 Camilla Pellegrini1
Camilla Pellegrini1 Chiara Pirazzini1
Chiara Pirazzini1 Claudia Sala2
Claudia Sala2 Luisa Sambati1,3Igor Yusipov4
Luisa Sambati1,3Igor Yusipov4 Alena Kalyakulina4
Alena Kalyakulina4 Francesco Ravaioli5Katarzyna M. Kwiatkowska5Danielle F. Durso6Mikhail Ivanchenko4
Francesco Ravaioli5Katarzyna M. Kwiatkowska5Danielle F. Durso6Mikhail Ivanchenko4 Daniela Monti7
Daniela Monti7 Raffaele Lodi1,3
Raffaele Lodi1,3 Claudio Franceschi4
Claudio Franceschi4 Pietro Cortelli1,3
Pietro Cortelli1,3 Paolo Garagnani5,8,9,10
Paolo Garagnani5,8,9,10 Maria Giulia Bacalini1*
Maria Giulia Bacalini1*- 1Istituto di Ricovero e Cura a Carattere Scientifico Istituto delle Scienze Neurologiche di Bologna, Bologna, Italy
- 2Department of Physics and Astronomy, University of Bologna, Bologna, Italy
- 3Department of Biomedical and Neuromotor Sciences, University of Bologna, Bologna, Italy
- 4Institute of Information Technologies, Mathematics and Mechanics, Lobachevsky University, Nizhny Novgorod, Russia
- 5Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, Bologna, Italy
- 6Department of Neurology, University of Massachusetts Medical School, Worcester, MA, United States
- 7Department of Experimental and Clinical Biomedical Sciences “Mario Serio,” University of Florence, Florence, Italy
- 8Department of Laboratory Medicine, Clinical Chemistry, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
- 9Applied Biomedical Research Center, Policlinico S.Orsola-Malpighi Polyclinic, Bologna, Italy
- 10National Research Council of Italy Institute of Molecular Genetics “Luigi Luca Cavalli-Sforza,” Unit of Bologna, Bologna, Italy
Alzheimer's disease (AD) is characterized by specific alterations of brain DNA methylation (DNAm) patterns. Age and sex, two major risk factors for AD, are also known to largely affect the epigenetic profiles in brain, but their contribution to AD-associated DNAm changes has been poorly investigated. In this study we considered publicly available DNAm datasets of four brain regions (temporal, frontal, entorhinal cortex, and cerebellum) from healthy adult subjects and AD patients, and performed a meta-analysis to identify sex-, age-, and AD-associated epigenetic profiles. In one of these datasets it was also possible to distinguish 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) profiles. We showed that DNAm differences between males and females tend to be shared between the four brain regions, while aging differently affects cortical regions compared to cerebellum. We found that the proportion of sex-dependent probes whose methylation is modified also during aging is higher than expected, but that differences between males and females tend to be maintained, with only a few probes showing age-by-sex interaction. We did not find significant overlaps between AD- and sex-associated probes, nor disease-by-sex interaction effects. On the contrary, we found that AD-related epigenetic modifications are significantly enriched in probes whose DNAm varies with age and that there is a high concordance between the direction of changes (hyper or hypo-methylation) in aging and AD, supporting accelerated epigenetic aging in the disease. In summary, our results suggest that age-associated DNAm patterns concur to the epigenetic deregulation observed in AD, providing new insights on how advanced age enables neurodegeneration.
Introduction
Alzheimer's disease (AD) is a chronic neurodegenerative disease that leads to a progressive decay of cognitive abilities and self-sufficiency. Neuronal loss involves multiple brain regions that are progressively affected by the disease. Hippocampus and entorhinal cortex exhibit the earliest pathological changes, preceding the onset of clinical signs and cognitive impairment by several years, and later the disease spreads to the other brain regions (Braak and Braak, 1991; Van Hoesen et al., 1991; Scahill et al., 2002; Coupé et al., 2019).
Advanced age and female sex are the two major non-modifiable risk factors for AD (Hickman et al., 2016; Podcasy and Epperson, 2016; Fisher et al., 2018). More than 95% of cases of AD occur after 65 years of age (late onset AD), and AD prevalence increases exponentially between 65 and 85 years (Hebert et al., 1995; Kawas and Corrada, 2006). Two-thirds of clinically diagnosed cases of AD are women, and the fact that women live longer than man does not fully explain this sex bias for AD (Pike, 2017; Nebel et al., 2018).
The etiology and pathogenesis of AD are complex and likely result from the interplay between genetic and environmental factors during lifespan. In this scenario epigenetic modifications have attracted increased interest in the study of AD, as they integrate genetic background and environment and modulate genomic organization and gene expression. Epigenetic modifications regulate brain biology throughout development and lifetime, influencing neuronal plasticity, cognition, and behavior (Fagiolini et al., 2009), and deregulation of brain epigenetic patterns has been associated to the pathogenesis of neurological and psychiatric disorders (Landgrave-Gómez et al., 2015; Jaffe et al., 2016). Several studies in post-mortem AD brains have investigated the role of DNA methylation (DNAm), the best-characterized epigenetic modification, identifying a number of CpG sites that show robust changes in DNAm compared to non-demented controls (Lunnon et al., 2014; Gasparoni et al., 2018; Smith et al., 2018, 2019, 2020; Altuna et al., 2019; Lardenoije et al., 2019; Semick et al., 2019; Smith R. G. et al., 2020; Wei et al., 2020).
Interestingly, the two major non-modifiable AD risk factors mentioned above, i.e., sex and age, are also among the main biological variables that influence epigenetic patterns in most human tissues, including brain (Gilbert et al., 2019).
Genome-wide DNAm differences between males and females have been found in whole blood (Singmann et al., 2015) and have been related to the sex-biased risk of psychiatric diseases (Maschietto et al., 2017). A similar link has been reported also in brain (Xia et al., 2019) where sex-specific DNAm patterns are established early during prenatal development (Spiers et al., 2015; Perzel Mandell et al., 2020) and are at least in part maintained in the adulthood (Xu et al., 2014; Spiers et al., 2015), contributing to the profound differences in brain functions between males and females (McCarthy et al., 2009; Forger, 2016; Gegenhuber and Tollkuhn, 2019) and to the different onset of psychiatric disorders (Perzel Mandell et al., 2020).
DNAm patterns are largely remodeled during aging (Pal and Tyler, 2016), where a trend toward global loss of DNAm together with hypermethylation at specific loci is observed (Xiao et al., 2019). Although with some differences among brain regions (Hernandez et al., 2011; Horvath et al., 2015), age-associated epigenetic changes interest also the brain, likely contributing to the structural and functional alterations that can result in progressive cognitive decline and increased susceptibility to neurodegenerative disorders (Bishop et al., 2010; Lardenoije et al., 2015).
So far, only few studies have considered how sex and age interact during lifespan in shaping the epigenome. Data on whole blood indicate that sex-dependent DNAm is remodeled during aging (McCartney et al., 2019), and we suggested that these changes occur at different extent in human models of successful and unsuccessful aging (Yusipov et al., 2020). In mouse hippocampus and human frontal cortex, Masser et al. identified both CpGs in which sex-dependent DNAm is maintained during lifetime, and CpG sites that are differentially affected by aging in relation to sex (Masser et al., 2017). Interestingly, some studies employing epigenetic clocks, i.e., DNAm-based predictors of age, reported accelerated aging in whole blood from males compared to females (Horvath et al., 2016; Xiao et al., 2018; Tajuddin et al., 2019), and the same trend was observed also in brain (Horvath et al., 2016).
Collectively, the available data sustain the importance of sex and aging in shaping the brain epigenome, but so far only one study combined different datasets to identify reproducible sex-associated DNAm profiles (Xia et al., 2019). No study has systematically analyzed multiple datasets and brain regions to identify DNAm patterns resulting from the interaction of sex and age during lifespan, and most importantly no study has evaluated whether sex- and age-dependent DNAm can contribute to epigenetic deregulation in AD, despite the pivotal role of these two factors in AD etiology and pathogenesis.
To fill this gap, in the present paper we performed a meta-analysis of DNAm across sex, age, and AD considering publicly available datasets from different brain regions.
Materials and Methods
Datasets
To select DNAm datasets based on Infinium BeadChip technology, the Gene Expression Omnibus (GEO) repository (Clough and Barrett, 2016) was interrogated by the GEOmetadb Bioconductor package using the following search terms: “GPL13534,” “GPL21145,” to include only datasets based on the Illumina Infinium HumanMethylation450 and MethylationEPIC BeadChips; “sex,” “gender,” “female,” to include only datasets in which the information on the sex of the subjects was available; “age,” to include only datasets in which the information on the age of the subjects was available; “brain,” “cortex,” “gyrus,” “lobe,” “gray,” to select datasets in which brain samples were analyzed; “control,” “normal,” “non-tumor,” “health,” or “Alzheimer,” “AD,” “Braak,” to select datasets including healthy and AD subjects, respectively. We considered only datasets including more than 10 healthy subjects. As to June 30th 2020, only Illumina Infinium HumanMethylation450 datasets were retrieved.
For the meta-analysis of sex- and age-dependent DNAm in healthy subjects, we selected only datasets including at least 10 males and 10 females, having more than 19 years and spanning an age range of at least 30 years. We further considered only brain regions for which at least two datasets were available. This resulted in eight datasets covering four regions: Frontal cortex (FC), Temporal cortex (TC), Entorhinal cortex (ERC), Cerebellum (CRB) (Table 1).
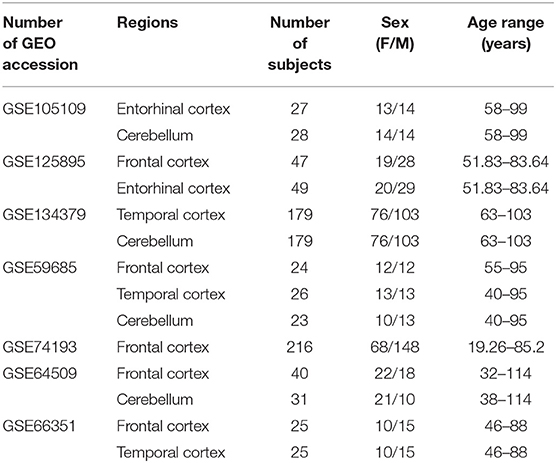
Table 1. Characteristics of the Infinium450k datasets including healthy subjects selected in the present study for the meta-analysis of sex- and age-associated DNAm.
For the meta-analysis of AD-associated methylation patterns, we selected only the datasets including subjects over 65 years of age with at least 3 males and 3 females in the control and AD groups. This resulted in eight datasets covering the same brain regions indicated above (Table 2).
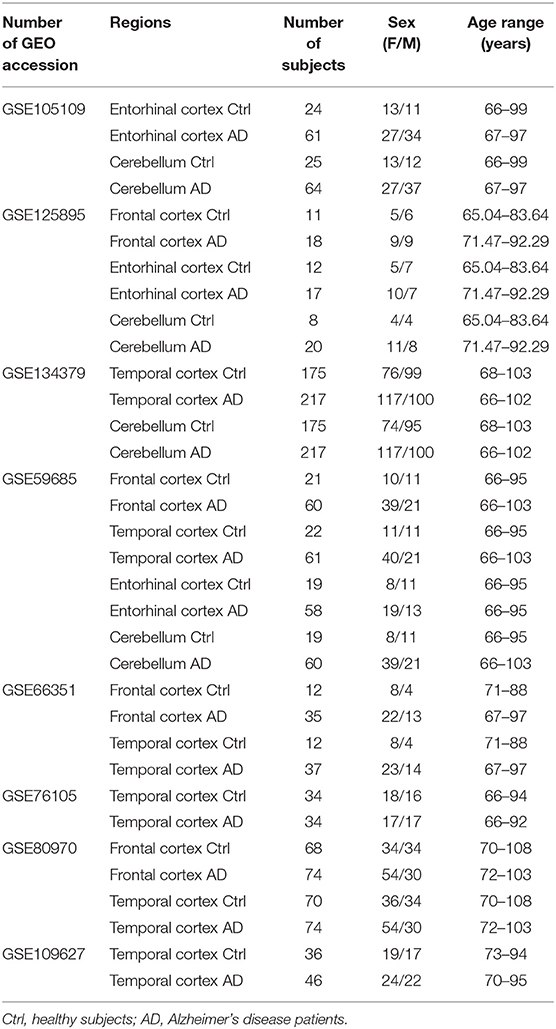
Table 2. Characteristics of the Infinium450k datasets investigated in the present study including AD patients and non-demented control subjects.
Pre-processing
As raw intensities files were not available for some datasets, all the analyses were performed on pre-processed methylation data downloaded from GEO. Potentially ambiguous probes (cross-reactive probes and probes including SNPs; Zhou et al., 2017) were excluded from the analyses. Probes mapping on sex chromosomes were removed, except when the comparison between AD and healthy controls was performed in males and females separately. GSE134379, GSE125895, GSE66351, and GSE76105 did not include probes mapping on sex chromosomes in the pre-processed data downloaded from GEO.
In each dataset, neuron/glia proportions were estimated using Horvath's calculator (Horvath, 2013) which implements the algorithm developed by Guintivano et al. (2013).
For the analysis of 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), and unmethylated cytosine (5uC) in the GSE105109 dataset, we considered only the samples for which both bisulfite (BS) and oxidative bisulfite (oxBS) were available. ERC included 25 healthy subjects (12 females and 13 males) and 57 AD (25 females and 32 males), while CRB included 28 healthy subjects (14 females and 14 males) and 63 AD (26 females and 37 males). OxBS beta values correspond to 5mC levels; 5hmC levels were calculated by subtracting oxBS beta values from BS beta values (BS-oxBS), while 5uC levels were calculated by subtracting BS beta values from 1 (1-BS; Lardenoije et al., 2019). Negative values returning from the difference BS-oxBS were set to a value close to zero (1 × 10−7; Ringh et al., 2019).
Differential Analysis and Meta-Analysis
To identify differentially methylated positions (DMPs), the lmFit function implemented in limma R package (Ritchie et al., 2015) was used to fit a linear model to each microarray probe, expressing DNAm as M-values. Association with age was calculated using age as a continuous value and correcting for sex and neuron/glia proportion. Association with sex was calculated using sex as a categorical value and correcting for age and neuron/glia proportion. Association with AD was calculated using AD as a categorical value and correcting for age, sex and neuron/glia proportion. The lmFit function was used also to calculate the interaction between sex and age, correcting for neuron/glia proportion, and between AD and sex, correcting for age and neuron/glia proportion. Effect sizes and standard errors were extracted from limma output. For each brain region, the results obtained in the different datasets were combined by inverse variance-weighted fixed-effects meta-analysis using METAL software (Willer et al., 2010). Finally, the p-values resulting from each meta-analysis were adjusted for multiple comparisons using the Benjamini-Hochberg (BH) procedure. Only probes with a BH-corrected p-value <0.01 and with concordant effect sizes between all the datasets included in each meta-analysis were retained as significant.
To identify DMPs specific for a certain brain region, we first selected the probes having a BH-corrected p-value <0.01 in one region and a BH-corrected p-value >0.01 in all the other regions; we further refined these lists by selecting the probes having large effect sizes (<5th percentile or >95th percentile) in the brain region under investigation and small absolute effect sizes (<0.1 for sex analysis; <0.001 for age analysis; <0.1 for AD analysis) in all the other regions.
Enrichment and Gene Ontology Analysis
Enrichment of genomic regions (islands, N- and S-shores and shelves, open sea regions) was calculated using Fisher exact test, as implemented in the fisher.test function from the stats R package (p-value <0.05). Enrichment of Gene Ontology (GO) terms was calculated using the methylgometh function implemented in the methylGSA R package (Ren and Kuan, 2019), and redundant significant GO terms (BH-corrected p-value <0.01) were removed by REViGO software (Supek et al., 2011).
Results
The selection criteria of publicly available DNAm datasets of healthy and AD human brains are described in Materials and Methods section, and the datasets included in the meta-analysis are reported in Tables 1, 2. An overview of the study design is reported in Supplementary Figure 1.
DNA Methylation Differences Across Sex
To identify sex-dependent differentially methylated positions (sDMPs) we performed an epigenome wide association study (EWAS) in each dataset and brain region separately, considering healthy subjects and correcting for age and estimated neuron/glia proportion (Section Materials and Methods). We then conducted a meta-analysis within each brain region.
We identified 4,860 sDMPs in FC, 1,985 sDMPs in TC, 159 sDMPs in ERC, and 2,322 sDMPs in CRB (Figures 1A–D, Supplementary Figure 2, and Supplementary File 1).
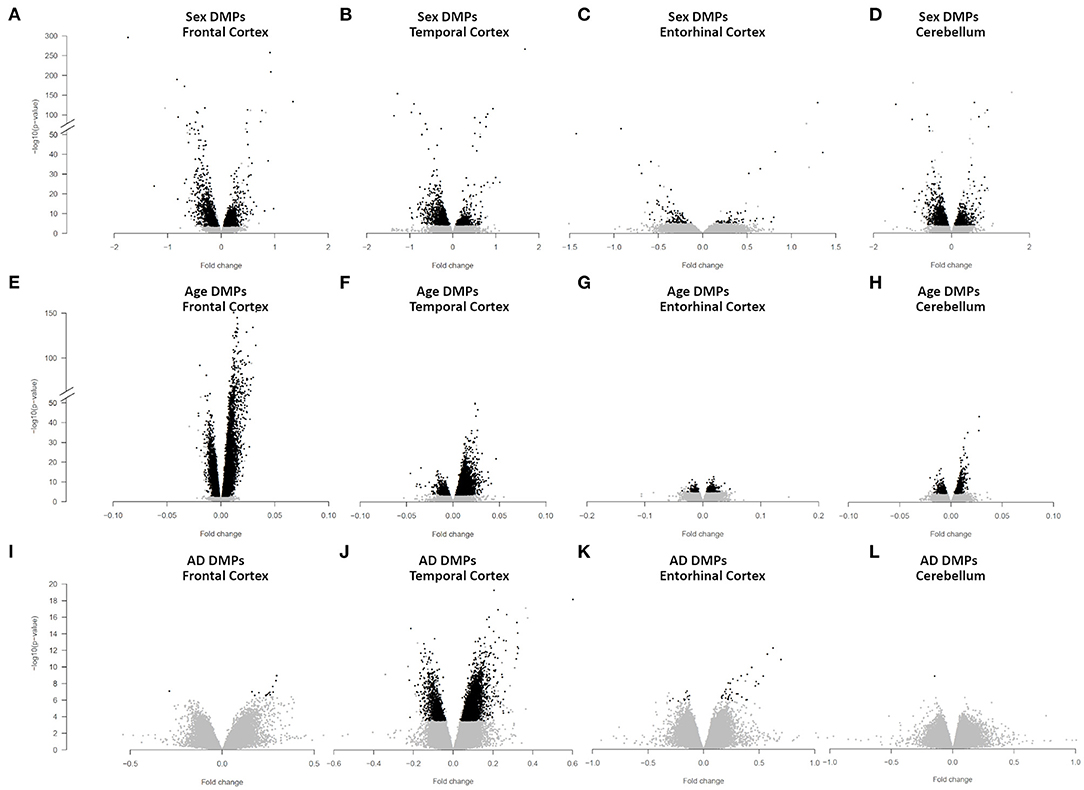
Figure 1. Sex-, age-, and AD-associated epigenetic changes in the four brain regions. Volcano plots of -log10(P-value) against effect sizes, resulting from the meta-analysis of: (i) sex-associated DMPs in FC (A), TC (B), ERC (C), and CRB (D); (ii) age-associated DMPs in FC (E), TC (F), ERC (G), and CRB (H); (iii) AD-associated DMPs in FC (I), TC (J), ERC (K), and CRB (L). Significant probes (BH-corrected p-value <0.01) are colored in black.
In FC, sDMPs were mainly hypermethylated in males compared to females (73% of hypermethylated probes) while the opposite was true for TC, ERC, and CRB (38, 33, and 36% of hypermethylated probes in TC, ERC, and CRB, respectively). When analyzing the genomic context of the sDMPs, we found that CpG islands were enriched in sDMPs in all the four brain regions, and that CpG island shores showed a similar trend (Supplementary File 2). Also the distribution of sDMPs across chromosomes was not random, with a trend toward enrichment in chromosome 19 in all the four brain regions. The enrichment analysis of GO terms did not reveal significant results except for FC, where the “homophilic cell adhesion via plasma membrane adhesion molecules” ontology was found (Supplementary File 2).
To investigate whether sex-dependent DNAm changes were consistent across brain regions, we evaluated the correlation of effect size values between FC, TC, ERC, and CRB (Figure 2A). The four brain regions were positively correlated each other. We next intersected the 4 sDMPs lists, identifying 77 common probes mapping in 57 genes (Figure 2D, Table 3, and Supplementary File 1).
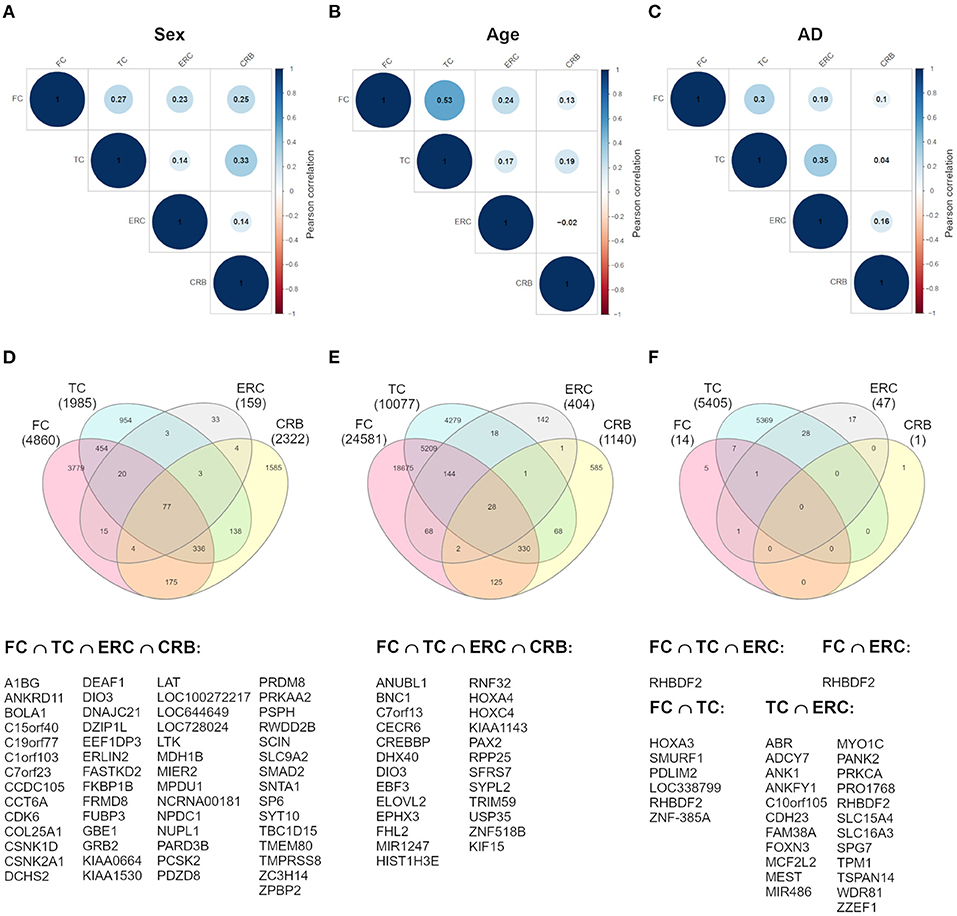
Figure 2. Cross-region analysis of sex-, age-, and AD-associated probes. (A–C) The correlation matrix plots show the magnitude of correlation among probes' effect sizes in the four brain regions, considering the results of the meta-analysis on sex- (A), age- (B), and AD- (C) associated probes. Positive and negative correlation values are indicated in blue and red, respectively. (D–F) The Venn diagrams display the number of significant DMPs shared between the four brain regions, considering sDMPs (D), aDMPs (E), and AD-DMPs (F). The genes in which the most shared probes map are reported below each diagram.
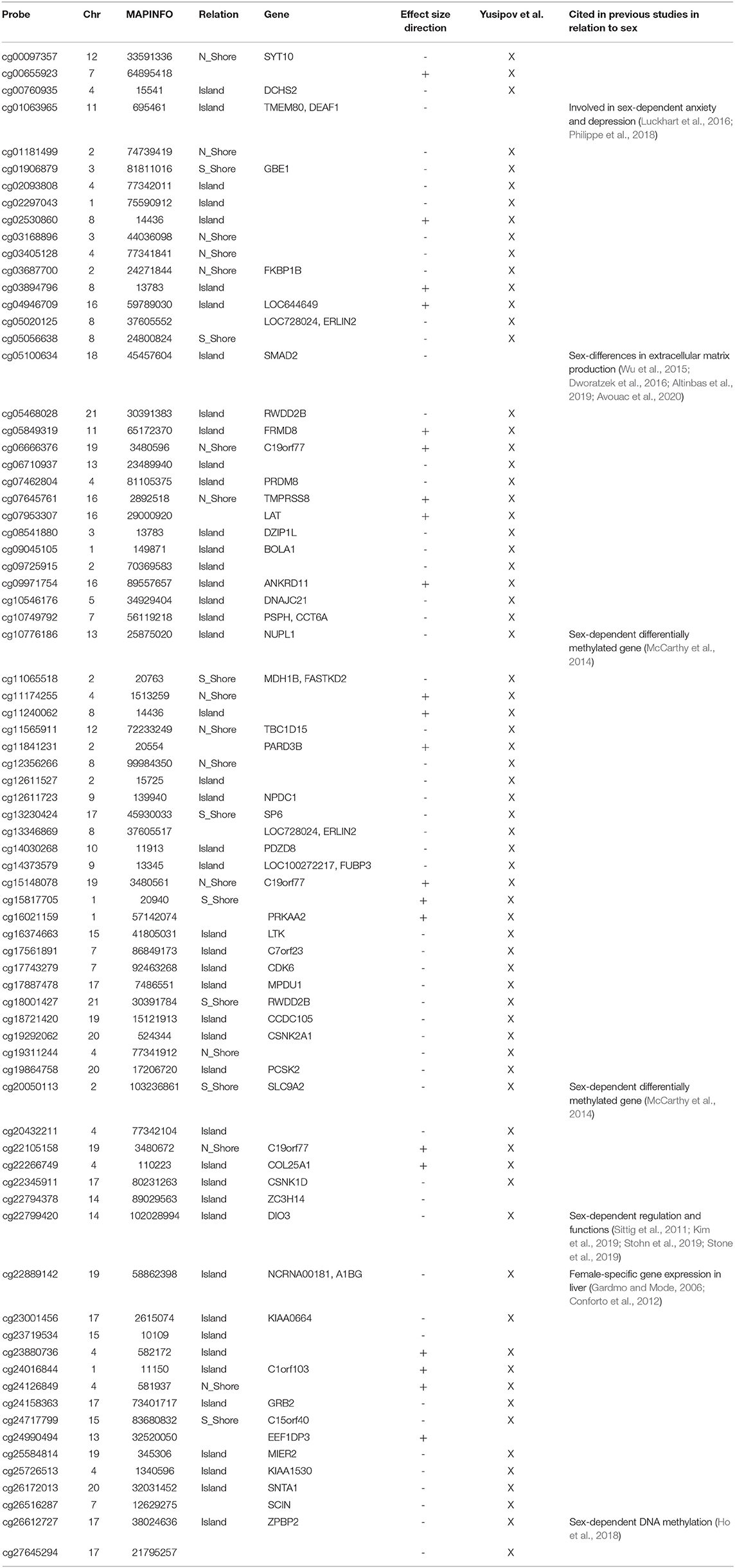
Table 3. sDMPs resulting from cross-region analysis.
All these probes showed concordant sex-dependent DNAm profiles in the four brain regions and most of them (73%) were hypomethylated in males. Furthermore, 93% of them were previously described to have sex-dependent DNAm also in whole blood (Yusipov et al., 2020).
On the other hand, we searched for probes having sex-related DNAm differences only in one brain region (region-specific sDMPs; Section Materials and Methods). We found 2, 4, 0, and 37 region-specific sDMPs in FC, TC, ERC, and CRB, respectively (Supplementary File 1). Interestingly five sDMPs specific for CRB mapped in Nuclear Enriched Abundant Transcript 1 (NEAT1) gene (Figure 3).
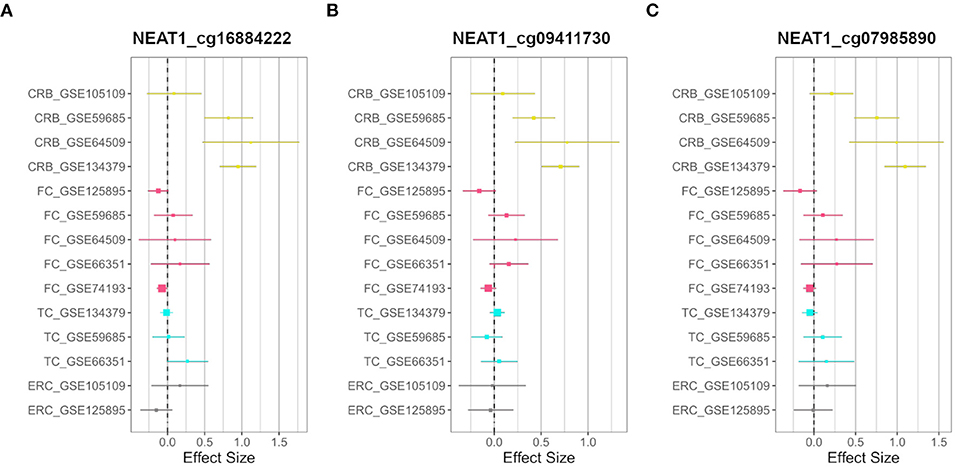
Figure 3. CRB-specific sex-associated DNAm of NEAT1 gene. Forest plots of three CRB-specific sDMPs mapping in NEAT1gene: (A) cg16884222, (B) cg09411730, (C) cg07985890. For each probe, effect sizes from the datasets used for our meta-analysis are reported, dividing them according to the four brain regions (CRB, yellow; FC, magenta; TC, cyan; ERC, gray).
DNA Methylation Changes Across Age
To identify age-dependent differentially methylated positions (aDMPs) we performed an EWAS in each dataset and brain region separately, considering healthy subjects and correcting for sex and estimated neuron/glia proportion (Section Materials and Methods). We then conducted a meta-analysis within each brain region.
We identified 24,581, 10,077, 404, and 1,140 aDMPs in FC, TC, ERC, and CRB, respectively (Figures 1E–H, Supplementary Figure 3E, and Supplementary File 3). In all brain regions, most of the aDMPs underwent hypermethylation with age (76, 88, 58, and 62% of hypermethylated aDMPs in FC, TC, ERC, and CRB, respectively). The genomic context of aDMPs was not consistent across the four brain regions, except for a significant under-representation in “open sea” regions (Supplementary File 4). Similarly, aDMPs were differently scattered across chromosomes in FC, TC, ERC, and CRB. GO enrichment analysis revealed several pathways involved in morphogenesis and developmental processes, with “pattern specification process” and “regionalization” common to FC, TC, and ERC (Supplementary File 4).
The analysis of correlation between the effect sizes revealed that age-associated changes were more similar between FC and TC compared to the other regions (Figure 2B). The intersection of the aDMPs from the 4 brain regions highlighted 28 common probes, all concordantly undergoing hypermethylation with age and mapping in 25 genes (Figure 2E and Table 4). Again, 93% of these probes were reported as age-associated also in while blood (Yusipov et al., 2020).
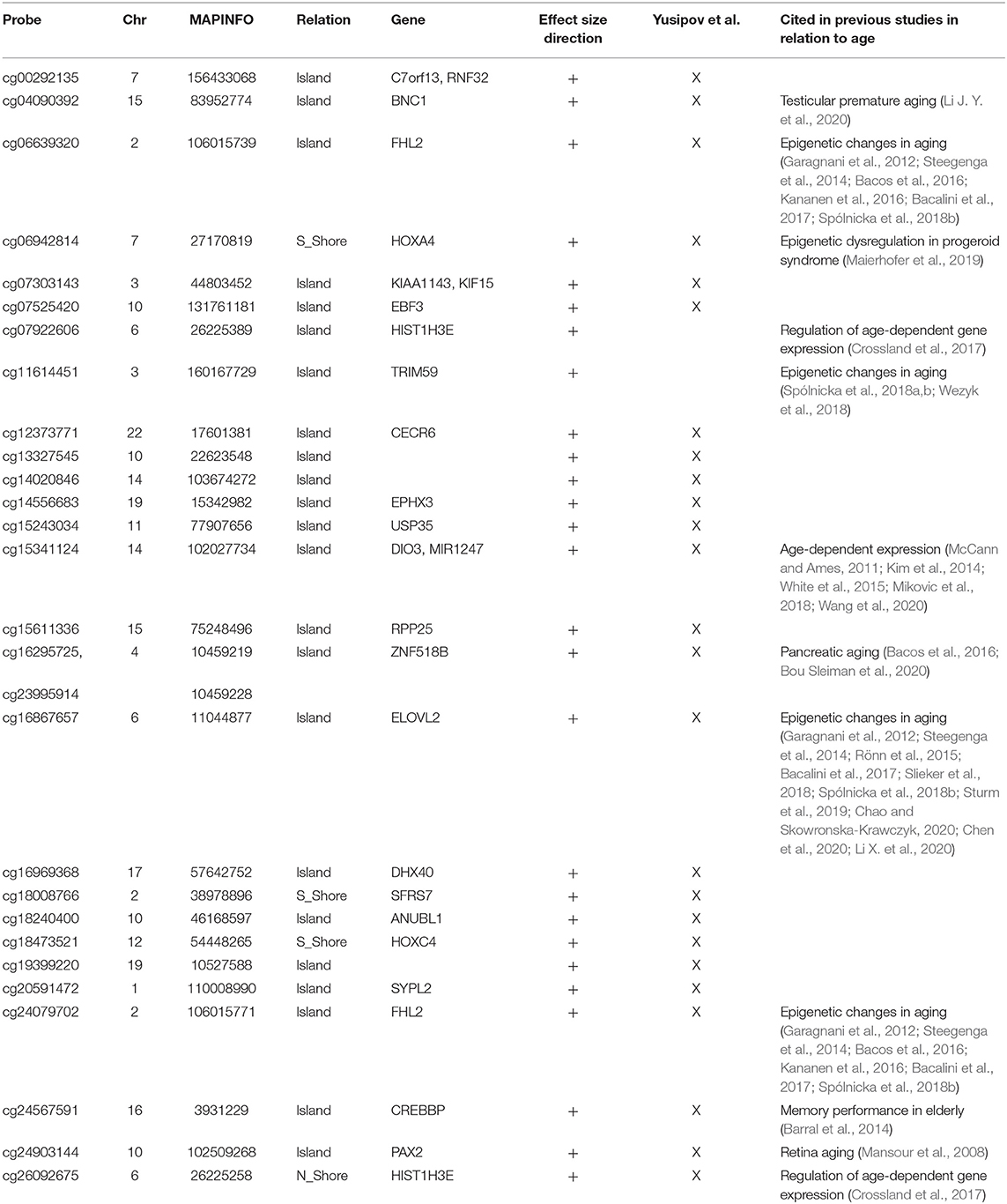
Table 4. aDMPs resulting from cross-region analysis.
The opposite analysis, i.e., the identification of region-specific aDMPs (section Materials and Methods), identified only one probe specific for FC (cg01725130), that maps in the body of Ras And Rab Interactor 3 (RIN3) gene (Supplementary File 2).
The Relation Between Age and Sex in Brain DNA Methylation
We then aimed at studying how sex-specific brain DNAm is modulated during aging.
First of all, we intersected sDMPs and aDMPs lists. In FC, we found 675 probes that change with sex and with age (s&aDMPs), corresponding to about 13% of all sDMPs identified. In TC s&aDMPs were 171, corresponding to 8.5% of sDMPs. In ERC we found only 2 s&aDMPs, while in CRB s&aDMPs were 19, corresponding to 4% of sDMPs (Figure 4 and Supplementary Files 1, 3). In all the four regions, the proportion of sDMPs changing with age (i.e., the proportion of s&aDMPs) was higher than expected (Fisher's Exact Test p-value <0.05; odds ratio of 2.6, 3.8, 13.0, and 3.0 in FC, TC, ERC, and CRB, respectively). In FC, TC, and CRB, most of the s&aDMPs were probes having higher DNAm levels in males respect to females and undergoing hypermethylation during aging. GO analysis revealed only one ontology enriched in FC (“homophilic cell adhesion via plasma membrane adhesion molecules”).
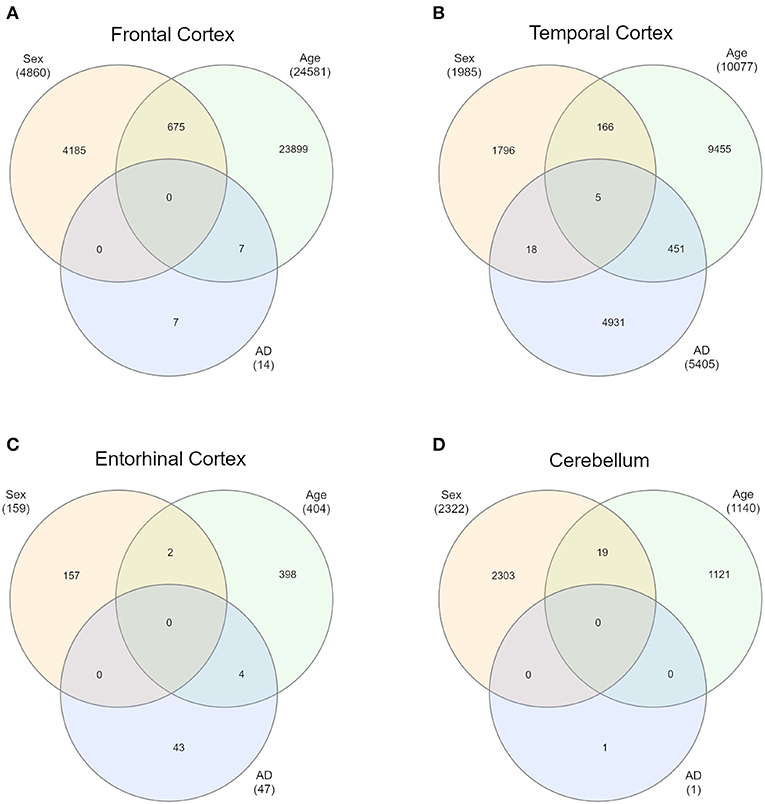
Figure 4. Intersections of sex-, age-, and AD-associated probes in each of the four brain regions. Venn diagrams depict the intersection between sDMPs, aDMPs, and AD-DMPs in FC (A), TC (B), ERC (C), and CRB (D).
The previous analysis identifies CpG probes whose DNAm varies according to both sex and age, but is not informative about possible differences in aging trajectories between males and females. To fulfill this point, we performed an age-by-sex interaction analysis in each dataset (Section Materials and Methods) and meta-analyzed the results for the four brain regions. Only 4, 4, 2, and 2 probes showed a significant age-by-sex interaction in FC, TC, ERC, and CRB, respectively (Supplementary File 5).
Brain DNA Methylation Changes Across AD
Then, we focused on brain DNAm datasets including late-onset AD patients and age-matched non-demented controls.
To identify differentially methylated positions associated with AD (AD-DMPs) we performed an EWAS in each dataset and brain region separately, correcting for age, sex, and estimated neuron/glia proportion (section Materials and Methods). We then conducted a meta-analysis within each brain region.
We identified 14 AD-DMPs in FC, 5405 in TC, 47 in ERC, and only 1 in CRB (Figures 1I–L, Supplementary Figure 4, and Supplementary File 6). In all brain regions most of AD-DMPs were hypermethylated in AD compared to controls (93, 80, 76, and 100% in FC, TC, ERC, and CRB, respectively). While in TC AD-DMPs were significantly under-represented in CpG islands and enriched in the other genomic contexts, a significant enrichment in CpG islands was found for AD-DMPs identified in FC (Supplementary File 7). GO analysis returned significant results only in TC, where pathways related to synapse organization and function were found (Supplementary File 7).
Correlation analysis of effect sizes between the four brain regions highlighted a distinctive pattern in CRB respect to FC, TC, and ERC, while the correlation was higher between TC and ERC (Figure 2C). Accordingly the intersection between AD-DMPs in the 4 brain regions did not return common probes, while 29 probes (mapping in 23 genes) and 8 probes (mapping in 6 genes) were identified by intersecting TC and ERC or FC and TC, respectively (Figure 2F and Table 5). The probe cg12163800, mapping in Rhomboid 5 Homolog 2 (RHBDF2) gene, was significantly hypermethylated in FC, TC, and ERC from AD patients. A comparison with AD-associated probes retrieved in a recent meta-analysis (Smith R. G. et al., 2020) is also reported in Table 5.
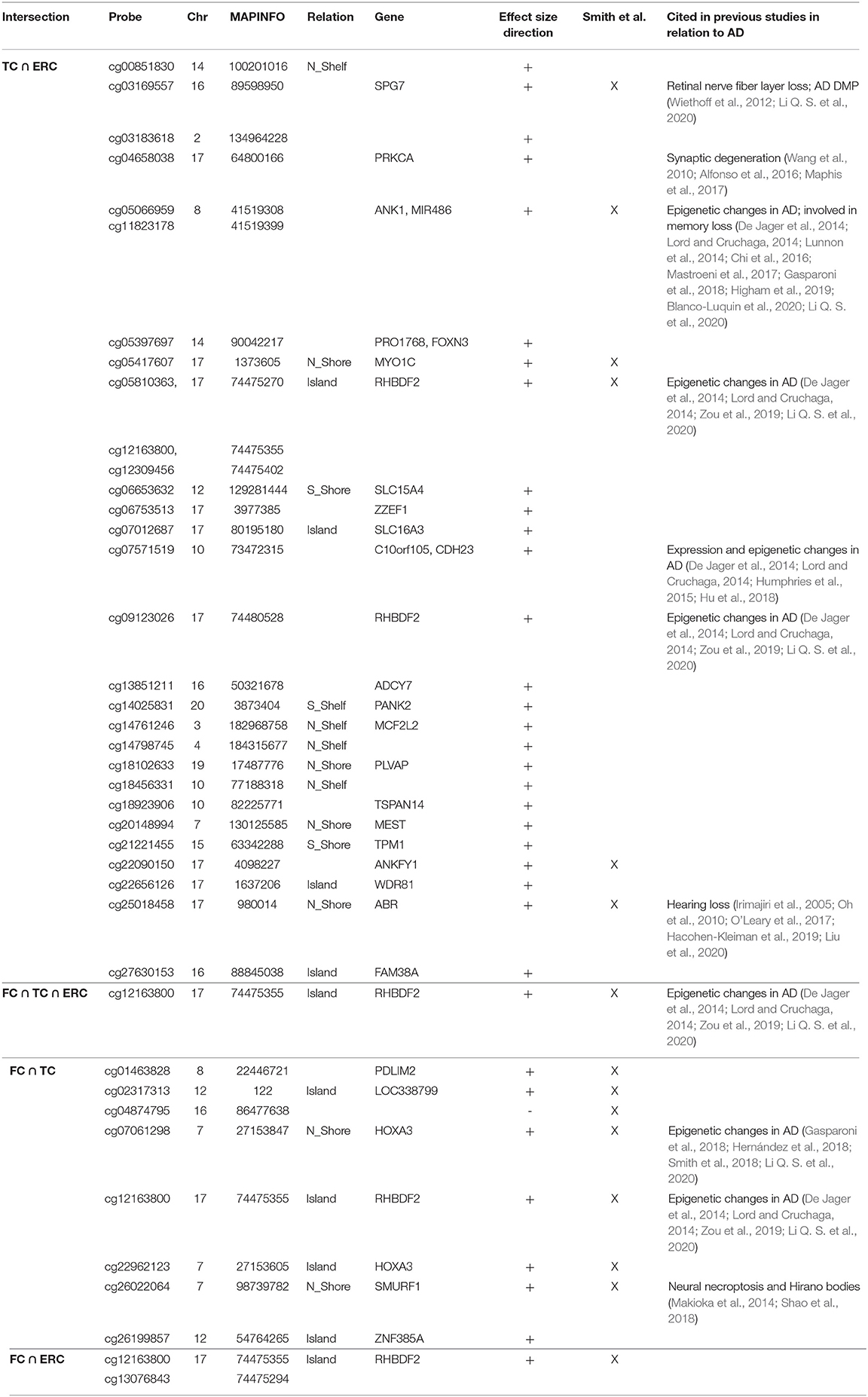
Table 5. AD-DMPs resulting from cross-region analysis.
Confirmation of Sex- and Age-Associated DNAm Changes in AD Subjects
We investigated whether the sDMPs and aDMPs identified above in the different brain regions from healthy controls were confirmed also in AD patients. To this aim, we evaluated their association with sex (correcting for age and estimated neuron/glia proportion) or with age (correcting for sex and estimated neuron/glia proportion) considering DNAm data from AD samples, and performed a meta-analysis in each brain region. The effect sizes obtained in AD were highly correlated with those previously obtained in healthy controls (Supplementary Figure 5). This correlation was slightly lower for aDMPs, which is expected considering that in most datasets the age range tends to be narrower for AD samples compared to healthy controls. Interestingly, also in AD samples we found an enrichment of sDMPs on chromosome 19 (data not shown). Collectively, these results indicate that sex- and age-dependent DNAm patterns are largely reproduced in AD samples.
The Relationship Between Sex- and Age-Associated DNAm Changes and AD Epigenetic Remodeling
We explored whether AD-associated DNAm changes were related to sex- and age-specific brain DNAm patterns occurring in physiological conditions, identified in the analyses described above.
In each brain region, we intersected the AD-DMPs and sDMPs in order to identify AD&sDMPs, i.e., probes that have basal differential DNAm between the two sexes and are also affected by AD. The intersection did not result in any probe for all the regions except that for TC, where we found 23 AD&sDMPs, mapping in 16 genes and corresponding to only 0.4% of AD-DMPs in TC (Fisher's Exact Test p-value >0.05; Figure 4 and Supplementary Files 1, 6). Moreover, AD-by-sex interaction analysis yielded no significant probes in any region.
To further explore the epigenetic relationship between sex and AD, we extended our analysis to probes located on sex chromosomes and focused on AD datasets in which their DNAm values were available (Section Materials and Methods). For each dataset, we considered males and females separately, we repeated the EWAS for AD-associated DNAm and we performed the meta-analysis within each brain region. We then searched for significant AD-DMPs located on the X or Y chromosomes. This analysis returned only few probes: four X-linked DMPs were found in TC when males with and without AD were compared, while one X-linked probe was found in male ERC (Supplementary File 6).
Similarly, we explored whether AD-DMPs occur in probes whose DNAm varies during physiological aging (AD&aDMPs). The intersection between AD-DMPs and aDMPs highlighted 7, 456, 4, and 0 probes in FC, TC, ERC, and CRB, respectively (Figure 4). The proportion of AD&aDMPs was higher than expected by chance in FC, TC, and ERC (Fisher's Exact Test p-value <0.05; odds ratio of 15.9, 3.8, and 95 in FC, TC, and ERC, respectively). We found that 87% of AD&aDMPs in TC are concordant for the effect size sign between aDMPs and AD-DMPs, while this percentage reached 100% in FC and ERC. Notably, the four AD&aDMPs found in ERC (cg11823178, cg03169557, cg25018458, and cg22090150) were also found in TC (Table 6). Also the intersection between AD&aDMPs in TC and FC returned four common probes (cg01463828, cg04874795, cg22962123, and cg07061298; Table 6). Figure 5 reports DNAm values of cg11823178 (ANK1) and cg22962123 (PDLIM2) in TC from GSE134379 dataset as an example of CpG sites displaying a positive association of DNAm with age and hypermethylated in AD.
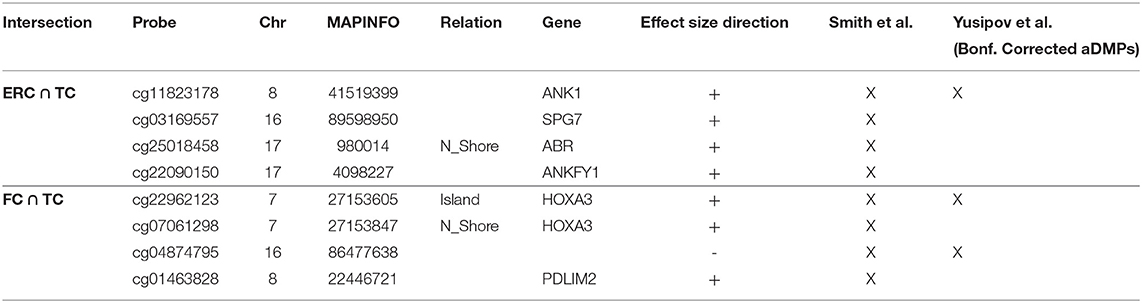
Table 6. AD&aDMPs resulting from cross-region intersections.
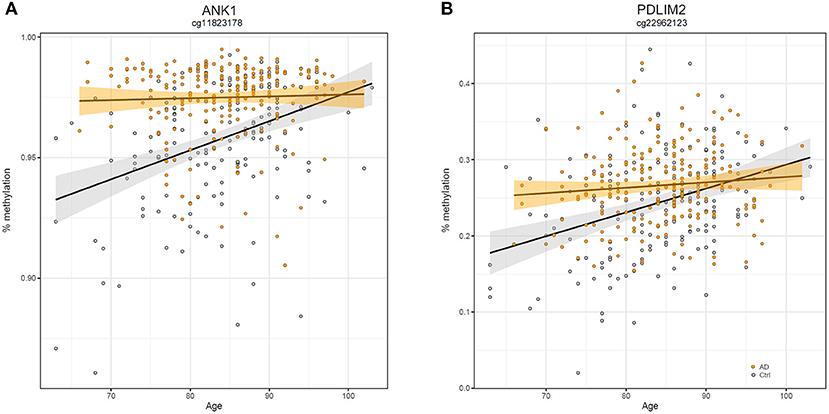
Figure 5. Scatter plots of ANK1 and PDLIM2 DNAm according to age and disease. Scatter plots of methylation values of cg11823178 within ANK1 (A) and of cg22962123 within PDLIM2 (B) in TC from GSE134379 dataset. Healthy subjects are colored in gray while AD patients are in orange. Regression lines and confidence intervals within each group are reported.
Finally, it is worth to note that TC is the only brain region in which we found probes at the intersection between aDMPs, sDMPs, and AD-DMPs (AD&a&sDMPs; Figure 4B). The five probes (cg20225999, cg03951603, cg08820801, cg22263793, cg10828284; Table 7) were all hypermethylated in males and with aging; three of them (cg20225999, cg08820801, cg10828284) were further hypermethylated in AD.

Table 7. Probes resulting from the intersection between aDMPs, sDMPs, and AD-DMPs in TC.
Contribution of 5-hydroxymethylcytosine to the Epigenetic Changes Across Sex, Age, and AD
All the analyses reported above are based on microarray data from BS converted DNA. BS treatment does not allow to distinguish between 5mC and 5hmC, another epigenetic mark which plays an important role especially in the brain (Kriaucionis and Heintz, 2009; Lunnon et al., 2016). On the contrary, the combination of BS with oxBS treatment allows discriminating the levels of 5mC, 5hmC, and 5uC in DNA (Booth et al., 2012). One of the datasets that we used in our meta-analysis for sex-, age-, and AD-associated epigenetic changes (GSE105109) includes microarray results from matched BS- and OxBS-treated ERC and CRB samples. We calculated the levels of 5mC, 5hmC, and 5uC (Section Materials and Methods) in this dataset and we analyzed them for the association with sex (in healthy subjects), with age (in healthy subjects), and with AD (comparing AD and healthy subjects). This analysis did not return any significant probe, neither in ERC nor in CRB. We then considered the lists of sDMPs, aDMPs, and AD-DMPs identified in the meta-analysis of ERC and CRB datasets, and used GSE105109 data to investigate the contribution of 5mC, 5hmC, and 5uC to the observed epigenetic changes. Supplementary Figure 6 reports the correlation between the effect size values resulting from the meta-analysis of sex, age, and AD, and the effect size values obtained in GSE105109 dataset using 5mC, 5hmC, and uC values in the association analysis. In both ERC and CRB, sDMPs and AD-DMPs showed high correlation between BS (5mC+5hmC) results and oxBS (5mC) results, while the correlation with 5hmC results was low. A similar trend was observed for aDMPs in CRB. This indicates that 5mC is the main contributor to the epigenetic changes observed for the sDMPs and the AD-DMPs in ERC and CRB, and for the aDMPs in CRB. On the contrary, for ERC aDMPs, BS-effect sizes were similarly correlated with 5mC- and 5hmC-effect sizes, indicating that both the epigenetic marks are remodeled during aging in this brain region. Furthermore, age-associated changes in 5mC and 5hmC were likely to involve different probes, as 5mC and 5hmC effect sizes were not clearly correlated.
Discussion
Sex and age are among the major risk factors for AD. In this paper, we performed a meta-analysis of DNAm changes that are associated to sex and aging in four brain regions (FC, TC, ERC, CRB) and we evaluated whether they contribute to the epigenetic alterations that have been widely described in AD. Our main findings are discussed in the following paragraphs.
Sex-Dependent DNAm Differences Tend to Be Shared Between Brain Regions, With Few Exceptions
To date some studies have reported DNAm sex differences in human brain, mainly focusing on frontal cortex (Xu et al., 2014; Spiers et al., 2015; Masser et al., 2017; Perzel Mandell et al., 2020) with few exceptions (Xia et al., 2019). Our meta-analysis confirms the presence of autosomic probes with differential methylation between males and females in all the brain regions. These probes preferentially map in CpG islands and shores suggesting their involvement in the regulation of sex-specific gene expression in brain (Xu et al., 2014). Surprisingly, in all the brain regions we found an enrichment of sDMPs in chromosome 19. This observation is difficult to be explained but a similar result was observed in a precedent study on sex-associated DNAm differences across childhood in whole blood (Suderman et al., 2017). Chromosome 19 has the highest content of CpG sites and genes in the genome (Grimwood et al., 2004), and seems to be involved in the process of X chromosome inactivation (Migeon et al., 2017).
Sex specific DNAm tended to be reproducible across the brain regions and 77 CpGs resulted from the cross-region intersection. Among them there are sDMPs mapping in genes that have been already associated to sex differences in brain physiology and pathology, like Par-3 Family Cell Polarity Regulator Beta (PARD3B) (Phillips et al., 2019), DEAF1 Transcription Factor (DEAF1) (Luckhart et al., 2016), and Iodothyronine Deiodinase 3 (DIO3) (Stohn et al., 2019) genes. Most of these 77 probes were previously reported as differentially methylated between males and females also in previous meta-analysis on blood (McCarthy et al., 2014; Yusipov et al., 2020).
In addition, we found few examples of sDMPs specific for a brain region. The most notable example is in cerebellum and maps in NEAT1. NEAT1 is a ubiquitously expressed long non-coding RNA (lncRNA) involved in a plethora of neurospecific processes such as brain development and aging (An et al., 2018; Pereira Fernandes et al., 2018; Salvatori et al., 2020). Recent transcriptomic studies on human central nervous system revealed altered NEAT1 levels in AD (Spreafico et al., 2018), PD (Simchovitz et al., 2019), and in schizophrenia (Katsel et al., 2019).
DNAm Tends to Be Differently Remodeled During Aging According to the Brain Region
Several studies have analyzed age-associated changes in DNAm in brain, both comparing fetal vs. adult brains and analyzing methylation profiles across adulthood (Hernandez et al., 2011; Horvath et al., 2012; Numata et al., 2012; Day et al., 2013; Jaffe et al., 2016; Gasparoni et al., 2018; Price et al., 2019). Our meta-analysis shows that during aging there is an increase in methylation at specific loci, accordingly to previously published data on blood (Xiao et al., 2016; Yusipov et al., 2020) and brain (Hernandez et al., 2011). As previously reported by Hernandez et al. (2011), also our results support the involvement of brain aDMPs in GO related to developmental processes and morphogenesis. Furthermore, our meta-analysis confirms and extends the observation that the epigenome is differently remodeled during aging across brain regions (Hernandez et al., 2011). In particular we observed that age-associated DNAm patterns are similar in TC and FC, while they are distinct in ERC and CRB. CRB was previously described to undergo a peculiar epigenetic aging, which was decelerated according to Horvath's epigenetic clock (Horvath et al., 2015).
The large fraction (93%) of the 28 aDMPs emerged from our cross region analysis was found also in aging studies on blood (Yusipov et al., 2020). Among them there are probes mapping in Four And A Half LIM Domains 2 (FHL2) and ELOVL Fatty Acid Elongase 2 (ELOVL2) genes, previously reported as age-associated in a large number of studies on several tissues (Garagnani et al., 2012; Slieker et al., 2018) including sorted neuron and glia cells (Gasparoni et al., 2018). According to what discussed above and to previous results (Bacalini et al., 2017; Slieker et al., 2018) the effect size of ELOVL2 probe cg16867657 was lower in CRB respect the other regions, but still significant in our meta-analysis. Elovl2 is an enzyme involved in the elongation of fatty acids and its functional role in aging has been recently suggested (Chao and Skowronska-Krawczyk, 2020).
Sites With Sex-Dependent DNAm Are Similarly Modulated During Aging in Males and Females
Previous studies in mice and humans suggested that, while sex-differences in DNAm at certain CpG sites are maintained during life, other CpG sites show sexually divergent aging patterns, i.e., they have a different response to aging in males and females (Masser et al., 2017). Our meta-analysis supports the fact that sDMPs have a high propensity to be modulated during aging, as the number of probes resulting from the intersection of sDMPs and aDMPs is higher than expected in all the four brain regions. However, we found only few probes with significant age-by-sex interaction, indicating similar rather than diverging changes in DNAm in males and females aging. The discrepancy between our results and previous findings can be due to different reasons: for example, while Masser et al. considered only one dataset including frontal cortex data, here we meta-analyzed several datasets using selective criteria of concordance between all datasets from the same brain region; furthermore, we applied a filtering step that removed potentially ambiguous probes, thus reducing the potential overlap with Masser's results. Our results are more similar to what reported by two independent studies in blood (McCartney et al., 2019; Yusipov et al., 2020) that showed that only a small fraction of CpGs have significant age-by-sex interaction. Further studies on larger cohorts are needed to better describe sex-dependent DNAm patterns during brain aging.
Epigenetic Changes in AD Are Enriched in Sites That Show Age-Dependent DNAm
A recent meta-analysis on EWAS studies identified 220 CpGs associated with AD neuropathology, shared by brain cortical cortex regions but not by CRB (Smith et al., 2019). The paper by Smith et al. included several datasets that we used also in our meta-analysis, with the exception of GSE125895 and GSE109627, while we did not have access to the ROS/MAP and RBD DNAm data. Furthermore, while Smith et al. considered the association with Braak stage, here we used the disease as a binary trait (affected/unaffected). Despite these differences, our results largely overlap with those previously reported. In particular, we did not find AD-related probes common to all the four brain regions that we investigated, with CRB DNAm less affected by the pathology. On the contrary, a subset of sites was shared between FC, TC, and ERC, and about 50% of these probes overlap with published data. These probes map within genes whose epigenetic deregulation has been largely documented in AD, including ANK1, RHBDF2, and HOXA3. On the contrary, we did not find any overlap when comparing our results on AD brain with CpG sites identified in AD patients' blood (Roubroeks et al., 2020), confirming that the pathology differently affects the two tissues as recently reported (Wei et al., 2020).
We did not find a significant overlap between AD-DMPs and sDMPs, nor significant interaction effects between sex and AD. Overall these results suggest that AD does not predominantly insist on autosomic sites with sex-specific DNAm. Similarly, when we repeated our analysis including probes on X and Y chromosomes (analyzing males and females separately) we found limited evidence of differential DNAm between AD and controls in sex chromosomes. Collectively these results suggest that no profound sex-associated DNAm remodeling occurs in AD. However, we cannot rule out that more subtle epigenetic differences exist, both on autosomes and sex chromosomes. It is possible that these differences did not emerge from our meta-analysis, due to the stringent selection criteria that we applied or to the small sample sizes when males and females were considered separately. Further studies should investigate possible epigenetic contributions to the different AD risk between the two sexes.
Conversely, our data show that in FC, TC and ERC, AD-related epigenetic modifications are significantly enriched in probes whose DNAm varies with age. Strikingly, we found a high concordance between the direction of DNAm changes (hyper or hypo-methylation) in AD&aDMPs, indicating that a subset of age-associated DNAm changes is exaggerated in AD. In TC, AD&aDMPs included probes mapping in ANK1, and it is worth to note that the down-regulation of Ank2 (ANK1 human ortholog gene) in Drosophila has been associated to memory loss, neuronal dysfunction and shortened lifespan in a recent report (Higham et al., 2019). Other interesting genes emerged from our analysis. Paraplegin (SPG7) mutation leads to shortened lifespan, environmental stress. and muscular and neuronal degeneration in Drosophila (Pareek et al., 2018). Mov10 Like RISC Complex RNA Helicase 1 (MOV10L1) is a putative germline-specific RNA helicase whose expression has been recently reported to be tightly correlated with brain development, aging and AD neurodegeneration (Skariah et al., 2017; Srinivasan et al., 2020).
Overall, these results support a geroscience view (Kennedy et al., 2014; Sierra, 2020) according to which AD can be considered a deviation of the physiological aging trajectories toward accelerated aging. Epigenetic age acceleration was previously reported in AD neurons, where a pronounced loss of CpH methylation was found at enhancers, similar to what observed in aging, and in bulk prefrontal cortex, where epigenetic age calculated by Horvath's clock was positively associated with neuritic plaques and amyloid load (Levine et al., 2015). It will be interesting to know whether similar results will be obtained using the recently published epigenetic clock optimized for brain tissues (Shireby et al., 2020).
Strengths, Limitations, and Conclusions
To the best of our knowledge, this is the first report in which sex-, age-, and AD-related DNAm changes are systematically assessed using the same analytical approach. We used stringent selection criteria that enabled to select only probes with concordant DNAm changes in the different datasets. Furthermore, we considered multiple brain regions and reported similarities and differences in their epigenetic remodeling. Previous studies showed that DNAm patterns differ between brain regions and that they may play a role in brain development and functional specialization (Ladd-Acosta et al., 2007; Rizzardi et al., 2019). These “baseline” DNAm differences can mediate disease mechanisms that are specific for certain brain regions (Rizzardi et al., 2019), and can be further modified across lifespan and in response to pathological conditions. Accordingly, brain areas are differently affected during aging and/or in AD onset and progression (Peters, 2006; Coupé et al., 2019), and also other molecular layers like transcriptomics and proteomics show region-specific changes (Patel et al., 2019; Xu et al., 2019).
On the other side, our study has some limitations. The datasets that we meta-analyzed largely vary in size and age range of the assessed subjects, an important aspect for the identification of aDMPs. Moreover, our meta-analysis included data on BS-treated DNA and it was therefore not possible to distinguish 5mC from 5hmC, an epigenetic modification that contributes to both brain function and neurodegeneration (Coppieters et al., 2014; Ellison et al., 2017; Lardenoije et al., 2019; Smith et al., 2019). The analysis of the dataset in which 5mC and 5hmC were distinguishable (thanks to the simultaneous analysis of BS- and oxBS-treated DNA) suggested that the contribution of 5hmC to sDMPs and AD-DMPs tended to be small. A recent study showed that in fetal brain autosomal 5hmC levels did not differ between males and females (Spiers et al., 2017), in accordance to our results. Global changes in 5hmC have been reported to occur in AD (Chouliaras et al., 2013; Condliffe et al., 2014; Coppieters et al., 2014), while microarray-based genome wide studies identified a limited set of CpG sites whose hmC levels are associated to the disease (Lardenoije et al., 2019; Smith et al., 2019). For example, Smith et al. reported that AD-associated hypermethylation of ANK1 detected on BS-treated DNA is not due to an increase in 5hmC levels, which on the contrary decreased in the disease (Smith et al., 2019). Similarly, in our analysis of AD in ERC most of the significant associations were due to changes in 5mC and we did not observe an evident co-variation of 5mC and 5hmC. On the contrary, when considering aDMPs in ERC we found a contribution of both 5mC and 5hmC, and the two epigenetic marks tended to involve different CpG sites. Age-associated increase in 5hmC levels has been previously reported (Chouliaras et al., 2012). Although potentially interesting, these results are based on only one dataset, and the analysis of the coordinated regulation of brain 5mC and 5hmC across sex, age, and AD deserves further studies. Another limitation of our study is that the datasets that we meta-analyzed were based on bulk brain tissues. Although all the analyses were corrected for neuron/glia proportions predicted from DNAm data, we cannot exclude that the observed sex-, age-, and AD-associated DNAm changes are at least in part driven by changes in brain cells composition that occur in physiological and pathological conditions. For example, Gasparoni et al. reported that ANK1 deregulation in AD is specific for glial cells (Gasparoni et al., 2018), a finding further supported by gene expression studies (Mastroeni et al., 2017), and that the epigenetic profiles of neurons and glia are differently modulated during aging. Notwithstanding, our results suggest that the (cell-specific) age-associated remodeling of DNAm is not just a confounding factor for the epigenetic deregulation observed in AD, but on the contrary, it is the predisposing milieu in which AD pathogenetic mechanisms are established.
In conclusion, we suggest that age-associated DNAm patterns concur to the epigenetic deregulation observed in AD, providing new insights on how advanced age enables neurodegeneration.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
CPe, CPi, CS, MI, DM, RL, CF, PC, PG, and MGB contributed to the conception and design of the study. CPe, CS, IY, KK, DD, and MGB organized the datasets. CPe, CPi, CS, FR, IY, KK, AK, and MGB performed the statistical analysis. MGB, CPe, and CPi wrote the manuscript. All authors contributed to manuscript revision, and read and approved the submitted version.
Funding
We acknowledge support by the grant of the Ministry of Education and Science of the Russian Federation Agreement No. 075-15-2019-871. This work was supported by the European Union (EU) H2020 Project PROPAG-AGEING (grant agreement 634821) and by the EU JPND ADAGE.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.639428/full#supplementary-material
Supplementary Figure 1. Overview of the meta-analysis. For simplicity, the age-by-sex and AD-by-sex interaction analyses are not reported, but they were performed using the same pipeline illustrated in this scheme.
Supplementary Figure 2. Manhattan plots of sDMPs in the four brain regions. The figure displays the Manhattan plots resulting from the meta-analysis of sex-associated probes in FC (A), TC (B), ERC (C), and CRB (D). Significant sDMPs are marked with dark color. Scale change across 50 is indicated by an axis break.
Supplementary Figure 3. Manhattan plots of aDMPs in the four brain regions. The figure displays the Manhattan plots resulting from the meta-analysis of age-associated probes in FC (A), TC (B), ERC (C), and CRB (D). Significant aDMPs are marked with dark color. Scale change across 50 is indicated by an axis break.
Supplementary Figure 4. Manhattan plots of AD-DMPs in the four brain regions. The figure displays the Manhattan plots resulting from the meta-analysis of AD-associated probes in FC (A), TC (B), ERC (C), and CRB (D). Significant AD-DMPs are marked with dark color.
Supplementary Figure 5. Confirmation of sDMPs and aDMPs in AD patients. The scatter plots report, for the sDMPs and aDMPs identified in each tissue in healthy subjects, the effect sizes obtained in healthy subjects against the effect sizes resulting from the meta-analysis in AD patients. Pearson Correlation coefficient is reported in each plot.
Supplementary Figure 6. Contribution of 5hmC to the epigenetic changes across sex, age, and AD. Correlation plots of the effect sizes of sDMPs, aDMPs, and AD-DMPs identified in ERC and CRB, calculated using BS values (5mC+5hmC), oxBS values (5mC), BS-oxBS values (5hmC), and 1-BS values (5uC). Absolute correlation values are reported.
Supplementary File 1. Significant sDMPs in the four brain regions. The tables report the lists of sDMPs for each brain region (FC, TC, ERC, and CRB). Probes resulting from the analysis of cross-region and region-specific sDMPs are indicated by a cross, together with the probes that are in common with aDMPs or AD-DMPs found in the same region.
Supplementary File 2. Enrichment analysis of sDMPs. The tables report: (1) the results of Fisher's test on genomic distribution of sDMPs for each brain region, considering genomic context and chromosomal location. Significant results (p-value <0.05) are colored in green or red if depleted or enriched, respectively. (2) the results of GO pathway enrichment analysis, after REViGO filtering. Only the significant results (adjusted p-value <0.01) for each brain region are reported.
Supplementary File 3. Significant aDMPs in the four brain regions. The tables report the lists of aDMPs for each brain region (FC, TC, ERC, and CRB). Probes resulting from the analysis of cross-region and region-specific sDMPs are indicated by a cross, together with the probes that are in common with sDMPs or AD-DMPs found in the same region.
Supplementary File 4. Enrichment analysis of aDMPs. The tables report: (1) the results of Fisher's test on genomic distribution of aDMPs for each brain region, considering genomic context and chromosomal location. Significant results (p-value <0.05) are colored in green or red if depleted or enriched, respectively. (2) the results of GO pathway enrichment analysis, after REViGO filtering. Only the significant results (adjusted p-value <0.01) for each brain region are reported.
Supplementary File 5. Probes with significant age-by-sex interaction in the four brain regions. The tables report the lists of probes with significant age-by-sex interaction in each brain region (FC, TC, ERC, and CRB).
Supplementary File 6. Significant AD-DMPs in the four brain regions. The tables report the lists of AD-DMPs for each brain region (FC, TC, ERC, and CRB). Probes resulting from the analysis of cross-region and region-specific AD-DMPs are indicated by a cross, together with the probes that are in common with sDMPs or aDMPs found in the same region. The sheet “Sex Chromosomes” reports the significant AD-DMPs identified on sex chromosomes when analyzing males and females separately.
Supplementary File 7. Enrichment analysis of AD-DMPs. The tables report: (1) the results of Fisher's test on genomic distribution of AD-DMPs for each brain region, considering genomic context and chromosomal location. Significant results (p-value <0.05) are colored in green or red if depleted or enriched, respectively. (2) The results of GO pathway enrichment analysis, after REViGO filtering. Only the significant results (adjusted p-value <0.01) for each brain region are reported.
Abbreviations
DNAm, DNA methylation; AD, Alzheimer's disease; DMPs, differentially methylated positions; EWAS, epigenome-wide association study; GO, gene ontology; sDMPs, sex-associated differentially methylated positions; aDMPs, age-associated differentially methylated positions; s&aDMPs, sex-, and age-associated differentially methylated positions; AD&aDMPs, late onset Alzheimer's disease-specific age-associated differentially methylated positions; AD&sDMPs, late onset Alzheimer's disease-specific sex-associated variably methylated positions; AD&a&sDMPs, late onset Alzheimer's disease-specific sex- and age-associated variably methylated positions; 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; 5uC, unmethylated cytosine; BS, bisulfite; oxBS, oxidative bisulfite.
References
Alfonso, S. I., Callender, J. A., Hooli, B., Antal, C. E., Mullin, K., Sherman, M. A., et al. (2016). Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer's disease. Sci Signal. 9:ra47. doi: 10.1126/scisignal.aaf6209
Altinbas, L., Bormann, N., Lehmann, D., Jeuthe, S., Wulsten, D., Kornak, U., et al. (2019). Assessment of bones deficient in fibrillin-1 microfibrils reveals pronounced sex differences. Int. J. Mol. Sci. 20:6059. doi: 10.3390/ijms20236059
Altuna, M., Urdánoz-Casado, A., Sánchez-Ruiz de Gordoa, J., Zelaya, M. V., Labarga, A., Lepesant, J. M. J., et al. (2019). DNA methylation signature of human hippocampus in Alzheimer's disease is linked to neurogenesis. Clin. Epigenetics 11:91. doi: 10.1186/s13148-019-0672-7
An, H., Williams, N. G., and Shelkovnikova, T. A. (2018). NEAT1 and paraspeckles in neurodegenerative diseases: a missing lnc found? Noncoding RNA Res. 3, 243–252. doi: 10.1016/j.ncrna.2018.11.003
Avouac, J., Pezet, S., Gonzalez, V., Baudoin, L., Cauvet, A., Ruiz, B., et al. (2020). Estrogens counteract the profibrotic effects of TGF-β and their inhibition exacerbates experimental dermal fibrosis. J. Invest. Dermatol. 140, 593.e7–601.e7. doi: 10.1016/j.jid.2019.07.719
Bacalini, M. G., Deelen, J., Pirazzini, C., De Cecco, M., Giuliani, C., Lanzarini, C., et al. (2017). Systemic age-associated DNA hypermethylation of ELOVL2 gene: in vivo and in vitro evidences of a cell replication process. J. Geront. A Biol. Sci. Med. Sci. 72, 1015–1023. doi: 10.1093/gerona/glw185
Bacos, K., Gillberg, L., Volkov, P., Olsson, A. H., Hansen, T., Pedersen, O., et al. (2016). Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat. Commun. 7:11089. doi: 10.1038/ncomms11089
Barral, S., Reitz, C., Small, S. A., and Mayeux, R. (2014). Genetic variants in a 'cAMP element binding protein' (CREB)-dependent histone acetylation pathway influence memory performance in cognitively healthy elderly individuals. Neurobiol. Aging 35, 2881.e7–2881.e10. doi: 10.1016/j.neurobiolaging.2014.06.024
Bishop, N. A., Lu, T., and Yankner, B. A. (2010). Neural mechanisms of ageing and cognitive decline. Nature 464, 529–535. doi: 10.1038/nature08983
Blanco-Luquin, I., Acha, B., Urdánoz-Casado, A., Sánchez-Ruiz De Gordoa, J., Vicuña-Urriza, J., Roldán, M., et al. (2020). Early epigenetic changes of Alzheimer's disease in the human hippocampus. Epigenetics 15, 1083–1092. doi: 10.1080/15592294.2020.1748917
Booth, M. J., Branco, M. R., Ficz, G., Oxley, D., Krueger, F., Reik, W., et al. (2012). Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336, 934–937. doi: 10.1126/science.1220671
Bou Sleiman, M., Jha, P., Houtkooper, R., Williams, R. W., Wang, X., and Auwerx, J. (2020). The gene-regulatory footprint of aging highlights conserved central regulators. Cell Rep. 32:108203. doi: 10.1016/j.celrep.2020.108203
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Chao, D. L., and Skowronska-Krawczyk, D. (2020). ELOVL2: not just a biomarker of aging. Transl. Med. Aging. 4, 78–80. doi: 10.1016/j.tma.2020.06.004
Chen, D., Chao, D. L., Rocha, L., Kolar, M., Nguyen Huu, V. A., Krawczyk, M., et al. (2020). The lipid elongation enzyme ELOVL2 is a molecular regulator of aging in the retina. Aging Cell. 19:e13100. doi: 10.1111/acel.13100
Chi, S., Song, J. H., Tan, M. S., Zhang, W., Wang, Z. X., Jiang, T., et al. (2016). Association of single-nucleotide polymorphism in ANK1 with late-onset Alzheimer's disease in Han Chinese. Mol. Neurobiol. 53, 6476–6481. doi: 10.1007/s12035-015-9547-x
Chouliaras, L., Mastroeni, D., Delvaux, E., Grover, A., Kenis, G., Hof, P. R., et al. (2013). Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer's disease patients. Neurobiol. Aging 34, 2091–2099. doi: 10.1016/j.neurobiolaging.2013.02.021
Chouliaras, L., van den Hove, D. L., Kenis, G., Keitel, S., Hof, P. R., van Os, J., et al. (2012). Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Curr. Alzheimers Res. 9, 536–544. doi: 10.2174/156720512800618035
Clough, E., and Barrett, T. (2016). The gene expression omnibus database. Methods Mol. Biol. 1418, 93–110. doi: 10.1007/978-1-4939-3578-9_5
Condliffe, D., Wong, A., Troakes, C., Proitsi, P., Patel, Y., Chouliaras, L., et al. (2014). Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer's disease brain. Neurobiol. Aging 35, 1850–1854. doi: 10.1016/j.neurobiolaging.2014.02.002
Conforto, T. L., Zhang, Y., Sherman, J., and Waxman, D. J. (2012). Impact of CUX2 on the female mouse liver transcriptome: activation of female-biased genes and repression of male-biased genes. Mol. Cell. Biol. 32, 4611–4627. doi: 10.1128/MCB.00886-12
Coppieters, N., Dieriks, B. V., Lill, C., Faull, R. L., Curtis, M. A., and Dragunow, M. (2014). Global changes in DNA methylation and hydroxymethylation in Alzheimer's disease human brain. Neurobiol. Aging 35, 1334–1344. doi: 10.1016/j.neurobiolaging.2013.11.031
Coupé, P., Manjón, J. V., Lanuza, E., and Catheline, G. (2019). Lifespan changes of the human brain in Alzheimer's disease. Sci. Rep. 9:3998. doi: 10.1038/s41598-019-39809-8
Crossland, H., Atherton, P. J., Strömberg, A., Gustafsson, T., and Timmons, J. A. (2017). A reverse genetics cell-based evaluation of genes linked to healthy human tissue age. FASEB J. 31, 96–108. doi: 10.1096/fj.201600296rrr
Day, K., Waite, L. L., Thalacker-Mercer, A., West, A., Bamman, M. M., Brooks, J. D., et al. (2013). Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol. 14:R102. doi: 10.1186/gb-2013-14-9-r102
De Jager, P. L., Srivastava, G., Lunnon, K., Burgess, J., Schalkwyk, L. C., Yu, L., et al. (2014). Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 17, 1156–1163. doi: 10.1038/nn.3786
Dworatzek, E., Baczko, I., and Kararigas, G. (2016). Effects of aging on cardiac extracellular matrix in men and women. Proteomics Clin. Appl. 10, 84–91. doi: 10.1002/prca.201500031
Ellison, E. M., Bradley-Whitman, M. A., and Lovell, M. A. (2017). Single-base resolution mapping of 5-hydroxymethylcytosine modifications in hippocampus of Alzheimer's disease subjects. J. Mol. Neurosci. 63, 185–197. doi: 10.1007/s12031-017-0969-y
Fagiolini, M., Jensen, C. L., and Champagne, F. A. (2009). Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol. 19, 207–212. doi: 10.1016/j.conb.2009.05.009
Fisher, D. W., Bennett, D. A., and Dong, H. (2018). Sexual dimorphism in predisposition to Alzheimer's disease. Neurobiol. Aging 70, 308–324. doi: 10.1016/j.neurobiolaging.2018.04.004
Forger, N. G. (2016). Epigenetic mechanisms in sexual differentiation of the brain and behaviour. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20150114. doi: 10.1098/rstb.2015.0114
Garagnani, P., Bacalini, M. G., Pirazzini, C., Gori, D., Giuliani, C., Mari, D., et al. (2012). Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 11, 1132–1134. doi: 10.1111/acel.12005
Gardmo, C., and Mode, A. (2006). In vivo transfection of rat liver discloses binding sites conveying GH-dependent and female-specific gene expression. J. Mol. Endocrinol. 37, 433–441. doi: 10.1677/jme.1.02116
Gasparoni, G., Bultmann, S., Lutsik, P., Kraus, T. F. J., Sordon, S., Vlcek, J., et al. (2018). DNA methylation analysis on purified neurons and glia dissects age and Alzheimer's disease-specific changes in the human cortex. Epigenet. Chromatin. 11:41. doi: 10.1186/s13072-018-0211-3
Gegenhuber, B., and Tollkuhn, J. (2019). Sex differences in the epigenome: a cause or consequence of sexual differentiation of the brain? Genes 10:432. doi: 10.3390/genes10060432
Gilbert, T. M., Zürcher, N. R., Catanese, M. C., Tseng, C. J., Di Biase, M. A., Lyall, A. E., et al. (2019). Neuroepigenetic signatures of age and sex in the living human brain. Nat. Commun. 10:2945. doi: 10.1038/s41467-019-11031-0
Grimwood, J., Gordon, L. A., Olsen, A., Terry, A., Schmutz, J., Lamerdin, J., et al. (2004). The DNA sequence and biology of human chromosome 19. Nature 428, 529–535. doi: 10.1038/nature02399
Guintivano, J., Aryee, M. J., and Kaminsky, Z. A. (2013). A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 8, 290–302. doi: 10.4161/epi.23924
Hacohen-Kleiman, G., Yizhar-Barnea, O., Touloumi, O., Lagoudaki, R., Avraham, K. B., Grigoriadis, N., et al. (2019). Atypical auditory brainstem response and protein expression aberrations related to ASD and hearing loss in the adnp haploinsufficient mouse brain. Neurochem. Res. 44, 1494–1507. doi: 10.1007/s11064-019-02723-6
Hebert, L. E., Scherr, P. A., Beckett, L. A., Albert, M. S., Pilgrim, D. M., Chown, M. J., et al. (1995). Age-specific incidence of Alzheimer's disease in a community population. JAMA 273, 1354–1359. doi: 10.1001/jama.1995.03520410048025
Hernandez, D. G., Nalls, M. A., Gibbs, J. R., Arepalli, S., van der Brug, M., Chong, S., et al. (2011). Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 20, 1164–1172. doi: 10.1093/hmg/ddq561
Hernández, H. G., Sandoval-Hernández, A. G., Garrido-Gil, P., Labandeira-Garcia, J. L., Zelaya, M. V., Bayon, G. F., et al. (2018). Alzheimer's disease DNA methylome of pyramidal layers in frontal cortex: laser-assisted microdissection study. Epigenomics 10, 1365–1382. doi: 10.2217/epi-2017-0160
Hickman, R. A., Faustin, A., and Wisniewski, T. (2016). Alzheimer disease and its growing epidemic: risk factors, biomarkers, and the urgent need for therapeutics. Neurol. Clin. 34, 941–953. doi: 10.1016/j.ncl.2016.06.009
Higham, J. P., Malik, B. R., Buhl, E., Dawson, J. M., Ogier, A. S., Lunnon, K., et al. (2019). Alzheimer's disease associated genes ankyrin and tau cause shortened lifespan and memory loss in Drosophila. Front. Cell. Neurosci. 13:260. doi: 10.3389/fncel.2019.00260
Ho, B., Greenlaw, K., Al Tuwaijri, A., Moussette, S., Martínez, F., Giorgio, E., et al. (2018). X chromosome dosage and presence of SRY shape sex-specific differences in DNA methylation at an autosomal region in human cells. Biol. Sex Differ. 9:10. doi: 10.1186/s13293-018-0169-7
Horvath, S. (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14:R115. doi: 10.1186/gb-2013-14-10-r115
Horvath, S., Gurven, M., Levine, M. E., Trumble, B. C., Kaplan, H., Allayee, H., et al. (2016). An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 17:171. doi: 10.1186/s13059-016-1030-0
Horvath, S., Mah, V., Lu, A. T., Woo, J. S., Choi, O. W., Jasinska, A. J., et al. (2015). The cerebellum ages slowly according to the epigenetic clock. Aging 7, 294–306. doi: 10.18632/aging.100742
Horvath, S., Zhang, Y., Langfelder, P., Kahn, R. S., Boks, M. P., van Eijk, K., et al. (2012). Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 13:R97. doi: 10.1186/gb-2012-13-10-r97
Hu, H., Li, H., Li, J., Yu, J., and Tan, L. (2018). Genome-wide association study identified ATP6V1H locus influencing cerebrospinal fluid BACE activity. BMC Med. Genet. 19:75. doi: 10.1186/s12881-018-0603-z
Humphries, C. E., Kohli, M. A., Nathanson, L., Whitehead, P., Beecham, G., Martin, E., et al. (2015). Integrated whole transcriptome and DNA methylation analysis identifies gene networks specific to late-onset Alzheimer's disease. J. Alzheimers Dis. 44, 977–987. doi: 10.3233/JAD-141989
Irimajiri, R., Golob, E. J., and Starr, A. (2005). Auditory brain-stem, middle- and long-latency evoked potentials in mild cognitive impairment. Clin. Neurophysiol. 116, 1918–1929. doi: 10.1016/j.clinph.2005.04.010
Jaffe, A. E., Gao, Y., Deep-Soboslay, A., Tao, R., Hyde, T. M., Weinberger, D. R., et al. (2016). Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat. Neurosci. 19, 40–47. doi: 10.1038/nn.4181
Kananen, L., Marttila, S., Nevalainen, T., Jylhävä, J., Mononen, N., Kähönen, M., et al. (2016). Aging-associated DNA methylation changes in middle-aged individuals: the Young Finns study. BMC Genomics. 17:103. doi: 10.1186/s12864-016-2421-z
Katsel, P., Roussos, P., Fam, P., Khan, S., Tan, W., Hirose, T., et al. (2019). The expression of long noncoding RNA NEAT1 is reduced in schizophrenia and modulates oligodendrocytes transcription. NPJ Schizophr. 5:3. doi: 10.1038/s41537-019-0071-2
Kawas, C. H., and Corrada, M. M. (2006). Alzheimer's and dementia in the oldest-old: a century of challenges. Curr. Alzheimer Res. 3, 411–419. doi: 10.2174/156720506779025233
Kennedy, B. K., Berger, S. L., Brunet, A., Campisi, J., Cuervo, A. M., Epel, E. S., et al. (2014). Geroscience: linking aging to chronic disease. Cell 159, 709–713. doi: 10.1016/j.cell.2014.10.039
Kim, J. Y., Park, Y. K., Lee, K. P., Lee, S. M., Kang, T. W., Kim, H. J., et al. (2014). Genome-wide profiling of the microRNA-mRNA regulatory network in skeletal muscle with aging. Aging 6, 524–544. doi: 10.18632/aging.100677
Kim, S., Cho, Y. H., Won, S., Ku, J. L., Moon, H. B., Park, J., et al. (2019). Maternal exposures to persistent organic pollutants are associated with DNA methylation of thyroid hormone-related genes in placenta differently by infant sex. Environ. Int. 130:104956. doi: 10.1016/j.envint.2019.104956
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Ladd-Acosta, C., Pevsner, J., Sabunciyan, S., Yolken, R. H., Webster, M. J., Dinkins, T., et al. (2007). DNA methylation signatures within the human brain. Am. J. Hum. Genet. 81, 1304–1315. doi: 10.1086/524110
Landgrave-Gómez, J., Mercado-Gómez, O., and Guevara-Guzmán, R. (2015). Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 9:58. doi: 10.3389/fncel.2015.00058
Lardenoije, R., Iatrou, A., Kenis, G., Kompotis, K., Steinbusch, H. W. M., Mastroeni, D., et al. (2015). The epigenetics of aging and neurodegeneration. Progr. Neurobiol. 131, 21–64. doi: 10.1016/j.pneurobio.2015.05.002
Lardenoije, R., Roubroeks, J. A. Y., Pishva, E., Leber, M., Wagner, H., Iatrou, A., et al. (2019). Alzheimer's disease-associated (hydroxy)methylomic changes in the brain and blood. Clin. Epigenetics 11:164. doi: 10.1186/s13148-019-0755-5
Levine, M. E., Lu, A. T., Bennett, D. A., and Horvath, S. (2015). Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer's disease related cognitive functioning. Aging 7, 1198–1211. doi: 10.18632/aging.100864
Li, J. Y., Liu, Y. F., Xu, H. Y., Zhang, J. Y., Lv, P. P., Liu, M. E., et al. (2020). Basonuclin 1 deficiency causes testicular premature aging: BNC1 cooperates with TAF7L to regulate spermatogenesis. J. Mol. Cell Biol. 12, 71–83. doi: 10.1093/jmcb/mjz035
Li, Q. S., Sun, Y., and Wang, T. (2020). Epigenome-wide association study of Alzheimer's disease replicates 22 differentially methylated positions and 30 differentially methylated regions. Clin. Epigenetics 12:149. doi: 10.1186/s13148-020-00944-z
Li, X., Wang, J., Wang, L., Feng, G., Li, G., Yu, M., et al. (2020). Impaired lipid metabolism by age-dependent DNA methylation alterations accelerates aging. Proc. Natl. Acad. Sci. U.S.A. 117, 4328–4336. doi: 10.1073/pnas.1919403117
Liu, Y., Fang, S., Liu, L. M., Zhu, Y., Li, C. R., Chen, K., et al. (2020). Hearing loss is an early biomarker in APP/PS1 Alzheimer's disease mice. Neurosci. Lett. 717:134705. doi: 10.1016/j.neulet.2019.134705
Lord, J., and Cruchaga, C. (2014). The epigenetic landscape of Alzheimer's disease. Nat. Neurosci. 17, 1138–1140. doi: 10.1038/nn.3792
Luckhart, C., Philippe, T. J., Le François, B., Vahid-Ansari, F., Geddes, S. D., Béïque, J.-C., et al. (2016). Sex-dependent adaptive changes in serotonin-1A autoreceptor function and anxiety in Deaf1-deficient mice. Mol. Brain 9:77. doi: 10.1186/s13041-016-0254-y
Lunnon, K., Hannon, E., Smith, R. G., Dempster, E., Wong, C., Burrage, J., et al. (2016). Variation in 5-hydroxymethylcytosine across human cortex and cerebellum. Genome Biol. 17:27. doi: 10.1186/s13059-016-0871-x
Lunnon, K., Smith, R., Hannon, E., De Jager, P. L., Srivastava, G., Volta, M., et al. (2014). Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer's disease. Nat. Neurosci. 17, 1164–1170. doi: 10.1038/nn.3782
Maierhofer, A., Flunkert, J., Oshima, J., Martin, G. M., Poot, M., Nanda, I., et al. (2019). Epigenetic signatures of Werner syndrome occur early in life and are distinct from normal epigenetic aging processes. Aging Cell. 18:e12995. doi: 10.1111/acel.12995
Makioka, K., Yamazaki, T., Takatama, M., Ikeda, M., and Okamoto, K. (2014). Immunolocalization of Smurf1 in Hirano bodies. J. Neurol. Sci. 336, 24–28. doi: 10.1016/j.jns.2013.09.028
Mansour, H., Chamberlain, C. G., Weible, M. W. II, Hughes, S., Chu, Y., and Chan-Ling, T. (2008). Aging-related changes in astrocytes in the rat retina: imbalance between cell proliferation and cell death reduces astrocyte availability. Aging Cell. 7, 526–540. doi: 10.1111/j.1474-9726.2008.00402.x
Maphis, N. M., Jiang, S., Binder, J., Wright, C., Gopalan, B., Lamb, B. T., et al. (2017). Whole genome expression analysis in a mouse model of tauopathy identifies MECP2 as a possible regulator of tau pathology. Front. Mol. Neurosci. 10:69. doi: 10.3389/fnmol.2017.00069
Maschietto, M., Bastos, L. C., Tahira, A. C., Bastos, E. P., Euclydes, V. L., Brentani, A., et al. (2017). Sex differences in DNA methylation of the cord blood are related to sex-bias psychiatric diseases. Sci. Rep. 7:44547. doi: 10.1038/srep44547
Masser, D. R., Hadad, N., Porter, H. L., Mangold, C. A., Unnikrishnan, A., Ford, M. M., et al. (2017). Sexually divergent DNA methylation patterns with hippocampal aging. Aging Cell. 16, 1342–1352. doi: 10.1111/acel.12681
Mastroeni, D., Sekar, S., Nolz, J., Delvaux, E., Lunnon, K., Mill, J., et al. (2017). ANK1 is up-regulated in laser captured microglia in Alzheimer's brain; the importance of addressing cellular heterogeneity. PLoS ONE 12:e0177814. doi: 10.1371/journal.pone.0177814
McCann, J. C., and Ames, B. N. (2011). Adaptive dysfunction of selenoproteins from the perspective of the triage theory: why modest selenium deficiency may increase risk of diseases of aging. FASEB J. 25, 1793–1814. doi: 10.1096/fj.11-180885
McCarthy, M. M., Auger, A. P., Bale, T. L., De Vries, G. J., Dunn, G. A., Forger, N. G., et al. (2009). The epigenetics of sex differences in the brain. J. Neurosci. 29, 12815–12823. doi: 10.1523/JNEUROSCI.3331-09.2009
McCarthy, N. S., Melton, P. E., Cadby, G., Yazar, S., Franchina, M., Moses, E. K., et al. (2014). Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns. BMC Genomics 15:981. doi: 10.1186/1471-2164-15-981
McCartney, D. L., Zhang, F., Hillary, R. F., Zhang, Q., Stevenson, A. J., Walker, R. M., et al. (2019). An epigenome-wide association study of sex-specific chronological ageing. Genome Med. 12:1. doi: 10.1186/s13073-019-0693-z
Migeon, B. R., Beer, M. A., and Bjornsson, H. T. (2017). Embryonic loss of human females with partial trisomy 19 identifies region critical for the single active X. PLoS ONE 12:e0170403. doi: 10.1371/journal.pone.0170403
Mikovic, J., Sadler, K., Butchart, L., Voisin, S., Gerlinger-Romero, F., Della Gatta, P., et al. (2018). MicroRNA and long non-coding RNA regulation in skeletal muscle from growth to old age shows striking dysregulation of the callipyge locus. Front. Genet. 9:548. doi: 10.3389/fgene.2018.00548
Nebel, R. A., Aggarwal, N. T., Barnes, L. L., Gallagher, A., Goldstein, J. M., Kantarci, K., et al. (2018). Understanding the impact of sex and gender in Alzheimer's disease: a call to action. Alzheimers Dement. 14, 1171–1183. doi: 10.1016/j.jalz.2018.04.008
Numata, S., Ye, T., Hyde, T. M., Guitart-Navarro, X., Tao, R., Wininger, M., et al. (2012). DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 90, 260–272. doi: 10.1016/j.ajhg.2011.12.020
Oh, D., Han, S., Seo, J., Lee, J. R., Choi, J., Groffen, J., et al. (2010). Regulation of synaptic Rac1 activity, long-term potentiation maintenance, and learning and memory by BCR and ABR Rac GTPase-activating proteins. J. Neurosci. 30, 14134–14144. doi: 10.1523/JNEUROSCI.1711-10.2010
O'Leary, T. P., Shin, S., Fertan, E., Dingle, R. N., Almuklass, A., Gunn, R. K., et al. (2017). Reduced acoustic startle response and peripheral hearing loss in the 5xFAD mouse model of Alzheimer's disease. Genes Brain Behav. 16, 554–563. doi: 10.1111/gbb.12370
Pal, S., and Tyler, J. K. (2016). Epigenetics and aging. Sci. Adv. 2:e1600584. doi: 10.1126/sciadv.1600584
Pareek, G., Thomas, R. E., and Pallanck, L. J. (2018). Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration. Cell Death Dis. 9:304. doi: 10.1038/s41419-018-0365-8
Patel, H., Dobson, R. J. B., and Newhouse, S. J. (2019). A meta-analysis of Alzheimer's disease brain transcriptomic data. J. Alzheimers Dis. 68, 1635–1656. doi: 10.3233/JAD-181085
Pereira Fernandes, D., Bitar, M., Jacobs, F. M. J., and Barry, G. (2018). Long non-coding RNAs in neuronal aging. Noncoding RNA 4:12. doi: 10.3390/ncrna4020012
Perzel Mandell, K. A., Price, A. J., Wilton, R., Collado-Torres, L., Tao, R., Eagles, N. J., et al. (2020). Characterizing the dynamic and functional DNA methylation landscape in the developing human cortex. Epigenetics 2020, 1–13. doi: 10.1080/15592294.2020.1786304
Philippe, T. J., Vahid-Ansari, F., Donaldson, Z. R., Le François, B., Zahrai, A., Turcotte-Cardin, V., et al. (2018). Loss of MeCP2 in adult 5-HT neurons induces 5-HT1A autoreceptors, with opposite sex-dependent anxiety and depression phenotypes. Sci Rep. 8:5788. doi: 10.1038/s41598-018-24167-8
Phillips, O. R., Onopa, A. K., Hsu, V., Ollila, H. M., Hillary, R. P., Hallmayer, J., et al. (2019). Beyond a binary classification of sex: an examination of brain sex differentiation, psychopathology, and genotype. J. Am. Acad. Child Adolesc. Psychiatry 58, 787–798. doi: 10.1016/j.jaac.2018.09.425
Pike, C. J. (2017). Sex and the development of Alzheimer's disease. J. Neurosci. Res. 95, 671–680. doi: 10.1002/jnr.23827
Podcasy, J. L., and Epperson, C. N. (2016). Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 18, 437–446. doi: 10.31887/DCNS.2016.18.4/cepperson
Price, A. J., Collado-Torres, L., Ivanov, N. A., Xia, W., Burke, E. E., Shin, J. H., et al. (2019). Divergent neuronal DNA methylation patterns across human cortical development reveal critical periods and a unique role of CpH methylation. Genome Biol. 20:196. doi: 10.1186/s13059-019-1805-1
Ren, X., and Kuan, P. F. (2019). methylGSA: a bioconductor package and Shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics 35, 1958–1959. doi: 10.1093/bioinformatics/bty892
Ringh, M. V., Hagemann-Jensen, M., Needhamsen, M., Kular, L., Breeze, C. E., Sjöholm, L. K., et al. (2019). Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine 46, 290–304. doi: 10.1016/j.ebiom.2019.07.006
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Rizzardi, L. F., Hickey, P. F., Rodriguez DiBlasi, V., Tryggvadóttir, R., Callahan, C. M., Idrizi, A., et al. (2019). Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat. Neurosci. 22, 307–316. doi: 10.1038/s41593-018-0297-8
Rönn, T., Volkov, P., Gillberg, L., Kokosar, M., Perfilyev, A., Jacobsen, A. L., et al. (2015). Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum. Mol. Genet. 24, 3792–3813. doi: 10.1093/hmg/ddv124
Roubroeks, J. A. Y., Smith, A. R., Smith, R. G., Pishva, E., Ibrahim, Z., Sattlecker, M., et al. (2020). An epigenome-wide association study of Alzheimer's disease blood highlights robust DNA hypermethylation in the HOXB6 gene. Neurobiol. Aging 95, 26–45. doi: 10.1016/j.neurobiolaging.2020.06.023
Salvatori, B., Biscarini, S., and Morlando, M. (2020). Non-coding RNAs in nervous system development and disease. Fron. Cell Dev. Biol. 8:273. doi: 10.3389/fcell.2020.00273
Scahill, R. I., Schott, J. M., Stevens, J. M., Rossor, M. N., and Fox, N. C. (2002). Mapping the evolution of regional atrophy in Alzheimer's disease: unbiased analysis of fluid-registered serial MRI. Proc. Natl. Acad. Sci. U.S.A. 99, 4703–4707. doi: 10.1073/pnas.052587399
Semick, S. A., Bharadwaj, R. A., Collado-Torres, L., Tao, R., Shin, J. H., Deep-Soboslay, A., et al. (2019). Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer's disease. Acta Neuropathol. 137, 557–569. doi: 10.1007/s00401-019-01966-5
Shao, L., Liu, X., Zhu, S., Liu, C., Gao, Y., and Xu, X. (2018). The role of Smurf1 in neuronal necroptosis after lipopolysaccharide-induced neuroinflammation. Cell. Mol. Neurobiol. 38, 809–816. doi: 10.1007/s10571-017-0553-6
Shireby, G. L., Davies, J. P., Francis, P. T., Burrage, J., Walker, E. M., Neilson, G. W. A., et al. (2020). Recalibrating the epigenetic clock: implications for assessing biological age in the human cortex. Brain 143, 3763–3775. doi: 10.1101/2020.04.27.063719
Sierra, F. (2020). Editorial: geroscience and the role of aging in the etiology and management of Alzheimer's disease. J. Prev. Alzheimers Dis. 7, 2–3. doi: 10.14283/jpad.2019.49
Simchovitz, A., Hanan, M., Niederhoffer, N., Madrer, N., Yayon, N., Bennett, E. R., et al. (2019). NEAT1 is overexpressed in Parkinson's disease substantia nigra and confers drug-inducible neuroprotection from oxidative stress. FASEB J. 33, 11223–11234. doi: 10.1096/fj.201900830R
Singmann, P., Shem-Tov, D., Wahl, S., Grallert, H., Fiorito, G., Shin, S.-Y., et al. (2015). Characterization of whole-genome autosomal differences of DNA methylation between men and women. Epigenetics Chromatin. 8:43. doi: 10.1186/s13072-015-0035-3
Sittig, L. J., Shukla, P. K., Herzing, L. B., and Redei, E. E. (2011). Strain-specific vulnerability to alcohol exposure in utero via hippocampal parent-of-origin expression of deiodinase-III. FASEB J. 25, 2313–2324. doi: 10.1096/fj.10-179234
Skariah, G., Seimetz, J., Norsworthy, M., Lannom, M. C., Kenny, P. J., Elrakhawy, M., et al. (2017). Mov10 suppresses retroelements and regulates neuronal development and function in the developing brain. BMC Biol. 15:54. doi: 10.1186/s12915-017-0387-1
Slieker, R. C., Relton, C. L., Gaunt, T. R., Slagboom, P. E., and Heijmans, B. T. (2018). Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin. 11:25. doi: 10.1186/s13072-018-0191-3
Smith, A. R., Smith, R. G., Pishva, E., Hannon, E., Roubroeks, J. A. Y., Burrage, J., et al. (2019). Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer's disease. Clin. Epigenetics 11:52. doi: 10.1186/s13148-019-0636-y
Smith, A. R., Wheildon, G., and Lunnon, K. (2020). A five-year update on epigenome-wide association studies of DNA modifications in Alzheimer's disease: progress, practicalities and promise. Neuropathol. Appl. Neurobiol. 46, 641–653. doi: 10.1111/nan.12650
Smith, R. G., Hannon, E., De Jager, P. L., Chibnik, L., Lott, S. J., Condliffe, D., et al. (2018). Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer's disease neuropathology. Alzheimers Dement. 14, 1580–1588. doi: 10.1016/j.jalz.2018.01.017
Smith, R. G., Pishva, E., Shireby, G., Smith, A. R., Roubroeks, J. A. Y., Hannon, E., et al. (2020). Meta-analysis of epigenome-wide association studies in Alzheimer's disease highlights novel differentially methylated loci across cortex. bioRxiv. 2020:2020.02.28.957894. doi: 10.1101/2020.02.28.957894
Spiers, H., Hannon, E., Schalkwyk, L. C., Bray, N. J., and Mill, J. (2017). 5-hydroxymethylcytosine is highly dynamic across human fetal brain development. BMC Genomics 18:738. doi: 10.1186/s12864-017-4091-x
Spiers, H., Hannon, E., Schalkwyk, L. C., Smith, R., Wong, C. C., O'Donovan, M. C., et al. (2015). Methylomic trajectories across human fetal brain development. Genome Res. 25, 338–352. doi: 10.1101/gr.180273.114
Spólnicka, M., Pośpiech, E., Adamczyk, J. G., Freire-Aradas, A., Pepłońska, B., Zbieć-Piekarska, R., et al. (2018a). Modified aging of elite athletes revealed by analysis of epigenetic age markers. Aging 10, 241–252. doi: 10.18632/aging.101385
Spólnicka, M., Pośpiech, E., Pepłońska, B., Zbieć-Piekarska, R., Makowska, Z., Pieta, A., et al. (2018b). DNA methylation in ELOVL2 and C1orf132 correctly predicted chronological age of individuals from three disease groups. Int. J. Legal Med. 132, 1–11. doi: 10.1007/s00414-017-1636-0
Spreafico, M., Grillo, B., Rusconi, F., Battaglioli, E., and Venturin, M. (2018). Multiple layers of CDK5R1 regulation in Alzheimer's disease implicate long non-coding RNAs. Int. J. Mol. Sci. 19:2022. doi: 10.3390/ijms19072022
Srinivasan, K., Friedman, B. A., Etxeberria, A., Huntley, M. A., van der Brug, M. P., Foreman, O., et al. (2020). Alzheimer's patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 31:107843. doi: 10.1016/j.celrep.2020.107843
Steegenga, W. T., Boekschoten, M. V., Lute, C., Hooiveld, G. J., de Groot, P. J., Morris, T. J., et al. (2014). Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age 36:9648. doi: 10.1007/s11357-014-9648-x
Stohn, J. P., Martinez, M. E., St Germain, D. L., and Hernandez, A. (2019). Adult onset of type 3 deiodinase deficiency in mice alters brain gene expression and increases locomotor activity. Psychoneuroendocrinology 110:104439. doi: 10.1016/j.psyneuen.2019.104439
Stone, G., Choi, A., Meritxell, O., Gorham, J., Heydarpour, M., Seidman, C. E., et al. (2019). Sex differences in gene expression in response to ischemia in the human left ventricular myocardium. Hum. Mol. Genet. 28, 1682–1693. doi: 10.1093/hmg/ddz014
Sturm, G., Cardenas, A., Bind, M. A., Horvath, S., Wang, S., Wang, Y., et al. (2019). Human aging DNA methylation signatures are conserved but accelerated in cultured fibroblasts. Epigenetics 14, 961–976. doi: 10.1080/15592294.2019.1626651
Suderman, M., Simpkin, A., Sharp, G., Gaunt, T., Lyttleton, O., McArdle, W., et al. (2017). Sex-associated autosomal DNA methylation differences are wide-spread and stable throughout childhood. bioRxiv. 2017:118265. doi: 10.1101/118265
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6:e21800. doi: 10.1371/journal.pone.0021800
Tajuddin, S. M., Hernandez, D. G., Chen, B. H., Noren Hooten, N., Mode, N. A., Nalls, M. A., et al. (2019). Novel age-associated DNA methylation changes and epigenetic age acceleration in middle-aged African Americans and whites. Clin. Epigenetics 11:119. doi: 10.1186/s13148-019-0722-1
Van Hoesen, G. W., Hyman, B. T., and Damasio, A. R. (1991). Entorhinal cortex pathology in Alzheimer's disease. Hippocampus 1, 1–8. doi: 10.1002/hipo.450010102
Wang, L., Sheng, Y., Xu, W., Sun, M., Lv, S., Yu, J., et al. (2020). Mechanism of thyroid hormone signaling in skeletal muscle of aging mice. Endocrine. doi: 10.1007/s12020-020-02428-9. [Epub ahead of print].
Wang, X., Chen, Y., and Lu, L. (2010). Genetic regulatory network analysis for app based on genetical genomics approach. Exp. Aging Res. 36, 79–93. doi: 10.1080/03610730903418729
Wei, X., Zhang, L., and Zeng, Y. (2020). DNA methylation in Alzheimer's disease: in brain and peripheral blood. Mech. Ageing Dev. 191:111319. doi: 10.1016/j.mad.2020.111319
Wezyk, M., Spólnicka, M., Pośpiech, E., Pepłońska, B., Zbieć-Piekarska, R., Ilkowski, J., et al. (2018). Hypermethylation of TRIM59 and KLF14 influences cell death signaling in familial Alzheimer's disease. Oxid. Med. Cell. Longev. 2018:6918797. doi: 10.1155/2018/6918797
White, R. R., Milholland, B., MacRae, S. L., Lin, M., Zheng, D., and Vijg, J. (2015). Comprehensive transcriptional landscape of aging mouse liver. BMC Genomics 16:899. doi: 10.1186/s12864-015-2061-8
Wiethoff, S., Zhour, A., Schöls, L., and Fischer, M. D. (2012). Retinal nerve fibre layer loss in hereditary spastic paraplegias is restricted to complex phenotypes. BMC Neurol. 12:143. doi: 10.1186/1471-2377-12-143
Willer, C. J., Li, Y., and Abecasis, G. R. (2010). METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. doi: 10.1093/bioinformatics/btq340
Wu, Q., Fukuda, K., Weinstein, M., Graff, J. M., and Saga, Y. (2015). SMAD2 and p38 signaling pathways act in concert to determine XY primordial germ cell fate in mice. Development 142, 575–586. doi: 10.1242/dev.119446
Xia, Y., Dai, R., Wang, K., Jiao, C., Zhang, C., Xu, Y., et al. (2019). Sex-differential DNA methylation and associated regulation networks in human brain implicated in the sex-biased risks of psychiatric disorders. Mol Psychiatry. doi: 10.1038/s41380-019-0416-2. [Epub ahead of print].
Xiao, F.-H., Kong, Q.-P., Perry, B., and He, Y.-H. (2016). Progress on the role of DNA methylation in aging and longevity. Brief. Funct. Genomics 15, 454–459. doi: 10.1093/bfgp/elw009
Xiao, F.-H., Wang, H.-T., and Kong, Q.-P. (2019). Dynamic DNA methylation during aging: a “prophet” of age-related outcomes. Front. Genet. 10:107. doi: 10.3389/fgene.2019.00107
Xiao, F. H., Chen, X. Q., He, Y. H., and Kong, Q. P. (2018). Accelerated DNA methylation changes in middle-aged men define sexual dimorphism in human lifespans. Clin. Epigenetics 10:133. doi: 10.1186/s13148-018-0573-1
Xu, H., Wang, F., Liu, Y., Yu, Y., Gelernter, J., and Zhang, H. (2014). Sex-biased methylome and transcriptome in human prefrontal cortex. Hum. Mol. Genet. 23, 1260–1270. doi: 10.1093/hmg/ddt516
Xu, J., Patassini, S., Rustogi, N., Riba-Garcia, I., Hale, B. D., Phillips, A. M., et al. (2019). Regional protein expression in human Alzheimer's brain correlates with disease severity. Commun. Biol. 2:43. doi: 10.1038/s42003-018-0254-9
Yusipov, I., Bacalini, M. G., Kalyakulina, A., Krivonosov, M., Pirazzini, C., Gensous, N., et al. (2020). Age-related DNA methylation changes are sex-specific: a comprehensive assessment. bioRxiv. 2020.01.15.905224. doi: 10.1101/2020.01.15.905224
Zhou, W., Laird, P. W., and Shen, H. (2017). Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45:e22. doi: 10.1093/nar/gkw967
Keywords: DNA methylation, Alzheimer's disease, brain, sex, aging
Citation: Pellegrini C, Pirazzini C, Sala C, Sambati L, Yusipov I, Kalyakulina A, Ravaioli F, Kwiatkowska KM, Durso DF, Ivanchenko M, Monti D, Lodi R, Franceschi C, Cortelli P, Garagnani P and Bacalini MG (2021) A Meta-Analysis of Brain DNA Methylation Across Sex, Age, and Alzheimer's Disease Points for Accelerated Epigenetic Aging in Neurodegeneration. Front. Aging Neurosci. 13:639428. doi: 10.3389/fnagi.2021.639428
Received: 08 December 2020; Accepted: 05 February 2021;
Published: 11 March 2021.
Edited by:
Franca Rosa Guerini, Fondazione Don Carlo Gnocchi Onlus (IRCCS), ItalyReviewed by:
Fu-Hui Xiao, Chinese Academy of Sciences, ChinaMassimo Santoro, Fondazione Don Carlo Gnocchi Onlus (IRCCS), Italy
Copyright © 2021 Pellegrini, Pirazzini, Sala, Sambati, Yusipov, Kalyakulina, Ravaioli, Kwiatkowska, Durso, Ivanchenko, Monti, Lodi, Franceschi, Cortelli, Garagnani and Bacalini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Giulia Bacalini, bWFyaWFnaXVsaWEuYmFjYWxpbmlAYXVzbC5ib2xvZ25hLml0