4
Zhu, X., Mcshea, A., Harris, P. L., Raina, A. K., Castellani, R. J., Funk, J. O., et al. (2004). Elevated expression of a regulator of the G2/M phase of the cell cycle, neuronal CIP-1-associated regulator of cyclin B, in Alzheimer’s disease. J. Neurosci. Res. 75, 698–703. doi: 10.1002/jnr.20028
Zhu, X., Raina, A. K., Perry, G., and Smith, M. A. (2006). Apoptosis in Alzheimer disease: a mathematical improbability. Curr. Alzheimer Res. 3, 393–396. doi: 10.2174/156720506778249470
Keywords: Alzheimer’s disease, U1 snRNP, cytoplasmic redistribution, cell cycle reentry, inflammation
Citation: Bai B (2018) U1 snRNP Alteration and Neuronal Cell Cycle Reentry in Alzheimer Disease. Front. Aging Neurosci. 10:75. doi: 10.3389/fnagi.2018.00075
Received: 29 December 2017; Accepted: 06 March 2018;
Published: 23 March 2018.
Edited by:
Antonio Camins, Universitat de Barcelona, SpainReviewed by:
Daniel Ortuño-Sahagún, Universidad de Guadalajara, MexicoZhexing Wen, Emory University School of Medicine, United States
Copyright © 2018 Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing Bai, YmIwMDAwNEBvdXRsb29rLmNvbQ==