 Caitlin M. Carroll
Caitlin M. Carroll Shannon L. Macauley
Shannon L. Macauley- Section of Gerontology and Geriatric Medicine, Department of Internal Medicine, Wake Forest School of Medicine, Winston-Salem, NC, United States
Alzheimer’s disease (AD) is the most common form of dementia and affects over 45 million people worldwide. Both type-2-diabetes (T2D), a metabolic condition associated with aging, and disrupted sleep are implicated in the pathogenesis of AD, but how sleep and metabolism interact to affect AD progression remains unclear. In the healthy brain, sleep/wake cycles are a well-coordinated interaction between metabolic and neuronal activity, but when disrupted, are associated with a myriad of health-related issues, including metabolic syndrome, cardiovascular disease, T2D, and AD. Therefore, this review will explore our current understanding of the relationship between metabolism, sleep, and AD-related pathology to identify the causes and consequences of disease progression in AD. Moreover, sleep disturbances and metabolic dysfunction could serve as potential therapeutic targets to mitigate the increased risk of AD in individuals with T2D or offer a novel approach for treating AD.
Introduction
Alzheimer’s disease (AD) is severe neurodegenerative disorder characterized by the accumulation of extracellular amyloid plaques and intracellular neurofibrillary tau tangles (NFT), with detectable pathology occurring 15–20 years prior to the onset of clinical symptoms [as reviewed in Holtzman et al. (2011)]. Deposition of amyloid-beta (Aβ) into amyloid plaques is one of the initial changes observed in presymptomatic AD, with tau hyperphosphorylation, aggregation, and NFT formation to follow in spatially distinct regions (Bateman et al., 2012). Although changes in cerebral amyloid burden occurs early and is necessary for an AD diagnosis, the development of tau pathology, both temporally and spatially, more closely aligns with neurodegeneration and clinical symptoms. In general, disease development is multifaceted and influenced by a variety of genetic, environmental, and lifestyle components. Brain hypometabolism, hyperexcitability, oxidative stress, mitochondrial dysfunction, brain insulin resistance, and inflammation all contribute to AD pathophysiology, yet it is unclear whether they are a cause or consequence of Aβ and tau pathology (Musiek and Holtzman, 2015). Type-2-diabetes (T2D), a metabolic disease also associated with aging, is characterized by hyperglycemia, hyperinsulinemia, insulin resistance, and beta cell dysfunction. Sleep, on the other hand, is a complex homeostatic mechanism tied to altered consciousness and decreased responsiveness to sensory stimuli that relies heavily on the tight coupling between cerebral metabolism and neuronal activity. Both T2D and sleep disorders are implicated in the pathogenesis of AD, but the exact role sleep and metabolism play remains unclear (Ott et al., 1999; Stanley et al., 2016). In this review, we summarize the current understanding of the interplay between metabolism, sleep, and AD pathophysiology with an emphasis on disrupted sleep as a modifiable lifestyle characteristic contributing to increased risk of both AD and T2D.
Glucose Metabolism and Neuronal Activity in AD
The relationship between metabolic activity and neuronal activity is central to both healthy brain function as well as the pathogenesis of AD. First, neuroimaging studies confirmed that prior to the hypometabolism typically described in preclinical stages of AD, brain regions most vulnerable to Aβ and tau accumulation are uniquely reliant on glucose for normal brain function and display increased glucose consumption at a young age (Edison et al., 2007; Mosconi et al., 2008; Vaishanavi et al., 2010; Vlassenko et al., 2010; Oh et al., 2016; Hanseeuw et al., 2017; Adams et al., 2018). Aerobic glycolysis is an emerging biomarker of network vulnerability in AD where brain regions prone to Aβ deposition utilize glucose beyond what is necessary for energy production, even though sufficient concentrations of oxygen exist for oxidative phosphorylation and ATP generation (Vlassenko et al., 2010, 2018; Yao et al., 2011; Vlassenko and Raichle, 2015; Harris et al., 2016). Biosynthesis, synaptic plasticity, synaptic remodeling, memory formation, and other key processes utilize glucose in this way and are essential for healthy brain function. In AD, brain regions predisposed to Aβ deposition markedly overlap with the brain’s default mode network, a network characterized by highly variant resting-state blood oxygen level dependent fMRI signals, high resting-state functional connectivity, high aerobic glycolysis, and high neuronal activity (Jagust and Mormino, 2011). In amyloid-positive, cognitively normal adults, elevated default mode network connectivity patterns are associated with low tau burden, while decreased connectivity is associated with increased tau burden, introducing a pattern of hyper- and hypoconnectivity along the preclinical-AD trajectory (Schultz et al., 2017). Recent studies demonstrate that individuals with high amyloid burden and lower aerobic glycolysis also have higher tau burden, another core pathological feature of AD (Vlassenko et al., 2018). Therefore, an interesting relationship emerges that links brain regions that are both metabolically and neuronally active with regions susceptible to AD pathology. This places regional glucose metabolism in the aging brain at the forefront of AD pathogenesis.
In a metabolically healthy brain, neuronal activity evokes a localized vascular response to increase cerebral blood flow and deliver enough oxygen and glucose to sustain periods of increased activity. Therefore, regional glucose consumption and glucose concentration in the brain’s interstitial fluid (ISF) are closely tied to changes in neuronal activity. However, it is still unclear how alterations in glucose availability and glycemic variability can modify the relationship between neuronal and metabolic activity to influence AD-related pathology and dementia. Elevated blood glucose levels, or hyperglycemia, are associated with an increased prevalence of dementia and AD, faster progression from mild cognitive impairment (MCI) to AD, and increased rates of Aβ accumulation (Whitmer et al., 2009; Aung et al., 2012; Crane et al., 2013; Lin and Sheu, 2013; Yaffe et al., 2013; Morris et al., 2014; Gomez et al., 2018). Similarly, repeated episodes of hypoglycemia that typically accompany treatment of T2D are also associated with increased risk of dementia and dementia of an Alzheimer’s type. In preclinical models, inducing peripheral hyperglycemia in rodents increases hippocampal ISF glucose, Aβ, and lactate levels. This effect is exacerbated in mice with significant Aβ pathology, demonstrating fluctuations in peripheral metabolism not only alter cerebral metabolism and Aβ release but amyloid plaques impact the brain’s response to glycemic changes (Macauley et al., 2015). Moreover, fluctuations in ISF lactate accompany hyperglycemia and represent changes in neuronal activity (Pellerin and Magistretti, 1994). Evidence from the astrocyte neuron lactate shuttle (ANLS) demonstrates glutamate release from neurons stimulates glycolysis within astrocytes, causing astrocytes to shuttle the glycolytic end-product, lactate, to neurons as fuel for ATP generation. Thus, preferential utilization of lactate over glucose occurs during periods of heightened neuronal activity and relative changes in the pool of ISF lactate represents changes in excitatory neurotransmission. Indeed, previous studies show that Aβ is released in an activity-dependent manner via changes in synaptic vesicle exocytosis (Cirrito et al., 2005) and increased neuronal activity is coupled to ISF lactate and Aβ levels. Moreover, regional differences in Aβ levels and subsequent plaque formation mirrors regional patterns of neuronal activity, where hyperexcitability is associated with increased plaque aggregation (Busche et al., 2008; Bero et al., 2011). Although tau is a cytoplasmic protein and pathological tau accumulates intraneuronally, tau is found in the ISF and cerebrospinal fluid (CSF) demonstrating it can be released from neurons. Historically, extracellular tau was thought to be released from dead or dying cells, but recent studies demonstrate, similar to Aβ, tau is released during neuronal activity under normal conditions (Pooler et al., 2013; Yamada et al., 2014), and accumulates in periods of hyperactivity (Huijbers et al., 2019). Extracellular tau monomers and aggregates can be taken up by neighboring neurons where tau pathology can propagate and spread through network connectivity (Clavaguera et al., 2009; Lehmann et al., 2013; Wu et al., 2016; Franzmeier et al., 2019). Therefore, mechanisms that alter neuronal excitability also modulate the levels of Aβ and tau in the extracellular space and over time, can promote or attenuate Aβ and tau aggregation. This has been shown in clinical epilepsy populations (Mackenzie and Miller, 1994; Tai et al., 2016) and APOE4 targeted replacement mice, a genetic knock-in model of AD (Klein et al., 2014; Nuriel et al., 2017), where network hyperexcitability leads to increased accumulation of AD pathology. Taken together, these data support a reciprocal relationship between metabolic activity, neuronal activity, and AD-related proteins.
Individuals with T2D have a 2–4-fold increased risk of developing AD (Ott et al., 1999). Although the relationship between T2D and AD is complex, alterations in glucose metabolism and glucose homeostasis are central to both diseases. As a disease associated with aging, T2D is characterized by hyperglycemia, hyperinsulinemia, and insulin resistance (Ott et al., 1999). While disrupted glucose regulation is correlated with cerebral hyperexcitability and accumulation of pathology, disrupted insulin signaling and insulin resistance also have strong associations with MCI, dementia, and AD (Craft, 2007; Talbot et al., 2012). Further, there is evidence that insulin resistance may be an early biomarker of AD risk in cognitively normal individuals, as greater insulin resistance is correlated with decreased cerebral glucose uptake and poor memory performance (Baker et al., 2011). Peripheral hyperinsulinemia leads to increased circulating Aβ and CSF Aβ42 in cognitively normal adults (Watson et al., 2003; Fishel et al., 2005) and increased hyperphosphorylated tau protein expression in mice (Freude et al., 2005). Further, in rodent models, hyperinsulinemic-euglycemic clamps raised serum insulin levels and modestly impacted ISF Aβ levels without detectable changes in insulin levels or signaling pathways in the brain (Stanley et al., 2016). Further, chronic hyperinsulinemia is thought to lead to lower insulin levels in the brain, which could explain why treating with insulin has been shown to cause memory improvements (Craft et al., 1996; Kern et al., 2001; Rerger et al., 2008). These studies suggest that both abnormal peripheral glucose metabolism and insulin resistance, akin to that seen in T2D, can accelerate AD pathogenesis through alterations in both neuronal and metabolic activity.
Sleep Architecture and Metabolic Regulation
Sleep is a conserved behavior that occurs in two distinct stages – non-rapid eye movement (NREM) and rapid eye movement (REM). NREM sleep is characterized by large, slow waves that propagate across the entire cortex, producing synchronized periods of neuronal activation and silence (Steriade et al., 1993). It is hypothesized that this slow wave activity (SWA) of NREM sleep is restorative in nature; a price paid for neuronal activity and synaptic strengthening that occurs during waking, and therefore representative of the homeostatic need for sleep (Vyazovskiy et al., 2009). SWA peaks early in the night and increases following periods of extended wakefulness and decreases across sleep periods as well as following daytime naps (Werth et al., 1996; Achermann and Borbley, 2003). This is correlated to the patterns of neuronal activity across the sleep/wake cycle where firing rates are high during waking and lower across sleep, therefore indicating a relationship between synaptic activity and sleep (Vyazovskiy et al., 2009). Further, periods of heightened neuronal activity and synaptic strengthening cause local increases in SWA, while stimulus deprivation or pharmacological depotentiation of synapses causes a reduction in SWA, further demonstrating SWA as a marker for homeostatic sleep need (Huber et al., 2006, 2007; Carroll et al., 2019).
Glucose and lactate also show consistent patterns across the sleep/wake cycle closely related to neuronal activity. Glucose concentrations in the brain increase during both NREM sleep, which is thought to aid in restoration and replenishment of glucose stores, and waking, indicative of increased metabolic functioning. While glucose cannot accurately measure sleep propensity because of these increases across both states, it is useful as a biomarker for cerebral metabolism (Dash et al., 2012b). Lactate concentrations in the brain increase with heightened neuronal activity during wakefulness and decrease across the night in correlation with both SWA and glutamate concentration, suggesting a shift toward astrocytic glycolysis and lactate utilization in periods of increased neuronal activity. Because of this tight correlation with neuronal activity, lactate is hypothesized to be an accurate biomarker for homeostatic sleep (Dash et al., 2012b; Naylor et al., 2012).
In the periphery, blood glucose is homeostatically regulated to maintain euglycemia and prevent extreme fluctuations. Glucose tolerance reflects both hepatic gluconeogenesis and glucose utilization and it is closely related to insulin release from pancreatic beta cells, insulin sensitivity of peripheral tissues, or the ability of insulin to inhibit glucose production and increase glucose utilization. With decreased insulin sensitivity or insulin resistance, the body is unable to effectively combat hyperglycemia, allowing for consistent states of high blood glucose (Knutsson et al., 2007). Blood glucose is also regulated by a circadian rhythm where plasma glucose levels are higher in the evening than morning and glucose tolerance and insulin sensitivity are lowest at night (Van Cauter et al., 1997). As the brain utilizes the majority of glucose in the body, peripheral and cerebral glucose levels are tightly correlated and shifts in peripheral glucose levels tend to model shifts in patterns of neuronal activity across the sleep/wake cycle. As such, glucose utilization is increased during wakefulness to accommodate for high levels of neuronal activity (Nofzinger et al., 2002). ISF glucose also increases across NREM sleep, likely reflecting both a decrease in glucose utilization as well as a decrease in insulin sensitivity (Spiegel et al., 2009; Dash et al., 2012a). Further, increasing concentrations of glucose can promote slow wave sleep through increased activation of the sleep-promoting neurons in the ventrolateral preoptic nucleus (VLPO) (Varin et al., 2015), as well as inhibiting hypothalamic orexin neurons that typically promote arousal (Yamanaka et al., 2003). As described earlier, glucose dysregulation and insulin resistance are key components connecting AD and T2D, so dysregulation of normal sleep/wake rhythms could be mediating this relationship between the two diseases.
AD and Sleep
Disrupted sleep is commonly observed in individuals with symptomatic AD (Peter-Derex et al., 2015) and can be detected in the preclinical stage (Ju et al., 2013; Hahn et al., 2014; Lucey et al., 2019). Moreover, sleep fragmentation, poor sleep quality, and excessive daytime sleepiness have been shown to increase risk of cognitive impairment and eventual dementia in clinical populations (Blackwell et al., 2013; Lim et al., 2013a; Spira et al., 2015, 2018). One of the most common causes of sleep disturbances in older adults is sleep disordered breathing (SDB), largely due to the high prevalence of obstructive sleep apnea (OSA) in the elderly (Andrade et al., 2018). In OSA, the airway is blocked during sleep, causing difficulty breathing, reduced blood oxygen saturation (e.g., hypoxemia), recurrent arousals, and sleep fragmentation (Yaffe et al., 2011; Osorio et al., 2015; Sharma et al., 2018; Ju et al., 2019). Cross-sectional and prospective studies in humans both demonstrate individuals with OSA are at increased risk for cognitive impairment, dementia, and changes in AD-related biomarkers (Yaffe et al., 2011; Ju et al., 2019). Conversely, treatment for OSA with continuous positive airway pressure (CPAP) reduces sleep fragmentation, hypoxia, and normalizes CSF Aβ/tau levels. In rodent models, acute sleep disruptions, as modeled in mice via chronic intermittent hypoxia, causes impaired spatial learning and memory (Ward et al., 2009), as well as increased Aβ and tau pathology (Gao et al., 2013; Shiota et al., 2013). This established a bidirectional relationship between sleep, cognition, and AD pathogenesis, which is further supported by the general characteristics of sleep and resulting fluctuations in Aβ and tau. As Aβ and tau are released in an activity dependent manner, the levels of Aβ and tau within the ISF of the brain parallel the diurnal rhythm of neuronal activity across the sleep/wake cycle. ISF Aβ and tau are closely correlated with wakefulness, with increases during sleep deprivation in both mice and humans (Kang et al., 2009; Lucey et al., 2017; Holth et al., 2019). Further, sleep has also been associated with the clearance of Aβ/tau from the ISF into the CSF and blood (Xie et al., 2013). Any situation that disrupts sleep, therefore, will alter Aβ and tau production and clearance. Decreased sleep spindle density, for example, has been connected to early neuronal dysfunction, particularly as it relates to tau release (Kam et al., 2019). Similarly, sleep deprivation increases Aβ and tau levels acutely and can lead to increased plaques, tangles, and cognitive decline in chronic conditions (Kang et al., 2009; Spira et al., 2013; Ju et al., 2017; Lucey et al., 2017; Zhu et al., 2018; Holth et al., 2019). Restoring sleep, however, has been shown to reduce ISF Aβ and tangle density (Kang et al., 2009; Lim et al., 2013b; Tabuchi et al., 2015). While the exact mechanism is unknown, increased neuronal activity could be playing a role in increasing AD pathology, as chronic sleep deprivation has been shown to increase intrinsic neuronal activity (Tabuchi et al., 2015). Because of the strong diurnal pattern of neuronal activity across the sleep/wake cycle, lactate, a biomarker for neuronal activity, is also used as a biomarker for sleep (Naylor et al., 2012). The diurnal oscillation of lactate, therefore, mirrors that of neuronal activity and Aβ, providing evidence that neuronal activity, mediated by the sleep/wake cycle, is responsible for driving AD pathology accumulation, particularly during sleep deprivation (Roh et al., 2012). While it is unknown what causes the initial disruption in sleep, degeneration of certain sleep-related nuclei, including the VLPO and intermediate nucleus, has been associated with sleep loss in AD (Lim et al., 2014). Moreover, recent work has shown individuals with MCI and AD have elevated levels of orexin in the CSF, which could be contributing to the overall increase in arousal seen in these individuals (Gabelle et al., 2017). This initial disruption could activate a cascade of accumulating sleep debt and pathology accumulation and explains the epidemiological evidence connecting sleep disturbances and AD.
Sleep Disruption and Metabolic Disturbances
Poor sleep quality and quantity is also a common feature of individuals with T2D (Spiegel et al., 1985; Yaggi et al., 2006), and can increase the risk of developing T2D by 2–3-fold (Kawakami et al., 2004). Hypoglycemia alone has been shown to activate orexin neurons in the hypothalamus, causing increased arousal in both mice and men (Tkacs et al., 2007). This effect is ablated in individuals with T2D, where hypoglycemia episodes failed to increase arousal, leading to poorer glycemic control and suggesting the presence of a dysfunctional autonomic arousal system (Banarer and Cryer, 2003). Currently, research suggests that most sleep disturbances related to T2D are largely the result of SDB. OSA is strongly associated with obesity and is likely caused by changes in the upper airway structure in humans (Young et al., 1993). There is a well-defined correlation between OSA and increased risk for T2D due to the presence of decrease glucose tolerance and insulin sensitivity, with a positive correlation between severity of OSA and poorer glucose regulation (Punjabi et al., 2004; Bostros et al., 2009; Aronsohn et al., 2010; Zhang et al., 2018). Similarly, in mice, chronic intermittent hypoxia results in insulin resistance and decreased glucose tolerance (Jiyori et al., 2007). Sleep deprivation, independent of any chronic sleep condition, is also known to disrupt glucose homeostasis. Specifically, sleep restriction causes reductions in glucose tolerance and insulin sensitivity, two components that contribute to increased risk for T2D and are present in early AD (Spiegel et al., 1999). While the mechanisms are not fully understood, there is evidence that sleep fragmentation can lead to altered glucose regulation through decreased cerebral glucose metabolism (Wu et al., 1991), alterations in endocrine function (Van Cauter et al., 2008; Spiegel et al., 2009), and increased inflammatory processes directly tied to T2D risk (Patel et al., 2009). Further, sleep deprivation also increases the expression of ANLS-associated genes, specifically within astrocytes, indicating SWA can regulate neuro-metabolic coupling and any disruption to this process can dysregulate cerebral metabolism independent of pathology (Petit et al., 2013). Selective disruption of slow wave sleep, which is prevalent in both aging and OSA, causes decreased insulin sensitivity and glucose tolerance, suggesting that slow wave sleep plays a specific role in maintaining glucose levels and the risk for T2D (Tasali et al., 2008). Similarly, improving sleep is thought to mitigate some of these metabolic impairments. Administration of orexins, a neuropeptide implicated in regulating both sleep/wake cycles and energy homeostasis, lowered fasting blood glucose levels in streptozotocin(stz)-induced diabetic mice (Tsuneki et al., 2002). In clinical trials, use of a CPAP, the main treatment option for OSA and other sleep breathing disorders, can improve glucose tolerance and lower hemoglobin A1C levels, a long-term biomarker of blood glucose levels (Babu et al., 2005; Salord et al., 2016).
Circadian Control of Metabolism and Impact on Sleep
It is worth noting that the circadian system controls the timing of sleep, as mutations in circadian clock genes which alter clock function are associated with sleep phenotypes in mice and humans (Hsu et al., 2015). Fragmentation of normal circadian timing of activity has been observed in preclinical AD and worsens in symptomatic AD in humans (Ju et al., 2013; Musiek et al., 2013; Peter-Derex et al., 2015). Loss of neurons in the suprachiasmatic nucleus (SCN), the master circadian clock of the body, has been described in AD and correlates with loss of circadian rhythms in activity (Swaab et al., 1985; Wang et al., 2015). Thus, degeneration of the circadian system, and the SCN in particular, may be a cause of sleep disruption in AD. Circadian disruption is associated with increased AD risk in humans, and can accelerate Aβ plaque formation in mice (Tranah et al., 2011; Kress et al., 2018). Moreover, the circadian system has strong influences on glucose metabolism, as poor circadian function is a major risk factor for the development of metabolic syndrome and T2D [Bass and Takahashi, 2010; as reviewed in Knutsson and Kempe (2014)]. In mice, a considerable literature links the circadian clock to glucose homeostasis, and mice with clock gene deletions develop obesity and hyperglycemia (Rudic et al., 2004; Turek et al., 2005; Bass and Takahashi, 2010; Marcheva et al., 2010). Thus, circadian rhythm dysfunction is another factor potentially linking sleep, metabolism, and AD.
Current Therapeutic Options
As previously discussed, treatment of OSA with CPAP to promote increased sleep efficiency can improve glucose tolerance and overall metabolic health (Babu et al., 2005; Salord et al., 2016) as well as AD biomarkers (Ju et al., 2019). Other treatments for T2D have also shown promising effect to alleviating some of the negative impacts on sleep. Metformin, the antihyperglycemic drug and first-line treatment for T2D, reduces hepatic gluconeogenesis, plasma insulin, and insulin resistance (Viollet et al., 2012). Further, individuals treated with metformin show improved sleep quality and quantity, suggesting that maintaining glucose homeostasis is essential for efficient sleep, although the exact mechanisms are not yet understood (Kaibaf et al., 2013). Treatment with insulin therapies has been linked to worse subjective sleep quality, potentially though hyperactivity in the sympathetic nervous system, although this treatment type has been widely understudied in terms of the effects on sleep (Song et al., 2013). Similarly, treating sleep disturbances has been shown to improve aberrant glucose metabolism. Orexins are critical for the regulation of glucose homeostasis and maintenance of the diurnal rhythm via influence over hepatic gluconeogenesis, making the timing modulation of the orexin system crucial. Administration of dual orexin receptor antagonist (DORA) drugs in accordance with the orexin rhythm can improve sleep and glucose tolerance, likely though suppression of hepatic gluconeogenesis rather than an effect on insulin production or sensitivity (Tsuneki et al., 2016). Additionally, results from Merck’s Phase 3 Clinical Trial DORA, suvorexant, improved sleep quality and quantity in individuals with AD. Sleep loss is known to promote the release of Aβ and tau, potentially suggesting that treating these sleep problems could slow disease progression, although no studies of this nature have been done (Herring et al., 2019). While more research on the subject is needed, sleep represents a modifiable aspect of these two diseases and offers a potential therapeutic benefit in delaying the onset of AD and mitigating the increased risk of AD among individuals with T2D by restoring glucose homeostasis.
Conclusion
This review outlines the interactions between sleep, altered glucose metabolism, and Alzheimer’s disease and suggests the emergence of a feedforward cascade where sleep dysfunction and metabolic impairment contribute to the progression of AD pathology (Figure 1). Similarly, we provide evidence of AD pathology leading to further disruptions to sleep and metabolic health. Therefore, exploring the timing and mechanisms that lead to the emergence of sleep disturbances in both conditions warrants further study as it represents a potential therapeutic treatment option for resolving peripheral glucose intolerance and insulin resistance in individuals with T2D and potentially slowing AD progression.
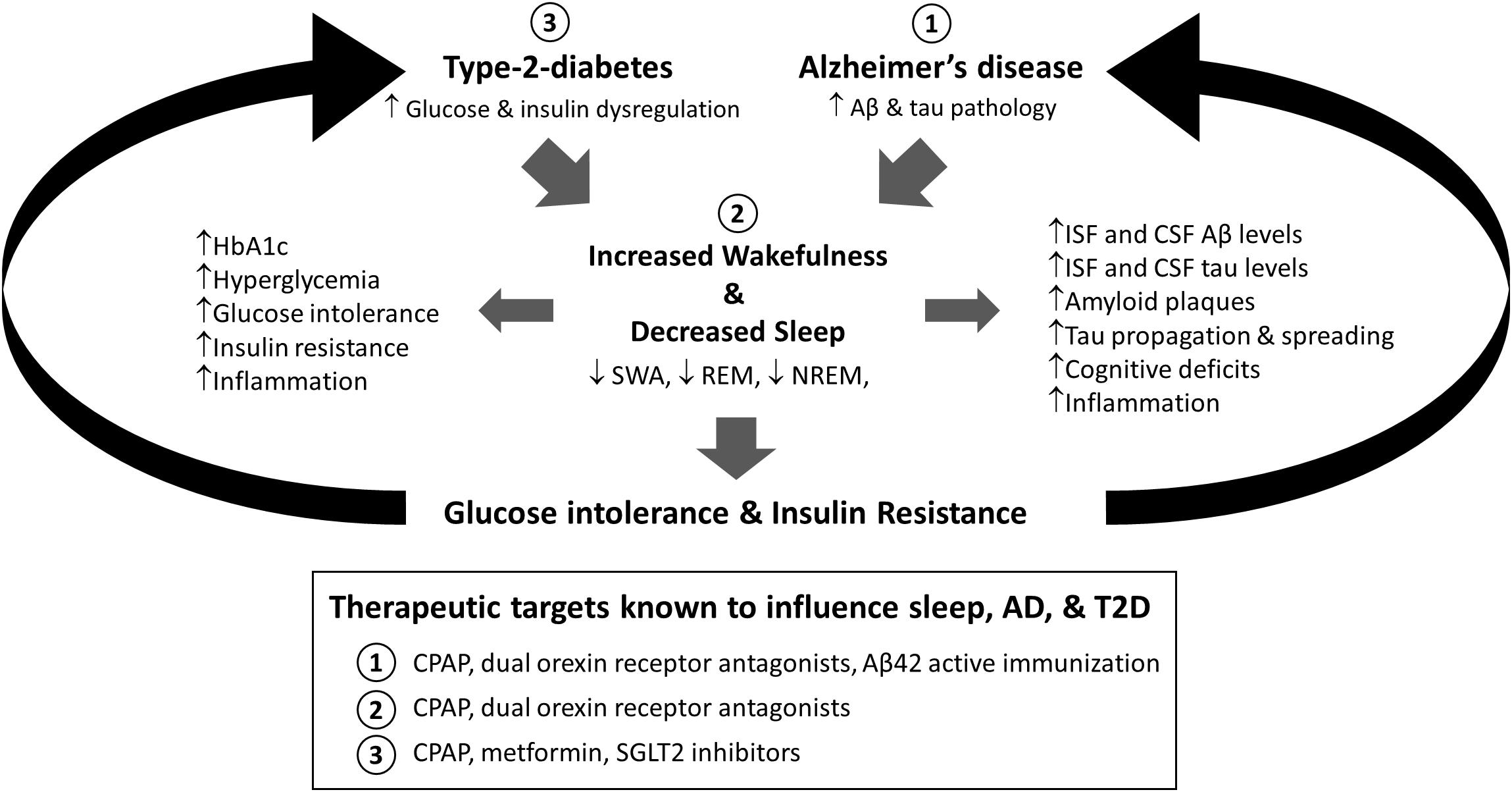
Figure 1. Proposed model for the relationship between sleep, type-2-diabetes (T2D), and Alzheimer’s disease (AD). Both alterations in glucose and insulin homeostasis associated with T2D and Aβ and tau aggregation found in AD are associated with increased sleep disruption. Moreover, sleep deprivation is associated with increased AD-related pathology, including Aβ and tau pathology, cognitive deficits, and inflammation, as well as diabetes related pathology, including metabolic dysregulation, glucose intolerance, and insulin resistance. Therefore, sleep is a modifiable risk factor in T2D and AD that could bidirectionally impact disease progression. Moreover, targeting sleep, AD, or T2D has been shown to modify these relationships (Box: 1, 2, 3). HbA1c = Hemoglobin A1c; SWA = slow wave activity; REM = rapid eye movement sleep; NREM = non-rapid eye movement sleep; Aβ = amyloid-beta; ISF = interstitial fluid; CSF = cerebrospinal fluid; CPAP = continuous positive airway pressure; SGLT2 = sodium glucose cotransporter 2.
Author Contributions
CC and SM conceived of the idea and equally contributed to the writing of the manuscript.
Funding
We would like to acknowledge the following grants: 1K01AG050719 (SM), R01AG061805 (SM), NCDRC Pilot Award (SM), and the Wake Forest University Alzheimer’s Disease Research Center (P30 AG049638), which is funded by the National Institute on Aging.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank their colleagues for the careful review and critique of this manuscript.
References
Achermann, P., and Borbley, A. A. (2003). Mathematical models of sleep regulation. Front. Biosci. 8:s683–s693.
Adams, J. N., Lockhart, S. N., Li, L., and Jagust, W. J. (2018). Relationships between tau and glucose metabolism reflect Alzheimer’s disease pathology in cognitively normal older adults. Cereb. Cortex 29, 1997–2009. doi: 10.1093/cercor/bhy078
Andrade, A. G., Babu, O. M., Varga, A. W., and Osorio, R. S. (2018). The Relationship between obstructive sleep apnea and Alzheimer’s Disease. J. Alzheimers Dis. 64(Suppl. 1), S255–S270. doi: 10.3233/JAD-179936
Aronsohn, R. S., Whitmore, H., van Cauter, E., and Tasali, E. (2010). Impact of untreated obstructive sleep apnea on glucose control in type 2 diabetes. Am. J. Respir. Crit. Care. Med. 181, 507–513. doi: 10.1164/rccm.200909-1423OC
Aung, P. P., Strachan, M. W., Frier, B. M., Butcher, I., Deary, I. J., and Price, J. F. (2012). Severe hypoglycaemia and late-life cognitive ability in older people with type 2 diabetes: the edinburgh type 2 diabetes study. Diabetic. Med. 29, 328–336. doi: 10.1111/j.1464-5491.2011.03505.x
Babu, A. R., Herdegen, J., Fogelfeld, L., Shott, S., and Mazzone, T. (2005). Type 2 diabetes, glycemic control, and continuous positive airway pressure in obstructive sleep apnea. Arch. Intern. Med. 165, 447–452. doi: 10.1001/archinte.165.4.447
Baker, L. D., Cross, D. J., Minoshima, S., Belongia, D., Watson, G. S., and Craft, S. (2011). Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early Type 2 diabetes. Arch. Neurol. 68, 51–57. doi: 10.1001/archneurol.2010.225
Banarer, S., and Cryer, P. E. (2003). Sleep-related hypoglycemia-assocaited autonomic failure in type 1 diabetes. Diabetes 52, 1195–1203. doi: 10.2337/diabetes.52.5.1195
Bass, J., and Takahashi, J. S. (2010). Circadian integration of metabolism and energetics. Science 330, 1349–1354. doi: 10.1126/science.1195027
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Bero, A. W., Yan, P., Roh, J. E., Cirrito, J. R., Stewart, F. R., Raichle, M. E., et al. (2011). Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat. Neurosci. 14, 750–756. doi: 10.1038/nn.2801
Blackwell, T., Yaffe, K., Redline, S., Ensrud, K. E., Stefanick, M. L., Laffan, A., et al. (2013). Association of sleep characteristics and cognition in older community-dwelling men: the MrOS sleep study. Sleep 34, 1347–1356. doi: 10.5665/SLEEP.1276
Bostros, N., Concato, J., Mohsenin, V., Selim, B., Doctor, K., and Yaggi, H. K. (2009). Obstructive sleep apnea as a risk factor for Type 2 diabetes. Am. J. Med. 122, 1122–1127. doi: 10.1016/j.amjmed.2009.04.026
Busche, M. A., Eichhoff, G., Adelsberger, H., Abramowski, D., Widerhold, K. H., Haass, C., et al. (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. doi: 10.1126/science.1162844
Carroll, C. M., Hsiang, H., Snyder, S., Forsberg, J., and Dash, M. B. (2019). Cortical zeta-inhibitory peptide injection reduces local sleep need. Sleep 42, zsz028. doi: 10.10933/sleep/zsz028
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell. Biol. 11, 909–913. doi: 10.1038/ncb1901
Craft, S. (2007). Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr. Alzheimer Res. 4, 147–152. doi: 10.2174/156720507780362137
Craft, S., Newcomer, J., Kanne, S., Dagogo-Jack, S., Cryer, P., Sheline, Y., et al. (1996). Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 17, 123–130. doi: 10.1016/0197-4580(95)02002-0
Crane, P. K., Walker, R., Hubbard, R. A., Li, G., Nathan, D. M., Zheng, H., et al. (2013). Glucose levels and risk of dementia. N. Engl. J. Med. 369, 540–548. doi: 10.1056/NEJMoa1215740
Dash, M. B., Bellesi, M., Tononi, G., and Cirelli, C. (2012a). Sleep/wake dependent changes in cortical glucose concentrations. J. Neurochem. 124, 79–89. doi: 10.1111/jnc.12063
Dash, M. B., Tononi, G., and Cirelli, C. (2012b). Extracellular levels of lactate, but not oxygen, reflect sleep homeostasis in the rat cerebral cortex. Sleep 35, 909–919. doi: 10.5665/sleep.1950
Edison, P., Archer, H. A., Hinz, R., Hammers, A., Pavese, N., Tai, Y. F., et al. (2007). Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurol 68, 501–508. doi: 10.1212/01.wnl.0000244749.20056.d4
Fishel, M. A., Watson, G. S., Montine, T. J., Wang, Q., Green, P. S., Kulstad, J., et al. (2005). Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch. Neurol. 62, 1539–1544. doi: 10.1001/archneur.62.10.noc50112
Franzmeier, N., Rubinski, A., Neitzel, J., Kim, Y., Damm, A., Na, D. L., et al. (2019). Functional connectivity associated with tau levels in ageing, Alzheimer’s, and small vessel disease. Brain 142, 1093–1097. doi: 10.1093/brain/awz026
Freude, S., Plum, L., Scnitker, J., Leeser, U., Udelhoven, M., Krone, W., et al. (2005). Peripheral hyperinsulinemia promotes tau phosphorylation in vivo. Diabetes 54, 3343–3348. doi: 10.2337/diabetes.54.12.3343
Gabelle, A., Jaussent, I., Hirtz, C., Vialaret, J., Navucet, S., Grasselli, C., et al. (2017). Cerebrospinal fluid levels of orexin-A and histamine, and sleep profil within the Alzheimer process. Neurobiol. Aging 53, 59–66. doi: 10.1016/j.neurobiolaging.2017.01.011
Gao, L., Tian, S., Gao, H., and Xu, Y. (2013). Hypoxia increases Aβ-induced tau phosphorylation by calpain and promotes behavioral consequences in AD transgenic mice. J. Mol. Neurosci. 51, 138–147. doi: 10.1007/s12031-013-9966-y
Gomez, G., Beason-Held, L. L., Bligel, M., An, Y., Wong, D. F., Studenski, S., et al. (2018). Metabolic syndroms and amyloid accumulation in the aging brain. J. Alzheimers Dis. 65, 629–639. doi: 10.3233/JAD-180297
Hahn, E. A., Wang, H. X., Andel, R., and Fratiglioni, L. (2014). A change in sleep pattern may predict Alzheimer disease. Am. J. Geriatr. Psychiatry 11, 1262–1271. doi: 10.1016/j.jagp.2013.04.015
Hanseeuw, B. J., Betensky, R. A., Schultz, A. P., Papp, K. V., Mormino, E. C., Sepulcre, J., et al. (2017). Fluorodeoxyglucose metabolism associated with tau0amyloid interaction predicts memory decline. Ann. Neurol. 81, 583–596. doi: 10.1002/ana.24910
Harris, R. A., Trindale, L., Lone, A., Singh, O., Macauley, S. L., Stanley, M., et al. (2016). Aerobic glycolysis in the frontal cortex correlated with memry performance in wild-type mice but not the APP/PS1 mouse model of cerebral amyloidosis. J. Neurosci. 36, 1871–1878.
Herring, W. J., Ceesay, P., Snyder, E., Bliwise, D., Budd, K., Hutzelmann, J., et al. (2019). Clinical polysomnography trial of suvorexant for treating insomnia in Alzheimer’s Disease (P3.6-022). Neurology 92(15 Suppl.), 3.6–022.
Holth, J. K., Fritschi, S. K., Wang, C., Pedersen, N. P., Cirrito, J. R., Mahan, T. E., et al. (2019). The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884. doi: 10.1126/science.aav2546
Holtzman, D. M., Morris, J. C., and Goate, A. M. (2011). Alzheimer’s Disease: the challenge of the second century. Sci. Transl. Med. 3:77sr1.
Hsu, P. K., Ptacek, L. J., and Fu, Y. H. (2015). Genetics of human sleep behavioral phenotypes. Methods. Enzymol. 552, 309–324. doi: 10.1016/bs.mie.2014.10.046
Huber, R., Esser, S. K., Ferrarelli, F., Massimini, M., Peterson, M. J., and Tononi, G. (2007). TMS-induced cortical potentiation during wakefulness locally increases slow wave activity during sleep. PLoS One 3:e276. doi: 10.1371/journal.pone.0000276
Huber, R., Ghilardi, M. F., Massimini, M., Ferrarelli, F., Riedner, B. A., Peterson, M. J., et al. (2006). Arm immobilization causes cortical plastic changes and locally decreases sleep slow wave activity. Nat. Neurosci. 9, 1169–1173. doi: 10.1038/nn1758
Huijbers, W., Schultz, A. P., Papp, K. V., LaPoint, M. P., Hanseeuw, B., Chhatwal, J. P., et al. (2019). Tau accumulation in clinically normal older adults is associated with hippocampal hyperactivity. J. Neurosci. 39, 548–556. doi: 10.1523/JNEUROSCI.1397-18.2018
Jagust, W. J., and Mormino, E. C. (2011). Lifespan brain activity, β-amyloid, and Alzheimer’s disease. Trends. Cogn. Sci. 15, 520–526. doi: 10.1016/j.tics.2011.09.004
Jiyori, N., Alonso, L. C., Li, J., Sanders, M. H., Garcia-Ocana, A., O’Doherty, R. M., et al. (2007). Intermittent hypoxia causes insulin resistance in lean mice independent of autonomic activity. Am. J. Respir. Crit. Care Med. 175, 851–857. doi: 10.1164/rccm.200610-1527OC
Ju, Y. E. S., McLeland, J. S., Toedebusch, C. D., Xiong, C., Fagan, A. M., Duntley, S. P., et al. (2013). Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 70, 587–593. doi: 10.1001/jamaneurol.2013.2334
Ju, Y. E. S., Ooms, S. J., Sutphen, C., Macauley, S. L., Zangrilli, M. A., Jerome, G., et al. (2017). Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 140, 2104–2111. doi: 10.1093/brain/awx148
Ju, Y. E. S., Zangrilli, M. A., Finn, M. B., Fagan, A. M., and Holtzman, D. M. (2019). Obstructive sleep apena treatment, slow wave activity, and amyloid-β. Ann. Neurol. 85, 291–295. doi: 10.1002/ana.25408
Kaibaf, F., Fendre, S., Basille-Fantinato, A., Diouf, M., Rose, D., Jounieaux, V., et al. (2013). The relationship between metformin therapy and sleep quantity and quality in patients with type 2 diabetes referred for potential sleep disorders. Diabetic. Med. 31, 577–580. doi: 10.1111/dme.12362
Kam, K., Parekh, A., Sharma, R. A., Andrade, A., Lewin, M., Castillo, B., et al. (2019). Sleep oscillation-specific associations with Alzheimer’s disease CSF biomarkers: novel roles for sleep spindles and tau. Mol. Neurodegener. 14:10. doi: 10.1186/s13024-019-0309-5
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kawakami, N., Takatsuka, N., and Shimizu, H. (2004). Sleep disturbance and onset of Type 2 diabetes. Diabetes. Care 27, 282–283. doi: 10.2337/diacare.27.1.282
Kern, W., Peters, A., Fruehwald-Schultes, B., Deininger, E., Born, J., and Fehm, H. L. (2001). Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 74, 270–280. doi: 10.1159/000054694
Klein, R. C., Acheson, S. K., Mace, B. E., Sullivan, P. M., and Moore, S. D. (2014). Altered neurotransmission in the lateral amygdala in aged human apoE4 targeted replacement mice. Neurobiol. Aging 35, 2046–2052. doi: 10.1016/j.neurobiolaging.2014.02.019
Knutsson, A., and Kempe, A. (2014). Shift work and diabetes–a systematic review. Chronobiol Int. 31, 1146–1151. doi: 10.3109/07420528.2014.957308
Knutsson, K. L., Spiegel, K., Peney, P., and Van Cauter, E. (2007). The metabolic consequences of sleep deprivation. Sleep. Med. Rev. 11, 163–178. doi: 10.1016/j.smrv.2007.01.002
Kress, G. J., Liao, F., Dimitry, J., Cedeno, M. R., FitzGerald, G. A., Holtzman, D. M., et al. (2018). Regulation of amyloid-beta dynamics and pathology by the circadian clock. J. Exp. Med. 215, 1059–1068. doi: 10.1084/jem.20172347
Lehmann, M., Madison, C. M., Ghosh, P. M., Seeley, W. W., Mormino, E., Greicius, M. D., et al. (2013). Intrinsic connectivity networks in healthy subjects explain clinical variability in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 110, 11606–11611. doi: 10.1073/pnas.1221536110
Lim, A. S., Kowgier, M., Yu, L., Buchman, A. S., and Bennett, D. A. (2013a). Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 36, 1027–1032. doi: 10.5665/sleep.2802
Lim, A. S., Yu, L., Kowgier, M., Schneider, J. A., Buchman, A. S., and Bennett, D. A. (2013b). Modifications of the relationship of the apolipoprotein E ε4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 70, 1544–1551. doi: 10.1001/jamaneurol.2013.4215
Lim, A. S. P., Ellison, B. A., Wang, J. L., Yu, L., Schneider, J. A., Buchman, A. S., et al. (2014). Sleep is related to neuron nembers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain 137, 2847–2861. doi: 10.1093/brain/awu22
Lin, C. H., and Sheu, W. H. (2013). Hypoglycaemic episodes and risk of dementia in diabetes mellitus: 7-year follow-up study. J. Intern. Med. 273, 102–110. doi: 10.1111/joim.12000
Lucey, B. P., Fagan, A. M., Holtzman, D. M., Morris, J. C., and Bateman, R. J. (2017). Diurnal oscillation of CSF Aβ and other biomarkers. Mol. Neurodeneger. 12:36. doi: 10.1186/s13024-017-0161-4
Lucey, B. P., McCullough, A., Landsness, E. C., Toedebusch, C. D., McLeland, J. S., Zaza, A. M., et al. (2019). Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Tranls. Med. 11:eaau6550. doi: 10.1126/scitranslmed.aau6550
Macauley, S. L., Stanley, M., Caesar, E. E., Yamada, S. A., Raichle, M. E., Perez, R., et al. (2015). Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Invest. 125, 2463–2467. doi: 10.1172/JCI79742
Mackenzie, I. R., and Miller, L. A. (1994). Senile plaques in temporal lobe epilepsy. Acta Neurpathol. 87, 504–510.
Marcheva, B., Ramsey, K. M., Buhr, E. D., Kobayashi, Y., Su, H., Ko, C. H., et al. (2010). Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466, 627–631. doi: 10.1038/nature09253
Morris, J. K., Vidoni, E. D., Honea, R. A., Burns, J. M., and Alzheimer’s Disease Neuroimaging Initiative. (2014). Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol. Aging 35, 585–589. doi: 10.1016/j.neurobiolaging.2013.09.033
Mosconi, L., Pupi, A., and De Leon, M. J. (2008). Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1147, 180–195. doi: 10.1196/annals.1427.007
Musiek, E. S., and Holtzman, D. M. (2015). Three dimensions of the amyloid hypothesis: time, space, and ‘wingmen’. Nat. Neurosci. 18, 800–806. doi: 10.1038/nn.4018
Musiek, E. S., Lim, M. M., Yang, G., Bauer, A. Q., Qi, L., Lee, Y., et al. (2013). Circadian clock proteins regulation neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 123, 5389–5400. doi: 10.1172/JCI70317
Naylor, E., Ailon, D. V., Barrett, B. S., Wilson, G. S., Johnson, D. A., Harmon, H. P., et al. (2012). Lactate as a biomarker for sleep. Sleep 35, 1209–1222. doi: 10.5665/sleep.2072
Nofzinger, E. A., Buysse, D. J., Miewald, J. M., Meltzer, C. C., Price, J. C., Sembrat, R. C., et al. (2002). Human regional cerebral glucose metabolism during non-rapid eye movement sleep in relation to waking. Brain 125, 1105–1115. doi: 10.1093/brain/awf103
Nuriel, T., Angulo, S. L., Khan, U., Ashok, A., Chen, Q., Figueroa, H. Y., et al. (2017). Neuronal hyperacitivty due to lost of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun. 8:1464. doi: 10.1038/s41467-017-01444-0
Oh, H., Madison, C., Baker, S., Rabinovici, G., and Jagust, W. (2016). Dynamic relationships between age, amyloid-β deposition, and glucose metabolism link to the regional vulnerability of Alzheimer’s disease. Brain 139, 2275–2289. doi: 10.1093/brain/aww108
Osorio, R. S., Gumb, T., Pirraglia, E., Varga, A. W., Lu, S., Lim, J., et al. (2015). Sleep-disordered breathing advances cognitive decline in the elderly. Neurology 84, 1964–1971. doi: 10.1212/WNL.0000000000001566
Ott, A., Stolk, R. P., van Harskamp, F., Pols, H. A., Hofman, A., and Breteler, M. M. (1999). Diabetes mellitus and the risk of dementia: the rotterdam study. Neurol 53, 1937–1942. doi: 10.1212/WNL.53.9.1937
Patel, S. R., Zhu, X., Storfer-Isser, A., Mehra, R., Jenny, N. S., Tracy, R., et al. (2009). Sleep duration and biomarkers of inflammation. Sleep 32, 200–204. doi: 10.1093/sleep/32.2.200
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629. doi: 10.1073/pnas.91.22.10625
Peter-Derex, L., Yammine, P., Bastuji, H., and Croisile, B. (2015). Sleep and Alzheimer’s disease. Sleep. Med. Rev. 19, 29–38. doi: 10.1016/j.smrv.2014.03.007
Petit, J., Gyger, J., Burlet-Godinot, S., Fiumelli, H., Martin, J., and Magistretti, P. J. (2013). Genes involved in the astrocyte-neuron lactate shuttle (ANLS) are specifically regulated in cortical astrocytes following sleep deprivation in mice. Sleep 36, 1445–1458. doi: 10.5665/sleep.3034
Pooler, A. M., Phillips, E. C., Lau, D. H. W., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO 14, 389–394. doi: 10.1038/embor.2013.15
Punjabi, N. M., Shahar, E., Redline, S., Gottlieb, D. J., Givelber, R., and Resnick, H. E. (2004). Sleep-disordered breathing, glucose intolerance, and insulin resistance: the sleep heart health study. Amer. J. Epidemiol. 160, 521–530. doi: 10.1093/aje/kwh261
Rerger, M. A., Watson, G. S., Green, P. S., Wilkinson, C. W., and Baker, D. (2008). Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 70, 440–448. doi: 10.1212/01.WNL.0000265401.62434.36
Roh, J. H., Huang, Y., Bero, A. W., Kasten, T., Stewart, F. R., Bateman, R. J., et al. (2012). Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 4, 150ra122. doi: 10.1126/scitranslmed.3004291
Rudic, R. D., McNamara, P., Curtis, A. M., Boston, R. C., Panda, S., Hogenesch, J. B., et al. (2004). BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2:e377. doi: 10.1371/journal.pbio.0020377
Salord, N., Fortuna, A. M., Monasterio, C., Gasa, M., Pérez, A., Bonsignore, M. R., et al. (2016). A randomized controlled trial of continuous positive airway pressure on glucose tolerance in obese patients with Obstructive Sleep Apnea. Sleep 39, 35–41. doi: 10.5665/sleep.5312
Schultz, A. P., Chhatwal, J. P., Hedden, T., Mormino, E. C., Hanseeuw, B. J., Sepulcre, J., et al. (2017). Phases of hyperconnectivity and hypoconnectivity in the default mode and salience networks track with amyloid and tau in clinically normal individuals. J. Neurosci. 37, 4323–4331. doi: 10.1523/JNEUROSCI.3263-16.2017
Sharma, R. A., Varga, A. W., Babu, O. M., Pirraglia, E., Kam, K., Parekh, A., et al. (2018). Obstructive sleep apnea severity affects amyloid burden in cognitively normal elderly. a longitudinal study. Am. J. Respir. Crit. Care Med. 197, 933–943. doi: 10.1164/rccm.201704-0704OC
Shiota, S., Takekawa, H., Matsumoto, S. E., Takeda, K., Nurwidya, F., Toshioka, Y., et al. (2013). Chronic intermittent hypoxia/reoxygenation facilitate amyloid-β generation in mice. J. Alzheimers Dis. 37, 325–333. doi: 10.3233/JAD-130419
Song, Y., Ye, X., Ye, L., Li, B., Wang, L., and Hua, Y. (2013). Disturbed subjective sleep in chinese females with type 2 diabetes on insulin therapy. PLoS. One 8:e54951. doi: 10.1371/journal.pone.0054951
Spiegel, K., Knutsson, K., Leprout, R., Tasali, E., and Van Cauter, E. (1985). Sleep loss: a novel risk factor for insulin resistance and type 2 diabetes. J. Appl. Physiol. 99, 2008–2019. doi: 10.1152/japplphysiol.00660.2005
Spiegel, K., Leproult, R., and Van Cauter, E. (1999). Impact of sleep debt on metabolic and endocrine function. Lancet 354, 1435–1439. doi: 10.1016/S0140-6736(99)01376-8
Spiegel, K., Tasali, E., Leproult, R., and Van Cauter, E. (2009). Effects of poor and short sleep on glucose metabolism and obesity risk. Nat. Rev. Endocrinol. 5, 253–261. doi: 10.1038/nrendo.2009.23
Spira, A. P., An, Y., Wu, M. N., Owusu, J. T., Simonsick, E. M., Bligel, M., et al. (2018). Excessive daytime sleepiness and napping in cognitively normal adults: associations with subsequence amyloid deposition measured by PiB PET. Sleep 41, zsy152. doi: 10.1093/sleep/zsy152
Spira, A. P., Chen-Edinboro, L. P., Wu, M. N., and Yaffe, K. (2015). Impact of sleep on the risk of cognitive decline and dementia. Curr. Opin. Psychiatry 27, 478–483. doi: 10.1097/YCO.0000000000000106
Spira, A. P., Gamaldo, A. A., An, Y., Wu, M. N., Simonsick, E. M., Bligel, M., et al. (2013). Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA. Neurol. 70, 1537–1543. doi: 10.1001/jamaneurol.2013.4258
Stanley, M., Macauley, S. L., and Holtzman, D. M. (2016). Changes in insulin and insulin signaling in Alzheimer’s disease: cause or consequence? J. Exp. Med. 213, 1375–1385. doi: 10.1084/jem.20160493
Steriade, M., McCormick, D. A., and Sejnowski, T. J. (1993). Thalamocortical oscillations in the sleeping and aroused brain. Science 262, 679–685. doi: 10.1126/science.8235588
Swaab, D. F., Fliers, E., and Partiman, T. S. (1985). The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 342, 37–44.
Tabuchi, M., Lone, S. R., Liu, S., Liu, Q., Zhang, J., Spira, A. P., et al. (2015). Sleep interacts with aβ to modulate intrinsic neuronal excitability. Curr. Biol. 25, 702–712. doi: 10.1016/j.cub.2015.01.016
Tai, X. Y., Koepp, M., Duncan, J. S., Fox, N., Thompson, P., Baxendale, S., et al. (2016). Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 139, 2441–2455. doi: 10.1093/brain/aww187
Talbot, K., Wang, H. W., Kazi, K., Han, L. Y., Bakashi, K. P., Stucky, A., et al. (2012). Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest 122, 1316–1338. doi: 10.1172/JCI59903
Tasali, E., Leproult, R., Ehrmann, D. A., and van Cauter, E. (2008). Slow-wave sleep and the risk of type 2 diabetes in humans. PNAS 105, 1044–1049. doi: 10.1073/pnas.0706446105
Tkacs, N. V., Pan, Y., Sawhney, G., Mann, G. L., and Morrison, A. R. (2007). Hypoglycemia activates arousal-related neurons and increases wake time in adult rats. Physiol. Behav. 91, 240–249. doi: 10.1016/j.physbeh.2007.03.003
Tranah, G. J., Blackwell, T., Stone, K. L., Ancoli-Israel, S., Paudel, M. L., Ensrud, K. E., et al. (2011). Circadian activity rhythms and risk of incident dementia and mild cognitive impairment in older women. Ann. Neurol. 70, 722–732. doi: 10.1002/ana.22468
Tsuneki, H., Kon, K., Ito, H., Yamazaki, M., Takahara, S., Toyooka, N., et al. (2016). Timed inhibition of orexin system by suvorexant improved sleep and glucose metabolism in type 2 diabetic db/db mice. Endocrinl 157, 4146–4157. doi: 10.1210/en.2016-1404
Tsuneki, H., Sugihara, Y., Honda, R., Wada, T., Sasaoka, T., and Kimura, I. (2002). Reduction of blood glucose level by orexins in fasting normal and streptozotocin-diabetic mice. Eur. J. Pharmacol. 448, 245–252. doi: 10.1016/S0014-2999(02)01936-2
Turek, F. W., Joshu, C., Kohsaka, A., Lin, E., Ivanova, G., McDearmon, E., et al. (2005). Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045. doi: 10.1126/science.1108750
Vaishanavi, S. N., Vlassenko, A. G., Rundle, M. M., Snyder, A. Z., Mintun, M. A., and Raichle, M. E. (2010). Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. U.S.A. 107, 17757–17762.
Van Cauter, E., Polonsky, K. S., and Scheen, A. J. (1997). Roles of circadian rhythmicity and sleep in human glucose regulation. Endocr. Rev. 18, 716–738.
Van Cauter, E., Spiegel, K., Tasali, E., and Leproult, R. (2008). Metabolic consequences of sleep and sleep loss. Sleep Med. 9, S23–S28. doi: 10.1016/S1389-9457(08)70013-3
Varin, C., Rancillac, A., Geoffroy, H., Arthaud, S., Fort, P., and Gallopin, T. (2015). Glucose induces slow-wave sleep by exiting the sleep-promoting neurons in the ventrolateral preoptic nucleus: a new link between sleep and metabolism. J. Neurosci. 35, 9900–9911. doi: 10.1523/JNEUROSCI.0609-15.2015
Viollet, B., Guigas, B., Sanz Garcia, N., Leclerc, J., Foretz, M., and Andreelli, F. (2012). Cellular and molecular mechanisms of metformin: an overview. Clin. Sci. 122, 253–270. doi: 10.1042/CS20110386
Vlassenko, A. G., Gordon, B. A., Goyal, M. S., Su, Y., Blazey, T. M., Durbin, T. J., et al. (2018). Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiol. Aging 67, 95–98. doi: 10.1016/j.neurobiolaging.2018.03.014
Vlassenko, A. G., and Raichle, M. E. (2015). Brain aerobic glycolysis functions and Alzheimer’s disease. Clin. Transl. Imaging. 3, 27–37. doi: 10.1007/s40336-014-0094-7
Vlassenko, A. G., Vaishnavi, S. N., Couture, L., Sacco, D., Shannon, B. J., Mach, R. H., et al. (2010). Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc. Natl. Acad. Sci. U.S.A. 107, 17763–17767. doi: 10.1073/pnas
Vyazovskiy, V. V., Olcese, U., Lazimy, Y. M., Faraguna, U., Esser, S. K., Williams, J. C., et al. (2009). Cortical firing and sleep homeostasis. Neuron 63, 865–878. doi: 10.1016/j.neuron.2009.08.024
Wang, J. L., Lim, A. S., Chiang, W. Y., Hsieh, W. H., Lo, M. T., Schneider, J. A., et al. (2015). Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann. Neurol. 2015, 317–322. doi: 10.1002/ana.24432
Ward, C. P., McCoy, J. G., McKenna, J. T., Connolly, N. P., McCarley, R. W., and Strecker, R. E. (2009). Spatial learning and memory deficits following exposure to 24 h of sleep fragmentation or intermittent hypoxia in a rat model of obstructive sleep apnea. Brain Res. 1294, 128–137. doi: 10.1015/j.brainres.2009.7.064
Watson, G. S., Peskind, E. R., Asthana, S., Purganan, K., Wait, C., Chapman, D., et al. (2003). Insulin increases CSF Aβ42 levels in normal older adults. Neurol 60, 1899–1903. doi: 10.1212/01.WNL.0000065916.25128.25
Werth, E., Dijk, D. J., Achermann, P., and Borbley, A. A. (1996). Dynamics of the sleep EEG after an early evening nap: experimental data and simulations. Am. J. Physiol Reg. I. 271, R501–R510. doi: 10.1152/ajpregu.1996.271.3.R501
Whitmer, R. A., Karter, A. J., Yaffe, K., Quesenberry, C. P., and Selby, J. V. (2009). Hypoglycemic episodes and risk of dementia in older patients with Type 2 diabetes mellitus. JAMA 301, 1565–1572. doi: 10.1001/jama.2009.460
Wu, J. C., Gillan, J. C., Buchsbaum, M. S., Hershey, T., Hazlett, E., Sicotte, N., et al. (1991). The effect of sleep deprivation in cerebral glucose metabolic rate in normal humans assessed with positron emission tomography. Sleep 14, 155–162. doi: 10.1093/sleep/14.2.155
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., and Rilett, K. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagaraian, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 343, 373–377. doi: 10.1126/science.1241224
Yaffe, K., Falvey, C. M., Hamilton, N., Harris, T. B., Simonsick, E. M., Strotmeyer, E. S., et al. (2013). Association between hypoglycemia and dementia in a biracial cohort of older adults with diabetes mellitus. JAMA. Intern. Med. 173, 1300–1306. doi: 10.1001/jamainternmed.2013.6176
Yaffe, K., Laffan, A. M., Harrison, S. L., Redline, S., Spira, A. P., Ensrud, K. E., et al. (2011). Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306, 613–619. doi: 10.1001/jama.2011.1115
Yaggi, H. K., Araujo, A. B., and McKinlay, J. B. (2006). Sleep duration as a risk factor for the development of type 2 diabetes. Diabetes. Care. 29, 657–661. doi: 10.2337/diacare.29.03.06.dc05-0879
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem/20131685
Yamanaka, A., Beuckmann, C. T., Willie, J. T., Hara, J., Tsuijno, N., Mieda, M., et al. (2003). Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38, 701–713.
Yao, J., Rettberg, J. R., Klosinski, L. P., Cadenas, E., and Brinton, R. D. (2011). Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol. Aspects. Med. 32, 247–257. doi: 10.1016/j.mam.2011.10.005
Young, T., Palta, M., Dempsey, J., Skatrud, J., Weber, S., and Badr, S. (1993). The occurrence of sleep-disordered breathing among middle-aged adults. N. Engl. J. Med. 328, 1230–1235. doi: 10.1056/NEJM199304293281704
Zhang, Y., Xing, H., Gang, X., Guo, W., Li, Z., and Wang, G. (2018). Impaired glucose metabolism of parients with obstructive sleep apnea and Type 2 diabetes. J. Diabetes Res. 23, 2714392. doi: 10.1155/2018/6714392
Keywords: Alzheimer’s disease, type-2-diabetes, sleep, glucose, insulin, metabolism
Citation: Carroll CM and Macauley SL (2019) The Interaction Between Sleep and Metabolism in Alzheimer’s Disease: Cause or Consequence of Disease? Front. Aging Neurosci. 11:258. doi: 10.3389/fnagi.2019.00258
Received: 07 June 2019; Accepted: 30 August 2019;
Published: 20 September 2019.
Edited by:
Ignacio Torres-Aleman, Spanish National Research Council (CSIC), SpainReviewed by:
Rebecca L. Cunningham, University of North Texas Health Science Center, United StatesMerina Varghese, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2019 Carroll and Macauley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shannon L. Macauley, c21hY2F1bGVAd2FrZWhlYWx0aC5lZHU=