 María Björk Birkisdóttir1,2,3
María Björk Birkisdóttir1,2,3 Lisanne J. Van’t Sant1
Lisanne J. Van’t Sant1 Renata M. C. Brandt4
Renata M. C. Brandt4 Sander Barnhoorn4Jan H. J. Hoeijmakers2,3,4,5
Sander Barnhoorn4Jan H. J. Hoeijmakers2,3,4,5 Wilbert P. Vermeij2,3*
Wilbert P. Vermeij2,3* Dick Jaarsma1*
Dick Jaarsma1*- 1Department of Neuroscience, Erasmus MC, Rotterdam, Netherlands
- 2Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands
- 3Oncode Institute, Utrecht, Netherlands
- 4Department of Molecular Genetics, Erasmus MC Cancer Institute, Erasmus University Medical Center Rotterdam, Rotterdam, Netherlands
- 5Faculty of Medicine, CECAD, Institute for Genome Stability in Aging and Disease, University of Cologne, Cologne, Germany
Dietary restriction (DR) is a universal anti-aging intervention, which reduces age-related nervous system pathologies and neurological decline. The degree to which the neuroprotective effect of DR operates by attenuating cell intrinsic degradative processes rather than influencing non-cell autonomous factors such as glial and vascular health or systemic inflammatory status is incompletely understood. Following up on our finding that DR has a remarkably large beneficial effect on nervous system pathology in whole-body DNA repair-deficient progeroid mice, we show here that DR also exerts strong neuroprotection in mouse models in which a single neuronal cell type, i.e., cerebellar Purkinje cells, experience genotoxic stress and consequent premature aging-like dysfunction. Purkinje cell specific hypomorphic and knock-out ERCC1 mice on DR retained 40 and 25% more neurons, respectively, with equal protection against P53 activation, and alike results from whole-body ERCC1-deficient mice. Our findings show that DR strongly reduces Purkinje cell death in our Purkinje cell-specific accelerated aging mouse model, indicating that DR protects Purkinje cells from intrinsic DNA-damage-driven neurodegeneration.
Introduction
Dietary restriction (DR, also known as caloric restriction) is a long-term dietary intervention that consists of a daily 15 to 40% reduction of food intake without malnutrition, and that is well known as a universal anti-aging intervention increasing health-and lifespan (Le Couteur and Simpson, 2018; Lee et al., 2021; Green et al., 2022). DR reduces age-related decline in motor and cognitive function, as well as nervous system pathologies associated with normal aging and age-related neurodegenerative diseases (Mattison et al., 2017; Xie et al., 2020). While the benefits of DR as an anti-aging intervention are well established, the mechanisms by which aging is counteracted are still largely unresolved (Speakman and Mitchell, 2011; Lee et al., 2021). On the one hand, aging itself is a complex multifactorial phenomenon with numerous detrimental processes occurring at cellular (accumulation of damaged macromolecules and intracellular debris), organ (e.g., impaired tissue renewal), as well as whole organism level (changes in hormonal, serological and inflammatory status; Lopez-Otin et al., 2013; Pluvinage and Wyss-Coray, 2020). On the other hand, DR affects numerous intra- and intercellular processes, which include nutrient sensing, metabolic, hormonal and immunoregulatory pathways (Speakman and Mitchell, 2011; Green et al., 2022). Thus, a major challenge in understanding the anti-aging mechanisms of DR lies in connecting the pathways modulated by DR with the variety of aging processes that operate distinctively across tissues and cell types (Speakman and Mitchell, 2011; Ma et al., 2020; Schaum et al., 2020; Vougioukalaki et al., 2022).
We have previously found that DR has a remarkably strong beneficial effect on life-and health span in progeroid Ercc1Δ/− mice, in which defective DNA repair results in chronic genome instability and numerous features of multimorbidity and accelerated aging (Vermeij et al., 2016a). These data show that DR may alleviate the detrimental effects of impaired DNA repair, and indicate that DR protects against genotoxic stress, which is one of the driving factors of aging (Lopez-Otin et al., 2013). Most notably, DR strongly reduced the dramatic, progressive neurodegeneration (Vermeij et al., 2016a). Since Ercc1Δ/− mice develop systemic multimorbidity, with severely compromised functions of vital organs, blood vessels and immune system (Vermeij et al., 2016a,b; Vougioukalaki et al., 2022), a key question remained whether DR is neuroprotective indirectly via local and/or systemic components (e.g., by reducing inflammation), or by directly lowering cell-intrinsic DNA-damage-driven neurotoxicity. This question is particularly relevant for the central nervous system in view of increasing evidence favoring a central role of systemic factors in nervous system aging and neurodegeneration (Pluvinage and Wyss-Coray, 2020). Therefore, we examined the effect of DR in a mouse model in which DNA repair deficiency drives accelerated aging only in one single neuronal cell type, i.e., cerebellar Purkinje cells. These neurons are severely affected, but largely protected by DR, in whole-body Ercc1Δ/− mice (Birkisdottir et al., 2021), and degenerate in human aging (Andersen et al., 2003) as well as in various genome instability disorders (Shiloh, 2020; Kwak et al., 2021).
Materials and Methods
Ethic statements
Animal experiments were performed according to institutional guidelines as overseen by the Animal Welfare Board of the Erasmus MC, following Dutch and EU legislation. Prior to the start of the experiments, a project license for the animal experiments performed for this study was obtained from the Dutch national authority and filed under no. AVD101002015273 (DEC no. 139-12-13, 139-12-18, 15-273-120, 15-273-159).
Mice
Ercc1∆/−, Pcp2-Ercc1Δ/f, and Pcp2-Ercc1f/− were generated as previously described (de Waard et al., 2010; de Graaf et al., 2013; Vermeij et al., 2016a; Van't Sant et al., 2021). To obtain Ercc1∆/− mice, we crossed Ercc1∆/+ with Ercc1+/− mice (both in pure FVB or C57BL6J backgrounds respectively) to yield Ercc1∆/− offspring with a genetically uniform C57BL6J/FVB F1 hybrid background (Weeda et al., 1997; Vermeij et al., 2016a). In these mice one allele is fully inactivated whereas the other shows reduced activity, caused by a seven amino acids truncation. Because the Ercc1∆/− mice were smaller, food was administered within the cages, and water bottles with long nozzles were used from around 2 weeks of age.
For the generation of Purkinje specific Pcp2-Cre+/Ercc1∆/f mice (named Pcp2-Ercc1Δ/f in this study), Pcp2-Cre+/Ercc1∆/+ mice in C57Bl6/J background were generated by crossing female Pcp2-Cre+ mice with male Ercc1∆/+ mice. Subsequently, female Pcp2-Cre+/Ercc1∆/+ were crossed with male Ercc1f/f mice in FVB background to yield Pcp2-Cre+/Ercc1∆/f in a uniform C57BL6J/FVB F1 hybrid background. Pcp2-Cre+/Ercc1+/f and Pcp2-Cre−/Ercc1∆/f transgenic littermates that do not develop a degenerative phenotype served as controls (Van't Sant et al., 2021). Additional controls for this study were wild-type animals from the same C57BL6J/FVB F1 hybrid genetic background. Typical unfavorable characteristics, such as blindness in an FVB background or deafness in a C57BL6J background, do not occur in this hybrid background.
For initial characterization of cerebellar motor deficits and Purkinje cell degeneration two different Pcp2-Cre lines were used: Mpin Pcp2-Cre (Barski et al., 2000) and Jdhu Pcp2-Cre (Zhang et al., 2004). We used the Pcp2-Cre Mpin line for the DR experiments.The Pcp2-Cre Mpin line also was used to generate Pcp2-Ercc1−/f (Pcp2-Cre+/Ercc1−/f) mice. These were generated similarly as the above described Pcp2-Ercc1Δ/f mice, but by introducing an Ercc1 allele that is fully inactivated (−) rather than truncated (Δ) from the initial breedings (de Graaf et al., 2013).
Housing conditions and dietary regimens
Animals of DR experiments were randomly divided over sex-matched dietary groups and housed in individual ventilated cages under specific pathogen-free conditions. Each cohort consisted of four groups (experimental/control genotype and AL/DR diet) of at least four animals, thereof two males and two females. Animals of other behavioral experiments were group housed. Environment was controlled with a temperature of 20–22°C and 12 h light:12 h dark cycles. Food and water were offered ad libitum (AL) unless otherwise stated. Mice were weighed, visually inspected weekly, and scored blindly for gross morphological and motor abnormalities weekly. Animals were bred and maintained on AIN93G synthetic pellets (Research Diet Services B.V., Wijk bij Duurstede, Netherlands; gross energy content 4.9 kcal/g dry mass, digestible energy 3.97 kcal/g). The amount of DR was determined in a prior pilot study where food intake of the AL-fed mice was continuously monitored. Mice on average ate 3.0 g food per day, resulting in 2.1 g/day for 30% DR. Water was freely available. Food was given to the animals just before the start of the dark (active) period (ZT12). DR was initiated at 7 weeks of age with 10% food reduction, and food was gradually reduced to 30% DR from 9 weeks of age onward as previously published (Vermeij et al., 2016a). Our DR regimen in a room with altered day-night rhythm was specifically chosen not to disturb the biological clock, as this may influence the anti-aging effect of DR (Nelson and Halberg, 1986; Acosta-Rodriguez et al., 2022).
Behavioral assays
To examine cerebellar motor function, we used an accelerating rotarod, balance beam and Erasmus automated ladder test, largely following previously described protocols (Vinueza Veloz et al., 2015; Vermeij et al., 2016a; White et al., 2021) and further detailed in the Supplementary methods.
Histological procedures
Mice were anaesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde. Cerebella were carefully dissected out, post-fixed for 1 h in 4% paraformaldehyde and left in sucrose overnight. Subsequently the brain was embedded in 12% gelatin, rapidly frozen, and sectioned at 40 μm using a freezing microtome. Sections were processed free floating using single-, double-, and triple-labelling immunofluorescence. Primary antibodies (supplier; catalogue number, RRID number; dilution) used in this study were as follows: rabbit anti-Calbindin (Swant; C9638, AB_2314070; 1:20,000); mouse anti-Calbindin (Sigma Aldrich; C8666, AB_2313712; 1:20,000); goat anti-FOXP2 (Santa Cruz Biotechnology; sc21069, AB_2107124; 1:1000); rabbit anti-GFAP (DAKO; Z0334, AB_10013382; 1:8000); rat anti-LGALS3 (Cedarlane; CL8942AP, AB_10060357; 1:1000) rabbit anti-P53 (Leica; NCL-p53-CMP5, AB_2744683; 1:1000) and mouse anti-GM130 (BD Bioscience; 610,822, AB_398141; 1:200). Alexa488-, Cy3-, and Cy5-conjugated antibodies raised in donkey (Jackson ImmunoResearch) diluted at 1:200 were used as secondary antibodies for visualization. Immunofluorescence sections were analyzed and imaged using Zeiss LSM700, Leica TCS SP8, and Leica Stellaris 5 confocal microscopes, and a Zeiss Axio Imager D2 widefield microsope.
Stereological analysis of Purkinje cell numbers
Stereological quantification of Purkinje neurons was done as previously described (Birkisdottir et al., 2021) using the Optical Fractionator tool of a StereoInvestigator software package (MBF Bioscience), integrated in a Zeiss LSM700 confocal microscope setup (see Supplementary methods). Our estimates of the number of Purkinje cells in control mice (from 2.04*105 to 2.48*105; Supplementary Figure S3A) were consistent with stereological quantifications of other studies (Woodruff-Pak et al., 2010; Birkisdottir et al., 2021).
Quantitative histological analyses
To determine the number of P53+ Purkinje cells, we double stained every 8th serial coronal section (8–11 section/animal) for Calbindin and P53 (see histological procedures). Purkinje neurons with strongly P53-immunostained nucleus (Supplementary Figure S1G) were systematically counted throughout the whole series using a Zeiss Axio Imager D2 widefield fluorescence microscope equipped with a 40x EC Plan-NeoFluar lens. The total number of P53+ Purkinje cells/ animal was estimated by multiplying the number of counted cells by 8. To determine the proportion of P53+ cells, we used the Purkinje cell numbers from the stereological analyses.
Golgi apparatus morphological abnormalities were analyzed in the cohort of 40-week-old AL vs. DR treated Pcp2-Ercc1Δ/f mice. Coronal sections of cerebellum were double immunostained with Calbindin and the cis-Golgi protein GM130 (see histological procedure). Stacks of the Purkinje cell layer, 0.5–1 mm in length and 15 μm thick, were randomly sampled at high resolution in lobules of the vermis and paravermis using a Leica Stellaris 5 confocal microscope with a HC PL APO CS2 63x oil objective. Stacks were examined in ImageJ (Version 1.52) by a trained observer unaware of genotype and diet (DJ). Only Purkinje cells whose cell body was fully embedded in the stack were counted. In control Purkinje cells, GM130 staining appears as a ribbon-like structure that forms an extensive network throughout the cell body. In most Purkinje cells of Pcp2-Ercc1Δ/f mice GM130 staining showed a normal or near normal appearance. Purkinje cells with abnormal Golgi apparatus either showed fragmentation of the ribbon into smaller elements resulting in punctate GM130 staining, or compaction and redistribution of the Golgi ribbon to a small part of the cell body. Percentages of Purkinje cells with abnormal Golgi were determined by analyzing 100–200 Purkinje cells per AL animal (n = 4), and 500–700 Purkinje cells per DR animal.
To assess differences in astrocytosis, images of GFAP stained cerebellar tissue were acquired using a wide field microscope Zeiss Axio Imager D2 widefield microscopes, and mean intensities of the molecular layer of the cerebellum were measured using ImageJ.
Statistical analyses
Statistical analyses were performed using GraphPad Prism Software (San Diego, CA, United States). For the analysis of difference between Purkinje neurons, P53 and Golgi abnormalities, values of AL and DR animals of each cohort was compared using two-tailed unpaired t-test. One way ANOVA with Tukey’s multiple comparison was used for GFAP analysis, Purkinje cell amount and relative rescue analysis. GFAP intensity values of AL and DR animals of each cohort were compared using two-tailed unpaired t-test. Two-way repeated measure ANOVA was used for rotarod data and mixed effect model (REML) for bodyweight data, both with post-hoc Tukey’s Multiple comparison test. Graphs illustrate individual values, means and standard error (SE). Statistical analyses and results are summarized in Supplementary Table S1.
Results
We generated a Cre-lox-based Purkinje cell-specific ERCC1-deficient (Pcp2-Ercc1Δ/f) mouse model that carried a floxed Ercc1 allele (Ercc1f), a C-terminally truncated “delta” Ercc1 allele (Ercc1Δ; Vermeij et al., 2016b), and a Pcp2-Cre transgene (Mpin line) that drives Cre recombinase expression in postnatal Purkinje cells (Van't Sant et al., 2021). After recombination by Cre, the floxed Ercc1 allele becomes a null allele, resulting in Purkinje cells that have one null and one truncated Ercc1 allele, and that reproduce the severe (but incomplete) reduction in ERCC1/XPF nuclease function of whole body Ercc1Δ/− mice (Figure 1A), affecting nucleotide excision repair, inter-strand crosslink repair, and single-strand annealing of double-strand breaks (Vermeij et al., 2016b; Mulderrig and Garaycoechea, 2020; Apelt et al., 2021). Pcp2-Ercc1Δ/f mice had, unlike Ercc1Δ/− mice, the same weight as control littermates and did not show evidence of neurodegenerative changes outside the cerebellum, consistent with the remainder of the body and nervous system being repair proficient and healthy (Figure 1A; Supplementary Figures S1A,F). Pcp2-Ercc1Δ/f mice at 12 weeks showed control performance in cerebellar motor tests, but developed progressive deficits thereafter (Supplementary Figures S1B–D). The time of onset and severity of cerebellar motor abnormalities as well as the severity of Purkinje cell degeneration were the same as in Pcp2-Ercc1Δ/f mice generated with another Pcp2-Cre transgene (Jhdu line), indicating that the Purkinje cell degenerative phenotype is not influenced by the Cre line (Supplementary Figure S1). Since the BAC construct of the Jdhu Pcp2-Cre line may introduce extra copies of genes implicated in neuronal function, splicing and DNA repair (Pnpla6, Stxbp2, Pcp2, and Xab2; Zhang et al., 2004) we used the Pcp2-Cre Mpin line for the DR experiments.
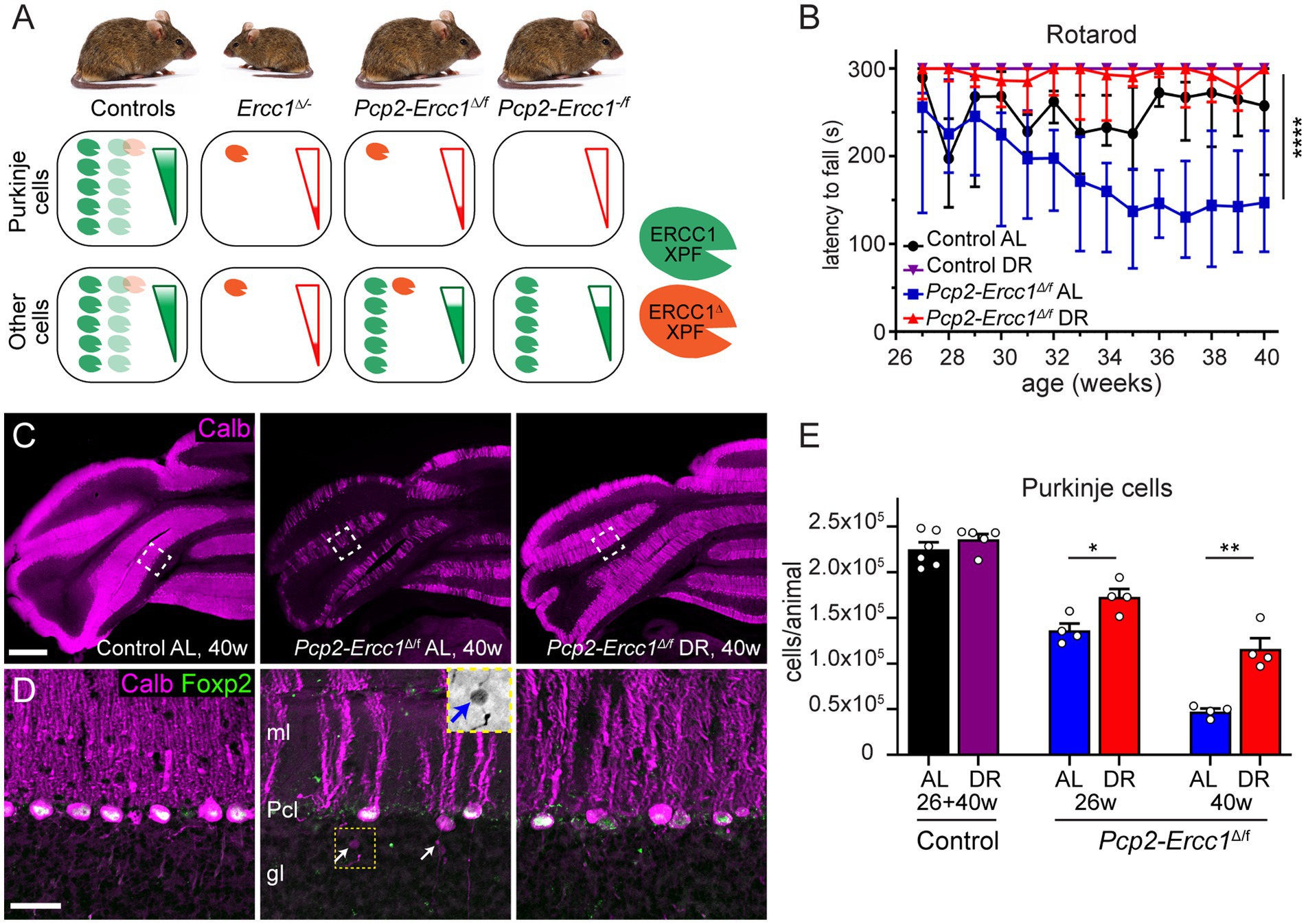
Figure 1. Dietary restriction reduces Purkinje cell loss in Purkinje cell specific ERCC1-deficient mice. (A) Schematic representation of Ercc1-XPF nuclease expression in Purkinje cells versus other cells in the Ercc1 mouse models of this study. The truncated Ercc1Δ allele results in strongly reduced levels of ERCC1Δ-XPF nuclease dimer (one red symbol) as compared to wild-type Ercc1+ allele (5 green symbols). Ercc1Δ/− mice have low mutant ERCC1 production in their whole body, whereas the hypomorphic mutation in the Pcp2-Ercc1Δ/f mice is restricted to Purkinje cells. In comparison, Pcp2-Ercc1−/f mice have no ERCC1 production in Purkinje cells, whereas the remainder of the body in both conditional mutants is repair-proficient due to the flox-allele that encodes wt protein. The genotype and ERCC1-XPF levels in littermates used as control animals for each cohort differ as indicated with light colored heterodimers (see methods). (B) Accelerating rotarod assay showing preserved performance in DR versus AL Pcp2-Ercc1Δ/f mice over time. Values represent medians with interquartile range (n = 4/group); ****p < 0.0001 (Dunn’s post-test; Pcp2-Ercc1Δ/f AL versus DR). (C,D) Low (C) and high magnification (D) confocal images of calbindin (magenta) and FOXP2 (green) immunostaining to outline Purkinje cells and their nucleus. Note, reduced loss of Purkinje cells in DR-vs AL-treated Pcp2-Ercc1Δ/f mice. Insert in middle panel in d shows an increased-contrast inverted-signal image of the axonal spheroid indicated by the yellow arrow in the same panel. gl, granular layer; ml, molecular layer; Pcl, Purkinje cell layer. (E) Bar graph with means ± SE of stereological quantification of Purkinje cells showing loss of Purkinje cells in 26-and 40-week-old Pcp2-Ercc1Δ/f mice and reduced loss in DR vs. AL animals. (n ≥ 4/group); *p < 0.05, **p < 0.01 (unpaired t-tests for each cohort). Scale bars: 500 μm (C), 25 μm (D).
We applied the same 30% DR regimen as used before in full-body Ercc1Δ/− mice starting with 10% restriction at the age of 7 weeks, 20% at 8 weeks, and 30% thereafter (Vermeij et al., 2016a). DR-treated Pcp2-Ercc1Δ/f animals showed the same weight loss as DR control mice (Supplementary Figure 2A), and displayed preserved performance in an accelerating rotarod test up to the age of 40 weeks (Figure 1B). In contrast, ad libitum (AL)-fed Pcp2-Ercc1Δ/f mice showed strongly declining rotarod performance till 35 weeks, after which the descent appears to stabilize (Figure 1B), concomitant with severe Purkinje cell degeneration throughout the cerebellar cortex as revealed by reduced immunohistological staining for calbindin that in the cerebellum is specifically expressed in Purkinje cells (Figures 1C,D; Supplementary Figures S2B). DR-treated Pcp2-Ercc1Δ/f mice at 40 weeks, instead, showed relatively preserved calbindin staining (Figures 1C,D; Supplementary Figure S2B), indicative of reduced Purkinje cell loss. We quantified Purkinje cells using a stereological approach (Andersen et al., 2003; Birkisdottir et al., 2021), and confirmed that Purkinje cell loss was significantly (p < 0.01) prevented by DR with 1.16 ± 0.12 *105 and 0.47 ± 0.03 *105 residual Purkinje cells in 40-week-old DR-and AL-Pcp2-Ercc1Δ/f mice, respectively. Additionally, Purkinje cells of DR-treated Pcp2-Ercc1Δ/f mice showed a lower frequency of morphological abnormalities, such as axonal spheroids (Figure 1D) and displayed less astrocytosis, indicated by GFAP staining, in the molecular layer consistent with reduced Purkinje cell degeneration (Figures 2A,B; de Waard et al., 2010; de Graaf et al., 2013). Next, we analyzed Golgi apparatus morphology of Purkinje cells, following up on our finding that in Ercc1Δ/− mice DR strongly reduced the frequency of spinal motor neurons with atypical Golgi, putatively representing a proxy of compromised neuronal health (de Waard et al., 2010; Vermeij et al., 2016a). Indeed, AL-fed Pcp2-Ercc1Δ/f mice showed a spectrum of abnormal Golgi in about 25% of residual Purkinje cells, while this was strongly diminished in DR-treated Pcp2-Ercc1Δ/f mice (Figures 2C,D). In sum, DR significantly reduces motor dysfunction and Purkinje cell pathology and loss in Pcp2-Ercc1Δ/f mice at 40 weeks.
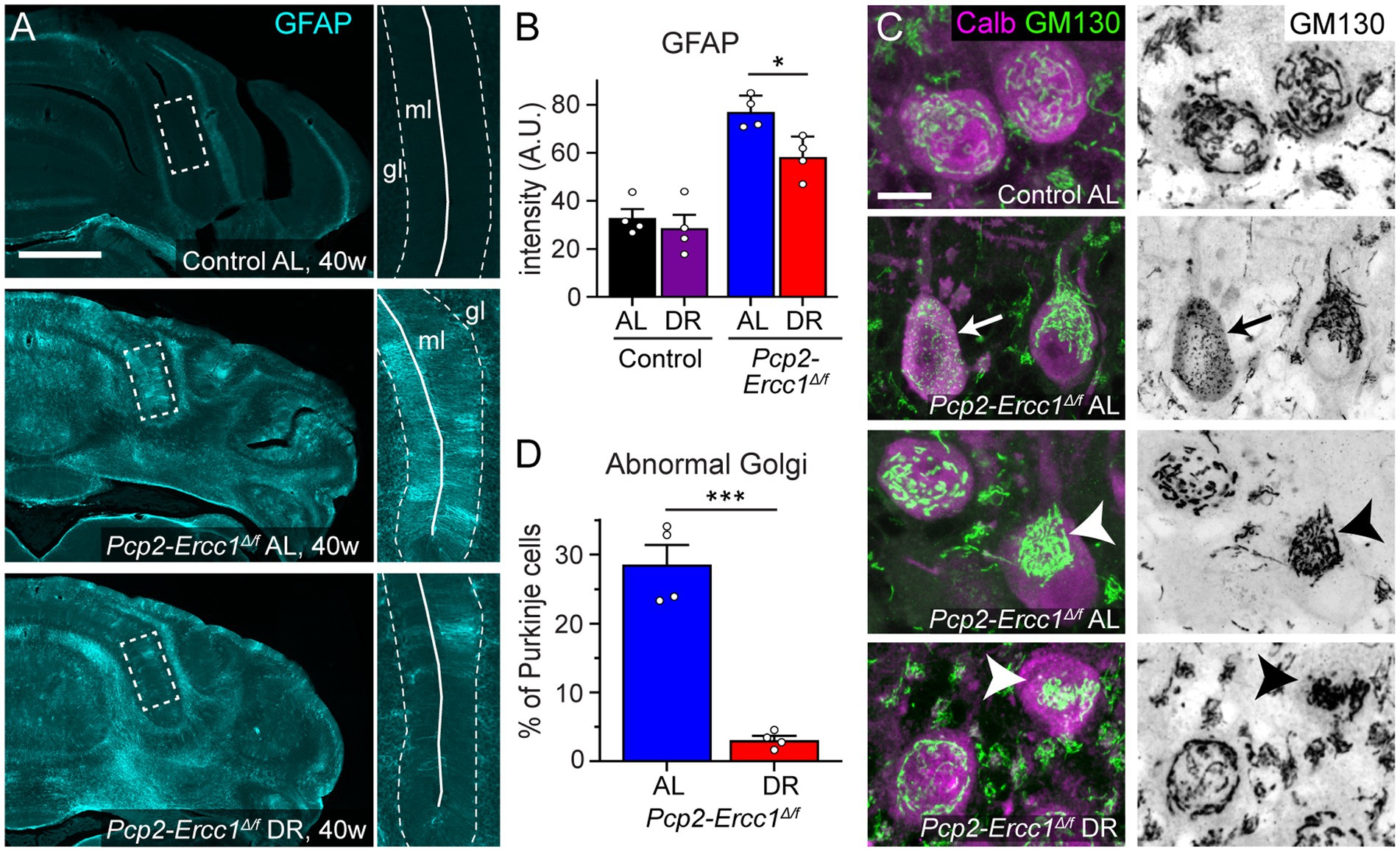
Figure 2. Dietary restriction reduces GFAP-immunostaining and Golgi apparatus abnormalities in Pcp2-Ercc1Δ/f mice. (A,B) Representative images and bar graph (means ± SE) of GFAP immunofluorescence illustrating increased GFAP immunostaining in Pcp2-Ercc1Δ/f cerebellum compared to control, and reduced GFAP-staining in the molecular layer (ml) of DR compared to AL Pcp2-Ercc1Δ/f cerebellum. (n = 4/group); *p < 0.05 (unpaired t-test). (C,D) Confocal images and bar graph (means ± SE) showing reduced amount of Golgi apparatus morphological abnormalities in 40 week old DR compared to AL Pcp2-Ercc1Δ/f Purkinje cells, examined using immunostaining of the cis-Golgi scaffold protein GM130. In control Purkinje cells the GM130 staining outlines a network of linear profiles throughout the cell body. In ERCC1-deficient mice 2 main types of changes occur in a subset of Purkinje cells: (1) fragmentation of linear profiles into smaller elements (arrow) and (2) more frequently, redistribution of unfragmented or fragmented Golgi stacks to part of the cell body (arrow heads). (n = 4/group); ***p < 0.001 (unpaired t-test). Scale bars: 500 μm (A), and 10 μm (C).
To robustly quantify the effect of DR on ERCC1-deficiency in Purkinje cells in time, we focused on stereological analysis of Purkinje cell numbers. At 8 weeks of age at the start of DR treatment, the number of Purkinje cells in Pcp2-Ercc1Δ/f mice appeared unaltered compared to controls of 8–40 weeks (2.28 ± 0.11 *105 vs. 2.27 ± 0.06 *105, respectively; Supplementary Figure S3). Accordingly, Pcp2-Ercc1Δ/f mice at 8 weeks showed no evidence of astrocytosis (Supplementary Figure S3D). At the age of 26 weeks, DR and AL mice showed 1.73 ± 0.09 * 105 and 1.36 ± 0.08 *105 remaining Purkinje cells, respectively (Figure 1E) revealing a significant protective effect of DR (p < 0.05). Using the mean value at 8 weeks (2.28 *105) as starting number of Purkinje cells in Pcp2-Ercc1Δ/f mice (Supplementary Figure S3B), we estimated the number of Purkinje cells that died in AL and DR animals at 26 and 40 weeks (= 2.28 *105- # of surviving Purkinje cells). Subsequently, these values were used to calculate a relative rescue of Purkinje cells by DR with the formula: (average loss in AL animals – loss in DR animal)/average loss in AL animals*100. This analysis indicated that DR attenuated Purkinje cell loss by about 40% at both ages (Figure 3A). This percentage appears somewhat smaller than the 59% estimated relative rescue of Purkinje cell loss by DR for full-body Ercc1Δ/− mice, examined at the age of 16 weeks, a few weeks before the progeroid mutants become moribund (Figure 3A; Birkisdottir et al., 2021), albeit the difference did not reach statistical significance (p = 0.289 and 0.188 for 26 and 40 weeks respectively). It should be noted, that systemic Ercc1Δ/− mice do not only differ from Pcp2-Ercc1Δ/f in regard of ubiquitous versus Purkinje-cell-specific ERCC1-deficiency, but also show earlier onset of ERCC1 cell deficiency (early development versus post-natal week 2), and already exhibit Purkinje cell loss at the onset of DR treatment at 8 weeks (Supplementary Figure S3B).
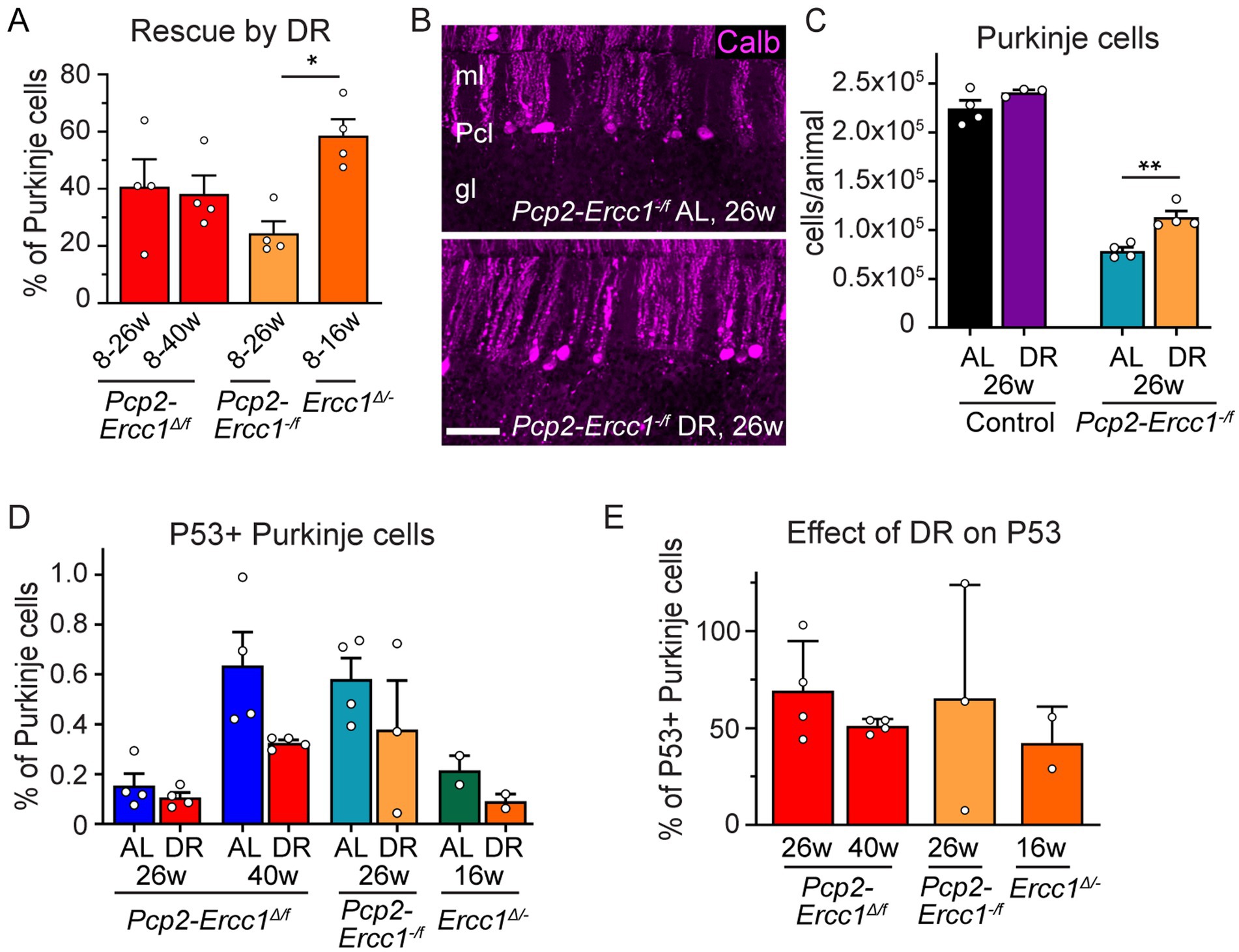
Figure 3. Differential protective effects of DR in Purkinje cell-specific and ubiquitous ERCC1-deficient mice. (A) Relative rescue of Purkinje cells by DR in Purkinje cell-specific (Pcp2-Ercc1Δ/f and Pcp2-Ercc1−/f) and global (Ercc1Δ/−) mice, calculated by dividing the difference in Purkinje cell loss between DR and AL animals by the loss showed in AL mice. (n = 4 per group); *p < 0.05 (One-way ANOVA; Tukey’s post-tests). (B,C) Representative confocal images (B) and bar graph (C) illustrating Purkinje cell degeneration in AL and DR Pcp2-Ercc1−/f mice. (n = 4 per group) **p < 0.01 (One-way ANOVA; Tukey’s post-test). (D) Bar graph of the proportion of Purkinje cells with P53-immunoreactive nuclei in ERCC1-deficient mouse mutants (n ≥ 2 per group). Impact of DR across all groups: p = 0.024 [2-way ANOVA F(1,16) = 6.019]; DR-AL pair comparisons: p = 0.0784 in AL vs. DR Pcp2-Ercc1Δ/f at 40 weeks; p > 0.4 in other AL-DR pairs (Sidak’s multiple comparison post-test). (E) Proportion of Purkinje cells with nuclear P53 accumulation expressed as % of mean AL values. Scale bar: 50 μm.
Hypomorphic Pcp2-Ercc1Δ/f and full-body Ercc1Δ/− mice still have some residual repair activity, which may contribute to the protective effect of DR. Therefore, we tested DR in Pcp2-Ercc1−/f mice that are fully ERCC1-deficient in Purkinje cells (Figure 1A). Pcp2-Ercc1−/f mice show unaltered numbers of Purkinje cells at 8 weeks (Supplementary Figure S3B), like Pcp2-Ercc1Δ/f mice and in accord with previous proteomic and neuropathological data (de Graaf et al., 2013). However, they display a larger (p < 0.0001) Purkinje cell loss at 26 weeks with 0.78 ± 0.07 *105 (vs. 1.36 ± 0.04 *105 for Pcp2-Ercc1Δ/f mice) residual Purkinje cells, and only sparse surviving Purkinje cells at 40 weeks (0.23 ± 0.03 *105; Supplementary Figures S3B,C), indicating a dose–response relationship between degree of repair deficiency and neuronal cell loss. As in Pcp2-Ercc1Δ/f mice, DR significantly (p < 0.01) reduced Purkinje cell loss in Pcp2-Ercc1−/f mice at 26 weeks of age (Figures 3B,C), with a relative rescue of about 25% compared to AL animals (Figure 3A). These data indicate that DR also is protective in Purkinje cells with complete loss of ERCC1, that experience more severe genotoxic stress than Ercc1Δ/− Purkinje cells (Figure 1A).
To get insight into the role of DNA damage we examined P53 as guardian of the genome. Previously, we found a decrease in P53-expressing cells in the neocortex and spinal cord of DR Ercc1Δ/− mice, supporting that DR reduces DNA damage load (Vermeij et al., 2016a). Here, we also observed an overall reduction [p = 0.024; 2-way ANOVA F(1,16) = 6.019] by DR on P53 positive Purkinje neurons, albeit the difference between each individual AL-DR pair not being statistically significant (Figure 3D). Across all models and cohorts, DR equally reduced the proportion of Purkinje cells with nuclear P53 accumulation by about 50% (Figure 3E).
Discussion
In this study we show that DR strongly reduces degeneration of ERCC1-deficient Purkinje cells, in mice that are unaffected in other cell types and tissues. The data imply that DR strongly reduces cell-intrinsic DNA-damage driven Purkinje cell degeneration. The data also indicate that reduced Purkinje cell degeneration by DR in whole-body ERCC1-deficient mice primarily results from cell intrinsic mechanisms rather than by restoring function of surrounding glia cells and vital organs or via systemic components. Our data support previous work indicating that DR may both reduce the levels of DNA damage and increase the ability of cells to cope with DNA damage (Heydari et al., 2007), and support the hypothesis that the anti-aging effect of DR in the nervous system, at least in part, is mediated by reducing genotoxic stress in neurons (Vermeij et al., 2016a; Xie et al., 2020).
Previously, we have shown that ERCC1-deficient mice develop multiple age-dependent neurological deficits and progressive neuropathology characterized by a diversity of degenerative changes throughout the nervous affecting both neurons and glia (de Waard et al., 2010; Borgesius et al., 2011; Vegh et al., 2012; Raj et al., 2014; Vermeij et al., 2016a). The nervous system pathology is consistent with a model where stochastic accumulation of DNA damage causes transcription stress, i.e., a variety of yet incompletely defined transcription problems and down-stream effects that can affect cell function in multiple ways, for instance by blocked or deregulated expression of essential genes or problems resulting from persistent transcription stalling (Vermeij et al., 2016b; Niedernhofer et al., 2018; Lans et al., 2019; Mulderrig and Garaycoechea, 2020; Apelt et al., 2021; Jia et al., 2021; Van't Sant et al., 2021). Here, we focused on stereological analysis of residual Purkinje cell numbers as the principal readout, representing an unequivocal quantitative readout that reflects the final outcome of multiple neurodegenerative pathways. In addition, we show that DR has impact on two additional readouts in Purkinje cells: it reduces the frequency of Purkinje cells expressing the genotoxic stress transcription factor P53, and Purkinje cells with abnormal Golgi apparatus morphologies. The varied perturbations in Golgi morphology in more than 20% of ERCC1-deficient Purkinje cells and their strongly reduced frequency in DR animals, are consistent with findings in spinal motor neurons in Ercc1Δ/− mice (de Waard et al., 2010; Vermeij et al., 2016a), and may reflect a diversity of degenerative and cell stress pathways including DNA damage signaling (Makhoul et al., 2019).
The identity of DNA lesions that accumulate in ERCC1-deficient nervous system and cause neuronal damage are yet poorly understood (Niedernhofer et al., 2018; Lans et al., 2019; Mulderrig et al., 2021). In our previous DR study with Ercc1Δ/− mice, analyses of liver transcriptomes showed a preferential reduction in expression of long genes in Ercc1Δ/− liver, which was attenuated by DR (Vermeij et al., 2016a). This is consistent with the notion that long genes have an increased likelihood of accumulating transcription stalling lesions (Lans et al., 2019; Noe Gonzalez et al., 2021), and display reduced damage in DR animals. Hence, analysis of gene-length-dependent transcriptional changes reflect a promising indirect readout for measuring transcription stress and the effect of DR in Ercc1Δ/− nervous system. Importantly, a bias of reduced expression of long genes also has been found in aged human nervous system (Vermeij et al., 2016a; Niedernhofer et al., 2018; Soheili-Nezhad et al., 2021), indicating that DNA-damage-induced transcription stress is an important factor contributing to nervous system aging and targeted by DR (Vermeij et al., 2016a). Future studies employing high resolution spatial transcriptomic approaches (Asp et al., 2020; Lewis et al., 2021) may uncover the relationships between transcriptional abnormalities and neuronal degeneration in aging and progeroid models.
What changes triggered by DR render Purkinje cells less vulnerable towards DNA repair deficiencies? DR has been shown to promote autophagy and to reduce ribosomal protein S6 phosphorylation in Purkinje cells, indicative of suppression of mTOR signalling (Alirezaei et al., 2010), one of the main pathways linked to DR benefits (Liu and Sabatini, 2020; Green et al., 2022). However, we recently showed that mTOR inhibition via rapamycin or genetic intervention fails to improve lifespan, neurological functioning, and Purkinje cell survival of Ercc1Δ/− mice (Birkisdottir et al., 2021), hence, pointing to alternative mechanisms. While a spectrum of other potential anti-aging compounds that have been proposed to reproduce aspects of the beneficial effects of DR by influencing nutrient sensing, inflammation, mitochondrial function, or glucose homeostasis, did not have an effect on survival and neurological function in Ercc1Δ/− mice, we found that two NAD+ precursors (nicotinamide riboside and nicotinic acid) induced small benefits in survival and neurological function (Alyodawi et al., 2019; Birkisdottir et al., 2022). Since DR may increase NAD+ levels and modulate a variety of NAD+-dependent mechanisms in energy and redox metabolism, as well as in DNA damage sensing, protein deacetylation or other enzymatic pathways requiring NAD+ (Covarrubias et al., 2020; Green et al., 2022), NAD+-dependent pathways may be part of the mechanisms mediating the beneficial effect of DR in ERCC1-deficient Purkinje cells. However, further studies are needed to dissect the mechanisms that underlie the strong beneficial effect of DR in ERCC1-deficient mice. Our Purkinje cell specific ERCC1-deficient mice provides a complementary model to global Ercc1Δ/− mice in these studies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The animal study was reviewed and approved by Animal Welfare Board of the Erasmus MC, ErasmusMC Rotterdam. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author contributions
WV and DJ conceptualized and designed the experiments. MB, LS, WV, and DJ wrote the manuscript and performed statistical analysis of data. JH provided critical advice, and contributed to writing and editing of the manuscript. RB, SB, and WV planned and performed mouse experiments. MB, LS, and DJ performed histopathological assessments. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge financial support of the National Institute of Health (NIH)/National Institute of Aging (NIA) (AG17242), European Research Council Advanced Grants Dam2Age (to JH), ONCODE supported by the Dutch Cancer Society, ADPS Longevity Research Award (to WV), Memorabel (ZonMW 733050810), BBoL (NWO-ENW 737.016.015), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation—Project-ID 73111208—SFB 829), Regiodeal Foodvalley (162135), and the European Joint Programme Rare Diseases (TC-NER RD20-113).
Acknowledgments
We are grateful to Elise D. Haasdijk for histological work; Y. van Loon and the animal caretakers for general assistance with mouse experiments.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.1095801/full#supplementary-material
References
Acosta-Rodriguez, V., Rijo-Ferreira, F., Izumo, M., Xu, P., Wight-Carter, M., Green, C. B., et al. (2022). Circadian alignment of early onset caloric restriction promotes longevity in male C57BL/6J mice. Science 376, 1192–1202. doi: 10.1126/science.abk0297
Alirezaei, M., Kemball, C. C., Flynn, C. T., Wood, M. R., Whitton, J. L., and Kiosses, W. B. (2010). Short-term fasting induces profound neuronal autophagy. Autophagy 6, 702–710. doi: 10.4161/auto.6.6.12376
Alyodawi, K., Vermeij, W. P., Omairi, S., Kretz, O., Hopkinson, M., Solagna, F., et al. (2019). Compression of morbidity in a progeroid mouse model through the attenuation of myostatin/activin signalling. J. Cachexia. Sarcopenia Muscle 10, 662–686. doi: 10.1002/jcsm.12404
Andersen, B. B., Gundersen, H. J., and Pakkenberg, B. (2003). Aging of the human cerebellum: a stereological study. J. Comp. Neurol. 466, 356–365. doi: 10.1002/cne.10884
Apelt, K., White, S. M., Kim, H. S., Yeo, J. E., Kragten, A., Wondergem, A. P., et al. (2021). ERCC1 mutations impede DNA damage repair and cause liver and kidney dysfunction in patients. J. Exp. Med. 218:e20200622. doi: 10.1084/jem.20200622
Asp, M., Bergenstrahle, J., and Lundeberg, J. (2020). Spatially resolved Transcriptomes-next generation tools for tissue exploration. BioEssays 42:e1900221. doi: 10.1002/bies.201900221
Barski, J. J., Dethleffsen, K., and Meyer, M. (2000). Cre recombinase expression in cerebellar Purkinje cells. Genesis 28, 93–98. doi: 10.1002/1526-968X(200011/12)28:3/4<93::AID-GENE10>3.0.CO;2-W
Birkisdottir, M. B., Jaarsma, D., Brandt, R. M. C., Barnhoorn, S., van Vliet, N., Imholz, S., et al. (2021). Unlike dietary restriction, rapamycin fails to extend lifespan and reduce transcription stress in progeroid DNA repair-deficient mice. Aging Cell 20:e13302. doi: 10.1111/acel.13302
Birkisdottir, M. B., van Galen, I., Brandt, R. M. C., Barnhoorn, S., van Vliet, N., van Dijk, C., et al. (2022). The use of progeroid DNA repair-deficient mice for assessing anti-aging compounds, illustrating the benefits of nicotinamide riboside. Front. Aging 3:1005322. doi: 10.3389/fragi.2022.1005322
Borgesius, N. Z., de Waard, M. C., van der Pluijm, I., Omrani, A., Zondag, G. C., van der Horst, G. T., et al. (2011). Accelerated age-related cognitive decline and neurodegeneration, caused by deficient DNA repair. J. Neurosci. 31, 12543–12553. doi: 10.1523/JNEUROSCI.1589-11.2011
Covarrubias, A. J., Kale, A., Perrone, R., Lopez-Dominguez, J. A., Pisco, A. O., Kasler, H. G., et al. (2020). Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2, 1265–1283. doi: 10.1038/s42255-020-00305-3
de Graaf, E. L., Vermeij, W. P., de Waard, M. C., Rijksen, Y., van der Pluijm, I., Hoogenraad, C. C., et al. (2013). Spatio-temporal analysis of molecular determinants of neuronal degeneration in the aging mouse cerebellum. Mol. Cell. Proteomics 12, 1350–1362. doi: 10.1074/mcp.M112.024950
de Waard, M. C., van der Pluijm, I., Zuiderveen Borgesius, N., Comley, L. H., Haasdijk, E. D., Rijksen, Y., et al. (2010). Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 120, 461–475. doi: 10.1007/s00401-010-0715-9
Green, C. L., Lamming, D. W., and Fontana, L. (2022). Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 23, 56–73. doi: 10.1038/s41580-021-00411-4
Heydari, A. R., Unnikrishnan, A., Lucente, L. V., and Richardson, A. (2007). Caloric restriction and genomic stability. Nucleic Acids Res. 35, 7485–7496. doi: 10.1093/nar/gkm860
Jia, N., Guo, C., Nakazawa, Y., van den Heuvel, D., Luijsterburg, M. S., and Ogi, T. (2021). Dealing with transcription-blocking DNA damage: repair mechanisms, RNA polymerase II processing and human disorders. DNA Repair (Amst) 106:103192. doi: 10.1016/j.dnarep.2021.103192
Kwak, Y. D., Shaw, T. I., Downing, S. M., Tewari, A., Jin, H., Li, Y., et al. (2021). Chromatin architecture at susceptible gene loci in cerebellar Purkinje cells characterizes DNA damage-induced neurodegeneration. Sci. Adv. 7:eabg6363. doi: 10.1126/sciadv.abg6363
Lans, H., Hoeijmakers, J. H. J., Vermeulen, W., and Marteijn, J. A. (2019). The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 20, 766–784. doi: 10.1038/s41580-019-0169-4
Le Couteur, D. G., and Simpson, S. J. (2018). 90th anniversary commentary: caloric restriction effects on aging. J. Nutr. 148, 1656–1659. doi: 10.1093/jn/nxy146
Lee, M. B., Hill, C. M., Bitto, A., and Kaeberlein, M. (2021). Antiaging diets: separating fact from fiction. Science 374:eabe7365. doi: 10.1126/science.abe7365
Lewis, S. M., Asselin-Labat, M. L., Nguyen, Q., Berthelet, J., Tan, X., Wimmer, V. C., et al. (2021). Spatial omics and multiplexed imaging to explore cancer biology. Nat. Methods 18, 997–1012. doi: 10.1038/s41592-021-01203-6
Liu, G. Y., and Sabatini, D. M. (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 21, 183–203. doi: 10.1038/s41580-019-0199-y
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cells 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
Ma, S., Sun, S., Geng, L., Song, M., Wang, W., Ye, Y., et al. (2020). Caloric restriction reprograms the single-cell transcriptional landscape of Rattus Norvegicus aging. Cells 180, 984–1001.e22. doi: 10.1016/j.cell.2020.02.008
Makhoul, C., Gosavi, P., and Gleeson, P. A. (2019). Golgi dynamics: the morphology of the mammalian Golgi apparatus in health and disease. Front. Cell Dev. Biol. 7:112. doi: 10.3389/fcell.2019.00112
Mattison, J. A., Colman, R. J., Beasley, T. M., Allison, D. B., Kemnitz, J. W., Roth, G. S., et al. (2017). Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 8:14063. doi: 10.1038/ncomms14063
Mulderrig, L., and Garaycoechea, J. I. (2020). XPF-ERCC1 protects liver, kidney and blood homeostasis outside the canonical excision repair pathways. PLoS Genet. 16:e1008555. doi: 10.1371/journal.pgen.1008555
Mulderrig, L., Garaycoechea, J. I., Tuong, Z. K., Millington, C. L., Dingler, F. A., Ferdinand, J. R., et al. (2021). Aldehyde-driven transcriptional stress triggers an anorexic DNA damage response. Nature 600, 158–163. doi: 10.1038/s41586-021-04133-7
Nelson, W., and Halberg, F. (1986). Meal-timing, circadian rhythms and life span of mice. J. Nutr. 116, 2244–2253. doi: 10.1093/jn/116.11.2244
Niedernhofer, L. J., Gurkar, A. U., Wang, Y., Vijg, J., Hoeijmakers, J. H. J., and Robbins, P. D. (2018). Nuclear genomic instability and aging. Annu. Rev. Biochem. 87, 295–322. doi: 10.1146/annurev-biochem-062917-012239
Noe Gonzalez, M., Blears, D., and Svejstrup, J. Q. (2021). Causes and consequences of RNA polymerase II stalling during transcript elongation. Nat. Rev. Mol. Cell Biol. 22, 3–21. doi: 10.1038/s41580-020-00308-8
Pluvinage, J. V., and Wyss-Coray, T. (2020). Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat. Rev. Neurosci. 21, 93–102. doi: 10.1038/s41583-019-0255-9
Raj, D. D., Jaarsma, D., Holtman, I. R., Olah, M., Ferreira, F. M., Schaafsma, W., et al. (2014). Priming of microglia in a DNA-repair deficient model of accelerated aging. Neurobiol. Aging 35, 2147–2160. doi: 10.1016/j.neurobiolaging.2014.03.025
Schaum, N., Lehallier, B., Hahn, O., Palovics, R., Hosseinzadeh, S., Lee, S. E., et al. (2020). Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602. doi: 10.1038/s41586-020-2499-y
Shiloh, Y. (2020). The cerebellar degeneration in ataxia-telangiectasia: a case for genome instability. DNA Repair (Amst) 95:102950. doi: 10.1016/j.dnarep.2020.102950
Soheili-Nezhad, S., van der Linden, R. J., Olde Rikkert, M., Sprooten, E., and Poelmans, G. (2021). Long genes are more frequently affected by somatic mutations and show reduced expression in Alzheimer's disease: implications for disease etiology. Alzheimers Dement. 17, 489–499. doi: 10.1002/alz.12211
Speakman, J. R., and Mitchell, S. E. (2011). Caloric restriction. Mol. Asp. Med. 32, 159–221. doi: 10.1016/j.mam.2011.07.001
Van't Sant, L. J., White, J. J., Hoeijmakers, J. H. J., Vermeij, W. P., and Jaarsma, D. (2021). In vivo 5-ethynyluridine (EU) labelling detects reduced transcription in Purkinje cell degeneration mouse mutants, but can itself induce neurodegeneration. Acta Neuropathol. Commun. 9:94. doi: 10.1186/s40478-021-01200-y
Vegh, M. J., de Waard, M. C., van der Pluijm, I., Ridwan, Y., Sassen, M. J., van Nierop, P., et al. (2012). Synaptic proteome changes in a DNA repair deficient ercc1 mouse model of accelerated aging. J. Proteome Res. 11, 1855–1867. doi: 10.1021/pr201203m
Vermeij, W. P., Dolle, M. E., Reiling, E., Jaarsma, D., Payan-Gomez, C., Bombardieri, C. R., et al. (2016a). Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537, 427–431. doi: 10.1038/nature19329
Vermeij, W. P., Hoeijmakers, J. H., and Pothof, J. (2016b). Genome integrity in aging: human syndromes, mouse models, and therapeutic options. Annu. Rev. Pharmacol. Toxicol. 56, 427–445. doi: 10.1146/annurev-pharmtox-010814-124316
Vinueza Veloz, M. F., Zhou, K., Bosman, L. W., Potters, J. W., Negrello, M., Seepers, R. M., et al. (2015). Cerebellar control of gait and interlimb coordination. Brain Struct. Funct. 220, 3513–3536. doi: 10.1007/s00429-014-0870-1
Vougioukalaki, M., Demmers, J., Vermeij, W. P., Baar, M., Bruens, S., Magaraki, A., et al. (2022). Different responses to DNA damage determine ageing differences between organs. Aging Cell 21:e13562. doi: 10.1111/acel.13562
Weeda, G., Donker, I., de Wit, J., Morreau, H., Janssens, R., Vissers, C. J., et al. (1997). Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 7, 427–439. doi: 10.1016/S0960-9822(06)00190-4
White, J. J., Bosman, L. W. J., Blot, F. G. C., Osorio, C., Kuppens, B. W., Krijnen, W., et al. (2021). Region-specific preservation of Purkinje cell morphology and motor behavior in the ATXN1[82Q] mouse model of spinocerebellar ataxia 1. Brain Pathol. 31:e12946. doi: 10.1111/bpa.12946
Woodruff-Pak, D. S., Foy, M. R., Akopian, G. G., Lee, K. H., Zach, J., Nguyen, K. P., et al. (2010). Differential effects and rates of normal aging in cerebellum and hippocampus. Proc. Natl. Acad. Sci. U. S. A. 107, 1624–1629. doi: 10.1073/pnas.0914207107
Xie, K., Kapetanou, M., Sidiropoulou, K., Bano, D., Gonos, E. S., Djordjevic, A. M., et al. (2020). Signaling pathways of dietary energy restriction and metabolism on brain physiology and in age-related neurodegenerative diseases. Mech. Ageing Dev. 192:111364. doi: 10.1016/j.mad.2020.111364
Keywords: cerebellum, DNA repair, nucleotide excision repair, transcription stress, dietary restriction, Progeria, Purkinje neurons, DNA damage
Citation: Birkisdóttir MB, Van’t Sant LJ, Brandt RMC, Barnhoorn S, Hoeijmakers JHJ, Vermeij WP and Jaarsma D (2023) Purkinje-cell-specific DNA repair-deficient mice reveal that dietary restriction protects neurons by cell-intrinsic preservation of genomic health. Front. Aging Neurosci. 14:1095801. doi: 10.3389/fnagi.2022.1095801
Edited by:
Christian Neri, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Jose Felix Moruno-Manchon, University of Texas Health Science Center at Houston, United StatesVivekananda Budamagunta, University of Florida, United States
Copyright © 2023 Birkisdóttir, Van’t Sant, Brandt, Barnhoorn, Hoeijmakers, Vermeij and Jaarsma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wilbert P. Vermeij, ✉ Vy5QLlZlcm1laWpAcHJpbnNlc21heGltYWNlbnRydW0ubmw=; Dick Jaarsma, ✉ RC5KYWFyc21hQGVyYXNtdXNtYy5ubA==