 Edoardo Nicolò Aiello1,2*†
Edoardo Nicolò Aiello1,2*† Sarah Feroldi2
Sarah Feroldi2 Giulia De Luca3Lucilla Guidotti4Eleonora Arrigoni2
Giulia De Luca3Lucilla Guidotti4Eleonora Arrigoni2 Ildebrando Appollonio5
Ildebrando Appollonio5 Federica Solca1
Federica Solca1 Laura Carelli1
Laura Carelli1 Barbara Poletti1†
Barbara Poletti1† Federico Verde1,6
Federico Verde1,6 Vincenzo Silani1,6†
Vincenzo Silani1,6† Nicola Ticozzi1,6
Nicola Ticozzi1,6- 1Department of Neurology and Laboratory of Neuroscience, IRCCS Istituto Auxologico Italiano, Milan, Italy
- 2Ph.D. Program in Neuroscience, School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 3Department of Brain and Behavioral Sciences, University of Pavia, Pavia, Italy
- 4Department of Psychology, University of Milano-Bicocca, Milan, Italy
- 5Neurology Section, School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy
- 6Department of Pathophysiology and Transplantation, “Dino Ferrari” Center, Università degli Studi di Milano, Milan, Italy
Background: This study aims at reviewing, within the framework of motor neuron disease-frontotemporal degeneration (MND-FTD)-spectrum disorders, evidence on the co-occurrence between primary progressive aphasia (PPA) and MND in order to profile such a complex at pathological, genetic and clinical levels.
Methods: This review was pre-registered (osf.io/ds8m4) and performed in accordance with the 2020 PRISMA guidelines. Case reports/series and group studies were included if addressing (1) progressive non-fluent aphasia (PNFA) or semantic dementia (SD) with MND or (2) MND patients with co-morbid PNFA/SD.
Results: Out of 546 initial records, 56 studies were included. As to case reports/series (N = 35), which included 61 PPA-MND patients, the following findings yielded: (1) PNFA is more frequent than SD in PPA-MND; (2) in PPA-MND, the most prevalent motor phenotypes are amyotrophic lateral sclerosis and predominant-upper MND, with bulbar involvement being ubiquitous; (3) extrapyramidal features are moderately frequent in PPA-MND; (4) PPA-MND patients usually display frontotemporal, left-greater-than-right involvement; (5) TDP-43-B is the typical pathological substrate of PPA-MND; (6) TBK1 mutations represent the most frequent genetic risk factors for PPA-MND.
As to group studies, including 121 patients, proportional meta-analytic procedures revealed that: (1) the lifetime prevalence of MND in PPA is 6%; (2) PPA occurs in 19% of patients with co-morbid MND and FTD; (3) MND is more frequent in PNFA (10%) than in SD patients (3%).
Discussion: Insights herewith delivered into the clinical, neuropathological and genetic features of PPA-MND patients prompt further investigations aimed at improving clinical practice within the MND-FTD spectrum.
Introduction
Due to the pathophysiological and genetic grounds shared by motor neuron diseases (MNDs) and frontotemporal degenerations (FTDs), these two disorders belong to the same nosological entity, i.e., the “MND-FTD spectrum” (Burrell et al., 2016). FTD-like cognitive and behavioral dysfunctions indeed occur in up to 50% of MND patients (Strong et al., 2017), 5–15% of whom showing a full-blown FTD (Montuschi et al., 2015). However, such a spectrum can be also “read backward” – i.e., as “FTD-MND” (Strong et al., 2017): evidence of upper (UMN) and/or lower motor neuron (LMN) dysfunction can be in fact detected in up to 30% of FTD patients (Burrell et al., 2011; Cerami et al., 2015; Gromicho et al., 2021).
Among FTDs, the behavioural variant (bvFTD) is the most prevalent phenotype associated with MNDs (Saxon et al., 2017a,b; Wagner et al., 2021): the co-occurrence of MND or UMN/LMN dysfunction and bvFTD has been indeed thoroughly explored (Ahmed et al., 2021). By contrast, little is still known about the pathology, genetics and clinical features of co-occurring primary progressive aphasia (PPA) and MND (Ulugut et al., 2021). In the light of the diagnostic (Strong et al., 2017; Suárez-González et al., 2021) and prognostic relevance (Garcin et al., 2009; Ahmed et al., 2020; De La Sablonnière et al., 2021) of language disorders in MND patients and of motor neuron (MN) dysfunction in PPA patients, the present study aims at systematically reviewing evidence on the co-occurrence between PPA and MND (hereafter referred to as “PPA-MND”).
Methods
This review was pre-registered (OSF Registries: osf.io/ds8m4) and performed in accordance with the 2020 revision of Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines (PRISMA) (Page et al., 2021).
Search
Records were searched for in Scopus and PubMed on August 8th, 2021 through the following string: (“motor neuron disease” OR “amyotrophic lateral sclerosis”) AND (“primary progressive aphasia” OR “progressive aphasia” OR “progressive non-fluent aphasia” OR “progressive non-fluent aphasia” OR “semantic dementia” OR “semantic variant” OR “agrammatic variant” OR “non-fluent variant” OR “non-fluent variant”). The string did not referred to logopenic-variant PPA (lvPPA), as being commonly underpinned by a neuropathology different from frontotemporal lobar degeneration (FTLD), i.e., that of Alzheimer’s disease (AD) (Roytman and Chiang, 2022).
Fields of search were the title, abstract and possibly key words. No date limit was set. Only peer-reviewed articles written in English, Italian, German, French or Spanish were considered. Records not indexed within online databases were not searched for. Further potentially relevant contributions were retrieved from the reference lists of included ones.
Inclusion and exclusion criteria
For a study to be included, it had to address either (1) progressive non-fluent aphasia (PNFA) or semantic dementia (SD) patients with MND or clinical/electromyographic evidence of MN dysfunction or (2) MND patients with co-morbid PNFA/SD.
Either Neary et al.’s (1998) or Gorno-Tempini et al.’s (2011) criteria were addressed for PNFA and SD diagnoses.
If neither of the two nosographic systems were referred to, a clinical diagnosis of PPA and/or a cluster of language deficits consistent with a progressive, either fluent or non-fluent aphasic syndrome was deemed as satisfactory for inclusion, based on a joint decision of two Authors expert in aphasiology (E.N.A. and S.F.).
Studies describing anarthric patients whose language was assessable only through writing were included only if an explicit mention to aphasia was made (Aiello et al., 2021).
Both case report/series and group studies were addressed as eligible. Abstracts, reviews, meta-analyses, research protocols and opinion papers were excluded.
Data collection and synthesis
Screening and eligibility checks were performed by two independent Authors (E.N.A. and S.F.), with a third independent Author supervising and solving disagreements (I.A.). Decisions during screening and eligibility stages were performed via Rayyan1. Data were extracted by four Authors (S.F., G.D.L., L.G., and E.A.) and independently checked by a third Author (E.N.A.).
The following outcomes were extracted: (1) age and sex; (2) PPA phenotype; (3) MND phenotype – according to Chiò et al.’s (2011) classification – or presence of MN dysfunction; (4) neuropsychological vs. motor onset; (5) survival; (6) clinical course, i.e., whether PPA or MND occurred first, and timespan between their occurrence; (7) bulbar signs at MND onset and their lifetime prevalence; (8) early bulbar involvement in “PPA-first” patients; (9) presence and type of extrapyramidal involvement; (10) cortical neuroradiological findings; (11) neuropathological findings; (12) genetics.
A qualitative synthesis of extracted data was performed by inserting them within pilot-tested tables. Additionally, by addressing studies including ≥30 patients, possibly eligible for prevalence/incidence estimation, a proportional meta-analytic procedure via the R package metafor (Viechtbauer, 2010) was performed, by addressing a random-effect model with I2 as the heterogeneity measure.
Results
Study selection process is shown in Supplementary Figure 1. Out of 546 initial unique records, 56 studies were included, of which 36 were case reports/series (Table 1; N = 61 patients) and 20 group studies (Table 2; N = 121 patients).
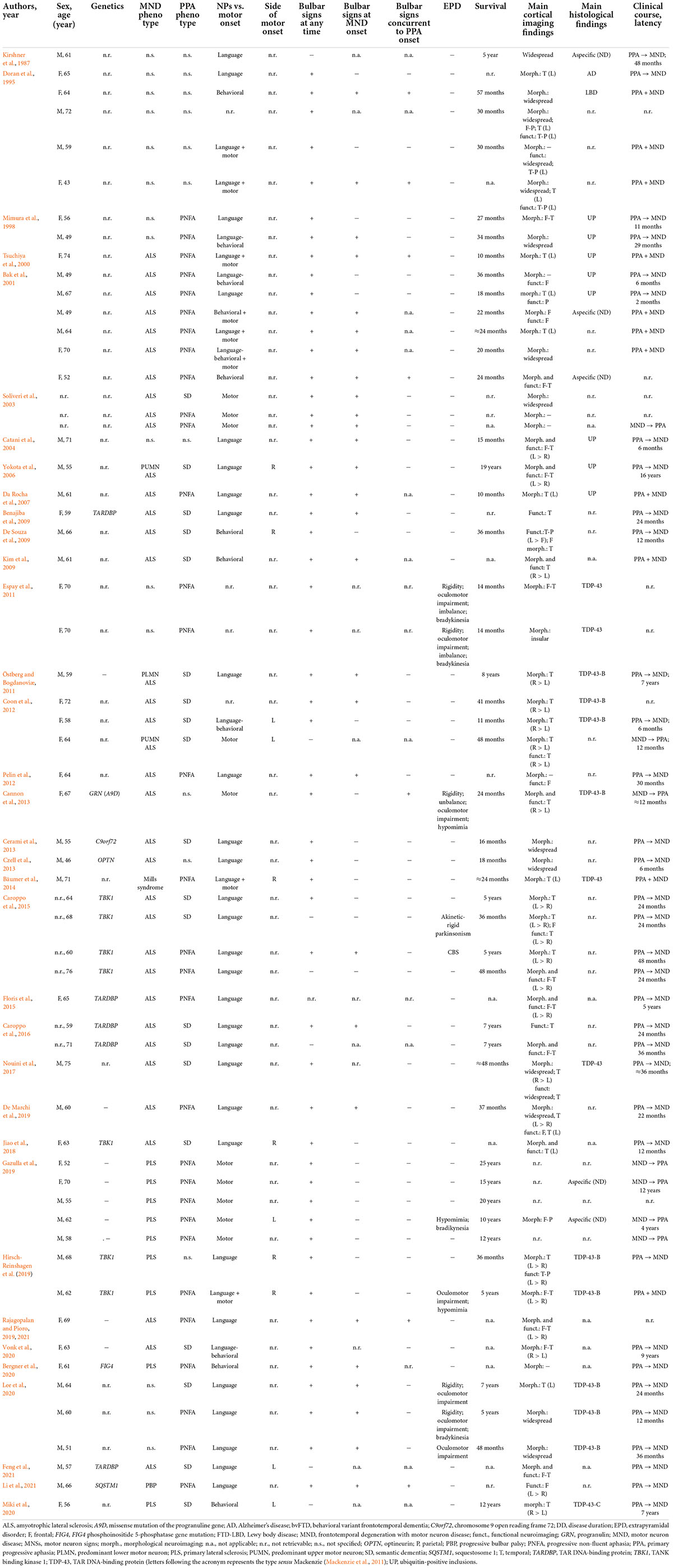
Table 1. Case reports and series.
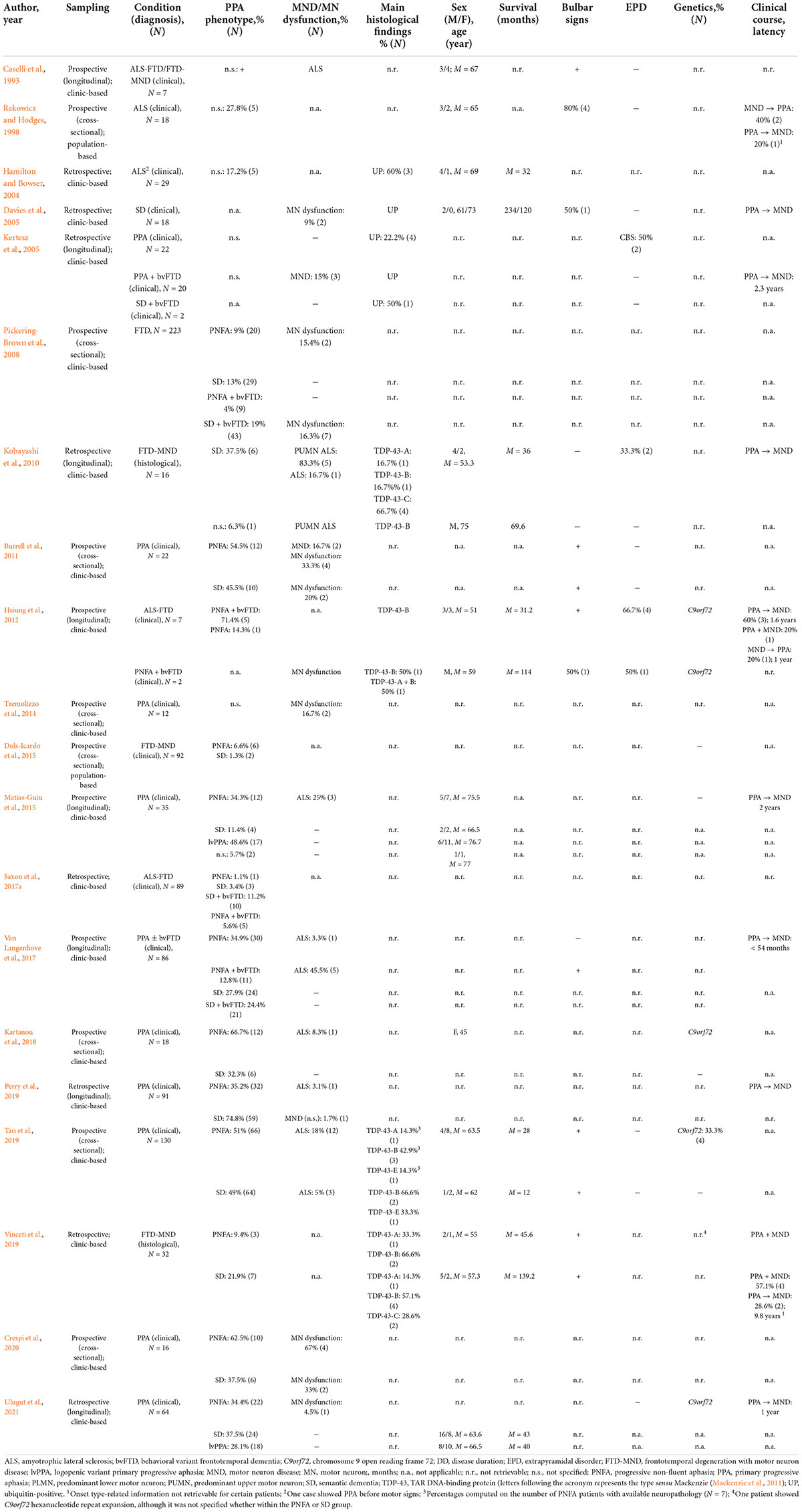
Table 2. Group studies.
Case reports and series
Within case reports and case series, 61 patients were described with PPA-MND (55.8% males; mean age: 62 ± 7.5 years, range = 43–76). Onset symptoms distributed as follows: only language: 50.9%; only motor: 17.5%; only behavioral: 10.5% language + motor: 10.5%; language + behavioral: 7%; motor + behavioral ± language: 3.5%. The median delay between the occurrence of PPA and MND/MN dysfunction was 24 months (mean: 40.5 ± 44), whereas that between MND/MN dysfunction and PPA was 12 months (mean: 24 ± 20.6), with 65.4% of patients showing PPA first. PNFA was more prevalent (60.8%) than SD (39.2%). Classical amyotrophic lateral sclerosis (ALS) was the most prevalent MND phenotype (70.2%), followed by predominant-UMN phenotypes (25.5%), including primary lateral sclerosis and Mills’ syndrome (Jaiser et al., 2019). The lifetime prevalence of bulbar signs was 88.3%, whilst that at MND onset was 56%. In 15.2% of “PPA first” patients, bulbar involvement preceded that of other bodily regions. The lifetime prevalence of extrapyramidal involvement was 16.4%. Survival was highly heterogeneous, with a median of 36.5 months (mean: 62.2 ± 64.6 months), ranging from 10 months to 25 years. Neuroradiological data were overall consistent with a left-predominant, frontotemporal, perisylvian involvement, except for 14% of patients showing SD with a selective right-sided temporal involvement. The most frequent post-mortem finding was, for studies before 2007 (N = 9 patients with available neuropathology), ubiquitin-positive inclusions (66.7%), while, for studies after 2007 (N = 17 patients with available neuropathology), TDP-43-B pathology [52.9%; sensu Mackenzie (Mackenzie et al., 2011)]. Among genetic-positive patients (N = 17), the most frequently mutated gene was TBK1 (41.2%), followed by TARDBP (29.4%).
Group studies
Group studies reporting PPA-MND patients addressed heterogeneous conditions falling under the FTD (N = 12), ALS-FTD (i.e., ALS as the primary diagnosis; N = 2) or FTD-MND (i.e., FTD as the primary diagnosis; N = 3) phenotypes (Strong et al., 2017) – except for two, which addressed ALS (Rakowicz and Hodges, 1998; Hamilton and Bowser, 2004), and another not specifying which the primary diagnosis was (either ALS-FTD or FTD-MND) (Caselli et al., 1993). Longitudinal analyses were detected in eight studies, focused either on PPA (N = 5) or ALS-FTD/FTD-MND (N = 3).
Seven eligible studies addressing PPA patients were entered into the proportional meta-analysis, yielding an estimate of lifetime prevalence of MND/MN dysfunction in PPA of 6% (CI 95% [3%, 9%]) (Supplementary Figure 2). When separately assessing PNFA and SD (N = 4 eligible studies), such a prevalence was of 10% in PNFA (CI 95% [3%, 17%]) and of 3% in SD (CI 95% [0%, 6%]) (Supplementary Figure 3). MND/MN dysfunction was never reported in lvPPA patients within the two studies also addressing this phenotype (Matias-Guiu et al., 2015; Ulugut et al., 2021).
As to the prevalence of PPA in ALS-FTD/FTD-MND, the 3 eligible, entered studies yielded an estimate of 19% (CI 95% [6%, 31%]) (Supplementary Figure 4) – and, when separately assessing PNFA and SD, of 7% for the former CI 95% [4%, 10%]) and of 11% for the latter (CI 95% [0%, 23%]), respectively (Supplementary Figure 5).
Besides prevalence- and phenotype-related information on PPA phenotypes, remaining data was often non-retrievable for individual patients, and thus could not be qualitatively summarized (Table 1). However, regarding neuropathology, it is worth mentioning that the large study by Tan et al. (2019), which addressed 130 PPA patients, of whom 11.5% had concurrent ALS, converged with case reports/series as to TDP-43-B being the most prevalent pathological substrate (50%). Moreover, with respect to genetics, it has to be noted that, within the same study, the sole, incidental finding in mutated patients (showing PNFA) was C9orf72 hexanucleotide repeat expansion (HRE) (Tan et al., 2019).
Discussion
The present study summarizes current evidence on the association between PPA and MND/MN dysfunction (i.e., PPA-MND) at clinical, pathological and genetic levels (Burrell et al., 2016).
Clinical phenotypes
The lifetime prevalence of MND/MN dysfunction in PPA patients was of 6%. Although no meta-analytic evidence on the topic is available for bvFTD, empirical studies suggest that, in such a phenotype, this prevalence would be similar (Cerami et al., 2015), or slightly higher (Tremolizzo et al., 2014; Ganapathy et al., 2020). Moreover, when separately addressing PPA phenotypes, such an estimate was higher for PNFA (10%) than for SD (3%) – this being explainable according to a corticofugal model of TDP-43-mediated neurodegeneration: indeed, PNFA primarily affects frontal-insular areas contiguous to motor regions, whereas SD mostly involves anterior temporal lobes, which are topographically distant from motor cortices (Braak et al., 2013; Brettschneider et al., 2013).
By contrast, the finding of MND/MN dysfunction never co-occurring with lvPPA was in line with the latter being commonly underpinned by a non-FTLD neuropathology (Roytman and Chiang, 2022).
The finding of 19% of ALS-FTD/FTD-MND patients presenting with PPA (with similar estimates for PNFA and SD – i.e., 7 and 11%, respectively) was likewise expected, given that PPA is known to be less commonly associated to MND than bvFTD (Saxon et al., 2017a,b; Lulé et al., 2019).
As to MND phenotypes, regardless of PPA variants, classical ALS was the most prevalent (70.2%), followed by predominant-UMN phenotypes (25.5%) – with only one patient (2.1%) showing a predominant-LMN phenotype. As to patients primarily diagnosed with FTD, such findings are unsurprising: indeed, it is unlikely that, in the presence of such a cortical pathology, UMNs are less involved than LMNs (McKenna et al., 2021). Conversely, as to primarily MND patients, these results align with recent evidence on extra-motor cortical burden being greater when UMNs are involved (Poletti et al., 2021; Sbrollini et al., 2021; Aiello et al., 2022; Maranzano et al., 2022).
The high lifetime prevalence of bulbar signs (88.3%), as well as the moderate proportion of bulbar-onset MNDs (56%), is then consistent with bulbar involvement being acknowledged, within the MND-FTD spectrum, as a risk factor for cognitive deficits (Yang et al., 2021), as well as with it being specifically linked to impairments of language as compared to other cognitive domains (Shellikeri et al., 2017).
As to extrapyramidal signs/syndromes, the present prevalence in PPA-MND (16.4%) overall resembles that previously detected in bvFTD (22.7%) (Padovani et al., 2007) and MND alone – ranging from 1.7 to 15.7% (McCluskey et al., 2014; Pupillo et al., 2015; Pasquini et al., 2022), being nevertheless lower than current estimates addressed to PPAs (i.e., up to 40%) (Kremen et al., 2011; Armstrong et al., 2013; Höglinger et al., 2017; Ulugut et al., 2021).
A relatively stable finding was patients’ age at onset/clinical referral, being among the fourth and seventh decade (62 years on average) – this overall aligning with the current epidemiological knowledge on PPA (Montembeault et al., 2018) and MND (Longinetti and Fang, 2019).
As to the median survival of PPA-MND patients – i.e., ≈3 years –, it was slightly lower than that of patients with PPA only (Montembeault et al., 2018), this agreeing with the fact that MN involvement represents a risk factor for a shorter survival in PPA patients (De La Sablonnière et al., 2021; El-Wahsh et al., 2021). Its high heterogeneity herewith found might be accounted for by the diversities across both PPA and MND phenotypes – since survival is longer in SD than in PNFA (Tastevin et al., 2021), and shorter in classical ALS as compared to atypical MND variants – such as predominant-UMN phenotypes (Turner and Talbot, 2020), which were relatively highly represented (25.5%) among PPA-MND patients.
Finally, PPA-MND patients frequently appeared (65.4%) to present with PPA first. However, a considerable, and extremely variable, delay between the two diseases was found – being higher for “PPA-first” patients (median: 24 months) than for “MND-first” ones (median: 12 months). Such findings partially align with a recent investigation (Gromicho et al., 2021) reporting a median delay of 1 year between the onset of bvFTD and MN dysfunction in FTD-MND patients.
Neuropathology
When referring to post-2007 studies reporting post-mortem data according to current TDP-43 classification systems (Mackenzie et al., 2011), the most common histological finding, regardless of PPA variants, was TDP-43-B (≈50% of cases), in line with this subtype being the pathological substrate of ALS-FTD/FTD-MND phenotypes (Mackenzie et al., 2011). However, group studies also showed a considerable heterogeneity of neuropathological features – including TDP-43-A and TDP-43-C, typical of PNFA and SD alone (Mackenzie et al., 2011), respectively, and the and the recently acknowledged TDP-43-E (Lee et al., 2017) – although not allowing inferences on these other substrates due to incomplete data or small sample sizes. Overall, it is likely that, when PPA co-occurs with MND, a one-to-one association between pathology and phenotype might not occur.
Genetics
TBK1 was the most frequently mutated gene in genetic-positive PPA-MND patients (41.2%), in accordance with evidence suggesting that loss of function mutations in this gene display a broad phenotypic heterogeneity - encompassing bvFTD, PPA (both PNFA and SD) (Le Ber et al., 2015; Lamb et al., 2019; Swift et al., 2021), MND (and, especially, predominant-UMN phenotypes) (Van Mossevelde et al., 2016; Gómez-Tortosa et al., 2017) and atypical parkinsonisms (Wilke et al., 2018; Seibert et al., 2021; Swift et al., 2021).
Also C-terminal missense mutations in the TARDBP gene were detected as a relatively frequent genetic underpinning of PPA-MND (29.4%), although with a disproportion toward SD (80%; PNFA: 20%) – this last finding being in line with several reports (Gelpi et al., 2014; González-Sánchez et al., 2018), also showing that SD is overrepresented in TARDBP carriers compared to other FTD phenotypes (Caroppo et al., 2016; van Rooij et al., 2020).
Conversely, C9orf72 HRE, which represents the major genetic determinant underlying the MND-FTD spectrum, appeared not to be ubiquitous – despite a large group study identifying it as the sole genetic feature of mutated PPA-MND patients showing PNFA (Tan et al., 2019). This aligns with evidence suggesting that C9orf72 HRE carriers are more represented among bvFTD – with or without comorbid MND – than PPA patients (Le Ber et al., 2013; Costa et al., 2020), as well as that this genetic form accounts mostly accounts for PNFA (Saracino et al., 2021).
Lastly, mutations in other genes, namely GNR, OPTN, FIG4, and SQSTM1, appeared to be extremely rare or incidental findings in PPA-MND.
Limitations
A number of limitations have to be acknowledged.
First, only two studies, biased by a small sample size, focused on the prevalence of PPA in MND patients – this preventing this study from providing generalizable estimates of the lifetime prevalence of PPA in MND patients.
Second, several outcomes proved to be irretrievable from group studies, due to the fact that the vast majority of them did not selectively focus on the conjunction between PPA and MND – and that, therefore, findings of interest were mostly incidental. Such a lack is the most obvious for neuroradiological data, which were indeed extracted for case reports/series only. Similarly, although to a lesser extent, data extracted from case reports/series also happened to be incomplete. Relatedly, this study lacks a formal quality assessment, which was not performed due to the fact itself that both case reports/series and group studies infrequently focused on the primary outcome of the present study – i.e., the conjunction between PPA and MND.
Third, the meta-analytic procedures performed within the present study included a very small number of studies, which, moreover, were markedly different from one another as to their design (i.e., cross-sectional vs. longitudinal, prospective vs. retrospective) and sample sizes. Such a diversity also prevented from reporting interpretable publication bias statistics. Thereupon, meta-analytic findings herewith reported must be approached with extreme caution, and respective estimates have to be interpreted as gross approximations of underlying parameters.
Author contributions
ENA: conceptualization, data synthesis, drafting, and revision. SF: conceptualization, data collection, data synthesis, drafting, and revision. GD, LG, and EA: data collection, drafting, and revision. IA, FS, LC, BP, FV, and VS: drafting, revision, and resources. NT: conceptualization, drafting, revision, and resources. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Italian Ministry of Health (Ricerca Corrente to IRCCS Istituto Auxologico Italiano, project 23C302) and IRCCS Istituto Auxologico Italiano covered publication fees.
Conflict of interest
VS received compensation for consulting services and/or speaking activities from AveXis, Cytokinetics, Italfarmaco, Liquidweb S.r.l., and Novartis Pharma AG, receives or has received research supports from the Italian Ministry of Health, AriSLA, and E-Rare Joint Transnational Call. He was in the Editorial Board of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration, European Neurology, American Journal of Neurodegenerative Diseases, Frontiers in Neurology. BP received compensation for consulting services and/or speaking activities from Liquidweb S.r.l. She was Associate Editor for Frontiers in Neuroscience. NT received compensation for consulting services from Amylyx Pharmaceuticals and Zambon Biotech SA. He was Associate Editor for Frontiers in Aging Neuroscience.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1003792/full#supplementary-material
Footnotes
References
Ahmed, R. M., Devenney, E. M., Strikwerda-Brown, C., Hodges, J. R., Piguet, O., and Kiernan, M. C. (2020). Phenotypic variability in ALS-FTD and effect on survival. Neurology 94, e2005–e2013. doi: 10.1212/WNL.0000000000009398
Ahmed, R. M., Hodges, J. R., and Piguet, O. (2021). “Behavioural variant frontotemporal dementia: recent advances in the diagnosis and understanding of the disorder,” in Frontotemporal Dementias: Emerging Milestones from the 21st Century, eds B. Ghetti, E. Buratti, B. Boeve, and R. Rademakers (Cham: Springer), 1–15. doi: 10.1007/978-3-030-51140-1_1
Aiello, E. N., Feroldi, S., Preti, A. N., Zago, S., and Appollonio, I. M. (2021). Dysgraphic features in motor neuron disease: a review. Aphasiology 1–26. doi: 10.1080/02687038.2021.1942774 [Epub ahead of print].
Aiello, E. N., Pain, D., Radici, A., Aktipi, K. M., Sideri, R., Appollonio, I., et al. (2022). Cognition and motor phenotypes in ALS: a retrospective study. Neurol. Sci. 43, 5397–5402. doi: 10.1007/s10072-022-06157-x
Armstrong, M. J., Litvan, I., Lang, A. E., Bak, T. H., Bhatia, K. P., Borroni, B., et al. (2013). Criteria for the diagnosis of corticobasal degeneration. Neurology 80, 496–503. doi: 10.1212/WNL.0b013e31827f0fd1
Bak, T. H., O’Donovan, D. G., Xuereb, J. H., Boniface, S., and Hodges, J. R. (2001). Selective impairment of verb processing associated with pathological changes in Brodmann areas 44 and 45 in the motor neurone disease–dementia–aphasia syndrome. Brain 124, 103–120. doi: 10.1093/brain/124.1.103
Bäumer, D., Butterworth, R., Menke, R. A., Talbot, K., Hofer, M., and Turner, M. R. (2014). Progressive hemiparesis (Mills syndrome) with aphasia in amyotrophic lateral sclerosis. Neurology 82, 457–458. doi: 10.1212/WNL.0000000000000090
Benajiba, L., Le Ber, I., Camuzat, A., Lacoste, M., Thomas-Anterion, C., Couratier, P., et al. (2009). TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 65, 470–473. doi: 10.1002/ana.21612
Bergner, C. G., Neuhofer, C. M., Funke, C., Biskup, S., von Gottberg, P., Bartels, C., et al. (2020). Case report: association of a variant of unknown significance in the fig4 gene with frontotemporal dementia and slowly progressing motoneuron disease: a case report depicting common challenges in clinical and genetic diagnostics of rare neuropsychiatric and neurologic disorders. Front. Neurosci. 14:559670. doi: 10.3389/fnins.2020.559670
Braak, H., Brettschneider, J., Ludolph, A. C., Lee, V. M., Trojanowski, J. Q., and Tredici, K. D. (2013). Amyotrophic lateral sclerosis—a model of corticofugal axonal spread. Nat. Rev. Neurol. 9, 708–714. doi: 10.1038/nrneurol.2013.221
Brettschneider, J., Del Tredici, K., Toledo, J. B., Robinson, J. L., Irwin, D. J., Grossman, M., et al. (2013). Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. doi: 10.1002/ana.23937
Burrell, J. R., Halliday, G. M., Kril, J. J., Ittner, L. M., Götz, J., Kiernan, M. C., et al. (2016). The frontotemporal dementia-motor neuron disease continuum. Lancet 388, 919–931. doi: 10.1016/S0140-6736(16)00737-6
Burrell, J. R., Kiernan, M. C., Vucic, S., and Hodges, J. R. (2011). Motor neuron dysfunction in frontotemporal dementia. Brain 134, 2582–2594. doi: 10.1093/brain/awr195
Cannon, A., Fujioka, S., Rutherford, N. J., Ferman, T. J., Broderick, D. F., Boylan, K. B., et al. (2013). Clinicopathologic variability of the GRN A9D mutation, including amyotrophic lateral sclerosis. Neurology 80, 1771–1777. doi: 10.1212/WNL.0b013e3182919059
Caroppo, P., Camuzat, A., De Septenville, A., Couratier, P., Lacomblez, L., Auriacombe, S., et al. (2015). Semantic and nonfluent aphasic variants, secondarily associated with amyotrophic lateral sclerosis, are predominant frontotemporal lobar degeneration phenotypes in TBK1 carriers. Alzheimers Dement. 1, 481–486. doi: 10.1016/j.dadm.2015.10.002
Caroppo, P., Camuzat, A., Guillot-Noel, L., Thomas-Antérion, C., Couratier, P., Wong, T. H., et al. (2016). Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol. Genet. 2:e80.
Caselli, R. J., Windebank, A. J., Petersen, R. C., Komori, T., Parisi, J. E., Okazaki, H., et al. (1993). Rapidly progressive aphasic dementia and motor neuron disease. Ann. Neurol. 33, 200–207. doi: 10.1002/ana.410330210
Catani, M., Piccirilli, M., Geloso, M. C., Cherubini, A., Finali, G., Pelliccioli, G., et al. (2004). Rapidly progressive aphasic dementia with motor neuron disease: a distinctive clinical entity. Dement. Geriatr. Cogn. Disord. 17, 21–28. doi: 10.1159/000074139
Cerami, C., Marcone, A., Crespi, C., Iannaccone, S., Marangoni, C., Dodich, A., et al. (2015). Motor neuron dysfunctions in the frontotemporal lobar degeneration spectrum: a clinical and neurophysiological study. J. Neurol. Sci. 351, 72–77. doi: 10.1016/j.jns.2015.02.039
Cerami, C., Marcone, A., Galimberti, D., Zamboni, M., Fenoglio, C., Serpente, M., et al. (2013). Novel evidence of phenotypical variability in the hexanucleotide repeat expansion in chromosome 9. J. Alzheimers Dis. 35, 455–462. doi: 10.3233/JAD-122302
Chiò, A., Calvo, A., Moglia, C., Mazzini, L., and Mora, G. (2011). Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J. Neurol. Neurosurg. Psychiatry 82, 740–746. doi: 10.1136/jnnp.2010.235952
Coon, E. A., Whitwell, J. L., Parisi, J. E., Dickson, D. W., and Josephs, K. A. (2012). Right temporal variant frontotemporal dementia with motor neuron disease. J. Clin. Neurosci. 19, 85–91. doi: 10.1016/j.jocn.2011.06.007
Costa, B., Manzoni, C., Bernal-Quiros, M., Kia, D. A., Aguilar, M., Alvarez, I., et al. (2020). C9orf72, age at onset, and ancestry help discriminate behavioral from language variants in FTLD cohorts. Neurology 95, e3288–e3302. doi: 10.1212/WNL.0000000000010914
Crespi, C., Dodich, A., Iannaccone, S., Marcone, A., Falini, A., Cappa, S. F., et al. (2020). Diffusion tensor imaging evidence of corticospinal pathway involvement in frontotemporal lobar degeneration. Cortex 125, 1–11. doi: 10.1016/j.cortex.2019.11.022
Czell, D., Andersen, P. M., Neuwirth, C., Morita, M., and Weber, M. (2013). Progressive aphasia as the presenting symptom in a patient with amyotrophic lateral sclerosis with a novel mutation in the OPTN gene. Amyotroph. Lateral Scler. Frontotemporal Degener. 14, 138–140. doi: 10.3109/21678421.2012.756525
Da Rocha, A., Valério, B. C. O., Buainain, R. P., Ferraz, M. E., Da Silva, C. J., and Maia, A. C. M. Jr., et al. (2007). Motor neuron disease associated with non-fluent rapidly progressive aphasia: case report and review of the literature. Eur. J. Neurol. 14, 971–975. doi: 10.1111/j.1468-1331.2007.01912.x
Davies, R. R., Hodges, J. R., Kril, J. J., Patterson, K., Halliday, G. M., and Xuereb, J. H. (2005). The pathological basis of semantic dementia. Brain 128, 1984–1995. doi: 10.1093/brain/awh582
De La Sablonnière, J., Tastevin, M., Lavoie, M., and Laforce, R. (2021). Longitudinal changes in cognition, behaviours, and functional abilities in the three main variants of primary progressive aphasia: a literature review. Brain Sci. 11:1209. doi: 10.3390/brainsci11091209
De Marchi, F., Tondo, G., Sarnelli, M. F., Corrado, L., Solara, V., D’Alfonso, S., et al. (2019). A case of progressive non-fluent aphasia as onset of amyotrophic lateral sclerosis with frontotemporal dementia. Int. J. Neurosci. 129, 719–721. doi: 10.1080/00207454.2018.1516657
De Souza, L. C., Sarazin, M., Samri, D., Kas, A., Lehericy, S., Levy, R., et al. (2009). Démence sémantique associée à une sclérose latérale amyotrophique. Rev. Neurol. 165, 278–281. doi: 10.1016/j.neurol.2008.06.008
Dols-Icardo, O., Nebot, I., Gorostidi, A., Ortega-Cubero, S., Hernández, I., Rojas-García, R., et al. (2015). Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 138:e400. doi: 10.1093/brain/awv175
Doran, M., Xuereb, J., and Hodges, J. R. (1995). Rapidly progressive aphasia with bulbar motor neuron disease; a clinical and neuropsychological study. Behav. Neurol. 8, 169–180.
El-Wahsh, S., Finger, E. C., Piguet, O., Mok, V., Rohrer, J. D., Kiernan, M. C., et al. (2021). Predictors of survival in frontotemporal lobar degeneration syndromes. J. Neurol. Neurosurg. Psychiatry 92, 425–433. doi: 10.1136/jnnp-2020-324349
Espay, A. J., Spina, S., Houghton, D. J., Murrell, J. R., de Courten-Myers, G. M., Ghetti, B., et al. (2011). Rapidly progressive atypical parkinsonism associated with frontotemporal lobar degeneration and motor neuron disease. J. Neurol. Neurosurg. Psychiatry 82, 751–753. doi: 10.1136/jnnp.2009.201608
Feng, F., Wang, H., Liu, J., Wang, Z., Xu, B., Zhao, K., et al. (2021). Genetic and clinical features of Chinese sporadic amyotrophic lateral sclerosis patients with TARDBP mutations. Brain Behav. 11:e2312. doi: 10.1002/brb3.2312
Floris, G., Borghero, G., Cannas, A., Di Stefano, F., Murru, M. R., Corongiu, D., et al. (2015). Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J. Neurol. 262, 375–384. doi: 10.1007/s00415-014-7575-5
Ganapathy, V. S., Chandra, S. R., Bharath, S., and Gs, R. (2020). Subclinical motor involvement in patients with behavioral variant frontotemporal dementia: a case control study. Asian J. Psychiatry 47:101821. doi: 10.1016/j.ajp.2019.10.001
Garcin, B., Lillo, P., Hornberger, M., Piguet, O., Dawson, K., Nestor, P. J., et al. (2009). Determinants of survival in behavioral variant frontotemporal dementia. Neurology 73, 1656–1661. doi: 10.1212/WNL.0b013e3181c1dee7
Gazulla, J., Ferrer, I., Izquierdo-Alvarez, S., Alvarez, S., Sánchez-Alcudia, R., Bestué-Cardiel, M., et al. (2019). Hereditary primary lateral sclerosis and progressive nonfluent aphasia. J. Neurol. 266, 1079–1090. doi: 10.1007/s00415-019-09235-x
Gelpi, E., van der Zee, J., Van Broeckhoven, C., and Sanchez-Valle, R. (2014). TARDBP mutation p. Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol. Appl. Neurobiol. 40, 225–230. doi: 10.1111/nan.12063
Gómez-Tortosa, E., Van der Zee, J., Ruggiero, M., Gijselinck, I., Esteban-Pérez, J., García-Redondo, A., et al. (2017). Familial primary lateral sclerosis or dementia associated with Arg573Gly TBK1 mutation. J. Neurol. Neurosurg. Psychiatry 88, 996–997. doi: 10.1136/jnnp-2016-315250
González-Sánchez, M., Puertas-Martin, V., Esteban-Pérez, J., Garcia-Redondo, A., Borrego-Hernandez, D., Mendez-Guerrero, A., et al. (2018). TARDBP mutation associated with semantic variant primary progressive aphasia, case report and review of the literature. Neurocase 24, 301–305. doi: 10.1080/13554794.2019.1581225
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Gromicho, M., Kuzma-Kozakiewicz, M., Szacka, K., Nieporecki, K., Andersen, P. M., Grosskreutz, J., et al. (2021). Motor neuron disease beginning with frontotemporal dementia: clinical features and progression. Amyotroph Lateral Scler. Frontotemporal Degener. 22, 508–516. doi: 10.1080/21678421.2021.1910309
Hamilton, R. L., and Bowser, R. (2004). Alzheimer disease pathology in amyotrophic lateral sclerosis. Acta Neuropathol. 107, 515–522. doi: 10.1007/s00401-004-0843-1
Hirsch-Reinshagen, V., Alfaify, O. A., Hsiung, G. Y. R., Pottier, C., Baker, M., and Perkerson, R. B. III, et al. (2019). Clinicopathologic correlations in a family with a TBK1 mutation presenting as primary progressive aphasia and primary lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 568–575. doi: 10.1080/21678421.2019.1632347
Höglinger, G. U., Respondek, G., Stamelou, M., Kurz, C., Josephs, K. A., Lang, A. E., et al. (2017). Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov. Disord. 32, 853–864. doi: 10.1002/mds.26987
Hsiung, G. Y. R., DeJesus-Hernandez, M., Feldman, H. H., Sengdy, P., Bouchard-Kerr, P., Dwosh, E., et al. (2012). Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain 135, 709–722. doi: 10.1093/brain/awr354
Jaiser, S. R., Mitra, D., Williams, T. L., and Baker, M. R. (2019). Mills’ syndrome revisited. J. Neurol. 266, 667–679. doi: 10.1007/s00415-019-09186-3
Jiao, B., Sun, Q., Yuan, Z., Wang, J., Zhou, L., Yan, X., et al. (2018). Rare TBK1 variants in patients with frontotemporal dementia and amyotrophic lateral sclerosis in a Chinese cohort. Transl. Neurodegener. 7:31. doi: 10.1186/s40035-018-0136-6
Kartanou, C., Karadima, G., Koutsis, G., Breza, M., Papageorgiou, S. G., Paraskevas, G. P., et al. (2018). Screening for the C9ORF72 repeat expansion in a greek frontotemporal dementia cohort. Amyotroph. Lateral Scler. Frontotemporal Degener. 19, 152–154. doi: 10.1080/21678421.2017.1400070
Kertesz, A., McMonagle, P., Blair, M., Davidson, W., and Munoz, D. G. (2005). The evolution and pathology of frontotemporal dementia. Brain 128, 1996–2005. doi: 10.1093/brain/awh598
Kim, S. H., Seo, S. W., Go, S. M., Suh, M. K., Chin, J., Jeong, J. H., et al. (2009). Semantic dementia combined with motor neuron disease. J. Clin. Neurosci. 16, 1683–1685. doi: 10.1016/j.jocn.2009.05.005
Kirshner, H. S., Tanridag, O., Thurman, L., and Whetsell, W. O. Jr. (1987). Progressive aphasia without dementia: two cases with focal spongiform degeneration. Ann. Neurol. 22, 527–532. doi: 10.1002/ana.410220413
Kobayashi, Z., Tsuchiya, K., Arai, T., Yokota, O., Yoshida, M., Shimomura, Y., et al. (2010). Clinicopathological characteristics of FTLD-TDP showing corticospinal tract degeneration but lacking lower motor neuron loss. J. Neurol. Sci. 298, 70–77. doi: 10.1016/j.jns.2010.08.013
Kremen, S. A., Mendez, M. F., Tsai, P. H., and Teng, E. (2011). Extrapyramidal signs in the primary progressive aphasias. Am. J. Alzheimers Dis. Dement. 26, 72–77. doi: 10.1177/1533317510391239
Lamb, R., Rohrer, J. D., Real, R., Lubbe, S. J., Waite, A. J., Blake, D. J., et al. (2019). A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Mol. Case Stud. 5:a003913. doi: 10.1101/mcs.a003913
Le Ber, I., Camuzat, A., Guillot-Noel, L., Hannequin, D., Lacomblez, L., Golfier, V., et al. (2013). C9ORF72 repeat expansions in the frontotemporal dementias spectrum of diseases: a flow-chart for genetic testing. J. Alzheimers Dis. 3, 485–499. doi: 10.3233/JAD-121456
Le Ber, I., De Septenville, A., Millecamps, S., Camuzat, A., Caroppo, P., Couratier, P., et al. (2015). TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol. Aging 36, 3116.e5–3116.e8. doi: 10.1016/j.neurobiolaging.2015.08.009
Lee, D. J., Bigio, E. H., Rogalski, E. J., and Mesulam, M. M. (2020). Speech and language presentations of FTLD-TDP type B neuropathology. J. Neuropathol. Exp. Neurol. 79, 277–283. doi: 10.1093/jnen/nlz132
Lee, E. B., Porta, S., Michael Baer, G., Xu, Y., Suh, E., Kwong, L. K., et al. (2017). Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 134, 65–78. doi: 10.1007/s00401-017-1679-9
Li, W., Gao, H., Dong, X., and Zheng, D. (2021). SQSTM1 variant in disorders of the frontotemporal dementia–amyotrophic lateral sclerosis spectrum: identification of a novel heterozygous variant and a review of the literature. J. Neurol. 268, 1351–1357. doi: 10.1007/s00415-020-10283-x
Longinetti, E., and Fang, F. (2019). Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr. Opin. Neurol. 32:771. doi: 10.1097/WCO.0000000000000730
Lulé, D. E., Aho-Özhan, H. E., Vázquez, C., Weiland, U., Weishaupt, J. H., Otto, M., et al. (2019). Story of the ALS-FTD continuum retold: rather two distinct entities. J. Neurol. Neurosurg. Psychiatry 90, 586–589. doi: 10.1136/jnnp-2018-318800
Mackenzie, I. R., Neumann, M., Baborie, A., Sampathu, D. M., Du Plessis, D., Jaros, E., et al. (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113. doi: 10.1007/s00401-011-0845-8
Maranzano, A., Poletti, B., Solca, F., Torre, S., Colombo, E., Faré, M., et al. (2022). Upper motor neuron dysfunction is associated with the presence of behavioural impairment in patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 29, 1402–1409. doi: 10.1111/ene.15243
Matias-Guiu, J. A., Cabrera-Martín, M. N., Moreno-Ramos, T., García-Ramos, R., Porta-Etessam, J., Carreras, J. L., et al. (2015). Clinical course of primary progressive aphasia: clinical and FDG-PET patterns. J. Neurol. 262, 570–577. doi: 10.1007/s00415-014-7608-0
McCluskey, L., Vandriel, S., Elman, L., Van Deerlin, V. M., Powers, J., Boller, A., et al. (2014). ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J. Neurol. Sci. 345, 118–124. doi: 10.1016/j.jns.2014.07.022
McKenna, M. C., Corcia, P., Couratier, P., Siah, W. F., Pradat, P. F., and Bede, P. (2021). Frontotemporal pathology in motor neuron disease phenotypes: insights from neuroimaging. Front. Neurol. 12:723450. doi: 10.3389/fneur.2021.723450
Miki, Y., Ling, H., Crampsie, S., Mummery, C. J., Rohrer, J. D., Jaunmuktane, Z., et al. (2020). Corticospinal tract degeneration and temporal lobe atrophy in frontotemporal lobar degeneration TDP-43 type C pathology. Neuropathol. Appl. Neurobiol. 46, 296–299. doi: 10.1111/nan.12582
Mimura, M., Tominaga, I., Kashima, H., Honda, M., Kosaka, K., and Kato, Y. (1998). Presenile non-Alzheimer dementia with motor neuron disease and laminar spongiform degeneration. Neuropathology 18, 19–26. doi: 10.1111/j.1440-1789.1998.tb00073.x
Montembeault, M., Brambati, S. M., Gorno-Tempini, M. L., and Migliaccio, R. (2018). Clinical, anatomical, and pathological features in the three variants of primary progressive aphasia: a review. Front. Neurol. 9:692. doi: 10.3389/fneur.2018.00692
Montuschi, A., Iazzolino, B., Calvo, A., Moglia, C., Lopiano, L., Restagno, G., et al. (2015). Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J. Neurol. Neurosurg. Psychiatry 86, 168–173. doi: 10.1136/jnnp-2013-307223
Neary, D., Snowden, J. S., Gustafson, L., Passant, U., Stuss, D., Black, S. A. S. A., et al. (1998). Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. doi: 10.1212/WNL.51.6.1546
Nouini, A., Gaspard, N., Fery, P., Slama, H., Kavec, M., Goldman, S., et al. (2017). Semantic dementia and motor neuron disease: case confirmation of TDP43 pathology associated with a predominant right temporal atrophy. EC Neurol. 6, 146–152.
Östberg, P., and Bogdanoviæ, N. (2011). Semantic dementia with lower motor neuron disease showing FTLD-TDP type 3 pathology (sensu Mackenzie). Neuropathology 31, 271–279. doi: 10.1111/j.1440-1789.2010.01154.x
Padovani, A., Agosti, C., Premi, E., Bellelli, G., and Borroni, B. (2007). Extrapyramidal symptoms in frontotemporal dementia: prevalence and clinical correlations. Neurosci. Lett. 422, 39–42. doi: 10.1016/j.neulet.2007.05.049
Page, M. J., Moher, D., Bossuyt, P. M., Boutron, I., Hoffmann, T. C., Mulrow, C. D., et al. (2021). PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ 372:160. doi: 10.1136/bmj.n160
Pasquini, J., Trogu, F., Morelli, C., Poletti, B., Girotti, F., Peverelli, S., et al. (2022). Parkinsonian syndromes in motor neuron disease: a clinical study. Front. Aging Neurosci. 14:917706. doi: 10.3389/fnagi.2022.917706
Pelin, Z., Küçükali, C. I, Kandemir, M., and Gencer, G. (2012). Primary progressive aphasia with motor neuron disease: a case report. J. Neurol. Sci. 29, 340–346.
Perry, D. C., Datta, S., Miller, Z. A., Rankin, K. P., Gorno-Tempini, M. L., Kramer, J. H., et al. (2019). Factors that predict diagnostic stability in neurodegenerative dementia. J. Neurol. 266, 1998–2009. doi: 10.1007/s00415-019-09362-5
Pickering-Brown, S. M., Rollinson, S., Du Plessis, D., Morrison, K. E., Varma, A., Richardson, A. M., et al. (2008). Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain 131, 721–731. doi: 10.1093/brain/awm331
Poletti, B., Solca, F., Carelli, L., Diena, A., Colombo, E., Torre, S., et al. (2021). Association of clinically evident eye movement abnormalities with motor and cognitive features in patients with motor neuron disorders. Neurology 97, e1835–e1846. doi: 10.1212/WNL.0000000000012774
Pupillo, E., Bianchi, E., Messina, P., Chiveri, L., Lunetta, C., Corbo, M., et al. (2015). Extrapyramidal and cognitive signs in amyotrophic lateral sclerosis: a population based cross-sectional study. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 324–330. doi: 10.3109/21678421.2015.1040028
Rajagopalan, V., and Pioro, E. P. (2019). Longitudinal 18F-FDG PET and MRI reveal evolving imaging pathology that corresponds to disease progression in a patient with ALS-FTD. Front. Neurol. 10:234. doi: 10.3389/fneur.2019.00234
Rajagopalan, V., and Pioro, E. P. (2021). Degeneration of gray and white matter differs between hypometabolic and hypermetabolic brain regions in a patient with ALS-FTD: a longitudinal MRI- PET multimodal study. Amyotroph. Lateral Scler. Frontotemporal Degener. 22, 127–132. doi: 10.1080/21678421.2020.1818784
Rakowicz, W. P., and Hodges, J. R. (1998). Dementia and aphasia in motor neuron disease: an underrecognised association? J. Neurol. Neurosurg. Psychiatry 65, 881–889. doi: 10.1136/jnnp.65.6.881
Roytman, M., and Chiang, G. C. (2022). “Logopenic variant primary progressive aphasia,” in Hybrid PET/MR Neuroimaging, eds A. M. Franceschi and D. Franceschi (Cham: Springer), 313–321. doi: 10.1007/978-3-030-82367-2_27
Saracino, D., Géraudie, A., Remes, A. M., Ferrieux, S., Noguès-Lassiaille, M., Bottani, S., et al. (2021). Primary progressive aphasias associated with C9orf72 expansions: another side of the story. Cortex 145, 145–159. doi: 10.1016/j.cortex.2021.09.005
Saxon, J. A., Harris, J. M., Thompson, J. C., Jones, M., Richardson, A. M., Langheinrich, T., et al. (2017a). Semantic dementia, progressive non-fluent aphasia and their association with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 88, 711–712. doi: 10.1136/jnnp-2016-314912
Saxon, J. A., Thompson, J. C., Jones, M., Harris, J. M., Richardson, A. M., Langheinrich, T., et al. (2017b). Examining the language and behavioural profile in FTD and ALS-FTD. J. Neurol. Neurosurg. Psychiatry 88, 675–680. doi: 10.1136/jnnp-2017-315667
Sbrollini, B., Preti, A. N., Zago, S., Papagno, C., Appollonio, I. M., and Aiello, E. N. (2021). Language impairment in motor neuron disease phenotypes different from classical amyotrophic lateral sclerosis: a review. Aphasiology 1–24. doi: 10.1080/02687038.2021.1959017 [Epub ahead of print].
Seibert, K., Smith, H., Lapins, A., Pytel, P., and Mastrianni, J. (2021). Corticobasal syndrome associated with a novel Lys694del variant of the TBK1 gene. Alzheimers Dement. 17:e054756.
Shellikeri, S., Karthikeyan, V., Martino, R., Black, S. E., Zinman, L., Keith, J., et al. (2017). The neuropathological signature of bulbar-onset ALS: a systematic review. Neurosci. Biobehav. Rev. 75, 378–392. doi: 10.1016/j.neubiorev.2017.01.045
Soliveri, P., Piacentini, S., Carella, F., Testa, D., Ciano, C., and Girotti, F. (2003). Progressive dysarthria: definition and clinical follow-up. Neurol. Sci. 24, 211–212. doi: 10.1007/s10072-003-0135-X
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., Mclaughlin, P., Snowden, J., et al. (2017). Amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 153–174. doi: 10.1080/21678421.2016.1267768
Suárez-González, A., Cassani, A., Gopalan, R., Stott, J., and Savage, S. (2021). When it is not primary progressive aphasia: a scoping review of spoken language impairment in other neurodegenerative dementias. Alzheimers Dement. 7:e12205. doi: 10.1002/trc2.12205
Swift, I. J., Bocchetta, M., Benotmane, H., Woollacott, I. O., Shafei, R., and Rohrer, J. D. (2021). Variable clinical phenotype in TBK1 mutations: Case report of a novel mutation causing primary progressive aphasia and review of the literature. Neurobiol. Aging 99, 100-e9–100.e15. doi: 10.1016/j.neurobiolaging.2020.08.014
Tan, R. H., Guennewig, B., Dobson-Stone, C., Kwok, J. B., Kril, J. J., Kiernan, M. C., et al. (2019). The underacknowledged PPA-ALS: a unique clinicopathologic subtype with strong heritability. Neurology 92, e1354–e1366. doi: 10.1212/WNL.0000000000007146
Tastevin, M., Lavoie, M., de la Sablonnière, J., Carrier-Auclair, J., and Laforce, R. Jr. (2021). Survival in the three common variants of primary progressive aphasia: a retrospective study in a tertiary memory clinic. Brain Sci. 11:1113. doi: 10.3390/brainsci11091113
Tremolizzo, L., Susani, E., Aliprandi, A., Salmaggi, A., Ferrarese, C., and Appollonio, I. (2014). Muscle ultrasonography for detecting fasciculations in frontotemporal dementia. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 546–550. doi: 10.3109/21678421.2014.913636
Tsuchiya, K., Ozawa, E., Fukushima, J., Yasui, H., Kondo, H., Nakano, I., et al. (2000). Rapidly progressive aphasia and motor neuron disease: a clinical, radiological, and pathological study of an autopsy case with circumscribed lobar atrophy. Acta Neuropathol. 99, 81–87. doi: 10.1007/PL00007411
Turner, M. R., and Talbot, K. (2020). Primary lateral sclerosis: diagnosis and management. Pract. Neurol. 20, 262–269. doi: 10.1136/practneurol-2019-002300
Ulugut, H., Stek, S., Wagemans, L. E., Jutten, R. J., Keulen, M. A., Bouwman, F. H., et al. (2021). The natural history of primary progressive aphasia: beyond aphasia. J. Neurol. 269, 1375–1385. doi: 10.1007/s00415-021-10689-1
Van Langenhove, T., Piguet, O., Burrell, J. R., Leyton, C., Foxe, D., Abela, M., et al. (2017). Predicting development of amyotrophic lateral sclerosis in frontotemporal dementia. J. Alzheimers Dis. 58, 163–170. doi: 10.3233/JAD-161272
Van Mossevelde, S., van der Zee, J., Gijselinck, I., Engelborghs, S., Sieben, A., Van Langenhove, T., et al. (2016). Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort. Brain 139, 452–467. doi: 10.1093/brain/awv358
van Rooij, J., Mol, M. O., Melhem, S., van der Wal, P., Arp, P., Paron, F., et al. (2020). Somatic TARDBP variants as a cause of semantic dementia. Brain 143, 3827–3841. doi: 10.1093/brain/awaa317
Viechtbauer, W. (2010). Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 36, 1–48. doi: 10.18637/jss.v036.i03
Vinceti, G., Olney, N., Mandelli, M. L., Spina, S., Hubbard, H. I., Santos-Santos, M. A., et al. (2019). Primary progressive aphasia and the FTD-MND spectrum disorders: clinical, pathological, and neuroimaging correlates. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 146–158. doi: 10.1080/21678421.2018.1556695
Vonk, J. M., Borghesani, V., Battistella, G., Younes, K., DeLeon, J., Welch, A., et al. (2020). Verbal semantics and the left dorsolateral anterior temporal lobe: a longitudinal case of bilateral temporal degeneration. Aphasiology 34, 865–885. doi: 10.1080/02687038.2019.1659935
Wagner, M., Lorenz, G., Volk, A. E., Brunet, T., Edbauer, D., Berutti, R., et al. (2021). Clinico-genetic findings in 509 frontotemporal dementia patients. Mol. Psychiatry 26, 5824–5832. doi: 10.1038/s41380-021-01271-2
Wilke, C., Baets, J., De Bleecker, J. L., Deconinck, T., Biskup, S., Hayer, S. N., et al. (2018). Beyond ALS and FTD: the phenotypic spectrum of TBK1 mutations includes PSP-like and cerebellar phenotypes. Neurobiol. Aging 62, 244.e9–244.e13. doi: 10.1016/j.neurobiolaging.2017.10.010
Yang, T., Hou, Y., Li, C., Cao, B., Cheng, Y., Wei, Q., et al. (2021). Risk factors for cognitive impairment in amyotrophic lateral sclerosis: a systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 92, 688–693. doi: 10.1136/jnnp-2020-325701
Yokota, O., Tsuchiya, K., Itoh, Y., Ishizu, H., Akiyama, H., Ikeda, M., et al. (2006). Frontotemporal lobar degeneration with ubiquitin pathology: an autopsy case presenting with semantic dementia and upper motor neuron signs with a clinical course of 19 years. Acta Neuropathol. 112, 739–749. doi: 10.1007/s00401-006-0149-6
Keywords: primary progressive aphasia, motor neuron disease, frontotemporal degeneration, amyotrophic lateral sclerosis, language
Citation: Aiello EN, Feroldi S, De Luca G, Guidotti L, Arrigoni E, Appollonio I, Solca F, Carelli L, Poletti B, Verde F, Silani V and Ticozzi N (2022) Primary progressive aphasia and motor neuron disease: A review. Front. Aging Neurosci. 14:1003792. doi: 10.3389/fnagi.2022.1003792
Received: 26 July 2022; Accepted: 24 August 2022;
Published: 08 September 2022.
Edited by:
Nilo Riva, San Raffaele Hospital (IRCCS), ItalyReviewed by:
Mamede De Carvalho, University of Lisbon, PortugalFrancesca Caso, San Raffaele Hospital (IRCCS), Italy
Copyright © 2022 Aiello, Feroldi, De Luca, Guidotti, Arrigoni, Appollonio, Solca, Carelli, Poletti, Verde, Silani and Ticozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Edoardo Nicolò Aiello, ZS5haWVsbG9AYXV4b2xvZ2ljby5pdA==
†ORCID: Edoardo Nicolò Aiello, orcid.org/0000-0001-8902-7733; Barbara Poletti, orcid.org/0000-0003-4398-2051; Vincenzo Silani, orcid.org/0000-0002-7698-3854