 Daoyuan Chen
Daoyuan Chen Qingxiu Chen1
Qingxiu Chen1- 1School of Bioengineering, Zunyi Medical University, Zhuhai, China
- 2Fujian Key Laboratory of Translational Research in Cancer and Neurodegenerative Diseases, School of Basic Medical Sciences, Institute of Basic Medicine, Fujian Medical University, Fuzhou, China
Human glutaminyl cyclase (hQC) is drawing considerable attention and emerging as a potential druggable target for Alzheimer's disease (AD) due to its close involvement in the pathology of AD via the post-translational pyroglutamate modification of amyloid-β. A recent phase 2a study has shown promising early evidence of efficacy for AD with a competitive benzimidazole-based QC inhibitor, PQ912, which also demonstrated favorable safety profiles. This finding has sparked new hope for the treatment of AD. In this review, we briefly summarize the discovery and evolution of hQC inhibitors, with a particular interest in classic Zinc binding group (ZBG)-containing chemicals reported in recent years. Additionally, we highlight several high-potency inhibitors and discuss new trends and challenges in the development of QC inhibitors as an alternative and promising disease-modifying therapy for AD.
1. Introduction
Alzheimer's disease (AD) is a complex neurodegenerative disease that is clinically characterized by progressive and irreversible dysfunction of language, memory, and cognition (Association, 2023). AD is the leading cause of dementia in the elderly, representing 60–80% of dementia cases globally (Association, 2023). Projections indicate that the number of people living with dementia around the world will sharply increase from 55 million to 139 million by 2050 (Association, 2023). However, for more than a century, only five drugs have been approved for the symptomatic treatment of AD, and these drugs are incapable of retarding or reversing disease progression. China and the United States have recently approved the mannan oligosaccharide GV-971 (Syed, 2020) and the anti-Aβ antibody aducanumab (Aduhelm) (Dhillon, 2021) as novel disease-modifying treatments for AD, respectively. Nevertheless, both treatments have been questioned for their limited clinical efficacy in clinical trials. Therefore, it is still urgently needed to develop new disease-modifying therapies for the early intervention of AD.
The deposition of senile plaques, dominantly consisting of β-amyloid proteins (Aβs), is one of the pathological hallmarks of AD brains. Full-length Aβ1 − 40/42 is generated through the amyloidogenic processing of the amyloid precursor protein (APP) mediated by the β-site APP cleaving enzyme (BACE) and γ-secretase complex (Chen et al., 2017). Compelling evidence showed that the highly hydrophobic and aggregation-prone Aβ1 − 40/42 plays an upstream role in the pathological progression of AD via inducing tau hyperphosphorylation, synaptic dysfunction, and neuroinflammation (Selkoe and Hardy, 2016; Lee et al., 2017). Hundreds of Aβ targeting or Aβ-related therapeutic strategies have thus been proposed in the past three decades (van Bokhoven et al., 2021), while unfortunately most of the interventions failed in clinical trials due to limited effects on cognition recovery or unfavorable safety profiles in AD patients.
Aβs in the senile plaques are highly diverse and heterogeneous due to various post-translational modifications (PTMs) such as truncations, oxidation, and pyroglutamation (Roher et al., 2017). The continuous failures have prompted researchers to reevaluate the role of PTMs of Aβ in the pathogenesis of AD (Grochowska et al., 2017; Roher et al., 2017). Among the PTMs, the pyroglutamation product pE3-Aβ has recently been shown to be closely involved in AD (Figure 1) and is gradually presumed to be a highly desirable biomarker and intervention target (Jawhar et al., 2011a; Bayer, 2022). pE3-Aβ is formed through the dehydration and cyclization of the Glu3 residue of the truncated Aβ3 − 40/42 under the catalytic action of human glutaminyl cyclase (hQC) (Figure 1) (Schilling et al., 2004; Cynis et al., 2008). It is noteworthy that the release of truncated Aβ3 − 40/42 is independent of BACE and may primarily relate to Meprins, members of the “astacin family” of metalloproteinases, which are able to cleave APP after the Ala2 at the N-terminus of the Aβ sequence (Stephan Schilling and Demuth, 2010).
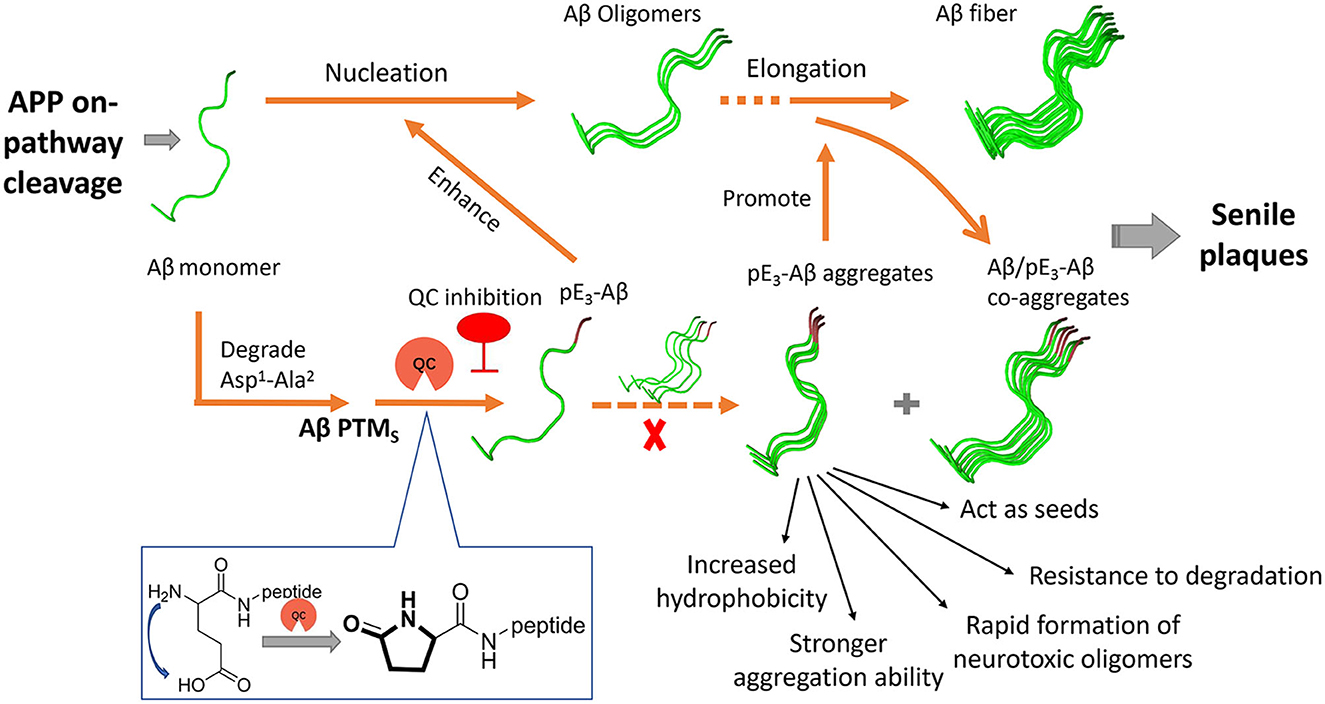
Figure 1. Generation and roles of pE3-Aβ in AD and schematic drawing of the QC inhibition approach.
pE3-Aβ constitutes a prominent fraction of the total Aβ species in AD brains (Harigaya et al., 2000; Wu et al., 2014) and the critical initiating role of pE3-Aβ in AD was supported by several lines of evidence (Figure 1) (Gunn et al., 2010; Nussbaum et al., 2012). First, pE3-Aβ has hundreds-fold higher aggregation ability (Schilling et al., 2006) and is much easier to maintain the neurotoxic oligomeric states compared with full-length Aβ (Lee et al., 2014; Gunn et al., 2016; Wulff et al., 2016). It can also act as seeds to accelerate Aβ assembly (Dammers et al., 2017b) and subsequently form denser, more stable, and cytotoxic Aβ/pE-Aβ copolymers than those of Aβ1 − 40/42 aggregates (Schilling et al., 2006; Nussbaum et al., 2012). Second, pE3-Aβ is perhaps more resistant to aminopeptidase due to the lactam ring in the N-terminus of pE3-Aβ (Gontsarova et al., 2008), which contributes to prolonged neurotoxicity in vivo. In addition, observations revealed that pE3-Aβ acts upstream of the neurotoxic Aβ cascade (Dammers et al., 2017a; Bayer, 2022). It progressively accumulates in the brain at the early stage of AD, even before full-length Aβ aggregation, and subsequently triggers neurodegeneration and ultimately exacerbates the severity of AD pathology and cognition. In a most recent study, donanemab, a pE3-Aβ-specific antibody developed by Eli Lilly, significantly cleared amyloid plaques and slowed down cognitive deterioration in patients with mild AD in a phase II trial (Mintun et al., 2021) and met all the primary and secondary endpoints in a phase III trial (TRAILBLAZER-ALZ 4), reducing brain amyloid plaque levels by 65.2% at 6 months compared to baseline (data were shared on 30 November 2022 at the Clinical Trials on Alzheimer's Disease conference). All this evidence strongly supports pE3-Aβ as an effective therapeutic target (Perez-Garmendia and Gevorkian, 2013).
QC is intimately associated with the pathology and severity of AD by paralleling the generation of pE-Aβ in the brain (Figure 1) (Morawski et al., 2014), and the deposition of pE-Aβ was found to be restricted to APP/QC co-expression areas (Hartlage-Rübsamen et al., 2018). Furthermore, the expression and enzymatic activity of QC are significantly elevated and are positively correlated with both the accumulation of pE-Aβ and cognition decline in the brains of AD subjects compared with those of age-matched controls (Valenti et al., 2013; Gunn et al., 2021). Besides, both QC knockout and treatment with QC inhibitors (QCIs) significantly rescue the behavioral phenotype and alleviate disease-like pathology in the AD mouse model (Schilling et al., 2008; Jawhar et al., 2011b). Hence, small molecule-based QCIs provide an alternative, promising, and cost-effective therapeutic approach apart from immunotherapy for early-stage AD treatment (Coimbra et al., 2019; Coimbra and Salvador, 2021; Xu et al., 2021). Recently, PQ912 (varoglutamstat), a QC competitive inhibitor developed by Probiodrug AG, passed the clinical phase IIa trial (Scheltens et al., 2018) and is regarded as the proof-of-concept validation of QC.
Over the past two decades, a number of QCIs, including both synthetic and natural compounds, have been discovered. Several reviews focusing on the function of QC and the development of QCIs have been published (Coimbra et al., 2019; Vijayan and Zhang, 2019; Coimbra and Salvador, 2021; Xu et al., 2021; Zhang et al., 2022), while new design and screening strategies have been applied in discovering new QCIs with unique structural characteristics in recent years. Hence, we reexamine the discovery and evolution of QC inhibitors, with a particular interest in classic Zinc binding group (ZBG)-containing chemicals. In addition, we highlight several representative high potent inhibitors as well as the challenges of QCIs as potential disease-modifying therapies for AD.
2. Brief functions and structural features of hQC
N-terminal pyroglutamation of proteins is ubiquitously found in a variety of organisms, including bacteria, plants, and animals, and two types of QCs with distinctive structures and catalytic sites have been identified and classified in these organisms so far. Type I QCs are mainly found in plants and bacteria, as exemplified by Papaya QC and Myxococcus xanthus QC, while type II QCs are primarily present in animals (Taudte et al., 2021), such as Drosophila melanogaster QC and human QC (hQC), which share substantial sequence identity and structural similarity (Koch et al., 2012b). Type I QC exhibits a five-bladed β-propeller structure composed of β-sheets and antiparallel β-strands, together with a Ca2+-binding motif in the active core (Carrillo et al., 2010), which significantly differs from the α/β topology and Zn2+-binding motif in the catalytic center of Type II hQCs (Taudte et al., 2021).
hQC, known as human glutaminyl-peptide acyltransferase (QPCT, EC2.3.2.5), belongs to the acyltransferase family and is abundant in the human brain and neuronal tissues. hQC is broadly expressed in various neurons, including urocortin-1 and cholinergic Edinger-Westphal neurons, as well as locus coeruleus and nucleus basalis Meynert neurons (Morawski et al., 2010). Normally, hQC promotes the maturation of neuropeptides or cytokines such as gonadotropin-releasing hormone (GnRH), thyrotropin-releasing hormone (TRH), and chemokine CCL-2 via catalyzing the cyclization of glutamine residue at the N-terminus of proteins (Cynis et al., 2011; Becker et al., 2016; Vijayan and Zhang, 2019). It was later revealed that QC only shows modest specificity for cyclization of their primary glutaminyl substrates (Seifert et al., 2009), it can also catalyze N-terminal glutamate cyclization (Schilling S. H. et al., 2003; Schilling et al., 2004), which thus provides a close link between QC and AD pathophysiology via the formation of pE-Aβ. Nevertheless, the enzymatic conversion has strikingly different condition preferences, with glutaminyl conversion occurring with an optimum pH of 8.0, whereas glutamyl conversion is favored at a pH of 6.0 (Schilling et al., 2004).
There are two isoforms of QC in humans, namely, the secretory QC (sQC, 361aa, encoded by the QPCT gene located at 2p22.2) and golgi-resident QC (gQC or isoQC, 382aa, encoded by the QPCTL gene located at 19p13.32). sQC is a secreted protein that contains a N-terminal secretion signal, while gQC contains a N-terminal anchor responsible for the retention within the Golgi complex. sQC and gQC share a sequence identity of >45%, have similar catalytic domain sizes, and catalyze the same enzymatic reaction (Stephan et al., 2009), making it uneasy to design iso-specific inhibitors. The discrepancy distribution of sQC and gQC results in the conversion of different substrates and even distinct physiological roles, which was suggested to be beneficial to the complementary function regulation of QC in a non-catalytic specificity manner (Coimbra and Salvador, 2021). As pE-Aβ in humans is mainly catalyzed by sQC rather than gQC in vivo, we will focus on the sQC inhibitors for the treatment of AD in this review.
The catalytic domain of sQC contains Zn2+ and approximately 330 amino acid residues, exhibiting a globular α/β-fold open-sandwich topology that comprises a central six-stranded β-sheet (among which two were antiparallel) surrounded by two and six α-helices on the opposite sides and flanked by two α-helices at one edge of the β-sheet (Huang et al., 2005; Xu et al., 2021). The catalytic domain has a hydrophobic entrance and a relatively narrow binding pocket. The essential Zn2+ is located at the bottom of the active pocket, coordinating with three conservative residues (Asp159, Glu202, and His330) and a water molecule to form a tetrahedral structure, which is necessary for catalysis (Huang et al., 2005). The loop domains near the active center of sQC have certain conformational variabilities that might be affected by N-linked glycosylation (Ruiz-Carrillo et al., 2011); meanwhile, the glycosylation has a limited impact on the overall structure and catalytic activity of sQC but may influence its solubility (Schilling et al., 2002; Ruiz-Carrillo et al., 2011). The crystal structure of hQC also revealed a unique hydrogen-bond network in the active site, formed by five highly conserved residues (Ser160, Glu201, Asp248, Asp305, and His319), within which Glu201 and Asp248 participate in binding to the substrate. When natural substrates or inhibitors enter the catalytic center, the carbonyl of glutamine, glutamate, or other metal binding groups can replace water molecules to coordinate with Zn2+ (Huang et al., 2008; Coimbra and Salvador, 2021), thereby catalyzing or inhibiting the cyclization of glutamine and glutamate of the substrates. The structural features, especially the mono-Zn2+ binding model, offer the most valuable guidance for the design and discovery of QCIs.
3. Development and evolution of QCIs
3.1. Rational design and experiment-based QCIs
A metal-chelating group has been initially considered an essential functional component for the construction of QCIs since the identification of hQC as a Zn2+-dependent metalloenzyme and the discovery that chelators such as imidazole and its analogs have weak QC inhibitory activity (Schilling S. et al., 2003; Schilling S. H. et al., 2003; Demuth et al., 2004a,b). Probiodrug AG (currently Vivoryon Therapeutics N.V.) was a pioneer in developing high-activity QCIs (Demuth et al., 2004a,b; Buchholz et al., 2008), and as early as 2006, the company contributed the foundational literature for the design and discovery of the first high potent imidazole-containing QC inhibitor via mimicking a tripeptide (Gln-Phe-Ala-NH2) substrate (Buchholz et al., 2006). The strategy has been proven to be highly efficient in generating a library of QCIs with Ki ranging from nanomolar to micromolar. In particular, the strongest inhibitor 1 (Table 1A), known as PBD150 or PQ50, had an excellent Ki of 60 nM (Buchholz et al., 2006). It was unexpected that PBD150 was approximately 19-fold more effective toward sQC than gQC, whereas the co-crystallization of PBD150-sQC complex revealed an almost identical binding mode as observed in PBD150-gQC complex, except for the slightly stronger hydrophobic interaction with Ile303 compared with that of Val324 in gQC (Huang et al., 2011). The binding properties of PBD150 to sQC in solution provide additional evidence that the conformation of PBD150 is susceptible to disruption through protein-protein interactions (Koch et al., 2012a). Surprisedly, replacing imidazole in PBD150 with 5-methyl imidazole leads to a stronger inhibitor 2 (Table 1A), with almost a 10-fold increase in the activity compared with PBD150 (Buchholz et al., 2009). However, the inhibitory activity of 3 (Table 1A) decreased to basal level when connecting with the two methoxy groups on the benzene ring (Tran et al., 2013). Subsequent studies regularly employed comparable substrate-mimicking approaches utilizing an alternative Aβ3 − 5 (Glu-Phe-Arg) or used PBD150 and 2 as lead compounds, leading to the identification of inhibitors with shared pharmacophores and an increasingly elucidated structure-activity relationship (SAR).
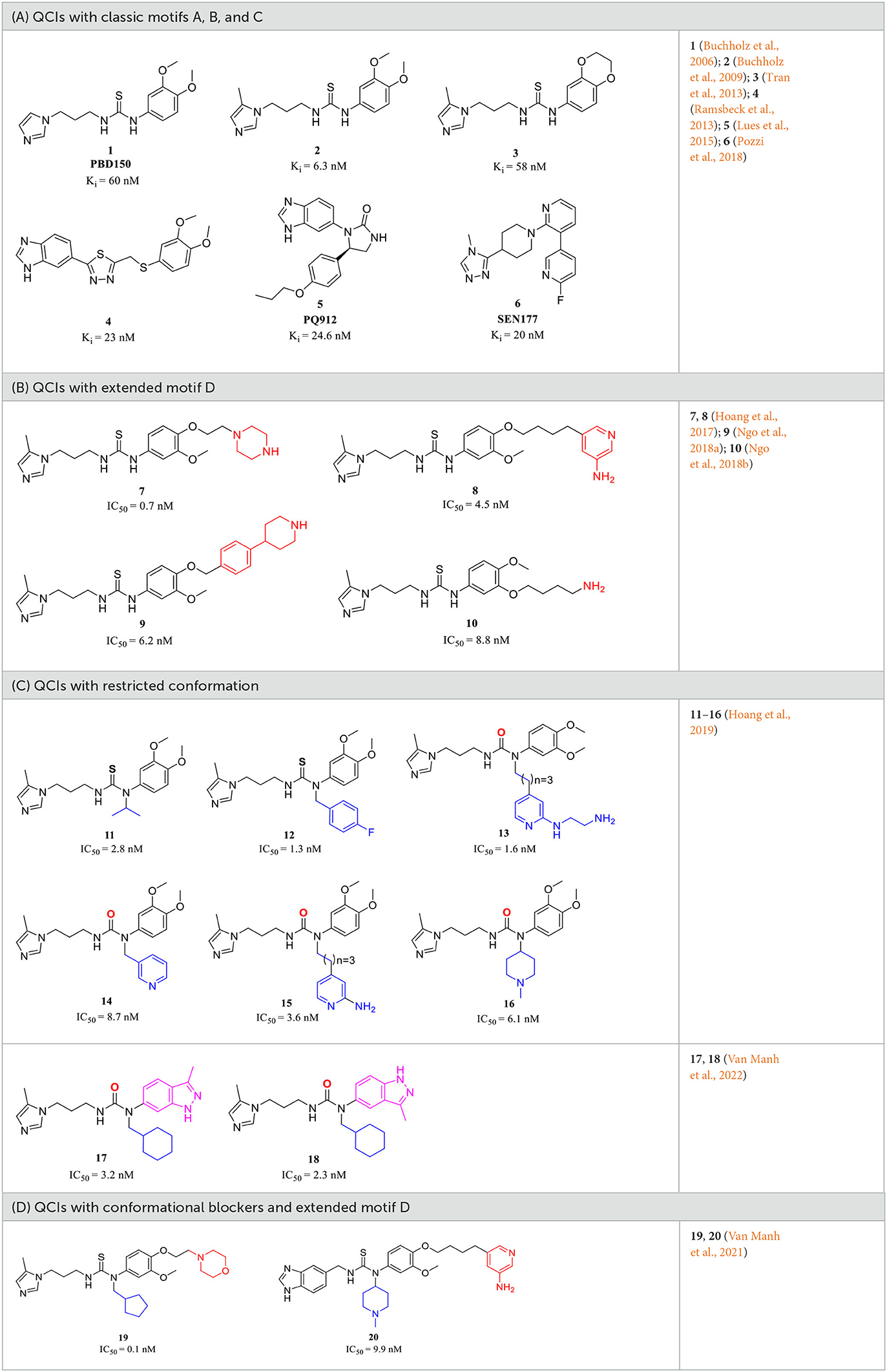
Table 1. Representative design and experiment-based QCIs.
The classic framework of these inhibitors consists of three crucial motifs A, B, and C (Figure 2). The motif A contains a zinc-binding group (ZBG) or general metal-binding group (MBG), with imidazole, benzimidazole, and triazole as the most common structure entities. The imidazole-based ZBG, particularly the 5-methyl imidazole, is commonly the first choice (Kumar et al., 2013), leading to the fact that the imidazole-based inhibitors constitute the vast majority of the total QCIs. Recently, hydrazides were identified as the most potent ZBG compared with other classic Zn-binders (Kupski et al., 2020), which offer another option for designing novel inhibitors. The motif B contains at least a hydrogen bond donor (HBD) or a hydrogen bond acceptor (HBA); it is usually peptide amide analogs such as urea, thiourea, and their derivatives. Both urea and thiourea contributed not only more than one HBD and HBA but also flexible bonding (Tran et al., 2019). The motif C is normally an aromatic ring opposite or close to the ZBG, mimicking the Phe2 residue of Aβ3 − 5, which participates in the π-π interaction with the benzyl side chain of the essential Phe325 of QC. Among these classic inhibitors, PBD150 (Buchholz et al., 2006), 5 (PQ912) (Lues et al., 2015), and 6 (SEN177) (Jimenez-Sanchez et al., 2015) were the most outstanding representatives of imidazole, benzimidazole, and triazole-based QCIs, respectively (Table 1A). These inhibitors exhibit favorable pharmacodynamic profiles and are widely used as positive controls in numerous studies. However, only PQ912 is undergoing clinical trials until now.
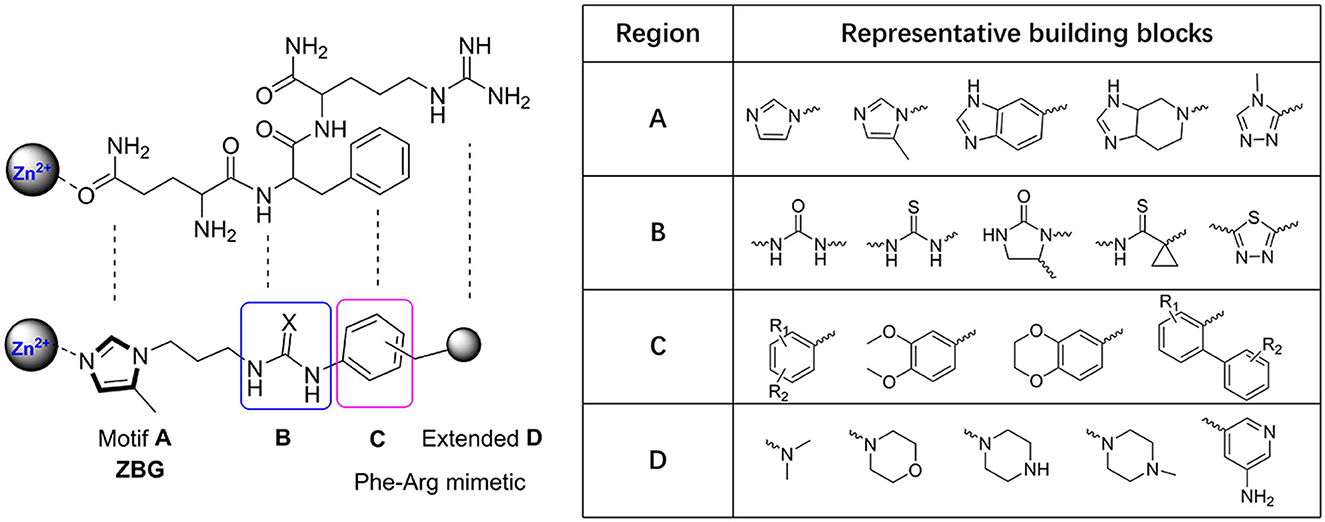
Figure 2. Essential pharmacophores of the classic QC inhibitor and its representative building blocks.
The Phe2 and Arg3 residues of Aβ3 − 5 both are deeply involved in the interaction with QC. Nevertheless, the significant role of the guanidine side chain of Arg3 was underestimated earlier. To further mimic the feature of the guanidine group and to improve the QC inhibitory activity, Jeewoo Lee et al. added a nitrogen-containing heterocyclic group as an extended motif D based on the scaffold of 2 (Hoang et al., 2017). The newly developed inhibitors displayed 5- to 40-fold activity increase compared with 2 (IC50=29.2 nM in this assay). Though 7 (IC50=0.7 nM for hQC, Table 1B) was the most potent candidate even among previously reported inhibitors, it was found to be inactive in an acute ICR mice model to study the in vivo pE3-Aβ40 lowering efficacy. Whereas compound 8 (IC50 = 4.5 nM for hQC, Table 1B) exhibited a prominent efficacy of lowing pE3-Aβ40 by 54.7%, significantly reducing the brain pE3-Aβ42 level of APP/PS1 mice, and restoring the cognitive function of 5×FAD mice. Based on these encouraging results, the authors then systematically studied the SAR of 7 and 8 by modifying the Arg-mimetic motif, leading to the discovery of 9 (IC50 = 6.2 nM, Table 1B) (Ngo et al., 2018a) and 10 (IC50 = 8.8 nM, Table 1B) (Ngo et al., 2018b). Molecular modeling studies demonstrated that all these inhibitors formed extra strong salt bridge interactions with the carboxylate residue of Glu327, supporting the necessity of the extended motif D in high potent QC inhibitor design.
The X-ray structure showed that the PBD150 resided in the active site of hQC with a bent Z-E conformation (Huang et al., 2011; Hoang et al., 2019), while the 2-aminopyridine of extended D in 8 can freely rotate in the active site as revealed by molecular modeling (Hoang et al., 2017), suggesting the possibility of improving binding potency and inhibitory activity by a conformational restriction. Jeewoo Lee et al. creatively incorporated a conformational blocker into the urea or thiourea nitrogen of motif B to induce the formation of bent Z-E conformers (Hoang et al., 2019). The strategy was proven to be effective as well. 24 inhibitors with various rigid blocks showed a significant activity enhancement with in vitro IC50 below 10 nM compared with PBD150 (evaluated IC50 = 29.2 nM in this assay). The 11, 12, and 13 have a remarkably low IC50 value of 2.8 nM, 1.3 nM, and 1.6 nM, respectively, while the in vivo QC inhibition efficacy of these compounds was much weaker than that of 14 (IC50 = 8.7 nM), 15 (IC50=3.6 nM), and 16 (IC50 = 6.1 nM) (Table 1C), which suppressed the generation of pE3-Aβ40 by more than 20% in an acute mouse model compared with a negative control. Among the selected inhibitors, 16 exhibited the most promising in vivo efficacy and druggable profiles, such as liver microsomal stability and up to 50-fold inhibitory selectivity against gQC. The molecular docking further demonstrated that 16 displayed a Z-E conformation at the active site of QC, as anticipated. The N-substituted piperidinyl blocker of 16 not only restricted the conformation but also formed additional hydrophobic interactions with Tyr299, Val302, and Ile303, which may be highly correlated with the high inhibitory activity and QC selectivity. Remarkably, the SAR indicates that the effect of conformational restriction was more marked in the urea series than that of thiourea. In their recent study, the 3,4-dimethoxyphenyl group of the urea series scaffold was replaced by indazole bio-isosteres, which were regarded as more metabolically stable. The representative 17 and 18 (Table 1D), both containing an N-cyclohexylurea blocker, displayed remarkable inhibitory IC50 values of 3.2 and 2.3 nM, respectively (Van Manh et al., 2022).
Inspired by the encouraging results of both mimetic-Arg motif D and conformational restriction strategies on the classic QC scaffold. A combination of the two approaches was rationally performed, leading to the discovery of 19 (Table 1D), the most potent QC inhibitor reported even to date, with a sub-nanomolar IC50 value of 0.1 nM and up to 290-fold inhibitory enhancement compared with PQ912. While similar to the denouement of 12, another weaker benzimidazole inhibitor 20 (IC50 = 9.9 nM, Table 1D) showed the most promising in vivo efficacy and selective profile with respect to its 21.5-fold sQC selectivity index toward gQC. Besides, 20 also has low toxicity and favorable pharmacokinetic properties, and it significantly improved the alternation behavior of mice in Y-maze tests as well.
Among these QCIs with high scaffold similarity, the common large polar groups such as urea and thiourea reduce the blood-brain barrier (BBB) permeability of the compounds, which may be the most likely reasons for the moderate or even inactive in vivo efficacy of the high potent inhibitors PBD150 (Brooks et al., 2015), 7, 11, 12, 19, etc. To ameliorate BBB permeability, Wu et al. tried to introduce a more hydrophobic biphenyl group in motif C to enhance molecular lipo-solubility as well as π-π stacking interaction and abandon the urea group in motif B (Li et al., 2017). The obtained 21 (Table 2A) exhibited potent inhibitory activity and significantly improved BBB permeability. Further assessments corroborated that 21 dramatically reduced the pE-Aβs level in cultured cells and in vivo and improved the behavior of B6C3-Tg AD mice. Interestingly, contrary to the commonly reported relationship, the SAR of DPCIs showed that 4-methyl substitution was better than that of 5-methyl substitution in imidazole. Although the activity of 21 was significantly decreased compared with the lead compound PBD150 due to the loss of the classic urea motif, the acquisition of SAR and the simple synthesis route of DPCIs still made it an ideal lead scaffold for further structural optimization.
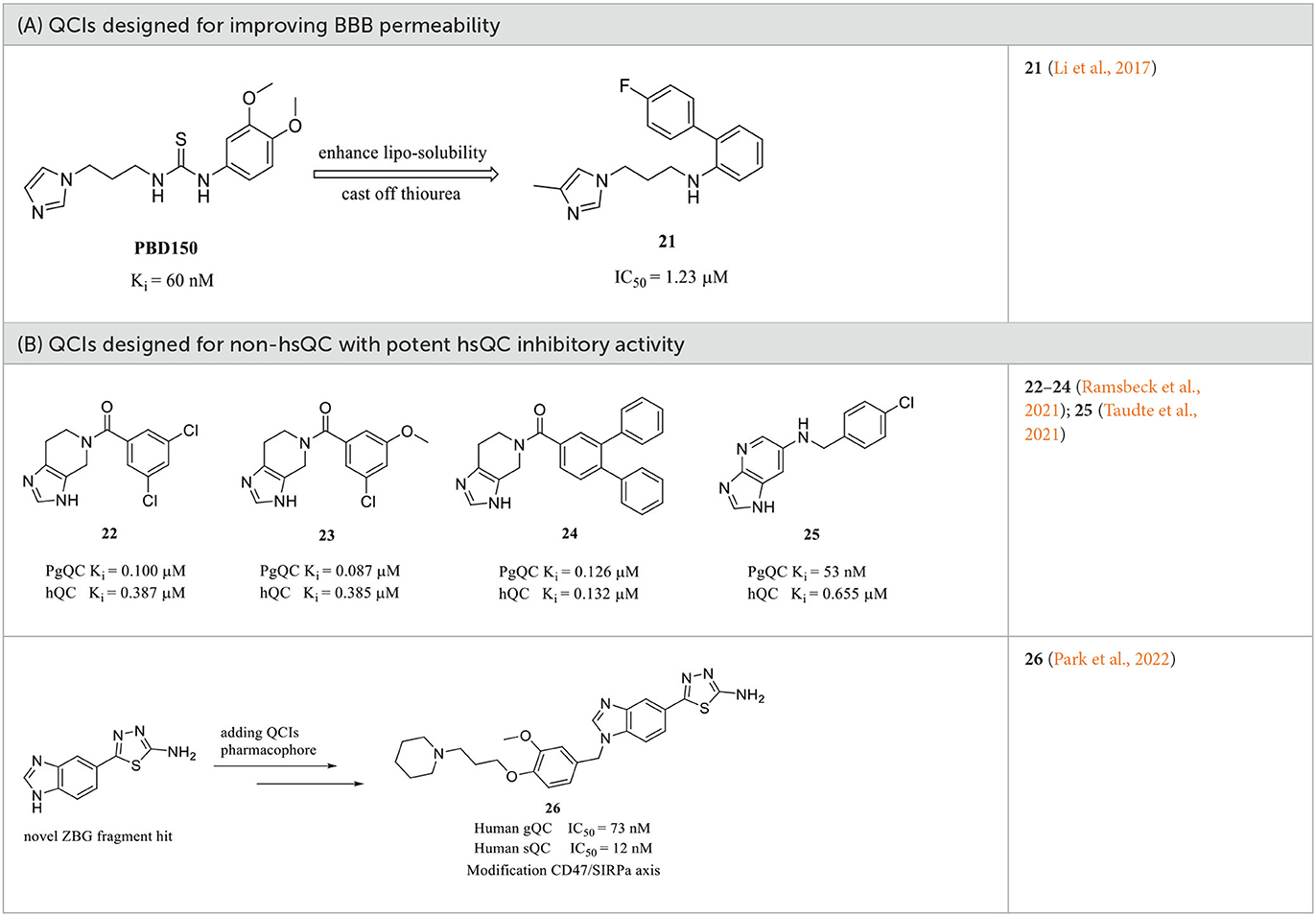
Table 2. Other representative QCIs.
3.2. Representative QCIs designed for non-hsQC with potent hsQC inhibitory activity
Although the structural differences in both the overall conformations and active cores are essential and convenient for the design of type I/II QC-specific inhibitors, some bacterial QCs, such as porphyromonas gingivalis QC (PgQC), show similar structures and enzymatic features to hQC and were thus characterized as Mammalian-like type II QCs (Lamers et al., 2021). Therefore, inhibitors designed for bacterial QCs may also bind to and suppress hQC activity. For example, PgQC inhibitors 22–24 (Table 2B) designed for the treatment of periodontitis are almost equally potent in suppressing hQC activity in vitro (Lamers et al., 2021; Ramsbeck et al., 2021). Meanwhile, another inhibitor 25 with imidazo[4,5-b]pyridin scaffold exhibited a significantly improved selectivity (>12) over PgQC (Taudte et al., 2021).
Human gQC was recently recognized as an important modulator of the CD47-SIRPα pathway via promoting pGlu formation on the N-terminus of CD47 (Logtenberg et al., 2019). The gQC blockade contributes to reducing the “do not eat me” immune signals of CD47 on tumor cells. Therefore, developing gQC inhibitors is regarded as a novel and promising strategy for cancer immunotherapy. In a most recent study, a novel ZBG (1H-benzimidazol-5-yl)-1,3,4-thiadiazol-2-amine was hit by a fragment identified through library screening, and an aromatic ring and alkylamine were further added as additional QC pharmacophores. The most potent gQC inhibitor 26 (Table 2B) showed an outstanding IC50 of 73 nM, while unluckily it has 6-fold stronger activity against sQC with an IC50 of 12 nM (Park et al., 2022), which further indicates that more attentions should be paid to the selectivity of inhibitors when developing QCIs for the treatment of AD.
3.3. Virtual screening-based QCIs
In addition to the various rational design and experimental-based QCI discoveries, virtual screening offers another efficient tool to advance the understanding of activity profiles, and the development of new QCIs (Kumar et al., 2013; Lin et al., 2019). Those screening strategies include fragment-based screening (Szaszko et al., 2017), QSAR modeling (Al-Attraqchi and Venugopala, 2020; Kumar et al., 2021), and pharmacophore-assisted high-throughput virtual screening (Lin et al., 2019). Katharigatta N. Venugopala et al. developed linear and non-linear 2D QSAR models and a partial least squares-based 3D model to help predict the activity of not yet synthesized compounds. Combined with ADME filtering and 2D-similarity search, potential QCIs 27–29 (Table 3) were identified from the ZINC database (Al-Attraqchi and Venugopala, 2020). Similarly, Ashwani Kumar et al. identified the structural features that are both positively and negatively responsible for the QC inhibitory activity based on a dataset of 125 QCIs for QSAR analysis via Monte Carlo modeling studies. The QSAR further supports the importance of 5-methy substituted imidazole and alkyl-substituted benzene in activity enhancement, as previous SAR revealed, and novel compounds 30–32 (Table 3) were then computationally designed and showed improved pKi and QC binding affinities (Kumar et al., 2021). The hits of the two studies actually inherited typical features of classic QCIs with imidazole or methyl-imidazole as ZBG and an aromatic group located in the opposite position, while the QC inhibitory activities were not experimentally evaluated and validated in vitro.
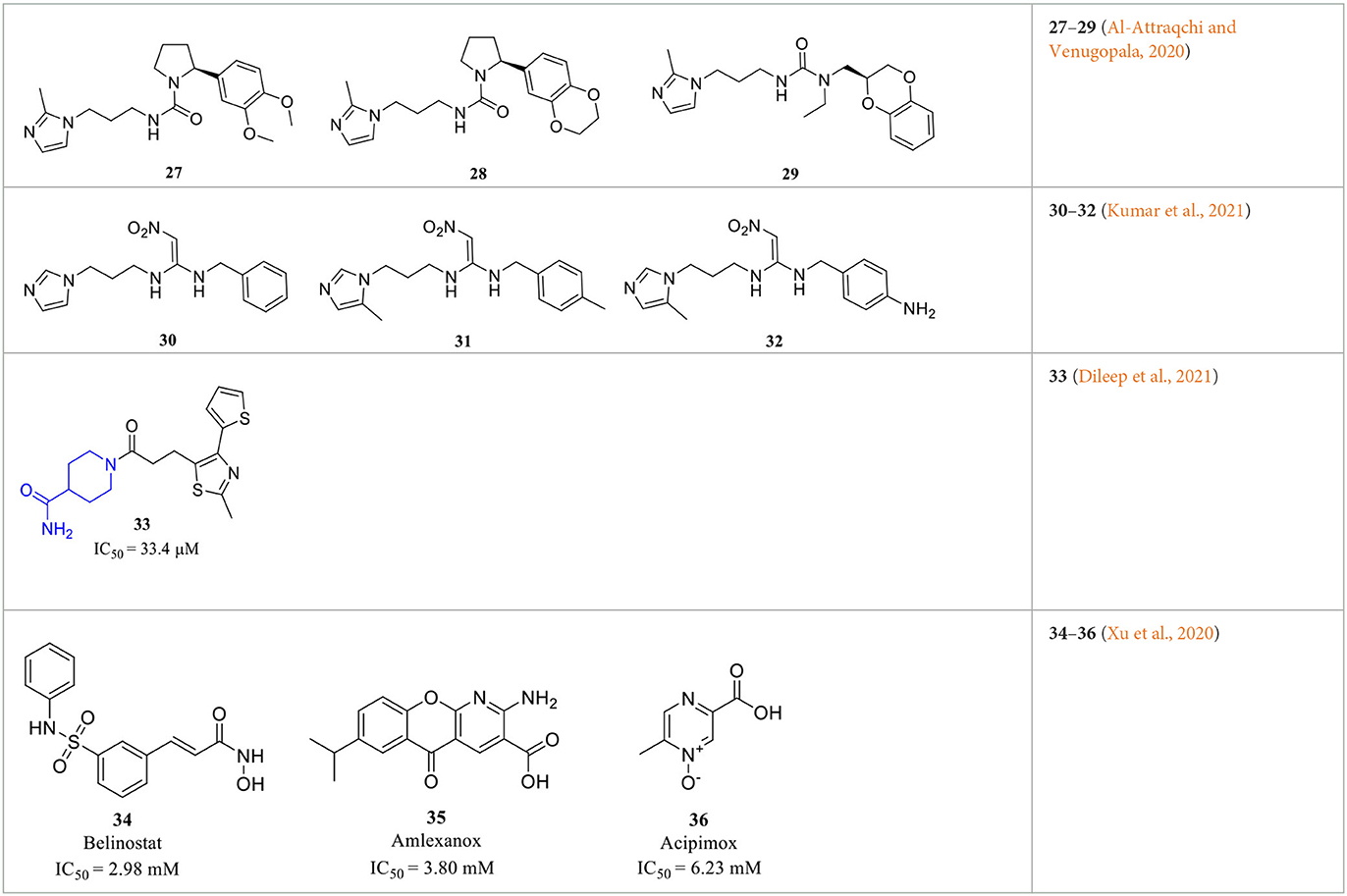
Table 3. Representative screening-based QCIs.
Combining activity evaluation with virtual screening will provide more convincing evidence. Kam Y. J. Zhang et al. reported a QC inhibitor 33 (Table 3) with a novel MBG moiety, peperidine-4-carboxamide, through a pharmacophore-assisted high-throughput virtual screening (Dileep et al., 2021). 33 showed moderate activity against QC with IC50 = 33.4 ± 5.1 μM, and docking, MD simulation, and crystallographic studies suggested that 33 anchors to the active site via a coordinate bond with Zn2+ located deeply in the active site cleft of the QC, while it lacks stacking interactions with Tyr299, Phe325, and Trp329, which are assumed to be critical for QC activity.
Wu et al. performed a less efficient but simple and direct approach for new scaffold QCIs discovery by repurposing FDA-approved drugs (Xu et al., 2020). Such a repurposing strategy is more likely to succeed since the drugs have been fully evaluated in both pre-clinical and clinical trials. The QC inhibitory evaluation of 1,621 drugs was performed at a concentration of 10 μM in vitro, and the top five compounds were highlighted with reasonable activity. Although the inhibitory activity was relatively weak with IC50 values at the millimolar level, only two drugs contained the imidazole group; the other three drugs 34–36 (Belinostat, Amlexanox, and Acipimox; Table 3) have completely different structures from the classic QCIs model, which may still offer insights for the design and discovery of novel QCIs with new structural features.
3.4. Natural product-based QCIs
Imidazole- and benzimidazole-based ZBGs have so far been the first choice for the design of QC inhibitors. Nevertheless, these ZBG groups are less selective and are likely to interact with various metalloproteins in vivo and thus increase the risk of side effects (Park et al., 2022). Natural products are an important resource for the discovery of new activity scaffolds, which may offer opportunities to overcome the potential drawbacks of classic QCIs with new pharmacophores (Table 4). The oleuropein aglycone (OLE, 37, Table 4), a natural phenol (secoiridoid) abundant in extra virgin olive oil, was found to be protective both in memory and behavioral performance of young and middle-aged TgCRND8 mice (Grossi et al., 2013). In an extended study of aged TgCRND8 mice showing increased pE3-Aβ42 deposits in the brain, OLE could also retard the growth of pE3-Aβ42 aggregates even in advanced and late stages of Aβ deposition (Luccarini et al., 2015). Several reviews summarized OLE as a QC inhibitor, while OLE certainly showed weak inhibitory activity at a concentration of 10 μM. Immunofluorescence staining and immunoblot analysis demonstrated that QC levels were significantly reduced in the brains of the OLE-fed Tg mice, suggesting that OLE is active against pE-Aβ generation by reducing QC expression rather than direct inhibition (Luccarini et al., 2015).
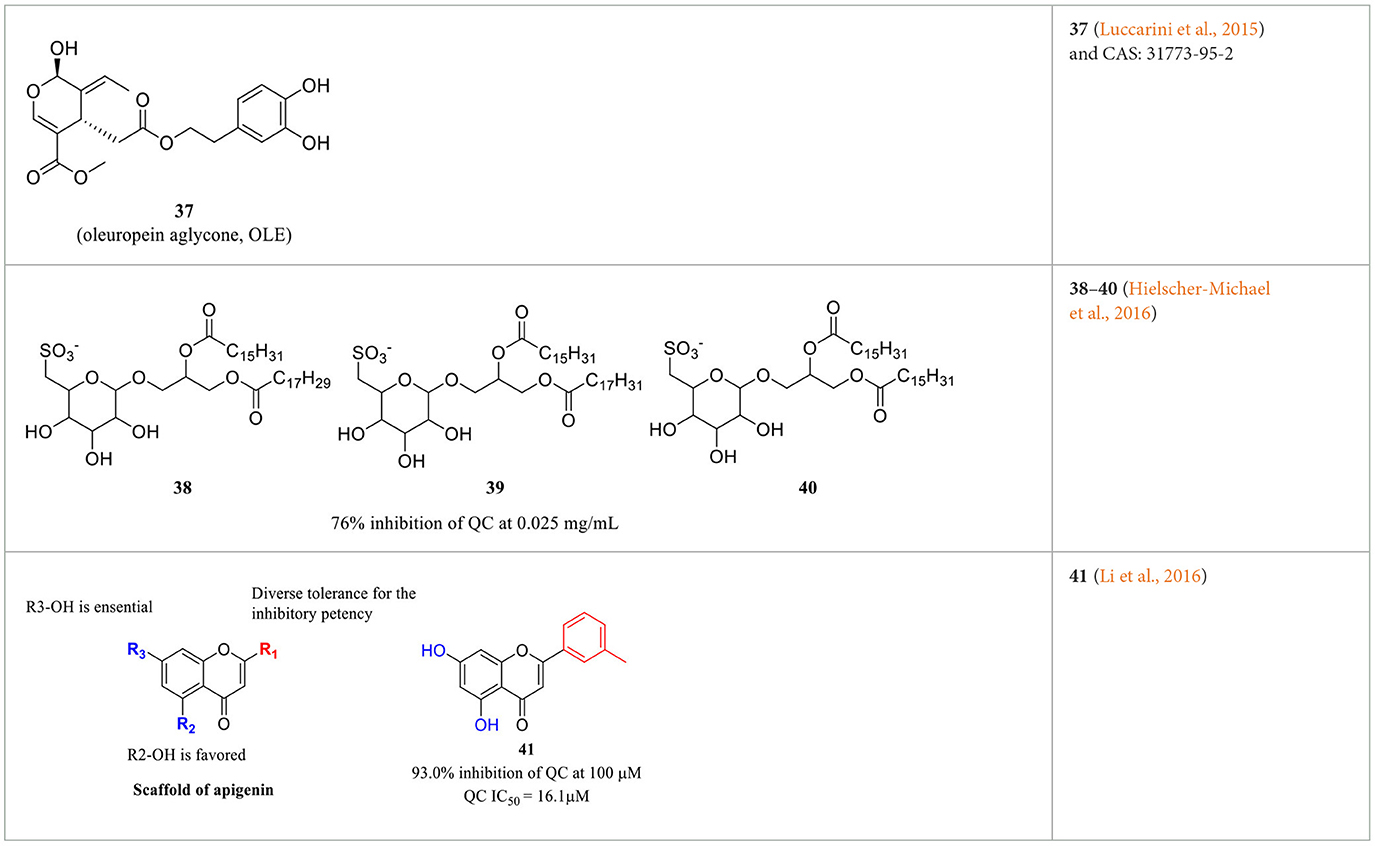
Table 4. Natural compounds with potential QC inhibitory activity.
Some algal extracts were preliminary reported to have positive effects on QC inhibition, while the bioactive chemical entities were not isolated and clearly identified by traditional methods, Wessjohann et al. identified three sulfolipid QC inhibitors (38–40, Table 4) from microalgae using a new “Reverse Metabolomics” technique including an activity-correlation analysis. The sulfolipids showed a noteworthy QC inhibition of 76% at a low concentration of 0.025 mg/ml, and the authors proposed that sulfolipids provide similar pharmacophore characteristics to PBD150, in which the negative sulfonate group and the polyhydroxy elements probably act as a ZBG and the glucose as the core scaffold. Interestingly, SODG (structure not shown), a lipid product used as a standard reference in the assay, was first shown to exhibit quite similar QC inhibition activity compared with sulfolipids (Hielscher-Michael et al., 2016). Unlike traditional natural product screening approaches, Wu et al. explored apigenin-based QCI discovery via chemical modification. A total of 40 apigenin derivatives belonging to the phenol-4′(R1), C5-OH(R2), and C7-OH(R3) modified series were synthesized and evaluated. The compound 41 (Table 4) has remarkable inhibitory potency with an IC50 value of 16.1±2 μM, and the SAR study indicated that the C7-OH was required for binding with Zn2+ and that the C5-OH was favored, whereas phenol-4′ was tolerant for the inhibitory activity. The essential role of C7-OH was further supported by the binding interaction with conservative Zn2+ via molecular docking. Although the activity of apigenin derivates was relatively weak compared with nanomolar level classic QCIs, the non-imidazole ZBG, acquisition of SAR, and simple synthesis route made it a potential lead scaffold for further optimization (Li et al., 2016).
4. QC inhibitor undergoing clinical trials
Hundreds of QC inhibitors have been revealed in the literature, and reports of novel, high-potency QC inhibitors have dramatically increased in recent years with the structure-activity relationships becoming clear, while only PQ912 is currently undergoing clinical trials in human subjects for AD treatment. PQ912 has a scaffold slightly different from the common classic QCIs (Table 1A). It is a heterocyclic competitive inhibitor with benzimidazole as the ZBG at position 1 of the imidazolidine-2-one. PQ912 has strong human, rat, and mouse QC inhibitory activity with Ki values ranging between 20 and 65 nM (Hoffmann et al., 2017). Preclinical studies revealed that PQ912 has an attractive drug-like profile and robust pharmacological therapeutic effects, both in vitro and in vivo (Hoffmann et al., 2017). PQ912 was considered safe and well tolerated with dose-proportional pharmacokinetics up to doses of 200 mg in the first-in-man phase 1 study (Lues et al., 2015). In the subsequent randomized, double-blind, placebo-controlled phase IIa trial (NCT 02389413), the safety, tolerability, and efficacy of higher doses of PQ912 (800 mg twice daily for 12 weeks) were carefully evaluated in biomarkers confirmed early AD patients (n = 120). PQ912 showed an acceptable safety and tolerability profile in a treatment regime of lower doses and slower titration (Scheltens et al., 2018). The treatment of PQ912 resulted in an average QC target occupancy of >90% in cerebrospinal fluid (CSF), an improvement of working memory, and a reduction of synaptotoxicity and neurogranin levels, as well as improvements in some other experimental endpoints (Scheltens et al., 2018). However, no PQ912 treatment effects were found on the composite scores of episodic memory, executive function, attention, or overall cognition (Scheltens et al., 2018). The results of the phase IIa study might indicate early beneficial effects of PQ912 on cognition by preventing the synaptotoxicity of pE-Aβ in the central nervous system and thus rescuing impaired synaptic functions. While episodic memory is the most impaired area of function in AD pathology, a relatively short intervention (12-week time period) by PQ912 was actually not expected to have an obvious clinical influence on episodic and long-term memory. To further test the efficacy (particularly on cognition and brain activity) of PQ912 as a disease modifier, a phase IIb program in participants with MCI and mild AD-VIVIAD was recently launched, and results are expected early in 2023 (Vijverberg et al., 2021).
5. New trends toward QCIs
Theoretically, QC inhibition can significantly decrease the formation of pE-Aβ while having little influence on the clearance of full-length Aβ and the existing pE-Aβ, which may still induce Aβ cascades and lead to the deposition of senile plaques. Hence, QC inhibition-based combination therapy and multi-target-directed ligands (MTDL) have drawn considerable attentions in recent years. The combination effects of PQ912 and a pE-Aβ specific antibody m6 on the formation and clearance of pE-Aβ in an AD mouse model were evaluated. The study showed that combination treatments resulted in significant reductions of total Aβ by 45–65% in the brain of AD mouse overexpressing both human amyloid precursor protein containing the Swedish and London mutations and human QC (hAPPsl × hQC), while single treatments at subtherapeutic levels only showed a moderate (16–41%) but statistically insignificant reduction in Aβ level. The additive effects of the combination of PQ912 and m6 on brain Aβ pathology were evaluated using a bliss independence model, and a combination index of ≈1 was determined (Hoffmann et al., 2021). The combination strategy may achieve a better therapeutic effect than a single treatment, even at a reduced dose for the individual drug.
Instead of drug combination treatment, Wu et al. in their recent study developed a new class of maleimide-DPCI hybrid QC/GSK-3β dual inhibitors by rationally combining the essential pharmacophores of QC and GSK-3β inhibitors. GSK-3β, namely, glycogen synthase kinase-3, is regarded as a critical pivotal kinase and a high-potential anti-AD target that links both Aβ and tau pathologies of AD. The most potent compounds 42–44 (Table 5) exhibited slightly enhanced QC inhibitory activity and similar GSK-3β inhibitory activity compared with individual control compounds DPCI-2 and SB-415286, respectively. The selected dual-target inhibitor 42 can dramatically reduce pE-Aβ accumulation and Tau hyperphosphorylation in the brains of 3 × Tg-AD mice. In addition, 42 also effectively attenuates cognitive deficits and decreases anxiety-like behavior in 3 × Tg mouse (Xie et al., 2023).

Table 5. Dual-target QCIs.
6. Perspective and conclusion
pE3-Aβ represents a highly desirable and abundant target due to its distinctive aggregation properties and neurotoxicity. As QC plays a key role in the conversion and formation of pE3-Aβ, QC inhibition is emerging as an alternative promising strategy apart from expensive active immune clearance to decrease the pathological toxicity of pE3-Aβ for the treatment of AD. Over the past two decades, a number of different scaffold QCIs have been designed and discovered, and the efficacy of several high potent inhibitors has been evaluated both in vitro and in vivo using different AD mice models. Moreover, PQ912 is regarded as the proof-of-concept validation of the QC target. However, there are still issues to be considered before QC inhibitors can effectively translate from bench to bedside.
First, the specificity of QC inhibitors, which includes the selective inhibition of hQC among various metalloproteins in vivo and selectivity toward the QC-Aβ pathway rather than other QC normal PTMs processes. Because QC is abundant in mammalian neuroendocrine tissues and is responsible for the maturation of numerous hormones and cytokines, non-selective QC inhibition may lead to wide and unpredictable side effects. In the phase IIa trial of PQ912, one-third (20/60) of subjects discontinued PQ912 treatment due to adverse events related to gastrointestinal disorders, skin disorders, etc. A broader battery of CSF biomarkers, including growth-associated protein 43 and pE-CCL2, have thus been set as exploratory endpoints to better monitor the potential adverse effects of the treatment in AD patients. Second, the pathological reversing effects of QC enzyme inhibitors remain questionable. Given that pE-Aβ acts as seeds to induce the formation of stable toxic heterogeneity polymers, QC inhibition can suppress but not completely prevent the pE-Aβ formation. The existing pE-Aβ may still trigger Aβ cascades. Fortunately, the QC-based combination strategy and MTDL strategy are drawing attention lately, which might be an ideal solution to enhance the additive effects. Last but not least, the pE-Aβ cascade is essentially an optimization of the original Aβ cascade hypothesis, in which the pE-Aβ replaces full-length Aβ serving as the core initiator in the progression of AD. In the context of endless clinical failures of Aβ-directed or related interventions and the controversy regarding the exact role of Aβ cascade in the pathogenesis of AD, how far will QC inhibition go remains blurred in clinical trials. Nevertheless, the encouraging effect of the pE-Aβ-targeting donanemab in clinical trials envisions the promising role of the pE-Aβ-related key protein QC as an alternative potential target for the novel disease-modifying treatment of AD.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was financially supported by the National Natural Science Foundation of China (No. 22007107), the Science and Technology Foundation of Guizhou Province (Qian Ke He J ZK [2022]585), the Science and Technology Fund Project of Guizhou Provincial Health Commission (gzwjkj2020-1-207), the Zhuhai Basic and Applied Basic Research Foundation (ZH22017003210074PWC), the Talent Project of Guizhou Science and Technology Cooperation Platform [2017]5733-041, and the Future Science and Technology Elite Project of Zunyi Medical University (ZYSE-2021-02).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Attraqchi, O. H. A., and Venugopala, K. N. (2020). 2D- and 3D-QSAR modeling of imidazole-based glutaminyl cyclase inhibitors. Curr. Comput. Aided Drug Des. 16, 682–697. doi: 10.2174/1573409915666190918150136
Association, A. (2023). 2023 Alzheimer's disease facts and figures. Alzheimers Dement. 19, 1598–1695. doi: 10.1002/alz.13016
Bayer, T. A. (2022). Pyroglutamate Abeta cascade as drug target in Alzheimer's disease. Mol. Psychiatry 27, 1880–1885. doi: 10.1038/s41380-021-01409-2
Becker, A., Eichentopf, R., Sedlmeier, R., Waniek, A., Cy nis, H., Koch, B., et al. (2016). IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol. Chem. 397, 45–55. doi: 10.1515/hsz-2015-0192
Brooks, A. F., Jackson, I. M., Shao, X., Kropog, G. W., Sherman, P., Quesada, C. A., et al. (2015). Synthesis and evaluation of [(11)C]PBD150, a radiolabeled glutaminyl cyclase inhibitor for the potential detection of Alzheimer's disease prior to amyloid beta aggregation. Medchemcomm 6, 1065–1068. doi: 10.1039/C5MD00148J
Buchholz, M., Hamann, A., Aust, S., Brandt, W., Bö hme, L., Hoffmann, T., et al. (2009). Inhibitors for human glutaminyl cyclase by structure based design and bioisosteric replacement. J. Med. Chem. 52, 7069–7080. doi: 10.1021/jm900969p
Buchholz, M., Heiser, U., Schilling, S., Niestroj, A. J., Zunkel, K., and Demuth, H. U. (2006). The first potent inhibitors for human glutaminyl cyclase: synthesis and structure-activity relationship. J. Med. Chem. 49, 664–677. doi: 10.1021/jm050756e
Buchholz, M., Heiser, U., and Sommer, R. (2008). Inhibitors of Glutaminyl Cyclase. Application filed by Probiodrug AG 2008-04-18, Application granted, 2012-05-29, Publication of US8188094B2.
Carrillo, D. R., Parthier, C., Jänckel, N., Grandke, J., St elter, M., Schilling, S., et al. (2010). Kinetic and structural characterization of bacterial glutaminyl cyclases from Zymomonas mobilis and Myxococcus xanthus. Biol. Chem. 391, 1419–1428. doi: 10.1515/bc.2010.130
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Coimbra, J. R., Sobral, P. J., Santos, A. E., Moreira, P. I., and Salvador, J. A. (2019). An overview of glutaminyl cyclase inhibitors for Alzheimer's disease. Future Med. Chem. 11, 3179–3194. doi: 10.4155/fmc-2019-0163
Coimbra, J. R. M., and Salvador, J. A. R. (2021). A patent review of glutaminyl cyclase inhibitors (2004-present). Expert Opin. Ther. Pat. 31, 809–836. doi: 10.1080/13543776.2021.1917549
Cynis, H., Hoffmann, T., Friedrich, D., Kehlen, A., G ans, K., Kleinschmidt, M., et al. (2011). The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. EMBO Mol. Med. 3, 545–558. doi: 10.1002/emmm.201100158
Cynis, H., Scheel, E., Saido, T. C., Schilling, S., and Demuth, H. U. (2008). Amyloidogenic processing of amyloid precursor protein: evidence of a pivotal role of glutaminyl cyclase in generation of pyroglutamate-modified amyloid-beta. Biochemistry 47, 7405–7413. doi: 10.1021/bi800250p
Dammers, C., Reiss, K., Gremer, L., Lecher, J., Zi ehm, T., Stoldt, M., et al. (2017a). Pyroglutamate-modified amyloid-beta(3-42) shows alpha-helical intermediates before amyloid formation. Biophys. J. 112, 1621–1633. doi: 10.1016/j.bpj.2017.03.007
Dammers, C., Schwarten, M., Buell, A. K., and Willbold, D. (2017b). Pyroglutamate-modified Abeta(3-42) affects aggregation kinetics of Abeta(1-42) by accelerating primary and secondary pathways. Chem. Sci. 8, 4996–5004. doi: 10.1039/C6SC04797A
Demuth, H. U., Heiser, U., Buchholz, M., Niestroj, A. J, and Schilling, S. (2004a). Inhibitors of Glutaminyl Cyclase, WO2004098591, PCT/EP2004/004773-Priority Data 60/468,014.
Demuth, H. U., Hoffmann, T., Niestroj, A. J., Schilling, S., and Heiser, U. (2004b). Use of Inhibitors of Glutaminyl Cyclases for Treatment and Prevention of Disease. 2004-05-05 Priority to US10/839,017, Prior Publication Data: US 2005/0058635 A1-2008-06-03 Application granted. Patent No.: US 7,381,537 B2.
Dhillon, S. (2021). Aducanumab: first approval. Drugs 81, 1437–1443. doi: 10.1007/s40265-021-01569-z
Dileep, K. V., Sakai, N., Ihara, K., Kato-Murayama, M., N akata, A., Ito, A., et al. (2021). Piperidine-4-carboxamide as a new scaffold for designing secretory glutaminyl cyclase inhibitors. Int. J. Biol. Macromol. 170, 415–423. doi: 10.1016/j.ijbiomac.2020.12.118
Gontsarova, A., Kaufmann, E., Tumani, H., Dressel, A., Man del, F., Wiesmuller, K. H., et al. (2008). Glutaminyl cyclase activity is a characteristic feature of human cerebrospinal fluid. Clin. Chim. Acta 389, 152–159. doi: 10.1016/j.cca.2007.12.010
Grochowska, K. M., Yuanxiang, P., Bar, J., Raman, R., B rugal, G., Sahu, G., et al. (2017). Posttranslational modification impact on the mechanism by which amyloid-beta induces synaptic dysfunction. EMBO Rep. 18, 962–981. doi: 10.15252/embr.201643519
Grossi, C., Rigacci, S., Ambrosini, S., Dami, T., Luccarini, I., Traini, C., et al. (2013). The polyphenol oleuropein aglycone protects TgCRND8 mice against Ass plaque pathology. PLoS ONE. 8, e71702. doi: 10.1371/journal.pone.0071702
Gunn, A. P., Masters, C. L., and Cherny, R. A. (2010). Pyroglutamate-abeta: role in the natural history of Alzheimer's disease. Int. J. Biochem. Cell Biol. 42, 1915–1918. doi: 10.1016/j.biocel.2010.08.015
Gunn, A. P., Wong, B. X., Johanssen, T., Griffith, J. C., M asters, C. L., Bush, A. I., et al. (2016). Amyloid-beta peptide Abeta3pE-42 induces lipid peroxidation, membrane permeabilization, and calcium influx in neurons. J. Biol. Chem. 291, 6134–6145. doi: 10.1074/jbc.M115.655183
Gunn, A. P., Wong, B. X., McLean, C., Fowler, C., Bar nard, P. J., Duce, J. A., et al. (2021). Increased glutaminyl cyclase activity in brains of Alzheimer's disease individuals. J. Neurochem. 156, 979–987. doi: 10.1111/jnc.15114
Harigaya, Y., Saido, T. C., Eckman, C. B., Prada, C. M., Shoji, M., and Younkin, S. G. (2000). Amyloid beta protein starting pyroglutamate at position 3 is a major component of the amyloid deposits in the Alzheimer's disease brain. Biochem. Biophys. Res. Commun. 276, 422–427. doi: 10.1006/bbrc.2000.3490
Hartlage-Rübsamen, M., Bluhm, A., Piechotta, A., Linnert, M., Rahfeld, J. U., Demuth, H. U., et al. (2018). Immunohistochemical evidence from APP-transgenic mice for glutaminyl cyclase as drug target to diminish pE-abeta formation. Molecules 23, 4. doi: 10.3390/molecules23040924
Hielscher-Michael, S., Griehl, C., Buchholz, M., Demuth, H. U., Arnold, N., and Wessjohann, L. A. (2016). Natural products from microalgae with potential against Alzheimer's disease: sulfolipids are potent glutaminyl cyclase inhibitors. Mar. Drugs 14(11). doi: 10.3390/md14110203
Hoang, V. H., Ngo, V. T. H., Cui, M., Manh, N. V., Tran, P. T., Ann, J., et al. (2019). Discovery of conformationally restricted human glutaminyl cyclase inhibitors as potent anti-Alzheimer's agents by structure-based design. J. Med. Chem. 62, 8011–8027. doi: 10.1021/acs.jmedchem.9b00751
Hoang, V. H., Tran, P. T., Cui, M., Ngo, V. T., Ann, J., Park, J., et al. (2017). Discovery of potent human glutaminyl cyclase inhibitors as anti-Alzheimer's agents based on rational design. J. Med. Chem. 60, 2573–2590. doi: 10.1021/acs.jmedchem.7b00098
Hoffmann, T., Meyer, A., Heiser, U., Kurat, S., Bö hme, L., Kleinschmidt, M., et al. (2017). Glutaminyl cyclase inhibitor PQ912 improves cognition in mouse models of Alzheimer's disease-studies on relation to effective target occupancy. J. Pharmacol. Exp. Ther. 362, 119–130. doi: 10.1124/jpet.117.240614
Hoffmann, T., Rahfeld, J. U., Schenk, M., Ponath, F., Ma kioka, K., Hutter-Paier, B., et al. (2021). Combination of the glutaminyl cyclase inhibitor PQ912 (Varoglutamstat) and the murine monoclonal antibody PBD-C06 (m6) shows additive effects on brain abeta pathology in transgenic mice. Int. J. Mol. Sci. 22, 21. doi: 10.3390/ijms222111791
Huang, K. F., Liaw, S. S., Huang, W. L., Chia, C. Y., Lo, Y. C., Chen, Y. L., et al. (2011). Structures of human Golgi-resident glutaminyl cyclase and its complexes with inhibitors reveal a large loop movement upon inhibitor binding. J. Biol. Chem. 286, 12439–12449. doi: 10.1074/jbc.M110.208595
Huang, K. F., Liu, Y. L., Cheng, W. J., Ko, T. P., and Wang, A. H. (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. Proc. Natl. Acad. Sci. USA. 102, 13117–13122. doi: 10.1073/pnas.0504184102
Huang, K. F., Wang, Y. R., Chang, E. C., Chou, T. L., and Wang, A. H. (2008). A conserved hydrogen-bond network in the catalytic centre of animal glutaminyl cyclases is critical for catalysis. Biochem. J. 411, 181–190. doi: 10.1042/BJ20071073
Jawhar, S., Wirths, O., and Bayer, T. A. (2011a). Pyroglutamate amyloid-beta (Abeta): a hatchet man in Alzheimer disease. J. Biol. Chem. 286, 38825–38832. doi: 10.1074/jbc.R111.288308
Jawhar, S., Wirths, O., Schilling, S., Graubner, S., Demuth, H. U., and Bayer, T. A. (2011b). Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J. Biol. Chem. 286, 4454–4460. doi: 10.1074/jbc.M110.185819
Jimenez-Sanchez, M., Lam, W., Hannus, M., Sonnichsen, B., Imari sio, S., Fleming, A., et al. (2015). siRNA screen identifies QPCT as a druggable target for Huntington's disease. Nat. Chem. Biol. 11, 347–354. doi: 10.1038/nchembio.1790
Koch, B., Buchholz, M., Wermann, M., Heiser, U., Schilling, S., and Demuth, H. U. (2012a). Probing secondary glutaminyl cyclase (QC) inhibitor interactions applying an in silico-modeling/site-directed mutagenesis approach: implications for drug development. Chem. Biol. Drug Des. 80, 937–946. doi: 10.1111/cbdd.12046
Koch, B., Kolenko, P., Buchholz, M., Carrillo, D. R., Par thier, C., Wermann, M., et al. (2012b). Crystal structures of glutaminyl cyclases (QCs) from Drosophila melanogaster reveal active site conservation between insect and mammalian QCs. Biochemistry 51, 7383–7392. doi: 10.1021/bi300687g
Kumar, A., Bagri, K., Nimbhal, M., and Kumar, P. (2021). In silico exploration of the fingerprints triggering modulation of glutaminyl cyclase inhibition for the treatment of Alzheimer's disease using SMILES based attributes in Monte Carlo optimization. J. Biomol. Struct. Dyn. 39, 7181–7193. doi: 10.1080/07391102.2020.1806111
Kumar, V., Gupta, M. K., Singh, G., and Prabhakar, Y. S. (2013). CP-MLR/PLS directed QSAR study on the glutaminyl cyclase inhibitory activity of imidazoles: rationales to advance the understanding of activity profile. J. Enzyme Inhib. Med. Chem. 28, 515–522. doi: 10.3109/14756366.2011.654111
Kupski, O., Funk, L. M., Sautner, V., Seifert, F., Worbs, B., Ramsbeck, D., et al. (2020). Hydrazides are potent transition-state analogues for glutaminyl cyclase implicated in the pathogenesis of Alzheimer's disease. Biochemistry 59, 2585–2591. doi: 10.1021/acs.biochem.0c00337
Lamers, S., Feng, Q., Cheng, Y., Yu, S., Sun, B., Lukman, M., et al. (2021). Structural and kinetic characterization of Porphyromonas gingivalis glutaminyl cyclase. Biol. Chem. 402, 759–768. doi: 10.1515/hsz-2020-0298
Lee, J., Gillman, A. L., Jang, H., Ramachandran, S., Kag an, B. L., Nussinov, R., et al. (2014). Role of the fast kinetics of pyroglutamate-modified amyloid-beta oligomers in membrane binding and membrane permeability. Biochemistry 53, 4704–4714. doi: 10.1021/bi500587p
Lee, S. J., Nam, E., Lee, H. J., Savelieff, M. G., and Lim, M. H. (2017). Towards an understanding of amyloid-beta oligomers: characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 46, 310–323. doi: 10.1039/C6CS00731G
Li, M., Dong, Y., Yu, X., Li, Y., Zou, Y., Zheng, Y., et al. (2017). Synthesis and evaluation of diphenyl conjugated imidazole derivatives as potential glutaminyl cyclase inhibitors for treatment of Alzheimer's disease. J. Med. Chem. 60, 6664–6677. doi: 10.1021/acs.jmedchem.7b00648
Li, M., Dong, Y., Yu, X., Zou, Y., Zh eng, Y., Bu, X., et al. (2016). Inhibitory effect of flavonoids on human glutaminyl cyclase. Bioorg. Med. Chem. 24, 2280–2286. doi: 10.1016/j.bmc.2016.03.064
Lin, W., Zheng, X., Fang, D., Zhou, S., Wu, W., and Zheng, K. (2019). Identifying hQC inhibitors of Alzheimer's disease by effective customized pharmacophore-based virtual screening, molecular dynamic simulation, and binding free energy analysis. Appl. Biochem. Biotechnol. 187, 1173–1192. doi: 10.1007/s12010-018-2780-9
Logtenberg, M. E. W., Jansen, J. H. M., Raaben, M., Toebes, M., Franke, K., Brandsma, A. M., et al. (2019). Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPalpha axis and a target for cancer immunotherapy. Nat. Med. 25, 612–619. doi: 10.1038/s41591-019-0356-z
Luccarini, I., Grossi, C., Rigacci, S., Coppi, E., Pugliese, A. M., Pantano, D., et al. (2015). Oleuropein aglycone protects against pyroglutamylated-3 amyloid-ß toxicity: biochemical, epigenetic and functional correlates. Neurobiol. Aging 36, 648–663. doi: 10.1016/j.neurobiolaging.2014.08.029
Lues, I., Weber, F., Meyer, A., Buhring, U., Hoffm ann, T., Kuhn-Wache, K., et al. (2015). A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimers Dement (N Y). 1, 182–195. doi: 10.1016/j.trci.2015.08.002
Mintun, M. A., Lo, A. C., Duggan Evans, C., Wessels, A. M., Ar dayfio, P. A., Andersen, S. W., et al. (2021). Donanemab in early Alzheimer's disease. N. Engl. J. Med. 384, 1691–1704. doi: 10.1056/NEJMoa2100708
Morawski, M., Hartlage-Rubsamen, M., Jager, C., Waniek, A., Schill ing, S., Schwab, C., et al. (2010). Distinct glutaminyl cyclase expression in Edinger-Westphal nucleus, locus coeruleus and nucleus basalis Meynert contributes to pGlu-Abeta pathology in Alzheimer's disease. Acta Neuropathol. 120, 195–207. doi: 10.1007/s00401-010-0685-y
Morawski, M., Schilling, S., Kreuzberger, M., Waniek, A., Ja ger, C., Koch, B., et al. (2014). Glutaminyl cyclase in human cortex: correlation with (pGlu)-amyloid-beta load and cognitive decline in Alzheimer's disease. J. Alzheimers. Dis. 39, 385–400. doi: 10.3233/JAD-131535
Ngo, V. T. H., Hoang, V. H., Tran, P. T., Ann, J., Cui, M., Park, G., et al. (2018a). Potent human glutaminyl cyclase inhibitors as potential anti-Alzheimer's agents: Structure-activity relationship study of Arg-mimetic region. Bioorg. Med. Chem. 26, 1035–1049. doi: 10.1016/j.bmc.2018.01.015
Ngo, V. T. H., Hoang, V. H., Tran, P. T., Van Manh, N., Ann, J., Kim, E., et al. (2018b). Structure-activity relationship investigation of Phe-Arg mimetic region of human glutaminyl cyclase inhibitors. Bioorg. Med. Chem. 26, 3133–3144. doi: 10.1016/j.bmc.2018.04.040
Nussbaum, J. M., Schilling, S., Cynis, H., Silva, A., Sw anson, E., Wangsanut, T., et al. (2012). Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature 485, 651–655. doi: 10.1038/nature11060
Park, E., Song, K. H., Kim, D., Lee, M., Van Manh, N., Kim, H., et al. (2022). 2-Amino-1,3,4-thiadiazoles as glutaminyl cyclases inhibitors increase phagocytosis through modification of CD47-SIRPalpha checkpoint. ACS Med. Chem. Lett. 13, 1459–1467. doi: 10.1021/acsmedchemlett.2c00256
Perez-Garmendia, R., and Gevorkian, G. (2013). Pyroglutamate-modified amyloid beta peptides: emerging targets for Alzheimer's disease immunotherapy. Curr. Neuropharmacol. 11, 491–498. doi: 10.2174/1570159X11311050004
Pozzi, C., Di Pisa, F., Benvenuti, M., and Mangani, S. (2018). The structure of the human glutaminyl cyclase-SEN177 complex indicates routes for developing new potent inhibitors as possible agents for the treatment of neurological disorders. J. Biol. Inorg. Chem. 23, 1219–1226. doi: 10.1007/s00775-018-1605-1
Ramsbeck, D., Buchholz, M., Koch, B., Böhme, L., Hoffm ann, T., Demuth, H. U., et al. (2013). Structure-activity relationships of benzimidazole-based glutaminyl cyclase inhibitors featuring a heteroaryl scaffold. J. Med. Chem. 56, 6613–6625. doi: 10.1021/jm4001709
Ramsbeck, D., Taudte, N., Janckel, N., Strich, S., Rahfeld, J. U., and Buchholz, M. (2021). Tetrahydroimidazo[4,5-c]pyridine-based inhibitors of porphyromonas gingivalis glutaminyl cyclase. Pharmaceuticals (Basel) 14(12). doi: 10.3390/ph14121206
Roher, A. E., Kokjohn, T. A., Clarke, S. G., Sierks, M. R., Maarouf, C. L., Serrano, G. E., et al. (2017). APP/abeta structural diversity and Alzheimer's disease pathogenesis. Neurochem. Int. 110, 1–13. doi: 10.1016/j.neuint.2017.08.007
Ruiz-Carrillo, D., Koch, B., Parthier, C., Wermann, M., Da mbe, T., Buchholz, M., et al. (2011). Structures of glycosylated mammalian glutaminyl cyclases reveal conformational variability near the active center. Biochemistry 50, 6280–6288. doi: 10.1021/bi200249h
Scheltens, P., Hallikainen, M., Grimmer, T., Duning, T., Gouw, A. A., Teunissen, C. E., et al. (2018). Safety, tolerability and efficacy of the glutaminyl cyclase inhibitor PQ912 in Alzheimer's disease: results of a randomized, double-blind, placebo-controlled phase 2a study. Alzheimers. Res. Ther. 10, 107. doi: 10.1186/s13195-018-0431-6
Schilling, S., Hoffmann, T., Manhart, S., Hoffmann, M., and Demuth, H. U. (2004). Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS Lett. 563, 191–196. doi: 10.1016/S0014-5793(04)00300-X
Schilling, S., Hoffmann, T., Rosche, F., Manhart, S., Wasternack, C., and Demuth, H. U. (2002). Heterologous expression and characterization of human glutaminyl cyclase: evidence for a disulfide bond with importance for catalytic activity. Biochemistry 41, 10849–10857. doi: 10.1021/bi0260381
Schilling, S., Lauber, T., Schaupp, M., Manhart, S., Sch eel, E., Bohm, G., et al. (2006). On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 45, 12393–12399. doi: 10.1021/bi0612667
Schilling, S., Niestroj, A. J., Rahfeld, J. U., Hoffmann, T., Wermann, M., Zunkel, K., et al. (2003). Identification of human glutaminyl cyclase as a metalloenzyme. Potent inhibition by imidazole derivatives and heterocyclic chelators. J. Biol. Chem. 278, 49773–49779. doi: 10.1074/jbc.M309077200
Schilling, S., Zeitschel, U., Hoffmann, T., Heiser, U., Fran cke, M., Kehlen, A., et al. (2008). Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer's disease-like pathology. Nat. Med. 14, 1106–1111. doi: 10.1038/nm.1872
Schilling, S. H., Torsten, N., Johannes, A., Demuth, H. U., and Heiser, U. (2003). Use of effectors of Glutaminyl and Glutamate Cyclases, WO2005039548. International Application No.PCT/EP2004/011630.
Seifert, F., Schulz, K., Koch, B., Manhart, S., Demuth, H. U., and Schilling, S. (2009). Glutaminyl cyclases display significant catalytic proficiency for glutamyl substrates. Biochemistry 48, 11831–11833. doi: 10.1021/bi9018835
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Stephan Schilling, D. S., and Demuth, H. U. (2010). Cleavage of β-Amyloid Precursor Protein. United States Patent Application Publication, US 20110152341A1.
Stephan, A., Wermann, M., von Bohlen, A., Koch, B., Cy nis, H., Demuth, H. U., et al. (2009). Mammalian glutaminyl cyclases and their isoenzymes have identical enzymatic characteristics. FEBS J. 276, 6522–6536. doi: 10.1111/j.1742-4658.2009.07337.x
Syed, Y. Y. (2020). Sodium oligomannate: first approval. Drugs 80, 441–444. doi: 10.1007/s40265-020-01268-1
Szaszko, M., Hajdu, I., Flachner, B., Dobi, K., Mag yar, C., Simon, I., et al. (2017). Identification of potential glutaminyl cyclase inhibitors from lead-like libraries by in silico and in vitro fragment-based screening. Mol. Divers. 21, 175–186. doi: 10.1007/s11030-016-9717-4
Taudte, N., Linnert, M., Rahfeld, J. U., Piechotta, A., Ram sbeck, D., Buchholz, M., et al. (2021). Mammalian-like type II glutaminyl cyclases in Porphyromonas gingivalis and other oral pathogenic bacteria as targets for treatment of periodontitis. J. Biol. Chem. 296, 100263. doi: 10.1016/j.jbc.2021.100263
Tran, P. T., Hoang, V. H., Lee, J., Hien, T. T. T., Tung, N. T., and Ngo, S. T. (2019). In vitro and in silico determination of glutaminyl cyclase inhibitors. RSC Adv. 9, 29619–29627. doi: 10.1039/C9RA05763C
Tran, P. T., Hoang, V. H., Thorat, S. A., Kim, S. E., Ann, J., Chang, Y. J., et al. (2013). Structure-activity relationship of human glutaminyl cyclase inhibitors having an N-(5-methyl-1H-imidazol-1-yl)propyl thiourea template. Bioorg. Med. Chem. 21, 3821–3830. doi: 10.1016/j.bmc.2013.04.005
Valenti, M. T., Bolognin, S., Zanatta, C., Donatelli, L., Innam orati, G., Pampanin, M., et al. (2013). Increased glutaminyl cyclase expression in peripheral blood of Alzheimer's disease patients. J. Alzheimers. Dis. 34, 263–271. doi: 10.3233/JAD-120517
van Bokhoven, P., de Wilde, A., Vermunt, L., Leferink, P. S., Hee tveld, S., Cummings, J., et al. (2021). The Alzheimer's disease drug development landscape. Alzheimers. Res. Ther. 13, 186–194. doi: 10.1186/s13195-021-00927-z
Van Manh, N., Hoang, V. H., Ngo, V. T. H., Ann, J., Jang, T. H., Ha, J. H., et al. (2021). Discovery of highly potent human glutaminyl cyclase (QC) inhibitors as anti-Alzheimer's agents by the combination of pharmacophore-based and structure-based design. Eur. J. Med. Chem. 226, 113819. doi: 10.1016/j.ejmech.2021.113819
Van Manh, N., Hoang, V. H., Ngo, V. T. H., Kang, S., Jeong, J. J., Ha, H. J., et al. (2022). Discovery of potent indazole-based human glutaminyl cyclase (QC) inhibitors as Anti-Alzheimer's disease agents. Eur. J. Med. Chem. 244, 114837. doi: 10.1016/j.ejmech.2022.114837
Vijayan, D. K., and Zhang, K. Y. J. (2019). Human glutaminyl cyclase: structure, function, inhibitors and involvement in Alzheimer's disease. Pharmacol. Res. 147, 104342. doi: 10.1016/j.phrs.2019.104342
Vijverberg, E. G. B., Axelsen, T. M., Bihlet, A. R., Henriksen, K., Weber, F., Fuchs, K., et al. (2021). Rationale and study design of a randomized, placebo-controlled, double-blind phase 2b trial to evaluate efficacy, safety, and tolerability of an oral glutaminyl cyclase inhibitor varoglutamstat (PQ912) in study participants with MCI and mild AD-VIVIAD. Alzheimers. Res. Ther. 13, 142. doi: 10.1186/s13195-021-00882-9
Wu, G., Miller, R. A., Connolly, B., Marcus, J., Renger, J., and Savage, M. J. (2014). Pyroglutamate-modified amyloid-beta protein demonstrates similar properties in an Alzheimer's disease familial mutant knock-in mouse and Alzheimer's disease brain. Neurodegener. Dis. 14, 53–66. doi: 10.1159/000353634
Wulff, M., Baumann, M., Thummler, A., Yadav, J. K., Hei nrich, L., Knupfer, U., et al. (2016). Enhanced fibril fragmentation of N-terminally truncated and pyroglutamyl-modified abeta peptides. Angew. Chem. Int. Ed Engl. 55, 5081–5084. doi: 10.1002/anie.201511099
Xie, Y., Chen, C., Lin, S., Yu, X., Ye, S., Chen, X., et al. (2023). Design, synthesis and anti-AD effects of dual inhibitor targeting glutaminyl cyclase/GSK-3beta. Eur. J. Med. Chem. 248, 115089. doi: 10.1016/j.ejmech.2023.115089
Xu, C., Wang, Y. N., and Wu, H. (2021). Glutaminyl cyclase, diseases, and development of glutaminyl cyclase inhibitors. J. Med. Chem. 64, 6549–6565. doi: 10.1021/acs.jmedchem.1c00325
Xu, C., Zou, H., Yu, X., Xie, Y., Cai, J., Shang, Q., et al. (2020). Repurposing FDA-approved compounds for the discovery of glutaminyl cyclase inhibitors as drugs against Alzheimer's disease. ChemistryOpen. doi: 10.1002/open.202000235
Keywords: Alzheimer's disease, human glutaminyl cyclase, amyloid-β, pyroglutamate modification, QC inhibitor, PQ912, PBD150
Citation: Chen D, Chen Q, Qin X, Tong P, Peng L, Zhang T and Xia C (2023) Development and evolution of human glutaminyl cyclase inhibitors (QCIs): an alternative promising approach for disease-modifying treatment of Alzheimer's disease. Front. Aging Neurosci. 15:1209863. doi: 10.3389/fnagi.2023.1209863
Received: 21 April 2023; Accepted: 10 July 2023;
Published: 03 August 2023.
Edited by:
Stephen D. Ginsberg, Nathan Kline Institute for Psychiatric Research, United StatesReviewed by:
Hans-Ulrich Demuth, Fraunhofer Institute for Cell Therapy and Immunology (IZI), GermanyErin Grace Reed-Geaghan, Northeast Ohio Medical University, United States
Copyright © 2023 Chen, Chen, Qin, Tong, Peng, Zhang and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Zhang, dGFvemhAZmptdS5lZHUuY24=; Chunli Xia, OTIzMDkwMzk2QHFxLmNvbQ==