 Jason A. Brandon
Jason A. Brandon Brandon C. Farmer
Brandon C. Farmer Holden C. Williams
Holden C. Williams Lance A. Johnson
Lance A. Johnson- Department of Physiology, University of Kentucky, Lexington, KY, United States
Apolipoprotein E4 (ApoE4) is the strongest genetic risk factor for late onset Alzheimer’s Disease (AD), and is associated with impairments in cerebral metabolism and cerebrovascular function. A substantial body of literature now points to E4 as a driver of multiple impairments seen in AD, including blunted brain insulin signaling, mismanagement of brain cholesterol and fatty acids, reductions in blood brain barrier (BBB) integrity, and decreased cerebral glucose uptake. Various neuroimaging techniques, in particular positron emission topography (PET) and magnetic resonance imaging (MRI), have been instrumental in characterizing these metabolic and vascular deficits associated with this important AD risk factor. In the current mini-review article, we summarize the known effects of APOE on cerebral metabolism and cerebrovascular function, with a special emphasis on recent findings via neuroimaging approaches.
Introduction
Apolipoprotein E (ApoE) plays an critical role in the metabolism of lipoproteins and redistribution of cholesterol, and has long been studied in relation to atherosclerosis and cardiovascular disease (Mahley and Rall, 2000; Eichner et al., 2002; Pendse et al., 2009). In the periphery, apoE is primarily produced by the liver, but is also expressed by a number of other tissues (Driscoll and Getz, 1984; Zechner et al., 1991). In the brain, apoE is primarily produced by astrocytes, and it plays a critical role in neuronal maintenance and repair (Xu et al., 1996, 2006; Mahley and Rall, 2000). In humans, there are three major isoforms of apoE: E2, E3 and E4 (Mahley and Rall, 2000). E3 is the major isoform expressed in humans, and the effects of E2 and E4 are typically compared to those of E3 to determine relative risk (Phillips, 2014). Importantly, APOE is the strongest genetic risk factor for late onset Alzheimer’s Disease (AD), with E4 conferring between a 3- (heterozygous) to 15-fold (homozygous) increase in risk of AD (Farrer et al., 1997; Raber et al., 2004). Conversely, E2 is associated with increased longevity and a decreased risk of AD (Farrer et al., 1997; Garatachea et al., 2014).
Normal synaptic function requires a multitude of energy-intensive processes, and a complex and intricately linked interplay between neurons and supporting glia is necessary to maintain efficient energy metabolism (Belanger et al., 2011). Metabolic dysfunction, as in the case of insulin resistance (IR) and type 2 diabetes, increases the risk of dementia and shares several pathological characteristics with AD, such as inflammation, increases in oxidative stress and vascular dysfunction (Craft, 2009; Walker and Harrison, 2015). Metabolic disorders also increase in incidence with age (Narayan et al., 2006) and a rapidly ageing demographic means the number of individuals suffering from both metabolic disorders and AD is expanding precipitously.
It is now well established that E4 is associated with various impairments in CNS metabolism, most notably decreased cerebral glucose uptake. A substantial body of literature now suggests that E4 carriage can in itself be viewed as a form of cerebral metabolic dysfunction. In the current mini-review article, we summarize important recent findings related to apoE’s role in modulating cerebral metabolism and cerebrovascular function, with a special emphasis on neuroimaging approaches (see Figure 1).
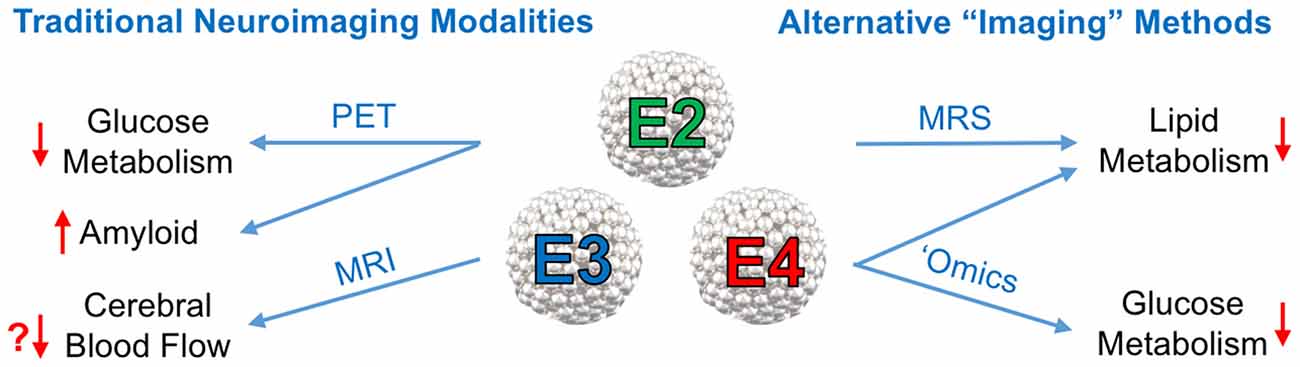
Figure 1. Neuroimaging approaches to study the effects of the Apolipoprotein E (ApoE) isoforms on cerebral metabolism. Left: traditional neuroimaging techniques, in particular positron emission topography (PET) and magnetic resonance imaging (MRI), have been invaluable in characterizing the metabolic and cerebrovascular deficits associated with APOE4, the strongest genetic risk factor for late onset Alzheimer’s disease (AD). 18F-Fluorodeoxyglucose positron emission topography (FDG-PET) imaging has consistently shown a pattern of brain glucose hypometabolism in individuals with E4, while amyloid PET imaging shows increased amyloid deposition. Some, but not all, blood oxygen level dependent (BOLD) functional magnetic resonance imaging (fMRI) studies in E4 individuals have shown decreased cerebral blood flow (CBF). Right: other alternative, or non-imaging methods of visualizing vascular and metabolic changes may prove instrumental in elucidating apoE isoform-specific effects on AD risk and progression. Magnetic resonance spectroscopy (MRS), metabolomics and lipidomics studies have implicated E4 in multiple pathways of lipid and glucose metabolism (effects of E4 denoted by red arrows).
APOE, Cerebral Glucose Metabolism, and Peripheral Glucose Regulation
18F-Fluorodeoxyglucose positron emission topography (FDG-PET) is commonly used to measure cerebral glucose metabolism. A reduction in cerebral metabolic rate of glucose (CMRglc), as measured by FDG-PET, is now considered one of the hallmarks of AD (Small et al., 2000). FDG-PET is able to differentiate AD from other types of dementia with a high degree of specificity due to specific regional patterns (Laforce and Rabinovici, 2011). Clinical AD symptoms essentially never occur without glucose hypometabolism, and the extent of the metabolic changes are strongly correlated with the severity of clinical symptoms (Grady et al., 1986; Haxby et al., 1990; Blass, 2002). Furthermore, recent evidence suggests that these alterations in glucose metabolism occur very early in the neurodegenerative process (Small et al., 1995; Reiman et al., 1996; de Leon et al., 2001; Mosconi et al., 2008).
Interestingly, a pattern of brain glucose hypometabolism regionally similar to that observed in AD has been described in individuals with E4 in a number of studies over the past two decades (reviewed in more detail here, Wolf et al., 2013). This pattern of decreased cerebral glucose metabolism is observed even in non-demented, cognitively normal E4 carriers (Small et al., 1995, 2000; Reiman et al., 1996, 2001) thereby lending support to this being an inherent biological feature of E4, rather than simply a byproduct of dementia (Reiman et al., 2004). Importantly, these metabolic deficits are present decades in advance of AD onset in E4 individuals—reductions in cerebral glucose utilization are observed in normal E4 individuals as young as their 20–30s (Reiman et al., 2004). This pattern of cerebral glucose hypometabolism in young E4 carriers is considered the earliest brain abnormality described to date in living individuals at risk for AD (Mosconi et al., 2008). Below, we summarize some more recent findings that may shed light on this well-established E4-associated phenomenon, including studies that look beyond FDG-PET imaging.
A recent study by Nielsen et al. (2017) examined how peripheral apoE levels affect cognition, gray matter volume (GMV) and cerebral glucose metabolism in an isoform-dependent manner. They showed that females not only have higher plasma levels of total apoE and apoE4 compared to males, but also see significant increases in the apoE3 isoform with age (Nielsen et al., 2017). In the same study, higher ratios of apoE4/apoE3 were negatively associated with CMRglc and GMV. These results may point toward an important role for peripheral apoE levels in modulating brain health and may offer insight into the higher risk of AD in women, particularly women with E4 (Altmann et al., 2014; Nielsen et al., 2017).
Several recent studies have explored the connection between E4, glucose metabolism and amyloid pathology. For example, by comparing FDG-PET data using β-amyloid as a continuous variable, Carbonell et al. (2016) showed that E4 and β-amyloid have a strong association with glucose hypometabolism during early AD stages. Cognitively normal E4 carriers have increased Aβ deposition, with a nonlinear relationship with age; albeit subtle effects on GMV and glucose metabolism compared to FDG-PET scans of other cognitively normal noncarriers (Gonneaud et al., 2016).
Other groups have recently begun to probe the relationship of peripheral glucose regulation and insulin sensitivity to cerebral metabolism. For example, Foley et al. (2016) showed that in E4 carriers, the degree of glucose dysregulation (measured by fasting blood glucose concentration and HbA1C) correlates with reduced cortical thickness; in fact, those diagnosed with diabetes demonstrated a level of cortical thinning comparable to that of preclinical AD. Additionally, impaired glycemia (defined here as a fasting glucose ≥100 mg/dL) and E4 genotype are independent risk factors for cerebral amyloid deposition in cortical regions, but do not appear to have an additive effect (Morris et al., 2016). E4 also confers a greater risk of age-related white matter hyperintensities in diabetics aged 73–76 years and acts as a predictor for progression of white matter hyperintensities (Cox et al., 2017). Finally, post-mortem studies of young adult E4 carriers showed upregulation of several transporters (GLUT1, GLUT3 and MCT2), metabolic enzymes (hexokinase, SCOT and AACS), and mitochondrial complexes I, II, IV, suggestive of inherent apoE-associated alterations in metabolism (Valla et al., 2010; Perkins et al., 2016).
Increasing evidence suggests that cognitive impairment resulting from IR and E4 share common neuropathological features and involve similar changes in metabolism and cerebrovascular function. For instance, the cerebrovascular pathology observed in diabetic patients and in individuals with E4 show significant overlap (Walker and Harrison, 2015) and both IR and E4 have been independently associated with brain glucose hypometabolism and reduced cerebral blood flow (CBF; Thambisetty et al., 2010; Filippini et al., 2011; Pallas and Larson, 1996; Chung et al., 2015). Additionally, these two risk factors appear to interact to impair cognition and drive neurodegeneration (Peila et al., 2002; Dore et al., 2009; Salameh et al., 2016; Johnson et al., 2017a,b). Along these lines, a number of recent studies directly implicate E4 in pathways of insulin signaling (Wolf et al., 2013). For example, in both human apoE mice and postmortem human brain tissue, E4 reduced the expression of insulin signaling proteins such as IRS1 and Akt (Ong et al., 2014; Keeney et al., 2015). In a mouse model of AD, E4 expression accelerated cognitive deficits and exaggerated impairments in insulin signaling (Chan et al., 2015, 2016). Importantly, a recent study by Zhao et al. (2017) showed that E4 directly impairs cerebral insulin signaling in an age-dependent manner, and that peripheral IR and E4 also acted synergistically to impair insulin signaling in the brain. Finally, E4+ individuals do not cognitively benefit from intranasal insulin administration, suggestive of brain IR (Reger et al., 2006; Hanson et al., 2015b). Together, these findings may suggest a metabolic dysregulation in early life stages preceding disease onset (Perkins et al., 2016).
APOE and Brain Lipid Metabolism
ApoE serves as the primary lipid carrier protein in the brain, carrying cholesterol synthesized from astrocytes to neurons in HDL-like lipoprotein particles. These lipid carrying lipoproteins have been shown to interact with Aβ (Sanan et al., 1994). In both transgenic AD mouse models and in post-mortem AD tissue, apoE and its corresponding cholesterol were shown to co-localize with Aβ plaques (Panchal et al., 2010; Lazar et al., 2013). Interestingly, lipid associated E4 has a higher Aβ binding affinity than the delipidated isoforms (Sanan et al., 1994). While this may suggest that cholesterol is involved in E4 driven AD pathology, the mechanism by which apoE-shuttled cholesterol interacts with Aβ is unclear.
In addition to APOE associated alterations in brain cholesterol, studies have shown that there is an isoform-dependent usage of fatty acids in both mice and humans. Arbones-Mainar et al. (2016) showed that E4 mice exhibit a metabolic shift toward fatty acid oxidation compared to controls using indirect calorimetry. In humans, it has been reported that E4 individuals β-oxidize uniformly labeled docosahexaenoic acid at higher rates than age- and disease-matched controls (Chouinard-Watkins et al., 2013). E4 expressing mice have also shown to have dysregulated fatty acid synthesis in the entorhinal cortex (Nuriel et al., 2017). Specifically, targeted metabolomics of E4 entorhinal cortices revealed significant changes in multiple glycerolipid and glycerophospholipid species, (Johnson et al., 2017a) as well as seven fatty acid species, (Nuriel et al., 2017) when compared to E3 mice. Thus, in addition to the glucose and insulin impairments described in the previous section, these studies point to a potential lipid mismanagement in E4 carriers which may contribute to AD pathogenesis.
APOE, Cerebral Blood Flow and Cerebral Amyloid Angiopathy
Similar to dynamic vasculature meeting metabolic needs through hyperemic action in skeletal muscle, a related theory has been posited in the brain as so called “neurovascular coupling”. These events are the observed increases in CBF to meet hyperactive neuronal activity. Initially thought to be a response to an oxygen deficit, there is now evidence suggesting direct action of various vasoactive agents on the local vasculature to modulate CBF. Neurovascular coupling is the basis for routinely used imaging platforms including functional magnetic resonance imaging (fMRI). The blood oxygen level dependent (BOLD) contrast response represents an fMRI signal comprised of both a blood flow and metabolic component, as each voxel reflects changes in deoxyhemoglobin and CBF.
Some studies suggest that cerebral hypoperfusion precedes, and possibly contributes to, the onset of dementia (Ruitenberg et al., 2005). Because metabolic rate and CBF are coupled, alterations in cerebral metabolism are likely to affect CBF (Koehler et al., 2009). However, it remains unclear whether cerebral hypoperfusion is a driver of cognitive decline, or whether the deficits simply reflect diminished metabolic demand due to aging and/or neurodegeneration. Given the importance of an efficient and responsive vascular system, it has been proposed that the accelerated AD pathogenesis associated with E4 may result from detrimental cerebrovasculature effects (Tai et al., 2016).
It is well established that CBF is decreased in AD patients (Celsis et al., 1997; Roher et al., 2012). However, both increased (Scarmeas et al., 2005; Thambisetty et al., 2010; Filippini et al., 2011) and decreased (Wierenga et al., 2013; Zlatar et al., 2014) CBF has been observed in individuals with E4, with results differing depending on age (Filippini et al., 2011). Reduced CBF in multiple brain regions has been observed in elderly E4 carriers relative to non-carriers (Filippini et al., 2011). Cognitively normal E4 individuals also show sharper age-related declines in regional CBF (Thambisetty et al., 2010) and APOE modifies the association between cognitive function and age-related changes in CBF (Wierenga et al., 2013). In addition to these differences in resting CBF, several studies have shown differences in functional activation, as measured by BOLD fMRI, in middle aged and older E4 individuals (Scarmeas et al., 2005). Functional differences in CBF have been noted in individuals as early as their 20s (Scarmeas et al., 2005) suggesting that E4-associated alterations in brain physiology occur early in life—in the absence of gross neuropathological changes and preceding cognitive impairments (Di Battista et al., 2016).
Evidence from studies using human apoE mice also highlight CBF as a potential link between E4 and impaired cognition. For example, Wiesmann et al. (2016) recently used a flow-sensitive MRI technique to show that 18-month old E4 mice have reduced CBF compared to WT mice. Using MRI, Lin et al. (2017) similarly showed that compared to WT mice, E4 mice have reduced CBF. Furthermore, they showed improvements in CBF in E4 mice following treatment with rapamycin, a pleiotropic compound with various metabolic effects, and provided evidence that the blood brain barrier (BBB) is involved in mediating these effects (Lin et al., 2017). Our own group recently showed that both diet-induced IR and E4 decreased cerebral blood volume (CBV) as measured with optical microangiography (Johnson et al., 2017b). We further demonstrated that an oral glucose gavage selectively improved cognitive performance in E4 mice with IR, and that this spike in blood glucose resulted in a significant increase in CBV. Interestingly, our results in this mouse model of human apoE mirrored a recent clinical research study, in which E4 carriers showed acute cognitive benefits from a high glycemic index meal (Hanson et al., 2015a). However, it should be noted that not all studies of CBF or CBV in human apoE mice have been in consensus. For example, a recent study using a steady-state gadolinium-enhanced fMRI technique showed that, compared to E3 mice, aged E4 mice were found to have higher CBV in the hippocampal formations (Nuriel et al., 2017).
BBB dysfunction can lead to impairments in microvascular function, and thus represents a potential pathway leading to neurodegeneration and AD (Zlokovic, 2013). In fact, multiple studies have linked APOE genotype with BBB function, with E4 leading to higher BBB permeability, decreased cerebral vascularization, thinner vessel walls and reduced CBF (Bell et al., 2012; Alata et al., 2015). Importantly, these E4-associated vascular defects were observed as early as 2 weeks of age (Bell et al., 2012) well preceding the neuronal and synaptic dysfunction that is observed in these mice in late age.
Apart from its effect on AD risk, E4 has also been independently linked to the development of cerebral amyloid angiopathy (CAA). A majority of AD brains show CAA, which is a result of amyloid deposition within the walls of small vessels in the leptomeninges and brain parenchyma. A recent study from Nielsen et al. (2017) showed increased incidence of CAA in ApoE4 postmortem brain tissues. Interestingly, they also found an association with E2, but this finding is not consistent across other studies (Rannikmae et al., 2014). CAA has been studied in the context of AD mouse models as well. Deletion of murine APOE in two mouse models of Aβ deposition resulted in abolishment of Aβ deposits in the brain parenchyma and cerebrovasculature (Holtzman et al., 2000). This deletion also resulted in less CAA-associated microhemorrhage (Fryer et al., 2005). These data demonstrate that apoE facilitates the formation of cerebrovascular plaques, which are pathological hallmarks of CAA. As compliance decreases with increased deposition of insoluble material, it is probable that cerebrovascular amyloid has substantial effects on the hemodynamics of the brain microvasculature. Could deposition of Aβ in the microvascular walls be a primary cause of the decreased CBF in E4 individuals? Questions such as these highlight the need for further studies examining CBF in the context of CAA.
Limitations of Cerebral Metabolic Imaging, Alternative “Imaging” Approaches and Future Directions
While they have provided an invaluable knowledge base, each brain imaging technology described above comes with its own set of limitations. Of particular importance in regards to the E4-associated phenomenon of cerebral glucose hypometabolism, are the limitations to biological interpretation of FDG-PET measures. Mainly that CMRglc as determined by FDG-PET is based on blood flow, 2-deoxyglucose (not glucose) transport out of the bloodstream, and phosphorylation of 2DG by the enzyme hexokinase. Thus, the process and its interpretation are restricted to the initial biochemical steps of glycolysis. In theory, the net rate of 2DG uptake is equal to the net rate of the entire glycolytic pathway at steady state (Reivich et al., 1969; Sokoloff et al., 1977; Sokoloff, 1977, 1984; Phelps et al., 1979). However, limited resolution means the cell type(s) responsible remain unknown, and there is no information on whether glucose is eventually converted to ATP in mitochondria, enters the pentose phosphate pathway, is stored as glycogen, or converted to lactate (Mosconi, 2013). Thus, future studies aimed at tracing glucose and other metabolites to their eventual fate will be critical in expanding our understanding of APOE’s effects on cerebral metabolism.
Outside of traditional PET- and MRI-based approaches, a few other imaging modalities have been applied to the question of APOE influences on cerebral metabolism. For example, magnetic resonance spectroscopy (MRS) has been used by several groups to examine common metabolite concentrations in control and AD individuals of various APOE genotypes, as well as human apoE mice. Results have been conflicting, with some groups showing increased choline/creatine and myo-inositol/creatine ratios in E4 carriers (Gomar et al., 2014; Riese et al., 2015) while others showed no APOE differences in measured metabolites (Kantarci et al., 2000; Suri et al., 2017). Finally, Dumanis et al. (2013) used MRS in human apoE mice to show a decrease in production of glutamate and increased levels of glutamine in E4 mice.
New applications of established imaging modalities are also providing novel insight into cerebral metabolism. For example, a recent study by Shokouhi et al. (2017) utilized a novel FDG-PET analysis, the regional FDG time correlation coefficient (rFTC) to sensitively measure longitudinal changes in metabolism in cognitively normal individuals. By capturing within-subject similarities between baseline and follow-up regional radiotracer distributions, they showed that rFTC decline was significantly steeper in E4 carriers compared to noncarriers (Shokouhi et al., 2017). PET can provide measurement of not only CMRglc, but also metabolic rate of oxygen (CMRO2), thereby allowing estimation of glucose metabolism outside of oxidative phosphorylation, or aerobic glycolysis. A series of articles by Raichle and colleagues have shed new light on the importance of cerebral rates of aerobic glycolysis by defining a new measure of aerobic glycolysis, the glycolytic index (GI), and applying this measure to pertinent studies of regional metabolic variability, amyloid deposition, and cognitive activation. Vaishnavi et al. (2010) showed strong regional variations in aerobic glycolysis, with two cortical regions (the default mode network and areas in the frontal and parietal cortex), showing the highest rate. Further, the areas of the normal brain that demonstrate the highest rates of aerobic glycolysis show near complete overlap with areas of the AD brain that preferentially accumulate amyloid, and it has thus been suggested that impairments in aerobic glycolysis may contribute to AD pathophysiology (Vlassenko et al., 2010). Finally, Shannon et al. (2016) combined fMRI and PET to examine the metabolic profile of activated brain areas before and after a task, and demonstrate that aerobic glycolysis is indeed enhanced in areas undergoing learning induced plasticity. Interestingly, declines in cerebral glucose utilization are greater than those in blood flow and oxygen consumption in the early stages of AD (Lying-Tunell et al., 1981; Hoyer et al., 1991; Fukuyama et al., 1994; Blass, 2002). This discrepancy in the initial stages of the disease may suggest that changes in aerobic glycolysis are an influential metabolic feature of early AD, and raises several important questions, including: Do differences in aerobic glycolysis underlie the hypometabolism observed in young E4 carriers?
Finally, other non-imaging methods of “visualizing” metabolic changes may prove instrumental in elucidating APOE-driven alterations in cerebral metabolism. For example, two groups have applied metabolomic analyses to brain tissue from human apoE expressing mice (Johnson et al., 2017a; Nuriel et al., 2017). Nuriel et al. (2017) used a mass spectrometry (MS) based metabolomics technique to report an E4-associated downregulation of several fatty acid species and an upregulation of multiple TCA-cycle metabolites, among other changes. Our own recent study applied an integrated ‘omics approach to provide insight into the metabolic pathways altered in E4 brains (Johnson et al., 2017a). Combining measures of DNA hydroxymethylation and MS-based metabolomics, we identified novel E4-associated alterations in multiple pathways, most notably purine metabolism, glutamate metabolism, and the pentose phosphate pathway (Johnson et al., 2017a).
Summary
Over the years, the neuropathological characterization of AD has expanded beyond the classic descriptions of amyloid and tau pathology to include metabolic and vascular dysfunction. Advances in brain imaging, most notably PET and MRI, have been invaluable in broadening our understanding of AD pathophysiology. Specifically, the metabolic and cerebrovascular dysfunction described by these studies includes reductions in brain glucose uptake, blunted insulin signaling, alterations in brain lipid metabolism, loss of BBB integrity and deficits in CBF. As reviewed above, many similar alterations have been observed in E4+ individuals, sometimes early in life and often even in the absence of cognitive impairment. Perhaps reflected in the sum of these findings is an inherent inability of E4+ individuals to efficiently regulate cerebral metabolism; although whether it occurs at the level of the BBB or cellular uptake, oxidative phosphorylation, aerobic glycolysis or elsewhere remains unclear. Future studies aimed at expanding the important knowledge base established by PET, MRI and other traditional imaging techniques will be critical in better understanding how E4 drives metabolic dysfunction in AD, and essential in identifying new therapeutic targets to correct these deficiencies in order to delay or prevent AD.
Author Contributions
JB, BF, HW and LJ wrote and edited the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grant No. P20GM103527. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding sources.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alata, W., Ye, Y., St-Amour, I., Vandal, M., and Calon, F. (2015). Human apolipoprotein E ε4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab. 35, 86–94. doi: 10.1038/jcbfm.2014.172
Altmann, A., Tian, L., Henderson, V. W., and Greicius, M. D., and Alzheimer’s Disease Neuroimaging Initiative Investigators. (2014). Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol. 75, 563–573. doi: 10.1002/ana.24135
Arbones-Mainar, J. M., Johnson, L. A., Torres-Perez, E., Garcia, A. E., Perez-Diaz, S., Raber, J., et al. (2016). Metabolic shifts toward fatty-acid usage and increased thermogenesis are associated with impaired adipogenesis in mice expressing human APOE4. Int. J. Obes. 40, 1574–1581. doi: 10.1038/ijo.2016.93
Belanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Bell, R. D., Winkler, E. A., Singh, I., Sagare, A. P., Deane, R., Wu, Z., et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. doi: 10.1038/nature11087
Blass, J. P. (2002). Alzheimer’s disease and Alzheimer’s dementia: distinct but overlapping entities. Neurobiol. Aging 23, 1077–1084. doi: 10.1016/s0197-4580(02)00036-2
Carbonell, F., Zijdenbos, A. P., McLaren, D. G., Iturria-Medina, Y., and Bedell, B. J. (2016). Modulation of glucose metabolism and metabolic connectivity by β-amyloid. J. Cereb. Blood Flow Metab. 36, 2058–2071. doi: 10.1177/0271678x16654492
Celsis, P., Agniel, A., Cardebat, D., Demonet, J. F., Ousset, P. J., and Puel, M. (1997). Age related cognitive decline: a clinical entity? A longitudinal study of cerebral blood flow and memory performance. J. Neurol. Neurosurg. Psychiatry 62, 601–608. doi: 10.1136/jnnp.62.6.601
Chan, E. S., Chen, C., Cole, G. M., and Wong, B. S. (2015). Differential interaction of Apolipoprotein-E isoforms with insulin receptors modulates brain insulin signaling in mutant human amyloid precursor protein transgenic mice. Sci. Rep. 5:13842. doi: 10.1038/srep13842
Chan, E. S., Shetty, M. S., Sajikumar, S., Chen, C., Soong, T. W., and Wong, B. S. (2016). ApoE4 expression accelerates hippocampus-dependent cognitive deficits by enhancing Aβ impairment of insulin signaling in an Alzheimer’s disease mouse model. Sci. Rep. 6:26119. doi: 10.1038/srep26119
Chouinard-Watkins, R., Rioux-Perreault, C., Fortier, M., Tremblay-Mercier, J., Zhang, Y., Lawrence, P., et al. (2013). Disturbance in uniformly 13C-labelled DHA metabolism in elderly human subjects carrying the apoE ε4 allele. Br. J. Nutr. 110, 1751–1759. doi: 10.1017/S0007114513001268
Chung, C. C., Pimentel, D., Jor’dan, A. J., Hao, Y., Milberg, W., and Novak, V. (2015). Inflammation-associated declines in cerebral vasoreactivity and cognition in type 2 diabetes. Neurology 85, 450–458. doi: 10.1212/WNL.0000000000001820
Cox, S. R., Ritchie, S. J., Dickie, D. A., Pattie, A., Royle, N. A., Corley, J., et al. (2017). Interaction of APOE ε4 and poor glycemic control predicts white matter hyperintensity growth from 73 to 76. Neurobiol. Aging 54, 54–58. doi: 10.1016/j.neurobiolaging.2017.02.014
Craft, S. (2009). The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch. Neurol. 66, 300–305. doi: 10.1001/archneurol.2009.27
de Leon, M. J., Convit, A., Wolf, O. T., Tarshish, C. Y., DeSanti, S., Rusinek, H., et al. (2001). Prediction of cognitive decline in normal elderly subjects with 2-[18F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc. Natl. Acad. Sci. U S A 98, 10966–10971. doi: 10.1073/pnas.191044198
Di Battista, A. M., Heinsinger, N. M., and Rebeck, G. W. (2016). Alzheimer’s disease genetic risk factor APOE-ε4 also affects normal brain function. Curr. Alzheimer Res. 13, 1200–1207. doi: 10.2174/1567205013666160401115127
Dore, G. A., Elias, M. F., Robbins, M. A., Elias, P. K., and Nagy, Z. (2009). Presence of the APOE ε4 allele modifies the relationship between type 2 diabetes and cognitive performance: the Maine-Syracuse Study. Diabetologia 52, 2551–2560. doi: 10.1007/s00125-009-1497-2
Driscoll, D. M., and Getz, G. S. (1984). Extrahepatic synthesis of apolipoprotein E. J. Lipid Res. 25, 1368–1379.
Dumanis, S. B., DiBattista, A. M., Miessau, M., Moussa, C. E., and Rebeck, G. W. (2013). APOE genotype affects the presynaptic compartment of glutamatergic nerve terminals. J. Neurochem. 124, 4–14. doi: 10.1111/j.1471-4159.2012.07908.x
Eichner, J. E., Dunn, S. T., Perveen, G., Thompson, D. M., Stewart, K. E., and Stroehla, B. C. (2002). Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am. J. Epidemiol. 155, 487–495. doi: 10.1093/aje/155.6.487
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Filippini, N., Ebmeier, K. P., MacIntosh, B. J., Trachtenberg, A. J., Frisoni, G. B., Wilcock, G. K., et al. (2011). Differential effects of the APOE genotype on brain function across the lifespan. Neuroimage 54, 602–610. doi: 10.1016/j.neuroimage.2010.08.009
Foley, J. M., Salat, D. H., Stricker, N. H., McGlinchey, R. E., Milberg, W. P., Grande, L. J., et al. (2016). Glucose dysregulation interacts with APOE-4 to potentiate temporoparietal cortical thinning. Am. J. Alzheimers Dis. Other Demen. 31, 76–86. doi: 10.1177/1533317515587084
Fryer, J. D., Simmons, K., Parsadanian, M., Bales, K. R., Paul, S. M., Sullivan, P. M., et al. (2005). Human apolipoprotein E4 alters the amyloid-β 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. 25, 2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005
Fukuyama, H., Ogawa, M., Yamauchi, H., Yamaguchi, S., Kimura, J., Yonekura, Y., et al. (1994). Altered cerebral energy metabolism in Alzheimer’s disease: a PET study. J. Nucl. Med. 35, 1–6.
Garatachea, N., Emanuele, E., Calero, M., Fuku, N., Arai, Y., Abe, Y., et al. (2014). ApoE gene and exceptional longevity: insights from three independent cohorts. Exp. Gerontol. 53, 16–23. doi: 10.1016/j.exger.2014.02.004
Gomar, J. J., Gordon, M. L., Dickinson, D., Kingsley, P. B., Ulug, A. M., Keehlisen, L., et al. (2014). APOE genotype modulates proton magnetic resonance spectroscopy metabolites in the aging brain. Biol. Psychiatry 75, 686–692. doi: 10.1016/j.biopsych.2013.05.022
Gonneaud, J., Arenaza-Urquijo, E. M., Fouquet, M., Perrotin, A., Fradin, S., de La Sayette, V., et al. (2016). Relative effect of APOE ε4 on neuroimaging biomarker changes across the lifespan. Neurology 87, 1696–1703. doi: 10.1212/WNL.0000000000003234
Grady, C. L., Haxby, J. V., Schlageter, N. L., Berg, G., and Rapoport, S. I. (1986). Stability of metabolic and neuropsychological asymmetries in dementia of the Alzheimer type. Neurology 36, 1390–1392. doi: 10.1212/WNL.36.10.1390
Hanson, A. J., Bayer, J. L., Baker, L. D., Cholerton, B., VanFossen, B., Trittschuh, E., et al. (2015a). Differential effects of meal challenges on cognition, metabolism, and biomarkers for apolipoprotein E ε4 carriers and adults with mild cognitive impairment. J. Alzheimers Dis. 48, 205–218. doi: 10.3233/JAD-150273
Hanson, A. J., Craft, S., and Banks, W. A. (2015b). The APOE genotype: modification of therapeutic responses in Alzheimer’s disease. Curr. Pharm. Des. 21, 114–120. doi: 10.2174/1381612820666141020164222
Haxby, J. V., Grady, C. L., Koss, E., Horwitz, B., Heston, L., Schapiro, M., et al. (1990). Longitudinal study of cerebral metabolic asymmetries and associated neuropsychological patterns in early dementia of the Alzheimer type. Arch. Neurol. 47, 753–760. doi: 10.1001/archneur.1990.00530070043010
Holtzman, D. M., Fagan, A. M., Mackey, B., Tenkova, T., Sartorius, L., Paul, S. M., et al. (2000). Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann. Neurol. 47, 739–747. doi: 10.1002/1531-8249(200006)47:6<739::aid-ana6>3.3.co;2-#
Hoyer, S., Nitsch, R., and Oesterreich, K. (1991). Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: a cross-sectional comparison against advanced late-onset and incipient early-onset cases. J. Neural Transm. Park. Dis. Dement. Sect. 3, 1–14. doi: 10.1007/bf02251132
Johnson, L. A., Torres, E. R., Impey, S., Stevens, J. F., and Raber, J. (2017a). Apolipoprotein E4 and insulin resistance interact to impair cognition and alter the epigenome and metabolome. Sci. Rep. 7:43701. doi: 10.1038/srep43701
Johnson, L. A., Torres, E. R., Weber Boutros, S., Patel, E., Akinyeke, T., Alkayed, N. J., et al. (2017b). Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J. Cereb. Blood Flow Metab. doi: 10.1177/0271678x17746186 [Epub ahead of print].
Kantarci, K., Jack, C. R. Jr., Xu, Y. C., Campeau, N. G., O’Brien, P. C., Smith, G. E., et al. (2000). Regional metabolic patterns in mild cognitive impairment and Alzheimer’s disease: a 1H MRS study. Neurology 55, 210–217. doi: 10.1212/wnl.55.2.210
Keeney, J. T., Ibrahimi, S., and Zhao, L. (2015). Human ApoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: evidence of the mechanism of neuroprotection by apoE2 and implications for Alzheimer’s disease prevention and early intervention. J. Alzheimers Dis. 48, 411–424. doi: 10.3233/JAD-150348
Koehler, R. C., Roman, R. J., and Harder, D. R. (2009). Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 32, 160–169. doi: 10.1016/j.tins.2008.11.005
Laforce, R. Jr., and Rabinovici, G. D. (2011). Amyloid imaging in the differential diagnosis of dementia: review and potential clinical applications. Alzheimers Res. Ther. 3:31. doi: 10.1186/alzrt93
Lazar, A. N., Bich, C., Panchal, M., Desbenoit, N., Petit, V. W., Touboul, D., et al. (2013). Time-of-flight secondary ion mass spectrometry (TOF-SIMS) imaging reveals cholesterol overload in the cerebral cortex of Alzheimer disease patients. Acta Neuropathol. 125, 133–144. doi: 10.1007/s00401-012-1041-1
Lin, A. L., Jahrling, J. B., Zhang, W., DeRosa, N., Bakshi, V., Romero, P., et al. (2017). Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J. Cereb. Blood Flow Metab. 37, 217–226. doi: 10.1177/0271678x15621575
Lying-Tunell, U., Lindblad, B. S., Malmlund, H. O., and Persson, B. (1981). Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 63, 337–350. doi: 10.1111/j.1600-0404.1981.tb00788.x
Mahley, R. W., and Rall, S. C. Jr. (2000). Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet. 1, 507–537. doi: 10.1146/annurev.genom.1.1.507
Morris, J. K., Vidoni, E. D., Wilkins, H. M., Archer, A. E., Burns, N. C., Karcher, R. T., et al. (2016). Impaired fasting glucose is associated with increased regional cerebral amyloid. Neurobiol. Aging 44, 138–142. doi: 10.1016/j.neurobiolaging.2016.04.017
Mosconi, L. (2013). Glucose metabolism in normal aging and Alzheimer’s disease: methodological and physiological considerations for PET studies. Clin. Transl. Imaging 1, 217–233. doi: 10.1007/s40336-013-0026-y
Mosconi, L., De Santi, S., Li, J., Tsui, W. H., Li, Y., Boppana, M., et al. (2008). Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 29, 676–692. doi: 10.1016/j.neurobiolaging.2006.12.008
Narayan, K. M., Boyle, J. P., Geiss, L. S., Saaddine, J. B., and Thompson, T. J. (2006). Impact of recent increase in incidence on future diabetes burden: U.S., 2005–2050. Diabetes Care 29, 2114–2116. doi: 10.2337/dc06-1136
Nielsen, H. M., Chen, K., Lee, W., Chen, Y., Bauer, R. J. III., Reiman, E., et al. (2017). Peripheral apoE isoform levels in cognitively normal APOE ε3/ε4 individuals are associated with regional gray matter volume and cerebral glucose metabolism. Alzheimers Res. Ther. 9:5. doi: 10.1186/s13195-016-0231-9
Nuriel, T., Angulo, S. L., Khan, U., Ashok, A., Chen, Q., Figueroa, H. Y., et al. (2017). Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun. 8:1464. doi: 10.1038/s41467-017-01444-0
Ong, Q. R., Chan, E. S., Lim, M. L., Cole, G. M., and Wong, B. S. (2014). Reduced phosphorylation of brain insulin receptor substrate and Akt proteins in apolipoprotein-E4 targeted replacement mice. Sci. Rep. 4:3754. doi: 10.1038/srep03754
Pallas, F., and Larson, D. F. (1996). Cerebral blood flow in the diabetic patient. Perfusion 11, 363–370. doi: 10.1177/026765919601100502
Panchal, M., Loeper, J., Cossec, J. C., Perruchini, C., Lazar, A., Pompon, D., et al. (2010). Enrichment of cholesterol in microdissected Alzheimer’s disease senile plaques as assessed by mass spectrometry. J. Lipid Res. 51, 598–605. doi: 10.1194/jlr.M001859
Peila, R., Rodriguez, B. L., Launer, L. J., and Honolulu-Asia Aging Study. (2002). Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the honolulu-asia aging study. Diabetes 51, 1256–1262. doi: 10.2337/diabetes.51.4.1256
Pendse, A. A., Arbones-Mainar, J. M., Johnson, L. A., Altenburg, M. K., and Maeda, N. (2009). Apolipoprotein E knock-out and knock-in mice: atherosclerosis, metabolic syndrome, and beyond. J. Lipid Res. 50, S178–S182. doi: 10.1194/jlr.r800070-jlr200
Perkins, M., Wolf, A. B., Chavira, B., Shonebarger, D., Meckel, J. P., Leung, L., et al. (2016). Altered energy metabolism pathways in the posterior cingulate in young adult apolipoprotein E ε4 carriers. J. Alzheimers Dis. 53, 95–106. doi: 10.3233/JAD-151205
Phelps, M. E., Huang, S. C., Hoffman, E. J., Selin, C., Sokoloff, L., and Kuhl, D. E. (1979). Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Ann. Neurol. 6, 371–388. doi: 10.1002/ana.410060502
Phillips, M. C. (2014). Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 66, 616–623. doi: 10.1002/iub.1314
Raber, J., Huang, Y., and Ashford, J. W. (2004). ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol. Aging 25, 641–650. doi: 10.1016/j.neurobiolaging.2003.12.023
Rannikmae, K., Kalaria, R. N., Greenberg, S. M., Chui, H. C., Schmitt, F. A., Samarasekera, N., et al. (2014). APOE associations with severe CAA-associated vasculopathic changes: collaborative meta-analysis. J. Neurol. Neurosurg. Psychiatry 85, 300–305. doi: 10.1136/jnnp-2013-306485
Reger, M. A., Watson, G. S., Frey, W. H. II., Baker, L. D., Cholerton, B., Keeling, M. L., et al. (2006). Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol. Aging 27, 451–458. doi: 10.1016/j.neurobiolaging.2005.03.016
Reiman, E. M., Caselli, R. J., Chen, K., Alexander, G. E., Bandy, D., and Frost, J. (2001). Declining brain activity in cognitively normal apolipoprotein E ε4 heterozygotes: a foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 98, 3334–3339. doi: 10.1073/pnas.061509598
Reiman, E. M., Caselli, R. J., Yun, L. S., Chen, K., Bandy, D., Minoshima, S., et al. (1996). Preclinical evidence of Alzheimer’s disease in persons homozygous for the ε4 allele for apolipoprotein E. N. Engl. J. Med. 334, 752–758. doi: 10.1056/nejm199603213341202
Reiman, E. M., Chen, K., Alexander, G. E., Caselli, R. J., Bandy, D., Osborne, D., et al. (2004). Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. U S A 101, 284–289. doi: 10.1073/pnas.2635903100
Reivich, M., Jehle, J., Sokoloff, L., and Kety, S. S. (1969). Measurement of regional cerebral blood flow with antipyrine-14C in awake cats. J. Appl. Physiol. 27, 296–300. doi: 10.1152/jappl.1969.27.2.296
Riese, F., Gietl, A., Zölch, N., Henning, A., O’Gorman, R., Kalin, A. M., et al. (2015). Posterior cingulate γ-aminobutyric acid and glutamate/glutamine are reduced in amnestic mild cognitive impairment and are unrelated to amyloid deposition and apolipoprotein E genotype. Neurobiol. Aging 36, 53–59. doi: 10.1016/j.neurobiolaging.2014.07.030
Roher, A. E., Debbins, J. P., Malek-Ahmadi, M., Chen, K., Pipe, J. G., Maze, S., et al. (2012). Cerebral blood flow in Alzheimer’s disease. Vasc. Health Risk Manag. 8, 599–611. doi: 10.2147/VHRM.S34874
Ruitenberg, A., den Heijer, T., Bakker, S. L., van Swieten, J. C., Koudstaal, P. J., Hofman, A., et al. (2005). Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann. Neurol. 57, 789–794. doi: 10.1002/ana.20493
Salameh, T. S., Rhea, E. M., Banks, W. A., and Hanson, A. J. (2016). Insulin resistance, dyslipidemia, and apolipoprotein E interactions as mechanisms in cognitive impairment and Alzheimer’s disease. Exp. Biol. Med. Maywood 241, 1676–1683. doi: 10.1177/1535370216660770
Sanan, D. A., Weisgraber, K. H., Russell, S. J., Mahley, R. W., Huang, D., Saunders, A., et al. (1994). Apolipoprotein E associates with β amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Invest. 94, 860–869. doi: 10.1172/jci117407
Scarmeas, N., Habeck, C. G., Hilton, J., Anderson, K. E., Flynn, J., Park, A., et al. (2005). APOE related alterations in cerebral activation even at college age. J. Neurol. Neurosurg. Psychiatry 76, 1440–1444. doi: 10.1136/jnnp.2004.053645
Shannon, B. J., Vaishnavi, S. N., Vlassenko, A. G., Shimony, J. S., Rutlin, J., and Raichle, M. E. (2016). Brain aerobic glycolysis and motor adaptation learning. Proc. Natl. Acad. Sci. U S A 113, E3782–E3791. doi: 10.1073/pnas.1604977113
Shokouhi, S., Riddle, W. R., and Kang, H. (2017). A new data analysis approach for measuring longitudinal changes of metabolism in cognitively normal elderly adults. Clin. Interv. Aging 12, 2123–2130. doi: 10.2147/CIA.S150859
Small, G. W., Ercoli, L. M., Silverman, D. H., Huang, S. C., Komo, S., Bookheimer, S. Y., et al. (2000). Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 97, 6037–6042. doi: 10.1073/pnas.090106797
Small, G. W., Mazziotta, J. C., Collins, M. T., Baxter, L. R., Phelps, M. E., Mandelkern, M. A., et al. (1995). Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273, 942–947. doi: 10.1001/jama.1995.03520360056039
Sokoloff, L. (1977). Relation between physiological function and energy metabolism in the central nervous system. J. Neurochem. 29, 13–26. doi: 10.1111/j.1471-4159.1977.tb03919.x
Sokoloff, L. (1984). Modeling metabolic processes in the brain in vivo. Ann. Neurol. 15, S1–S11. doi: 10.1002/ana.410150703
Sokoloff, L., Reivich, M., Kennedy, C., Des Rosiers, M. H., Patlak, C. S., Pettigrew, K. D., et al. (1977). The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J. Neurochem. 28, 897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x
Suri, S., Emir, U., Stagg, C. J., Near, J., Mekle, R., Schubert, F., et al. (2017). Effect of age and the APOE gene on metabolite concentrations in the posterior cingulate cortex. Neuroimage 152, 509–516. doi: 10.1016/j.neuroimage.2017.03.031
Tai, L. M., Thomas, R., Marottoli, F. M., Koster, K. P., Kanekiyo, T., Morris, A. W., et al. (2016). The role of APOE in cerebrovascular dysfunction. Acta Neuropathol. 131, 709–723. doi: 10.1007/s00401-016-1547-z
Thambisetty, M., Beason-Held, L., An, Y., Kraut, M. A., and Resnick, S. M. (2010). APOE ε4 genotype and longitudinal changes in cerebral blood flow in normal aging. Arch. Neurol. 67, 93–98. doi: 10.1001/archneurol.2009.913
Vaishnavi, S. N., Vlassenko, A. G., Rundle, M. M., Snyder, A. Z., Mintun, M. A., and Raichle, M. E. (2010). Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. U S A 107, 17757–17762. doi: 10.1073/pnas.1010459107
Valla, J., Yaari, R., Wolf, A. B., Kusne, Y., Beach, T. G., Roher, A. E., et al. (2010). Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE ε4 allele, the major late-onset alzheimer’s susceptibility gene. J. Alzheimers Dis. 22, 307–313. doi: 10.3233/JAD-2010-100129
Vlassenko, A. G., Vaishnavi, S. N., Couture, L., Sacco, D., Shannon, B. J., Mach, R. H., et al. (2010). Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl. Acad. Sci. U S A 107, 17763–17767. doi: 10.1073/pnas.1010461107
Walker, J. M., and Harrison, F. E. (2015). Shared neuropathological characteristics of obesity, type 2 diabetes and Alzheimer’s disease: impacts on cognitive decline. Nutrients 7, 7332–7357. doi: 10.3390/nu7095341
Wierenga, C. E., Clark, L. R., Dev, S. I., Shin, D. D., Jurick, S. M., Rissman, R. A., et al. (2013). Interaction of age and APOE genotype on cerebral blood flow at rest. J. Alzheimers Dis. 34, 921–935. doi: 10.3233/JAD-121897
Wiesmann, M., Zerbi, V., Jansen, D., Haast, R., Lütjohann, D., Broersen, L. M., et al. (2016). A dietary treatment improves cerebral blood flow and brain connectivity in aging apoE4 mice. Neural Plast. 2016:6846721. doi: 10.1155/2016/6846721
Wolf, A. B., Caselli, R. J., Reiman, E. M., and Valla, J. (2013). APOE and neuroenergetics: an emerging paradigm in Alzheimer’s disease. Neurobiol. Aging 34, 1007–1017. doi: 10.1016/j.neurobiolaging.2012.10.011
Xu, Q., Bernardo, A., Walker, D., Kanegawa, T., Mahley, R. W., and Huang, Y. (2006). Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci. 26, 4985–4994. doi: 10.1523/JNEUROSCI.5476-05.2006
Xu, P. T., Schmechel, D., Rothrock-Christian, T., Burkhart, D. S., Qiu, H. L., Popko, B., et al. (1996). Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol. Dis. 3, 229–245. doi: 10.1006/nbdi.1996.0023
Zechner, R., Moser, R., Newman, T. C., Fried, S. K., and Breslow, J. L. (1991). Apolipoprotein E gene expression in mouse 3T3–L1 adipocytes and human adipose tissue and its regulation by differentiation and lipid content. J. Biol. Chem. 266, 10583–10588.
Zhao, N., Liu, C. C., Van Ingelgom, A. J., Martens, Y. A., Linares, C., Knight, J. A., et al. (2017). Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron 96, 115.e5–129.e5. doi: 10.1016/j.neuron.2017.09.003
Zlatar, Z. Z., Wierenga, C. E., Bangen, K. J., Liu, T. T., and Jak, A. J. (2014). Increased hippocampal blood flow in sedentary older adults at genetic risk for Alzheimer’s disease. J. Alzheimers Dis. 41, 809–817. doi: 10.3233/JAD-132252
Keywords: ApoE, apolipoprotein E, cerebral, metabolism, brain, imaging, neurodegeneration, Alzheimer’s Disease (AD)
Citation: Brandon JA, Farmer BC, Williams HC and Johnson LA (2018) APOE and Alzheimer’s Disease: Neuroimaging of Metabolic and Cerebrovascular Dysfunction. Front. Aging Neurosci. 10:180. doi: 10.3389/fnagi.2018.00180
Received: 28 March 2018; Accepted: 25 May 2018;
Published: 14 June 2018.
Edited by:
Fahmeed Hyder, Yale University, United StatesReviewed by:
Ayman ElAli, CHU de Québec Research Center, CanadaIgnacio Torres-Aleman, Consejo Superior de Investigaciones Científicas (CSIC), Spain
Copyright © 2018 Brandon, Farmer, Williams and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lance A. Johnson, am9obnNvbi5sYW5jZUB1a3kuZWR1