 Qiujie Li1†
Qiujie Li1† Zhumei Bi1†
Zhumei Bi1† Weiming Liang1†Shan Yin1Huaicheng Li1Zhongyou Liang1Minyao Wu1Jieru Quan2Cheng Li3*
Weiming Liang1†Shan Yin1Huaicheng Li1Zhongyou Liang1Minyao Wu1Jieru Quan2Cheng Li3*- 1The First Affiliated Hospital of Guangxi University of Science and Technology, Guangxi University of Science and Technology, Liuzhou, Guangxi, China
- 2School of Economics and Management, Guangxi University of Science and Technology, Liuzhou, Guangxi, China
- 3Department of Geriatrics, Hezhou Guangji Hospital, Hospital, Hezhou, China
Objective: This meta-analysis aimed to investigate the effects of glucocerebrosidase gene (GBA) variations on the risk of Parkinson’s disease dementia (PDD) and to identify the relationship between GBA variations and PDD.
Method: A comprehensive search was performed to retrieve publications from PubMed, Cochrane Library, Embase and Web of Science up to March 19, 2025. The search terms included “glucocerebrosidase,” “Parkinson’s disease,” and “dementia.” After rigorous screening, cohort studies were included for meta-analysis.
Results: This meta-analysis revealed a significant overall association between the presence of GBA variation and an increased risk of dementia in PD patients (RR = 1.82, 95% CI: 1.52–2.18, p < 0.00001). When stratified by variant type, carriers of GBA mutations exhibited a similar elevation in dementia risk (RR = 1.82, 95% CI: 1.49–2.23, p < 0.00001), and carriers of GBA polymorphisms also demonstrated a heightened risk (RR = 1.82, 95% CI: 1.26–2.61, p = 0.001). Analysis of specific mutations revealed that the N370S variant was associated with an increase in dementia risk (RR = 1.54, 95% CI: 1.24–1.92, p < 0.0001), whereas the L444P variant conferred a stronger effect (RR = 2.17, 95% CI: 1.74–2.71, p < 0.00001). Additionally, the E326K polymorphism was also significantly associated with an increased risk of dementia (RR = 2.34, 95% CI: 1.88–2.91, p < 0.00001).
Conclusion: GBA variations are significant risk factors for PDD, with varying degrees of risk conferred by different variants. These findings underscore the critical role of GBA in the pathogenesis of PDD and highlight its potential as a key genetic risk factor.
Systematic review registration: https://www.crd.york.ac.uk/prospero/display_record.php?, Identifier CRD420251109378.
1 Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder, affecting millions globally. It is primarily characterized by motor symptoms such as bradykinesia, rigidity, tremor, and postural instability, resulting from the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (Kalia and Lang, 2015). However, PD is increasingly recognized as a complex disorder presenting a wide range of non-motor symptoms, including cognitive impairment, sleep disturbances, autonomic dysfunction, and psychiatric symptoms (Malec-Litwinowicz et al., 2014). Among these, Parkinson’s disease dementia (PDD) stands as one of the most debilitating non-motor complications, significantly impacting the quality of life for both patients and their caregivers, and contributing to increased morbidity and mortality (Aarsland and Kurz, 2010). PDD is defined by a decline in cognitive function, particularly in executive function, attention, and visuospatial skills, occurring within the context of established PD (Emre et al., 2007). The prevalence of PDD escalates with disease duration, affecting up to 80% of PD patients over the course of their illness (Hely et al., 2008). Dementia in Parkinson’s disease carries substantial adverse implications for quality of life, caregiver burden, and healthcare-related costs (Vossius et al., 2011).
Genetic factors play a pivotal role in the aetiology and progression of PD. While most PD cases are sporadic, a significant proportion, especially early-onset forms, have a genetic basis (Cacabelos, 2017). Over the past two decades, numerous genes have been identified as being associated with an elevated risk of PD, including SNCA, LRRK2, PARK7, PINK1, and GBA (Blauwendraat et al., 2020). These genetic discoveries have provided invaluable insights into the molecular pathways underpinning PD pathogenesis, such as alpha-synuclein aggregation, mitochondrial dysfunction, and lysosomal impairment (Schapira and Tolosa, 2010). Emerging evidence further suggests that certain genetic variations not only predispose individuals to PD but also influence the clinical phenotype and disease progression, including the development of cognitive decline and dementia (Lill, 2016; Agosta et al., 2013).
Deleterious mutations of GBA are defined as those associated with the onset of Gaucher disease and causative of PD in a heterozygous state, encompassing the common p.N370S and p.L444P (Beutler et al., 2005; Lesage et al., 2011). Sequence variants of exons with no identified relationships with PD in a heterozygous state are defined as GBA polymorphisms, including E326K, T369M, and E388K (Horowitz et al., 2011; Pankratz et al., 2012). GBA variations comprise the abovementioned GBA mutations and polymorphisms (Winder-Rhodes et al., 2013). These mutations are also considered an important risk factor for PD.
The glucocerebrosidase (GBA) gene, located on chromosome 1q21, encodes the lysosomal enzyme glucocerebrosidase (GCase). Mutations in GBA are well-established as the genetic cause of Gaucher disease, a lysosomal storage disorder (Rosenbloom and Weinreb, 2013). Crucially, GBA mutations are also recognized as the most common genetic risk factor for PD, with carriers exhibiting a significantly increased risk of developing the disease compared to non-carriers (Ye et al., 2023; Alcalay et al., 2012). Beyond its role in PD susceptibility, a growing body of research indicates that GBA variations are also strongly associated with an increased risk of developing PDD (Oftedal et al., 2023). The proposed mechanism involves reduced GCase activity, leading to the accumulation of its substrate, glucosylceramide, and subsequent lysosomal dysfunction. This, in turn, is thought to promote the aggregation and spread of alpha-synuclein, a hallmark pathological feature of PD and PDD (Jellinger, 2018).
Despite the accumulating evidence, studies investigating the association between GBA gene polymorphisms and mutations and the risk of dementia in PD patients have reported inconsistent findings. These discrepancies may arise from several factors, including differences in study populations, sample sizes, methodologies for assessing cognitive function, and the specific GBA variants analyzed (Riboldi and Di Fonzo, 2019). Some studies have identified a strong association between GBA mutations and PDD, while others have reported weaker or no significant links, particularly for certain polymorphisms or mild mutations (Gan-Or et al., 2018; Filippi et al., 2022). Given the clinical significance of identifying risk factors for PDD and the potential implications for personalized medicine, a comprehensive and systematic evaluation of the existing literature is warranted.
2 Materials and methods
2.1 Data sources and search strategy
This meta-analysis was conducted in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (Moher et al., 2009). The study has been registered at PROSPERO with the registration number CRD420251109378. A comprehensive search was performed across major electronic databases, including PubMed, EMBASE, Web of Science, and the Cochrane Library, from their inception up to March 19, 2025. The search strategy was developed using a combination of Medical Subject Headings (MeSH) terms and free-text keywords related to Parkinson’s disease, glucocerebrosidase, and dementia. The search technique adhered to the PICOS principle and utilized a blend of MeSH terms and unrestricted text phrases. The search strategy employed combined the terms “Parkinson’s disease,” “glucocerebrosidase” and “dementia.” No language restrictions were applied during the initial search. Additionally, the reference lists of identified relevant articles and review papers were manually screened to identify any additional eligible studies.
2.2 Inclusion and exclusion criteria
Inclusion criteria: (1) patients diagnosed with Parkinson’s disease according to established diagnostic criteria, such as UK Parkinson’s Disease Society Brain Bank (UKPDSBB) Criteria (Hughes et al., 1992); (2) Exposure: Patients with genetically confirmed GBA variations, including but not limited to common mutations such as N370S and L444P, and polymorphisms like E326K; (3) Outcome: The incidence of dementia in GBA variant carriers compared to non-carriers within the PD patient cohort; (4) Types of study: Cohort studies.
Exclusion criteria were: (1) Not relevant; (2) other types of articles, such as conference abstracts yearbook, case reports, publications, letters, meta-analyses, reviews, retrospective studies, pharmacological intervention, animal studies and protocols; (3) Full text unavailable; (4) Data duplication; (5) Data could not be extracted for meta-analysis; (6) Case-control study designs.
2.3 Selection of studies
Study selection and duplicate removal were conducted using EndNote (Version 20; Clarivate Analytics). Two independent reviewers performed the initial screening by removing duplicate records, evaluating titles and abstracts for relevance, and categorising each study as either included or excluded. Disagreements were resolved through discussion and consensus. In cases where consensus could not be reached, a third reviewer served as an arbitrator to make the final decision.
2.4 Data extraction
Data were extracted by two reviewers independently. The extracted data included: (1) Basic study information, including the first author, publication year, country, study design, sample size, and main outcomes; (2) Baseline characteristics of study subjects, including number of patients, male ratio of patients, age at onset, disease duration, GBA genotype, and groups; (3) The data analyzed included total carriers and dementia cases for each GBA variations including GBA polymorphisms, GBA mutations and specific subtypes N370S, L444P, E326K, alongside equivalent data for non-GBA variant carriers. For studies reporting multiple GBA variants, data for each variant was extracted separately where possible. In the absence of consensus between the two independent reviewers, a third reviewer assumed the position of a mediator.
2.5 Quality assessment
The methodological quality of the included observational cohort studies was assessed using the Newcastle–Ottawa Scale (NOS) (Stang, 2010). The NOS evaluates studies based on three broad perspectives: selection of the study groups, comparability of the groups, and ascertainment of either the exposure or outcome of interest. A study can be awarded a maximum of nine stars, with higher scores indicating better methodological quality. Studies with a score of 7 or higher were considered to be of high quality, 4–6 of moderate quality, and less than 4 of low quality.
2.6 Statistical analysis
All statistical analyses were performed using Review Manager (RevMan) software and Stata12.0 software. The primary outcome measure was the risk ratio (RR) and its corresponding 95% confidence interval (CI) for the association between GBA variations and the risk of dementia in PD patients.
Due to the anticipated clinical and methodological heterogeneity among the included studies, a random-effects model was employed for all meta-analyses, which accounts for both within-study and between-study variability. Heterogeneity across studies was assessed using Cochran’s Q test and quantified by the I2 statistic (Cumpston et al., 2022). An I2 value of 0 to 40% was considered to represent unimportant heterogeneity, 30 to 60% moderate heterogeneity, 50 to 90% substantial heterogeneity, and 75 to 100% considerable heterogeneity (Higgins et al., 2003). A p-value <0.10 for the Q test or an I2 > 50% indicated significant heterogeneity, in which case the random-effects model was retained. If I2 was <50%, a fixed-effects model would have been considered.
Analyses were conducted for: overall GBA variations, GBA gene mutations (including N370S and L444P), and GBA gene polymorphisms (including E326K). Publication bias was visually inspected using funnel plots for outcomes. Sensitivity analyses, were conducted to evaluate the robustness of the pooled estimates by sequentially removing one study at a time and re-calculating the overall effect size. To quantitatively assess publication bias, Egger’s regression test was performed for each outcome, with a p-value <0.05 indicating significant publication bias. Furthermore, to address potential sources of heterogeneity and provide more detailed insights, subgroup analyses were performed based on ethnicity (e.g., Asian, Caucasian, Oceanian) for overall GBA variations and GBA mutations. Second, subgroup analyses were conducted based on dementia diagnostic criteria (e.g., DSM-IV, MDS, MMSE, CDR, MoCA) for overall GBA variations, GBA mutations, GBA polymorphisms, and specific variants (N370S, L444P, E326K).
3 Results
3.1 Search results
A comprehensive search was performed to retrieve publications regarding the effects of GBA on PDD risk from PubMed, Cochrane Library, Embase, and Web of Science. A total of 865 records were identified through database searching and additional manual records. After removal of duplicates, 614 unique records were screened based on their titles and abstracts. Of these, 24 full-text articles were retrieved for detailed assessment. After a comprehensive inspection of the entire text, a total of 18 article (Malec-Litwinowicz et al., 2014; Agosta et al., 2013; Alcalay et al., 2012; Chen et al., 2023; Cilia et al., 2016; Davis et al., 2016; De Michele et al., 2023; Graham et al., 2020; Lunde et al., 2018; Malek et al., 2018; Mata et al., 2016; Moran et al., 2017; Oeda et al., 2015; Setó-Salvia et al., 2012; Simuni et al., 2020; Straniero et al., 2020; Szwedo et al., 2022; Yahalom et al., 2019) were chosen for inclusion in this meta-analysis. The detailed study selection process was illustrated in the flow diagram (Figure 1), which outlined the number of records identified, screened, and included at each stage of the review.
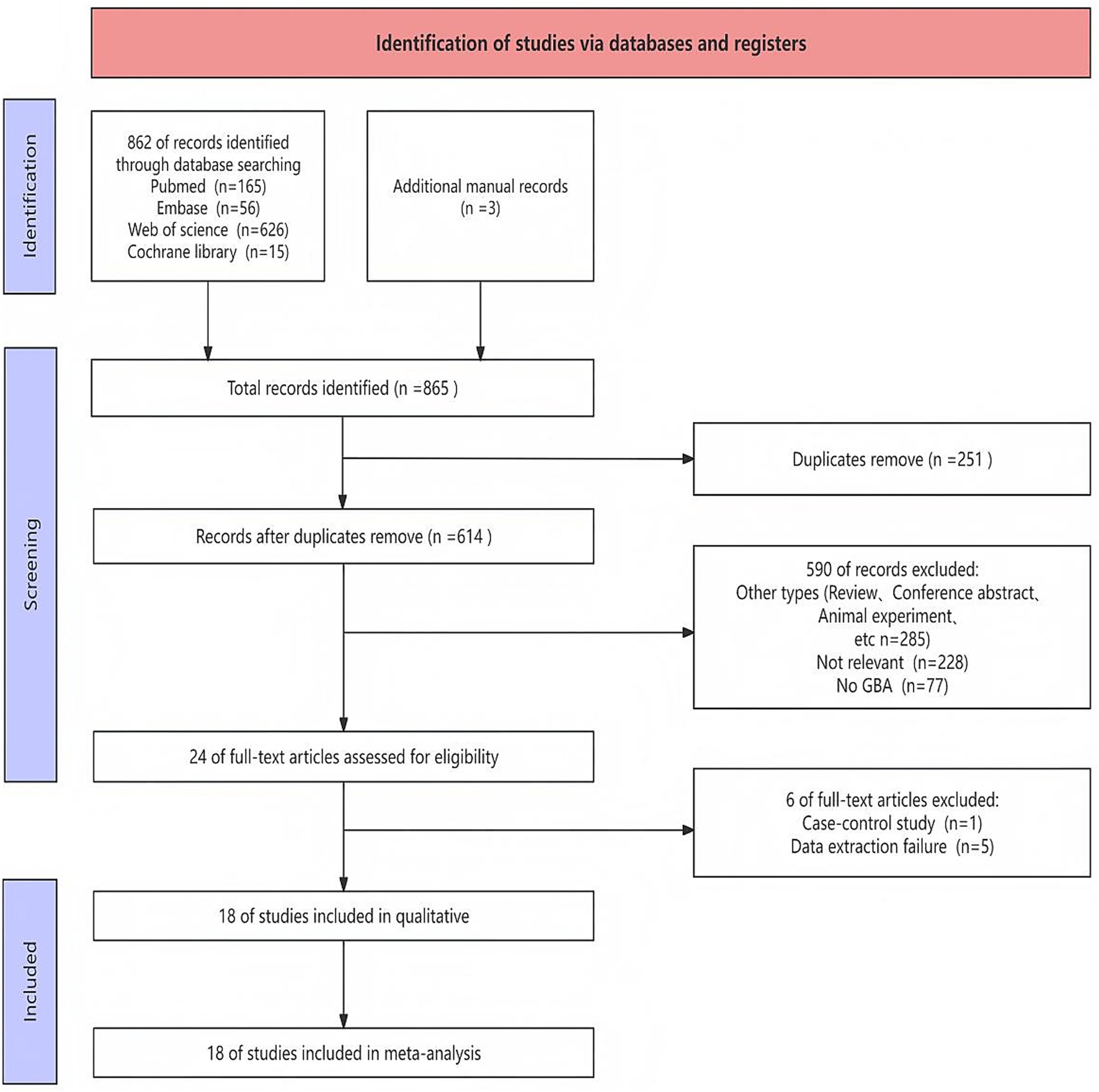
Figure 1. Flow chart of literature search strategies.
3.2 Basic characteristics and quality assessment
The studies were published between 2012 and 2023, and originated from various countries, including USA, UK, Norway, Italy, Israel, Japan, China, Poland, Spain, and New Zealand. A total of 13,175 patients were included. Eighteen studies investigated the effects of GBA variations on PDD risk, 16 addressed the effects of GBA mutations on PDD risk, five explored the effects of GBA polymorphisms on PDD risk, four investigated the effects of GBA p.L444P on PDD risk, four explored the effects of GBA p.N370S on PDD risk, and three studied the effects of GBA p.E326K on PDD risk. The diagnostic criteria for PD and PDD varied across studies but were generally consistent with established clinical guidelines. The quality assessment using the Newcastle–Ottawa Scale (NOS) revealed that all 18 studies were of high quality (NOS score ≥7) (Table 1). A summary of the characteristics of the included studies is presented in Table 2.
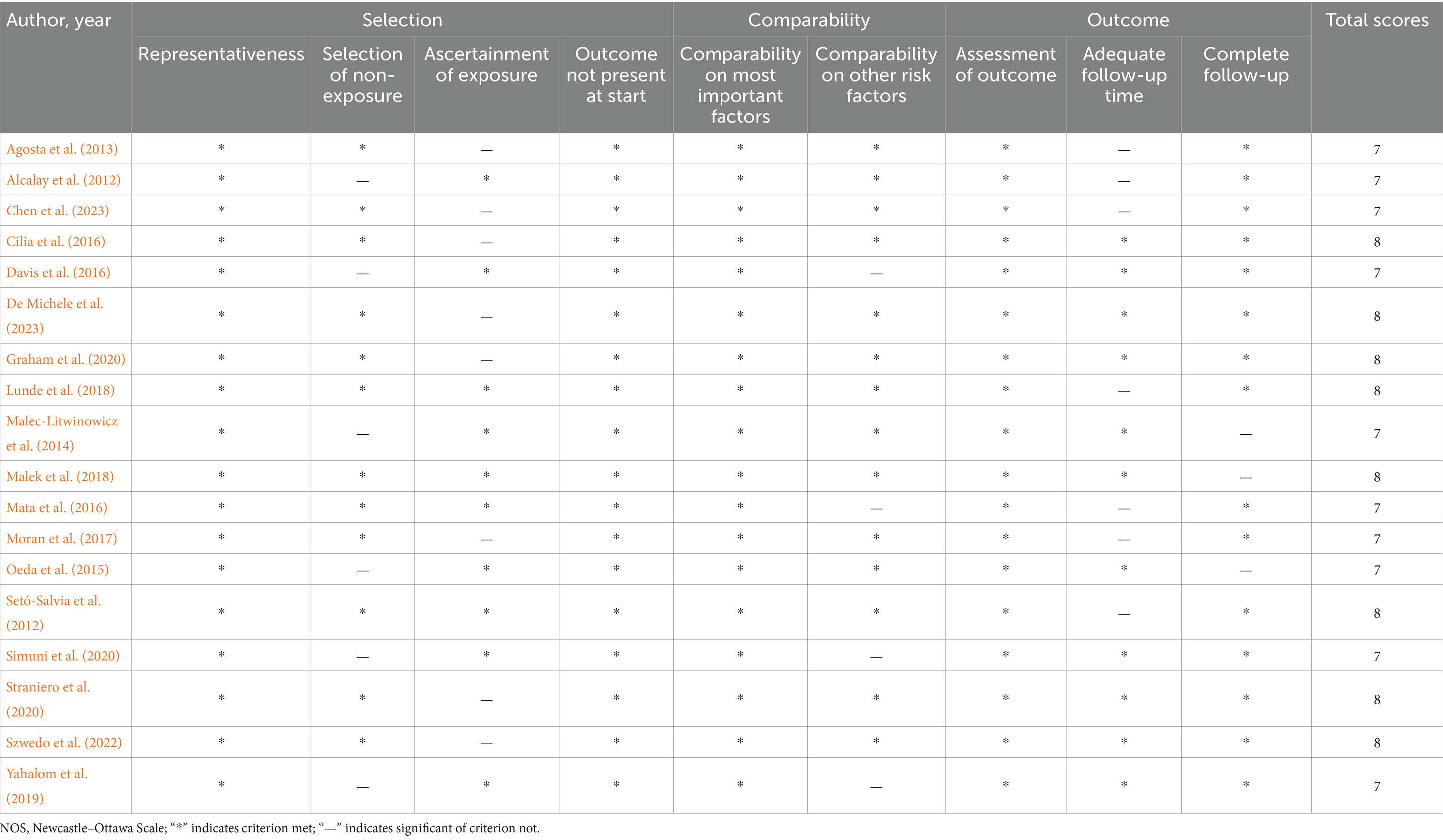
Table 1. Quality assessment according to the NOS scale.
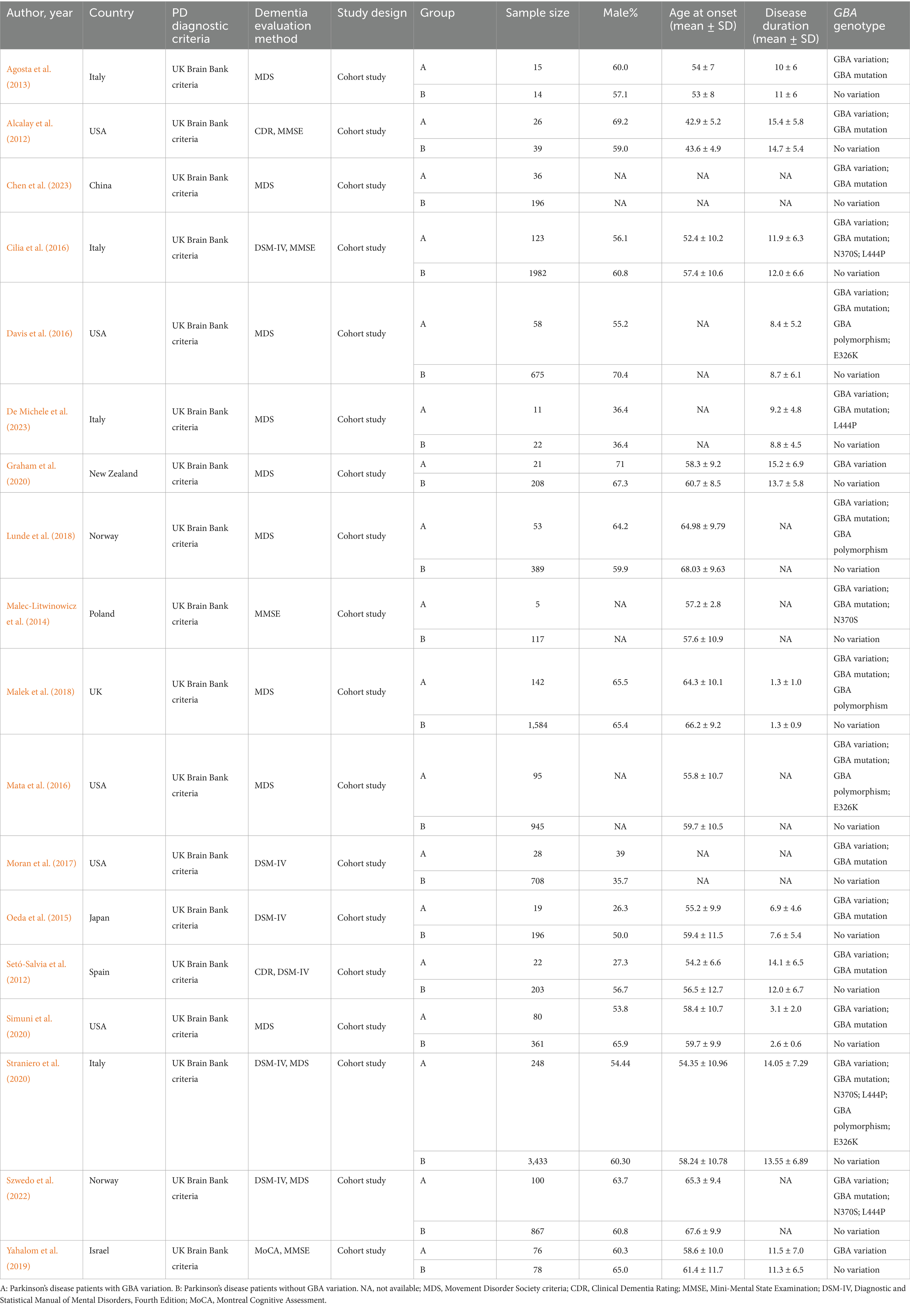
Table 2. Characteristics of included studies and patients.
3.3 Clinical outcomes
The meta-analysis results for clinical outcomes were consolidated and shown in Table 3.
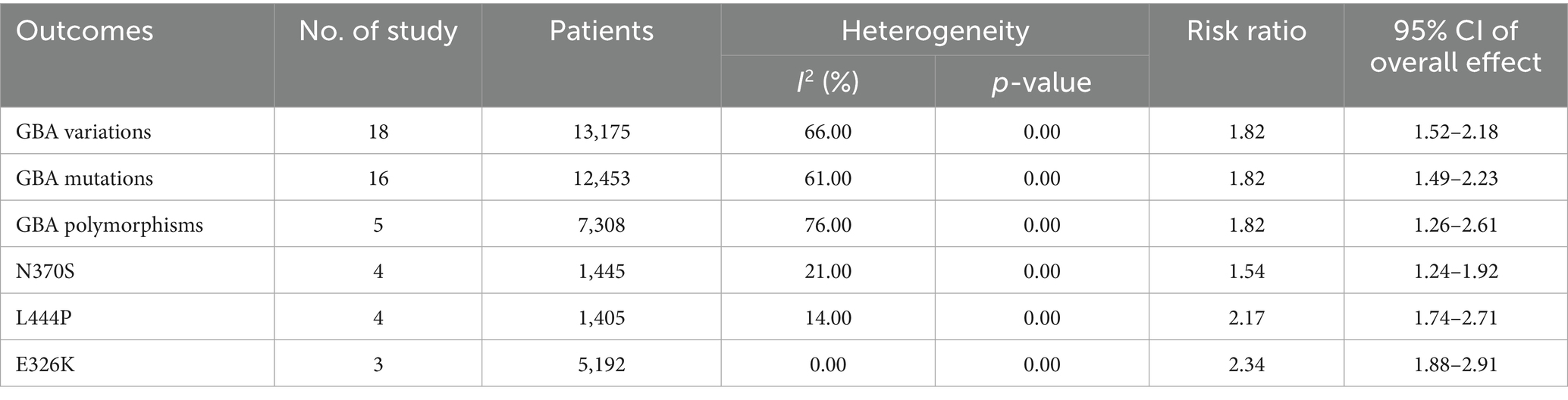
Table 3. The results of the meta-analysis.
3.3.1 Overall association of GBA variations with dementia risk
This meta-analysis, which synthesizes data from 18 studies, revealed a significant overall association between the presence of GBA variations and an increased risk of dementia in Parkinson’s disease patients (RR = 1.82, 95% CI: 1.52–2.18, p < 0.00001, I2 = 66%), indicating that PD patients carrying GBA variations have an higher risk of developing dementia compared to non-carriers. Substantial heterogeneity was observed across these studies, justifying the use of a random-effects model (Figure 2).
![Forest plot displaying studies comparing risk ratios of experimental versus control groups. Each study is listed with corresponding events, totals, weights, and risk ratios with confidence intervals. A diamond summarizes the overall effect. The plot shows log scale with a line at one, indicating no effect, and directional markers for favoring experimental or control. Overall effect size is 1.82 with confidence interval [1.52, 2.18]. Heterogeneity indicators are present.](https://www.frontiersin.org/files/Articles/1671760/fnagi-17-1671760-HTML/image_m/fnagi-17-1671760-g002.jpg)
Figure 2. Forest plot of the meta-analysis for the overall association of GBA variations with dementia risk in PD patients.
3.3.2 Association of GBA mutations with dementia risk
A subsequent subgroup analysis included 16 studies that investigated the association between GBA mutations and the risk of dementia in PD patients (RR = 1.82, 95% CI: 1.49–2.23, p < 0.00001, I2 = 61%), indicating that individuals with GBA mutations have a significantly increased risk of developing dementia. Heterogeneity was observed across these studies, suggesting some variability in effects among different mutation studies. This finding highlights the critical role of GBA mutations in the pathogenesis of Parkinson’s disease dementia (Figure 3).
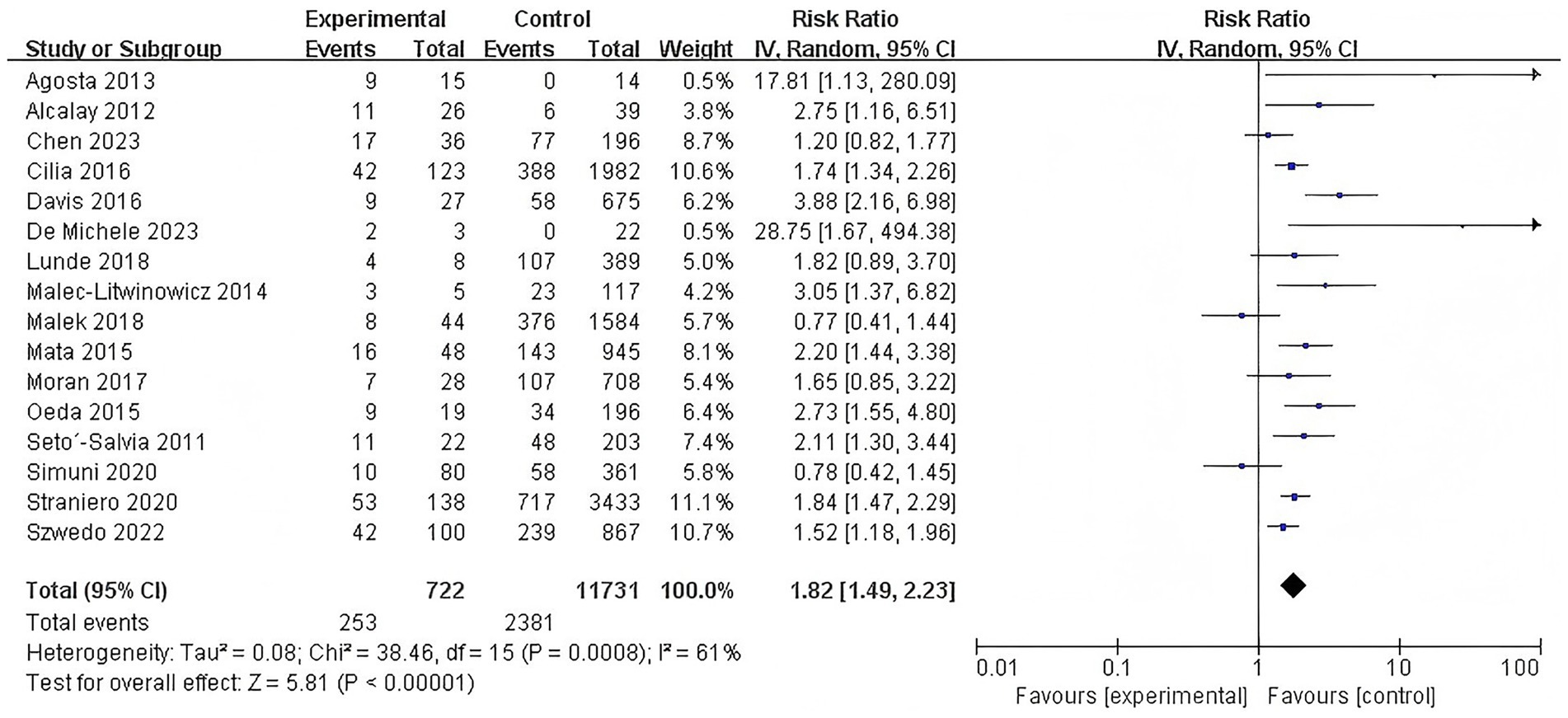
Figure 3. Forest plot of the meta-analysis for the association of GBA mutations with dementia risk in PD patients.
3.3.3 Association of GBA polymorphisms with dementia risk
Similarly, a separate subgroup analysis of studies examining GBA polymorphisms identified a significantly elevated risk of dementia among carriers (RR = 1.82, 95% CI: 1.26–2.61, p = 0.001, I2 = 76%). This finding underscores the important contribution of GBA polymorphisms to the genetic risk of PDD. However, there was substantial heterogeneity among the studies, suggesting considerable variability in the effect sizes across the included studies (Figure 4).
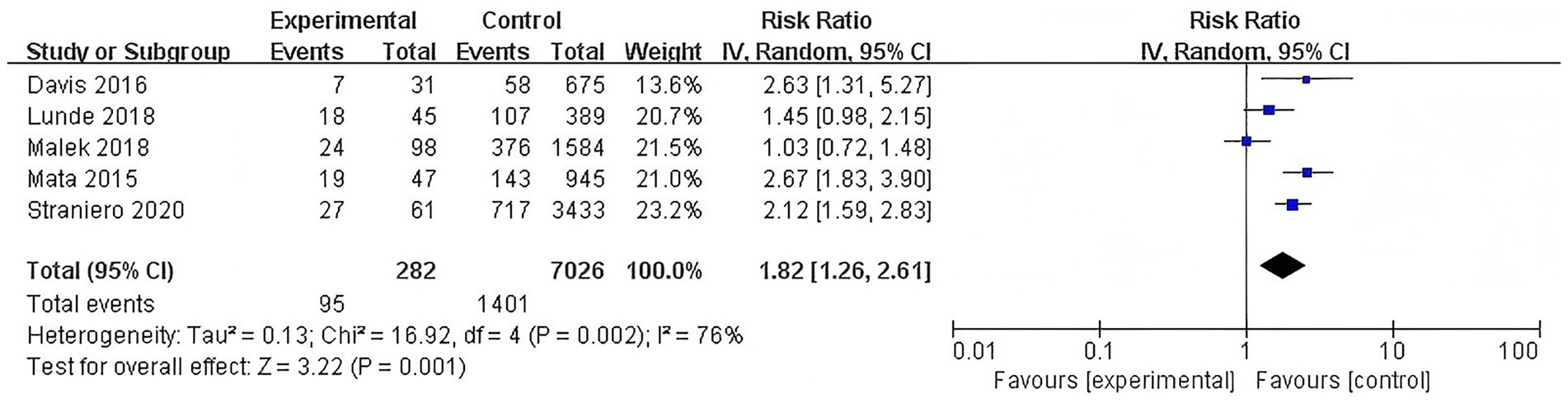
Figure 4. Forest plot of the meta-analysis for the association of GBA polymorphisms with dementia risk in PD patients.
3.3.4 Association of N370S mutation with dementia risk
Further investigation into specific variants demonstrated that the N370S mutation is significantly associated with an increased risk of dementia, accompanied by low heterogeneity (RR = 1.54, 95% CI: 1.24–1.92, p < 0.0001, I2 = 21%) (Figure 5).
![Forest plot showing a meta-analysis of four studies comparing experimental and control groups. Studies listed are Cilia 2016, Malec-Litwinowicz 2014, Straniero 2020, and Szwedo 2022. Risk ratios with 95% confidence intervals are presented. The overall risk ratio is 1.54 [1.24, 1.92], favoring the experimental group. Heterogeneity statistics are Tau² = 0.01, Chi² = 3.80, df = 3, and I² = 21%. The test for overall effect shows Z = 3.91 (P < 0.0001). Visual representation includes squares and a diamond indicating risk ratio positions.](https://www.frontiersin.org/files/Articles/1671760/fnagi-17-1671760-HTML/image_m/fnagi-17-1671760-g005.jpg)
Figure 5. Forest plot of the meta-analysis for the association of N370S mutation with dementia risk in PD patients.
3.3.5 Association of L444P mutation with dementia risk
This meta-analysis determined that the L444P mutation was associated with a substantial increase in dementia risk associated with this severe GBA mutation and low heterogeneity was observed (RR = 2.17, 95% CI: 1.74–2.71, p < 0.00001, I2 = 14%) (Figure 6).
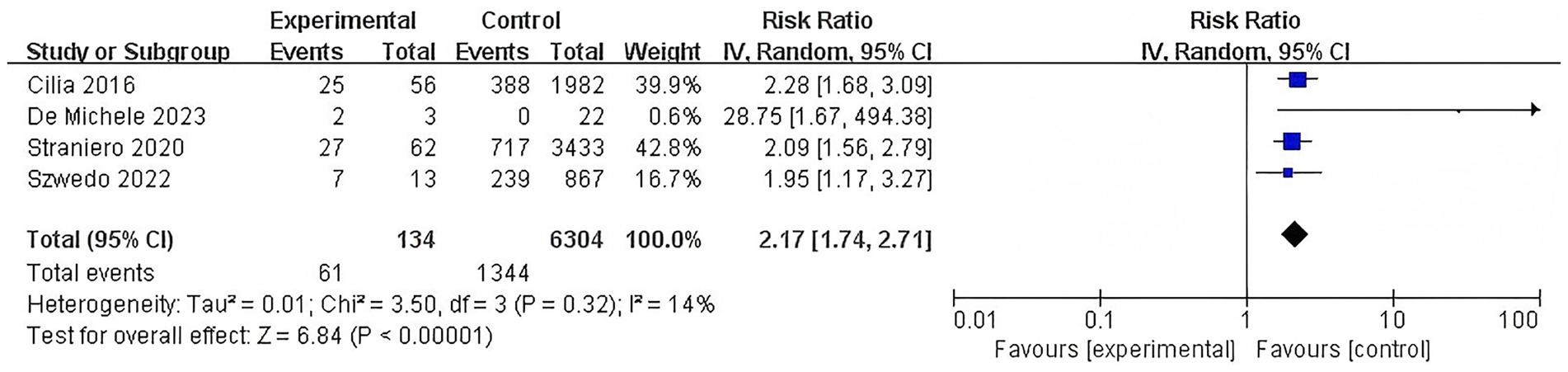
Figure 6. Forest plot of the meta-analysis for the association of L444P mutation with dementia risk in PD patients.
3.3.6 Association of E326K polymorphism with dementia risk
The E326K polymorphism was also significantly associated with an increased risk of dementia and no significant heterogeneity was detected for this subgroup (RR = 2.34, 95% CI: 1.88–2.91, p < 0.00001, I2 = 0%) (Figure 7).
![Forest plot illustrating a meta-analysis of three studies comparing experimental and control groups. Each study entry includes events, total participants, and weight. Risk ratios with 95% confidence intervals are shown. Total events are 53 for experimental and 918 for control. The overall effect size indicates a risk ratio of 2.34 [1.88, 2.91]. Heterogeneity is low with I² = 0 percent, and the test for overall effect is significant (Z = 7.63, P < 0.00001). A diamond at the bottom represents the pooled effect size favoring the experimental group.](https://www.frontiersin.org/files/Articles/1671760/fnagi-17-1671760-HTML/image_m/fnagi-17-1671760-g007.jpg)
Figure 7. Forest plot of the meta-analysis for the association of E326K polymorphism with dementia risk in PD patients.
3.4 Subgroup analyses
3.4.1 Subgroup analysis by ethnicity
For GBA mutations, subgroup analysis by ethnicity revealed an RR of 1.76 (95% CI, 0.79–3.94, p = 0.17, I2 = 82%) for Asian populations and an RR of 1.86 (95% CI, 1.48–2.34, p < 0.00001, I2 = 63%) for Caucasian populations. The overall pooled RR for GBA mutations across all ethnicities was 1.83 (95% CI, 1.48–2.27, p < 0.00001, I2 = 64%). Crucially, the test for subgroup differences indicated that there was no significant heterogeneity between the ethnicity subgroups (Chi2 = 0.02, df = 1, p = 0.90, I2 = 0%), suggesting that ethnicity does not significantly modify the association between GBA mutations and PDD risk (Supplementary Figure 1).
For overall GBA variations, subgroup analysis by ethnicity showed an RR of 1.76 (95% CI: 0.79–3.94, p = 0.17, I2 = 82%) for Asian populations, an RR of 1.84 (95% CI: 1.49–2.27, p < 0.00001, I2 = 70%) for Caucasian populations, and an RR of 2.12 (95% CI: 1.21–3.73, p = 0.009) for Oceanian populations. The overall pooled RR for GBA variations across all ethnicities was 1.83 (95% CI: 1.52–2.21, p < 0.00001, I2 = 68%). The analysis for subgroup differences confirmed that ethnicity was not a significant source of heterogeneity (Chi2 = 0.24, df = 2, p = 0.89, I2 = 0%) (Supplementary Figure 2).
3.4.2 Subgroup analysis by dementia diagnostic criteria
Subgroup analysis for GBA mutations based on dementia diagnostic criteria showed the following RRs: DSM-IV criteria (RR = 1.78, 95% CI: 1.56–2.02, p < 0.00001, I2 = 0%), MDS criteria (RR = 1.65, 95% CI: 1.23–2.23, p = 0.0010, I2 = 72%), CDR criteria (RR = 2.25, 95% CI: 1.48–3.44, p = 0.0002, I2 = 0%), and MMSE criteria (RR = 2.02, 95% CI: 1.44–2.82, p < 0.0001, I2 = 18%). The overall pooled RR for GBA mutations across all criteria was 1.80 (95% CI: 1.57–2.07, p < 0.00001, I2 = 52%). The test for subgroup differences revealed no significant impact of the diagnostic criteria used on the overall effect size (Chi2 = 1.87, df = 3, p = 0.60, I2 = 0%) (Supplementary Figure 3).
For overall GBA variations, subgroup analysis by dementia diagnostic criteria yielded these RRs: CDR criteria (RR = 2.25, 95% CI: 1.48–3.44, p = 0.0002, I2 = 0%), DSM-IV criteria (RR = 1.74, 95% CI: 1.55–1.96, p < 0.00001, I2 = 0%), MDS criteria (RR = 1.65, 95% CI: 1.29–2.11, p < 0.0001, I2 = 76%), MMSE criteria (RR = 2.07, 95% CI: 1.57–2.72, p < 0.00001, I2 = 12%), and MoCA criteria (RR = 2.74, 95% CI: 1.36–5.50, p = 0.005). The overall pooled RR for GBA variations across all criteria was 1.81 (95% CI: 1.59–2.06, p < 0.00001, I2 = 58%). Similarly, the test for subgroup differences indicated no significant influence of the diagnostic criteria on the risk estimate (Chi2 = 4.23, df = 4, p = 0.38, I2 = 5.3%) (Supplementary Figure 4).
For GBA polymorphisms, subgroup analysis by dementia diagnostic criteria showed an RR of 2.12 (95% CI: 1.59–2.83, p < 0.00001) for DSM-IV criteria and an RR of 1.82 (95% CI: 1.26–2.61, p = 0.001, I2 = 76%) for MDS criteria. The overall pooled RR was 1.86 (95% CI: 1.40–2.48, p < 0.0001, I2 = 72%). The test for subgroup differences was not significant (Chi2 = 0.43, df = 1, p = 0.51, I2 = 0%), suggesting consistent effects across the diagnostic criteria (Supplementary Figure 5).
For the N370S mutation, subgroup analysis by dementia diagnostic criteria showed an RR of 1.47 (95% CI: 1.22–1.78, p < 0.0001, I2 = 0%) for DSM-IV criteria, an RR of 1.52 (95% CI: 1.23–1.89, p = 0.0001, I2 = 0%) for MDS criteria, and an RR of 1.85 (95% CI: 0.81–4.24, p = 0.14, I2 = 71%) for MMSE criteria. The overall pooled RR was 1.50 (95% CI: 1.32–1.72, p < 0.00001, I2 = 0%). The test for subgroup differences indicated no significant heterogeneity across the different diagnostic criteria (Chi2 = 0.30, df = 2, p = 0.86, I2 = 0%) (Supplementary Figure 6).
For the L444P mutation, subgroup analysis by dementia diagnostic criteria showed an RR of 2.14 (95% CI: 1.76–2.60, p < 0.00001, I2 = 0%) for DSM-IV criteria, an RR of 2.16 (95% CI: 1.40–3.32, p = 0.0005, I2 = 40%) for MDS criteria, and an RR of 2.28 (95% CI: 1.68–3.09, p < 0.00001) for MMSE criteria. The overall pooled RR was 2.16 (95% CI: 1.88–2.47, p < 0.00001, I2 = 0%). The test for subgroup differences showed no significant impact of diagnostic criteria on the risk estimate (Chi2 = 0.12, df = 2, p = 0.94, I2 = 0%) (Supplementary Figure 7).
For the E326K polymorphism, subgroup analysis by dementia diagnostic criteria showed an RR of 2.12 (95% CI: 1.59–2.83, p < 0.00001) for DSM-IV criteria and an RR of 2.34 (95% CI: 1.88–2.91, p < 0.00001, I2 = 0%) for MDS criteria. The overall pooled RR was 2.26 (95% CI: 1.90–2.68, p < 0.00001, I2 = 0%). The test for subgroup differences was not significant (Chi2 = 0.28, df = 1, p = 0.60, I2 = 0%), indicating that the choice of diagnostic criteria did not introduce heterogeneity (Supplementary Figure 8).
3.5 Sensitivity analysis
To further evaluate the stability and robustness of the pooled estimates, sensitivity analysis was performed. This method involves systematically removing one study at a time from the meta-analysis and re-calculating the overall effect size. The purpose is to identify whether any single study disproportionately influences the overall pooled estimate, which could indicate a lack of robustness or the presence of an outlier study. The results of the sensitivity analysis for the overall association of GBA variations with dementia risk (Supplementary Figure 9), as well as for the GBA mutations (Supplementary Figure 10), GBA polymorphisms (Supplementary Figure 11), N370S mutation (Supplementary Figure 12), L444P mutation (Supplementary Figure 13), and E326K polymorphism (Supplementary Figure 14), consistently demonstrated that no single study had an undue influence on the respective pooled estimates. The recalculated effect sizes remained within a narrow range, and the statistical significance of the associations was maintained across all iterations. This consistency across the sensitivity analyses strongly confirms the stability and robustness of the findings, indicating that the conclusions are not driven by any single study and are reliable despite the observed heterogeneity.
3.6 Publication bias analysis
Publication bias was visually inspected using funnel plots for outcomes. For the overall association of GBA variations, GBA mutations and GBA polymorphisms, with dementia risk, the funnel plot (Supplementary Figures 15–17) suggested some asymmetry. This asymmetry could potentially indicate the presence of publication bias. The funnel plots of p.N370S, p.L444P and p.E326K are basically contralateral. Each score is scattered on both sides of the midline and is within the 95% CI, with no obvious missing angles. This suggests a small possibility in publication bias (Supplementary Figures 18–20).
To provide a quantitative assessment of publication bias, Egger’s regression test was performed. The results indicated no significant publication bias for N370S (t = 1.41, p = 0.293) (Supplementary Figure 21), L444P (t = 1.87, p = 0.203) (Supplementary Figure 22), E326K (t = 0.84, p = 0.553) (Supplementary Figure 23), GBA polymorphisms (t = 0.21, p = 0.850) (Supplementary Figure 24), GBA mutations (t = 1.36, p = 0.196) (Supplementary Figure 25), and overall GBA variations (t = 1.66, p = 0.117) (Supplementary Figure 26). These quantitative findings complement the visual inspection of funnel plots and further support the robustness of the meta-analysis results against publication bias.
4 Discussion
This meta-analysis provides compelling evidence that GBA gene variations, encompassing both mutations and polymorphisms, are significantly associated with an increased risk of dementia in patients with Parkinson’s disease. The findings demonstrate that PD patients carrying GBA variation have an approximately 82% higher risk of developing dementia compared to non-carriers. This robust association underscores the critical role of GBA in the pathogenesis of PDD and highlights its potential as a key genetic risk factor (Straniero et al., 2020). Furthermore, analyses revealed that both severe mutations (L444P), mild mutations (N370S), and even the common polymorphism (E326K) are independently associated with an elevated risk of PDD, with varying degrees of risk (Malec-Litwinowicz et al., 2014; De Michele et al., 2023; Mata et al., 2016; Setó-Salvia et al., 2012; Straniero et al., 2020). Notably, the L444P mutation showed the highest risk ratio, followed by the E326K polymorphism and the N370S mutation, suggesting a potential correlation between the severity of the GBA variant and the magnitude of dementia risk. This observation aligns with the understanding that different GBA variants may lead to varying degrees of GCase enzyme deficiency, thereby differentially impacting downstream pathological processes.
The comprehensive subgroup analyses performed in this study provide further insights into the influence of ethnicity and dementia diagnostic criteria on the observed associations. The consistent findings across different ethnic groups for GBA mutations and variations suggest a broad applicability of these genetic risk factors. Moreover, the varying risk ratios observed across different dementia diagnostic criteria highlight the importance of standardized diagnostic approaches in future research and clinical practice. These detailed subgroup analyses enhance the generalizability and clinical relevance of the findings, addressing potential sources of heterogeneity that could confound the overall estimates.
The underlying mechanisms linking GBA variations to PDD are complex and likely involve lysosomal dysfunction and altered alpha-synuclein homeostasis. The GBA gene encodes glucocerebrosidase, a lysosomal enzyme responsible for the hydrolysis of glucosylceramide to glucose and ceramide (Grabowski, 2012; Chatterjee and Krainc, 2023). Mutations in GBA lead to reduced GCase activity, resulting in the accumulation of its substrate within lysosomes (Oftedal et al., 2023). This lysosomal dysfunction is hypothesized to impair the clearance of alpha-synuclein, leading to its aggregation and the formation of Lewy bodies, which are neuropathological hallmarks of both PD and PDD (Lee et al., 2013; Do et al., 2019). The accumulation of misfolded alpha-synuclein can further exacerbate lysosomal dysfunction, creating a vicious cycle that contributes to neurodegeneration and cognitive decline (Cerri et al., 2018; Silva et al., 2025; Rocha et al., 2023; Smith and Schapira, 2022). Moreover, reduced GCase activity may also impact other cellular processes, including mitochondrial function, oxidative stress, and neuroinflammation, all of which are implicated in PD pathogenesis and PDD development (Smith and Schapira, 2022; Gegg and Schapira, 2018; Atashrazm et al., 2018). The varying risk levels observed for different GBA variants could be attributed to their differential impact on GCase activity and subsequent cellular consequences (Smith and Schapira, 2022). For instance, severe mutations like L444P may lead to a more profound reduction in GCase activity, resulting in a greater burden of alpha-synuclein pathology and a higher risk of dementia, compared to milder mutations or polymorphisms (Granek et al., 2023; Bendikov-Bar et al., 2011).
Recent advances in the understanding of PDD, particularly since 2020, have illuminated several interconnected mechanisms that extend beyond traditional models. Lysosomal dysfunction, directly linked to GBA mutations, remains a central theme. Impaired GCase activity leads to the accumulation of glycosphingolipids, which promotes α-synuclein aggregation and neuroinflammation (Calabresi et al., 2023). This has spurred the development of pharmacological chaperones and enzyme replacement therapies, which have shown promise in preclinical and early clinical settings (Pardo-Moreno et al., 2023). Mitochondrial dysfunction has also been identified as a critical factor. GBA mutations can indirectly impair mitochondrial function, leading to oxidative stress and contributing to neuronal damage. Consequently, therapeutic strategies targeting mitochondrial health, such as coenzyme Q10 supplementation and novel mitochondrial-targeted antioxidants, are under active investigation (Colca and Finck, 2022). Neuroinflammation, mediated by activated microglia and astrocytes, is increasingly recognized as a key driver of PDD progression. GBA mutations appear to exacerbate these neuroinflammatory responses. As such, immunomodulatory therapies targeting specific inflammatory pathways represent a promising new frontier in treatment development (Iarkov et al., 2021). Furthermore, the gut-brain axis has emerged as a significant area of research. Dysbiosis of the gut microbiota can influence neuroinflammation and α-synuclein pathology. Interventions such as probiotics, prebiotics, and faecal microbiota transplantation are being explored for their potential to modulate disease progression (Klann et al., 2021).
The findings of this meta-analysis have significant clinical implications. The identification of GBA gene variations as a strong risk factor for PDD suggests that genetic screening for GBA variants could be a valuable tool for assessing dementia risk in PD patients. Early identification of high-risk individuals could enable more targeted monitoring, earlier intervention strategies, and personalized management plans (Moore and Barker, 2014; Szwedo et al., 2025). For example, patients carrying GBA variants might benefit from more frequent cognitive assessments, or be prioritized for clinical trials investigating novel therapies aimed at improving lysosomal function or reducing alpha-synuclein aggregation (Zhang et al., 2019; Ciccaldo et al., 2025; Williams et al., 2024a; Williams et al., 2024b). Furthermore, understanding the specific GBA variants and their associated risk levels could help clinicians provide more accurate prognoses and counsel patients and their families more effectively regarding the potential trajectory of their disease (Menozzi et al., 2023). This personalized approach to care, informed by genetic insights, represents a significant step towards improving outcomes for PD patients at risk of developing dementia (Cook et al., 2021; Hill et al., 2022).
The clinical significance of these findings is substantial. The integration of GBA gene screening into routine clinical practice for PD patients could serve as a valuable tool for early risk assessment of dementia. This genetic information can help clinicians identify individuals at higher risk for PDD, enabling the implementation of more proactive and individualized management strategies, including targeted monitoring, early cognitive interventions, and personalized therapeutic approaches. Ultimately, understanding the genetic predisposition to PDD, particularly concerning GBA variations, holds promise for improving patient outcomes and advancing the development of precision medicine in Parkinson’s disease.
This meta-analysis builds upon previous research by providing a comprehensive and updated assessment of the association between GBA gene variations and the risk of Parkinson’s disease dementia. This study distinguishes itself from prior work, such as the 2020 meta-analysis by Zhang et al. (2020) which similarly concluded that GBA polymorphisms and mutations increase PDD risk.
Firstly, it represents the most comprehensive meta-analysis to date specifically focusing on the association between GBA gene polymorphisms and mutations and the risk of dementia in PD patients. By incorporating a larger and more recent dataset, it significantly enhances statistical power and the timeliness of the findings. Secondly, the systematic search strategy across multiple databases minimized the risk of publication bias, and the independent data extraction and quality assessment enhanced the reliability of the findings. This study quantitatively assesses publication bias using Egger’s regression test, complementing visual inspection of funnel plots to ensure the robustness of the pooled estimates. Thirdly, the use of a random-effects model appropriately accounted for the inherent heterogeneity across studies, providing a more conservative and generalizable estimate of the effect. Finally, the separate analyses for different GBA variants, coupled with refined methodological approaches such as detailed subgroup analyses based on ethnicity and various dementia diagnostic criteria, allowed for a detailed examination of their impact. This provides more granular insights into specific GBA variants and their differential effects on PDD risk, exploring potential sources of heterogeneity.
Furthermore, this meta-analysis acknowledges the existence of GBA variants beyond N370S, L444P, and E326K, including other Gaucher disease-related pathogenic mutations (e.g., D409H, R463C, RecNciI complex allele) and other polymorphisms (e.g., E388K, R120W, IVS10+1G>T), which may contribute to PD and PDD risk in specific populations. Future research should consider expanding genetic screening to include these less common but clinically relevant variants, potentially through whole-genome sequencing approaches. By addressing these aspects, this meta-analysis seeks to provide a more robust estimate of the association and to clarify whether different GBA variants confer varying degrees of risk for PDD, thereby offering valuable insights for clinical practice and future research directions.
Despite these strengths, this meta-analysis also has several limitations. The observed heterogeneity, particularly in the overall analysis, suggests that other factors not accounted for in the analysis may influence the association between GBA variations and PDD. These factors could include differences in patient demographics, disease duration, concomitant medications, and the specific diagnostic criteria used for PDD across studies. While analyses were performed for specific GBA variants, further stratification by other clinical or genetic factors was limited by the available data in the included studies. Additionally, the reliance on published data means that individual patient data could not be accessed, which would have allowed for more detailed analyses and adjustment for potential confounders. The visual inspection of funnel plots suggested some asymmetry, indicating a potential for publication bias. Although Egger’s regression tests did not indicate significant publication bias for individual variants and overall categories, this does not entirely rule out the possibility of bias, especially for subgroups with fewer studies. Future research should aim to address these limitations by conducting larger, prospective cohort studies with standardized methodologies for GBA genotyping, PDD diagnosis, and comprehensive collection of clinical and demographic data. Further mechanistic studies are also needed to fully elucidate the complex interplay between GBA dysfunction, alpha-synuclein pathology, and cognitive decline in PD (Moore and Barker, 2014).
In conclusion, this comprehensive meta-analysis provides robust evidence confirming that both mutations and polymorphisms in the GBA gene are significantly associated with an increased risk of dementia in patients with Parkinson’s disease. The findings highlight that different GBA variants confer varying degrees of risk, with severe mutations, mild mutations, and even common polymorphisms all contributing to an elevated likelihood of developing PDD. This study underscores the critical role of GBA in the genetic landscape of PDD and its potential as a predictive biomarker. The detailed subgroup analyses and quantitative assessment of publication bias further solidify these conclusions, offering a more refined understanding of GBA’s impact on PDD risk across diverse clinical and demographic contexts.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
QL: Methodology, Investigation, Conceptualization, Writing – review & editing, Supervision, Software, Project administration, Writing – original draft, Formal analysis, Data curation, Resources. ZB: Writing – review & editing, Supervision, Funding acquisition, Conceptualization, Resources, Data curation, Project administration. WL: Writing – original draft, Funding acquisition, Writing – review & editing, Validation, Methodology, Data curation, Project administration. SY: Conceptualization, Methodology, Writing – review & editing, Resources, Software. HL: Validation, Investigation, Writing – original draft, Formal analysis, Methodology. ZL: Conceptualization, Visualization, Project administration, Methodology, Writing – original draft, Investigation. MW: Data curation, Writing – review & editing, Visualization, Validation, Formal analysis. JQ: Project administration, Writing – review & editing, Methodology, Supervision, Investigation, Data curation. CL: Funding acquisition, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Key Laboratory Construction Project of Guangxi Health Commission (ZPZH2020007), the Guangxi Natural Science Foundation (AD22035199), the Liuzhou Natural Science Foundation (2022CAC0116) and the Scientific Research Foundation of Guangxi University of Science and Technology (21Z39).
Acknowledgments
Everyone who contributed significantly to this study has been listed.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnagi.2025.1671760/full#supplementary-material
References
Aarsland, D., and Kurz, M. W. (2010). The epidemiology of dementia associated with Parkinson disease. J. Neurol. Sci. 289, 18–22. doi: 10.1016/j.jns.2009.08.034
Agosta, F., Kostic, V. S., Davidovic, K., Kresojević, N., Sarro, L., Svetel, M., et al. (2013). White matter abnormalities in Parkinson’s disease patients with glucocerebrosidase gene mutations. Mov. Disord. 28, 772–778. doi: 10.1002/mds.25397
Alcalay, R. N., Caccappolo, E., Mejia-Santana, H., Tang, M., Rosado, L., Orbe Reilly, M., et al. (2012). Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78, 1434–1440. doi: 10.1212/WNL.0b013e318253d54b
Atashrazm, F., Hammond, D., Perera, G., Dobson-Stone, C., Mueller, N., Pickford, R., et al. (2018). Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 8:15446. doi: 10.1038/s41598-018-33921-x
Bendikov-Bar, I., Ron, I., Filocamo, M., and Horowitz, M. (2011). Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 46, 4–10. doi: 10.1016/j.bcmd.2010.10.012
Beutler, E., Gelbart, T., and Scott, C. R. (2005). Hematologically important mutations: Gaucher disease. Blood Cells Mol. Dis. 35, 355–364. doi: 10.1016/j.bcmd.2005.07.005
Blauwendraat, C., Nalls, M. A., and Singleton, A. B. (2020). The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178. doi: 10.1016/s1474-4422(19)30287-x
Cacabelos, R. (2017). Parkinson’s disease: from pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 18:551. doi: 10.3390/ijms18030551
Calabresi, P., Mechelli, A., Natale, G., Volpicelli-Daley, L., Di Lazzaro, G., and Ghiglieri, V. (2023). Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 14:176. doi: 10.1038/s41419-023-05672-9
Cerri, S., Ghezzi, C., Sampieri, M., Siani, F., Avenali, M., Dornini, G., et al. (2018). The exosomal/total α-synuclein ratio in plasma is associated with glucocerebrosidase activity and correlates with measures of disease severity in PD patients. Front. Cell. Neurosci. 12:125. doi: 10.3389/fncel.2018.00125
Chatterjee, D., and Krainc, D. (2023). Mechanisms of glucocerebrosidase dysfunction in Parkinson’s disease. J. Mol. Biol. 435:168023. doi: 10.1016/j.jmb.2023.168023
Chen, J., Zhao, D., Wang, Q., Chen, J., Bai, C., Li, Y., et al. (2023). Predictors of cognitive impairment in newly diagnosed Parkinson’s disease with normal cognition at baseline: a 5-year cohort study. Front. Aging Neurosci. 15:1142558. doi: 10.3389/fnagi.2023.1142558
Ciccaldo, M., Pérez-Carmona, N., Piovesana, E., Cano-Crespo, S., Ruano, A., Delgado, A., et al. (2025). A novel allosteric GCase modulator prevents Tau accumulation in GBA1WT and GBA1L444P/L444P cellular models. Sci. Rep. 15:17646. doi: 10.1038/s41598-025-02346-8
Cilia, R., Tunesi, S., Marotta, G., Cereda, E., Siri, C., Tesei, S., et al. (2016). Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann. Neurol. 80, 662–673. doi: 10.1002/ana.24777
Colca, J. R., and Finck, B. N. (2022). Metabolic mechanisms connecting Alzheimer’s and Parkinson’s diseases: potential avenues for novel therapeutic approaches. Front. Mol. Biosci. 9:929328. doi: 10.3389/fmolb.2022.929328
Cook, L., Schulze, J., Naito, A., and Alcalay, R. N. (2021). The role of genetic testing for Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 21:17. doi: 10.1007/s11910-021-01100-7
Cumpston, M. S., McKenzie, J. E., Welch, V. A., and Brennan, S. E. (2022). Strengthening systematic reviews in public health: guidance in the Cochrane handbook for systematic reviews of interventions, 2nd edition. J. Public Health 44, e588–e592. doi: 10.1093/pubmed/fdac036
Davis, M. Y., Johnson, C. O., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., et al. (2016). Association of GBA mutations and the E326k polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol. 73, 1217–1224. doi: 10.1001/jamaneurol.2016.2245
De Michele, G., Palmieri, G. R., Pane, C., Valente, E. M., Palmieri, I., Dello Iacovo, C. D. P., et al. (2023). Motor and non-motor features in Parkinson’s disease patients carrying GBA gene mutations. Acta Neurol. Belg. 123, 221–226. doi: 10.1007/s13760-022-02165-y
Do, J., McKinney, C., Sharma, P., and Sidransky, E. (2019). Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 14:36. doi: 10.1186/s13024-019-0336-2
Emre, M., Aarsland, D., Brown, R., Burn, D. J., Duyckaerts, C., Mizuno, Y., et al. (2007). Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 22, 1689–1707. doi: 10.1002/mds.21507
Filippi, M., Balestrino, R., Basaia, S., and Agosta, F. (2022). Neuroimaging in glucocerebrosidase-associated parkinsonism: a systematic review. Mov. Disord. 37, 1375–1393. doi: 10.1002/mds.29047
Gan-Or, Z., Liong, C., and Alcalay, R. N. (2018). GBA-associated Parkinson’s disease and other synucleinopathies. Curr. Neurol. Neurosci. Rep. 18:44. doi: 10.1007/s11910-018-0860-4
Gegg, M. E., and Schapira, A. H. V. (2018). The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 285, 3591–3603. doi: 10.1111/febs.14393
Grabowski, G. A. (2012). Gaucher disease and other storage disorders. Hematology Am. Soc. Hematol. Educ. Program 2012, 13–18. doi: 10.1182/asheducation-2012.1.13
Graham, O. E. E., Pitcher, T. L., Liau, Y., Miller, A. L., Dalrymple-Alford, J. C., Anderson, T. J., et al. (2020). Nanopore sequencing of the glucocerebrosidase (GBA) gene in a New Zealand Parkinson’s disease cohort. Parkinsonism Relat. Disord. 70, 36–41. doi: 10.1016/j.parkreldis.2019.11.022
Granek, Z., Barczuk, J., Siwecka, N., Rozpędek-Kamińska, W., Kucharska, E., and Majsterek, I. (2023). GBA1 gene mutations in α-synucleinopathies-molecular mechanisms underlying pathology and their clinical significance. Int. J. Mol. Sci. 24:2044. doi: 10.3390/ijms24032044
Hely, M. A., Reid, W. G., Adena, M. A., Halliday, G. M., and Morris, J. G. (2008). The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Mov. Disord. 23, 837–844. doi: 10.1002/mds.21956
Higgins, J. P., Thompson, S. G., Deeks, J. J., and Altman, D. G. (2003). Measuring inconsistency in meta-analyses. BMJ 327, 557–560. doi: 10.1136/bmj.327.7414.557
Hill, E. J., Robak, L. A., Al-Ouran, R., Deger, J., Fong, J. C., Vandeventer, P. J., et al. (2022). Genome sequencing in the Parkinson disease clinic. Neurol. Genet. 8:e200002. doi: 10.1212/nxg.0000000000200002
Horowitz, M., Pasmanik-Chor, M., Ron, I., and Kolodny, E. H. (2011). The enigma of the E326k mutation in acid Β-glucocerebrosidase. Mol. Genet. Metab. 104, 35–38. doi: 10.1016/j.ymgme.2011.07.002
Hughes, A. J., Daniel, S. E., Kilford, L., and Lees, A. J. (1992). Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 55, 181–184. doi: 10.1136/jnnp.55.3.181
Iarkov, A., Mendoza, C., and Echeverria, V. (2021). Cholinergic receptor modulation as a target for preventing dementia in Parkinson’s disease. Front. Neurosci. 15:665820. doi: 10.3389/fnins.2021.665820
Jellinger, K. A. (2018). Dementia with Lewy bodies and Parkinson’s disease-dementia: current concepts and controversies. J. Neural Transm. 125, 615–650. doi: 10.1007/s00702-017-1821-9
Kalia, L. V., and Lang, A. E. (2015). Parkinson’s disease. Lancet 386, 896–912. doi: 10.1016/s0140-6736(14)61393-3
Klann, E. M., Dissanayake, U., Gurrala, A., Farrer, M., Wagle Shukla, A., Ramirez-Zamora, A., et al. (2021). The gut-brain axis and its relation to Parkinson’s disease: a review. Front. Aging Neurosci. 13:782082. doi: 10.3389/fnagi.2021.782082
Lee, H. J., Cho, E. D., Lee, K. W., Kim, J. H., Cho, S. G., and Lee, S. J. (2013). Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 45:e22. doi: 10.1038/emm.2013.45
Lesage, S., Anheim, M., Condroyer, C., Pollak, P., Durif, F., Dupuits, C., et al. (2011). Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210. doi: 10.1093/hmg/ddq454
Lill, C. M. (2016). Genetics of Parkinson’s disease. Mol. Cell. Probes 30, 386–396. doi: 10.1016/j.mcp.2016.11.001
Lunde, K. A., Chung, J., Dalen, I., Pedersen, K. F., Linder, J., Domellöf, M. E., et al. (2018). Association of glucocerebrosidase polymorphisms and mutations with dementia in incident Parkinson’s disease. Alzheimers Dement. 14, 1293–1301. doi: 10.1016/j.jalz.2018.04.006
Malec-Litwinowicz, M., Rudzińska, M., Szubiga, M., Michalski, M., Tomaszewski, T., and Szczudlik, A. (2014). Cognitive impairment in carriers of glucocerebrosidase gene mutation in Parkinson disease patients. Neurol. Neurochir. Pol. 48, 258–261. doi: 10.1016/j.pjnns.2014.07.005
Malek, N., Weil, R. S., Bresner, C., Lawton, M. A., Grosset, K. A., Tan, M., et al. (2018). Features of GBA-associated Parkinson’s disease at presentation in the UK tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 89, 702–709. doi: 10.1136/jnnp-2017-317348
Mata, I. F., Leverenz, J. B., Weintraub, D., Trojanowski, J. Q., Chen-Plotkin, A., Van Deerlin, V. M., et al. (2016). GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 31, 95–102. doi: 10.1002/mds.26359
Menozzi, E., Schapira, A. H. V., Blandini, F., and Avenali, M. (2023). Who is at risk of Parkinson disease? Refining the preclinical phase of GBA1 and Lrrk2 variant carriers: a clinical, biochemical, and imaging approach. Curr. Neurol. Neurosci. Rep. 23, 121–130. doi: 10.1007/s11910-023-01259-1
Moher, D., Liberati, A., Tetzlaff, J., and Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6:e1000097. doi: 10.1371/journal.pmed.1000097
Moore, S. F., and Barker, R. A. (2014). Predictors of Parkinson’s disease dementia: towards targeted therapies for a heterogeneous disease. Parkinsonism Relat. Disord. 20, S104–S107. doi: 10.1016/s1353-8020(13)70026-9
Moran, E. E., Wang, C., Katz, M., Ozelius, L., Schwartz, A., Pavlovic, J., et al. (2017). Cognitive and motor functioning in elderly glucocerebrosidase mutation carriers. Neurobiol. Aging 58, 239.e1–239.e7. doi: 10.1016/j.neurobiolaging.2017.06.010
Oeda, T., Umemura, A., Mori, Y., Tomita, S., Kohsaka, M., Park, K., et al. (2015). Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol. Aging 36, 3306–3313. doi: 10.1016/j.neurobiolaging.2015.08.027
Oftedal, L., Maple-Grødem, J., Dalen, I., Tysnes, O. B., Pedersen, K. F., Alves, G., et al. (2023). Association of CSF glucocerebrosidase activity with the risk of incident dementia in patients with Parkinson disease. Neurology 100, e388–e395. doi: 10.1212/wnl.0000000000201418
Pankratz, N., Beecham, G. W., DeStefano, A. L., Dawson, T. M., Doheny, K. F., Factor, S. A., et al. (2012). Meta-analysis of Parkinson’s disease: identification of a novel locus, Rit2. Ann. Neurol. 71, 370–384. doi: 10.1002/ana.22687
Pardo-Moreno, T., García-Morales, V., Suleiman-Martos, S., Rivas-Domínguez, A., Mohamed-Mohamed, H., Ramos-Rodríguez, J. J., et al. (2023). Current treatments and new, tentative therapies for Parkinson’s disease. Pharmaceutics 15:770. doi: 10.3390/pharmaceutics15030770
Riboldi, G. M., and Di Fonzo, A. B. (2019). GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8:364. doi: 10.3390/cells8040364
Rocha, S. M., Kirkley, K. S., Chatterjee, D., Aboellail, T. A., Smeyne, R. J., and Tjalkens, R. B. (2023). Microglia-specific knock-out of NF-κB/IKK2 increases the accumulation of misfolded α-synuclein through the inhibition of p62/sequestosome-1-dependent autophagy in the rotenone model of Parkinson’s disease. Glia 71, 2154–2179. doi: 10.1002/glia.24385
Rosenbloom, B. E., and Weinreb, N. J. (2013). Gaucher disease: a comprehensive review. Crit. Rev. Oncog. 18, 163–175. doi: 10.1615/critrevoncog.2013006060
Schapira, A. H., and Tolosa, E. (2010). Molecular and clinical prodrome of Parkinson disease: implications for treatment. Nat. Rev. Neurol. 6, 309–317. doi: 10.1038/nrneurol.2010.52
Setó-Salvia, N., Pagonabarraga, J., Houlden, H., Pascual-Sedano, B., Dols-Icardo, O., Tucci, A., et al. (2012). Glucocerebrosidase mutations confer a greater risk of dementia during Parkinson’s disease course. Mov. Disord. 27, 393–399. doi: 10.1002/mds.24045
Silva, I., Silva, M. F. D., Dutra, T. S. A., Souza, S. O., Araújo-Júnior, J. X., Leite, A. C. R., et al. (2025). Alpha-synuclein aggregation in Parkinson’s disease. Adv. Protein Chem. Struct. Biol. 146, 35–75. doi: 10.1016/bs.apcsb.2024.11.002
Simuni, T., Brumm, M. C., Uribe, L., Caspell-Garcia, C., Coffey, C. S., Siderowf, A., et al. (2020). Clinical and dopamine transporter imaging characteristics of leucine rich repeat kinase 2 (LRRK2) and glucosylceramidase beta (GBA) Parkinson’s disease participants in the Parkinson’s progression markers initiative: a cross-sectional study. Mov. Disord. 35, 833–844. doi: 10.1002/mds.27989
Smith, L., and Schapira, A. H. V. (2022). GBA variants and Parkinson disease: mechanisms and treatments. Cells 11:1261. doi: 10.3390/cells11081261
Stang, A. (2010). Critical evaluation of the Newcastle–Ottawa Scale for the assessment of the quality of nonrandomized studies in meta-analyses. Eur. J. Epidemiol. 25, 603–605. doi: 10.1007/s10654-010-9491-z
Straniero, L., Asselta, R., Bonvegna, S., Rimoldi, V., Melistaccio, G., Soldà, G., et al. (2020). The SPID-GBA study: sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol. Genet. 6:e523. doi: 10.1212/nxg.0000000000000523
Szwedo, A. A., Dalen, I., Lawson, R. A., Yarnall, A. J., Pedersen, K. F., Macleod, A. D., et al. (2025). Dementia risk prediction in early Parkinson’s disease: Validation and genetic integration of the Montreal Parkinson risk of dementia scale (MoPaRDS). J. Parkinsons Dis. 15, 868–878. doi: 10.1177/1877718x251329857
Szwedo, A. A., Dalen, I., Pedersen, K. F., Camacho, M., Bäckström, D., Forsgren, L., et al. (2022). GBA and APOE impact cognitive decline in Parkinson’s disease: a 10-year population-based study. Mov. Disord. 37, 1016–1027. doi: 10.1002/mds.28932
Vossius, C., Larsen, J. P., Janvin, C., and Aarsland, D. (2011). The economic impact of cognitive impairment in Parkinson’s disease. Mov. Disord. 26, 1541–1544. doi: 10.1002/mds.23661
Williams, D., Glasstetter, L. M., Jong, T. T., Chen, T., Kapoor, A., Zhu, S., et al. (2024a). High-throughput screening for small-molecule stabilizers of misfolded glucocerebrosidase in Gaucher disease and Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 121:e2406009121. doi: 10.1073/pnas.2406009121
Williams, D., Glasstetter, L. M., Jong, T. T., Kapoor, A., Zhu, S., Zhu, Y., et al. (2024b). Development of quantitative high-throughput screening assays to identify, validate, and optimize small-molecule stabilizers of misfolded β-glucocerebrosidase with therapeutic potential for Gaucher disease and Parkinson’s disease. bioRxiv. Available online at: https://doi.org/10.1101/2024.03.22.586364. [Epub ahead of preprint]
Winder-Rhodes, S. E., Evans, J. R., Ban, M., Mason, S. L., Williams-Gray, C. H., Foltynie, T., et al. (2013). Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136, 392–399. doi: 10.1093/brain/aws318
Yahalom, G., Greenbaum, L., Israeli-Korn, S., Fay-Karmon, T., Livneh, V., Ruskey, J. A., et al. (2019). Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: risk estimates and genotype-phenotype correlations. Parkinsonism Relat. Disord. 62, 179–184. doi: 10.1016/j.parkreldis.2018.12.014
Ye, H., Robak, L. A., Yu, M., Cykowski, M., and Shulman, J. M. (2023). Genetics and pathogenesis of Parkinson’s syndrome. Annu. Rev. Pathol. 18, 95–121. doi: 10.1146/annurev-pathmechdis-031521-034145
Zhang, Y., Chen, J., Xu, C., Feng, J., and Li, J. (2020). Effects of glucocerebrosidase gene polymorphisms and mutations on the risk of Parkinson’s disease dementia: a meta-analysis. Neurosci. Lett. 714:134544. doi: 10.1016/j.neulet.2019.134544
Zhang, Y., Wu, Q., Zhang, L., Wang, Q., Yang, Z., Liu, J., et al. (2019). Caffeic acid reduces A53T α-synuclein by activating JNK/Bcl-2-mediated autophagy in vitro and improves behaviour and protects dopaminergic neurons in a mouse model of Parkinson’s disease. Pharmacol. Res. 150:104538. doi: 10.1016/j.phrs.2019.104538
Keywords: glucocerebrosidase, Parkinson’s disease, dementia, meta-analysis, gene variations
Citation: Li Q, Bi Z, Liang W, Yin S, Li H, Liang Z, Wu M, Quan J and Li C (2025) Effects of glucocerebrosidase gene variations on the risk of Parkinson’s disease dementia: a meta-analysis. Front. Aging Neurosci. 17:1671760. doi: 10.3389/fnagi.2025.1671760
Edited by:
Roberta Marongiu, Cornell University, United StatesReviewed by:
Laetitia Francelle, Northwestern University, United StatesMicol Avenali, University of Pavia, Italy
Copyright © 2025 Li, Bi, Liang, Yin, Li, Liang, Wu, Quan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheng Li, Y2hlbmdsaTIwMjAwM0AxNjMuY29t
†These authors share first authorship