 Anna Bartoletti-Stella1†
Anna Bartoletti-Stella1† Martina Tarozzi1†
Martina Tarozzi1† Giacomo Mengozzi2Francesca Asirelli3Laura Brancaleoni2,4Nicola Mometto5Michelangelo Stanzani-Maserati2
Giacomo Mengozzi2Francesca Asirelli3Laura Brancaleoni2,4Nicola Mometto5Michelangelo Stanzani-Maserati2 Simone Baiardi1,2Simona Linarello6Marco Spallazzi7Roberta Pantieri2Elisa Ferriani8
Simone Baiardi1,2Simona Linarello6Marco Spallazzi7Roberta Pantieri2Elisa Ferriani8 Paolo Caffarra9
Paolo Caffarra9 Rocco Liguori2,10
Rocco Liguori2,10 Piero Parchi2,10
Piero Parchi2,10 Sabina Capellari2,10*
Sabina Capellari2,10*- 1Department of Experimental Diagnostic and Specialty Medicine (DIMES), University of Bologna, Bologna, Italy
- 2IRCCS Istituto delle Scienze Neurologiche di Bologna, Bellaria Hospital, Bologna, Italy
- 3Department of Medical Science and Surgery (DIMEC), University of Bologna, Bologna, Italy
- 4Neurologia e Rete Stroke Metropolitana, Ospedale Maggiore, Bologna, Italy
- 5UOC Neurologia, Ospedale Guglielmo da Saliceto, Piacenza, Italy
- 6Programma Cure Intermedie - Azienda USL di Bologna, Bologna, Italy
- 7U.O. di Neurologia, Azienda Ospedaliero-Universitaria, Parma, Italy
- 8UOC Psicologia Clinica Ospedaliera, Ospedale Bellaria, Azienda USL di Bologna, Bologna, Italy
- 9Unità di Neuroscienze, Università di Parma, Parma, Italy
- 10Department of Biomedical and NeuroMotor Sciences (DIBINEM), University of Bologna, Bologna, Italy
Early-onset Alzheimer’s disease (EOAD) is the most common form of early-onset dementia. Although three major genes have been identified as causative, the genetic contribution to the disease remains unsolved in many patients. Recent studies have identified pathogenic variants in genes representing a risk factor for developing Alzheimer’s disease (AD) and in causative genes for other degenerative dementias as responsible for EOAD. To study them further, we investigated a panel of candidate genes in 102 Italian EOAD patients, 45.10% of whom had a positive family history and 21.74% with a strong family history of dementia. We found that 10.78% of patients carried pathogenic or likely pathogenic variants, including a novel variant, in PSEN1, PSEN2, or APP, and 7.84% showed homozygosity for the ε4 APOE allele. Additionally, 7.84% of patients had a moderate risk allele in PSEN1, PSEN2, or TREM2 genes. Besides, we observed that 12.75% of our patients carried only a variant in genes associated with other neurodegenerative diseases. The combination of these variants contributes to explain 46% of cases with a definite familiarity and 32% of sporadic forms. Our results confirm the importance of extensive genetic screening in EOAD for clinical purposes, to select patients for future treatments and to contribute to the definition of overlapping pathogenic mechanisms between AD and other forms of dementia.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia in the elderly and is associated with environmental and genetic components. Approximately 10% of patients with AD have an early onset disease (<65 years, EOAD) (Cacace et al., 2016) in which the heritability is between 92 and 100% (Wingo et al., 2012). Conversely, late onset AD (LOAD) is genetically more complex with heritability estimates of 58–70% (Gatz et al., 2006; Wingo et al., 2012). Still, the genetic factors identified account only for a portion of the genetic basis of the disease. To date, only about 33% of the genetic variance in sporadic AD is accounted for by common variants, and ultra-rare, rare and low-frequency variants that typically have a more harmful impact on protein function may be significant to the ‘missing heritability’ of AD (Khani et al., 2022). A proportion of 35–60% of EOAD patients have at least one affected first-degree relative (Campion et al., 1999; Brickell et al., 2006), while in 10–15%, the inheritance is autosomal dominant. Pathogenic variants in presenilin 1 (PSEN1), presenilin 2 (PSEN2) and amyloid precursor protein (APP) genes, the main autosomal dominant genetic causes, explain 5–10% of EOAD and about 50% of familial forms (Cacace et al., 2016). New data suggest that a mix of common and rare variants may cause unexplained cases that follow a non-Mendelian pattern of inheritance (Mrdjen et al., 2019). On the other hand, sporadic LOAD is considered a complex trait for which approximately 40 disease-associated genes/loci have been reported, exerting moderate to high pathogenic effects (Kunkle et al., 2019; Bellenguez et al., 2020), which have often been confirmed in EOAD (Cochran et al., 2019; Lacour et al., 2019). Among these, the ε4 allele of the apolipoprotein E (APOE) gene is not only a major genetic risk factor for LOAD, increasing the risk of disease by 3-fold in heterozygous and 15-fold in homozygous carriers (Genin et al., 2011), but also for EOAD. In these patients, the risk of disease increases more significantly than in LOADs in both ε4 homozygous and heterozygous carriers with a positive family history (Cochran et al., 2019). Furthermore, truncating and pathogenic variants in the ATP binding cassette subfamily A member 7 (ABCA7), Sortilin-related receptor 1, (SORL1), and Triggering receptor expressed on myeloid cells 2, (TREM2) genes were shown to act in a Mendelian mode in EOAD (Bellenguez et al., 2017). Additionally, a role in the disease was demonstrated for variants in genes involved in other types of neurodegenerative dementia (Cacace et al., 2016; Bartoletti-Stella et al., 2018, 2020; Sassi et al., 2018; Bonvicini et al., 2019; Giau et al., 2019; Wang et al., 2019; Park et al., 2020; Tarozzi et al., 2022), supporting the hypothesis of overlapping molecular mechanisms and a shared genetic basis. It is also likely that variants specific to individual populations, and thus difficult to detect with genome-wide ass1ociation study (GWAS) approaches unless the population is homogenous, may explain at least a portion of the missing heritability of EOAD. In addition, other inheritance patterns should be considered, e.g., autosomal recessive loci might cause EOAD (Moreno-Grau et al., 2021).
To investigate the role of rare variants in a well-characterized EOAD population of Italian origin, we analyzed 102 EOAD patients by a Next-Generation Sequencing (NGS) multigene panel covering causal and risk factor genes for AD and genes related to other forms of dementia.
Materials and methods
Participants
The EOAD consecutive unrelated patients referred to the Cognitive Disorders and Dementia Center of the UOC Clinica Neurologica, IRCCS Institute of Neurological Sciences of Bologna, from 2004 to 2019, either as outpatients, inpatients, or sent for genetic analysis, were recruited.
The AD was diagnosed according to the 2011 NIA-AA and International Working Group 2 criteria (McKhann et al., 2011; Dubois et al., 2014). All patients had evidence of AD pathophysiological process as defined by the presence of a characteristic AD CSF biomarker profile, calculated using in-house cutoff values [phosphorylated (p)-tau/Aβ42 ratio > 0.108 and total Aβ42/Aβ40 ratio < 0.68] as reported in Abu-Rumeileh et al. (2018). The strength of a patient’s family history was quantified with the modified Goldman score (Goldman et al., 2005) as reported in Cochran et al. (2019). Briefly, S1 Score: at least three people in two generations affected by EOAD, with one being a first-degree relative of the other two; S1.5 is the same as S1 but LOAD instead of EOAD; S2: at least three relatives with AD without complete autosomal dominant inheritance; S3: a single first- or second-degree family member affected with EOAD; S3.5 same as S3 but LOAD instead of EOAD. We considered patients with strong familiarity those with S1 and S1.5 scores, and with moderate family history if linked to S2, S3 or S3.5 scores.
Next generation sequencing
Genomic DNA from peripheral blood was isolated using the Maxwell 16 extractor (Promega, Madison, WI, United States) and quantified using the Quantus Fluorometer (Promega) with QuantiFluor double-stranded DNA system. Genetic screening was performed by Next Generation Sequencing (NGS) multigene panels, by using either one of the following panels: amplicon-based Illumina panel (Bartoletti-Stella et al., 2018) and probe-based Illumina panel (Truseq Neurodegeneration Illumina). Sequencing was performed on a MiSeq or NextSeq 500 sequencer using Illumina V2 reagent kit, with 2 × 150 bp paired end read cycles. Sequencing data were analyzed with an in-house bioinformatic pipeline: trimming and quality assessment of raw reads was performed with Trimmomatic (Bolger et al., 2014), mapping was performed with Burrows-Wheeler Aligner (Li and Durbin, 2009) using bwa-mem algorithm on the reference genome GRCh37/Hg19. Variant calling was performed with Strelka2 (Kim et al., 2018). Variant filtration and depth of coverage analysis were performed using Genome Analysis Toolkit (GATK) v4 (McKenna et al., 2010).
Variant classification
Variants annotation and selection were performed with BaseSpace Variant Interpreter (Illumina, CA, United States). Variants [single-nucleotide variants (SNV) and small indels] in the coding region or in the flanking 7 bp were filtered and selected with the following criteria: (i) sequence read depth at least 10× (ii) for heterozygous variants, an allelic balance value in the range of 0.30 and 0.70 (iii) Minor Allele Frequency (MAF) in the European population reported on the Genome Aggregation Database (GnomAD) (Karczewski et al., 2020) < 1%. Selected variants were classified according to the American College of Medical Genetics and Genomics guidance for the interpretation of sequence variants (Richards et al., 2015). Those reported in ClinVar (Landrum et al., 2018) or HGMD (Stenson et al., 2003) databases were classified accordingly as known disease-causing variants (Pathogenic) or variants of uncertain significance (VUS). To predict the pathogenicity of never reported variants, we performed several in silico analyses. The functional consequences of missense variants were predicted by four in silico models: Polyphen2 (Adzhubei et al., 2013), M-CAP (Jagadeesh et al., 2016), CADD v1.4 (Kircher et al., 2014), and MutationTaster (Schwarz et al., 2014), intronic splicing variants by: NetGene2 (Hebsgaard et al., 1996), MaxEntScan and Human Splicing Finder1, while silent variants by MutationTaster (Schwarz et al., 2014), CADD (Kircher et al., 2014), and FATHMM XF2. Allele frequencies were compared with those reported in the Genome Aggregation Database (GnomAD v2.1.1). Variant calling files (VCF) related to the analyzed genomic regions were reported in the Supplementary File.
Copy number variation analysis in Alzheimer’s disease causative genes
A preliminary in silico copy number variation analysis was performed on the sequencing data using the CNVkit (Talevich et al., 2016). Results for APP, PSEN1, and PSEN2 genes were validated using a Multiplex Ligation-dependent Probe Amplification (MLPA) assay (MRC Holland). The results of the MLPA analysis were analyzed with Coffalyzer.net.
APOE genotyping
Genotyping of APOE was performed by restriction fragment length polymorphism according to Wenham et al. (1991).
Clinical classification of variants
According to Cochran et al. (2019), pathogenic and likely pathogenic variants were returned as “diagnostic.” Variants classified in ClinVar or HGMD database as of uncertain significance (VUS) identified in AD-Causative genes or AD-risk factor genes (TREM2, ABCA7, and SORL1) were considered as risk factor alleles. We considered contributor of disease variants reported in ClinVar or HGMD as uncertain significance if found in fEOAD and if enriched in our AD cohort than GnomAD European non-finish population (v.2.1.1), assessed with Fisher’s exact Test and Benjamini–Hochberg false discovery rate correction, p-value < 0.05.
Statistical analysis
Variant zygosity was extracted from the VCF files and allele frequencies of our dataset were compared with those reported in GnomAD. Statistical significance (p < 0.05) of variant allele frequencies between our dataset and those reported in GnomAD for the European (non-Finnish) population was assessed with Fisher’s exact Test and Benjamini–Hochberg false discovery rate correction. All tables report only adjusted p-values.
Results
Study population
The cohort included 102 patients diagnosed with EOAD; in one the diagnosis was neuropathologically confirmed (0.98%), in 72 was defined as “probable” (70.59%), and in 29 as “possible” (28.43%) with evidence of the AD pathophysiological process (McKhann et al., 2011). According to the International Working Group 2 criteria, a “typical” AD phenotype was found in 58 individuals (56.8%), the frontal variant in 15 (14.7%), the logopenic variant in 14 (13.7%), and the posterior variant in 15 (14.7%). The mean of age at onset (AAO) was 56.88 ± 5.84 years, while 10 patients presented the first symptoms before the age of 51. Forty-nine patients were male (48.04%), 46 showed a positive family history (45.10%), of whom 10 were classified as having a strong family history (21.74% of all family cases) (Table 1). Dimensionality reduction plots performed with Principal Component Analysis (PCA) and t-distributed stochastic neighbor embedding (t-SNE, Jaccard similarity used as metric, right panel) show an overall homogenous genetic background in our EOAD cohort, with no confounders caused by the geographical origin of the patients (Supplementary Figure 1).
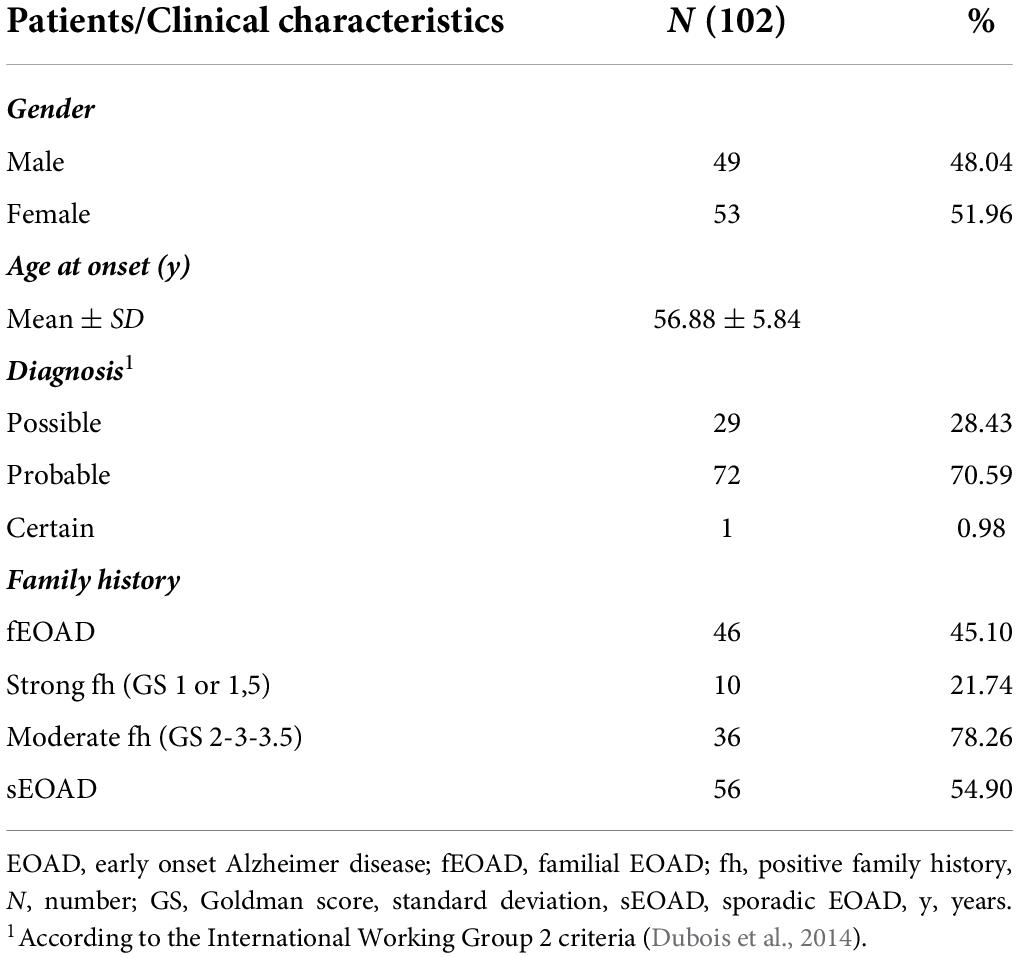
Table 1. Clinical features of study population.
Analysis of causative genes: APP, PSEN1, and PSEN2
We found 17 different rare variants (MAF < 0.01) in the AD causative genes APP, PSEN1, and PSEN2 (Table 2). Nine (8.82%) patients carried a “diagnostic” variant; seven had a positive family history, although a strong familiarity was established only in two. All identified variants had been previously reported, with the only exception of p.Leu85Phe in the PSEN1 gene (Table 2), which was not even present in the GnomAD. The carrier had a posterior variant of AD, with positive familial history. Two patients (AD#101 and AD#043) carried two variants, one in the APP and PSEN1 genes, and the second in the PSEN1 and PSEN2 genes. No CNV were identified in AD-causative gene. Six rare variants were previously reported in the ClinVar or HGMD database as likely benign or of uncertain significance (APP p.Phe435=, PSEN1 p.Arg35Gln, PSEN2 p.Arg62His, p.Arg71Trp, p.Met174Val, p.Ser236=) (Table 2).
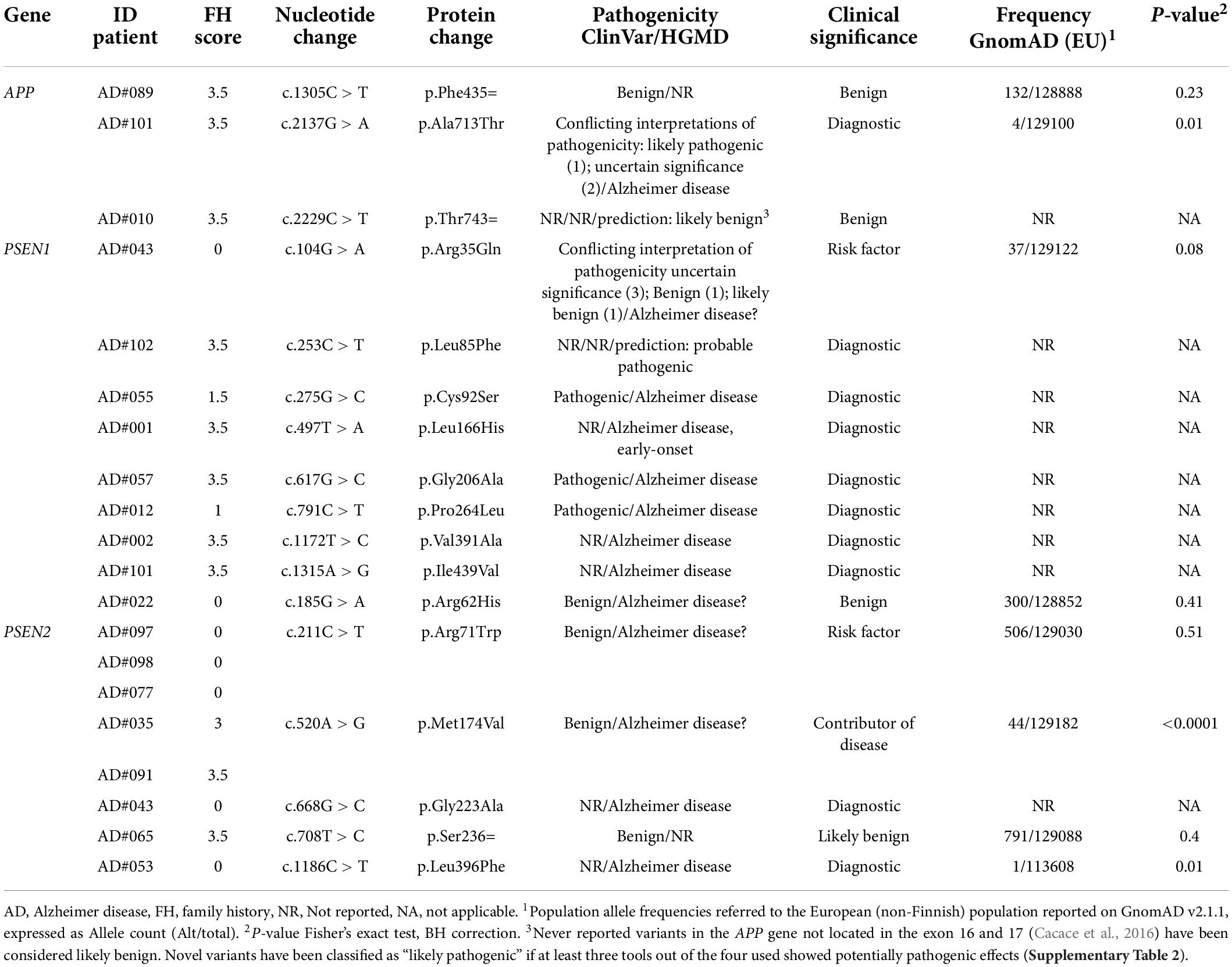
Table 2. Rare variants in AD-causative genes identified in this study.
APOE genotype
Eight patients (7.84%), all negative for variants in AD causative genes, carried the APOE ε4/ε4 genotype (Cochran et al., 2019). Seven of them had a positive family history, while familiarity was strong in two. Thirty-two (31.37%) patients showed heterozygosity for the APOE ε4 allele: thirty carried the APOE ε3/ε4 and two the APOE ε2/ε4 genotype. The frequency of heterozygosity for the APOE ε4 allele was not increased in patients with a positive family history.
Variants in TREM2 gene
Like APOE ε4, pathogenic variants in TREM2 also increase a person’s odds of developing late-onset AD from three- to 12-fold (Wolfe et al., 2019).
We identified five different variants in this gene, all singletons, in four patients (Table 3). One patient (AD#026) carried two variants. The variants discovered were not strictly deleterious: four were previously reported in the ClinVar database as benign, and one as VUS.

Table 3. Rare variants in AD-risk gene TREM2 identified in this study.
Variants in dementia-associated genes
Given the common mechanisms previously reported between AD and other neurodegenerative diseases (Bartoletti-Stella et al., 2018; Park et al., 2020), we also collected data on likely pathogenic rare variants in genes that play a role in other dementias.
After applying the filtering criteria, we identified 33 different rare variants in 12 genes (Table 4). Variants previously reported as pathogenic or likely pathogenic were identified in patients with motor neuron disease (FUS p.Gly144_Tyr149del, DCTN1 p.Arg997Trp, OPTN p. Gln314Leu and c.1401 + 4A > G, SQSTM1 p.Pro392Leu) and amyotrophic lateral sclerosis (ALS)-Charcot-Marie-Tooth disease type 4 (FIG4 p.Gln823Ter). Five previously unreported variants were classified as likely pathogenic (Table 5) by in silico prediction. These variants map to the CCNF, DCTN1, and NOTCH3 genes, which are involved in the ALS/frontotemporal dementia (FTD) spectrum (Supplementary Table 2). In addition, NOTCH3 variants have been previously associated with AD risk (Patel et al., 2019).
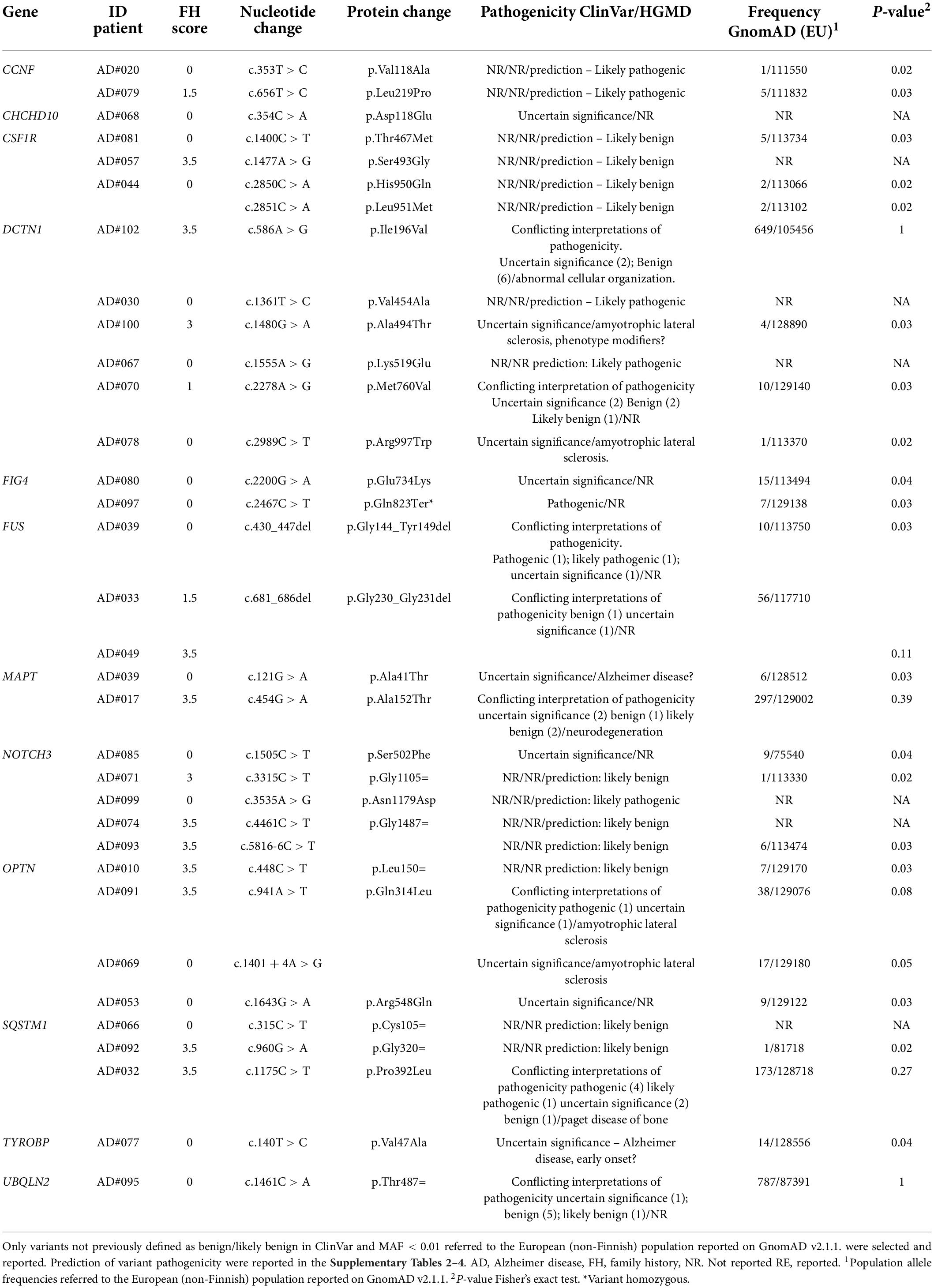
Table 4. Rare variants in other dementia causative genes identified in this study.
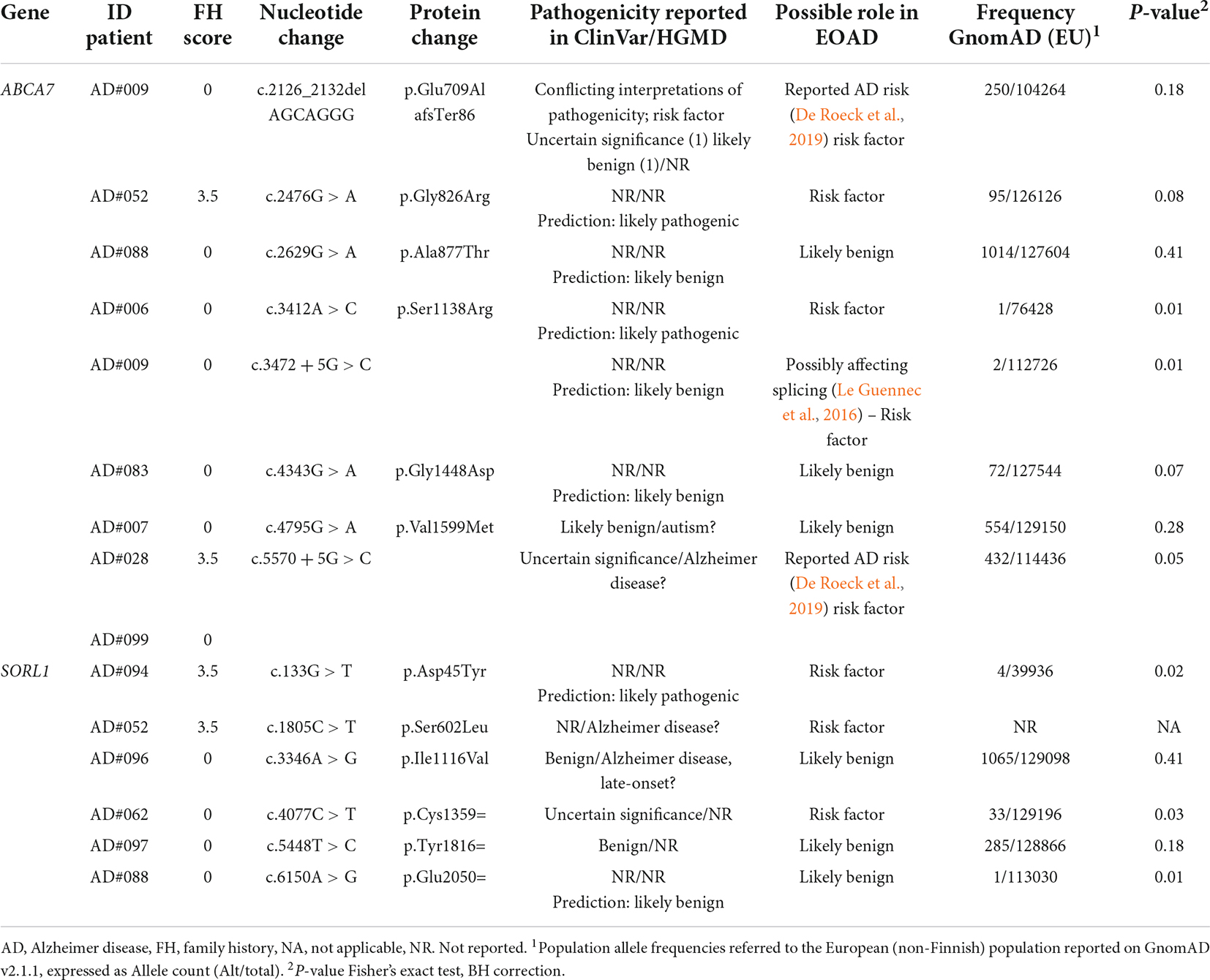
Table 5. Rare variants in AD-risk genes ABAC7 and SORL1 identified in this study.
Among the VUS, two were identified in the MAPT gene. Although MAPT pathogenic variants are typically associated with FTD (Cruts et al., 2012), these variants have already been reported in patients with AD (Cochran et al., 2019). One of them (p.Ala152Thr) has been identified as a risk factor for several neurodegenerative diseases, including AD (Sydow et al., 2016). The other MAPT variant (p.Ala41Thr) occurred in a patient also carrying a pathogenic mutation in the FUS gene (AD#039). Due to the heterogeneity of genetic factors contributing to neurodegeneration, pathogenic/likely pathogenic variants and VUS have been considered as weak allele risk factors (Dilliott et al., 2021). Carriers of these variants were equally distributed between the familial and sporadic groups (Fisher exact test p-value = 1).
Missing heritability: Role of rare variants in other Alzheimer’s disease-risk factor genes
Rare pathogenic variants in AD causative genes, homozygosity for APOE ε4/ε4, risk factor alleles and likely pathogenic variants in genes related to other forms of neurodegenerative dementias explain 46% of familial and 32% of sporadic patients in our cohort (Figure 1). In recent years, more than 40 AD-associated genes/loci have been identified by GWAS, and subsequent sequencing projects have highlighted the role of rare variants in these genes in EOAD (Bellenguez et al., 2020; Wightman et al., 2021). In a pilot cohort of EOADs without causative variants (n = 30) we analyzed 34 genes (Supplementary Table 1), identified as susceptibility factors for AD, for the presence of rare and possibly pathogenic variants.
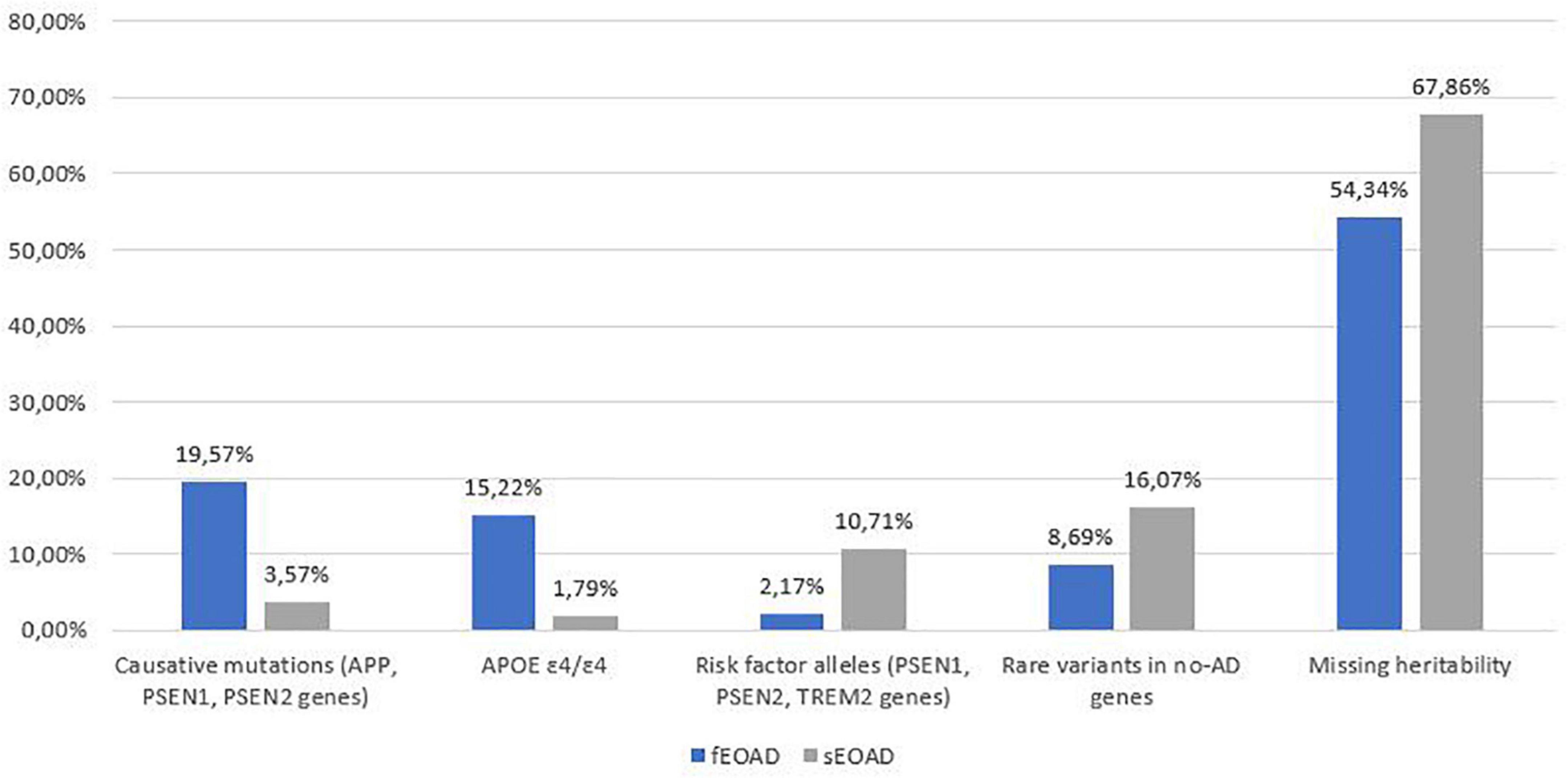
Figure 1. Summary of genetic variants identified in the 102 AD patients, divided into familial (fEOAD) and sporadic patients (sEOAD). Causative mutations included diagnostic and strong contributor alleles in APP, PSEN1, and PSEN2 genes, moderate risk alleles included variant not strictly pathogenic in APP, PSEN1, PSEN2, and TREM2 genes, rare variants in no-AD genes included pathogenic/likely pathogenic/VUS (uncertain significant variants or with major evidence of pathogenicity in ClinVar and/or HGMD) variants in genes causative for other type of dementia (Supplementary Table 1).
Most of the variants identified were in the ABCA7 (n = 8; one in two patients) and SORL1 (n = 6) genes (Table 5). Of these, two in the ABCA7 gene were previously reported PTV (protein truncating variants) (Table 5). Among them, the c.2126_2132delAGCAGG p.Glu709AlafsTer86 variant was classified as a contributor to the disease (De Roeck et al., 2019). We also found three rare missense variants in the coding sequence of SORL1, one of which (p.Asp45Tyr) was classified as a likely pathogenic variant. Among the possibly pathogenic variants in ABCA7 and SORL1, five had a positive association in our cohort compared to the frequency in the European population (Table 5). The remaining missense or silent variants were considered as rare benign variants (Table 5 and Supplementary Tables 2–4).
Additionally, 26 rare variants of potential interest (Table 6) were identified in 17 of the 32 other AD susceptibility genes analyzed (Supplementary Table 1). All identified variants were singleton except for the FERMT2 p.Thr513Met variant. Only three variants were reported in ClinVar or HGMD databases, all classified as VUS. Variants in the BIN1 gene (p.Asn232Lys, and c.1462-3C > T) were linked to myopathy, while the variant in PTK2B was associated with Parkinson’s disease. As for the remaining variants, eight were classified as likely pathogenic by in silico analyses (Table 6 and Supplementary Tables 2–4).
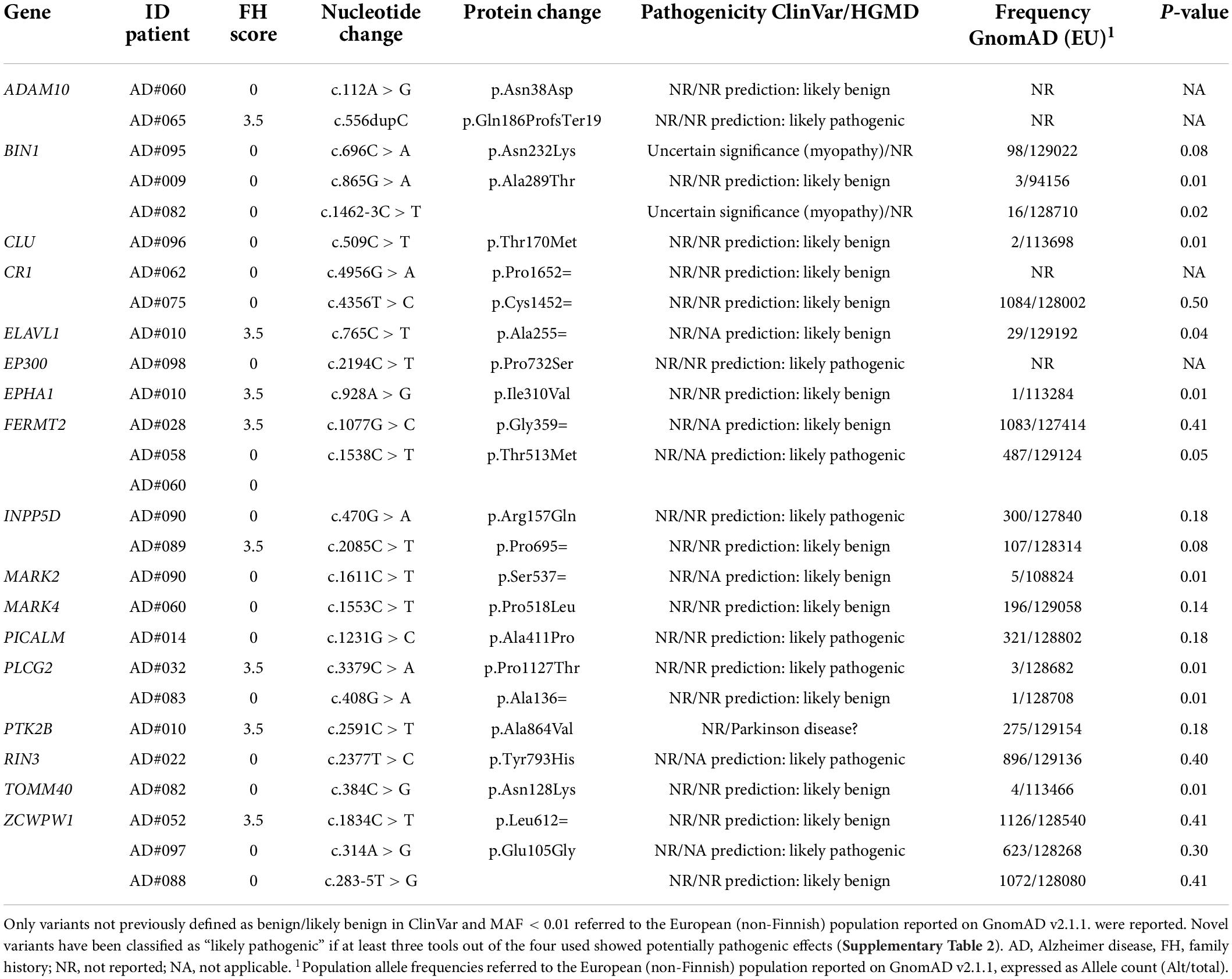
Table 6. Rare variants in GWAS genes identified in this study.
In conclusion, 63% of the examined patients presented at least one likely pathogenic variant or a rare VUS in AD-susceptibility genes (Tables 5, 6), suggesting that they might also confer risk for the development of EOAD. Familial and sporadic patients did not show a different distribution of these variants.
Discussion
Clarifying the genetic and molecular basis of EOAD and its clinical variability is crucial for improving diagnostic screening and developing more effective, and possibly preventive, disease modifying treatments.
Since a concern in genetic studies of AD patients is that at least some variants are private or specific to certain populations, we analyzed a well-characterized single-center cohort of 102 patients by a NGS panel including AD causative genes, risk factors and genes involved in other types of dementia.
We found a pathogenic mutation in one of the three AD-causing genes in 8.82% of patients, in agreement with the expected maximum of 11% reported in the literature (Mendez, 2019). These include a novel, likely pathogenic variant, PSEN1 c.253C > T p.Leu85Phe. According to Guerreiro algorithm (Guerreiro et al., 2010), this variant can be defined as probably pathogenic: it maps to the first transmembrane domain, is defined as damaging by in silico tools (Supplementary Table 2), a causative mutation in the same codon (p.Leu85Pro) has been described in patients with EOAD (Ataka et al., 2004), and the residue is conserved in PSEN2 (p.Leu91). PSEN2 mutations are considered very rare but have the highest frequency in Spain and Italy (Cai et al., 2015). In agreement, we identified four possibly pathogenic missense variants in this gene, two possibly pathogenic missense variants, p.Gly223Ala and p.Leu396Phe, both in patients with no family history of AD, and two rare variants, p.Met174Val and p.Arg71Trp, which have a questionable classification. These variants were initially described as possibly pathogenic (Guerreiro et al., 2010) but subsequently found in healthy controls. Thus, they are currently classified as non-pathogenic (Alzforum Mutation database3). We demonstrated a positive association with the risk of developing EOAD for the PSEN2 p.Met174Val variant, compared with the non-Finnish European allele frequency reported in GnomAD, having found it in two patients with EOAD and positive family history. This variants could be considered a contributing factor to the disease.
Given the previously reported common mechanisms between AD and other dementias (Bartoletti-Stella et al., 2018; Giau et al., 2019), we also collected data on likely pathogenic rare variants in genes implicated in other dementias. Of the 33 rare variants identified in 12 genes, 10 were pathogenic or likely pathogenic, nine VUS, equally distributed between the familial and sporadic EOAD (Table 4). Most of them were in genes causal for the ALS/FTD continuum. Thus, our study supports the view that AD and other neurodegenerative diseases might represent shades of the same disease spectrum, and that extended genetic testing of causative genes for other degenerative dementias should be offered to patients diagnosed with EOAD (Bartoletti-Stella et al., 2018Giau et al., 2019). In agreement with this hypothesis, Spina et al., 2021, found at least one non-AD neuropathological diagnosis in 98% of patients with EOAD. Due to the heterogeneity of genetic factors contributing to neurodegeneration, and the equal distribution between sporadic and familial forms, we consider pathogenic/hypothetical pathogenic variants and variants with uncertain significance in these genes to be weak risk factors (Dilliott et al., 2021).
Concerning the main known risk factors for AD, homozygosity for APOE ε4 allele has previously been reported to be associated with a significantly increased risk of EOAD than of LOAD, regardless of family history (van Duijn et al., 1994; BLACKER et al., 1997; Cochran et al., 2019); in agreement, it reaches 7.84% in our dataset. This evidence confirms previous data suggesting that APOE may strongly influence AD risk at younger ages, but as age increases, the effect of APOE is reduced, and other risk variants start to play a more significant role in AD risk (Bellou et al., 2020). These include variants in ABCA7, SORL1, and TREM2 gene. Regarding TREM2, a significant exome-wide association between the p.Arg47His variant and the risk of EOAD has been reported while rare TREM2 variants have been associated with Aβ deposition, Aβ uptake by microglia, and increased tau in CSF (Gratuze et al., 2018). Together, these data suggest a connection with EOAD neuropathologic findings, and both the amyloid and tau hypotheses of AD (Ayodele et al., 2021). Furthermore, in patients with autosomal-dominant AD, CSF Aβ42 decline, cortical atrophy and cognitive impairment were lower in patients with high soluble receptor stub (Morenas-Rodríguez et al., 2022). In the TREM2 coding region we found five rare VUS (one patient carried two variants), two of which (p.Arg136Gln, p.Thr223Ile) showed a higher allele frequency in EOAD than the European population reported on GnomAD (Table 3). The p.Arg136Gln variant causes the substitution of an arginine residue required for optimal binding of extracellular ligands, and other known missense variants were found in familial and early onset forms of the disease (Sirkis et al., 2016). In agreement with a previous study (Bellenguez et al., 2017), we considered these variants as risk factor/weak contributor to the disease.
Our results are therefore consistent with previous findings and strengthen the potential role of these rare variants as risk factors.
Recent meta-analyses and GWAS have shown a fivefold increased risk, similar to that of APOE-ε4 carriers, of developing EOAD with rare variants in SORL1 (Ayodele et al., 2021) and a study that used the Exome Aggregation Consortium (ExAC; ExAC browser is not available anymore, and it is now part of the gnomAD) database established that pathogenic SORL1 variants increase AD risk by 12-fold, as well as causing an earlier age of onset (58.6 ± 5.2 years) (Holstege et al., 2017). Moreover, protein-truncating variants in SORL1 were observed exclusively in AD patients, and are highly penetrant, whereas two SORL1 missense mutations (p.R1303C and p.G1732A) and a splice site variant (c.3050-2A > G) have been shown to segregate with disease in families affected by autosomal dominant AD (Holstege et al., 2017). Among the six rare variants in the SORL1 gene identified in our cohort, p.Asp45Tyr and p.Ser402Leu were found in two familial patients. The variant p.Asp45Tyr was never previously reported and classified as possibly pathogenic.
In addition, we identified five unreported missense variants in ABCA7, (Table 2), a gene containing risk factors for developing both EOAD and LOAD due to its role in amyloid clearance and decreasing Aβ production through interference with APP processing (Satoh et al., 2015; De Roeck et al., 2017; Sleegers and Van Broeckhoven, 2019). In addition, ABCA7 has been shown to provide a greater predisposition to develop AD than the APOE ε4 allele in African American adults. To date, four autosomal dominant AD families are known in which rare ABCA7 PTC and missense variants segregated with the disease (Hoogmartens et al., 2021). Although our variants are missense, they may likely act as essential contributors to EOAD susceptibility, albeit with variable penetrance (Khani et al., 2022).
Lastly, in EOAD without causative mutations or APOE ε4/ε4, we analyzed 34 genes previously identified as susceptibility factors for AD. We found an average of 0.6 rare variants in each patient in 14 of them, also confirming in EOAD patients a high amount of risk factors.
Overall, the extended genetic analysis of a well-defined cohort of Italian EOAD patients showed that in cases without dominant pathogenic variants in PSEN1, PSEN2 and APP, there is an enrichment for multiple pathogenic/likely pathogenic variants in genes associated with risk factors for AD, covering 38% of our cohort, supporting the extreme genetic heterogeneity of EOAD. Independently of family history, a high proportion of EOAD patients carried genetic risk factors, suggesting oligogenic determinism, particularly in individuals without familiarity. Therefore, our data also highlight the role of rare variants especially in sporadic EOAD, each exerting a moderate to high pathogenic effect and corroborate an extended analysis to identify variants implicated in the disease.
We also confirmed enrichment of APOE ε4/ε4 homozygosity and rare pathogenic/likely pathogenic variants in SORL1, TREM2, and ABCA7 (Khani et al., 2022). Additionally, we found a substantial proportion of pathogenic variants in several autosomal dominant genes, causal in other dementias or previously identified as risk factors for AD, most often in patients with positive familiarity, providing evidence to the hypothesis that many different rare mutations usually detected only in a single family or in small populations could be causal in familial EOAD.
In summary, our study describes new pathogenic variants in AD linked genes, PSEN1, ABCA7, SORL1, and contributes to disentangle the broad genetic landscape of Italian EOAD. The results suggest that a systematic application of comprehensive genetic assessments could aid in the interpretation of early-onset dementia cases by providing a molecular basis to their phenotypic heterogeneity and potentially in enabling future personalized medicine approaches.
Data availability statement
The datasets presented in this study can be found in online repositories and the study is deposited in the European Nucleotide Archive (ENA, https://www.ebi.ac.uk/ena/) EMBLEBI repository, accession number PRJEB55143 (ERP140024).
Ethics statement
Ethical approval of the study was obtained by the ethical board Area Vasta Emilia Centro, (AVEC). The patients/participants provided their written informed consent to participate in this study.
Author contributions
AB-S and MT: manuscript drafting and revising, data collection, analysis, and interpretation of data. GM, FA, LB, NM, MS-M, SB, SL, MS, RP, EF, PC, RL, and PP: data collection and revision of the manuscript. SC: study concept and design, drafting and revising the manuscript, analysis and interpretation of data, and study supervision. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Italian Ministry of Research RFO and by Fondazione del Monte.
Acknowledgments
The authors acknowledge the patients and their families; the Italian Ministry of Research RFO and Fondazione del Monte.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2022.969817/full#supplementary-material
Footnotes
- ^ www.umd.be/HSF/
- ^ https://fathmm.biocompute.org.uk/fathmm-xf/
- ^ https://www.alzforum.org/mutations
References
Abu-Rumeileh, S., Mometto, N., Bartoletti-Stella, A., Polischi, B., Oppi, F., Poda, R., et al. (2018). Cerebrospinal fluid biomarkers in patients with frontotemporal dementia spectrum: a single-center study. J. Alzheimers Dis. 66, 551–563. doi: 10.3233/JAD-180409
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 7:20. doi: 10.1002/0471142905.hg0720s76
Ataka, S., Tomiyama, T., Takuma, H., Yamashita, T., Shimada, H., Tsutada, T., et al. (2004). A novel presenilin-1 mutation (Leu85Pro) in early-onset Alzheimer disease with spastic paraparesis. Arch. Neurol. 61, 1773–1776. doi: 10.1001/archneur.61.11.1773
Ayodele, T., Rogaeva, E., Kurup, J. T., Beecham, G., and Reitz, C. (2021). Early-onset Alzheimer’s disease: what is missing in research? Curr. Neurol. Neurosci. Rep. 21:4. doi: 10.1007/s11910-020-01090-y
Bartoletti-Stella, A., Baiardi, S., Stanzani-Maserati, M., Piras, S., Caffarra, P., Raggi, A., et al. (2018). Identification of rare genetic variants in Italian patients with dementia by targeted gene sequencing. Neurobiol. Aging 66, 180.e23–180.e31. doi: 10.1016/j.neurobiolaging.2018.02.006
Bartoletti-Stella, A., De Pasqua, S., Baiardi, S., Bartolomei, I., Mengozzi, G., Orio, G., et al. (2020). Characterization of novel progranulin gene variants in Italian patients with neurodegenerative diseases. Neurobiol. Aging 97, .e7–.e145. doi: 10.1016/j.neurobiolaging.2020.05.004
Bellenguez, C., Charbonnier, C., Grenier-Boley, B., Quenez, O., Le Guennec, K., Nicolas, G., et al. (2017). Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 59, 220.e1–220.e9. doi: 10.1016/j.neurobiolaging.2017.07.001
Bellenguez, C., Grenier-Boley, B., and Lambert, J. C. (2020). Genetics of Alzheimer’s disease: where we are, and where we are going. Curr. Opin. Neurobiol. 61, 40–48. doi: 10.1016/j.conb.2019.11.024
Bellou, E., Baker, E., Leonenko, G., Bracher-Smith, M., Daunt, P., Menzies, G., et al. (2020). Age-dependent effect of APOE and polygenic component on Alzheimer’s disease. Neurobiol. Aging 93, 69–77. doi: 10.1016/j.neurobiolaging.2020.04.024
BLACKER, D., Haines, J. L., Rodes, L., Terwedow, H., Go, R. C., Harrell, L. E., et al. (1997). ApoE-4 and age at onset of Alzheimer’s disease: The NIMH genetics initiative. Neurology 48, 139–147. doi: 10.1212/wnl.48.1.139
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonvicini, C., Scassellati, C., Benussi, L., Di Maria, E., Maj, C., Ciani, M., et al. (2019). Next generation sequencing analysis in early onset dementia patients. J. Alzheimers Dis. 67, 243–256. doi: 10.3233/JAD-180482
Brickell, K. L., Steinbart, E. J., Rumbaugh, M., Payami, H., Schellenberg, G. D., Van Deerlin, V., et al. (2006). Early-onset Alzheimer disease in families with late-onset Alzheimer disease: a potential important subtype of familial Alzheimer disease. Arch. Neurol. 63, 1307–1311. doi: 10.1001/archneur.63.9.1307
Cacace, R., Sleegers, K., and Van Broeckhoven, C. (2016). Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 12, 733–748. doi: 10.1016/j.jalz.2016.01.012
Cai, Y., An, S. S., and Kim, S. (2015). Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 10, 1163–1172. doi: 10.2147/CIA.S85808
Campion, D., Dumanchin, C., Hannequin, D., Dubois, B., Belliard, S., Puel, M., et al. (1999). Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am. J. Hum. Genet. 65, 664–670. doi: 10.1086/302553
Cochran, J. N., McKinley, E. C., Cochran, M., Amaral, M. D., Moyers, B. A., Lasseigne, B. N., et al. (2019). Genome sequencing for early-onset or atypical dementia: high diagnostic yield and frequent observation of multiple contributory alleles. Cold Spring Harb. Mol. Case Stud. 5:a003491. doi: 10.1101/mcs.a003491
Cruts, M., Theuns, J., and Van Broeckhoven, C. (2012). Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 33, 1340–1344. doi: 10.1002/humu.22117
De Roeck, A., Van Broeckhoven, C., and Sleegers, K. (2019). The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. 138, 201–220. doi: 10.1007/s00401-019-01994-1
De Roeck, A., Van den Bossche, T., van der Zee, J., Verheijen, J., De Coster, W., Van Dongen, J., et al. (2017). Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset Alzheimer’s disease. Acta Neuropathol. 134, 475–487. doi: 10.1007/s00401-017-1714-x
Dilliott, A. A., Abdelhady, A., Sunderland, K. M., Farhan, S. M. K., Abrahao, A., Binns, M. A., et al. (2021). Contribution of rare variant associations to neurodegenerative disease presentation. NPJ Genom. Med. 6:80. doi: 10.1038/s41525-021-00243-3
Dubois, B., Feldman, H. H., Jacova, C., Hampel, H., Molinuevo, J. L., Blennow, K., et al. (2014). Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629. doi: 10.1016/S1474-4422(14)70090-0
Gatz, M., Reynolds, C. A., Fratiglioni, L., Johansson, B., Mortimer, J. A., Berg, S., et al. (2006). Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. doi: 10.1001/archpsyc.63.2.168
Genin, E., Hannequin, D., Wallon, D., Sleegers, K., Hiltunen, M., Combarros, O., et al. (2011). APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16:903. doi: 10.1038/mp.2011.52.-7
Giau, V. V., Bagyinszky, E., Yang, Y. S., Youn, Y. C., An, S. S. A., and Kim, S. Y. (2019). Genetic analyses of early-onset Alzheimer’s disease using next generation sequencing. Sci. Rep. 9:8368.
Goldman, J. S., Farmer, J. M., Wood, E. M., Johnson, J. K., Boxer, A., Neuhaus, J., et al. (2005). Comparison of family histories in FTLD subtypes and related tauopathies. Neurology 65, 1817–1819. doi: 10.1212/01.wnl.0000187068.92184.63
Gratuze, M., Leyns, C. E. G., and Holtzman, D. M. (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13:66. doi: 10.1186/s13024-018-0298-9
Guerreiro, R. J., Baquero, M., Blesa, R., Boada, M., Brás, J. M., Bullido, M. J., et al. (2010). Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging. 31, 725–731. doi: 10.1016/j.neurobiolaging.2008.06.012
Hebsgaard, S. M., Korning, P. G., Tolstrup, N., Engelbrecht, J., Rouzé, P., and Brunak, S. (1996). Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 24, 3439–3452. doi: 10.1093/nar/24.17.3439
Holstege, H., van der Lee, S. J., Hulsman, M., Wong, T. H., van Rooij, J. G., Weiss, M., et al. (2017). Characterization of pathogenic SORL1 genetic variants for association with Alzheimer’s disease: a clinical interpretation strategy. Eur. J. Hum. Genet. 25, 973–981. doi: 10.1038/ejhg.2017.87
Hoogmartens, J., Cacace, R., and Van Broeckhoven, C. (2021). Insight into the genetic etiology of Alzheimer’s disease: a comprehensive review of the role of rare variants. Alzheimers Dement. 13:e12155. doi: 10.1002/dad2.12155
Jagadeesh, K. A., Wenger, A. M., Berger, M. J., Guturu, H., Stenson, P. D., Cooper, D. N., et al. (2016). M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 48, 1581–1586. doi: 10.1038/ng.3703
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Khani, M., Gibbons, E., Bras, J., and Guerreiro, R. (2022). Challenge accepted: uncovering the role of rare genetic variants in Alzheimer’s disease. Mol. Neurodegener. 17:3. doi: 10.1186/s13024-021-00505-9
Kim, S., Scheffler, K., Halpern, A. L., Bekritsky, M. A., Noh, E., Källberg, M., et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. doi: 10.1038/s41592-018-0051-x
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., and Shendure, J. A. (2014). general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Lacour, M., Quenez, O., Rovelet-Lecrux, A., Salomon, B., Rousseau, S., Richard, A. C., et al. (2019). Causative mutations and genetic risk factors in sporadic early onset Alzheimer’s disease before 51 years. J. Alzheimers Dis. 71, 227–243. doi: 10.3233/JAD-190193
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. doi: 10.1093/nar/gkx1153
Le Guennec, K., Nicolas, G., Quenez, O., Charbonnier, C., Wallon, D., Bellenguez, C., et al. (2016). ABCA7 rare variants and Alzheimer disease risk. Neurology 86, 2134–2137. doi: 10.1212/WNL.0000000000002627
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Mendez, M. F. (2019). Early-onset alzheimer disease and its variants. Continuum 25, 34–51. doi: 10.1212/CON.0000000000000687
Morenas-Rodríguez, E., Li, Y., Nuscher, B., Franzmeier, N., Xiong, C., Suárez-Calvet, M., et al. (2022). Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: a longitudinal observational study. Lancet Neurol. 21, 329–341. doi: 10.1016/S1474-4422(22)00027-8
Moreno-Grau, S., Fernández, M. V., de Rojas, I., Garcia-González, P., Hernández, I., Farias, F., et al. (2021). Long runs of homozygosity are associated with Alzheimer’s disease. Transl. Psychiatry 11:142. doi: 10.1038/s41398-020-01145-1
Mrdjen, D., Fox, E. J., Bukhari, S. A., Montine, K. S., Bendall, S. C., and Montine, T. J. (2019). The basis of cellular and regional vulnerability in Alzheimer’s disease. Acta Neuropathol. 138, 729–749. doi: 10.1007/s00401-019-02054-4
Park, J. E., Kim, H. J., Kim, Y. E., Jang, H., Cho, S. H., Kim, S. J., et al. (2020). Analysis of dementia-related gene variants in APOE ε4 noncarrying Korean patients with early-onset Alzheimer’s disease. Neurobiol. Aging 85, 155.e5–155.e8. doi: 10.1016/j.neurobiolaging.2019.05.009
Patel, D., Mez, J., Vardarajan, B. N., Staley, L., Chung, J., Zhang, X., et al. (2019). Association of rare coding mutations with alzheimer disease and other dementias among adults of european ancestry. JAMA Netw. Open 2:e191350. doi: 10.1001/jamanetworkopen.2019.1350
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sassi, C., Nalls, M. A., Ridge, P. G., Gibbs, J. R., Lupton, M. K., Troakes, C., et al. (2018). Mendelian adult-onset leukodystrophy genes in Alzheimer’s disease: Critical influence of CSF1R and NOTCH3. Neurobiol. Aging 66, 179.e17–179.e179. doi: 10.1016/j.neurobiolaging.2018.01.015
Satoh, K., Abe-Dohmae, S., Yokoyama, S., St George-Hyslop, P., and Fraser, P. E. (2015). ATP-binding cassette transporter A7 (ABCA7) loss of function alters Alzheimer amyloid processing. J. Biol. Chem. 290, 24152–24165. doi: 10.1074/jbc.M115.655076
Schwarz, J. M., Cooper, D. N., Schuelke, M., and Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362. doi: 10.1038/nmeth.2890
Sirkis, D. W., Bonham, L. W., Aparicio, R. E., Geier, E. G., Ramos, E. M., Wang, Q., et al. (2016). Rare TREM2 variants associated with Alzheimer’s disease display reduced cell surface expression. Acta Neuropathol. Commun. 4:98. doi: 10.1186/s40478-016-0367-7
Sleegers, K., and Van Broeckhoven, C. (2019). Novel Alzheimer’s disease risk genes: exhaustive investigation is paramount. Acta Neuropathol. 138, 171–172. doi: 10.1007/s00401-019-02041-9
Spina, S., La Joie, R., Petersen, C., Nolan, A. L., Cuevas, D., Cosme, C., et al. (2021). Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain 144, 2186–2198. doi: 10.1093/brain/awab099
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S., et al. (2003). Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–581. doi: 10.1002/humu.10212
Sydow, A., Hochgräfe, K., Könen, S., Cadinu, D., Matenia, D., Petrova, O., et al. (2016). Age-dependent neuroinflammation and cognitive decline in a novel Ala152Thr-Tau transgenic mouse model of PSP and AD. Acta Neuropathol. Commun. 4:17. doi: 10.1186/s40478-016-0281-z
Talevich, E., Shain, A. H., Botton, T., and Bastian, B. C. (2016). CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 12:e1004873. doi: 10.1371/journal.pcbi.1004873
Tarozzi, M., Bartoletti-Stella, A., Dall’Olio, D., Matteuzzi, T., Baiardi, S., Parchi, P., et al. (2022). Identification of recurrent genetic patterns from targeted sequencing panels with advanced data science: a case-study on sporadic and genetic neurodegenerative diseases. BMC Med. Genomics 15:26. doi: 10.1186/s12920-022-01173-4
van Duijn, C. M., de Knijff, P., Cruts, M., Wehnert, A., Havekes, L. M., Hofman, A., et al. (1994). Apolipoprotein E4 allele in a population-based study of early-onset Alzheimer’s disease. Nat. Genet. 7, 74–78. doi: 10.1038/ng0594-74
Wang, G., Zhang, D. F., Jiang, H. Y., Fan, Y., Ma, L., Shen, Z., et al. (2019). Mutation and association analyses of dementia-causal genes in Han Chinese patients with early-onset and familial Alzheimer’s disease. J. Psychiatr. Res. 113, 141–147. doi: 10.1016/j.jpsychires.2019.03.026
Wenham, P. R., Price, W. H., and Blandell, G. (1991). Apolipoprotein E genotyping by one-stage PCR. Lancet 337, 1158–1159. doi: 10.1016/0140-6736(91)92823-k
Wightman, D. P., Jansen, I. E., Savage, J. E., Shadrin, A. A., Bahrami, S., Holland, D., et al. (2021). A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282. doi: 10.1038/s41588-021-00921-z
Wingo, T. S., Lah, J. J., Levey, A. I., and Cutler, D. J. (2012). Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 69, 59–64. doi: 10.1001/archneurol.2011.221
Keywords: Alzheimer’s disease, early onset Alzheimer disease, next generation sequencing, genetic heterogeneity, mutation screening
Citation: Bartoletti-Stella A, Tarozzi M, Mengozzi G, Asirelli F, Brancaleoni L, Mometto N, Stanzani-Maserati M, Baiardi S, Linarello S, Spallazzi M, Pantieri R, Ferriani E, Caffarra P, Liguori R, Parchi P and Capellari S (2022) Dementia-related genetic variants in an Italian population of early-onset Alzheimer’s disease. Front. Aging Neurosci. 14:969817. doi: 10.3389/fnagi.2022.969817
Received: 15 June 2022; Accepted: 02 August 2022;
Published: 05 September 2022.
Edited by:
Robert Petersen, Central Michigan University, United StatesReviewed by:
Valentina Bessi, University of Florence, ItalyValerio Napolioni, University of Camerino, Italy
Anastasia Gurinovich, Tufts Medical Center, United States
Copyright © 2022 Bartoletti-Stella, Tarozzi, Mengozzi, Asirelli, Brancaleoni, Mometto, Stanzani-Maserati, Baiardi, Linarello, Spallazzi, Pantieri, Ferriani, Caffarra, Liguori, Parchi and Capellari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabina Capellari, c2FiaW5hLmNhcGVsbGFyaUB1bmliby5pdA==
†These authors have contributed equally to this work and share first authorship