 Natalia Salvadores
Natalia Salvadores Cristian Gerónimo-Olvera1,2
Cristian Gerónimo-Olvera1,2- 1Center for Integrative Biology, Faculty of Sciences, Universidad Mayor, Santiago, Chile
- 2Fondap Geroscience Center for Brain Health and Metabolism, Santiago, Chile
- 3Buck Institute for Research on Aging, Novato, CA, United States
Alzheimer’s disease (AD) represents the most common age-related neurodegenerative disorder, affecting around 35 million people worldwide. Despite enormous efforts dedicated to AD research over decades, there is still no cure for the disease. Misfolding and accumulation of Aβ and tau proteins in the brain constitute a defining signature of AD neuropathology, and mounting evidence has documented a link between aggregation of these proteins and neuronal dysfunction. In this context, progressive axonal degeneration has been associated with early stages of AD and linked to Aβ and tau accumulation. As the axonal degeneration mechanism has been starting to be unveiled, it constitutes a promising target for neuroprotection in AD. A comprehensive understanding of the mechanism of axonal destruction in neurodegenerative conditions is therefore critical for the development of new therapies aimed to prevent axonal loss before irreversible neuronal death occurs in AD. Here, we review current evidence of the involvement of Aβ and tau pathologies in the activation of signaling cascades that can promote axonal demise.
Introduction
Alzheimer’s disease (AD) is an adult-onset neurodegenerative disorder and the leading cause of dementia in aged people. While most AD cases are sporadic, less than 5% of the cases are caused by mutations in the amyloid precursor protein (APP) gene or presenilin 1 or 2 genes, leading to excessive production and accumulation of amyloid-β peptide (Aβ; Citron et al., 1992; Hendriks et al., 1992; Mullan et al., 1992; Suzuki et al., 1994; Harvey et al., 2003; Goate, 2006). Many risk factors have been associated with the development of sporadic AD, including the apolipoprotein E ɛ4 allele (Agosta et al., 2009), female gender (Koran et al., 2017), cardiovascular disease risk factors (Samieri et al., 2018), traumatic brain injury (TBI; LoBue et al., 2017) and aging, which is the most important one (Oh et al., 2014). Currently, there is no cure for AD and available treatments can only modestly and briefly alleviate symptoms (Alzheimer’s Association, 2020). The progression of AD, from alterations that include only changes in biomarkers but without the involvement of cognitive decline, to changes that indeed translate into cognitive impairment, follows a continuum that comprises three phases: preclinical AD, mild cognitive impairment (MCI), and dementia due to AD. Clinical symptoms of the disease include deficits in short term memory and language difficulties, as well as behavioral symptoms such as personality changes and depression. Progressive cognitive decline characterized by severe memory loss occurs as the disease progresses. Basic vital functions -such as swallowing- are altered at later stages of the disease, leading to death (Lopez and Dekosky, 2008; Alzheimer’s Association, 2020).
Protein misfolding and accumulation are prominent hallmarks of the disease, with Aβ plaques and tau tangles being the neuropathological signature of AD brains (Wang et al., 2016). Numerous studies have demonstrated that these toxic structures do not act independently and that rather, the neurodegenerative process in AD depends on the interaction between both Aβ and tau (Götz et al., 2001; Lewis et al., 2001; Rapoport et al., 2002; King et al., 2006; Roberson et al., 2007; Hurtado et al., 2010; Ittner et al., 2010; Vossel et al., 2010; Nussbaum et al., 2012; Zempel et al., 2013; Wang et al., 2016). Progressive build-up of these abnormal aggregates is associated with synaptic disruption (Shankar et al., 2008; Koffie et al., 2009; Moreno et al., 2011; Zempel et al., 2013; Rajmohan and Reddy, 2017; Pickett et al., 2019) and neuronal loss (Kadowaki et al., 2005; Jawhar et al., 2012; DeVos et al., 2017; Fu et al., 2017), leading to atrophy of specific brain regions (Spires-Jones and Hyman, 2014; Ferreira et al., 2017; Ten Kate et al., 2018).
Remarkably, the evidence indicates that accumulation of Aβ and tau is slow and that initiates more than two decades before clinical symptoms appear (Jack et al., 2009; Braak et al., 2011; Bateman et al., 2012; Villemagne et al., 2013), which has important diagnostic and therapeutic implications. In this context of early pathological changes during AD, axonal degeneration constitutes a common, initial event in several neurodegenerative conditions (Salvadores et al., 2017). Supporting evidence comes from imaging analyses of individuals with MCI -which are subjects at risk of developing AD- showing a significant decrease in white matter volume. Importantly, these results suggest that atrophy due to disruption of white matter fibers might contribute to memory decline (Kalus et al., 2006; Stoub et al., 2006; Rogalski et al., 2009; Ihara et al., 2010; Bozzali et al., 2011). Additionally, the use of diffusion tensor imaging to examine the microstructural integrity of white matter has revealed a pattern of alterations characteristically observed in axon-related pathologies. These changes correlate with cognitive impairment and are consistent with loss of brain connectivity (Huang and Auchus, 2007; Power et al., 2019).
Acute axonal degeneration, as the result of a traumatic lesion in the central nervous system, is a rapid process that takes hours to days depending on the organism and degenerative stimuli (Court and Coleman, 2012). However, in the case of neurodegenerative diseases, the degeneration of axons might take much longer periods (Lingor et al., 2012). Indeed, histopathological analyses using a mouse model of AD showed that, despite extreme dystrophy, axons maintain continuity throughout the disease course for several months (Adalbert et al., 2009). Moreover, computer modeling using data on neuron loss and neurofibrillary tangle (NFT) formation on AD brains, revealed that NFT bearing neurons can survive for up to 20 years (Morsch et al., 1999). This evidence suggests that neurodegeneration is a slow process in AD, thus providing the opportunity to target degenerating axons as an early therapeutic intervention. Compelling experimental and pathological studies have demonstrated that neurons in AD follow a dying-back pattern of neurodegeneration, where axonal terminals and then axons progressively degenerate toward the neuronal cell body (Bell and Claudio Cuello, 2006; Kalus et al., 2006; Stoub et al., 2006; Huang and Auchus, 2007; Adalbert et al., 2009; Rogalski et al., 2009; Gilley et al., 2011; Nishioka et al., 2019). Despite the difference in timing, axonal degeneration in the context of both acute lesions and neurodegenerative diseases, share morphologic features that include axonal swelling, microtubule disruption, and fragmentation of neuronal processes (Wang et al., 2012), suggesting that they correspond to similar processes. Recent studies have shed light into the mechanisms that govern acute axonal degeneration, revealing the dependence of NAD+ in this process, as well as the involvement of mitochondrial dysfunction and necroptosis activation (Barrientos et al., 2011; Osterloh et al., 2012; Neukomm et al., 2017; Hernández et al., 2018; Arrázola et al., 2019; Ko et al., 2020; Loreto et al., 2020; Oñate et al., 2020). Notably, all these pathways are associated to the hallmarks of aging (Kennedy et al., 2014; Sun et al., 2016; Deepa et al., 2018; Haas, 2019; Lautrup et al., 2019; Royce et al., 2019; McReynolds et al., 2020). In the context of AD, different pathogenic pathways have been shown to contribute to axon demise, including calcium signaling imbalance, mitochondrial dysfunction, alterations in axonal transport, and increased oxidative stress (Bamburg and Bloom, 2009; Yu et al., 2009; Ye et al., 2012; Cioffi et al., 2019; Guo et al., 2020). As stated above, the accumulation of misfolded proteins constitutes a salient feature of AD neuropathology and a large body of evidence linking Aβ and tau pathologies with the disruption of axons has been published. In this review, we will present the pathways that contribute to the mechanism of acute axonal degeneration. Then, we will critically evaluate the evidence associating Aβ and tau pathologies with the disruption of axons in AD.
Cellular Mechanisms Associated with Axonal Degeneration
Wallerian Degeneration
The degeneration of axons corresponds to a process activated in response to several stimuli including chemotherapy drugs, infection, inflammation, toxins, and mechanical injury, among others. Recent advances in the study of the molecular mechanisms that govern axon demise have contributed to uncovering essential components of the axon degeneration program. A schematic representation of the steps associated with axonal degeneration is presented in Figure 1.
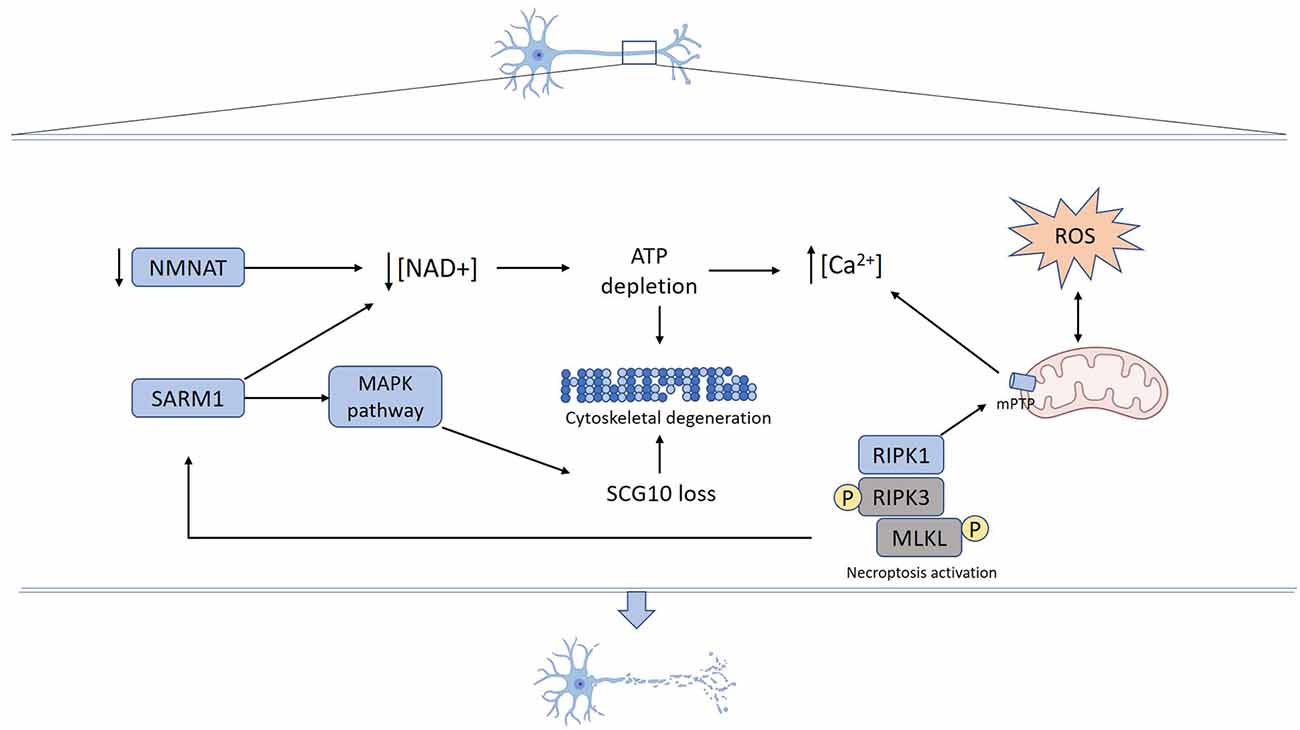
Figure 1. Mechanisms underlying axon degeneration. Mechanical or toxic injury lead to nicotinamide mononucleotide adenylyltransferase (NMNAT) axonal loss and SARM1 activation resulting in NAD+ depletion. Activated SARM1 promotes NAD+ destruction and NMAT loss decreases NAD+ synthesis. Reduced axonal NAD+ levels lead to energy failure and ATP depletion. SARM1 also activates MAPK signaling pathways, which promotes SCG10 proteolysis. Increased reactive oxygen species (ROS) production promotes mPTP formation that also can be triggered by necroptosis activation. Energy failure derived from both NAD+ depletion and mitochondrial damage contributes to calcium unbalance, ROS production, and mPTP formation. Cumulative activation of mechanisms and structural damage ultimately result in cytoskeleton fragmentation and axon degeneration.
Initial studies performed by August Waller to study axonal degeneration following nerve transection (Waller, 1850) led to the discovery of an ordered process in which three distinctive phases are typically observed. Initially, a latent period of about 36 h occurs, where the distal injured nerve fiber remains intact. A rapid phase then takes place, where the cytoskeleton is disrupted, and axons undergo fragmentation, which in vivo is associated with glial activation (Catenaccio et al., 2017). Finally, axonal disintegration and myelin degradation occurs, followed by macrophage infiltration and clearance of cell debris. This process is known as Wallerian degeneration (Coleman, 2005; Court and Coleman, 2012).
NAD+ Metabolism in Axonal Degeneration
More than a 100 years later, studies performed on the Wallerian degeneration slow (Wlds) mutant mice uncovered that upon axotomy, denucleated axons actively execute their own destruction, which is mediated by an evolutionarily conserved signaling pathway (Lunn et al., 1989; Perry et al., 1990, 1991; Lyon et al., 1993). Moreover, the discovery of the Wlds strain supported the idea that axonal degeneration and cell body death are two events regulated by different molecular mechanisms (Lunn et al., 1989; Deckwerth and Johnson, 1994). In Wlds mice, the distal denucleated axon remains functional for 3–4 weeks after nerve injury, suggesting that the Wlds gene confers a protective effect intrinsic to the axon. Wlds phenotype is caused by the overexpression of a chimeric Wlds gene, encoding the full-length nicotinamide mononucleotide adenylyltransferase (NMNAT1) and a short region of a ubiquitin assembly protein (UFD2; Conforti et al., 2000). The two components of the Wlds gene suggest the involvement of the ubiquitin-proteasome system (UPS) and NAD+ metabolism in the process of axon degeneration. Although genetic and pharmacological inhibition of UPS activity delays axon degeneration, overexpression of NMNAT1 alone can prevent axonal degeneration (Zhai et al., 2003; Araki et al., 2004). In injured axons, NAD+ levels decrease, and preventing this axonal NAD+ decline by exogenous application of NAD+ protects axons from degeneration (Wang et al., 2005). Since NAD+ is essential for glucose-dependent ATP production, reduced levels of NAD+ impair axonal energy production that contributes to axon degeneration (Gerdts et al., 2016). These data suggest that NAD+ metabolism plays a crucial role in axon degeneration.
SARM1 Mediates Axonal Loss
Mammals harbor three different isoforms of NMNAT proteins (NMNAT 1–3) that differ in subcellular localization and kinetic activity. NMNAT2 is the most labile isoform and it is constantly replenished in axons through fast axonal transport. Upon axotomy, NMNAT2 fails to be transported toward the axons and its levels rapidly drop in axons before Wallerian degeneration occurs (Gilley and Coleman, 2010; Gerdts et al., 2016). Thus, specific depletion of NMNAT2 is sufficient to induce Wallerian-like degeneration of uninjured axons. Moreover, the overexpression of NMNAT3, which is predominantly located in mitochondria, confers axonal protection after injury (Sasaki et al., 2006). The loss of NAD+ is suppressed in sterile alpha and TIR motif-containing 1 (SARM1) knockout (ko) axons both in vitro and in vivo, suggesting that SARM1 is a key mediator of axon destruction (Gerdts et al., 2013, 2015; Gilley et al., 2015). SARM1 contains a C-terminal Toll-interleukin receptor domain, which dimerizes and mediates the rapid breakdown of NAD+ (Gerdts et al., 2016). Axonal defects and embryonic lethality observed in Nmnat2 ko mice are suppressed by Sarm1 ablation, as Nmnat2/Sarm1 double-ko mice are healthy. This indicates a relationship between NMNAT2 and SARM1 in the control of NAD+ metabolism and axon degeneration. It has been hypothesized that loss of NMNAT2 induces a decline in NAD+ levels, which in turn might lead to SARM1-dependent NAD+ destruction, and consequently to a disastrous loss of NAD+ in the axon (Gerdts et al., 2016).
Although current data indicates that NAD+ depletion occurs downstream SARM1 activation after injury, additional evidence suggests the participation of other signaling pathways in axonal degeneration. Forced dimerization of the SARM1 TIR domain which results in NAD+ depletion and axon degeneration, also triggers MAPK signaling activation. Moreover, in damaged axons, SARM1 is required for activation of MAPK and this signaling disrupts axonal energy homeostasis leading to ATP depletion (Yang et al., 2015). Thus, the deletion of mitogen-activated protein kinase kinase 12 (MAP3K12), also known as DLK, significantly delays the degeneration of distal axons (Miller et al., 2009). DLK signals through the downstream target of MAPK JNK and pharmacological inhibition of JNK leads to axon protection similar to Dlk ablation. Although Dlk ablation confers axon protection, this protective effect is weaker than the one resulting from overexpression of Nmnat and Wlds genes. Also, after injury activation of two additional members of the MAPK cascade has been identified, MEKK4 (MAP3k4) and MLK2 (MAP3K10), which promote axon degeneration. Genetic ablation of all three MAP3Ks leads to robust axonal protection (Yang et al., 2015). All these MAP3Ks converge in the activation of the JNK pathway, which induces UPS-dependent degradation of SCG10 (stathmin 2) after axon injury (Shin et al., 2012).
The Role of Mitochondria in Axonal Degeneration
As aforementioned, a crucial role of NMNAT in the axonal degeneration cascade has been proven. Supporting studies demonstrated that NMNAT3 overexpression prevents axonal loss mediated by oxidative damage induced by reactive oxygen species (ROS) exposure (Press and Milbrandt, 2008). Additional evidence supported this data and suggested that mitochondrial localization of NMNAT activity has a key role in NMNAT-mediated axonal protection (Yahata et al., 2009). Prompted by this evidence and considering the involvement of mitochondrial permeability transition (mPT) on neurodegenerative conditions (Forte et al., 2007; Du et al., 2008; Martin et al., 2009), Barrientos et al. (2011) sought to determine the role of the mPT pore (mPTP) on the mechanism of axonal degeneration. The researchers showed that degeneration of axons induced by vincristine or nerve transection—in ex vivo and in vitro models—was associated with activation of the mPTP and targeting the mPTP component Cyclophilin D (CypD), either by pharmacological or genetic means, significantly delayed axonal disintegration (Barrientos et al., 2011). These results identify the mPTP as a key effector of axonal degeneration, as well as a potential target to prevent axonal loss triggered by both mechanical and toxic stimuli. Further studies revealed that upon axonal injury, mPTP formation is mediated by calcium release from the axonal endoplasmic reticulum, constituting an early step in the mechanism of axonal degeneration (Villegas et al., 2014). Moreover, many studies have demonstrated that following axotomy, an increase in intra-axonal Ca2+ occurs, constituting a common step to activate the axonal degeneration cascade (George et al., 1995; Adalbert et al., 2012; Avery et al., 2012; Mishra et al., 2013; Vargas et al., 2015). Besides mitochondrial calcium overload, mPTP opening can also be triggered by oxidative stress (Brockemeier et al., 1992). In accordance, in vivo work performed in both C. elegans and mice, recognized ROS as key intermediates in the mechanism of axonal degeneration, as increasing the anti-oxidative capacity of the neuron efficiently prevented axon demise and functional loss triggered by the hyperactivated degenerin channel MEC-4d (Calixto et al., 2012). Moreover, in vivo studies carried out in a mouse model of Charcot-Marie-Tooth 2A disease demonstrated an uncoupling between ATP and ROS production in axonal mitochondria. Similarly, in vivo-induced demyelination triggered reduced levels of ATP, along with increased ROS production in axonal mitochondria. These data suggest that mitochondrial ATP and ROS imbalances may contribute to axonal degeneration (van Hameren et al., 2019).
Necroptosis Involvement in the Mechanism of Axonal Demise
Mitochondrial dysfunction, ROS production, and intracellular calcium increase, which as mentioned above are key events that mediate axonal degeneration, have also been associated with activation of the necroptosis signaling pathway (Vandenabeele et al., 2010). To test the involvement of necroptosis in the mechanism of axonal degeneration, Hernández et al. (2018) used an in vitro model of glutamate-induced excitotoxicity in hippocampal neurons seeded in microfluidic devices. The authors showed that axonal degeneration proceeds by necroptosis, which involved the activation of the mPTP as well as calcium dyshomeostasis in axons. Pharmacological inhibition of the necroptotic kinase RIPK1 using Nec-1s, or genetic downregulation of Ripk3 or Mlkl, significantly prevented axonal degeneration and neuronal death (Hernández et al., 2018). In the same line, it was also demonstrated that axonal demise induced by mechanical and toxic stimuli in vitro—by axotomy or vincristine, respectively—is dependent on RIPK1, as Nec-1s prevented the degeneration of axons under those conditions. This protective effect was also observed by genetic inhibition of Ripk3 or Mlkl. Notably, the loss of the electrophysiological nerve function was also prevented by blocking the necroptotic machinery (Arrázola et al., 2019). Furthermore, investigating the mechanisms of axonal degeneration in in vitro and in vivo models of Parkinson’s disease, we also uncover the role of the necroptosis signaling pathway in this process. Neurons treated with 6-OHDA exhibited significant axonal degeneration, as well as elevated expression levels of necroptotic markers, which was prevented by Nec-1s or the pMLKL inhibitor GW80. Similarly, intracerebral injection of 6-OHDA in mice triggered axonal degeneration, which was accompanied by RIPK3 and pMLKL upregulation. Nec-1s administration decreased axonal degeneration and improved motor performance of 6-OHDA injected mice, and similar results were obtained by Ripk3 or Mlkl ko (Oñate et al., 2020). Together, these studies confirm that necroptosis activation takes place to mediate the degeneration of axons under different pro-degenerative stimuli, a process we have named necroaxoptosis (Oñate et al., 2020). Recently, a link between necroptosis and SARM1 was discovered using a neuroinflammatory model of glaucoma. TNF-α was injected into the vitreous cavity of wild type mice, triggering axonal and cell body loss, which was prevented in the SARM1 ko mice. Increased levels of necroptosis markers in optic nerves of both wild type and SARM1 ko mice suggested that SARM1 functions downstream of necroptosis mediate axonal degeneration. Additional experiments where necroptosis was induced by direct MLKL dimerization, showed that necroptosis-mediated calcium influx, loss of mitochondrial potential, and axon degeneration were blocked in SARM1 ko axons, integrating previous steps of the axonal degenerative pathway (Press and Milbrandt, 2008; Barrientos et al., 2011; Villegas et al., 2014; Arrázola et al., 2019; Ko et al., 2020). Therefore, necroaxoptosis seems to be a common mechanism for axonal degeneration after a variety of insults.
The Role of Protein Misfolding in Axonal Degeneration in AD
Axonal degeneration and dysfunction are prominent features of AD brains. White matter alterations revealed by in vivo imaging can be observed at the early stages of the disease, even in MCI patients. Importantly, these changes correlate with clinical measures of cognitive disability (Kalus et al., 2006; Stoub et al., 2006; Rogalski et al., 2009; Ihara et al., 2010; Bozzali et al., 2011). Accumulation of misfolded proteins is also an early pathological signature during AD, and cumulative evidence documenting a link between these two neuropathological events has been published, which is the focus of this section. A schematic representation of the signaling cascades triggered by Aβ and tau that contribute to axonal degeneration is presented in Figure 2.
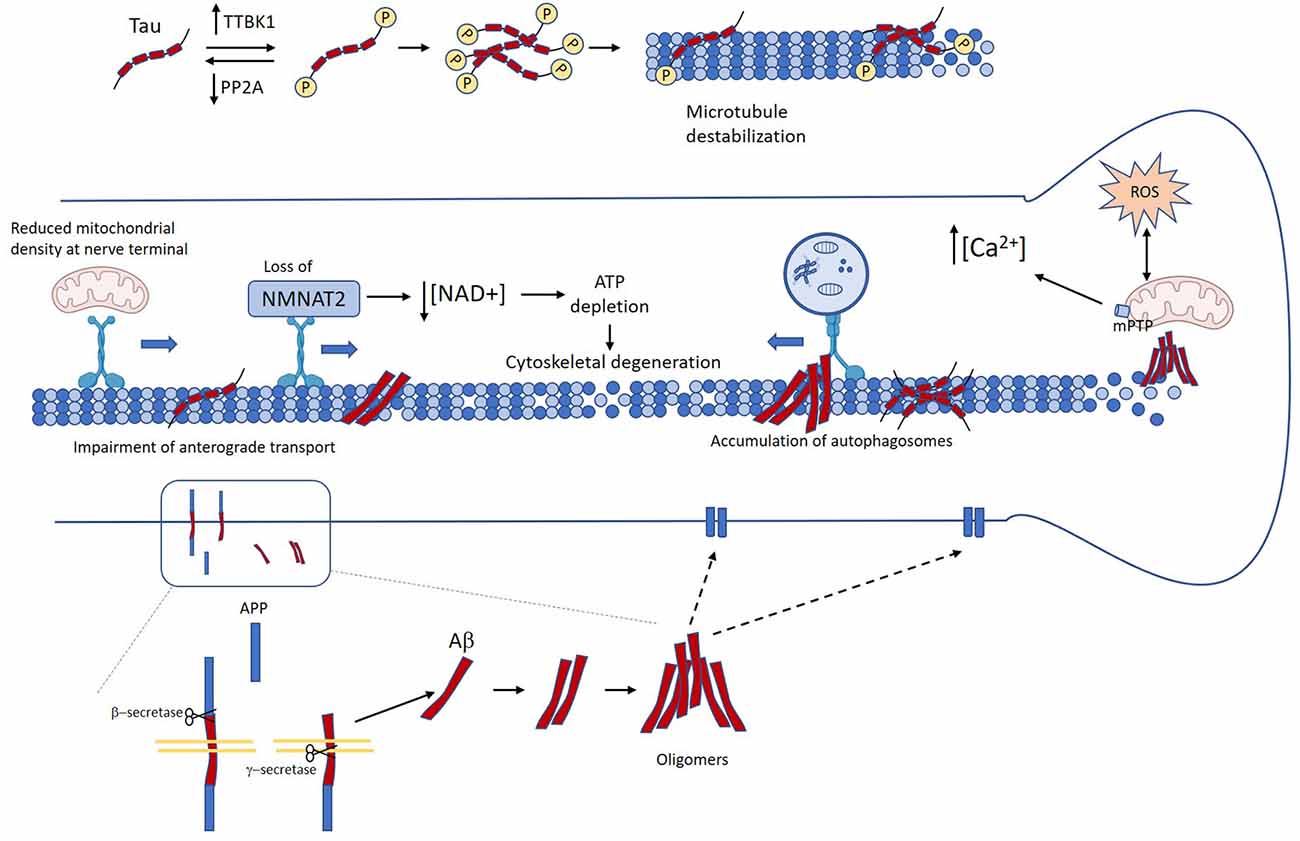
Figure 2. Mechanisms linking Tau and amyloid-β (Aβ) in Alzheimer’s disease (AD) to axon degeneration. Unbalance between kinases and phosphatases leads to the accumulation of abnormally phosphorylated tau, which in turn induces detachment form microtubules and microtubule destabilization (Köpke et al., 1993). Several age-related factors contribute to the accumulation of Aβ oligomers. Accumulation of pathological tau and Aβ promote axonal transport impairment (Calkins and Reddy, 2011; Tang et al., 2012; Wang et al., 2015; Sadleir et al., 2016; Zhang et al., 2018). This in turn causes loss of axonal NMAT2, decreased mitochondrial density at the nerve terminal, and autophagy flux impairment (Gilley and Coleman, 2010; Ljungberg et al., 2011; Ali et al., 2016). Neuron loss and neurofibrillary tangle (NFT) and Aβ oligomers also promote mitochondrial dysfunction, oxidative stress, and calcium dyshomeostasis (Stamer et al., 2002; Chee et al., 2005; Cieri et al., 2018; Mata, 2018; Albensi, 2019). All these events drive the failure of critical mechanisms for axonal functioning and maintenance that lead to loss of axonal homeostasis, and ultimately axon degeneration.
Axonal Disruption Associated With Tau Pathology
The first report describing tau pathology showed that neurofibrillary changes in the form of NFT and neuropil threads (NT) exhibit a characteristic distribution pattern affecting vulnerable brain regions such as the cerebral cortex and hippocampus (Braak and Braak, 1997). These lesions begin with misfolded phospho-tau in the proximal axon and then spreads into the somatodendritic compartment (Braak and Del Tredici, 2011). Deterioration of the cytoskeleton in individual neurons reveals a sequence of changes occurring in neuronal processes suggesting that the disruption of microtubules containing tau may cause the degeneration of axons (Kowall and Kosik, 1987; Braak et al., 1994; Braak and Del Tredici, 2011). Moreover, analysis of hippocampal regions showed that tau inclusions within dystrophic neurites correlate with several measures of the mini-mental state examination, suggesting that these pathological lesions contribute to cognitive dysfunction (Ghoshal et al., 2002).
The Role of Tau-Mediated Axonal Transport Disruption in Axonal Degeneration
Due to their polarized nature, neurons rely on an efficient axonal transport system for delivering proteins, lipids, and organelles from the cell body to the axon and synapses. The proper function of axonal transport depends on the correct assembly and functioning of all components including microtubules and motor proteins. Therefore, alterations in axonal transport render neurons vulnerable to the loss of synapses and can trigger axonal degeneration (Mandelkow et al., 2003). Indeed, chemical interventions that directly or indirectly affect axonal transport result in a dying-back form of axonal degeneration (Fukuda et al., 2017). Disruption of axonal transport as well as morphological alterations of the axons occur early in the course of AD and can be detected even a year before other neuropathological abnormalities develop, including amyloid deposition (Stokin, 2005). Defects in axonal transport have been extensively studied in the context of AD and it has been suggested to have a causative role in the disease (Muresan and Muresan, 2009; Vicario-Orri et al., 2014).
In AD, pathological changes associated with tau begin as the granular accumulation of phosphorylated tau in the cytoplasm, axon, and dendrites. Then, tau gradually aggregates to form NT and NFT, which affect the neuronal process that eventually undergoes degeneration (Braak et al., 1994; Braak and Del Tredici, 2011). Tau is a microtubule-associated protein involved in microtubules dynamics, which function as a track for axonal transport (Gao et al., 2018). It has been hypothesized that an imbalance in intracellular signaling causes excessive tau phosphorylation and its subsequent detachment from microtubules. This, in turn, promotes microtubule destabilization and impairment of axonal transport. Several studies have shown that tau overexpression in neurons increases tau phosphorylation and inhibits axonal transport (Stamer et al., 2002; Mandelkow et al., 2003; Chee et al., 2005; Thies and Mandelkow, 2007). As a result of tau-dependent axonal transport inhibition, organelles and vesicles are reduced in cell processes, which lead to energy depletion, decreased oxidative defense, and dying-back of neurites (Stamer et al., 2002; Mandelkow et al., 2003). The NAD+ biosynthetic enzyme NMNAT2, a well-known pro-survival factor that inhibits axonal degeneration after injury, depends on constant replenishment by anterograde axonal transport (Gilley and Coleman, 2010). Interestingly, in human AD brains as well as in rTg4510 mice, NMNAT2 expression is reduced, suggesting a possible mechanism by which tau pathology can contribute to axonal degeneration (Ljungberg et al., 2011; Ali et al., 2016). Thus, disruption of axonal transport-associated to abnormal tau phosphorylation might lead to decreased axonal NMNAT2 levels. The unbalance of the NAD+/NMN index due to loss of NMNAT2 could lead to SARM1-dependent NAD+ destruction in the axon and ultimately axon degeneration. However, this remains to be established.
Also, to regulate microtubule dynamics, tau modulates the function of the motor proteins dynein and kinesin, modifying the axonal transport of proteins and organelles. Enhanced tau phosphorylation also causes synaptic dysfunction as a consequence of energy depletion, associated with a reduced number of mitochondria in the pre-synapsis (Chee et al., 2005). This reduced density of mitochondria in axons and presynaptic terminals directly affects the energy status and calcium buffering. Additionally, it has been shown that tau may directly affect the endoplasmic reticulum-mitochondria interactions and therefore calcium handling (Cieri et al., 2018). Calcium unbalance may lead to activation of calcium-dependent proteases, calpains, which are implicated in the granular disintegration of the axonal cytoskeleton, a hallmark of Wallerian degeneration. Indeed, calpains are abnormally activated in AD brains, and they have been implicated in the development of tau pathology (Mahaman et al., 2019).
Phosphorylation plays a critical role in the regulation of tau functions, including the regulation of microtubule stabilization and assembly. Phosphorylation of tau is increased in AD brains, suggesting an unbalance in tau-associated kinases and phosphatases (Köpke et al., 1993). The expression of tau-tubulin kinase1 (TTBK1), a brain-specific tau kinase, is significantly up-regulated in the frontal neocortical region of the AD brain and colocalizes with NFT-positive neurons (Sato et al., 2006, 2008). Additionally, transgenic mice expressing human TTBK1 show increased tau phosphorylation and significant axonal degeneration in the entorhinal cortex (Sato et al., 2008; Ikezu et al., 2020). On the other hand, the activity of PP2A, the main tau phosphatase, is reduced in AD brains, and the inhibition of PP2A activity in mice models of AD result in tau pathology and cognitive impairment (Wang et al., 2010; Louis et al., 2011; Braithwaite et al., 2012). Strikingly, PP2A activity can be regulated by NMNAT2, hence reduced NMNAT2 expression observed in AD might down-regulate PP2A activity resulting in tau hyperphosphorylation (Cheng et al., 2013). Thus, in AD pathogenesis the unbalance between kinases and phosphatases may lead to abnormally hyperphosphorylated tau, which disrupts axonal transport, triggering axonal degeneration.
Axons depend on the constant remodeling of damaged proteins and organelles to maintain their correct functioning and connectivity (Maday and Holzbaur, 2016). This remodeling depends on homeostatic/degradation systems such as the UPS and autophagy (Korhonen and Lindholm, 2004; Kulkarni and Maday, 2018). Acetylation of tau inhibits the degradation of phosphorylated tau by the UPS, and the accumulation of acetylated tau has been identified in AD brains (Min et al., 2010, 2015). Moreover, the loss of Sirtuin-1, which deacetylate tau, is closely associated with the accumulation of tau in the cerebellar cortex (Julien et al., 2009). Although the UPS is responsible for the degradation of up to 80–90% of proteins including tau, misfolded proteins and aggregates are too large to be processed through the proteasome barrel and impede UPS function by physical occlusion (David et al., 2002; Chung et al., 2019). However, in vitro studies have demonstrated that tau aggregates can be degraded by autophagy. Ultrastructural analysis of post-mortem brain samples from AD patients showed the accumulation of autophagic vesicles within swollen and dystrophic neurites. Interestingly, autophagic vesicles were found more frequently in neurons bearing NFT (Nixon et al., 2005). Inducing autophagy by pharmacological interventions in different models of AD results in lower tau accumulation and better cognitive performance (Li et al., 2017). These observations suggest that autophagy impairment is associated with the deposition of pathological tau and contributes to neuronal demise in AD. Transport of autophagic vesicles loaded with unfolded proteins and organelles from distal axonal domains relies on a well-functioning retrograde transport to reach the soma, where autophagy degradation occurs (Maday and Holzbaur, 2014; Kulkarni and Maday, 2018). Dysregulations of tau observed in AD can impair dynein-retrograde axonal transport (Wang et al., 2015), which may lead to autophagy disruption and accumulation of autophagic vesicles within axons. Ultimately all these events might cause the loss of axonal homeostasis that gradually dye back to the soma.
Oxidative stress is another event associated with axonal degeneration in AD at the early stages of the pathologic process (Alavi Naini and Soussi-Yanicostas, 2015). Increased susceptibility to oxidative stress is linked to tau hyperphosphorylation, which leads to peroxisome depletion in neurites due to the inhibition of microtubule transport (Stamer et al., 2002). On the other hand, oxidative stress leads to increased tau phosphorylation in neuronal cultures and animal models of AD (Melov et al., 2007; Su et al., 2010). Indeed, antioxidant treatment reduces oxidative stress, tau pathology, and improves cognitive performance in 3xTg-AD mice (Clausen et al., 2012). An imbalance between pro-oxidants and antioxidants at the early stages of AD leads to increased oxidative stress, which may promote dysregulation of tau phosphorylation. This in turn might promote disruption of axonal transport that exacerbates oxidative stress and tau phosphorylation. Thus, tau phosphorylation and oxidative stress interplay is a key component of a vicious circle that plays a crucial role in the pathological process of AD. Then, ROS production might trigger mitochondrial dysfunction and necroptosis activation, two well-established mechanisms of axonal degeneration.
Analysis of postmortem brain samples suggests that tau pathology begins before the formation of NFT, as the accumulation of hyperphosphorylated tau is observed in young AD patients (Braak et al., 2011). Several lines of evidence suggest that abnormal phosphorylation of soluble tau causes synapse loss, impaired synaptic function, disrupted axonal transport, and cognitive deficits (DeKosky and Scheff, 1990; Callahan and Coleman, 1995; Mandelkow et al., 2003; Thies and Mandelkow, 2007; Hoover et al., 2010). Therefore, it has been hypothesized that NFT occurs as a protective cellular response, where NFT might scavenge the toxic monomeric or oligomeric tau. However, these tau aggregates might sequester other cell components or even cause an axonal clogging that ultimately leads to axonal degeneration (Mandelkow et al., 2003). Thus, tau pathology in AD likely initiates with a signaling dysregulation leading to hyperphosphorylation of tau, which causes microtubule destabilization and tau mislocalization. These events may drive the failure of critical mechanisms for axonal functioning and maintenance that lead to loss of axonal homeostasis. Finally, phospho-tau assembles into NFT, and the aggregation of these tau oligomers eventually contributes to axonal degeneration.
Evidence Linking Aβ to Axonal Disruption
Aβ-Related Axonal Dystrophy
In the first report describing the neuropathology of the postmortem brain of an AD patient, the appearance of altered neuronal processes (referred to as dystrophic neurites) was described (Alzheimer, 1907; article translated in Stelzmann et al., 1995). Subsequently, numerous studies examining AD brain tissue, as well as transgenic animal models of the disease, have revealed an intimate association between dystrophic neurites (i.e., axons and dendrites) and Aβ deposition (reviewed in Spires and Hyman, 2004; Woodhouse et al., 2005; Bell and Claudio Cuello, 2006; Mokhtar et al., 2013). Dystrophic neurites are defined as abnormal neuronal processes, characterized by the presence of thick, tortuous, as well as swollen segments (Su et al., 1993). The morphology of these irregular neuronal processes, which correspond to regions with cytoskeletal alteration and organelle accumulation, may change as the disease progresses (Vickers et al., 1996; Su et al., 1998; Woodhouse et al., 2009; Sanchez-Varo et al., 2012).
Although most research in AD has focused on the study of the loss of dendrites and synapses associated with Aβ pathology, as there is compelling evidence linking these alterations with cognitive decline (Davies et al., 1987; DeKosky and Scheff, 1990; Terry et al., 1991; Scheff et al., 2006; Bastrikova et al., 2008; Jackson et al., 2019), Aβ-related dystrophic axons are widespread in diseased brains and several studies suggest that they contribute to synaptic damage (Adalbert et al., 2009; Sanchez-Varo et al., 2012).
In vivo evidence of the temporal course of Aβ-related axonal pathology underwent by different AD mouse models—using two-photon imaging—has revealed that the environment around plaques is not uniform and that rather, it promotes a continuous remodeling, suggesting that axonal dystrophies associated with Aβ plaques are highly plastic structures, with a morphology that varies over time (Tsai et al., 2004; Blazquez-Llorca et al., 2017). Notably, although a re-growth phenomenon has been observed, were newly formed axonal segments were visualized in dystrophic axons, the elimination rates of neurites near amyloid aggregates are significantly higher than the formation rates, thereby leading to a progressive net loss over time (Tsai et al., 2004). As the formation of dystrophic axons seems to be a very dynamic process, with alterations appearing and disappearing within short periods, it has been suggested that early treatment to prevent amyloid accumulation may induce the recovery of the neuronal networks (Blazquez-Llorca et al., 2017). In agreement, different research groups have demonstrated that Aβ clearance by immunotherapy can attenuate Aβ-related axonal degeneration in AD transgenic models (Lombardo et al., 2003; Brendza et al., 2005; Liu et al., 2011).
Axonal Transport Disruption Associated With Aβ
Although there is a general notion that the development of dystrophic axons occurs as a consequence of nearby amyloid deposition, a growing body of evidence supports the converse view of an axonal origin of Aβ substrates for extracellular plaque formation, at sites were dystrophies are formed. Thus, the cause and effect relationship between amyloid plaques and axon damage remains a matter of debate.
Altered axonal transport constitutes a typical feature seen in the brain of TBI patients (who are at high risk of developing AD), as well as in TBI animal models (Choe, 2016). Immunohistochemical assessment of TBI brain samples shows increased levels of intra-axonal APP at swelling sites, and this has been attributed to the neuroprotective functions of this protein, as well as the products of its proteolytic processing (Plummer et al., 2016). However, there is well-documented evidence showing that widespread diffuse Aβ plaque deposition occurs in TBI survivors, and this has also been demonstrated in animal models of TBI (Johnson et al., 2012; Shishido et al., 2016; Edwards et al., 2017; Abu Hamdeh et al., 2018). Importantly, longitudinal studies performed to these individuals have revealed that NFT and amyloid plaque pathologies persist, and are associated with cognitive impairment (Johnson et al., 2012). Moreover, compelling studies have demonstrated that alterations in APP transport can promote the local release of Aβ, which can further impair axonal transport (Rodrigues et al., 2012; Mórotz et al., 2019). Thus, just as alterations in axonal transport can lead to amyloid pathology, there is also extensive evidence that Aβ itself can cause microtubule-based transport defects (Pigino et al., 2009; Zhao et al., 2010; Calkins and Reddy, 2011; Tang et al., 2012; Cruz et al., 2018; Zhang et al., 2018). In this line, and considering the role of microtubules in axonal transport, several studies have focused on the effects of Aβ on the integrity of the microtubule network (Fifre et al., 2006; Gevorkian et al., 2008; Silva et al., 2011; Mota et al., 2012; Pianu et al., 2014; Wang et al., 2018; Gao et al., 2019). For instance, Sadleir et al. (2016) hypothesized that presynaptic dystrophies were triggered by Aβ-mediated microtubule disruption. Using live-cell imaging of primary neurons, the authors observed that exposure to Aβ oligomers causes microtubule depolymerization, neuritic beading, and altered axonal trafficking. These data were validated in brain tissue from an AD mouse model as well as in human AD samples, where dystrophic axons and terminals in the proximity to Aβ deposits, displayed aberrant localization of tubulin, as well as evidence of decreased lysosomal function and autophagic vesicles accumulation, suggesting alterations in the microtubule-based transport. Notably, elevated β-secretase-1 and APP levels were also observed in peri-plaque dystrophies, which caused local Aβ generation that may further exacerbate extracellular amyloid pathology (Sadleir et al., 2016). Together, this evidence points towards a mechanism in which Aβ-mediated axonal defects, and the contribution of axonal dystrophies to Aβ plaque formation, are not isolated processes. Rather, a vicious circle seems to occur, where axons in the vicinity of amyloid deposits undergo alterations that further enhance Aβ accumulation.
As discussed above, defects in axonal transport occur early in the onset of AD. Considering the architecture of axons, transport along this structure is critical to preserve its integrity, as a failure of intra-axonal trafficking can lead to deprivation of cargoes that are essential for axon survival and can also impair the communication with the cell body or other cells (Coleman, 2011; Kanaan et al., 2013). Accordingly, disruptions on this vital cellular process can eventually lead to axonal degeneration (Ferri et al., 2003; Coleman, 2005, 2011; Pigino et al., 2009; Kanaan et al., 2013). In this regard, Aβ-mediated disturbances in microtubule-based cellular transport block the trafficking of vital cargoes to synapses. For example, many works reporting impairment of mitochondrial transport induced by Aβ have been published (Rui et al., 2006; Calkins and Reddy, 2011; Kim et al., 2012; Umeda et al., 2015; Zhang et al., 2018). Mitochondria are required at pre- and postsynaptic terminals for the correct function of neurotransmission, as it has essential roles in ATP production and buffering synaptic calcium (Li et al., 2004; Sheng and Cai, 2012). Thereby, the abnormal function of axonal transport induced by Aβ can block mitochondria trafficking, triggering synaptic alterations, which can initiate retrograde degeneration of axons. However, the exact mechanism that mediates this process remains unclear. As previously mentioned, alterations in NMNAT2 are associated with AD (Ali et al., 2016). Although a direct link between Aβ and NMNAT2 has not been proven, it is likely that, in the course of events leading to the activation of the axon death cascade, tau -which indeed has been related to NMNAT2 downregulation (Ljungberg et al., 2011)- acts downstream Aβ.
Aβ and the Mechanism of Axonal Degeneration
As previously stated, white matter alterations, reflecting axonal degeneration and dysfunction, are present not only in AD brains but also in MCI patients. Growing data from in vivo imaging studies have documented a relationship between such white matter abnormalities and Aβ deposition (Collins-Praino et al., 2014; Hoy et al., 2017; Schilling et al., 2018; Vipin et al., 2019; Weaver et al., 2019; Caballero et al., 2020). Importantly, recent longitudinal studies on the association between white matter integrity and amyloid pathology in cognitively normal individuals, revealed that Aβ aggregates build-up in white matter fibers known to be affected in AD, in an age-dependent manner (Vipin et al., 2019; Caballero et al., 2020). These studies underscore the impact of Aβ load on early white matter alterations in normal aged subjects at risk of AD. For this reason, a thorough understanding of the signaling that mediates axonal degeneration is crucial, as this would allow the identification of potential therapeutic targets.
Several lines of investigation have demonstrated that rather than senile plaques, Aβ oligomers are the most cytotoxic form of the aggregates and that plaques might serve as reservoirs from which oligomers can diffuse, reaching cellular targets in their vicinity. Aβ oligomers can exert toxicity through several different pathways, and many of them are involved in the process of axonal degeneration, including mitochondrial dysfunction (Swerdlow, 2018; Albensi, 2019), oxidative stress (Butterfield and Boyd-Kimball, 2018; Cheignon et al., 2018) and calcium dyshomeostasis (Mata, 2018; Popugaeva et al., 2018; Wang and Zheng, 2019). As discussed, previous work by our group demonstrated that axonal degeneration triggered by distinct insults is dependent on mitochondrial dysfunction and activation of the mPTP (Barrientos et al., 2011). In this line, Aβ oligomers were shown to target the mPTP regulator cyclophilin D (CypD) in a transgenic mouse model of AD as well as in AD brain samples. In this animal model, CypD deficiency prevented Aβ-induced mitochondrial swelling and permeability transition, improved calcium buffering capacity, and decreased mitochondrial ROS (Du et al., 2008, 2011). in vitro, the absence of CypD protected neurons from Aβ- and oxidative stress-mediated cell death. Remarkably, inhibiting the mPTP by genetic ablation of CypD improved cognitive and synaptic function in the transgenic AD model (Du et al., 2008, 2011). Furthermore, Aβ-induced impairment of axonal mitochondrial trafficking depends on CypD-mediated mPTP activation. Genetic deletion of CypD suppressed Aβ-mediated activation of the p38/MAPK signaling pathway, reversed axonal mitochondrial alterations, improved synaptic function, and prevented synapse loss (Guo et al., 2013). These studies shed light on the mechanism by which Aβ triggers mitochondrial dysfunction, which may promote axonal degeneration in AD.
As mentioned, both oxidative stress and calcium dyshomeostasis have shown to mediate axonal degeneration, and these two events have been largely studied in the context of Aβ-mediated neurodegeneration. However, most of the works have focused on the neuron as a whole, instead of studying the subcellular mechanisms that are activated in response to Aβ. Thus, to comprehend the exact pathways that trigger the loss of axonal structures, the use of compartmentalized microfluidic devices, which allow the isolation of axons from cell bodies and dendrites, represent a useful tool. In this regard, Song et al. (2006) used a compartmented chamber to study the differential effects of Aβ in somas and axons of sympathetic neurons. The researchers demonstrated that exposure of axons to Aβ triggered a caspase-independent mechanism of degeneration, which lead to nuclear apoptosis, as caspase inhibitors prevented apoptosis but did not protect neurons from axonal degeneration. Interestingly, the treatment of axons with calpastatin to inhibit calpains, which have been shown to mediate axonal cytoskeleton disintegration during Wallerian degeneration, not only protected axons from degeneration but also prevented nuclear apoptosis (Song et al., 2006). Using the same approach, another research group found that exposure of axons from hippocampal neurons to Aβ oligomers activated intra-axonal translation and induced local ATF4 synthesis. Retrograde transport of the protein to the soma induced ATF4-dependent gene expression and cell death. Similarly, intracerebral injection of Aβ in mice led to ATF4 translation in cholinergic axons, which was required for the retrograde transmission of Aβ-induced neurodegeneration (Baleriola et al., 2014; Walker et al., 2018).
As described here, recent studies have unveiled that axonal degeneration depends on necroaxoptosis activation (Hernández et al., 2018; Arrázola et al., 2019; Ko et al., 2020; Oñate et al., 2020). Although necroaxoptosis has not been demonstrated to occur in the context of AD, the evidence of necroptosis activation in AD brains (Caccamo et al., 2017) suggests that—similar to the findings in PD (Oñate et al., 2020)—this process may indeed contribute to axonal demise in AD.
Synergistic Contribution of Aβ and Tau to Axonal Degeneration
Mounting data has proven not only a synergistic but also a dependent neurotoxic effect of Aβ and tau pathologies (Bloom, 2014). To in vivo assess the pathological mechanism of Aβ and tau interaction on axonal degeneration, Nishioka et al. (2019) used a transgenic tau model and injected Aβ aggregates into the axonal terminals of retinal ganglion cells. Diffusion tensor imaging revealed a progressive white matter loss that correlated with the histopathological observation of retrograde axonal degeneration. Moreover, Aβ exposure triggered tau phosphorylation that preceded axon loss, and treatment with the microtubule-stabilizing compound Epothilone D inhibited tau phosphorylation and prevented axonal degeneration (Nishioka et al., 2019). This work emphasizes the cross-talk between Aβ and tau proteins that trigger the degeneration of axons and represents the first study to in vivo demonstrate the influence of this interaction in the mechanism of axonal degeneration. However, the exact signaling pathway activated downstream Aβ and tau interaction, and responsible for the disintegration of the axonal cytoskeleton, has not yet been explored.
Previous works have shown that reductions in tau can prevent Aβ-induced neurodegeneration and cognitive alterations in AD mouse models (Roberson et al., 2007; Ittner et al., 2010). Considering both the evidence of Aβ-mediated axonal transport deficits, as well as the interaction between tau and Aβ to induce neuronal alterations, Vossel et al. (2010) studied the effects of Aβ on axonal transport of mitochondria and TrkA, in hippocampal neurons from tau deficient and wild type mice. The researchers found that Aβ-dependent inhibition of axonal transport of mitochondria and TrkA was prevented in tau−/− neurons, stressing that a dependent degenerative effect between both proteins might take place in AD (Vossel et al., 2010). In line with this, in vitro and in vivo experiments showed that Aβ trimers derived from postmortem human AD brains, induced conformational changes in tau that led to reduced expression of the kinesin-1 light chain and increased intra-axonal APP, suggesting disruption of axonal transport (Sherman et al., 2016).
Although axonal transport deficits associated with misfolded amyloid and tau have been largely studied in AD brains and models, the signaling cascades acting downstream these alterations that might activate axonal degeneration pathways are not clearly defined. Thus, the evidence demonstrates that axonal degeneration is an early event in AD, where Aβ and tau pathologies interplay contributes to the degenerative process. Nevertheless, besides the reduced NMNAT2 levels found in AD brains, there is no evidence of the involvement of key molecules that govern Wallerian degeneration—such as necroptosis-associated proteins or SARM1—in the context of AD. Moreover, whereas Wallerian degeneration proceeds rapidly, the evidence indicates that axonal degeneration in AD rather constitutes an extended process that may take months or even years. Hence, it is possible that, although some common players are indeed involved, a mechanism different from the one activated during Wallerian degeneration mediates axonal degeneration in AD.
Concluding Remarks
Aging is considered the main risk factor contributing to the development of neurodegenerative diseases, including AD. The age-dependent decrease of homeostatic systems such as chaperones, degradation systems, and antioxidant defense considerably contributes to the accumulation of misfolded proteins in the brain. Although deposition of Aβ and tau has been the center of AD drug development research, many clinical trials on Aβ and tau clearing have been conducted, with disappointing outcomes (Congdon and Sigurdsson, 2018; Huang et al., 2020). As discussed in this review, a large body of evidence has uncovered a link between the development of Aβ and tau pathologies and the process of axonal degeneration. Nonetheless, a clear mechanism that integrates the known signaling cascades that activate axonal death, with the pathways that have been shown to mediate axonal degeneration in the context of AD, remains elusive. Many reports have documented that significant decreases in NAD+ occur during aging (Lautrup et al., 2019; McReynolds et al., 2020) and supplementation of this metabolite has been suggested as a potential treatment for age-related diseases including AD (Braidy et al., 2018; Hou et al., 2018). Despite the importance of NAD+ in the mechanism of axonal degeneration, its role in axon demise in AD is undefined. Furthermore, whether necroaxoptosis is activated in AD, is still unknown. Axonal loss in AD appears to be a slow process that initiates early during disease progression, thus providing a window for therapeutic intervention. An in-depth understanding of the mechanism that governs axonal degeneration is critical for the development of new therapies that allow to halt axonal loss and therefore prevent cell death and cognitive decline in AD.
Author Contributions
NS planned and structured the manuscript, researched, and wrote the review. CG-O researched and wrote the review. FC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Center for Integrative Biology, Universidad Mayor, FONDECYT-1150766, Geroscience Center for Brain Health, and Metabolism (FONDAP-15150012; FC), FONDECYT Postdoctoral fellowship N° 3180341 (NS), FONDECYT Postdoctoral fellowship N° 3190608 (CG-O).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abu Hamdeh, S., Waara, E. R., Möller, C., Söderberg, L., Basun, H., Alafuzoff, I., et al. (2018). Rapid amyloid-β oligomer and protofibril accumulation in traumatic brain injury. Brain Pathol. 28, 451–462. doi: 10.1111/bpa.12532
Adalbert, R., Morreale, G., Paizs, M., Conforti, L., Walker, S. A., Roderick, H. L., et al. (2012). Intra-axonal calcium changes after axotomy in wild-type and slow Wallerian degeneration axons. Neuroscience 225, 44–54. doi: 10.1016/j.neuroscience.2012.08.056
Adalbert, R., Nogradi, A., Babetto, E., Janeckova, L., Walker, S. A., Kerschensteiner, M., et al. (2009). Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132, 402–416. doi: 10.1093/brain/awn312
Agosta, F., Vossel, K. A., Miller, B. L., Migliaccio, R., Bonasera, S. J., Filippi, M., et al. (2009). Apolipoprotein E ε4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc. Natl. Acad. Sci. U S A 106, 2018–2022. doi: 10.1073/pnas.0812697106
Alavi Naini, S. M., and Soussi-Yanicostas, N. (2015). Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid. Med. Cell. Longev. 2015:151979. doi: 10.1155/2015/151979
Albensi, B. C. (2019). Dysfunction of Mitochondria: Implications for Alzheimer’s Disease. 1st Edn. Elsevier Inc. doi: 10.1016/bs.irn.2019.03.001
Ali, Y. O., Allen, H. M., Yu, L., Li-Kroeger, D., Bakhshizadehmahmoudi, D., Hatcher, A., et al. (2016). NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 14:e1002472. doi: 10.1371/journal.pbio.1002472
Alzheimer, A. (1907). Über eine Eigenartige Erkankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie under Psychisch Gerichtliche Medizin 64, 146–148.
Alzheimer’s Association. (2020). 2020 Alzheimer’s disease facts and figures. Alzheimers Dement. doi: 10.1002/alz.12068 [Epub ahead of print].
Araki, T., Sasaki, Y., and Milbrandt, J. (2004). Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010–1013. doi: 10.1126/science.1098014
Arrázola, M. S., Saquel, C., Catalán, R. J., Barrientos, S. A., Hernandez, D. E., Martínez, N. W., et al. (2019). Axonal degeneration is mediated by necroptosis activation. J. Neurosci. 39, 3832–3844. doi: 10.1523/JNEUROSCI.0881-18.2019
Avery, M. A., Rooney, T. M., Pandya, J. D., Wishart, T. M., Gillingwater, T. H., Geddes, J. W., et al. (2012). Wlds prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr. Biol. 22, 596–600. doi: 10.1016/j.cub.2012.02.043
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Bamburg, J. R., and Bloom, G. S. (2009). Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskeleton 66, 635–649. doi: 10.1002/cm.20388
Barrientos, S. A., Martinez, N. W., Yoo, S., Jara, J. S., Zamorano, S., Hetz, C., et al. (2011). Axonal degeneration is mediated by the mitochondrial permeability transition pore. J. Neurosci. 31, 966–978. doi: 10.1523/JNEUROSCI.4065-10.2011
Bastrikova, N., Gardner, G. A., Reece, J. M., Jeromin, A., and Dudek, S. M. (2008). Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc. Natl. Acad. Sci. U S A 105, 3123–3127. doi: 10.1073/pnas.0800027105
Bateman, R. J., Xiong, C., Benzinger, T. L. S., Fagan, A. M., Goate, A., Fox, N. C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367, 795–804. doi: 10.1056/NEJMoa1202753
Bell, K. F. S., and Claudio Cuello, A. (2006). Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 545, 11–21. doi: 10.1016/j.ejphar.2006.06.045
Blazquez-Llorca, L., Valero-Freitag, S., Rodrigues, E. F., Merchán-Pérez, Á., Rodríguez, J. R., Dorostkar, M. M., et al. (2017). High plasticity of axonal pathology in Alzheimer’s disease mouse models. Acta Neuropathol. Commun. 5:14. doi: 10.1186/s40478-017-0415-y
Bloom, G. S. (2014). Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Bozzali, M., Padovani, A., Caltagirone, C., and Borroni, B. (2011). Regional grey matter loss and brain disconnection across Alzheimer disease evolution. Curr. Med. Chem. 18, 2452–2458. doi: 10.2174/092986711795843263
Braak, F., Braak, H., and Mandelkow, E.-M. (1994). A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567. doi: 10.1007/BF00293315
Braak, H., and Braak, E. (1997). Frequency of stages of alzheimer-related lesions in different age categories. Neurobiol. Aging 18, 351–357. doi: 10.1016/s0197-4580(97)00056-0
Braak, H., and Del Tredici, K. (2011). The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 121, 171–181. doi: 10.1007/s00401-010-0789-4
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in alzheimer disease: Age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/nen.0b013e318232a379
Braidy, N., Grant, R., and Sachdev, P. S. (2018). Nicotinamide adenine dinucleotide and its related precursors for the treatment of Alzheimer’s disease. Curr. Opin. Psychiatry 31, 160–166. doi: 10.1097/yco.0000000000000394
Braithwaite, S. P., Stock, J. B., Lombroso, P. J., and Nairn, A. C. (2012). Protein phosphatases and Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 106, 343–379. doi: 10.1016/B978-0-12-396456-4.00012-2
Brendza, R. P., Bacskai, B. J., Cirrito, J. R., Simmons, K. A., Skoch, J. M., Klunk, W. E., et al. (2005). Anti-Aβ antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J. Clin. Invest. 115, 428–433. doi: 10.1172/JCI23269
Brockemeier, K. M., Carpenter-Deyo, L., Reed, D. J., and Pfeiffer, D. R. (1992). Cyclosporin A protects hepatocytes subjected to high Ca2+ and oxidative stress. FEBS Lett. 304, 192–194. doi: 10.1016/0014-5793(92)80616-o
Butterfield, D., and Boyd-Kimball, D. (2018). Oxidative stress, amyloid-β peptide and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s disease. J. Alzheimers Dis. 62, 1345–1367. doi: 10.3233/JAD-170543
Caballero, M. Á. A., Song, Z., Rubinski, A., Duering, M., Dichgans, M., Park, D. C., et al. (2020). Age-dependent amyloid deposition is associated with white matter alterations in cognitively normal adults during the adult life span. Alzheimers Dement. 16, 651–661. doi: 10.1002/alz.12062
Caccamo, A., Branca, C., Piras, I. S., Ferreira, E., Huentelman, M. J., Liang, W. S., et al. (2017). Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246. doi: 10.1038/nn.4608
Calixto, A., Jara, J. S., and Court, F. A. (2012). Diapause formation and downregulation of insulin-like signaling via DAF-16/FOXO delays axonal degeneration and neuronal loss. PLoS Genet. 8:e1003141. doi: 10.1371/journal.pgen.1003141
Calkins, M. J., and Reddy, P. H. (2011). Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 1812, 507–513. doi: 10.1016/j.bbadis.2011.01.007
Callahan, L. M., and Coleman, P. D. (1995). Neurons bearing neurofibrillary tangles are responsible for selected synaptic deficits in Alzheimer’s disease. Neurobiol. Aging 16, 311–314. doi: 10.1016/0197-4580(95)00035-d
Catenaccio, A., Llavero Hurtado, M., Diaz, P., Lamont, D. J., Wishart, T. M., and Court, F. A. (2017). Molecular analysis of axonal-intrinsic and glial-associated co-regulation of axon degeneration. Cell Death Dis. 8:e3166. doi: 10.1038/cddis.2017.489
Chee, F. C., Mudher, A., Cuttle, M. F., Newman, T. A., MacKay, D., Lovestone, S., et al. (2005). Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 20, 918–928. doi: 10.1016/j.nbd.2005.05.029
Cheignon, C., Tomas, M., Bonnefont-Rousselot, D., Faller, P., Hureau, C., and Collin, F. (2018). Oxidative stress and the amyloid β peptide in Alzheimer’s disease. Redox Biol. 14, 450–464. doi: 10.1016/j.redox.2017.10.014
Cheng, X. S., Zhao, K. P., Jiang, X., Du, L. L., Li, X. H., Ma, Z. W., et al. (2013). Nmnat2 attenuates tau phosphorylation through activation of PP2A. J. Alzheimers Dis. 36, 185–195. doi: 10.3233/jad-122173
Choe, M. C. (2016). The pathophysiology of concussion. Curr. Pain Headache Rep. 20:42. doi: 10.1007/s11916-016-0573-9
Chung, K. M., Hernández, N., Sproul, A. A., and Yu, W. H. (2019). Alzheimer’s disease and the autophagic-lysosomal system. Neurosci. Lett. 697, 49–58. doi: 10.1016/j.neulet.2018.05.017
Cieri, D., Vicario, M., Vallese, F., D’Orsi, B., Berto, P., Grinzato, A., et al. (2018). Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca2+ handling. Biochim. Biophys. Acta 1864, 3247–3256. doi: 10.1016/j.bbadis.2018.07.011
Cioffi, F., Adam, R. H. I., and Broersen, K. (2019). Molecular mechanisms and genetics of oxidative stress in Alzheimer’s disease. J. Alzheimers Dis. 72, 981–1017. doi: 10.3233/JAD-190863
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the β-amyloid precursor protein in familial Alzheimer’s disease increases β-protein production. Nature 360, 672–674. doi: 10.1038/360672a0
Clausen, A., Xu, X., Bi, X., and Baudry, M. (2012). Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer’s disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J. Alzheimers Dis. 30, 183–208. doi: 10.3233/JAD-2012-111298
Coleman, M. (2005). Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 6, 889–898. doi: 10.1038/nrn1788
Coleman, M. (2011). Molecular Signaling. How Do Axons Die? 1st Edn. Elsevier Inc. doi: 10.1016/B978-0-12-380860-8.00005-7
Collins-Praino, L. E., Francis, Y. I., Griffith, E. Y., Wiegman, A. F., Urbach, J., Lawton, A., et al. (2014). Soluble amyloid beta levels are elevated in the white matter of Alzheimer’s patients, independent of cortical plaque severity. Acta Neuropathol. Commun. 2:83. doi: 10.1186/s40478-014-0083-0
Conforti, L., Tarlton, A., Mack, T. G. A., Mi, W., Buckmaster, E. A., Wagner, D., et al. (2000). A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (Wld(s)) mouse. Proc. Natl. Acad. Sci. U S A 97, 11377–11382. doi: 10.1073/pnas.97.21.11377
Congdon, E. E., and Sigurdsson, E. M. (2018). Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415. doi: 10.1038/s41582-018-0013-z
Court, F. A., and Coleman, M. P. (2012). Mitochondria as a central sensor for axonal degenerative stimuli. Trends Neurosci. 35, 364–372. doi: 10.1016/j.tins.2012.04.001
Cruz, E., Kumar, S., Yuan, L., Arikkath, J., and Batra, S. K. (2018). Intracellular amyloid beta expression leads to dysregulation of the mitogen-activated protein kinase and bone morphogenetic protein-2 signaling axis. PLoS One 13:e0191696. doi: 10.1371/journal.pone.0191696
David, D. C., Layfield, R., Serpell, L., Narain, Y., Goedert, M., and Spillantini, M. G. (2002). Proteasomal degradation of tau protein. J. Neurochem. 83, 176–185. doi: 10.1046/j.1471-4159.2002.01137.x
Davies, C. A., Mann, D. M. A., Sumpter, P. Q., and Yates, P. O. (1987). A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 78, 151–164. doi: 10.1016/0022-510x(87)90057-8
Deckwerth, T. L., and Johnson, E. M. (1994). Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis). Dev. Biol. 165, 63–72. doi: 10.1006/dbio.1994.1234
Deepa, S. S., Unnikrishnan, A., Matyi, S., Hadad, N., and Richardson, A. (2018). Necroptosis increases with age and is reduced by dietary restriction. Aging Cell 17:e12770. doi: 10.1111/acel.12770
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
DeVos, S. L., Miller, R. L., Schoch, K. M., Holmes, B. B., Kebodeaux, C. S., Wegener, A. J., et al. (2017). Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 9:eaag0481. doi: 10.1126/scitranslmed.aag0481
Du, H., Guo, L., Fang, F., Chen, D., A Sosunov, A., McKhann, M. G., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Du, H., Guo, L., Zhang, W., Rydzewska, M., and Yan, S. (2011). Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 32, 398–406. doi: 10.1016/j.neurobiolaging.2009.03.003
Edwards, G., Moreno-Gonzalez, I., and Soto, C. (2017). Amyloid β and tau pathology following repetitive mild traumatic brain injury. Biochem. Biophys. Res. Commun. 483, 1137–1142. doi: 10.1016/j.bbrc.2016.07.123
Ferreira, D., Verhagen, C., Hernández-Cabrera, J. A., Cavallin, L., Guo, C. J., Ekman, U., et al. (2017). Distinct subtypes of Alzheimer’s disease based on patterns of brain atrophy: Longitudinal trajectories and clinical applications. Sci. Rep. 7:46263. doi: 10.1038/srep46263
Ferri, A., Sanes, J. R., Coleman, M. P., Cunningham, J. M., and Kato, A. C. (2003). Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr. Biol. 13, 669–673. doi: 10.1016/s0960-9822(03)00206-9
Fifre, A., Sponne, I., Koziel, V., Kriem, B., Yen Potin, F. T., Bihain, B. E., et al. (2006). Microtubule-associated protein MAP1A, MAP1B and MAP2 proteolysis during soluble amyloid β-peptide-induced neuronal apoptosis: Synergistic involvement of calpain and caspase-3. J. Biol. Chem. 281, 229–240. doi: 10.1074/jbc.m507378200
Forte, M., Gold, B. G., Marracci, G., Chaudhary, P., Basso, E., Johnsen, D., et al. (2007). Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. U S A 104, 7558–7563. doi: 10.1073/pnas.0702228104
Fu, H., Rodriguez, G. A., Herman, M., Emrani, S., Nahmani, E., Barrett, G., et al. (2017). Tau pathology induces excitatory neuron loss, grid cell dysfunction and spatial memory deficits reminiscent of early Alzheimer’s disease. Neuron 93, 533.e5–541.e5. doi: 10.1016/j.neuron.2016.12.023
Fukuda, Y., Li, Y., and Segal, R. A. (2017). A mechanistic understanding of axon degeneration in chemotherapy-induced peripheral neuropathy. Front. Neurosci. 11:481. doi: 10.3389/fnins.2017.00481
Gao, Q., Fang, Y., Zhang, S., Wong, H. S. H., Chan, Y. E., Wong, S. S. M., et al. (2019). Dynamic effect of beta-amyloid 42 on cell mechanics. J. Biomech. 86, 79–88. doi: 10.1016/j.jbiomech.2019.01.046
Gao, Y., Tan, L., Yu, J.-T., and Tan, L. (2018). Tau in Alzheimer’s disease: mechanisms and therapeutic strategies. Curr. Alzheimer Res. 15, 283–300. doi: 10.2174/1567205014666170417111859
George, E., Glass, J., and Griffin, J. (1995). Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J. Neurosci. 15, 6445–6452. doi: 10.1523/jneurosci.15-10-06445.1995
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2015). SARM1 activation triggers axon degeneration locally via NAD destruction. Science. 348, 453–457. doi: 10.1126/science.1258366
Gerdts, J., Summers, D. W., Milbrandt, J., and DiAntonio, A. (2016). Axon self-destruction: new links among SARM1, MAPKs and NAD+ metabolism. Neuron 89, 449–460. doi: 10.1016/j.neuron.2015.12.023
Gerdts, J., Summers, D. W., Sasaki, Y., DiAntonio, A., and Milbrandt, J. (2013). Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 33, 13569–13580. doi: 10.1523/jneurosci.1197-13.2013
Gevorkian, G., Gonzalez-Noriega, A., Acero, G., Ordoñez, J., Michalak, C., Munguia, M. E., et al. (2008). Amyloid-β peptide binds to microtubule-associated protein 1B (MAP1B). Neurochem. Int. 52, 1030–1036. doi: 10.1016/j.neuint.2007.10.020
Ghoshal, N., García-Sierra, F., Wuu, J., Leurgans, S., Bennett, D. A., Berry, R. W., et al. (2002). Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease. Exp. Neurol. 177, 475–493. doi: 10.1006/exnr.2002.8014
Gilley, J., Adalbert, R., and Coleman, M. P. (2011). Modelling early responses to neurodegenerative mutations in mice. Biochem. Soc. Trans. 39, 933–938. doi: 10.1042/bst0390933
Gilley, J., and Coleman, M. P. (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 8:e1000300. doi: 10.1371/journal.pbio.1000300
Gilley, J., Orsomando, G., Nascimento-Ferreira, I., and Coleman, M. P. (2015). Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 10, 1974–1981. doi: 10.1016/j.celrep.2015.02.060
Goate, A. (2006). Segregation of a missense mutation in the 889 amyloid β-protein precursor gene with familial Alzheimer’s disease. J. Alzheimers Dis. 9, 341–347. doi: 10.3233/jad-2006-9s338
Götz, J., Chen, F., Van Dorpe, J., and Nitsch, R. M. (2001). Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science 293, 1491–1495. doi: 10.1126/science.1062097
Guo, L., Du, H., Yan, S., Wu, X., McKhann, G. M., Chen, J. X., et al. (2013). Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One 8:e54914. doi: 10.1371/journal.pone.0054914
Guo, W., Stoklund Dittlau, K., and Van Den Bosch, L. (2020). Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin. Cell Dev. Biol. 99, 133–150. doi: 10.1016/j.semcdb.2019.07.010
Haas, R. H. (2019). Mitochondrial dysfunction in aging and diseases of aging. Biology 8:48. doi: 10.3390/biology8020048
Harvey, R. J., Skelton-Robinson, M., and Rossor, M. N. (2003). The prevalence and causes of dementia in people under the age of 65 years. J. Neurol. Neurosurg. Psychiatry 74, 1206–1209. doi: 10.1136/jnnp.74.9.1206
Hendriks, L., van Duijn, C. M., Cras, P., Cruts, M., Van Hul, W., van Harskamp, F., et al. (1992). Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β-amyloid precursor protein gene. Nat. Genet. 1, 218–221. doi: 10.1016/0197-4580(92)90390-j
Hernández, D. E., Salvadores, N. A., Moya-Alvarado, G., Catalán, R. J., Bronfman, F. C., and Court, F. A. (2018). Axonal degeneration induced by glutamate excitotoxicity is mediated by necroptosis. J. Cell Sci. 131:jcs214684. doi: 10.1242/jcs.214684
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Hou, Y., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U S A 115, E1876–E1885. doi: 10.1073/pnas.1718819115
Hoy, A. R., Ly, M., Carlsson, C. M., Okonkwo, O. C., Zetterberg, H., Blennow, K., et al. (2017). Microstructural white matter alterations in preclinical Alzheimer’s disease detected using free water elimination diffusion tensor imaging. PLoS One 12:e0173982. doi: 10.1371/journal.pone.0173982
Huang, J., and Auchus, A. P. (2007). Diffusion tensor imaging of normal appearing white matter and its correlation with cognitive functioning in mild cognitive impairment and Alzheimer’s disease. Ann. N Y Acad. Sci. 1097, 259–264. doi: 10.1196/annals.1379.021
Huang, L. K., Chao, S. P., and Hu, C. J. (2020). Clinical trials of new drugs for Alzheimer disease. J. Biomed. Sci. 27:18. doi: 10.1186/s12929-019-0609-7
Hurtado, D. E., Molina-Porcel, L., Iba, M., Aboagye, A. K., Paul, S. M., Trojanowski, J. Q., et al. (2010). Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 177, 1977–1988. doi: 10.2353/ajpath.2010.100346
Ihara, M., Polvikoski, T. M., Hall, R., Slade, J. Y., Perry, R. H., Oakley, A. E., et al. (2010). Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. 119, 579–589. doi: 10.1007/s00401-009-0635-8
Ikezu, S., Ingraham Dixie, K. L., Koro, L., Watanabe, T., Kaibuchi, K., and Ikezu, T. (2020). Tau-tubulin kinase 1 and amyloid-β peptide induce phosphorylation of collapsin response mediator protein-2 and enhance neurite degeneration in Alzheimer disease mouse models. Acta Neuropathol. Commun. 8:12. doi: 10.1186/s40478-020-0890-4
Ittner, L. M., Ke, Y. D., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jack, C. R., Lowe, V. J., Weigand, S. D., Wiste, H. J., Senjem, M. L., Knopman, D. S., et al. (2009). Serial PIB and MRI in normal, mild cognitive impairment and Alzheimers disease: Implications for sequence of pathological events in Alzheimers disease. Brain 132, 1355–1365. doi: 10.1093/brain/awp062
Jackson, J., Jambrina, E., Li, J., Marston, H., Menzies, F., Phillips, K., et al. (2019). Targeting the synapse in Alzheimer’s disease. Front. Neurosci. 13:735. doi: 10.3389/fnins.2019.00735
Jawhar, S., Trawicka, A., Jenneckens, C., Bayer, T. A., and Wirths, O. (2012). Motor deficits, neuron loss and reduced anxiety coinciding with axonal degeneration and intraneuronal Aβ aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 196.e29–196.e40. doi: 10.1016/j.neurobiolaging.2010.05.027
Johnson, V. E., Stewart, W., and Smith, D. H. (2012). Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 22, 142–149. doi: 10.1111/j.1750-3639.2011.00513.x
Julien, C., Tremblay, C., Émond, V., Lebbadi, M., Salem, N. Jr., Bennett, D. A., et al. (2009). Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 48–58. doi: 10.1097/nen.0b013e3181922348
Kadowaki, H., Nishitoh, H., Urano, F., Sadamitsu, C., Matsuzawa, A., Takeda, K., et al. (2005). Amyloid β induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 12, 19–24. doi: 10.1038/sj.cdd.4401528
Kalus, P., Slotboom, J., Gallinat, J., Mahlberg, R., Cattapan-Ludewig, K., Wiest, R., et al. (2006). Examining the gateway to the limbic system with diffusion tensor imaging: The perforant pathway in dementia. NeuroImage 30, 713–720. doi: 10.1016/j.neuroimage.2005.10.035
Kanaan, N. M., Pigino, G. F., Brady, S. T., Lazarov, O., Binder, L. I., and Morfini, G. A. (2013). Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol. 44–53. doi: 10.1007/3-211-26392-6_151
Kennedy, B. K., Berger, S. L., Brunet, A., Campisi, J., Cuervo, A. M., Epel, E. S., et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. doi: 10.1016/j.cell.2014.10.039
Kim, C., Choi, H., Jung, E. S., Lee, W., Oh, S., Jeon, N. L., et al. (2012). HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS One 7:e42983. doi: 10.1371/journal.pone.0042983
King, M. E., Kan, H. M., Baas, P. W., Erisir, A., Glabe, C. G., and Bloom, G. S. (2006). Tau-dependent microtubule disassembly initiated by prefibrillar β-amyloid. J. Cell Biol. 175, 541–546. doi: 10.1083/jcb.200605187
Ko, K. W., Milbrandt, J., and DiAntonio, A. (2020). SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 219:e201912047. doi: 10.1083/jcb.201912047
Koffie, R. M., Meyer-Luehmann, M., Hashimoto, T., Adams, K. W., Mielke, M. L., Garcia-Alloza, M., et al. (2009). Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U S A 106, 4012–4017. doi: 10.1073/pnas.0811698106
Köpke, E., Tung, Y. C., Shaikh, S., Alonso, A. C., Iqbal, K., and Grundke-Iqbal, I. (1993). Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384.
Koran, M. E. I., Wagener, M., and Hohman, T. J. (2017). Sex differences in the association between AD biomarkers and cognitive decline. Brain Imaging Behav. 11, 205–213. doi: 10.1007/s11682-016-9523-8
Korhonen, L., and Lindholm, D. (2004). The ubiquitin proteasome system in synaptic and axonal degeneration: a new twist to an old cycle. J. Cell Biol. 165, 27–30. doi: 10.1083/jcb.200311091
Kowall, N. W., and Kosik, K. S. (1987). Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann. Neurol. 22, 639–643. doi: 10.1002/ana.410220514
Kulkarni, V. V., and Maday, S. (2018). Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol. 78, 298–310. doi: 10.1002/dneu.22562
Lautrup, S., Sinclair, D. A., Mattson, M. P., and Fang, E. F. (2019). NAD+ in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655. doi: 10.1016/j.cmet.2019.09.001
Lewis, J., Dickson, D. W., Lin, W. L., Chisholm, L., Corral, A., Jones, G., et al. (2001). Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293, 1487–1491. doi: 10.1126/science.1058189
Li, Q., Liu, Y., and Sun, M. (2017). Autophagy and Alzheimer’s disease. Cell. Mol. Neurobiol. 37, 377–388. doi: 10.1007/s10571-016-0386-8
Li, Z., Okamoto, K. I., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Lingor, P., Koch, J. C., Tönges, L., and Bähr, M. (2012). Axonal degeneration as a therapeutic target in the CNS. Cell Tissue Res. 349, 289–311. doi: 10.1007/s00441-012-1362-3
Liu, Y., Lee, M. K., James, M. M., Price, D. L., Borchelt, D. R., Troncoso, J. C., et al. (2011). Passive (Amyloid-β) immunotherapy attenuates monoaminergic axonal degeneration in the AβPPswe/PS1dE9 mice. J. Alzheimers Dis. 23, 271–279. doi: 10.3233/JAD-2010-101602
Ljungberg, M. C., Ali, Y. O., Zhu, J., Wu, C.-S., Oka, K., Zhai, R. G., et al. (2011). CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum. Mol. Genet. 21, 251–267. doi: 10.1093/hmg/ddr492
LoBue, C., Wadsworth, H., Wilmoth, K., Clem, M., Hart, J., Womack, K. B., et al. (2017). Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin. Neuropsychol. 31, 85–98. doi: 10.1080/13854046.2016.1257069
Lombardo, J. A., Stern, E. A., McLellan, M. E., Kajdasz, S. T., Hickey, G. A., Bacskai, B. J., et al. (2003). Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J. Neurosci. 23, 10879–10883. doi: 10.1523/JNEUROSCI.23-34-10879.2003
Lopez, O. L., and Dekosky, S. T. (2008). Clinical symptoms in Alzheimer’s disease. Handb. Clin. Neurol. 89, 207–216. doi: 10.1016/S0072-9752(07)01219-5
Loreto, A., Hill, C. S., Hewitt, V. L., Orsomando, G., Angeletti, C., Gilley, J., et al. (2020). Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol. Dis. 134:104678. doi: 10.1101/683342
Louis, J. V., Martens, E., Borghgraef, P., Lambrecht, C., Sents, W., Longin, S., et al. (2011). Mice lacking phosphatase PP2A subunit PR61/B’δ (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3β. Proc. Natl. Acad. Sci. U S A 108, 6957–6962. doi: 10.1073/pnas.1018777108
Lunn, E. R., Perry, V. H., Brown, M. C., Rosen, H., and Gordon, S. (1989). Absence of wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1, 27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x
Lyon, M. F., Ogunkolade, B. W., Brown, M. C., Atherton, D. J., and Perry, V. H. (1993). A gene affecting Wallerian nerve degeneration maps distally on mouse chromosome 4. Proc. Natl. Acad. Sci. U S A 90, 9717–9720. doi: 10.1073/pnas.90.20.9717
Maday, S., and Holzbaur, E. L. F. (2014). Autophagosome Biogenesis in Primary Neurons Follows an Ordered and Spatially Regulated Pathway. Dev. Cell 30, 71–85. doi: 10.1016/j.devcel.2014.06.001
Maday, S., and Holzbaur, E. L. F. (2016). Compartment-specific regulation of autophagy in primary neurons. J. Neurosci. 36, 5933–5945. doi: 10.1523/jneurosci.4401-15.2016
Mahaman, Y. A. R., Huang, F., Kessete Afewerky, H., Maibouge, T., Ghose, B., and Wang, X. (2019). Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med. Res. Rev. 39, 608–630. doi: 10.1002/med.21534
Mandelkow, E.-M., Stamer, K., Vogel, R., Thies, E., and Mandelkow, E. (2003). Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 24, 1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007
Martin, L. J., Gertz, B., Pan, Y., Price, A. C., Molkentin, J. D., and Chang, Q. (2009). The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 218, 333–346. doi: 10.1016/j.expneurol.2009.02.015
Mata, A. M. (2018). Functional interplay between plasma membrane Ca2+-ATPase, amyloid β-peptide and tau. Neurosci. Lett. 663, 55–59. doi: 10.1016/j.neulet.2017.08.004
McReynolds, M. R., Chellappa, K., and Baur, J. A. (2020). Age-related NAD+ decline. Exp. Gerontol. 134:110888. doi: 10.1016/j.exger.2020.110888
Melov, S., Adlard, P. A., Morten, K., Johnson, F., Golden, T. R., Hinerfeld, D., et al. (2007). Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2, e536–e536. doi: 10.1371/journal.pone.0000536
Miller, B. R., Press, C., Daniels, R. W., Sasaki, Y., Milbrandt, J., and DiAntonio, A. (2009). A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 12, 387–389. doi: 10.1038/nn.2290
Min, S.-W., Chen, X., Tracy, T. E., Li, Y., Zhou, Y., Wang, C., et al. (2015). Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154–1162. doi: 10.1038/nm.3951
Min, S.-W., Cho, S.-H., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W. W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966. doi: 10.1016/j.neuron.2010.08.044
Mishra, B., Carson, R., Hume, R. I., and Collins, C. A. (2013). Sodium and potassium currents influence wallerian degeneration of injured Drosophila axons. J. Neurosci. 33, 18728–18739. doi: 10.1523/jneurosci.1007-13.2013
Mokhtar, S. H., Bakhuraysah, M. M., Cram, D. S., and Petratos, S. (2013). The beta-amyloid protein of Alzheimer’s disease: communication breakdown by modifying the neuronal cytoskeleton. Int. J. Alzheimers Dis. 2013:910502. doi: 10.1155/2013/910502
Moreno, H., Choi, S., Yu, E., Brusco, J., Avila, J., Moreira, J. E., et al. (2011). Blocking effects of human tau on squid giant synapse transmission and its prevention by T-817 MA. Front. Synaptic Neurosci. 3:3. doi: 10.3389/fnsyn.2011.00003
Mórotz, G. M., Glennon, E. B., Greig, J., Lau, D. H. W., Bhembre, N., Mattedi, F., et al. (2019). Kinesin light chain-1 serine-460 phosphorylation is altered in Alzheimer’s disease and regulates axonal transport and processing of the amyloid precursor protein. Acta Neuropathol. Commun. 7:200. doi: 10.1186/s40478-019-0857-5
Morsch, R., Simon, W., and Coleman, P. D. (1999). Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197. doi: 10.1097/00005072-199902000-00008
Mota, S., L. Ferreira, I., Pereira, C., R. Oliveira, C., and Cristina Rego, A. (2012). Amyloid-beta peptide 1–42 causes microtubule deregulation through N-methyl-D-aspartate receptors in mature hippocampal cultures. Curr. Alzheimer Res. 9, 844–856. doi: 10.2174/156720512802455322
Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B., et al. (1992). A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1, 345–347. doi: 10.1038/ng0892-345
Muresan, V., and Muresan, Z. (2009). Is abnormal axonal transport a cause, a contributing factor or a consequence of the neuronal pathology in Alzheimer’s disease? Future Neurol. 4, 761–773. doi: 10.2217/fnl.09.54
Neukomm, L. J., Burdett, T. C., Seeds, A. M., Hampel, S., Coutinho-Budd, J. C., Farley, J. E., et al. (2017). Axon death pathways converge on axundead to promote functional and structural axon disassembly. Neuron 95, 78–91. doi: 10.1016/j.neuron.2017.06.031
Nishioka, C., Liang, H. F., Barsamian, B., and Sun, S. W. (2019). Amyloid-beta induced retrograde axonal degeneration in a mouse tauopathy model. Neuroimage 189, 180–191. doi: 10.1016/j.neuroimage.2019.01.007
Nixon, R. A., Wegiel, J., Kumar, A., Yu, W. H., Peterhoff, C., Cataldo, A., et al. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122. doi: 10.1093/jnen/64.2.113
Nussbaum, J. M., Schilling, S., Cynis, H., Silva, A., Swanson, E., Wangsanut, T., et al. (2012). Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655. doi: 10.1038/nature11060
Oh, H., Madison, C., Villeneuve, S., Markley, C., and Jagust, W. J. (2014). Association of gray matter atrophy with age, β-amyloid and cognition in aging. Cereb. Cortex 24, 1609–1618. doi: 10.1093/cercor/bht017
Oñate, M., Catenaccio, A., Salvadores, N., Saquel, C., Martinez, A., Moreno-Gonzalez, I., et al. (2020). The necroptosis machinery mediates axonal degeneration in a model of Parkinson disease. Cell Death Differ. 27, 1169–1185. doi: 10.1038/s41418-019-0408-4
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. doi: 10.1126/science.1223899
Perry, V. H., Brown, M. C., and Lunn, E. R. (1991). Very slow retrograde and wallerian degeneration in the CNS of C57BL/Ola mice. Eur. J. Neurosci. 3, 102–105. doi: 10.1111/j.1460-9568.1991.tb00815.x
Perry, V. H., Brown, M. C., Lunn, E. R., Tree, P., and Gordon, S. (1990). Evidence that very slow wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur. J. Neurosci. 2, 802–808. doi: 10.1111/j.1460-9568.1990.tb00472.x
Pianu, B., Lefort, R., Thuiliere, L., Tabourier, E., and Bartolini, F. (2014). The Aβ1–42 peptide regulates microtubule stability independently of tau. J. Cell Sci. 127, 1117–1127. doi: 10.1242/jcs.143750
Pickett, E. K., Herrmann, A. G., McQueen, J., Abt, K., Dando, O., Tulloch, J., et al. (2019). Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 29, 3592.e5–3604.e5. doi: 10.1016/j.celrep.2019.11.044
Pigino, G., Morfini, G., Atagi, Y., Deshpande, A., Yu, C., Jungbauer, L., et al. (2009). Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc. Natl. Acad. Sci. U S A 106, 5907–5912. doi: 10.1073/pnas.0901229106
Plummer, S., Van Den Heuvel, C., Thornton, E., Corrigan, F., and Cappai, R. (2016). The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis. 7, 163–179. doi: 10.14336/ad.2015.0907
Popugaeva, E., Pchitskaya, E., and Bezprozvanny, I. (2018). Dysregulation of intracellular calcium signaling in Alzheimer’s disease. Antioxidants Redox Signal. 29, 1176–1188. doi: 10.1089/ars.2018.7506
Power, M. C., Su, D., Wu, A., Reid, R. I., Jack, C. R., Knopman, D. S., et al. (2019). Association of white matter microstructural integrity with cognition and dementia. Neurobiol. Aging 83, 63–72. doi: 10.1016/j.neurobiolaging.2019.08.021
Press, C., and Milbrandt, J. (2008). Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J. Neurosci. 28, 4861–4871. doi: 10.1523/jneurosci.0525-08.2008
Rajmohan, R., and Reddy, P. H. (2017). Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimers Dis. 57, 975–999. doi: 10.3233/jad-160612
Rapoport, M., Dawson, H. N., Binder, L. I., Vitek, M. P., and Ferreira, A. (2002). Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. U S A 99, 6364–6369. doi: 10.1073/pnas.092136199
Roberson, E. D., Scearce-Levie, K., Palop, J. J., Yan, F., Cheng, I. H., Wu, T., et al. (2007). Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316, 750–754. doi: 10.1126/science.1141736
Rodrigues, E. M., Weissmiller, A. M., and Goldstein, L. S. B. (2012). Enhanced β-secretase processing alters APP axonal transport and leads to axonal defects. Hum. Mol. Genet. 21, 4587–4601. doi: 10.1093/hmg/dds297
Rogalski, E. J., Murphy, C. M., Detoledo-Morrell, L., Shah, R. C., Moseley, M. E., Bammer, R., et al. (2009). Changes in parahippocampal white matter integrity in amnestic mild cognitive impairment: a diffusion tensor imaging study. Behav. Neurol. 21, 51–61. doi: 10.1155/2009/408037
Royce, G. H., Brown-Borg, H. M., and Deepa, S. S. (2019). The potential role of necroptosis in inflammaging and aging. Geroscience 41, 795–811. doi: 10.1007/s11357-019-00131-w
Rui, Y., Tiwari, P., Xie, Z., and Zheng, J. Q. (2006). Acute impairment of mitochondrial trafficking by β-amyloid peptides in hippocampal neurons. J. Neurosci. 26, 10480–10487. doi: 10.1523/jneurosci.3231-06.2006
Sadleir, K. R., Kandalepas, P. C., Buggia-Prévot, V., Nicholson, D. A., Thinakaran, G., and Vassar, R. (2016). Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 132, 235–256. doi: 10.1007/s00401-016-1558-9
Salvadores, N., Sanhueza, M., Manque, P., and Court, F. A. (2017). Axonal degeneration during aging and its functional role in neurodegenerative disorders. Front. Neurosci. 11:451. doi: 10.3389/fnins.2017.00451
Samieri, C., Perier, M. C., Gaye, B., Proust-Lima, C., Helmer, C., Dartigues, J. F., et al. (2018). Association of cardiovascular health level in older age with cognitive decline and incident dementia. JAMA 320, 657–664. doi: 10.1001/jama.2018.11499
Sanchez-Varo, R., Trujillo-Estrada, L., Sanchez-Mejias, E., Torres, M., Baglietto-Vargas, D., Moreno-Gonzalez, I., et al. (2012). Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol. 123, 53–70. doi: 10.1007/s00401-011-0896-x
Sasaki, Y., Araki, T., and Milbrandt, J. (2006). Stimulation of nicotinamide adenine dinucleotide biosynthetic pathways delays axonal degeneration after axotomy. J. Neurosci. 26, 8484–8491. doi: 10.1523/JNEUROSCI.2320-06.2006
Sato, S., Cerny, R. L., Buescher, J. L., and Ikezu, T. (2006). Tau-tubulin kinase 1 (TTBK1), a neuron-specific tau kinase candidate, is involved in tau phosphorylation and aggregation. J. Neurochem. 98, 1573–1584. doi: 10.1111/j.1471-4159.2006.04059.x
Sato, S., Xu, J., Okuyama, S., Martinez, L. B., Walsh, S. M., Jacobsen, M. T., et al. (2008). Spatial learning impairment, enhanced CDK5/p35 activity and downregulation of NMDA receptor expression in transgenic mice expressing tau-tubulin kinase 1. J. Neurosci. 28, 14511–14521. doi: 10.1523/jneurosci.3417-08.2008
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Schilling, L. P., Pascoal, T. A., Zimmer, E. R., Mathotaarachchi, S., Shin, M., Roberto, C., et al. (2018). Regional amyloid- β load and white matter abnormalities contribute to hypometabolism in Alzheimer’s dementia. Mol. Neurobiol. 56, 4916–4924. doi: 10.1007/s12035-018-1405-1
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: Impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Sherman, M. A., Lacroix, M., Amar, F., Larson, M. E., Forster, C., Aguzzi, A., et al. (2016). Soluble conformers of Aβ and tau alter selective proteins governing axonal transport. J. Neurosci. 36, 9647–9658. doi: 10.1523/JNEUROSCI.1899-16.2016
Shin, J. E., Miller, B. R., Babetto, E., Cho, Y., Sasaki, Y., Qayum, S., et al. (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. U S A 109, E3696–E3705. doi: 10.1073/pnas.1216204109
Shishido, H., Kishimoto, Y., Kawai, N., Toyota, Y., Ueno, M., Kubota, T., et al. (2016). Traumatic brain injury accelerates amyloid-β deposition and impairs spatial learning in the triple-transgenic mouse model of Alzheimer’s disease. Neurosci. Lett. 629, 62–67. doi: 10.1016/j.neulet.2016.06.066
Silva, D. F. F., Esteves, A. R., Arduino, D. M., Oliveira, C. R., and Cardoso, S. M. (2011). Amyloid-β-induced mitochondrial dysfunction impairs the autophagic lysosomal pathway in a tubulin dependent pathway. J. Alzheimers Dis. 26, 565–581. doi: 10.3233/JAD-2011-110423
Song, M. S., Saavedra, L., and de Chaves, E. I. P. (2006). Apoptosis is secondary to non-apoptotic axonal degeneration in neurons exposed to Aβ in distal axons. Neurobiol. Aging 27, 1224–1238. doi: 10.1016/j.neurobiolaging.2005.06.007
Spires, T. L., and Hyman, B. T. (2004). Neuronal structure is altered by amyloid plaques. Rev. Neurosci. 15, 267–278. doi: 10.1515/revneuro.2004.15.4.267
Spires-Jones, T. L., and Hyman, B. T. (2014). The intersection of amyloid β and tau at synapses in Alzheimer’s disease. Neuron 82, 756–771. doi: 10.1016/j.neuron.2014.05.004
Stamer, K., Vogel, R., Thies, E., Mandelkow, E., and Mandelkow, E.-M. (2002). Tau blocks traffic of organelles, neurofilaments and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 156, 1051–1063. doi: 10.1083/jcb.200108057
Stelzmann, R. A., Norman Schnitzlein, H., and Reed Murtagh, F. (1995). An english translation of alzheimer’s 1907 paper, “über eine eigenartige erkankung der hirnrinde”. Clin. Anat. 8, 429–431.
Stokin, G. B. (2005). Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307, 1282–1288. doi: 10.1126/science.1105681
Stoub, T. R., DeToledo-Morrell, L., Stebbins, G. T., Leurgans, S., Bennett, D. A., and Shah, R. C. (2006). Hippocampal disconnection contributes to memory dysfunction in individuals at risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. U S A 103, 10041–10045. doi: 10.1073/pnas.0603414103
Su, J. H., Cummings, B. J., and Cotman, C. W. (1993). Identification and distribution of axonal dystrophic neurites in Alzheimer’s disease. Brain Res. 625, 228–237. doi: 10.1016/0006-8993(93)91063-x
Su, J. H., Cummings, B. J., and Cotman, C. W. (1998). Plaque biogenesis in brain aging and Alzheimer’s disease. II. Progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol. 96, 463–471. doi: 10.1007/s004010050920
Su, B., Wang, X., Lee, H., Tabaton, M., Perry, G., Smith, M. A., et al. (2010). Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 468, 267–271. doi: 10.1016/j.neulet.2009.11.010
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028.
Suzuki, N., Cheung, T., Cai, X., Odaka, A., Otvos, L., Eckman, C., et al. (1994). An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264, 1336–1340. doi: 10.1126/science.8191290
Swerdlow, R. H. (2018). Mitochondria and mitochondrial cascades in Alzheimer’s disease. J. Alzheimers Dis. 62, 1403–1416. doi: 10.3233/JAD-170585
Tang, Y., Scott, D. A., Das, U., Edland, S. D., Radomski, K., Koo, E. H., et al. (2012). Early and selective impairments in axonal transport kinetics of synaptic cargoes induced by soluble amyloid β-protein oligomers. Traffic 13, 681–693. doi: 10.1111/j.1600-0854.2012.01340.x
Ten Kate, M., Dicks, E., Visser, P. J., Van Der Flier, W. M., Teunissen, C. E., Barkhof, F., et al. (2018). Atrophy subtypes in prodromal Alzheimer’s disease are associated with cognitive decline. Brain 141, 3443–3456. doi: 10.1093/brain/awy264
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Thies, E., and Mandelkow, E.-M. (2007). Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 27, 2896–2907. doi: 10.1523/jneurosci.4674-06.2007
Tsai, J., Grutzendler, J., Duff, K., and Gan, W. B. (2004). Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183. doi: 10.1038/nn1335
Umeda, T., Ramser, E. M., Yamashita, M., Nakajima, K., Mori, H., Silverman, M. A., et al. (2015). Intracellular amyloid β oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol. Commun. 3:51. doi: 10.1186/s40478-015-0230-2.
van Hameren, G., Campbell, G., Deck, M., Berthelot, J., Gautier, B., Quintana, P., et al. (2019). In vivo real-time dynamics of ATP and ROS production in axonal mitochondria show decoupling in mouse models of peripheral neuropathies. Acta Neuropathol. Commun. 7:86. doi: 10.1186/s40478-019-0740-4
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi: 10.1038/nrm2970
Vargas, M. E., Yamagishi, Y., Tessier-Lavigne, M., and Sagasti, A. (2015). Live imaging of calcium dynamics during axon degeneration reveals two functionally distinct phases of calcium influx. J. Neurosci. 35, 15026–15038. doi: 10.1523/jneurosci.2484-15.2015
Vicario-Orri, E., Opazo, C. M., and Muñoz, F. J. (2014). The pathophysiology of axonal transport in Alzheimer’s disease. J. Alzheimers Dis. 43, 1097–1113. doi: 10.3233/JAD-141080
Vickers, J. C., Chin, D., Edwards, A. M., Sampson, V., Harper, C., and Morrison, J. (1996). Dystrophic neurite formation associated with age-related β amyloid deposition in the neocortex: Clues to the genesis of neurofibrillary pathology. Exp. Neurol. 141, 1–11. doi: 10.1006/exnr.1996.0133
Villegas, R., Martinez, N. W., Lillo, J., Pihan, P., Hernandez, D., Twiss, J. L., et al. (2014). Calcium release from intra-axonal endoplasmic reticulum leads to axon degeneration through mitochondrial dysfunction. J. Neurosci. 34, 7179–7189. doi: 10.1523/jneurosci.4784-13.2014
Villemagne, V. L., Burnham, S., Bourgeat, P., Brown, B., Ellis, K. A., Salvado, O., et al. (2013). Amyloid β deposition, neurodegeneration and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 12, 357–367. doi: 10.1016/S1474-4422(13)70044-9
Vipin, A., Ng, K. K., Ji, F., Shim, H. Y., Lim, J. K. W., Pasternak, O., et al. (2019). Amyloid burden accelerates white matter degradation in cognitively normal elderly individuals. Hum. Brain Mapp. 40, 2065–2075. doi: 10.1002/hbm.24507
Vossel, K. A., Zhang, K., Brodbeck, J., Daub, A. C., Sharma, P., Finkbeiner, S., et al. (2010). Tau reduction prevents Aβ-induced axonal transport deficits by blocking activation of GSK3β. Science 330, 198–198. doi: 10.1083/jcb.201407065
Walker, C. A., Randolph, L. K., Matute, C., Alberdi, E., Baleriola, J., and Hengst, U. (2018). Aβ1-42 triggers the generation of a retrograde signaling complex from sentinel mRNAs in axons. EMBO Rep. 19:e45435. doi: 10.15252/embr.201745435
Waller, A. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lond. 140, 423–429. doi: 10.1098/rstl.1850.0021
Wang, L., Benzinger, T. L., Su, Y., Christensen, J., Friedrichsen, K., Aldea, P., et al. (2016). Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol. 73, 1070–1077. doi: 10.1001/jamaneurol.2016.2078
Wang, X., Blanchard, J., Kohlbrenner, E., Clement, N., Linden, R. M., Radu, A., et al. (2010). The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 24, 4420–4432. doi: 10.1096/fj.10-158477
Wang, L., Cao, J., Shi, Z., Fan, W., Liu, H., Deng, J., et al. (2018). Experimental study on the neurotoxic effect of β-amyloid on the cytoskeleton of PC12 cells. Int. J. Mol. Med. 41, 2764–2770. doi: 10.3892/ijmm.2018.3467
Wang, J. T., Medress, Z. A., and Barres, B. A. (2012). Axon degeneration: molecular mechanisms of a self-destruction pathway. J. Cell Biol. 196, 7–18. doi: 10.1083/jcb.201108111
Wang, Z. X., Tan, L., and Yu, J. T. (2015). Axonal transport defects in Alzheimer’s disease. Mol. Neurobiol. 51, 1309–1321. doi: 10.1007/s12035-014-8810-x
Wang, J., Zhai, Q., Chen, Y., Lin, E., Gu, W., McBurney, M. W., et al. (2005). A local mechanism mediates NAD-dependent protection of axon degeneration . J. Cell Biol. 170, 349–355. doi: 10.1083/jcb.200504028
Wang, X., and Zheng, W. (2019). Ca2+ homeostasis dysregulation in Alzheimer’s disease: a focus on plasma membrane and cell organelles. FASEB J. 33, 6697–6712. doi: 10.1096/fj.201801751R
Weaver, N. A., Doeven, T., Barkhof, F., Biesbroek, J. M., Groeneveld, O. N., Kuijf, H. J., et al. (2019). Cerebral amyloid burden is associated with white matter hyperintensity location in specific posterior white matter regions. Neurobiol. Aging 84, 225–234. doi: 10.1016/j.neurobiolaging.2019.08.001
Woodhouse, A., Vickers, J. C., Adlard, P. A., and Dickson, T. C. (2009). Dystrophic neurites in TgCRND8 and Tg2576 mice mimic human pathological brain aging. Neurobiol. Aging 30, 864–874. doi: 10.1016/j.neurobiolaging.2007.09.003
Woodhouse, A., West, A. K., Chuckowree, J. A., Vickers, J. C., and Dickson, T. C. (2005). Does β-amyloid plaque formation cause structural injury to neuronal processes? Neurotox. Res. 7, 5–15. doi: 10.1007/BF03033772
Yahata, N., Yuasa, S., and Araki, T. (2009). Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J. Neurosci. 29, 6276–6284. doi: 10.1523/jneurosci.4304-08.2009
Yang, J., Wu, Z., Renier, N., Simon, D. J., Uryu, K., Park, D. S., et al. (2015). Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176. doi: 10.1016/j.cell.2014.11.053
Ye, X., Tai, W., and Zhang, D. (2012). The early events of Alzheimer’s disease pathology: from mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol. Aging 33, 1122.e1–1122.e10. doi: 10.1016/j.neurobiolaging.2011.11.004
Yu, J. T., Chang, R. C. C., and Tan, L. (2009). Calcium dysregulation in Alzheimer’s disease: from mechanisms to therapeutic opportunities. Prog. Neurobiol. 89, 240–255. doi: 10.1016/j.pneurobio.2009.07.009
Zempel, H., Luedtke, J., Kumar, Y., Biernat, J., Dawson, H., Mandelkow, E., et al. (2013). Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32, 2920–2937. doi: 10.1038/emboj.2013.207
Zhai, Q., Wang, J., Kim, A., Liu, Q., Watts, R., Hoopfer, E., et al. (2003). Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron 39, 217–225. doi: 10.1016/s0896-6273(03)00429-x
Zhang, L., Trushin, S., Christensen, T. A., Tripathi, U., Hong, C., Geroux, R. E., et al. (2018). Differential effect of amyloid beta peptides on mitochondrial axonal trafficking depends on their state of aggregation and binding to the plasma membrane. Neurobiol. Dis. 114, 1–16. doi: 10.1016/j.nbd.2018.02.003
Keywords: Alzheheimer’s disease, protein misfolding, axonal degeneration, amyloid β, tau, necroaxoptosis
Citation: Salvadores N, Gerónimo-Olvera C and Court FA (2020) Axonal Degeneration in AD: The Contribution of Aβ and Tau. Front. Aging Neurosci. 12:581767. doi: 10.3389/fnagi.2020.581767
Received: 09 July 2020; Accepted: 09 September 2020;
Published: 15 October 2020.
Edited by:
Rodrigo Morales, University of Texas Health Science Center at Houston, United StatesReviewed by:
Shuji Wakatsuki, National Center of Neurology and Psychiatry (Japan), JapanAlessio Vagnoni, King’s College London, United Kingdom
Copyright © 2020 Salvadores, Gerónimo-Olvera and Court. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felipe A. Court, ZmVsaXBlLmNvdXJ0QHVtYXlvci5jbA==