 Xuewen Xiao
Xuewen Xiao Hui Liu1
Hui Liu1 Weiwei Zhang
Weiwei Zhang Bin Jiao
Bin Jiao- 1Department of Neurology, Xiangya Hospital, Central South University, Changsha, China
- 2National Clinical Research Center for Geriatric Disorders, Central South University, Changsha, China
- 3Engineering Research Center of Hunan Province in Cognitive Impairment Disorders, Central South University, Changsha, China
- 4Hunan International Scientific and Technological Cooperation Base of Neurodegenerative and Neurogenetic Diseases, Changsha, China
- 5Key Laboratory of Hunan Province in Neurodegenerative Disorders, Central South University, Changsha, China
The strategies of classifying APP, PSEN1, and PSEN2 variants varied substantially in the previous studies. We aimed to re-evaluate these variants systematically according to the American college of medical genetics and genomics and the association for molecular pathology (ACMG-AMP) guidelines. In our study, APP, PSEN1, and PSEN2 variants were collected by searching Alzforum and PubMed database with keywords “PSEN1,” “PSEN2,” and “APP.” These variants were re-evaluated based on the ACMG-AMP guidelines. We compared the number of pathogenic/likely pathogenic variants of APP, PSEN1, and PSEN2. In total, 66 APP variants, 323 PSEN1 variants, and 63 PSEN2 variants were re-evaluated in our study. 94.91% of previously reported pathogenic variants were re-classified as pathogenic/likely pathogenic variants, while 5.09% of them were variants of uncertain significance (VUS). PSEN1 carried the most prevalent pathogenic/likely pathogenic variants, followed by APP and PSEN2. Significant statistically difference was identified among these three genes when comparing the number of pathogenic/likely pathogenic variants (P < 2.2 × 10–16). Most of the previously reported pathogenic variants were re-classified as pathogenic/likely pathogenic variants while the others were re-evaluated as VUS, highlighting the importance of interpreting APP, PSEN1, and PSEN2 variants with caution according to ACMG-AMP guidelines.
Introduction
Being the most common neurodegenerative disease, Alzheimer’s disease (AD) is hallmarked by insidious cognitive impairment. It is estimated that 50 million individuals are affected by dementia worldwide and AD accounts for 50–60% of dementia. With the coming of an aging society, the number of AD is increasing rapidly (Saez-Atienzar and Masliah, 2020). To date, only three causative genes have been identified in the pathogenesis of AD, including amyloid precursor protein (APP), presenilin1 (PSEN1), and presenilin2 (PSEN2). APP encodes a protein called amyloid-β protein precursor, whose proteolysis generates amyloid-β(Aβ), a key component of amyloid plaque. Additionally, presenilin-1 and presenilin-2 are encoded by PSEN1 and PSEN2, respectively. Both of them are subunits of γ-secretase and associated with either the increase of Aβ or the raised ratio of Aβ42 over Aβ40 (Loy et al., 2014), causing the formation of amyloid plaques and leading to the development of AD (Sun et al., 2017).
The diagnosis criteria of AD are evolving rapidly. Currently, Aβ deposition, pathologic tau, and neurodegeneration [AT(N)] classification system is used to define AD based on biomarker evidence. However, the methods of obtaining biomarkers are expensive or invasive. The identification of pathogenic mutations is still of vital importance in the diagnosis of AD (Jack et al., 2018). The Dominantly Inherited Alzheimer Network (DIAN), funded by the National Institute on Aging (NIA), collected over 450 individuals with 90 different mutations in PSEN1, PSEN2, and APP. DIAN constitutes a strong impact in AD research because it is remarkably helpful in the understanding of the disease’s natural history (Morris et al., 2012). In the DIAN study, the classification rate between the mutation-carriers group and normal controls is approximately 80% using biomarkers with machine learning (Castillo-Barnes et al., 2020). The DIAN study is not only important for discovering disease trajectories (Luckett et al., 2021), but also for drug trials (Bateman et al., 2017). All the results can be well established only through the correct diagnosis of AD.
Currently, the most frequent cause of AD is variants of PSEN1. Variants in APP are responsible for the second common cause of AD. By contrast, variants in PSEN2 leading to AD are relatively rare1. Nevertheless, although these three causative genes have been widely investigated, the interpretations of variants remain complex (Hsu et al., 2020). The strategies of classifying variants varied substantially in the previous studies (Denham et al., 2019). Furthermore, the age of onset and clinical manifestations differed among patients with different mutations (Ryan et al., 2016). Subsequently, timely genetic testing and correct classification are fundamental in the diagnosis and treatment of AD patients.
In 2015, the American college of medical genetics and genomics and the association for molecular pathology (ACMG-AMP) issued a guideline for classifying variants based on typical types of variant evidence (e.g., population data, computational data, functional data, segregation data, etc.) (Richards et al., 2015). In our study, to classify these variants systematically and scientifically, we re-evaluate APP, PSEN1, and PSEN2 variants according to the ACMG-AMP guidelines. Variants of uncertain significance (VUS) are defined by the criteria for benign and pathogenic are contradictory or the lack of criteria to be classified as pathogenic or benign. Our study re-assessed the APP, PSEN1, and PSEN2 variants as well as compared the mutation spectrum of these three genes, which may have important implications in the molecular diagnosis and treatment of AD.
Materials and Methods
Systematic Search
We re-analyzed APP, PSEN1, and PSEN2 variants from the Alzforum database (see text footnote 1) and searched related literature using PubMed2 with the keywords “PSEN1,” “PSEN2,” or “APP.” All the studies included either clinical characteristics or functional data about these variants. All these variants were re-evaluated by two independent investigators according to the ACMG-AMP guidelines (Richards et al., 2015). Permission was obtained from Alzforum to re-analyze the variants in APP, PSEN1, and PSEN2 genes.
Analysis of Variant Frequency
According to the ACMG-AMP guideline, if a variant doesn’t exist in a large general population or a control cohort, it can be considered as pathogenic moderate criterion 2 (PM2). In our study, the variant frequency was searched using Exome Sequencing Project (ESP6500) (Auer et al., 2016), 1,000 Genomes Project (Auton et al., 2015), and the ExAC Browser (Lek et al., 2016). Given that APP, PSEN1 and PSEN2 are inherited in an autosomal dominant mode in AD, therefore, if a variant was absent from these databases, pathogenic moderate criterion 2 (PM2) can be applied (Li and Wang, 2017). Given that the APP, PSEN1, and PSEN2 are not fully penetrant, whereas benign interpretation (BS2) can be established only when the penetrance is 100% at an early age in healthy controls. Subsequently, the BS2 is not applied in the classification of APP, PSEN1, and PSEN2 variants (Table 1).
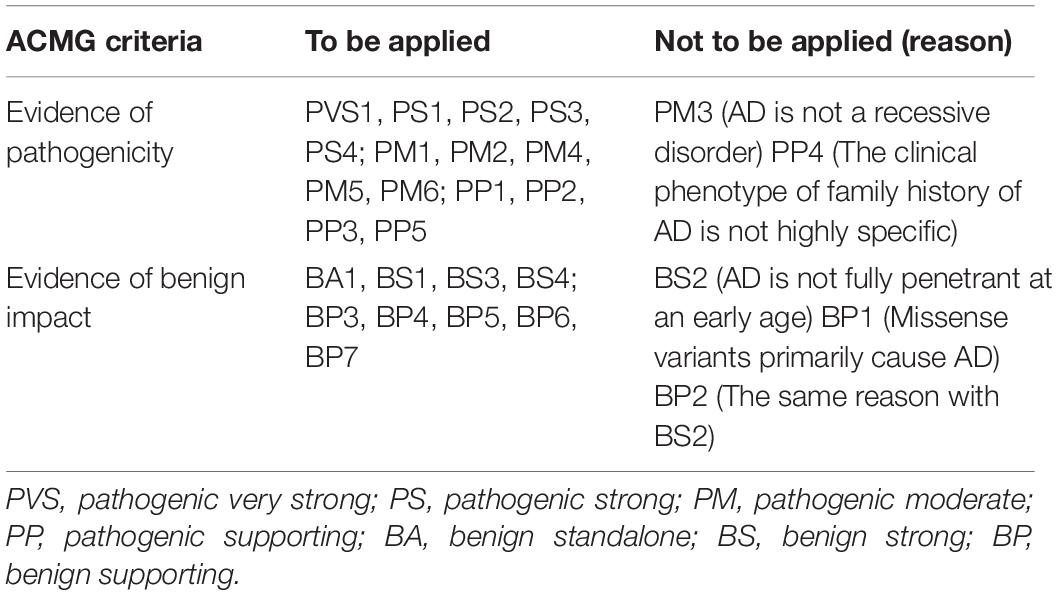
Table 1. Relevance of ACMG criteria in AD.
In silico Evidence
The pathogenicity of variants was also predicted using multiple in silico genomic tools, including SIFT (Ng and Henikoff, 2003), Polyphen-2 (Adzhubei et al., 2010), LRT (Chun and Fay, 2009), MutationTaster (Schwarz et al., 2010), MutationAssessor (Reva et al., 2011), FATHMM (Shihab et al., 2013), PROVEAN (Choi et al., 2012), CADD (Kircher et al., 2014), REVEL (Ioannidis et al., 2016), and Reve (Li et al., 2018b). For missense variants, we used VarCards3, an integrated genetic database, to get the in silico prediction results of variants (Li et al., 2018a). For splicing variants, GeneSplicer and Human Splicing Finder were applied to predict the pathogenicity of variants. If all of the genomic tools supported the damaging of variants, then pathogenic supporting criterion 3 (PP3) was established. However, if the prediction results were conflicting, then PP3 would not be used.
Analysis of Functional Studies
The increased amount of Aβ or raised ratio of Aβ42 over Aβ40 was considered as the key events in the pathogenesis of AD (Selkoe and Hardy, 2016). Consequently, pathogenic strong criterion 2 (PS3) could be applied if a variant leads to elevated total Aβ production or increased Aβ42/Aβ40 ratio in well-established in vitro functional studies. The functional data were excluded when there is conflicting evidence among studies. Besides, if functional studies showed that a variant exerts no effect on Aβ production and Aβ42/Aβ40 ratio, then benign strong criterion 3 (BS3) would be taken into consideration.
Statistical Analyses
We performed a Chi-square test to compare the number of pathogenic/likely pathogenic variants of APP, PSEN1, and PSEN2 genes using the SPSS 20.0 test (SPSS, Chicago, IL, United States). Besides, the number of pathogenic/likely pathogenic variants was analyzed between the transmembrane domain and non-transmembrane domain in APP, PSEN1, and PSEN2 separately. In the pathogenic/likely pathogenic variants, each variant type of PSEN1, PSEN2, and APP was compared, respectively, by Fisher test. A p-value < 0.05 was considered statistically significant.
Results
Summary of Variants
A total of 452 variants of APP, PSEN1, and PSEN2 were collected, in which 66 APP variants, 323 PSEN1 variants, and 63 PSEN2 variants were re-analyzed in our study (Figure 1 and Supplementary Table 1). PSEN1 was the most common gene in patients with AD (323/452, 71.24%). According to the ACMG-AMP guidelines, 89.16% PSEN1 variants (288/323) were re-classified as pathogenic/likely pathogenic variants, followed by APP gene, in which 46.97% variants (31/66) were re-considered pathogenic/likely pathogenic variants. The least pathogenic or likely pathogenic variants came from the PSEN2 gene, among them, only 20.63% of variants (13/63) fulfilled the criteria for pathogenicity/likely pathogenicity (Table 2). Significant differences were observed among these three genes when comparing the number of pathogenic/likely pathogenic variants (P < 2.20 × 10–16). 317 previously reported pathogenic variants (94.91%) were re-evaluated as pathogenic/likely pathogenic variants, and 17 variants (5.09%) VUS variants. Moreover, the number of pathogenic/likely pathogenic variants in PSEN1 was more than that of APP (P = 1.78 × 10–15) as well as PSEN2 (P < 2.20 × 10–16) (Figure 2). There are more pathogenic/likely pathogenic variants in APP than in PSEN2 (P = 3.00 × 10–3). Additionally, In the PSEN1 pathogenic/likely pathogenic variants, 262 missense, 15 indel, six CNV, two splicing, and one frameshift variant were identified. Seven missense, five frameshift, and one splicing variant were found in the PSEN2 pathogenic/likely pathogenic variants. APP pathogenic/likely pathogenic variants consisted of 29 missense and one indel variant. In the pathogenic/likely pathogenic variants, missense variants are more common in PSEN1 than those in PSEN2 (P = 6.47 × 10–4). No difference was observed in the number of missense variants between PSEN1 and APP (P = 0.49). More frameshift variants were identified in PSEN2 than those in PSEN1 (P = 3.92 × 10–7). The splicing variant exhibited no difference between PSEN1 and PSEN2 (P = 0.13). Also, the indel variants in PSEN1 were similar to those in APP (P = 1.00).
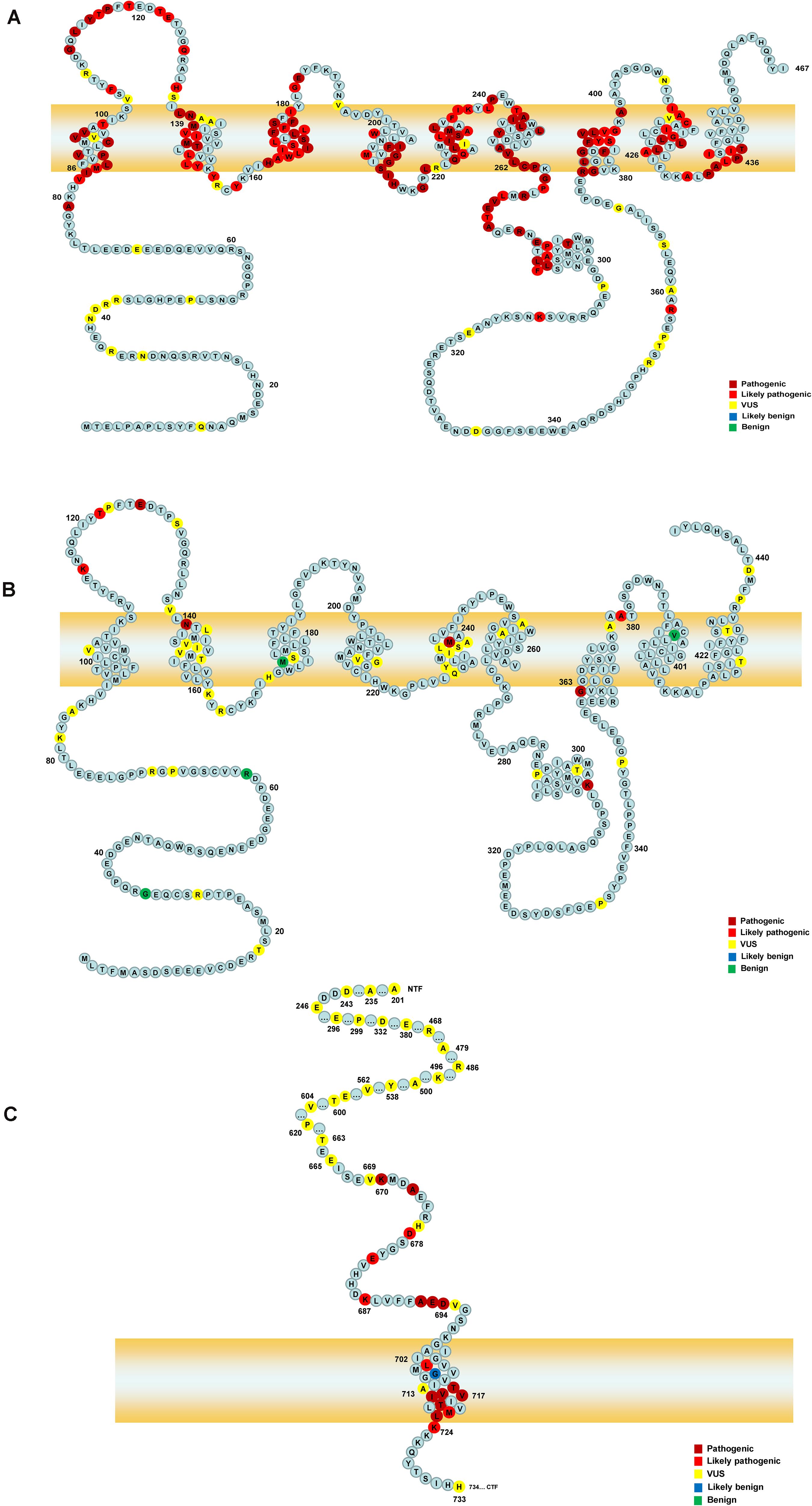
Figure 1. The schematic of PSEN1, PSEN2, and APP proteins. Panels (A,B) represent the full-length amino acid sequence of PSEN1 and PSEN2, respectively. Panel (C) shows a part of the amino acid sequence of APP. Each circle with colors represents the likelihood of pathogenicity. Crimson circles: Pathogenic variants; Red circles: Likely pathogenic variants; Yellow circles: VUS variants; Blue circles: Likely benign circles; Green circles: Benign variants.
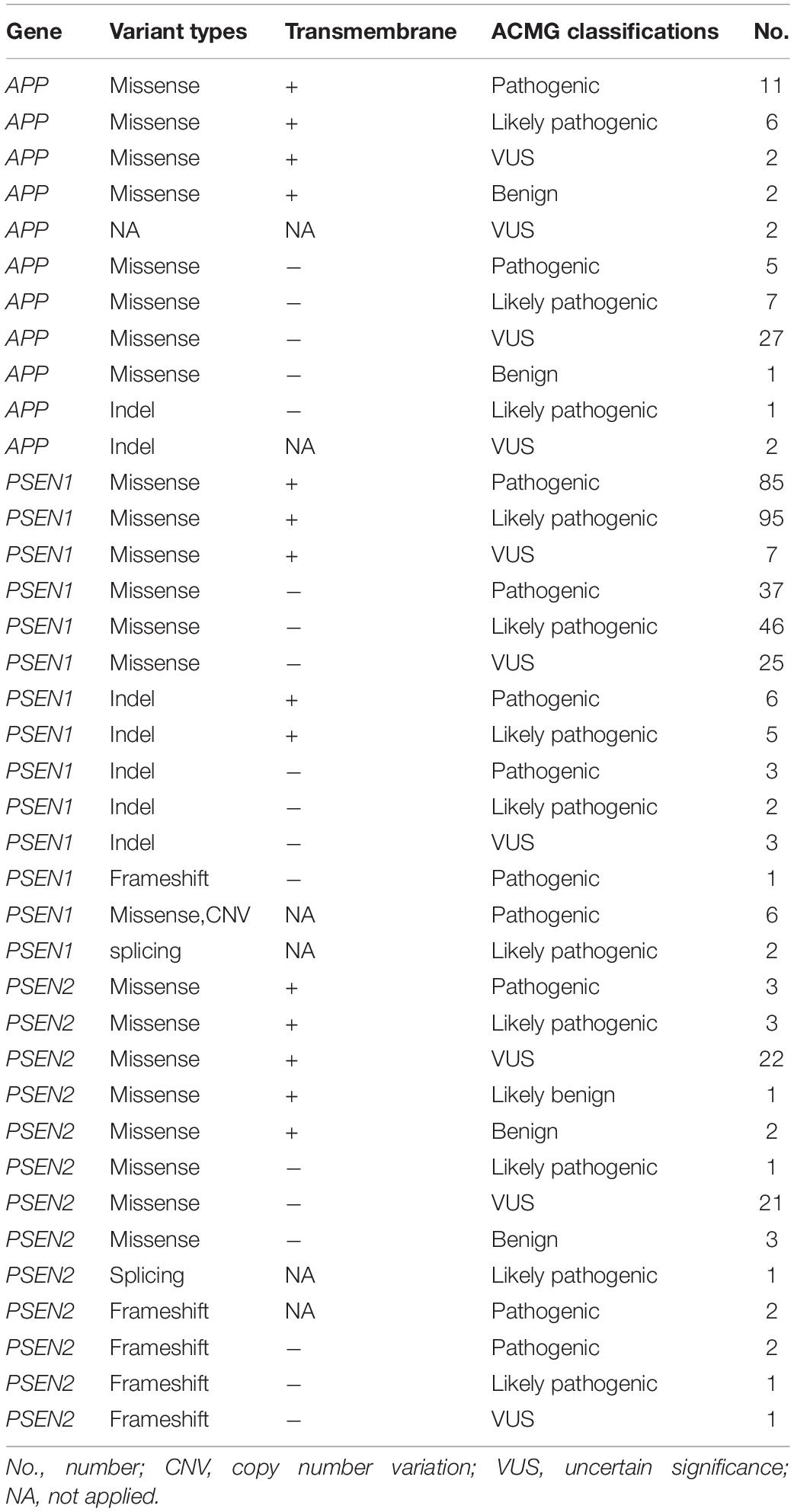
Table 2. ACMG classifications of three pathogenic variants in AD.
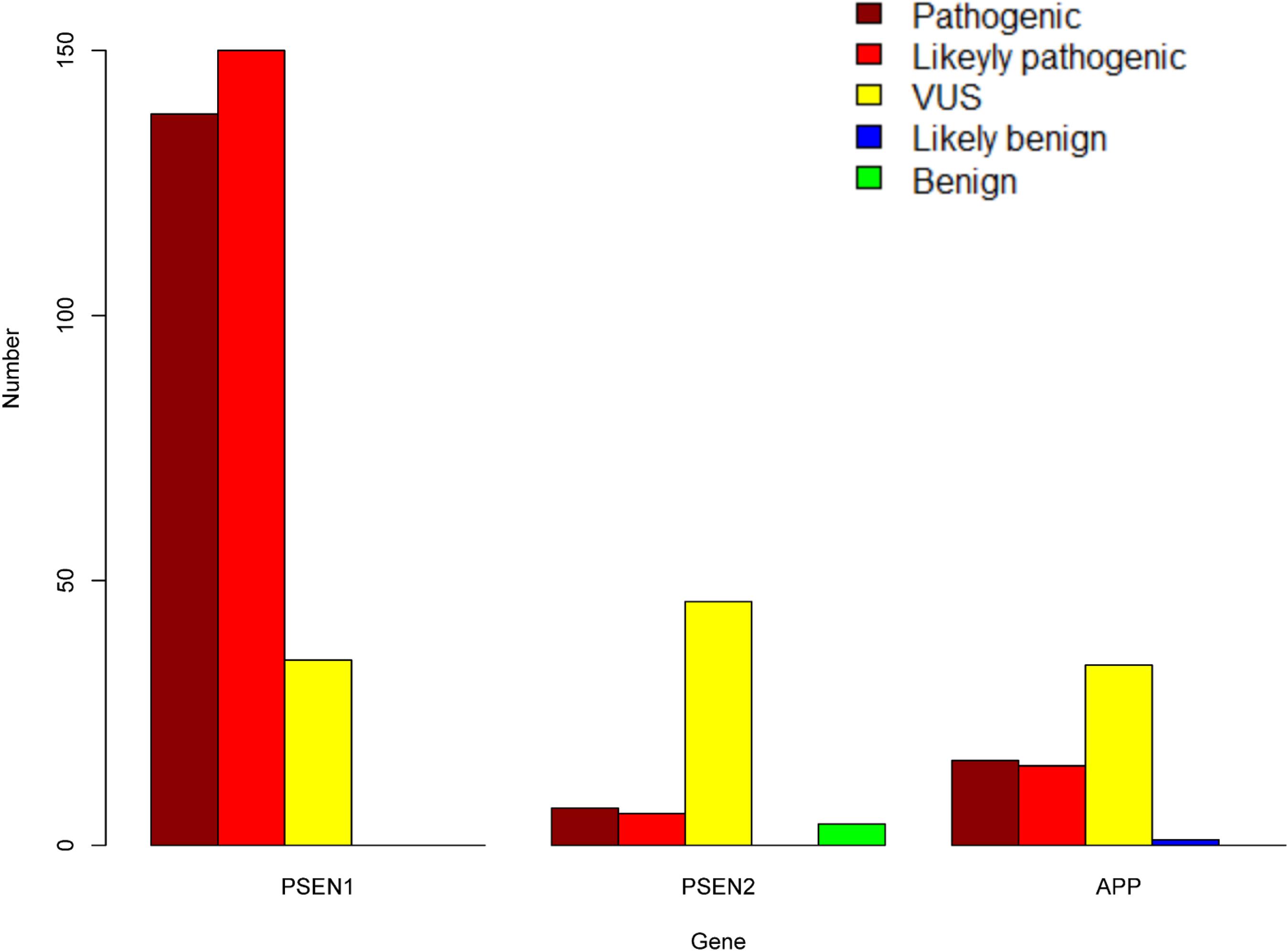
Figure 2. The number of pathogenic, likely pathogenic, VUS, likely benign, and benign variants in PSEN1, PSEN2, and APP.
PSEN1
323 PSEN1 variants were collected in our study, among them, according to the ACMG-AMP guidelines, 138 variants (42.72%) were re-classified as pathogenic variants, 150 variants (46.44%) likely pathogenic variants, and 35 variants (10.84%) VUS variants. 97.21% of previously reported pathogenic variants (279/287) were re-interpreted as pathogenic/likely pathogenic variants. PSEN1 variants were located in exon 7 (22.29%, n = 72), exon 5 (18.89%, n = 61), exon 8 (13.00%, n = 42),exon 4 (10.84%, n = 35), exon 6 (10.53%, n = 34), exon 11 (9.29%, n = 30), exon 12 (7.12%, n = 23), exon 10 (3.10%, n = 10), exon 9 (1.55%, n = 5), intron 8, exon 9 (1.86%, n = 6), intron 8/11 (0.62%, n = 2), exon 3 (0.31%, n = 1), exons 9–10, introns 8–10 (0.31%, n = 1), and intron 4 (0.31%, n = 1). Most of PSEN1 variants (91.95%) were found in exon 7, exon 5, exon 8, exon 6, exon 4, exon 11, and exon 12. No variants were detected in exon 1 and exon 2. There are five types of variants: 295 missense variants (91.33%), 19 indel variants (5.88%), two splicing variants (1.86%), six copy number variation (CNV) (0.62%), and one frameshift variant (0.31%). 198 variants (62.26%) were located in the transmembrane domain and 112 (35.22%) variants in the non-transmembrane domain, while eight variants (2.52%) were not applied. When it comes to the variants in the transmembrane domain, 191 variants were re-classified as pathogenic/likely pathogenic variants and eight variants VUS variants. In contrast, 89 variants in the non-transmembrane domain were re-interpreted as pathogenic/likely pathogenic variants, while 28 variants were VUS variants. The number of pathogenic/likely pathogenic variants differed significantly between transmembrane domain and non-transmembrane domain in PSEN1 gene (P = 2.04 × 10–7).
PSEN2
We re-analyzed 63 PSEN2 variants in our study. Seven variants (11.11%) were re-classified as pathogenic variants, six variants (9.52%) were likely pathogenic variants, 46 variants (73.02%) were VUS variants, and four variants (6.35%) were benign. Only 50% of previously reported pathogenic variants (8/16) fulfilled the criteria for pathogenicity/likely pathogenicity. With regard to variant type, 56 missense variants (88.89%), six frameshift variants (9.52%), and one splicing variant (1.59%) were identified. These variants belonged to exon 5 (28.57%, n = 18), exon 7 (23.81%, n = 15), exon 4 (14.29%, n = 9), exon 3 (4.76%, n = 3), exon 6 (4.76%, n = 3), exon 10 (4.76%, n = 3), exon 11 (4.76%, n = 3), exon 12 (6.35%, n = 4), exon 8 (1.59%, n = 1), exon 9 (1.59%, n = 1), intron 11/12 (3.17%, n = 2), and intron 9/12 (1.59%, n = 1). No variants were detected in exon 1, exon 2, and exon 3. 31 variants were located in the transmembrane domain and 29 variants in the non-transmembrane domain, while three variants couldn’t be determined. Six variants (20.00%) were re-classified as pathogenic/likely pathogenic variants in the transmembrane domain and four variants (14.29%) were pathogenic/likely pathogenic in the non-transmembrane domain. The number of pathogenic/likely pathogenic variants exhibited no significant difference between the transmembrane domain and non-transmembrane domain (P = 0.73).
APP
A total of 66 APP variants were re-evaluated in our study. 16 variants (24.24%) were re-classified as pathogenic variants, 15 likely pathogenic variants (22.73%), 34 VUS variants (51.52%), and one likely benign variant (1.51%) based on the ACMG-AMP guidelines. 96.77% of the previously pathogenic variants (30/31) were re-evaluated as pathogenic/likely pathogenic variants. 61 APP variants (92.42%) were missense variants, three variants (4.55%) were indel variants and two variants (3.03%) were located in UTR. These variants were located in exon 17 (45.45%, n = 30), exon 16 (18.18%, n = 12), exon 14 (7.58%, n = 5), exon 6 (4.55%, n = 3), exon 7 (4.55%, n = 3), exon 11 (4.55%, n = 3), exon 12 (3.03%, n = 2), exon 13 (3.03%, n = 2), exon 5 (1.52%, n = 1), exon 9 (1.52%, n = 1), 3′UTR (4.55%, n = 3), and intron 17 (1.52%, n = 1). No variants were found in exon 1, exon 2, exon 3, exon 4, exon 8, exon 10, exon 15, and exon 18. 21 variants were located in the transmembrane domain and 41 variants in the non-transmembrane domain. 17 variants were re-interpreted as pathogenic/likely pathogenic variants in the transmembrane domain while 14 variants were pathogenic/likely pathogenic in the non-transmembrane domain. The number of pathogenic/likely pathogenic variants showed a significant difference between the transmembrane domain and non-transmembrane domain (P = 1.01 × 10–3).
Discussion
In our study, for the first time, all APP, PSEN1, and PSEN2 variants were re-evaluated systematically on the basis of the ACMG-AMP guidelines. We found that 94.91% of previously reported pathogenic variants were re-evaluated as pathogenic/likely pathogenic variants, and the others were VUS variants. The most prevalent pathogenic/likely pathogenic variants were located in the PSEN1 gene, followed by the APP gene and the PSEN2 gene. Our study may have important implications in the molecular diagnosis of AD.
Thanks to the rapid development of sequencing technology, an increasing number of studies investigated the APP, PSEN1, and PSEN2 genetic spectrum worldwide, demonstrating their significant role in AD pathogenesis (Sassi et al., 2014; Gao et al., 2019). Nevertheless, the classification of variants remains a challenge (Lanoiselée et al., 2017). The heterogeneity of clinical data and the difference in the approaches used for variant interpretation are the major challenges in classifying variants (Denham et al., 2019). The APP, PSEN1, and PSEN2 genes have a high rate of rare variants, and the appropriate classification of them is essential in the correct diagnosis and treatment of AD. Furthermore, a high degree of variation in interpreting these variants existed in the previous studies, which may impose negative effects on clinical practice and scientific research. Consequently, re-evaluation of the APP, PSEN1, and PSEN2 variants is particularly relevant (Denham et al., 2019). The ACMG-AMP revised standards and guidelines to interpret variants using detailed criteria, such as variant types and population frequency (Richards et al., 2015). The ACMG-AMP guidelines are feasible and standard in classifying variants, which were widely applied in variant classification (Pakhrin et al., 2018; Peng et al., 2018). In some recent AD genetic screening studies, the variants of APP, PSEN1, and PSEN2 were also classified based on the ACMG-AMP guidelines (Xu et al., 2018; Jiang et al., 2019). However, to date, no study has evaluated all of the variants in APP, PSEN1 and PSEN2 reported previously. Consequently, in our study, all of the reported variants in these genes were re-classified according to the ACMG-AMP guidelines.
Our study indicated that most of the previously reported pathogenic variants (317/334) in APP, PSEN1, and PSEN2 were still re-classified as pathogenic/likely pathogenic variants, whereas 17 variants were re-evaluated as VUS variants, including one variant in the APP, eight variants in the PSEN1 and eight variants in the PSEN2. There are several reasons why the previously reported pathogenic variants were re-classified as VUS variants. Firstly, despite the extensive support evidence of pathogenicity of some variants, they were classified as VUS because some in silico prediction tools disagreed on the damaging effects of variants. Secondly, the existence of evidence of benign impact argued against the pathogenicity of variants. A few variants showed no damaging effect on Aβ production, arguing against their pathogenicity. Thirdly, a few variants were interpreted as VUS since they lacked enough evidence of pathogenicity. Take PSEN2 T122R for example, it fulfilled the criteria of PM2, PM5, and PP3. However, it was classified as VUS since there was no other evidence of pathogenicity. We identified that 25.44% (115/452) of APP, PSEN1, and PSEN2 variants were classified as VUS. To assess the VUS variants more accurately, increased collaborations, genetic data sharing, and well-established functional studies may be of great importance in the discovery and classification of VUS.
We demonstrated that the PSEN1 gene possessed the highest number of pathogenic/likely pathogenic variants, followed by the APP gene and the PSEN2 gene, which was consistent with our previous study. In 404 Chinese AD pedigrees, the most common mutated gene is PSEN1, also followed by APP and PSEN2 (Jia et al., 2020). Similarly, DIAN collected autosomal dominant AD globally and re-analyzed the variants’ pathogenicity using available information, showing that two PSEN1, one APP, and one PSEN2 are likely pathogenic variants (Hsu et al., 2018). 38 different PSEN1 mutations and six APP mutations were identified in patients with autosomal dominant familial Alzheimer’s disease (Ryan et al., 2016). 76 PSEN1, 6 PSEN2, and 6 APP symptomatic mutation carriers were recruited to characterize neuroimaging biomarkers change in DIAN (Gordon et al., 2018). Currently, 265 mutation carriers were included in DIAN, including 202 PSEN1, 22 PSEN2, and 43 APP mutation carriers (Castillo-Barnes et al., 2020; Luckett et al., 2021). In another France AD whole-exome sequencing study, three PSEN1 and one PSEN2 likely causative variants were identified (Nicolas et al., 2016). In short, PSEN1 is the most common pathogenic gene while APP or PSEN2 owns relatively few pathogenic variants in AD.
Our re-analysis found that most PSEN1 variants are located in exon 7, exon 5, exon 8, exon 6, exon 4, exon 11, and exon 12, most PSEN2 variants in exon 5, and exon 7, and most APP variants in exon 14, exon 16, exon 17, which was consistent with previous studies (Raux et al., 2005; Barber et al., 2016). In the Asain population, variants in exon 4, exon 7, exon 11, exon 14, and exon 17 of the APP, exon 4, exon 5, exon 6, exon 7, exon 8, and exon 12 of the PSEN1, as well as exon 5, exon 6, and exon 7 of the PSEN2 gene were discovered between 2009 and 2018 (Giau et al., 2019). In the French population, PSEN1 variants are located in exon 4, exon 5, exon 6, exon 7, exon 8, exon 9, exon 10, exon 11, and exon 12. Additionally, variants in exon 6 and exon 9 in the PSEN2 gene as well as variants in exon 17 were identified in AD patients (Lanoiselée et al., 2017). Generally, most of the pathogenic AD mutations are located in exons 16–17 of the APP, exons 3–12 of PSEN1, and exons 3–12 of PSEN2 genes (An et al., 2016). These results suggested the above exons are variant hotspots and needed to be given priority when performing DNA sequencing (Zhao and Liu, 2017). Moreover, in the pathogenic/likely pathogenic variants, missense variants are more common in PSEN1 than those in PSEN2. More frameshift variants were identified in PSEN2 than those in PSEN1. These results indicated that the importance of considering these variant type when interpreting variants’ pathogenicity.
Besides, we demonstrated that the number of pathogenic/likely pathogenic variants in the transmembrane domain is significantly higher than that of the non-transmembrane domain in PSEN1 and APP, whereas PSEN2 exhibited no difference. Wolfe et al. (1999) indicated that transmembrane aspartate of PSEN1 plays an essential role in γ-secretase activity. The APP transmembrane domain contains the γ-secretase site and it can bind with PSEN1, resulting in the production of Aβ (Esselens et al., 2012). PSEN1 shares a 60% sequence homology with PSEN2 with highly conserved transmembrane, and both of them are the sub-units of γ-secretase (Escamilla-Ayala et al., 2020). However, PSEN1 lacks the sorting motif found in PSEN2 and expresses broadly at the cell surface and endosomes (Kanatsu et al., 2014; Sannerud et al., 2016). The differences in subcellular localization between PSEN1 and PSEN2 may explain why the variants in the PSEN2 transmembrane domain are less likely to be pathogenic (Park et al., 2015).
In this study, the increased amount of Aβ or ratio of Aβ42 over Aβ40 in well-established studies was considered a piece of strong evidence of pathogenicity-PS3. It is noteworthy that prominent Aβ42 deposition not only causes senile plaques in the cortex but also may result in amyloid angiopathy in AD patients (Lemere et al., 1996). Despite an increasing number of functional works (Chen et al., 2015; Zhang et al., 2020), the absence of functional data is a major challenge in re-analyzing variants of APP, PSEN1, and PSEN2. Meanwhile, the lack of families’ sequencing results is also an important confounding factor because the de novo or co-segregation of a variant cannot be determined (Chen et al., 2012; Dobricic et al., 2012). Of note, only a small proportion of AD patients are caused by APP, PSEN1, and PSEN2. AD is a complex disease that risk genes may interact with environmental factors (Kunkle et al., 2019; Hsu et al., 2020). Thus, variants in AD should be classified carefully as some previously reported pathogenic variants may be risk ones rather than disease-causing in the pathogenesis of AD.
Taken together, in the current study, according to ACMG-AMP guidelines, we systematically re-analyzed the variants in APP, PSEN1, and PSEN2. We found that most of the previously pathogenic variants were re-classified as pathogenic/likely pathogenic variants, whereas a small proportion of previously pathogenic variants were interpreted as VUS variants. PSEN1 possessed the highest number of pathogenic/likely pathogenic variants, followed by APP and PSEN2. These findings may underscore the importance of classifying APP, PSEN1, and PSEN2 variants with caution.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
XX and BJ: study design, acquisition of data, analysis, interpretation of data, and drafting/revising the manuscript. HL, XL, WZ, and SZ: acquisition of data. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Key R&D Program of China (No. 2020YFC2008500) and the National Natural Science Foundation of China (No. 82071216).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2021.695808/full#supplementary-material
Supplementary Table 1 | The ACMG-AMP classifications of three genes in AD.
Footnotes
- ^ https://www.alzforum.org/mutations, accessed in March 2021
- ^ http://ncbi.nlm.nih.gov/pubmed
- ^ http://varcards.biols.ac.cn/
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
An, S. S., Park, S. A., Bagyinszky, E., Bae, S. O., Kim, Y. J., Im, J. Y., et al. (2016). A genetic screen of the mutations in the Korean patients with early-onset Alzheimer’s disease. Clin. Interv. Aging 11, 1817–1822. doi: 10.2147/cia.S116724
Auer, P. L., Reiner, A. P., Wang, G., Kang, H. M., Abecasis, G. R., Altshuler, D., et al. (2016). Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project. Am. J. Hum. Genet. 99, 791–801. doi: 10.1016/j.ajhg.2016.08.012
Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Barber, I. S., García-Cárdenas, J. M., Sakdapanichkul, C., Deacon, C., Zapata Erazo, G., Guerreiro, R., et al. (2016). Screening exons 16 and 17 of the amyloid precursor protein gene in sporadic early-onset Alzheimer’s disease. Neurobiol. Aging 39, .e221–.e227. doi: 10.1016/j.neurobiolaging.2015.12.011
Bateman, R. J., Benzinger, T. L., Berry, S., Clifford, D. B., Duggan, C., Fagan, A. M., et al. (2017). The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement. 13, 8–19. doi: 10.1016/j.jalz.2016.07.005
Castillo-Barnes, D., Su, L., Ramírez, J., Salas-Gonzalez, D., Martinez-Murcia, F. J., Illan, I. A., et al. (2020). Autosomal Dominantly Inherited Alzheimer Disease: Analysis of genetic subgroups by Machine Learning. Inf. Fusion 58, 153–167. doi: 10.1016/j.inffus.2020.01.001
Chen, W. T., Hong, C. J., Lin, Y. T., Chang, W. H., Huang, H. T., Liao, J. Y., et al. (2012). Amyloid-beta (Aβ) D7H variant increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies. PLoS One 7:e35807. doi: 10.1371/journal.pone.0035807
Chen, W. T., Hsieh, Y. F., Huang, Y. J., Lin, C. C., Lin, Y. T., Liu, Y. C., et al. (2015). G206D variant of Presenilin-1 Reduces Pen2 Interaction, Increases Aβ42/Aβ40 Ratio and Elevates ER Ca(2+) Accumulation. Mol. Neurobiol. 52, 1835–1849. doi: 10.1007/s12035-014-8969-1
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R., and Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688. doi: 10.1371/journal.pone.0046688
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Denham, N. C., Pearman, C. M., Ding, W. Y., Waktare, J., Gupta, D., Snowdon, R., et al. (2019). Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J. Cardiovasc. Electrophysiol. 30, 118–127. doi: 10.1111/jce.13740
Dobricic, V., Stefanova, E., Jankovic, M., Gurunlian, N., Novakovic, I., Hardy, J., et al. (2012). Genetic testing in familial and young-onset Alzheimer’s disease: variant spectrum in a Serbian cohort. Neurobiol. Aging 33, .e1487–.e1412. doi: 10.1016/j.neurobiolaging.2011.12.007
Escamilla-Ayala, A., Wouters, R., Sannerud, R., and Annaert, W. (2020). Contribution of the Presenilins in the cell biology, structure and function of γ-secretase. Semin. Cell Dev. Biol. 105, 12–26. doi: 10.1016/j.semcdb.2020.02.005
Esselens, C., Sannerud, R., Gallardo, R., Baert, V., Kaden, D., Serneels, L., et al. (2012). Peptides based on the presenilin-APP binding domain inhibit APP processing and Aβ production through interfering with the APP transmembrane domain. Faseb J. 26, 3765–3778. doi: 10.1096/fj.11-201368
Gao, Y., Ren, R. J., Zhong, Z. L., Dammer, E., Zhao, Q. H., Shan, S., et al. (2019). variant profile of APP, PSEN1, and PSEN2 in Chinese familial Alzheimer’s disease. Neurobiol. Aging 77, 154–157. doi: 10.1016/j.neurobiolaging.2019.01.018
Giau, V. V., Bagyinszky, E., Youn, Y. C., An, S. S. A., and Kim, S. (2019). APP, PSEN1, and PSEN2 variants in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 20:19. doi: 10.3390/ijms20194757
Gordon, B. A., Blazey, T. M., Su, Y., Hari-Raj, A., Dincer, A., Flores, S., et al. (2018). Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol. 17, 241–250. doi: 10.1016/s1474-4422(18)30028-0
Hsu, S., Gordon, B. A., Hornbeck, R., Norton, J. B., Levitch, D., Louden, A., et al. (2018). Discovery and validation of autosomal dominant Alzheimer’s disease variants. Alzheimers Res. Ther. 10:67. doi: 10.1186/s13195-018-0392-9
Hsu, S., Pimenova, A. A., Hayes, K., Villa, J. A., Rosene, M. J., Jere, M., et al. (2020). Systematic validation of variants of unknown significance in APP, PSEN1 and PSEN2. Neurobiol. Dis. 139:104817. doi: 10.1016/j.nbd.2020.104817
Ioannidis, N. M., Rothstein, J. H., Pejaver, V., Middha, S., McDonnell, S. K., Baheti, S., et al. (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 99, 877–885. doi: 10.1016/j.ajhg.2016.08.016
Jack, C. R. Jr., Bennett, D. A., Blennow, K., Carrillo, M. C., Dunn, B., Haeberlein, S. B., et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14, 535–562. doi: 10.1016/j.jalz.2018.02.018
Jia, L., Fu, Y., Shen, L., Zhang, H., Zhu, M., Qiu, Q., et al. (2020). PSEN1, PSEN2, and APP variants in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimers Dement 16, 178–191. doi: 10.1002/alz.12005
Jiang, B., Zhou, J., Li, H. L., Chen, Y. G., Cheng, H. R., Ye, L. Q., et al. (2019). variant screening in Chinese patients with familial Alzheimer’s disease by whole-exome sequencing. Neurobiol. Aging 76, e215–.e215. doi: 10.1016/j.neurobiolaging.2018.11.024
Kanatsu, K., Morohashi, Y., Suzuki, M., Kuroda, H., Watanabe, T., Tomita, T., et al. (2014). Decreased CALM expression reduces Aβ42 to total Aβ ratio through clathrin-mediated endocytosis of γ-secretase. Nat. Commun. 5:3386. doi: 10.1038/ncomms4386
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Lanoiselée, H. M., Nicolas, G., Wallon, D., Rovelet-Lecrux, A., Lacour, M., Rousseau, S., et al. (2017). APP, PSEN1, and PSEN2 variants in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 14:e1002270. doi: 10.1371/journal.pmed.1002270
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lemere, C. A., Lopera, F., Kosik, K. S., Lendon, C. L., Ossa, J., Saido, T. C., et al. (1996). The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat. Med. 2, 1146–1150. doi: 10.1038/nm1096-1146
Li, J., Shi, L., Zhang, K., Zhang, Y., Hu, S., Zhao, T., et al. (2018a). VarCards: an integrated genetic and clinical database for coding variants in the human genome. Nucleic Acids Res. 46, D1039–D1048. doi: 10.1093/nar/gkx1039
Li, J., Zhao, T., Zhang, Y., Zhang, K., Shi, L., Chen, Y., et al. (2018b). Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res. 46, 7793–7804. doi: 10.1093/nar/gky678
Li, Q., and Wang, K. (2017). InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 100, 267–280. doi: 10.1016/j.ajhg.2017.01.004
Loy, C. T., Schofield, P. R., Turner, A. M., and Kwok, J. B. (2014). Genetics of dementia. Lancet 383, 828–840. doi: 10.1016/s0140-6736(13)60630-3
Luckett, P. H., McCullough, A., Gordon, B. A., Strain, J., Flores, S., Dincer, A., et al. (2021). Modeling autosomal dominant Alzheimer’s disease with machine learning. Alzheimers Dement 2021, 12259. doi: 10.1002/alz.12259
Morris, J. C., Aisen, P. S., Bateman, R. J., Benzinger, T. L., Cairns, N. J., Fagan, A. M., et al. (2012). Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin. Investig. 2, 975–984. doi: 10.4155/cli.12.93
Ng, P. C., and Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Nicolas, G., Wallon, D., Charbonnier, C., Quenez, O., Rousseau, S., Richard, A. C., et al. (2016). Screening of dementia genes by whole-exome sequencing in early-onset Alzheimer disease: input and lessons. Eur. J. Hum. Genet. 24, 710–716. doi: 10.1038/ejhg.2015.173
Pakhrin, P. S., Xie, Y., Hu, Z., Li, X., Liu, L., Huang, S., et al. (2018). Genotype-phenotype correlation and frequency of distribution in a cohort of Chinese Charcot-Marie-Tooth patients associated with GDAP1 variants. J. Neurol. 265, 637–646. doi: 10.1007/s00415-018-8743-9
Park, H. J., Ran, Y., Jung, J. I., Holmes, O., Price, A. R., Smithson, L., et al. (2015). The stress response neuropeptide CRF increases amyloid-β production by regulating γ-secretase activity. Embo J. 34, 1674–1686. doi: 10.15252/embj.201488795
Peng, Y., Ye, W., Chen, Z., Peng, H., Wang, P., Hou, X., et al. (2018). Identifying SYNE1 Ataxia With Novel variants in a Chinese Population. Front. Neurol. 9:1111. doi: 10.3389/fneur.2018.01111
Raux, G., Guyant-Maréchal, L., Martin, C., Bou, J., Penet, C., Brice, A., et al. (2005). Molecular diagnosis of autosomal dominant early onset Alzheimer’s disease: an update. J. Med. Genet. 42, 793–795. doi: 10.1136/jmg.2005.033456
Reva, B., Antipin, Y., and Sander, C. (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39:e118. doi: 10.1093/nar/gkr407
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Ryan, N. S., Nicholas, J. M., Weston, P. S. J., Liang, Y., Lashley, T., Guerreiro, R., et al. (2016). Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol. 15, 1326–1335. doi: 10.1016/s1474-4422(16)30193-4
Saez-Atienzar, S., and Masliah, E. (2020). Cellular senescence and Alzheimer disease: the egg and the chicken scenario. Nat. Rev. Neurosci. 21, 433–444. doi: 10.1038/s41583-020-0325-z
Sannerud, R., Esselens, C., Ejsmont, P., Mattera, R., Rochin, L., Tharkeshwar, A. K., et al. (2016). Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell 166, 193–208. doi: 10.1016/j.cell.2016.05.020
Sassi, C., Guerreiro, R., Gibbs, R., Ding, J., Lupton, M. K., Troakes, C., et al. (2014). Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol. Aging 35, .e2881–.e2881. doi: 10.1016/j.neurobiolaging.2014.06.002
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Shihab, H. A., Gough, J., Cooper, D. N., Stenson, P. D., Barker, G. L., Edwards, K. J., et al. (2013). Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65. doi: 10.1002/humu.22225
Sun, L., Zhou, R., Yang, G., and Shi, Y. (2017). Analysis of 138 pathogenic variants in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. U S A 114, E476–E485. doi: 10.1073/pnas.1618657114
Wolfe, M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T., and Selkoe, D. J. (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398, 513–517. doi: 10.1038/19077
Xu, Y., Liu, X., Shen, J., Tian, W., Fang, R., Li, B., et al. (2018). The Whole Exome Sequencing Clarifies the Genotype- Phenotype Correlations in Patients with Early-Onset Dementia. Aging Dis. 9, 696–705. doi: 10.14336/ad.2018.0208
Zhang, S., Cai, F., Wu, Y., Bozorgmehr, T., Wang, Z., Zhang, S., et al. (2020). A presenilin-1 variant causes Alzheimer disease without affecting Notch signaling. Mol. Psychiatry 25, 603–613. doi: 10.1038/s41380-018-0101-x
Keywords: Alzheimer’s disease, ACMG-AMP guidelines, APP, PSEN1, PSEN2, re-evaluation
Citation: Xiao X, Liu H, Liu X, Zhang W, Zhang S and Jiao B (2021) APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-evaluation According to ACMG Guidelines. Front. Aging Neurosci. 13:695808. doi: 10.3389/fnagi.2021.695808
Received: 15 April 2021; Accepted: 31 May 2021;
Published: 18 June 2021.
Edited by:
Nicola Ticozzi, University of Milan, ItalyReviewed by:
Francesca Luisa Conforti, University of Calabria, ItalyDiego Castillo-Barnes, University of Granada, Spain
Copyright © 2021 Xiao, Liu, Liu, Zhang, Zhang and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Jiao, NDAxMTA3MEBjc3UuZWR1LmNu