 Yong Peng
Yong Peng Hong Jin1,2
Hong Jin1,2 Shu Liu
Shu Liu- 1Neurology Department, The First Affiliated Hospital of Hunan Traditional Chinese Medical College, Zhuzhou, Hunan, China
- 2Neurology Department, The Third Affiliated Hospital of Hunan University of Chinese Medicine, Zhuzhou, Hunan, China
Alzheimer’s disease (AD) is the most common chronic neurodegenerative disease worldwide. It causes cognitive dysfunction, such as aphasia and agnosia, and mental symptoms, such as behavioral abnormalities; all of which place a significant psychological and economic burden on the patients’ families. No specific drugs are currently available for the treatment of AD, and the current drugs for AD only delay disease onset and progression. The pathophysiological basis of AD involves abnormal deposition of beta-amyloid protein (Aβ), abnormal tau protein phosphorylation, decreased activity of acetylcholine content, glutamate toxicity, autophagy, inflammatory reactions, mitochondria-targeting, and multi-targets. The US Food and Drug Administration (FDA) has approved five drugs for clinical use: tacrine, donepezil, carbalatine, galantamine, memantine, and lecanemab. We have focused on the newer drugs that have undergone clinical trials, most of which have not been successful as a result of excessive clinical side effects or poor efficacy. Although aducanumab received rapid approval from the FDA on 7 June 2021, its long-term safety and tolerability require further monitoring and confirmation. In this literature review, we aimed to explore the possible pathophysiological mechanisms underlying the occurrence and development of AD. We focused on anti-Aβ and anti-tau drugs, mitochondria-targeting and multi-targets, commercially available drugs, bottlenecks encountered in drug development, and the possible targets and therapeutic strategies for future drug development. We hope to present new concepts and methods for future drug therapies for AD.
1. Introduction
Alzheimer’s disease (AD) is a chronic progressive disease with a hidden onset, unknown etiology, and long-term course. It is characterized mainly by cognitive dysfunction, such as aphasia and agnosia, and mental symptoms, such as hallucinations, delusions, and behavioral abnormalities, which significantly reduce the quality of life of older people (Alzheimer’s Association, 2022). The number of patients with AD worldwide is expected to exceed 150 million by 2050, according to the 2022 Alzheimer’s Disease Facts and Figures report (Man et al., 2023). Despite extensive research, the etiology of AD is complex, and its pathogenesis remains unknown. The primary hypotheses are abnormal deposition of beta-amyloid protein (Aβ), abnormal phosphorylation of tau protein, and nervous system inflammation, among others. Unfortunately, no drugs that can block AD progression are currently available.
Five drugs, Tacrine, Donepezil, Carbalatine, Galanthamine, and Memantine, have been approved by the Food and Drug Administration (FDA) for clinical use. Recently, FDA also approved Lecanemab. The first four types of drugs are acetylcholinesterase inhibitors (AChEIs), which can inhibit the activity of acetylcholinesterase (AChE) to prevent the degradation of acetylcholine in the synaptic gap, increasing the cholinergic effects, maintaining neuronal activity, and improving memory and learning abilities. Memantine is an N-methyl-D-aspartate (NMDA) receptor antagonist that can reduce the neurotoxicity of excitatory amino acids in the synaptic cleavage and reduce neuronal apoptosis. However, these drugs only manage symptoms and delay the onset of AD but do not cure it. Lecanemab (BAN2401), an IgG1 monoclonal antibody, was well tolerated during the trial, although some participants experienced ARIA-E (Swanson et al., 2021). In a multicenter, double-blind, 18-month Phase III trial, Lecanemab reduced amyloid markers in patients with early AD; longer trials are required to determine the effectiveness and safety of this drug (van Dyck et al., 2023).
Several drugs are undergoing clinical trials for AD, but unfortunately, many have been terminated because of poor efficacy or large adverse reactions. Existing clinical trials have focused on two pathological features of AD: amyloid plaques (Aβ), tau protein, mitochondria-targeting, and multi-targets. Therefore, we classified AD’s future treatment strategies into four main aspects: resistance against Aβ or anti tau protein treatment, mitochondria-targeting, and multi-targets agents.
2. Brief introduction of the physiological and pathological basis of AD
There were nine major mechanisms of the physiological and pathological basis of AD, such as Aβ deposition (Basisty et al., 2020), abnormal phosphorylation of tau protein (Jouanne et al., 2017; Hampel et al., 2019; Jo et al., 2020), decreasing acetylcholine activity (Cho et al., 2019; Babic Leko et al., 2021), glutamate toxicity (Ogbodo et al., 2022), autophagy (Reddy and Oliver, 2019; Luo et al., 2020), inflammatory response (Nho et al., 2019), neurovascular mechanism and mitochondrial hypothesis (Reddy and Reddy, 2017; Shevtsova et al., 2017, 2021; Wilkins and Morris, 2017; Liu et al., 2019), as well as “multi-target” agents (Makhaeva et al., 2019).
2.1. Aβ deposition
Beta-amyloid protein is an important biomarker used in the diagnosis of AD. Aβ plaques are formed by the hydrolysis of amyloid precursor protein (APP) through α, β, and γ secretory enzymes. Specifically, a portion of APP is cleaved by β-site APP-cleaving enzyme-1 (BACE-1) to produce a membrane-bound carbon terminal 99 amino acid fragment known as C99. C99 is then cleaved by γ-secretase to form Aβ1-40 and Aβ42. While Aβ monomers are typically soluble in small amounts and have no neurotoxicity, Aβ1–40 and Aβ42 are neurotoxic because of modulation by γ-secretase. They are more likely to accumulate into oligomers, which are eventually deposited in areas such as the olfactory cortex, hippocampus, and other areas of the cortex to form amyloid plaques. Ultimately, Aβ1–40 and Aβ42 lead to synaptic dysfunction, neuronal death, and cognitive decline (Basisty et al., 2020).
2.2. Abnormal phosphorylation of tau protein
Tau protein is a soluble microtubule-associated protein that combines with other tubules to form microtubules that coordinate various cellular functions. Abnormal phosphorylated tau protein forms neurofibrillary tangles (NFTs) that are deposited in the cytoplasm, rendering it unable to perform normal biological functions such as maintaining microtubule stability, reducing dissociation, and inducing microtubule bunching (Jouanne et al., 2017). It is also closely associated with cognitive decline. Protein kinases and phosphatases regulate tau protein phosphorylation. Studies have shown that Aβ can affect the activity of glycogen synthase kinase 3β (GSK-3β) and other protein kinases and the stability of the PP system, thus inducing tau protein deposition (Hampel et al., 2019). A study of 107 participants using Tau positron emission tomography (PET) scans found that mild cognitive decline in precursor AD was mainly related to abnormal tau protein accumulation in the medial and infratemporal cortex (Jo et al., 2020).
2.3. Acetylcholine activity was decreased
Acetylcholine is a neurotransmitter closely related to cognitive functions in the brain, such as learning and memory. The severity of AD is positively correlated with the degree of cholinergic deficiency. Cholinergic deficiency in patients with AD affects the blood–brain barrier (BBB), reducing neuronal excitability and weakening memory and learning functions. The nucleus basalis of Meynert (NbM) is the cerebral cortex’s main source of cholinergic innervation. Extensive literature has shown that in the early stages of AD, patients have a significant loss of “large cell neurons” in the NbM and degeneration of nerve fibers (Ch4) in cholinergic NbM neurons. The degree of nerve fiber degeneration is associated with cognitive deficits (Cho et al., 2019). The number of Ch4 neurons is reduced by 80% in patients with AD compared with healthy controls (Babic Leko et al., 2021).
2.4. Glutamate toxicity
Glutamic acid is an excitatory amino acid in the nervous system that is involved in synaptic transmission, structural differentiation, learning, memory, and other neuronal functions. Ionotropic glutamate receptors (iGluRs) and metabotropic glutamate receptors (mGluRs) are distributed throughout the postsynaptic membranes of neurons. In patients with AD, the expression levels of vesicular glutamate transporters (VGLUT) 1 and 2 in the cerebral cortex are decreased, and the process of converting glutamic acid to glutamine is blocked, leading to excessive accumulation of glutamic acid between synapses. This accumulation acts on the anti-N-methyl-D-aspartate receptor (NMDAR), ultimately increasing Ca2+ concentration, neuroexcitatory toxicity, and neuronal apoptosis (Ogbodo et al., 2022).
2.5. Autophagy
Autophagy is a lysosome-mediated process that prevents abnormal protein aggregation and cell aging; it is essential for eliminating harmful substances in the body (Reddy and Oliver, 2019). Mitochondria play a crucial role in providing large amounts of adenosine triphosphate (ATP) for normal neuronal function. The removal of damaged mitochondria because of aging is vital for the maintenance of cellular homeostasis. Studies have shown that abnormal mitochondrial autophagy can lead to abnormal accumulation of the Aβ42 protein, even before the onset of the pathological symptoms of AD. Moreover, abnormal deposition of the Aβ42 protein has toxic effects on mitochondrial autophagy, inhibiting its normal function by suppressing key enzymes involved in mitochondrial metabolism (Luo et al., 2020).
2.6. Inflammatory response
Neuroinflammatory responses play a critical role in AD progression. Acute inflammation protects against brain injury and microglial cells act as phagocytes of the immune system. However, when the phagocytic capacity of microglia reaches maximum, their continuous activation results in the loss of their ability to clear Aβ plaques. Subsequently, the continuous deposition of Aβ plaques contributes to learning and memory dysfunction in patients with AD. Additionally, studies have shown that intestinal microbial disorders are closely associated with the occurrence of AD. Intestinal microbial disorders cause an increase in deoxycholic acid, which is deposited in the brain through the BBB, leading to apoptosis, reactive oxygen species (ROS) generation, inflammation, and neurodegeneration (Nho et al., 2019).
2.7. Mitochondrial hypothesis
The hypothesis of mitochondria-targeting drugs on AD included were as follows: (1) an improvement of the energy deficit related to neurodegeneration, including mitochondrial bioenergetics stimulants, mitochondrial biogenesis activators, and neuroprotectors and (2) increase in the resistance of mitochondria to the opening of mitochondrial permeability transition (MPT) pores (Shevtsova et al., 2021). There are some early signs in the early stage of AD and mild cognitive impairment (MCI), such as (1) the decrease in glucose consumption and disruption of mitochondrial bioenergetics (Chételat et al., 2003; Bachurin et al., 2018) and (2) disruption of glucose transport through BBB (Delbarba et al., 2016; Kuehn, 2020).
Alzheimer’s disease might be called “type 3 diabetes” because insulin resistance increases the risk of dementia (Kandimalla et al., 2017; Neth and Craft, 2017). Insulin is associated with the brain’s energy metabolism; insulin receptors are widely expressed in the brain’s temporal lobe and hippocampus, which control memory and language (Watson and Craft, 2003). Also, the insulin-sensitive glucose transporter GLUT4 is important for memory and cognitive functions, which is expressed in the brain area, particularly in the hippocampus. Finally, additional glucose supply contributes to the activation of brain bioenergetics.
The activity and expression of the mitochondrial respiratory chain (mRC) decreased in early AD and its animal model (Yao et al., 2009). An Aβ-induced mitochondrial dysfunction of AD model was made by a transgenic Caenorhabditis elegans strain, which expressed human Aβ peptide specifically in neurons (GRU102). It showed that alterations in the tricarboxylic acid (TCA) cycle metabolism; reduced activity of a rate-limiting TCA cycle enzyme, i.e., alpha-ketoglutarate dehydrogenase (α-KGD); and low-level Aβ expression in GRU102 result in increasing protein carbonyl content, specifically in mitochondria. Moreover, metformin (an anti-diabetes drug) recovered Aβ-induced metabolic defects, reduced protein aggregation, and normalized the lifespan of GRU102 (Teo et al., 2019). Furthermore, metformin decreased the blood glucose level, α-KGD activity, and formation of Àβ aggregates. It can even extend the life span of C. elegans, enhancing the mRC activity and mitochondrial fission (Wang et al., 2019).
2.8. “Multi-target” agents
Multiple pathogenic factors (e.g., Aβ, metal ions, metal-bound Aβ, and ROS) are found in the brain of patients with AD. One of the modern approaches for creating multitarget agents for AD treatment is polypharmacophore design—building hybrid molecules that are conjugates of two or more different pharmacophores linked together with spacers (Bolognesi and Cavalli, 2016; Han et al., 2018).
Alzheimer’s disease is a multifactorial neurodegenerative disease; therefore, logically multi-target drugs would be the best choice (Makhaeva et al., 2019). To date, there are five pharmacophores that demonstrate multi-target effects on AD, which are γ-carbolines, carbazoles, tetrahydrocarbazoles, phenothiazines, and aminoadamantanes. Biological activity of these compounds include inhibitory potency against AChE, butyrylcholinesterase (BChE), anti-carboxylesterase and anti-aggregation activities, and binding to the two sites of the NMDA subtype of the glutamate receptor to conduct potential cognition enhancement and neuroprotection against mitochondrial triggers of cell death (Makhaeva et al., 2019). There were selective BChE inhibitors (conjugates of γ-carbolines and phenothiazine I, γ-carbolines and carbazoles II, and aminoadamantanes and carbazoles III) as well as inhibitors of both cholinesterases (conjugates of γ-carbolines and methylene blue IV and bis-γ-carbolines with ditriazole-containing spacers V). These compounds exhibit combined potential for cognition enhancement, neuroprotection, and disease modification. Moreover, none of the conjugates exhibited high potency against carboxylesterase (CaE), thereby precluding potential drug–drug interactions through CaE inhibition (Makhaeva et al., 2019; Figure 1).
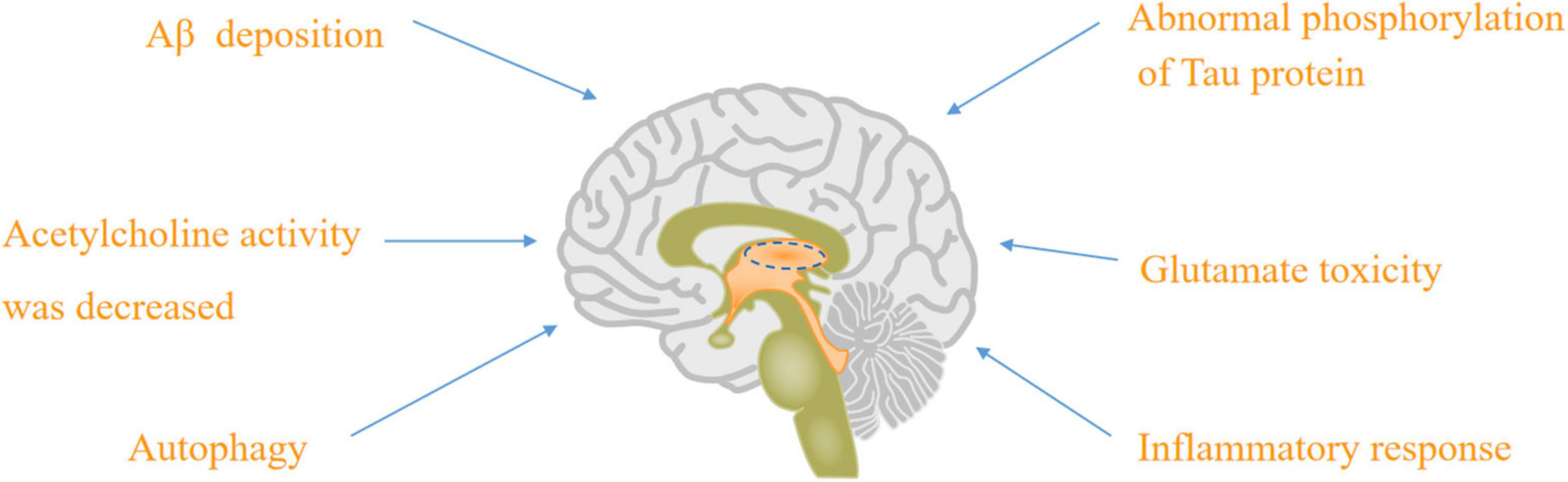
Figure 1. The physiological and pathological basis of AD: Aβ deposition, abnormal phosphorylation of tau protein, acetylcholine activity was decreased, glutamate toxicity, autophagy, and inflammatory response.
3. Therapeutic strategies for developing anti-AD drugs
The FDA has currently approved single-target drugs such as AChEI and NMDAR antagonists. There is a growing interest in developing multi-target drugs (Athar et al., 2021) that can address various aspects of AD pathology, including anti-Aβ deposition, tau protein phosphorylation, oxidative stress, and mitochondrial autophagy dysfunction. Many of these drugs are currently undergoing clinical trials.
3.1. Upcoming AD drugs targeting Aβ
The above FDA-approved therapies are only intended to ameliorate symptoms. Therefore, disease-modifying therapies are required to slow, modify, and control AD progression. The main mechanism of action of anti-Aβ drugs is to reduce the Aβ production, prevent its deposition, and accelerate its clearance.
3.1.1. β-Secretase inhibitor
β-Secretase inhibitors primarily reduce the production of amyloid beta. However, clinical trials of inhibitors targeting β-site APP-cleaving enzyme 1 (BACE1, also known as β-secretase 1) have largely been unsuccessful. Verubecestat, Lanabecestat, and Atabecestat are some of the molecules from the acyl guanidine class that have successfully reached the later stages of clinical trials. Nevertheless, they all failed to reach the market because of toxicity or a lack of clinical efficacy (Patel et al., 2022).
Verubecestat (MK-8931) was the first compound to enter Phase III trials because of its ability to cross the BBB and improve bioavailability. However, two Phase III clinical trials, EPOCH and APECS, were terminated prematurely after the drug failed to improve cognitive decline in participants and increased adverse events (AE) (Doggrell, 2019; Egan et al., 2019; Moussa-Pacha et al., 2020; Patel et al., 2022).
Lanabecestat (AZD3293) was studied in two Phase II/III and Phase III trials: AMARANTH and DAYBREAK-ALZ. These trials were designed to demonstrate its ability to slow the progression of mild AD. However, it was not found to slow cognitive decline in patients with mild AD at the mid-stage of the trial. Therefore, the trial was terminated prematurely (Wessels et al., 2020).
Atabecestat (JNJ-54861911), a potent BACE1 inhibitor, reduces the Aβ production in treating AD. Two Phase I trials, NCT01978548 and NCT02360657, showed an average 67 and 90% reduction in Aβ1-40 in the cerebrospinal fluid (CSF) of patients with early AD who received daily doses of 10 and 50 mg atabecestat for 4 weeks (Timmers et al., 2018). However, in a Phase II/III randomized, double-blind, placebo-controlled study, the trial was stopped early because of serious liver-related AE, and cognitive deterioration was found to be reversible after a 6-month follow-up of patients with AD (Sperling et al., 2021).
The BACE-1 inhibitor, Umibecestat (CNP520), has high selectivity and brain penetration, and animal toxicology studies have shown that it has a sufficiently safe range without AEs such as hair loss, cardiovascular damage, or liver toxicity (Neumann et al., 2018). However, trials of umibecestat at doses of 15 and 50 mg were stopped in two clinical prevention studies after the participants showed deterioration in the Neuropsychological State Cognition Test, even displaying significant brain shrinkage and weight loss (Vormfelde et al., 2020).
Elenbecestat (E2609) is another BACE-1 inhibitor that is an aminothiazine derivative. It has been shown to reduce the Aβ level in CSF (Roberts et al., 2021). In preclinical studies, without evidence of hypopigmentation, Elenbecestat reduced Aβ protein levels in rat and guinea pig brains, CSF, and plasma (Moriyama et al., 2017; Hsiao et al., 2019). In an elenbecestat healthy volunteer Phase I study (E2609-A001-002), it showed that Aβ decreased at 50 mg and increased at 100 and 400 mg (ClinicalTrials.gov.NCT01511783). This result was supported by an elenbecestat Phase II study (E2609-G000-201), which showed that CSF Aβ decreased at 50 mg in patients with MCI and early mild AD. Unfortunately, two elenbecestat global Phase III studies (E2609-G000-301 or MissionAD1) (ClinicalTrials.gov.NCT02956486) and (E2609-G000-302 or MissionAD2) were terminated because of an unfavorable risk-benefit ratio (Miranda et al., 2021).
3.1.2. γ-Secretase inhibitors
Semagacestat (LY450139) is a non-selective small-molecule γ-secretase inhibitor that targets the same mechanism as that of β-secretase inhibitors, aiming to reduce the deposition of Aβ amyloid protein. Two single-dose (140 mg), open-label, randomized crossover Phase III clinical trials showed that the clinical efficacy of semagacestat was independent of the preparation, food, and administration time. Additionally, the drug was well tolerated during the trial, and no safety concerns were reported (Willis et al., 2012). However, in a later Phase III trial (NCT00594568), the trial was terminated because of weight loss in patients treated with semagacestat and significantly higher rates of AEs, such as skin cancer and infection, than in the placebo group (Doody et al., 2013; Henley et al., 2014). Similarly, in a Phase II clinical trial of avagacestat in patients with mild-to-moderate AD, the trial was terminated because of the development of AEs such as brain microbleeds, diabetes, and skin cancer (Pinheiro and Faustino, 2019).
3.1.3. Drugs that enhance Aβ clearance (immunotherapy)
The two main types of immunotherapeutic drugs that can enhance the immune clearance of pathogens are active immunity (achieved through vaccination) and passive immunity (achieved through the administration of monoclonal antibodies).
3.1.4. Active immunity
The first anti-Aβ vaccine (AN1792) demonstrated the success of active immunotherapy in eliminating Aβ plaques, which could also be maintained for up to 14 years. However, in the Phase IIa clinical trial, approximately 6% of patients with AD treated with AN1792 developed meningoencephalitis (ME), leading to the termination of the trial. ME production may be related to the T-cell immune response (Nicoll et al., 2019).
A novel vaccine called ACC-001 was developed to avoid harmful T-cell responses and accelerate the clearance of Aβ plaques to address this issue (Pride et al., 2008). Phase II clinical trials of ACC-001 in patients with mild and moderate AD indicated that the vaccine had tolerable safety, regardless of whether the QS-21 adjuvant was used (Pasquier et al., 2016). Additionally, it was found that ACC-001 + QS-21 produced higher anti-Aβ antibody titers than the control group without QS-21 (Hull et al., 2017).
However, CAD106, an anti-Aβ vaccine containing peptide Aβ1-6, was terminated in another study of an AD prevention program because of abnormal changes in cognitive function, brain volume, and body weight in participants (Ohtake et al., 2017). In contrast, in Phase I clinical trials, ABvac40, the first active vaccine targeting the C-terminal of Aβ40, has shown good safety and tolerability (Lacosta et al., 2018).
3.1.5. Passive immunity
Bapineuzumab is a humanized monoclonal antibody that specifically targets Aβ and aims to reduce the abnormal deposition of Aβ plaques. In a Phase II study, treatment-emergent adverse events (TEAEs), including agitation and urinary tract infections, were reported in patients with severe AD. Bapineuzumab also causes amyloid-related imaging abnormalities (ARIA) with effusion or edema (ARIA-E) and ferriflavin deposition (Salloway et al., 2018). However, in two Phase III trials (NCT00575055 and NCT00574132), bapineuzumab did not significantly improve cognitive function in patients with AD (Salloway et al., 2014).
In contrast, gantenerumab is an IgG monoclonal antibody that accelerates the clearance of Aβ plaques through Fc receptor-mediated phagocytosis. A PET substudy clinical trial showed that a 1,200 mg dose of gantenerumab could stably clear Aβ plaques (Klein et al., 2019). No serious adverse events were reported after large-volume subcutaneous injection of gantenerumab (Portron et al., 2020). This drug can potentially reverse the pathology of amyloid plaques significantly and may alter the course of the disease by slowing or stopping its clinical progression (Bateman et al., 2022).
Crenezumab (RO5490245) is an IgG4 antibody with a high affinity for Aβ plaque oligomers and can be administrated at higher doses. No serious adverse events were reported in the GP29523 or GP40201 studies (Dolton et al., 2021). Two other Phase III multicenter trials were halted during mid-stage reviews because of the lack of clinical effectiveness of crenezumab (Ostrowitzki et al., 2022).
Ponezumab (PF-04360365), an IgG2 monoclonal antibody, was well tolerated in the trial but did not significantly affect Aβ deposition (Landen et al., 2017). In a double-blind, placebo-controlled Phase III trial of solanezumab at a dose of 400 mg every 4 weeks in patients with mild AD, no significant improvement in cognitive decline was observed, and cognitive decline preceded dysfunction in patients with mild AD throughout the trial. These findings could provide new insights into the prevention and treatment of AD at an early stage (Honig et al., 2018; Liu-Seifert et al., 2018).
Donanemab is currently in Phase III trials for the treatment of early AD. In the four Donanemab studies, 228 participants receiving Donanemab and 168 participants receiving placebo had low baseline levels of complete amyloid clearance. It was also found that in the Donanemab group, Tau accumulation was slower, and Donanemab was associated with ARIA-related AEs during the trial (Mintun et al., 2021; Rashad et al., 2022; Shcherbinin et al., 2022).
Aducanumab, a monoclonal antibody that targets soluble and insoluble Aβ aggregates (IgG1) and selectively binds to Aβ, received accelerated approval from the US FDA on 7 June 2021. It was the first new drug for the treatment of AD for 20 years, following the approval of the US FDA. It is currently under regulatory review in Japan and Europe to assess its safety and tolerability for long-term use (Dhillon, 2021). However, this drug can significantly increase the incidence of ARIA (Cummings et al., 2021; Table 1 and Figure 2).
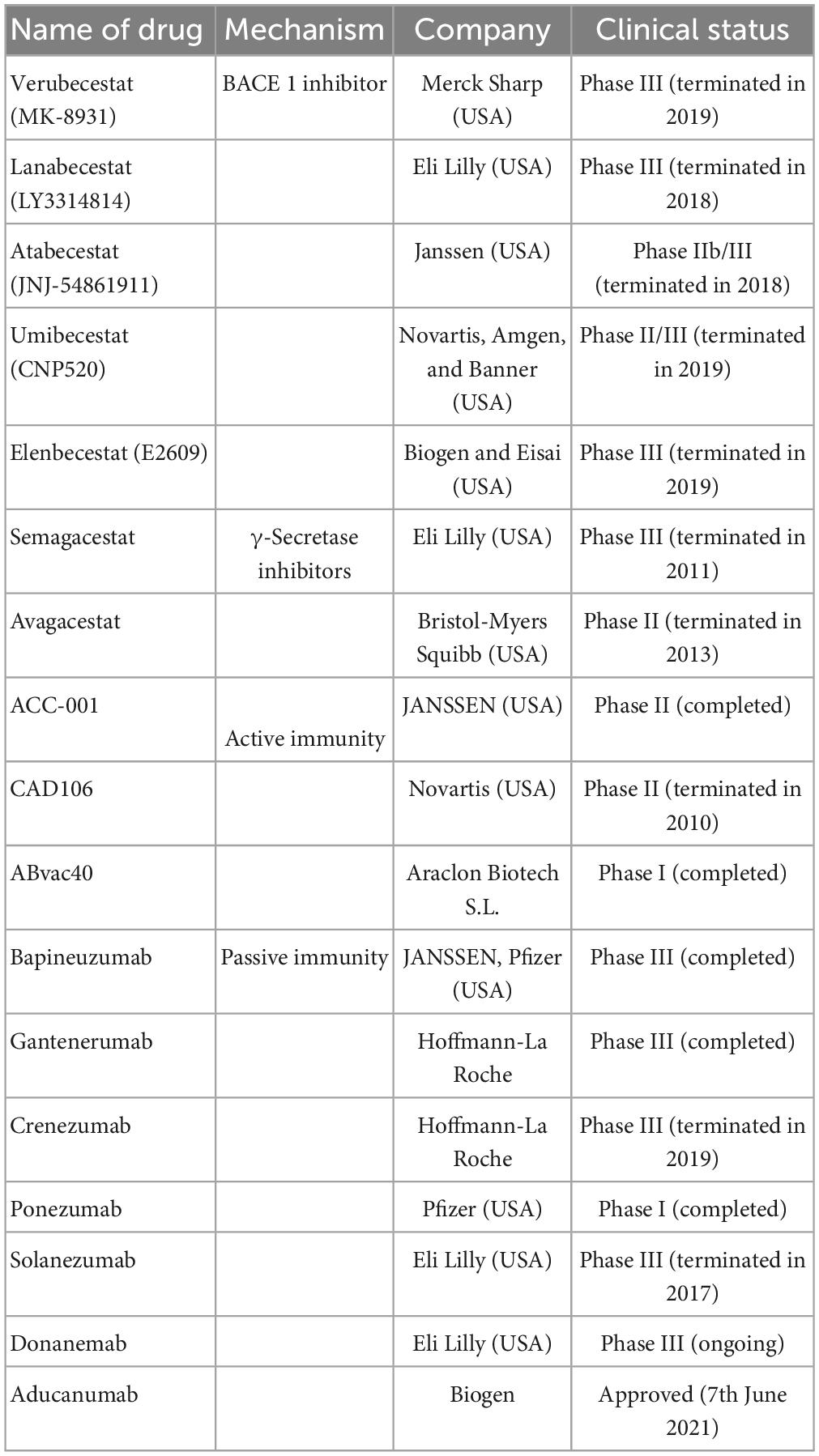
Table 1. The anti-Aβ drugs are currently undergoing clinical trials or just approved including name of drugs, mechanism, company, and clinical trials.
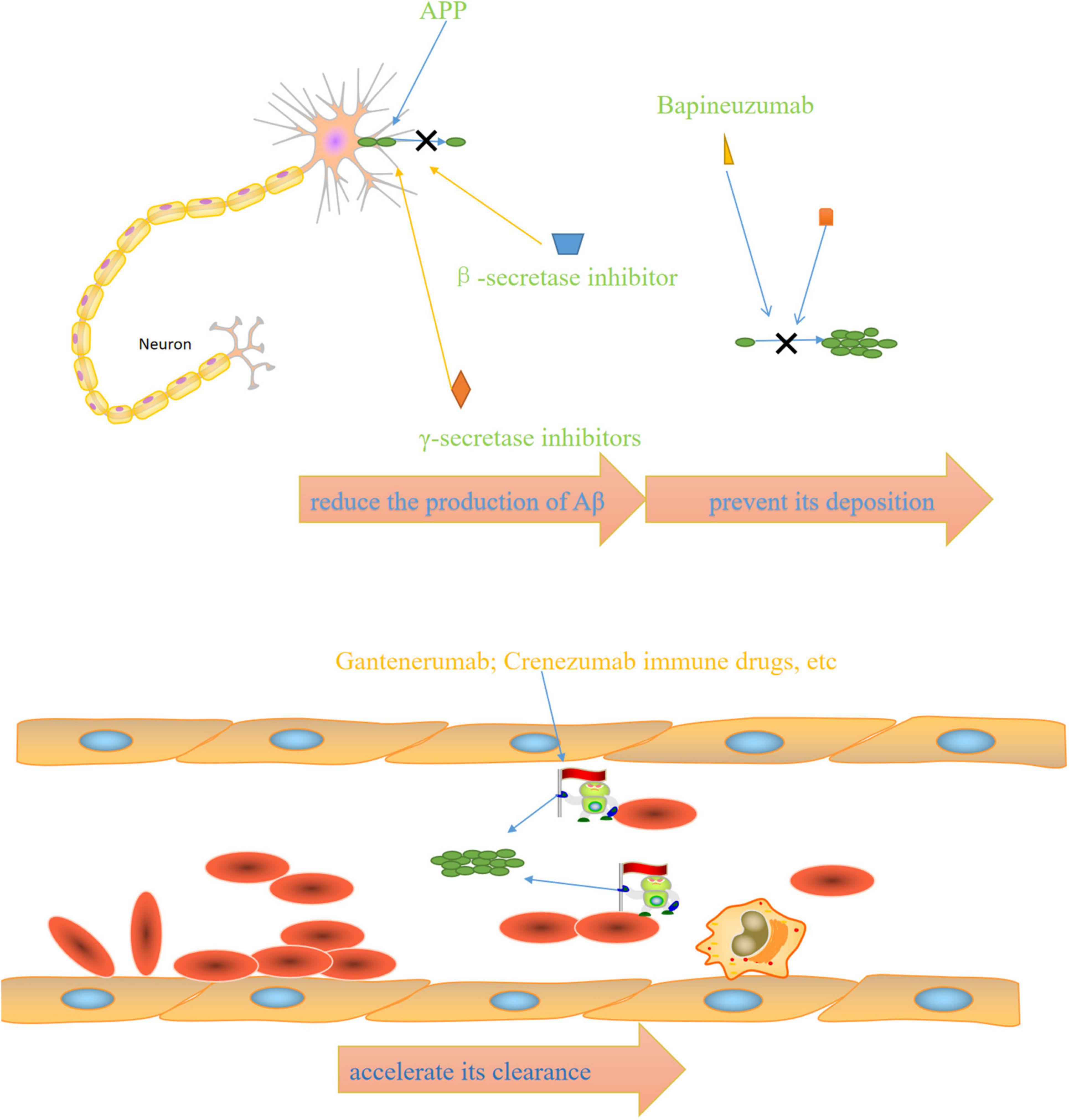
Figure 2. Mechanisms of anti-AD drugs: β-secretase inhibitors and γ-secretase inhibitors reduce the production of Aβ, bapineuzumab and other drugs prevent its deposition, immune drugs such as gantenerumab and crenezumab accelerate its clearance.
3.2. Anti-tau drugs
The role of the tau protein is not fully understood, but studies have shown that it plays an important role in the assembly and stabilization of cytoskeletal microtubules. Abnormal hyperphosphorylation of Tau (p-tau) reduces its affinity with bound microtubules, and Tau’s abnormal phosphorylation leads to the aggregation and formation of NFT. The treatment of anti-tau drugs mainly includes three aspects: preventing tau hyperphosphorylation and aggregation, stabilizing microtubules, and accelerating tau clearance.
3.2.1. GSK-3β inhibitor
Tau phosphorylation is regulated by protein kinase and phosphatase. Among these, GSA-3β is associated with p-tau production and subsequent neuronal degeneration in AD (Wegmann et al., 2021). GSK-3β inhibitors can prevent tau hyperphosphorylation. Studies have shown that GSK-3β can reduce abnormal Tau phosphorylation and amyloid protein production in vitro and in vivo, a promising disease-modifying therapy for AD. Tideglusib, a thiadiazolone that irreversibly inhibits GSK-3β and reduces tau phosphorylation, did not show any clinical benefit in a double-blind, placebo-controlled Phase II trial demonstrating the clinical efficacy of GSK-3 inhibitors in AD and is subject to further study (Lovestone et al., 2015). Lithium was first used in psychiatry and was discovered by Australian psychiatrist John Cade in 1949 and has been widely used to treat manic episodes. In recent years, Lithium has been found to be an inhibitor of GSK3, involved in glucose metabolism, cell signaling and proliferation, and glial cell function regulation. Lithium can prevent amyloid formation and Tau hyperphosphorylation. There have been some case reports as well as case control studies showing that Lithium can reduce the symptoms of AD. However, clinically available Lithium has serious side effects (SAE) with long-term use. It requires constant monitoring of Lithium concentrations in the blood; safer and more effective Lithium is needed for clinical use (Haussmann et al., 2021; Hu et al., 2022; Muronaga et al., 2022; Luca and Luca, 2023).
3.2.2. Tau aggregation inhibitor
Tau accumulation is associated with neuron loss. Tau aggregation inhibitors such as methylthioninium chloride (methblue) and hydromethanesulfonate (LMTM) can reduce Tau accumulation.
Methylthioninium chloride (methylene blue) is also a drug with a long history of use, primarily in malaria, methemogenemia, and carbon monoxide poisoning, as well as histological dyes. Methylthioninium chloride failed to show clinical benefit for AD in a 24-week Phase II study (Tucker et al., 2018). LMTM is a compound with a higher bioavailability and lower toxicity than methylthioninium chloride. In one Phase III trial involving mild to moderate AD, LMTM failed to slow cognitive or functional decline, and another phase III trial involving healthy older people with mild to moderate AD is still being conducted (Seripa et al., 2016; Hashweh et al., 2020).
TRx-0014 (Rember) was completed in a Phase II study of patients with mild to moderate AD.
It showed improvements in the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog) over 24 weeks, as well as in the Alzheimer’s Disease Cooperative Study–Clinical Global Impression of Change scale (ADCS-CGIC), the MMSE, and cerebral blood flow assessed by HMPAO-SPECT. Unfortunately, TRx-0014 showed no statistically significant effect on cognition in patients with mild AD (NCT00515333) (Wischik et al., 2015).
A Phase II trial of TRx0237 (LMT/hydromethylthionine) in mild-moderate AD (NCT01626391) was terminated early for administrative reasons (ClinicalTrials.gov.NCT01626391). Moreover, two Phase III studies [(NCT01689246)/the European Union Clinical Trials Registry (2012-002866-11), (NCT01689233) and the European Union Clinical Trials Registry (21012-002847-28)] confirmed that TRx0237 improved the cognition in patients with mild to moderate AD, such as changes in ADAS-Cog and Alzheimer’s Disease Cooperative Study– Activities of Daily Living Inventory (ADCS-ADL) (Gauthier et al., 2016; Wilcock et al., 2018). A Phase II/III trial of TRx0237 Monotherapy in participants with AD (LUCIDITY/NCT03446001) was completed recently. The use of TRx0237 reflected as improvements in ADAS-Cog11 and ADCS-ADL23 (ClinicalTrials.gov.NCT03446001).
3.2.3. Stable microtubules
Davunetide (alternative names: NAP or NAPVSIPQ or A-L108 or CP201) is an eight amino-acid peptide derived from the neuroprotective fragment of activity-dependent neuroprotective protein (ADNP) (Gozes et al., 2009). Davunetide is a Src homology 3 (SH3) domain-ligand association site responsible for controlling signaling pathways regulating the cytoskeleton, direct microtubule end-binding protein interaction facilitating microtubule dynamics, and Tau microtubule interaction at the microtubule end-binding protein site EB1 and EB3 (Gozes and Shazman, 2023). Davunetide may contribute to the progression of several CNS disorders, such as Autism, Schizophrenia, AD (Idan-Feldman et al., 2011; Sragovich et al., 2017, 2019), and Progressive Supranuclear Palsy (Dale et al., 2020; VandeVrede et al., 2020). A placebo-controlled, ascending-dose 12 weeks Phase I study in participants with amnestic MCI (AL-108-21) was completed in 2013, showing that davunetide was generally safe and well tolerated. However, it failed to detect a statistically significant difference between the treatment groups on the composite cognitive memory score of efficacy data (Morimoto et al., 2013).
Epothilones are derived from Sorangium cellulosum and inhibit tubulin depolymerization, thus leading to the death of cancer cells (Ye et al., 2023). Moreover, Epothilone D can bind to tau protein, thus effectively preventing nerve injury and improving cognitive performance in mouse models of AD (Brunden et al., 2010; Guo et al., 2020). Epothilone D (KOS-862) showed manageable toxicity, favorable PK profile, and the suggestion of clinical activity in Phase I clinical study of patients with advanced solid tumors and lymphoma (Konner et al., 2012) and in a Phase III trial in patients with advanced or metastatic breast cancer (Vahdat, 2008). Unfortunately, there were no publications regarding clinical trials of Epothilone D therapy in AD.
3.2.4. Active immunity
AADvac1 is a peptide that contains one of the epitopes of antibody DC8E8 (294KDNIKHVPGGGS305). AADvac1 is conjugated to keyhole limpet hemocyanin (KLH) along with aluminum hydroxide as an adjuvant. AADvac-1 therapy in patients with mild-to-moderate AD was completed in Phase I trials of (NCT01850238), without aberrant immune response or microhemorrhages (Novak et al., 2017, 2019); similar results were found in a follow-up study of 72 weeks (NCT02031198) (Novak et al., 2018). Moreover, cognitive decline (ADAS-cog11 value) in patients with mild-to-moderate AD was significantly reduced by AADvac1 (Novak et al., 2018). These results were confirmed in an AADvac-1 Phase II clinical trial (NCT02579252) (ClinicalTrials.gov.NCT02579252).
ACI-35 is a liposomal-anchored 16-amino acid tetra-palmitoylated phospho-tau peptide (393VYKSPVVSGDTSPRHL408) (Theunis et al., 2013). ACI-35 decreased soluble and insoluble Tau in tau-transgenic mouse models (Theunis et al., 2013).
3.2.5. Passive immunity
Gosuranemab is a humanized mouse monoclonal antibody (IPN002), which recognizes a phosphorylated epitope in the N-terminal region of Tau consisting of amino acid residues 15AGTYGLGDRK24 and targets extracellular Tau (Qureshi et al., 2018). Gosuranemab was found to be safe and well-tolerated in Phase 1 trials (NCT02460094) conducted on patients with PSP. Additionally, it demonstrated a reduction in unbound N-terminal Tau in CSF (Boxer et al., 2019). Unfortunately, AD biomarkers such as total Tau and ptau181 were not reduced by Gosuranemab (Tatebe et al., 2017; Yang et al., 2018). A Phase II clinical trial of Gosuranemab (TANGO trial, NCT03352557) is ongoing, with a completion date of 2024.
Tilavonemab (ABBV-8E12/C2N8E12/HJ8.5) is the humanized anti-Tau IgG4 antibody, which was found to be safe and tolerable as IV injections in Phase I trials (NCT02494024) (West et al., 2017). A Phase II trial of Tilavonemab for early AD (NCT02880956) found that Tilavonemab was tolerated generally well but found non-significant efficacy in treating patients with early AD (Florian et al., 2023).
Zagotenemab (LY3303560, MC-1 IgG1) is a humanized antibody that recognizes a conformational Tau epitope with a primary epitope located in the N-terminal region (Alam et al., 2017). Two Phase I trials of zagotenemab in healthy volunteers and patients with mild to moderate AD (NCT02754830 and NCT03019536) have been completed. However, no reports were released for unknown reasons (ClinicalTrials.gov.NCT02754830 and ClinicalTrials.gov.NCT03019536). Recently, a Phase II trial of zagotenemab (NCT03518073) was completed, showing that zagotenemab improves the clinical characteristics of patients with early AD. However, the trial showed an SAE occurrence of approximately 17% (ClinicalTrials.gov.NCT03518073).
Semorinemab (RO7105705) targets maximum binding across different extracellular Tau species, which was confirmed by preclinical studies in mouse models (Lee et al., 2016). Phase 1 Semorinemab (NCT02820896) studies have been completed; however, the corresponding report has not been made available. The study details can be found at https://beta.clinicaltrials.gov/study/NCT02820896. Recently, two ongoing Phase II trials have been completed: one involving participants with prodromal/probable AD (TAURIEL trial, NCT03289143) and another involving participants with moderate AD (NCT03828747). Both trials showed improvement in the clinical characteristics of patients with AD (Teng et al., 2022; ClinicalTrials.gov.NCT03828747; Table 2).
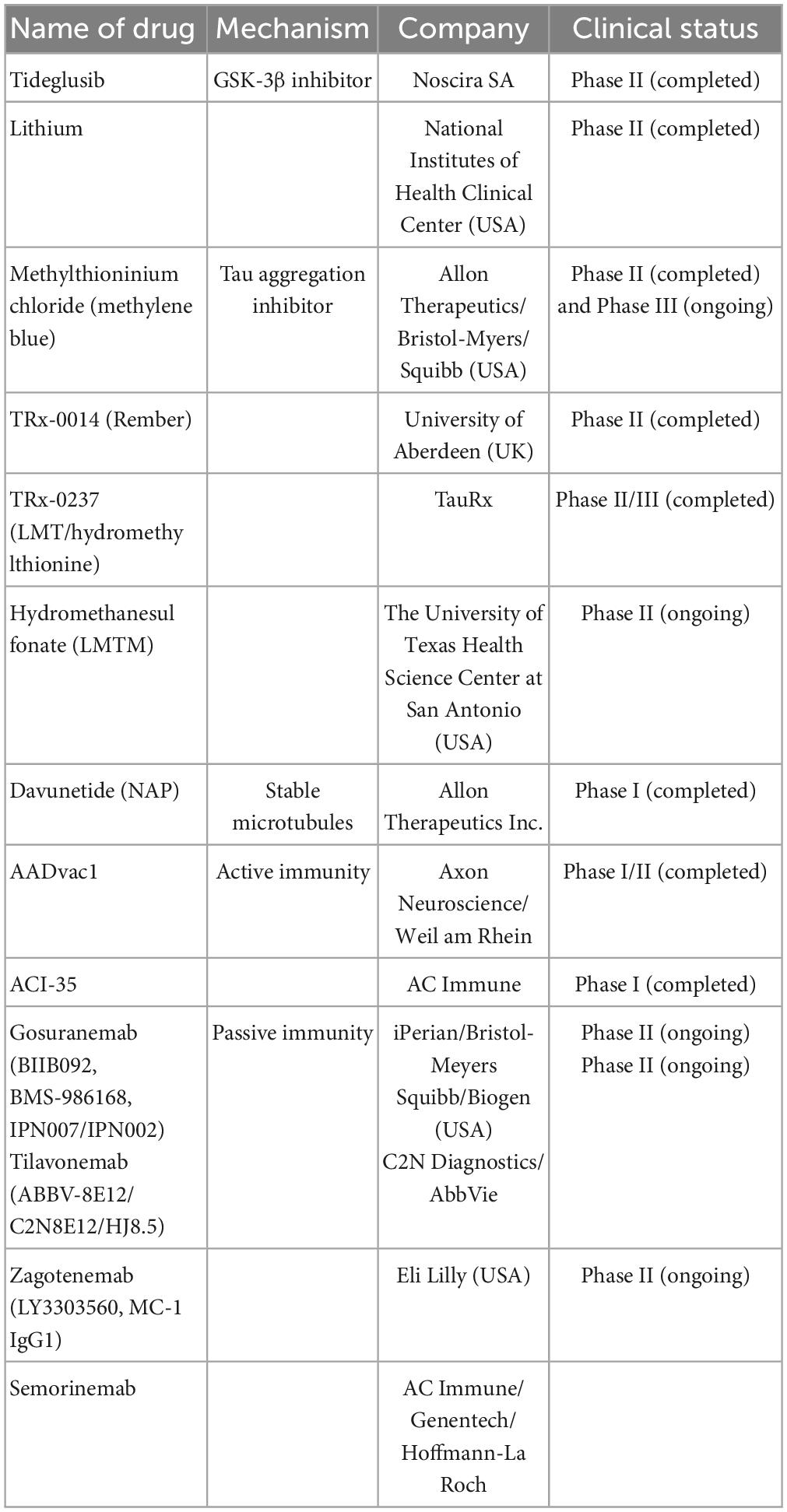
Table 2. The anti-tau drugs are currently undergoing clinical trials including name of drugs, mechanism, company, and clinical trials.
3.3. Mitochondria-targeting drugs on AD
3.3.1. Insulin
Phase II/III clinical trials on insulin for AD (NCT01767909) have been completed. However, no cognitive or functional benefits were observed with a 12-months period of intranasal insulin treatment, although no clinically important AE was associated with the treatment (Craft et al., 2020).
3.3.2. Mitochondrial enhancers
“Mitochondrial enhancers” therapy in the early stages of AD included coenzyme Q (CoQ) and its synthetic analog, idebenone, which stimulate the mitochondrial electron transport chain activity, increase ATP production, and exhibit antioxidant- and free-radical-scavenging activity. Studies showed that the oxidized/total CoQ ratio was increased in the CSF of patients with AD (Isobe et al., 2009; Orsucci et al., 2011). Similar results were also observed in animal models, including older animals and diabetic rats (Yang et al., 2016). Clinical trials on these drugs (NCT00117403) have been completed, and they failed to show statistically significant efficacy (Gutzmann et al., 2002; Thal et al., 2003; Salloway et al., 2021).
Another mitochondrial enhancer, methylene blue, is a member of the phenothiazines family, which interacts with mitochondria and induces an alternative electron transfer to cytochrome oxidase, thus increasing its activity and possessing antioxidant properties (Tucker et al., 2018). Methylene blue is also a multi-target drug on several biotargets, such as mitochondria, membrane-associated transporters, and ion channels (Saitow and Nakaoka, 1997) and the activity of cholinergic, monoaminergic, or glutamatergic synaptic neurotransmission (Ramsay et al., 2007; Vutskits et al., 2008; Shevtsova et al., 2021). Unfortunately, a compound of methylene blue [Leuco-methylthioninium bis (hydromethanesulfonate; LMTM)] has failed to show a statistically significant positive effect in phase III clinical trials (NCT01689246) on AD (Gauthier et al., 2016). However, another phase III clinical trial showed that LMTM improved cognitive function, brain atrophy, and blood glucose in patients with AD (Wilcock et al., 2018). Thus, further evidence is needed to support its efficacy.
Mitochondrial dysfunction can be recovered through mitochondrial biogenesis, which includes peroxisome proliferator-activated receptor (PPAR) and transcription coactivators such as PPARγ coactivator-1 (PGC-1) family, nuclear transcription factors including nuclear respiratory factors 1 (NRF-1) and 2 (NRF-2), and the mitochondrial transcription factor (ÒFAM). The impairment of PGC-1α-mediated mitochondrial biogenesis appears in patients with AD (Qin et al., 2009) and AD model-3xTg mouse (Singulani et al., 2020).
Peroxisome proliferator-activated receptor (α, β/δ, γ) agonist, bezafibrate, decreases the tau protein level and microglia activation, enhances mitochondrial biogenesis, and improves behavioral characteristics in P301s transgenic mice (Dumont et al., 2012). Unfortunately, there were no clinical trial reports on Bezafibrate for AD treatment in ClinicalTrials.gov.
In mouse models, other PPAR-γ agonists such as thiazolidinediones, pioglitazone, and rosiglitazone showed improvement in memory. Rosiglitazone improved cognitive functions only in a small group of patients with MCI. However, some extensive clinical trials (REFLECT-2 and REFLECT-3) did not show a statistically significant efficacy (Harrington et al., 2011; Shevtsova et al., 2021). Recently, multiple trials on rosiglitazone therapy for AD, including a Phase IIb (NCT00334568) and Phase III [(AVA105640; NCT00428090), (AVA102677; NCT00550420); (study AVA10267, NCT00348309); (study AVA102670; NCT00348140)] trials were reported. These reports showed that six protein-predictive biomarkers (IL6, IL10, CRP, TNF, FABP-3, and PPY) could accurately classify 100% of rosiglitazone treatment responders. Unfortunately, this report did not mention any improvement in the cognitive function of patients with AD (O’Bryant et al., 2021).
3.3.3. Mitochondrial permeability transition inhibitors
Mitochondrial permeability transition inhibitors can prevent neurodegenerative processes and can be considered potential neuroprotectors. MPT inhibitors include modulators of mitochondrial calcium homeostasis and antioxidants. The MPT pore is considered a complex consisting of poly-R-3-hydroxybutyrate, polyphosphates, and calcium cations (PHB/polyp/Ca2+ complex) (Pavlov et al., 2005). An experiment showed that a decrease in the polyphosphate level increases the calcium retention capacity of mitochondria and reduces the probability of Ñà2+-induced MPT pore opening (Abramov et al., 2007).
Cyclophilin D (peptidyl-prolyl cis–trans isomerase, PPIase) might be a master regulator of mitochondrial function, such as the mitochondrial redox status, presence of inorganic phosphate, and state of respiratory chain components, including complex I of the MRC, creatine kinase, and translocator protein (TSPO) peripheral benzodiazepine receptor (Azarashvili et al., 2015; Bernardi et al., 2015; Gutiérrez-Aguilar and Baines, 2015; Porter and Beutner, 2018). Cyclophilin family members have co-operations. For example, cyclophilin D is regulated by cyclosporin A through calcium. Cyclophilin D interacts with cardiolipin to release cytochrome C from mitochondria via tau protein-441, α-synuclein, and β-amyloid oligomers (Camilleri et al., 2013). There were some specific inhibitors, including cyclosporin À, alisporivir (Debio025) (Schiavone et al., 2017), N-methyl-4-isoleucine-cyclosporin (NIM811) (Springer et al., 2018), low-molecular-weight cyclophilin D ligands (4-aminobenzenesulfonamide derivative C-9) (Valasani et al., 2014, 2016), and cyclophilin D-independent MPT inhibitors (imidazole, thiadiazole, urea derivatives, N-phenylbenzamides, cinnamic anilides, and isoxazoles) (Valasani et al., 2016). Unfortunately, there are no clinical trials or animal model reports regarding these compounds (Shevtsova et al., 2021).
Dimebon prevents the opening of MPT pores and is considered a treatment for AD. Dimebon showed strong neuroprotective and cognition-enhancing effects in different animal models (Bachurin et al., 2001). Phase II clinical trials (NCT00377715) showed that Dimebon had a strong beneficial effect on memory and cognition in patients with AD (Doody et al., 2008). Unfortunately, this result was not supported by a phase III trial conducted in multiple centers, possibly because of the involvement of a heterogeneous population with various neuropathologies unrelated to AD symptoms (MacKay et al., 2010).
Melatonin and its precursor, N-acetylserotonin (NAS), not only have the receptor-defined hormonal effect but also act as antioxidants and are accumulated in mitochondria. They stimulate the MRC, inhibit the MPT, and possess significant neuroprotective potential (Pandi-Perumal et al., 2013; Zhou et al., 2014; Tarocco et al., 2019; Shevtsova et al., 2021). Melatonin and NAS affect MPT induction conditions and have multimodal capabilities, such as regulators of endogenous and local MPT and synaptic and neuronal viability (Tarocco et al., 2019). One clinical trial in Japan showed that melatonin significantly prolonged sleep time (Asayama et al., 2003). Unfortunately, there was no significant improvement in sleep or agitation in two different clinical trials on melatonin treatment for AD in the USA (Singer et al., 2003; Gehrman et al., 2009). Interestingly, a significant improvement in cognitive performance, such as IADL and MMSE, was observed in a 24-weeks clinical trial on prolonged-release melatonin (PRM, also called Piromelatine) therapy for patients with AD, particularly in those with insomnia comorbidity (Wade et al., 2014). However, a recently completed phase II clinical trial (ReCognition, NCT02615002) on Piromelatine therapy for AD showed no statistically significant improvement in cognitive functions (Schneider et al., 2022).
Translocator protein density has been used as a biomarker for neuroinflammation in AD (Chen and Guilarte, 2008). Recently it showed that TSPO binding was greater in patients with AD than in age-matched controls or patients with MCI who had a positive amyloid scan (Kreisl et al., 2013). It showed that increased TSPO binding might play a pathophysiological role in the transition from MCI to AD (Lyoo et al., 2015). In a clinical trial (NCT00613119), TSPO binding (VT/fP)1) was greater in patients with AD than in healthy controls in expected temporoparietal regions and 2) was not significantly different among the three groups in the cerebellum (Lyoo et al., 2015; Table 3).
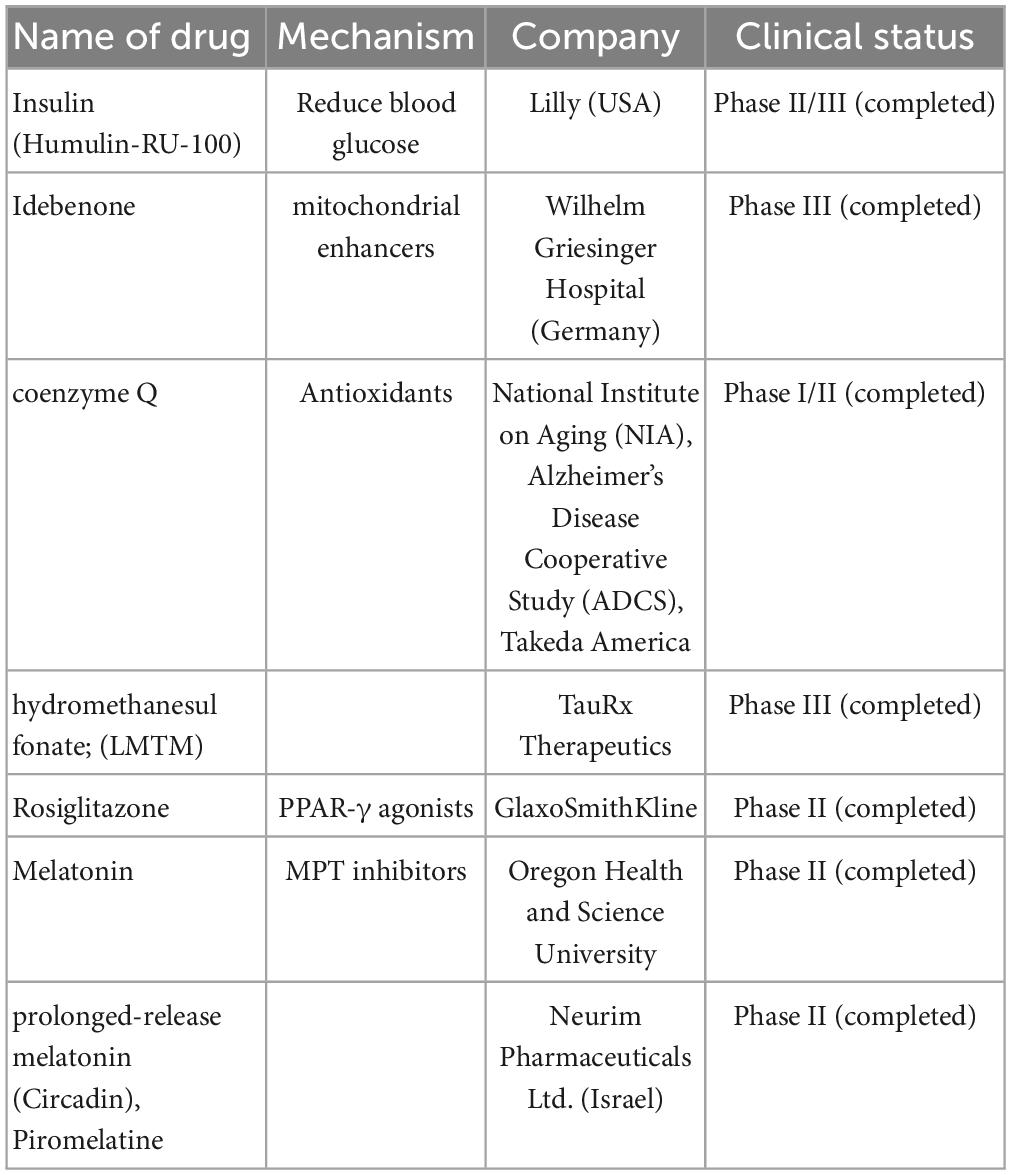
Table 3. Mitochondria-targeting drugs are currently undergoing clinical trials including name of drugs, mechanism, company, and clinical trials.
3.4. “Multi-target” agents on AD
3.4.1. γ-Carbolines
Dimebon is a compound of γ-carboline derivatives that conjugates with methylene blue (Bachurin et al., 2019; Makhaeva et al., 2019). Dimebon is a multitarget agent; its activities include protecting neurons from death, reducing the development of proteopathy, and increasing autophagy (Shevtsova et al., 2014; Skvortsova et al., 2018; Ustyugov et al., 2018). However, owing to a lack of statistically significant efficacy, the use of dimebon for AD was not confirmed through a phase II clinical trial (NCT00377715) (Doody et al., 2008).
3.4.2. Phenothiazine
Phenothiazine does not inhibit AChE and has a rather low anti-BChE activity, which was supported by molecular docking (Makhaeva et al., 2015). In a double transgenic mouse AD model, phenothiazine-based theranostic compounds inhibited Aβ aggregation and might act as imaging probes for amyloid plaques in AD on near-infrared fluorescent (NIRF) imaging (Dao et al., 2017). Unfortunately, there is no clinical trial report currently available on phenothiazine therapy for AD in the PubMed database.
3.4.3. Carbazoles
P7C3 is a neuroprotective aminopropyl carbazole identified on studies of postnatal hippocampal neurogenesis (Voorhees et al., 2018). P7C3 was named as it is the third compound (C3) of the seventh pool (P7) and has protective action on young hippocampal neurons in preventing neuron death; it has also been shown to inhibit cognitive decline in terminally aging rats (Pieper et al., 2010). Moreover, P7C3 molecules enhance the flux of nicotinamide adenine dinucleotide (NAD) in mammalian cells (Wang et al., 2014) and indirectly inhibit other critical cell death signaling events (Gu et al., 2017). P7C3 treatment shows neuroprotective effect in different animal models, such as amyotrophic lateral sclerosis (Tesla et al., 2012), Parkinson’s disease (De Jesús-Cortés et al., 2012, 2015; Naidoo et al., 2014; Gu et al., 2017), traumatic brain injury (Dutca et al., 2014; Yin et al., 2014; Vázquez-Rosa et al., 2020), psychological stress–related hippocampal cell death (Walker et al., 2015), peripheral nerve crush injury (Kemp et al., 2015), stroke (Kemp et al., 2015), and AD (TgF344-AD rat model) (Cohen et al., 2013; Voorhees et al., 2018). Unfortunately, there is no clinical trial report available on P7C3 therapy for AD in the PubMed database.
3.4.4. 5-HT
Idalopirdine is a novel selective 5-HT6 receptor antagonist that binds with ChEI, potentiates central acetyl choline levels and neuronal activity, and improves cognition in animal models (Herrik et al., 2016; Amat-Foraster et al., 2017).
A phase II, proof-of-concept (PoC) study of idalopirdine plus donepezil therapy for AD showed a significant improvement in cognitive performance of AD, such as in ADAS-cog and MMSE scores (Wilkinson et al., 2014). However, phase III development programs for idalopirdine therapy (“OLEX,” idalopirdine only and “MEMOLEX,” idalopirdine plus memantine) for AD showed no statistically significant efficacy.
AVN-101 is a very potent 5-HT7 receptor antagonist that blocks 5-HT6, 5-HT2A, and 5HT-2C receptors as well as histamine H1 and adrenergic 2A, 2B, and 2C receptors. AVN-101 shows good oral bioavailability, facilitates BBB permeability, and has a low toxicity and reasonable efficacy in animal models of CNS diseases (Ivachtchenko et al., 2016). Moreover, a phase I clinical study indicated that the AVN-101 is well tolerated (Ivachtchenko et al., 2016).
3.4.5. Tyrosine kinase inhibitor
Masitinib is an oral tyrosine kinase inhibitor that has demonstrated neuroprotective action in neurodegenerative diseases via inhibition of mast cell and microglia/macrophage activity, such as in cases of multiple sclerosis (AB07002) (Vermersch et al., 2022). Recently, a phase III clinical trial of masitinib therapy for AD (AB09004, NCT01872598) was completed, which demonstrated that masitinib causes significant improvement in ADAS-cog and ADCS-ADL scores (Dubois et al., 2023; Table 4).
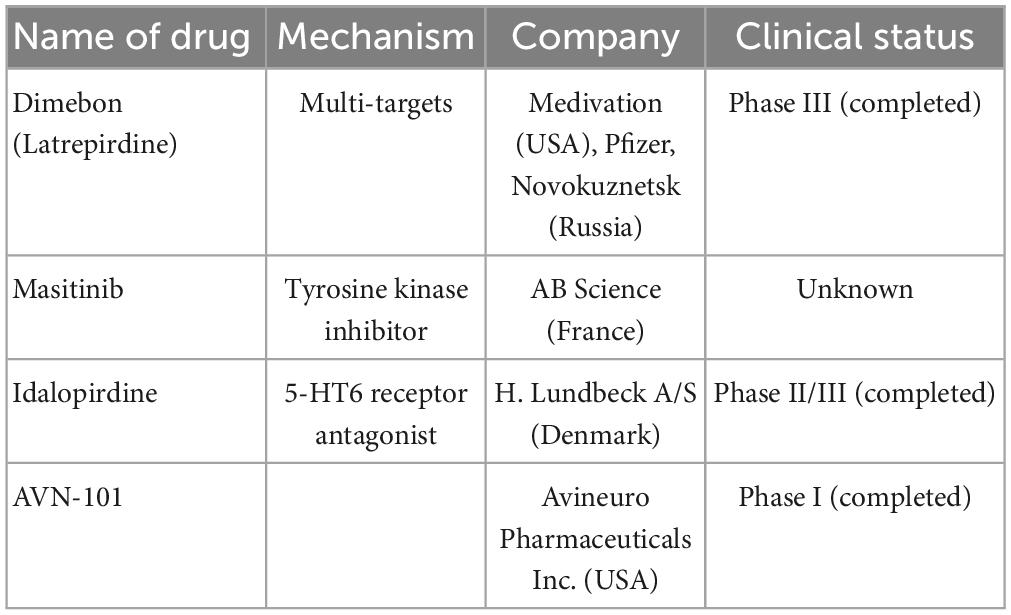
Table 4. Multi-targets drugs are currently undergoing clinical trials including name of drugs, mechanism, company, and clinical trials.
4. Discussion
Alzheimer’s disease is a neurodegenerative disease with increasing annual incidence. However, the pathogenesis of AD is complex, and its etiology has not been fully elucidated. Research and development of therapeutic drugs for AD are still in progress. Since AD has an insidious onset and slow disease progression, it can take up to 20 years from the onset of pathological changes to the appearance of clinically significant symptoms. Therefore, the early treatment of AD is crucial in controlling its progression. Anti-Aβ amyloid drugs are currently the focus of clinical trials; however, most clinical trials have been terminated because of AE and poor efficacy. For example, a clinical trial on aducanumab, donanemab, lecanemab, and other anti-Aβ drugs concluded that Aβ plaque clearance was closely related to the occurrence of ARIA-E (Wang et al., 2022). The high incidence of ARIAs suggests a need to clarify the early benefits of such interventions when conducting clinical trials (Loureiro et al., 2020). In clinical studies on donanemab, a trend toward slower Tau accumulation was observed. Thus, a future research direction would be to explore the relationship between reduced Aβ plaques and Tau levels, to achieve meaningful benefits for patients with AD. The accelerated FDA approval of aducanumab brought hope for AD drug development, and we look forward to more effective and economical treatments for patients with AD.
5. Conclusion
In conclusion, although there has not been a curative breakthrough in drug therapy for AD, progress is being made; new drugs with good efficacy, few adverse reactions, and economic feasibility will certainly be developed in the near future.
Author contributions
YP received funding support and developed the research hypothesis. YP, HJ, Y-hX, QC, S-yY, M-qD, and SL wrote the main manuscript. All authors jointly wrote the final manuscript as the end product.
Funding
This work was supported by the Scientific Research Project of Hunan Provincial Health Commission, China (No. C202303076574 to YP), Key Plans of Hunan Administration Traditional Chinese Medicine, China (No. A2023039 to YP), University-Hospital Joint-Fund of Hunan University of Chinese Medicine, China (No. 2022XYLH198 to YP), Fund for Creative Research Group of Affiliated First Hospital of Hunan Traditional Chinese Medical College, China (No. 2021B-003 to YP), and Technology Plan Project of Zhuzhou City, Hunan Province, China (No. 2021-009 to YP).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Aβ, beta-amyloid protein; Ach, acetylcholine; AChE, acetylcholinesterase; AChEI, acetylcholinesterase inhibitor; α -KGD, alpha-ketoglutarate dehydrogenase; AD, Alzheimer’s disease; ADAS-Cog, Alzheimer’s Disease Assessment Scale–Cognitive Subscale; ADCS-CGIC, Alzheimer’s Disease Cooperative Study–Clinical Global Impression of Change scale; ADCS-ADL, Alzheimer’s Disease Cooperative Study–Activities of Daily Living Inventory; AE, adverse effects; APP, amyloid precursor protein; ARIA, amyloid-related imaging abnormalities; ARIA-E, amyloid-related imaging abnormalities-edema; BACE-1, β-site amyloid precursor protein cleaving enzyme-1; BBB, blood–brain barrier; BChE, butyrylcholinesterase; C99, the membrane-bound carbon terminal 99 amino acid fragment; CaE, carboxylesterase; FDA, Food and Drug Administration; GSA-3β, glycogen synthase kinase 3β; iGluRs, ionotropic glutamate receptors; LMTM, leuco-methylthioninium bis (hydromethanesulfonate); mRC, mitochondrial respiratory chain; mGluRs, metabotropic glutamate receptors; ME, meningoencephalitis; MPT, mitochondrial permeability transition; MCI, mild cognitive impairment; NMDAR, anti-N-methyl-D-aspartate receptor; NbM, nucleus basalis of Meynert; NFTs, neurofibrillary tangles; NRF, nuclear respiratory factors; PGC-1, PPAR γ coactivator-1; PoC, proof-of-concept; PPAR, peroxisome proliferator-activated receptor; PPIase, peptidyl-prolyl cis–trans isomerase; ROS, reactive oxygen species; TCA, tricarboxylic acid; ÒFAM, the mitochondrial transcription factor; TEAEs, treatment-related adverse reactions; TSPO, translocator protein; VGLUT, vesicular glutamate transporter.
References
Alzheimer’s Association. (2022). 2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 18, 700–789.
Abramov, A. Y., Fraley, C., Diao, C. T., Winkfein, R., Colicos, M. A., Duchen, M. R., et al. (2007). Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc. Natl. Acad. Sci. U.S.A. 104, 18091–18096. doi: 10.1073/pnas.0708959104
Alam, R., Driver, D., Wu, S., Lozano, E., Key, S. L., Hole, J. T., et al. (2017). [O2–14–05]: preclinical characterization of an antibody [LY3303560] targeting aggregated tau. Alzheimer Dement. 13, 592–593.
Amat-Foraster, M., Leiser, S. C., Herrik, K. F., Richard, N., Agerskov, C., Bundgaard, C., et al. (2017). The 5-HT(6) receptor antagonist idalopirdine potentiates the effects of donepezil on gamma oscillations in the frontal cortex of anesthetized and awake rats without affecting sleep-wake architecture. Neuropharmacology 113, 45–59. doi: 10.1016/j.neuropharm.2016.09.017
Asayama, K., Yamadera, H., Ito, T., Suzuki, H., Kudo, Y., and Endo, S. (2003). Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J. Nippon Med. Schl. 70, 334–341.
Athar, T., Al Balushi, K., and Khan, S. A. (2021). Recent advances on drug development and emerging therapeutic agents for Alzheimer’s disease. Mol. Biol. Rep. 48, 5629–5645.
Azarashvili, T., Krestinina, O., Baburina, Y., Odinokova, I., Grachev, D., Papadopoulos, V., et al. (2015). Combined effect of G3139 and TSPO ligands on Ca(2+)-induced permeability transition in rat brain mitochondria. Arch. Biochem. Biophys. 587, 70–77. doi: 10.1016/j.abb.2015.10.012
Babic Leko, M., Hof, P. R., and Simic, G. (2021). Alterations and interactions of subcortical modulatory systems in Alzheimer’s disease. Prog. Brain Res. 261, 379–421. doi: 10.1016/bs.pbr.2020.07.016
Bachurin, S. O., Gavrilova, S. I., Samsonova, A., Barreto, G. E., and Aliev, G. (2018). Mild cognitive impairment due to Alzheimer disease: contemporary approaches to diagnostics and pharmacological intervention. Pharmacol. Res. 129, 216–226.
Bachurin, S. O., Makhaeva, G. F., Shevtsova, E. F., Boltneva, N. P., Kovaleva, N. V., Lushchekina, S. V., et al. (2019). Conjugates of methylene blue with γ-carboline derivatives as new multifunctional agents for the treatment of neurodegenerative diseases. Sci. Rep. 9:4873. doi: 10.1038/s41598-019-41272-4
Bachurin, S., Bukatina, E., Lermontova, N., Tkachenko, S., Afanasiev, A., Grigoriev, V., et al. (2001). Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Ann. N. Y. Acad. Sci. 939, 425–435. doi: 10.1111/j.1749-6632.2001.tb03654.x
Basisty, N., Holtz, A., and Schilling, B. (2020). Accumulation of “Old Proteins” and the critical need for MS-based protein turnover measurements in aging and longevity. Proteomics 20:e1800403. doi: 10.1002/pmic.201800403
Bateman, R. J., Cummings, J., Schobel, S., Salloway, S., Vellas, B., Boada, M., et al. (2022). Gantenerumab: an anti-amyloid monoclonal antibody with potential disease-modifying effects in early Alzheimer’s disease. Alzheimers Res. Ther. 14:178.
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Bolognesi, M. L., and Cavalli, A. (2016). Multitarget drug discovery and polypharmacology. Chem. Med. Chem. 11, 1190–1192.
Boxer, A. L., Qureshi, I., Ahlijanian, M., Grundman, M., Golbe, L. I., Litvan, I., et al. (2019). Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol. 18, 549–558. doi: 10.1016/S1474-4422(19)30139-5
Brunden, K. R., Zhang, B., Carroll, J., Yao, Y., Potuzak, J. S., Hogan, A. M., et al. (2010). Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J. Neurosci. 30, 13861–13866. doi: 10.1523/JNEUROSCI.3059-10.2010
Camilleri, A., Zarb, C., Caruana, M., Ostermeier, U., Ghio, S., Högen, T., et al. (2013). Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim. Biophys. Acta 1828, 2532–2543. doi: 10.1016/j.bbamem.2013.06.026
Chen, M. K., and Guilarte, T. R. (2008). Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol. Ther. 118, 1–17. doi: 10.1016/j.pharmthera.2007.12.004
Chételat, G., Desgranges, B., de la Sayette, V., Viader, F., Eustache, F., and Baron, J. C. (2003). Mild cognitive impairment: can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology 60, 1374–1377.
Cho, H., Choi, J. Y., Lee, H. S., Lee, J. H., Ryu, Y. H., Lee, M. S., et al. (2019). Progressive tau accumulation in Alzheimer disease: 2-year follow-up study. J. Nucl. Med. 60, 1611–1621.
ClinicalTrials.gov.NCT01511783. A Randomized, Double-Blind, Placebo-Controlled, Multiple Ascending Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of E2609 in Healthy Subjects. Available online at: https://www.clinicaltrials.gov/ct2/show/NCT01511783?term=E2609-A001-002&draw=2&rank=1
ClinicalTrials.gov.NCT01626391. Safety Study of TRx0237 in Patients Already Taking Medications for Mild and Moderate Alzheimer’s Disease. Available online at: https://beta.clinicaltrials.gov/search?term=NCT01626391
ClinicalTrials.gov.NCT02579252. 24 Months Safety and Efficacy Study of AADvac1 in Patients With Mild Alzheimer’s Disease (ADAMANT). Available online at: https://beta.clinicaltrials.gov/search?term=NCT02579252
ClinicalTrials.gov.NCT02754830. A Study of LY3303560 in Healthy Participants and Participants With Alzheimer’s Disease (AD). Available online at: https://beta.clinicaltrials.gov/search?term=NCT02754830
ClinicalTrials.gov.NCT02956486. A Placebo-Controlled, Double-Blind, Parallel-Group, 24 Month Study With an Open-Label Extension Phase to Evaluate the Efficacy and Safety of Elenbecestat (E2609) in Subjects With Early Alzheimer’s Disease. Available online at: https://beta.clinicaltrials.gov/study/NCT02956486?distance=50&term=E2609-G000-301&rank=1; E2609-G000-302
ClinicalTrials.gov.NCT03019536. A Study of LY3303560 in Participants With Mild Cognitive Impairment or Alzheimer’s Disease. Available online at: https://beta.clinicaltrials.gov/search?term=NCT03019536
ClinicalTrials.gov.NCT03446001. Safety and Efficacy of TRx0237 in Subjects With Alzheimer’s Disease Followed by Open-Label Treatment. Available online at: https://beta.clinicaltrials.gov/search?term=NCT03446001
ClinicalTrials.gov.NCT03518073. A Study of LY3303560 in Participants With Early Symptomatic Alzheimer’s Disease. Available online at: https://beta.clinicaltrials.gov/search?term=NCT03518073
ClinicalTrials.gov.NCT03828747. A Study of Semorinemab in Patients With Moderate Alzheimer’s Disease. Available online at: https://beta.clinicaltrials.gov/search?term=NCT03828747
Cohen, R. M., Rezai-Zadeh, K., Weitz, T. M., Rentsendorj, A., Gate, D., Spivak, I., et al. (2013). A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric aβ, and frank neuronal loss. J. Neurosci. 33, 6245–6256.
Craft, S., Raman, R., Chow, T. W., Rafii, M. S., Sun, C. K., Rissman, R. A., et al. (2020). Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: a randomized clinical trial. JAMA Neurol. 77, 1099–1109.
Cummings, J., Aisen, P., Apostolova, L. G., Atri, A., Salloway, S., and Weiner, M. (2021). Aducanumab: appropriate use recommendations. J. Prev. Alzheimers Dis. 8, 398–410.
Dale, M. L., Brumbach, B. H., Boxer, A. L., and Hiller, A. L. (2020). Associations between amantadine usage, gait, and cognition in PSP: a post-hoc analysis of the davunetide trial. Front. Neurol. 11:606925. doi: 10.3389/fneur.2020.606925
Dao, P., Ye, F., Liu, Y., Du, Z. Y., Zhang, K., Dong, C. Z., et al. (2017). Development of phenothiazine-based theranostic compounds that act both as inhibitors of β-amyloid aggregation and as imaging probes for amyloid plaques in Alzheimer’s disease. ACS Chem. Neurosci. 8, 798–806.
De Jesús-Cortés, H., Miller, A. D., Britt, J. K., DeMarco, A. J., De Jesús-Cortés, M., Stuebing, E., et al. (2015). Protective efficacy of P7C3-S243 in the 6-hydroxydopamine model of Parkinson’s disease. NPJ Parkinsons Dis. 1:15010. doi: 10.1038/npjparkd.2015.10
De Jesús-Cortés, H., Xu, P., Drawbridge, J., Estill, S. J., Huntington, P., Tran, S., et al. (2012). Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of Parkinson disease. Proc. Natl. Acad. Sci. U.S.A. 109, 17010–17015. doi: 10.1073/pnas.1213956109
Delbarba, A., Abate, G., Prandelli, C., Marziano, M., Buizza, L., Arce Varas, N., et al. (2016). Mitochondrial alterations in peripheral mononuclear blood cells from Alzheimer’s disease and mild cognitive impairment patients. Oxid. Med. Cell. Longev. 2016:5923938.
Doggrell, S. A. (2019). Lessons that can be learnt from the failure of verubecestat in Alzheimer’s disease. Expert Opin. pharmacother. 20, 2095–2099. doi: 10.1080/14656566.2019.1654998
Dolton, M. J., Chesterman, A., Moein, A., Sink, K. M., Waitz, A., Blondeau, K., et al. (2021). Safety, tolerability, and pharmacokinetics of high-volume subcutaneous crenezumab, with and without recombinant human hyaluronidase in healthy volunteers. Clin. Pharmacol. Ther. 110, 1337–1348. doi: 10.1002/cpt.2385
Doody, R. S., Gavrilova, S. I., Sano, M., Thomas, R. G., Aisen, P. S., Bachurin, S. O., et al. (2008). Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet 372, 207–215. doi: 10.1016/S0140-6736(08)61074-0
Doody, R. S., Raman, R., Farlow, M., Iwatsubo, T., Vellas, B., Joffe, S., et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 369, 341–350.
Dubois, B., López-Arrieta, J., Lipschitz, S., Doskas, T., Spiru, L., Moroz, S., et al. (2023). Masitinib for mild-to-moderate Alzheimer’s disease: results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimers Res. Ther. 15:39.
Dumont, M., Stack, C., Elipenahli, C., Jainuddin, S., Gerges, M., Starkova, N., et al. (2012). Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum. Mol. Genet. 21, 5091–5105. doi: 10.1093/hmg/dds355
Dutca, L. M., Stasheff, S. F., Hedberg-Buenz, A., Rudd, D. S., Batra, N., Blodi, F. R., et al. (2014). Early detection of subclinical visual damage after blast-mediated TBI enables prevention of chronic visual deficit by treatment with P7C3-S243. Investig. Ophthalmol. Vis. Sci. 55, 8330–8341. doi: 10.1167/iovs.14-15468
Egan, M. F., Mukai, Y., Voss, T., Kost, J., Stone, J., Furtek, C., et al. (2019). Further analyses of the safety of verubecestat in the phase 3 EPOCH trial of mild-to-moderate Alzheimer’s disease. Alzheimers Res. Ther. 11:68. doi: 10.1186/s13195-019-0520-1
Florian, H., Wang, D., Arnold, S. E., Boada, M., Guo, Q., Jin, Z., et al. (2023). Tilavonemab in early Alzheimer’s disease: results from a phase 2, randomized, double-blind study. Brain 146, 2275–2284. doi: 10.1093/brain/awad024
Gauthier, S., Feldman, H. H., Schneider, L. S., Wilcock, G. K., Frisoni, G. B., Hardlund, J. H., et al. (2016). Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388, 2873–2884.
Gehrman, P. R., Connor, D. J., Martin, J. L., Shochat, T., Corey-Bloom, J., and Ancoli-Israel, S. (2009). Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am. J. Geriatr. Psychiatry 17, 166–169.
Gozes, I., and Shazman, S. (2023). A novel davunetide (NAPVSIPQQ to NAPVSIPQE) point mutation in activity-dependent neuroprotective protein (ADNP) causes a mild developmental syndrome. Eur. J. Neurosci. doi: 10.1111/ejn.15920 [Epub ahead of print].
Gozes, I., Stewart, A., Morimoto, B., Fox, A., Sutherland, K., and Schmeche, D. (2009). Addressing Alzheimer’s disease tangles: from NAP to AL-108. Curr. Alzheimer Res. 6, 455–460.
Gu, C., Zhang, Y., Hu, Q., Wu, J., Ren, H., Liu, C. F., et al. (2017). P7C3 inhibits GSK3β activation to protect dopaminergic neurons against neurotoxin-induced cell death in vitro and in vivo. Cell Death Dis. 8:e2858.
Guo, B., Huang, Y., Gao, Q., and Zhou, Q. (2020). Stabilization of microtubules improves cognitive functions and axonal transport of mitochondria in Alzheimer’s disease model mice. Neurobiol. Aging 96, 223–232. doi: 10.1016/j.neurobiolaging.2020.09.011
Gutiérrez-Aguilar, M., and Baines, C. P. (2015). Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 1850, 2041–2047. doi: 10.1016/j.bbagen.2014.11.009
Gutzmann, H., Kühl, K. P., Hadler, D., and Rapp, M. A. (2002). Safety and efficacy of idebenone versus tacrine in patients with Alzheimer’s disease: results of a randomized, double-blind, parallel-group multicenter study. Pharmacopsychiatry 35, 12–18. doi: 10.1055/s-2002-19833
Hampel, H., Mesulam, M. M., Cuello, A. C., Khachaturian, A. S., Vergallo, A., Farlow, M. R., et al. (2019). Revisiting the cholinergic hypothesis in Alzheimer’s disease: emerging evidence from translational and clinical research. J. Prev. Alzheimers Dis. 6, 2–15.
Han, J., Lee, H. J., Kim, K. Y., Lee, S. J. C., Suh, J. M., Cho, J., et al. (2018). Tuning structures and properties for developing novel chemical tools toward distinct pathogenic elements in Alzheimer’s disease. ACS Chem. Neurosci. 9, 800–808. doi: 10.1021/acschemneuro.7b00454
Harrington, C., Sawchak, S., Chiang, C., Davies, J., Donovan, C., Saunders, A. M., et al. (2011). Rosiglitazone does not improve cognition or global function when used as adjunctive therapy to AChE inhibitors in mild-to-moderate Alzheimer’s disease: two phase 3 studies. Curr. Alzheimer Res. 8, 592–606.
Hashweh, N. N., Bartochowski, Z., Khoury, R., and Grossberg, G. T. (2020). An evaluation of hydromethylthionine as a treatment option for Alzheimer’s disease. Expert Opin. Pharmacother. 21, 619–627. doi: 10.1080/14656566.2020.1719066
Haussmann, R., Noppes, F., Brandt, M. D., Bauer, M., and Donix, M. (2021). Lithium: a therapeutic option in Alzheimer’s disease and its prodromal stages? Neurosci. Lett. 760:136044. doi: 10.1016/j.neulet.2021.136044
Henley, D. B., Sundell, K. L., Sethuraman, G., Dowsett, S. A., and May, P. C. (2014). Safety profile of semagacestat, a gamma-secretase inhibitor: identity trial findings. Curr. Med. Res. Opin. 30, 2021–2032. doi: 10.1185/03007995.2014.939167
Herrik, K. F., Mørk, A., Richard, N., Bundgaard, C., Bastlund, J. F., and de Jong, I. E. M. (2016). The 5-HT6 receptor antagonist idalopirdine potentiates the effects of acetylcholinesterase inhibition on neuronal network oscillations and extracellular acetylcholine levels in the rat dorsal hippocampus. Neuropharmacology 107, 351–363. doi: 10.1016/j.neuropharm.2016.03.043
Honig, L. S., Vellas, B., Woodward, M., Boada, M., Bullock, R., Borrie, M., et al. (2018). Trial of Solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 378, 321–330.
Hsiao, C. C., Rombouts, F., and Gijsen, H. J. M. (2019). New evolutions in the BACE1 inhibitor field from 2014 to 2018. Bioorganic Med. Chem. Lett. 29, 761–777. doi: 10.1016/j.bmcl.2018.12.049
Hu, W., Zhao, M., Lian, J., Li, D., Wen, J., and Tan, J. (2022). Lithium cholesterol sulfate: a novel and potential drug for treating Alzheimer’s disease and autism spectrum disorder. CNS Neurol. Disord. Drug Targets. doi: 10.2174/1871527321666220825114236 [Epub ahead of print].
Hull, M., Sadowsky, C., Arai, H., Le Prince Leterme, G., Holstein, A., Booth, K., et al. (2017). Long-term extensions of randomized vaccination trials of ACC-001 and QS-21 in mild to moderate Alzheimer’s disease. Curr. Alzheimer Res. 14, 696–708. doi: 10.2174/1567205014666170117101537
Idan-Feldman, A., Schirer, Y., Polyzoidou, E., Touloumi, O., Lagoudaki, R., Grigoriadis, N. C., et al. (2011). Davunetide (NAP) as a preventative treatment for central nervous system complications in a diabetes rat model. Neurobiol. Dis. 44, 327–339. doi: 10.1016/j.nbd.2011.06.020
Isobe, C., Abe, T., and Terayama, Y. (2009). Increase in the oxidized/total coenzyme Q-10 ratio in the cerebrospinal fluid of Alzheimer’s disease patients. Dement. Geriatr. Cogn. Disord. 28, 449–454. doi: 10.1159/000256209
Ivachtchenko, A. V., Lavrovsky, Y., and Okun, I. (2016). AVN-101: a multi-target drug candidate for the treatment of CNS disorders. J. Alzheimers Dis. 53, 583–620. doi: 10.3233/JAD-151146
Jo, T., Nho, K., Risacher, S. L., Saykin, A. J., and Alzheimer’s Neuroimaging Initiative (2020). Deep learning detection of informative features in tau PET for Alzheimer’s disease classification. BMC Bioinform. 21:496. doi: 10.1186/s12859-020-03848-0
Jouanne, M., Rault, S., and Voisin-Chiret, A. S. (2017). Tau protein aggregation in Alzheimer’s disease: an attractive target for the development of novel therapeutic agents. Eur. J. Med. Chem. 139, 153–167.
Kandimalla, R., Thirumala, V., and Reddy, P. H. (2017). Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 1078–1089.
Kemp, S. W. P., Szynkaruk, M., Stanoulis, K. N., Wood, M. D., Liu, E. H., Willand, M. P., et al. (2015). Pharmacologic rescue of motor and sensory function by the neuroprotective compound P7C3 following neonatal nerve injury. Neuroscience 284, 202–216. doi: 10.1016/j.neuroscience.2014.10.005
Klein, G., Delmar, P., Voyle, N., Rehal, S., Hofmann, C., Abi-Saab, D., et al. (2019). Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res. Ther. 11:101. doi: 10.1186/s13195-019-0559-z
Konner, J., Grisham, R. N., Park, J., O’Connor, O. A., Cropp, G., Johnson, R., et al. (2012). Phase I clinical, pharmacokinetic, and pharmacodynamic study of KOS-862 (Epothilone D) in patients with advanced solid tumors and lymphoma. Investig. New Drugs 30, 2294–2302. doi: 10.1007/s10637-011-9765-7
Kreisl, W. C., Lyoo, C. H., McGwier, M., Snow, J., Jenko, K. J., Kimura, N., et al. (2013). In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain 136, 2228–2238. doi: 10.1093/brain/awt145
Kuehn, B. M. (2020). In Alzheimer research, glucose metabolism moves to center stage. JAMA 323, 297–299.
Lacosta, A.-M., Pascual-Lucas, M., Pesini, P., Casabona, D., Pérez-Grijalba, V., Marcos-Campos, I., et al. (2018). Safety, tolerability and immunogenicity of an active anti-Aβ40 vaccine (ABvac40) in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase I trial. Alzheimers Res. Ther. 10:12.
Landen, J. W., Andreasen, N., Cronenberger, C. L., Schwartz, P. F., Borjesson-Hanson, A., Ostlund, H., et al. (2017). Ponezumab in mild-to-moderate Alzheimer’s disease: randomized phase II PET-PIB study. Alzheimers Dement. 3, 393–401. doi: 10.1016/j.trci.2017.05.003
Lee, S. H., Le Pichon, C. E., Adolfsson, O., Gafner, V., Pihlgren, M., Lin, H., et al. (2016). Antibody-mediated targeting of tau in vivo does not require effector function and microglial engagement. Cell Rep. 16, 1690–1700. doi: 10.1016/j.celrep.2016.06.099
Liu, P. P., Xie, Y., Meng, X. Y., and Kang, J. S. (2019). History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduction Targeted Ther. 4:29.
Liu-Seifert, H., Siemers, E., Sundell, K., Mynderse, M., Cummings, J., Mohs, R., et al. (2018). Analysis of the relationship of cognitive impairment and functional impairment in mild Alzheimer’s disease in EXPEDITION 3. J. Prev. Alzheimers Dis. 5, 184–187.
Loureiro, J. C., Pais, M. V., Stella, F., Radanovic, M., Teixeira, A. L., Forlenza, O. V., et al. (2020). Passive antiamyloid immunotherapy for Alzheimer’s disease. Curr. Opin. Psychiatry 33, 284–291.
Lovestone, S., Boada, M., Dubois, B., Hull, M., Rinne, J. O., Huppertz, H. J., et al. (2015). A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88.
Luca, A., and Luca, M. (2023). Lithium in Alzheimer’s disease: from prevention to treatment. Psychogeriatrics 23, 204–205.
Luo, F., Sandhu, A. F., Rungratanawanich, W., Williams, G. E., Akbar, M., Zhou, S., et al. (2020). Melatonin and autophagy in aging-related neurodegenerative diseases. Int. J. Mol. Sci. 21:7174. doi: 10.3390/ijms21197174
Lyoo, C. H., Ikawa, M., Liow, J. S., Zoghbi, S. S., Morse, C. L., Pike, V. W., et al. (2015). Cerebellum can serve as a pseudo-reference region in Alzheimer disease to detect neuroinflammation measured with PET radioligand binding to translocator protein. J. Nucl. Med. 56, 701–706. doi: 10.2967/jnumed.114.146027
MacKay, J., Harnett, S., and Machado, P. (2010). Pfizer and Medivation announce results from two phase 3 studies in Dimebon (Latrepirdine*) Alzheimer’s disease clinical development program; 2010. Available online at: https://www.pfizer.com/news/press-release/press-release-detail/pfizer_and_medivation_announce_results_from_two_phase_3_studies_in_dimebon_latrepirdine_alzheimer_s_disease_clinical_development_program
Makhaeva, G. F., Lushchekina, S. V., Boltneva, N. P., Sokolov, V. B., Grigoriev, V. V., Serebryakova, O. G., et al. (2015). Conjugates of γ-Carbolines and Phenothiazine as new selective inhibitors of butyrylcholinesterase and blockers of NMDA receptors for Alzheimer Disease. Sci. Rep. 5:13164. doi: 10.1038/srep13164
Makhaeva, G. F., Shevtsova, E. F., Boltneva, N. P., Lushchekina, S. V., Kovaleva, N. V., Rudakova, E. V., et al. (2019). Overview of novel multifunctional agents based on conjugates of γ-carbolines, carbazoles, tetrahydrocarbazoles, phenothiazines, and aminoadamantanes for treatment of Alzheimer’s disease. Chem. Biol. Interact. 308, 224–234. doi: 10.1016/j.cbi.2019.05.020
Man, V. H., He, X., Han, F., Cai, L., Wang, L., Niu, T., et al. (2023). Phosphorylation at Ser289 enhances the oligomerization of tau repeat R2. J. Chem. Inform. Model. 63, 1351–1361. doi: 10.1021/acs.jcim.2c01597
Mintun, M. A., Lo, A. C., Duggan Evans, C., Wessels, A. M., Ardayfio, P. A., Andersen, S. W., et al. (2021). Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 384, 1691–1704.
Miranda, A., Montiel, E., Ulrich, H., and Paz, C. (2021). Selective secretase targeting for Alzheimer’s disease therapy. J. Alzheimers Dis. 81, 1–17.
Morimoto, B. H., Schmechel, D., Hirman, J., Blackwell, A., Keith, J., and Gold, M. (2013). A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 35, 325–336. doi: 10.1159/000348347
Moriyama, T., Fukushima, T., Kokate, T., and Albala, B. (2017). [P3–037]: preclinical studies with elenbecestat, a novel BACE1 inhibitor, show no evidence of hypopigmentation. Alzheimers Dement. 13:944.
Moussa-Pacha, N. M., Abdin, S. M., Omar, H. A., Alniss, H., and Al-Tel, T. H. (2020). BACE1 inhibitors: current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 40, 339–384.
Muronaga, M., Terao, T., Kohno, K., Hirakawa, H., Izumi, T., and Etoh, M. (2022). Lithium in drinking water and Alzheimer’s dementia: epidemiological findings from national data base of Japan. Bipolar Disord. 24, 788–794. doi: 10.1111/bdi.13257
Naidoo, J., De Jesus-Cortes, H., Huntington, P., Estill, S., Morlock, L. K., Starwalt, R., et al. (2014). Discovery of a neuroprotective chemical, (S)-N-(3-(3,6-dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(-)-P7C3-S243], with improved druglike properties. J. Med. Chem. 57, 3746–3754.
Neth, B. J., and Craft, S. (2017). Insulin resistance and Alzheimer’s disease: bioenergetic linkages. Front. Aging Neurosci. 9:345. doi: 10.3389/fnagi.2017.00345
Neumann, U., Ufer, M., Jacobson, L. H., Rouzade-Dominguez, M. L., Huledal, G., Kolly, C., et al. (2018). The BACE-1 inhibitor CNP 520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 10:e9316. doi: 10.15252/emmm.201809316
Nho, K., Kueider-Paisley, A., MahmoudianDehkordi, S., Arnold, M., Risacher, S. L., Louie, G., et al. (2019). Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: relationship to neuroimaging and CSF biomarkers. Alzheimers dement. 15, 232–244. doi: 10.1016/j.jalz.2018.08.012
Nicoll, J. A. R., Buckland, G. R., Harrison, C. H., Page, A., Harris, S., Love, S., et al. (2019). Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain 142, 2113–2126.
Novak, P., Schmidt, R., Kontsekova, E., Kovacech, B., Smolek, T., Katina, S., et al. (2018). FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res. Ther. 10:108. doi: 10.1186/s13195-018-0436-1
Novak, P., Schmidt, R., Kontsekova, E., Zilka, N., Kovacech, B., Skrabana, R., et al. (2017). Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 16, 123–134. doi: 10.1016/S1474-4422(16)30331-3
Novak, P., Zilka, N., Zilkova, M., Kovacech, B., Skrabana, R., Ondrus, M., et al. (2019). AADvac1, an active immunotherapy for Alzheimer’s disease and non Alzheimer tauopathies: an overview of preclinical and clinical development. J Prev Alzheimers Dis 6, 63–69. doi: 10.14283/jpad.2018.45
O’Bryant, S. E., Zhang, F., Petersen, M., Johnson, L., Hall, J., and Rissman, R. A. (2021). A precision medicine approach to treating Alzheimer’s disease using rosiglitazone therapy: a biomarker analysis of the REFLECT trials. J. Alzheimers Dis. 81, 557–568. doi: 10.3233/JAD-201610
Ogbodo, J. O., Agbo, C. P., Njoku, U. O., Ogugofor, M. O., Egba, S. I., Ihim, S. A., et al. (2022). Alzheimer’s disease: pathogenesis and therapeutic interventions. Curr. Aging Sci. 15, 2–25.
Ohtake, Y., Kong, W., Hussain, R., Horiuchi, M., Tremblay, M. L., Ganea, D., et al. (2017). Protein tyrosine phosphatase σ regulates autoimmune encephalomyelitis development. Brain Behav. Immun. 65, 111–124. doi: 10.1016/j.bbi.2017.05.018
Orsucci, D., Mancuso, M., Ienco, E. C., LoGerfo, A., and Siciliano, G. (2011). Targeting mitochondrial dysfunction and neurodegeneration by means of coenzyme Q10 and its analogues. Curr. Med. Chem. 18, 4053–4064. doi: 10.2174/092986711796957257
Ostrowitzki, S., Bittner, T., Sink, K. M., Mackey, H., Rabe, C., Honig, L. S., et al. (2022). Evaluating the safety and efficacy of crenezumab vs placebo in adults with early alzheimer disease: two phase 3 randomized placebo-controlled trials. JAMA Neurol. 79, 1113–1121. doi: 10.1001/jamaneurol.2022.2909
Pandi-Perumal, S. R., BaHammam, A. S., Brown, G. M., Spence, D. W., Bharti, V. K., Kaur, C., et al. (2013). Melatonin antioxidative defense: therapeutical implications for aging and neurodegenerative processes. Neurotoxicity Res. 23, 267–300. doi: 10.1007/s12640-012-9337-4
Pasquier, F., Sadowsky, C., Holstein, A., Leterme Gle, P., Peng, Y., Jackson, N., et al. (2016). Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J. Alzheimers Dis. 51, 1131–1143.
Patel, S., Bansoad, A. V., Singh, R., and Khatik, G. L. (2022). BACE1: a key regulator in Alzheimer’s disease progression and current development of its inhibitors. Curr. Neuropharmacol. 20, 1174–1193. doi: 10.2174/1570159X19666211201094031
Pavlov, E., Zakharian, E., Bladen, C., Diao, C. T., Grimbly, C., Reusch, R. N., et al. (2005). A large, voltage-dependent channel, isolated from mitochondria by water-free chloroform extraction. Biophys. J. 88, 2614–2625. doi: 10.1529/biophysj.104.057281
Pieper, A. A., Xie, S., Capota, E., Estill, S. J., Zhong, J., Long, J. M., et al. (2010). Discovery of a proneurogenic, neuroprotective chemical. Cell 142, 39–51.
Pinheiro, L., and Faustino, C. (2019). Therapeutic strategies targeting amyloid-beta in Alzheimer’s disease. Curr. Alzheimer Res. 16, 418–452.
Porter, G. A. Jr., and Beutner, G. (2018). Cyclophilin D, somehow a master regulator of mitochondrial function. Biomolecules 8:176. doi: 10.3390/biom8040176
Portron, A., Jordan, P., Draper, K., Muenzer, C., Dickerson, D., van Iersel, T., et al. (2020). A phase I study to assess the effect of speed of injection on pain, tolerability, and pharmacokinetics after high-volume subcutaneous administration of gantenerumab in healthy volunteers. Clin. Ther. 42, 108–120.e1. doi: 10.1016/j.clinthera.2019.11.015
Pride, M., Seubert, P., Grundman, M., Hagen, M., Eldridge, J., and Black, R. S. (2008). Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener. Dis. 5, 194–196. doi: 10.1159/000113700
Qin, W., Haroutunian, V., Katsel, P., Cardozo, C. P., Ho, L., Buxbaum, J. D., et al. (2009) PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 66, 352–361.
Qureshi, I. A., Tirucherai, G., Ahlijanian, M. K., Kolaitis, G., Bechtold, C., and Grundman, M. (2018). A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement. 4, 746–755. doi: 10.1016/j.trci.2018.10.007
Ramsay, R. R., Dunford, C., and Gillman, P. K. (2007). Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br. J. Pharmacol. 152, 946–951. doi: 10.1038/sj.bjp.0707430
Rashad, A., Rasool, A., Shaheryar, M., Sarfraz, A., Sarfraz, Z., Robles-Velasco, K., et al. (2022). Donanemab for Alzheimer’s disease: a systematic review of clinical trials. Healthcare 11:32. doi: 10.3390/healthcare11010032
Reddy, A. P., and Reddy, P. H. (2017). Mitochondria-targeted molecules as potential drugs to treat patients with Alzheimer’s disease. Progr. Mol. Biol. Transl. Sci. 146, 173–201. doi: 10.1016/bs.pmbts.2016.12.010
Reddy, P. H., and Oliver, D. M. (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8:488.
Roberts, C., Kaplow, J., Giroux, M., Krause, S., and Kanekiyo, M. (2021). Amyloid and APOE status of screened subjects in the Elenbecestat MissionAD phase 3 program. J. Prev. Alzheimers Dis. 8, 218–223. doi: 10.14283/jpad.2021.4
Saitow, F., and Nakaoka, Y. (1997). The photodynamic action of methylene blue on the ion channels of Paramecium causes cell damage. Photochem. Photobiol. 65, 902–907. doi: 10.1111/j.1751-1097.1997.tb01941.x
Salloway, S., Farlow, M., McDade, E., Clifford, D. B., Wang, G., Llibre-Guerra, J. J., et al. (2021). A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med. 27, 1187–1196.
Salloway, S., Marshall, G. A., Lu, M., and Brashear, H. R. (2018). Long-term safety and efficacy of bapineuzumab in patients with mild-to-moderate Alzheimer’s disease: a phase 2, open-label extension study. Curr. Alzheimer Res. 15, 1231–1243. doi: 10.2174/1567205015666180821114813
Salloway, S., Sperling, R., Fox, N. C., Blennow, K., Klunk, W., Raskind, M., et al. (2014). Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333.
Schiavone, M., Zulian, A., Menazza, S., Petronilli, V., Argenton, F., Merlini, L., et al. (2017). Alisporivir rescues defective mitochondrial respiration in Duchenne muscular dystrophy. Pharmacol. Res. 125, 122–131. doi: 10.1016/j.phrs.2017.09.001
Schneider, L. S., Laudon, M., Nir, T., Caceres, J., Ianniciello, G., Capulli, M., et al. (2022). A polymorphism cluster at the 2q12 locus may predict response to Piromelatine in patients with mild Alzheimer’s disease. J. Prev. Alzheimers Dis. 9, 247–254. doi: 10.14283/jpad.2021.61
Seripa, D., Solfrizzi, V., Imbimbo, B. P., Daniele, A., Santamato, A., Lozupone, M., et al. (2016). Tau-directed approaches for the treatment of Alzheimer’s disease: focus on leuco-methylthioninium. Expert Rev. Neurother. 16, 259–277. doi: 10.1586/14737175.2016.1140039
Shcherbinin, S., Evans, C. D., Lu, M., Andersen, S. W., Pontecorvo, M. J., Willis, B. A., et al. (2022). Association of amyloid reduction after Donanemab treatment with tau pathology and clinical outcomes: the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol. 79, 1015–1024.
Shevtsova, E. F., Maltsev, A. V., Vinogradova, D. V., Shevtsov, P. N., and Bachurin, S. O. (2021). Mitochondria as a promising target for developing novel agents for treating Alzheimer’s disease. Med. Res. Rev. 41, 803–827.
Shevtsova, E. F., Vinogradova, D. V., Kireeva, E. G., Reddy, V. P., Aliev, G., and Bachurin, S. O. (2014). Dimebon attenuates the Aβ-induced mitochondrial permeabilization. Curr. Alzheimer Res. 11, 422–429.
Shevtsova, E. F., Vinogradova, D. V., Neganova, M. E., Avila-Rodriguez, M., Ashraf, G. M., Barreto, G. E., et al. (2017). Mitochondrial permeability transition pore as a suitable target for neuroprotective agents against Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 16, 677–685. doi: 10.2174/1871527316666170424114444
Singer, C., Tractenberg, R. E., Kaye, J., Schafer, K., Gamst, A., Grundman, M., et al. (2003). A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep 26, 893–901.
Singulani, M. P., Pereira, C. P. M., Ferreira, A. F. F., Garcia, P. C., Ferrari, G. D., Alberici, L. C., et al. (2020). Impairment of PGC-1α-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp. Gerontol. 133:110882.
Skvortsova, V. I., Bachurin, S. O., Ustyugov, A. A., Kukharsky, M. S., Deikin, A. V., Buchman, V. L., et al. (2018). Gamma-carbolines derivatives as promising agents for the development of pathogenic therapy for proteinopathy. Acta Naturae 10, 59–62.
Sperling, R., Henley, D., Aisen, P. S., Raman, R., Donohue, M. C., Ernstrom, K., et al. (2021). Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical alzheimer disease: a truncated randomized phase 2b/3 clinical trial. JAMA Neurol. 78, 293–301. doi: 10.1001/jamaneurol.2020.4857
Springer, J. E., Visavadiya, N. P., Sullivan, P. G., and Hall, E. D. (2018). Post-injury treatment with NIM811 promotes recovery of function in adult female rats after spinal cord contusion: a dose-response study. J. Neurotrauma 35, 492–499. doi: 10.1089/neu.2017.5167
Sragovich, S., Malishkevich, A., Piontkewitz, Y., Giladi, E., Touloumi, O., Lagoudaki, R., et al. (2019). The autism/neuroprotection-linked ADNP/NAP regulate the excitatory glutamatergic synapse. Transl. Psychiatry 9:2. doi: 10.1038/s41398-018-0357-6
Sragovich, S., Merenlender-Wagner, A., and Gozes, I. (2017). ADNP plays a key role in autophagy: from autism to schizophrenia and Alzheimer’s Disease. Bioessays 39:1700054. doi: 10.1002/bies.201700054
Swanson, C. J., Zhang, Y., Dhadda, S., Wang, J., Kaplow, J., Lai, R. Y. K., et al. (2021). A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res. Ther. 13:80.
Tarocco, A., Caroccia, N., Morciano, G., Wieckowski, M. R., Ancora, G., Garani, G., et al. (2019). Melatonin as a master regulator of cell death and inflammation: molecular mechanisms and clinical implications for newborn care. Cell Death Dis. 10:317. doi: 10.1038/s41419-019-1556-7
Tatebe, H., Kasai, T., Ohmichi, T., Kishi, Y., Kakeya, T., Waragai, M., et al. (2017). Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener. 12:63. doi: 10.1186/s13024-017-0206-8
Teng, E., Manser, P. T., Pickthorn, K., Brunstein, F., Blendstrup, M., Sanabria Bohorquez, S., et al. (2022). Safety and efficacy of semorinemab in individuals with prodromal to mild alzheimer disease: a randomized clinical trial. JAMA Neurol. 79, 758–767. doi: 10.1001/jamaneurol.2022.1375
Teo, E., Ravi, S., Barardo, D., Kim, H. S., Fong, S., Cazenave-Gassiot, A., et al. (2019). Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. eLife 8:e50069. doi: 10.7554/eLife.50069
Tesla, R., Wolf, H. P., Xu, P., Drawbridge, J., Estill, S. J., Huntington, P., et al. (2012). Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 109, 17016–17021. doi: 10.1073/pnas.1213960109
Thal, L. J., Grundman, M., Berg, J., Ernstrom, K., Margolin, R., Pfeiffer, E., et al. (2003). Idebenone treatment fails to slow cognitive decline in Alzheimer’s disease. Neurology 61, 1498–1502. doi: 10.1212/01.wnl.0000096376.03678.c1
Theunis, C., Crespo-Biel, N., Gafner, V., Pihlgren, M., López-Deber, M. P., Reis, P., et al. (2013). Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One 8:e72301. doi: 10.1371/journal.pone.0072301
Timmers, M., Streffer, J. R., Russu, A., Tominaga, Y., Shimizu, H., Shiraishi, A., et al. (2018). Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: randomized, double-blind, placebo-controlled study. Alzheimers Res. Ther. 10:85. doi: 10.1186/s13195-018-0415-6
Tucker, D., Lu, Y., and Zhang, Q. (2018). From mitochondrial function to neuroprotection-an emerging role for methylene blue. Mol. Neurobiol. 55, 5137–5153. doi: 10.1007/s12035-017-0712-2
Ustyugov, A., Shevtsova, E., Ashraf, G. M., Tarasov, V. V., Bachurin, S. O., and Aliev, G. (2018). New therapeutic property of dimebon as a neuroprotective agent. Curr. Med. Chem. 25, 5315–5326.
Vahdat, L. T. (2008). Clinical studies with epothilones for the treatment of metastatic breast cancer. Semin. Oncol. 35, S22–S30.
Valasani, K. R., Sun, Q., Fang, D., Zhang, Z., Yu, Q., Guo, Y., et al. (2016). Identification of a small molecule cyclophilin D inhibitor for rescuing Aβ-mediated mitochondrial dysfunction. ACS Med. Chem. Lett. 7, 294–299.
Valasani, K. R., Vangavaragu, J. R., Day, V. W., and Yan, S. S. (2014). Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors. J. Chem. Inform. Model. 54, 902–912. doi: 10.1021/ci5000196
van Dyck, C. H., Swanson, C. J., Aisen, P., Bateman, R. J., Chen, C., Gee, M., et al. (2023). Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21.
VandeVrede, L., Dale, M. L., Fields, S., Frank, M., Hare, E., Heuer, H. W., et al. (2020). Open-label phase 1 futility studies of salsalate and young plasma in progressive supranuclear palsy. Mov. Disord. Clin. Pract. 7, 440–447. doi: 10.1002/mdc3.12940
Vázquez-Rosa, E., Shin, M. K., Dhar, M., Chaubey, K., Cintrón-Pérez, C. J., Tang, X., et al. (2020). P7C3-A20 treatment one year after TBI in mice repairs the blood-brain barrier, arrests chronic neurodegeneration, and restores cognition. Proc. Natl. Acad. Sci. U.S.A. 117, 27667–27675. doi: 10.1073/pnas.2010430117
Vermersch, P., Brieva-Ruiz, L., Fox, R. J., Paul, F., Ramio-Torrenta, L., Schwab, M., et al. (2022). Efficacy and safety of Masitinib in progressive forms of multiple sclerosis: a randomized, phase 3, clinical trial. Neurol. Neuroimmunol. Neuroinflamm. 9:e1148. doi: 10.1212/NXI.0000000000001148
Voorhees, J. R., Remy, M. T., Cintrón-Pérez, C. J., El Rassi, E., Khan, M. Z., Dutca, L. M., et al. (2018). (-)-P7C3-S243 protects a rat model of Alzheimer’s disease from neuropsychiatric deficits and neurodegeneration without altering amyloid deposition or reactive glia. Biol. Psychiatry 84, 488–498.
Vormfelde, S. V., Pezous, N., Lefevre, G., Kolly, C., Neumann, U., Jordaan, P., et al. (2020). A pooled analysis of three randomized phase I/IIa clinical trials confirms absence of a clinically relevant effect on the QTc interval by Umibecestat. Clin. Transl. Sci. 13, 1316–1326. doi: 10.1111/cts.12832
Vutskits, L., Briner, A., Klauser, P., Gascon, E., Dayer, A. G., Kiss, J. Z., et al. (2008). Adverse effects of methylene blue on the central nervous system. Anesthesiology 108, 684–692.
Wade, A. G., Farmer, M., Harari, G., Fund, N., Laudon, M., Nir, T., et al. (2014). Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer’s disease: a 6-month, randomized, placebo-controlled, multicenter trial. Clin. Interv. Aging 9, 947–961. doi: 10.2147/CIA.S65625
Walker, A. K., Rivera, P. D., Wang, Q., Chuang, J. C., Tran, S., Osborne-Lawrence, S., et al. (2015). The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol. Psychiatry 20, 500–508. doi: 10.1038/mp.2014.34
Wang, D., Kowalewski, E. K., and Koch, G. (2022). Application of meta-analysis to evaluate relationships among ARIA-E Rate, amyloid reduction rate, and clinical cognitive response in amyloid therapeutic clinical trials for early Alzheimer’s disease. Ther. Innov. Regul. Sci. 56, 501–516. doi: 10.1007/s43441-022-00390-4
Wang, T., Zhao, L., Liu, M., Xie, F., Ma, X., Zhao, P., et al. (2014). Oral intake of hydrogen-rich water ameliorated chlorpyrifos-induced neurotoxicity in rats. Toxicol. Appl. Pharmacol. 280, 169–176. doi: 10.1016/j.taap.2014.06.011
Wang, Y., An, H., Liu, T., Qin, C., Sesaki, H., Guo, S., et al. (2019). Metformin Improves Mitochondrial Respiratory Activity through Activation of AMPK. Cell Rep. 29, 1511–1523.e5.
Watson, G. S., and Craft, S. (2003). The role of insulin resistance in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs 17, 27–45.
Wegmann, S., Biernat, J., and Mandelkow, E. (2021). A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 69, 131–138.
Wessels, A. M., Tariot, P. N., Zimmer, J. A., Selzler, K. J., Bragg, S. M., Andersen, S. W., et al. (2020). Efficacy and safety of Lanabecestat for treatment of early and mild Alzheimer Disease: the AMARANTH and DAYBREAK-ALZ randomized clinical trials. JAMA Neurol. 77, 199–209. doi: 10.1001/jamaneurol.2019.3988
West, T., Hu, Y., Verghese, P. B., Bateman, R. J., Braunstein, J. B., Fogelman, I., et al. (2017). Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J. Prev. Alzheimers Dis. 4, 236–241. doi: 10.14283/jpad.2017.36
Wilcock, G. K., Gauthier, S., Frisoni, G. B., Jia, J., Hardlund, J. H., Moebius, H. J., et al. (2018). Potential of low dose leuco-methylthioninium Bis(Hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s Disease: cohort analysis as modified primary outcome in a phase III clinical trial. J. Alzheimers Dis. 61, 435–457. doi: 10.3233/JAD-170560
Wilkins, H. M., and Morris, J. K. (2017). New therapeutics to modulate mitochondrial function in neurodegenerative disorders. Curr. Pharm. Des. 23, 731–752.
Wilkinson, D., Windfeld, K., and Colding-Jørgensen, E. (2014). Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 13, 1092–1099. doi: 10.1016/S1474-4422(14)70198-X
Willis, B. A., Zhang, W., Ayan-Oshodi, M., Lowe, S. L., Annes, W. F., Sirois, P. J., et al. (2012). Semagacestat pharmacokinetics are not significantly affected by formulation, food, or time of dosing in healthy participants. J. Clin. Pharmacol. 52, 904–913.
Wischik, C. M., Staff, R. T., Wischik, D. J., Bentham, P., Murray, A. D., Storey, J. M., et al. (2015). Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers Dis. 44, 705–720.
Yang, C. C., Chiu, M. J., Chen, T. F., Chang, H. L., Liu, B. H., and Yang, S. Y. (2018). Assay of plasma phosphorylated tau protein (Threonine 181) and total tau protein in early-stage Alzheimer’s disease. J. Alzheimers Dis. 61, 1323–1332. doi: 10.3233/JAD-170810
Yang, X., Zhang, Y., Xu, H., Luo, X., Yu, J., Liu, J., et al. (2016). Neuroprotection of coenzyme Q10 in neurodegenerative diseases. Curr. Top. Med. Chem. 16, 858–866.
Yao, J., Irwin, R. W., Zhao, L., Nilsen, J., Hamilton, R. T., and Brinton, R. D. (2009). Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 106, 14670–14675.
Ye, W., Liu, T., Zhang, W. M., Zhang, W., and Li, S. (2023). The improvement of epothilone D yield by the disruption of epoK gene in Sorangium cellulosum using TALEN system. Mol. Biotechnol. 65, 282–289. doi: 10.1007/s12033-022-00602-0
Yin, T. C., Britt, J. K., De Jesús-Cortés, H., Lu, Y., Genova, R. M., Khan, M. Z., et al. (2014). P7C3 neuroprotective chemicals block axonal degeneration and preserve function after traumatic brain injury. Cell Rep. 8, 1731–1740. doi: 10.1016/j.celrep.2014.08.030
Zhou, H., Wang, J., Jiang, J., Stavrovskaya, I. G., Li, M., Li, W., et al. (2014). N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury. J. Neurosci. 34, 2967–2978. doi: 10.1523/JNEUROSCI.1948-13.2014
Keywords: Alzheimer’s disease, β-amyloid protein, tau protein, mitochondria-targeting, multi-targets, clinical trials
Citation: Peng Y, Jin H, Xue Y-h, Chen Q, Yao S-y, Du M-q and Liu S (2023) Current and future therapeutic strategies for Alzheimer’s disease: an overview of drug development bottlenecks. Front. Aging Neurosci. 15:1206572. doi: 10.3389/fnagi.2023.1206572
Received: 16 April 2023; Accepted: 10 July 2023;
Published: 03 August 2023.
Edited by:
Woon-Man Kung, Chinese Culture University, TaiwanReviewed by:
Tadanori Hamano, University of Fukui, JapanSergey Bachurin, Institute of Physiologically Active Compounds (RAS), Russia
Copyright © 2023 Peng, Jin, Xue, Chen, Yao, Du and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong Peng, MTc3OTM0MjQ0NkBxcS5jb20=