
 Quentin Chevy1,2,3
Quentin Chevy1,2,3
- 1INSERM, UMR-839, Paris, France
- 2Université Pierre et Marie Curie, Paris, France
- 3Institut du Fer à Moulin, Paris, France
The K-Cl co-transporter KCC2 plays multiple roles in the physiology of central neurons and alterations of its function and/or expression are associated with several neurological conditions. By regulating intraneuronal chloride homeostasis, KCC2 strongly influences the efficacy and polarity of the chloride-permeable γ-aminobutyric acid (GABA) type A and glycine receptor (GlyR) mediated synaptic transmission. This appears particularly critical for the development of neuronal circuits as well as for the dynamic control of GABA and glycine signaling in mature networks. The activity of the transporter is also associated with transmembrane water fluxes which compensate solute fluxes associated with synaptic activity. Finally, KCC2 interaction with the actin cytoskeleton appears critical both for dendritic spine morphogenesis and the maintenance of glutamatergic synapses. In light of the pivotal role of KCC2 in the maturation and function of central synapses, it is of particular importance to understand the cellular and molecular mechanisms underlying its regulation. These include development and activity-dependent modifications both at the transcriptional and post-translational levels. We emphasize the importance of post-translational mechanisms such as phosphorylation and dephosphorylation, oligomerization, cell surface stability, clustering and membrane diffusion for the rapid and dynamic regulation of KCC2 function.
Introduction
Fast synaptic transmission relies on ion fluxes through ligand-gated channels. Therefore, the maintenance of transmembrane ionic gradients is critical to preserve synaptic efficacy. The electroneutral KCC2 co-transporter is the major chloride (Cl−) extruder in mature neurons (Payne et al., 1996; Williams et al., 1999; Karadsheh and Delpire, 2001; Uvarov et al., 2005). Unlike other KCC family members known to regulate cell volume in non-neuronal cells (Zeuthen and MacAulay, 2002; Zeuthen, 2010), KCC2 has been mostly studied for its role in maintaining low intracellular chloride concentration [Cl−]i in neurons [reviewed in (Ben-Ari, 2002; Blaesse et al., 2009)]. [Cl−]i influences the efficacy and polarity of synaptic transmission mediated by γ-aminobutyric acid (GABA) type A receptors (GABAARs) and glycine receptors (GlyRs) which both flux chloride ions. The spatio-temporal regulation of KCC2 mRNA and protein expression levels orchestrates the developmental shift in synaptic glycinergic and GABAergic transmission from depolarizing to hyperpolarizing or shunting in many but not all species (Rivera et al., 1999; Vanhatalo et al., 2005) and brain regions (Li et al., 2002; Vinay and Jean-Xavier, 2008). GABAA receptor-mediated depolarization appears as a critical determinant of early network activities (Cherubini et al., 2011), circuit formation (Ben-Ari, 2002; Akerman and Cline, 2006), neuronal migration (Bortone and Polleux, 2009), and synapse maturation (Aguado et al., 2003). Recent studies also indicate a critical role of KCC2 in both the formation (Li et al., 2007) and functional maintenance (Gauvain et al., 2011) of glutamatergic synapses. These mechanisms appear independent of KCC2 function but instead involve KCC2 interaction with submembrane cytoskeleton.
KCC2 expression and function are tightly regulated by neuronal activity (Fiumelli and Woodin, 2007). This regulation involves trophic factors such as brain-derived neurotrophic factor (BDNF), neuronal intrinsic activity or the activity of excitatory synapses (Kaila et al., 1997; Rivera et al., 2002, 2004; Woodin et al., 2003; Fiumelli et al., 2005; Wang et al., 2006a–c; Kitamura et al., 2008; Lee et al., 2011). In some conditions, neuronal activity may either upregulate KCC2 to strengthen synaptic inhibition in a homeostatic manner or, on the contrary, downregulate KCC2 possibly to enhance the gain of recently active excitatory synapses. In several pathological conditions associated with enhanced excitation, suppression of KCC2 is often observed and may contribute to further alter the balance of excitation and inhibition, leading to excitotoxicity, or anomalous activities such as seizures (Reid et al., 2001; Rivera et al., 2002, 2004; Huberfeld et al., 2007; Pathak et al., 2007; Wake et al., 2007; Li et al., 2008b; Shimizu-Okabe et al., 2011). Therefore, KCC2 appears to mediate a crosstalk between excitatory and inhibitory transmission. It is, therefore, crucial to understand the mechanisms that control its expression and activity. Neuronal activity modulates KCC2 activity through both transcriptional and post-translational modifications. Whereas transcriptional mechanisms may be involved in regional, developmental, and regulation of KCC2 activity in pathological conditions, post-translational mechanisms of KCC2 regulation occurs in a time scale compatible with a control by fast synaptic transmission (Woodin et al., 2003; Wang et al., 2006b; Fiumelli and Woodin, 2007; Wake et al., 2007; Kitamura et al., 2008; Chorin et al., 2011; Lee et al., 2011). Several recent and comprehensive reviews have addressed the biology of cation-chloride co-transporters (CCCs) both in neuronal (Blaesse et al., 2009) and non-neuronal cells (Gamba, 2005). Here, we will review the recent literature more specifically on (1) the subcellular distribution of KCC2 in relation with excitatory and inhibitory synapses, (2) its basic properties as well as its function at inhibitory and excitatory synapses, (3) its regulation during development and by neuronal activity. We will discuss the cellular and molecular mechanisms underlying its regulation.
Structure and Diversity of KCC2 Co-Transporters
The K-Cl co-transporter KCC2 is one of the nine cation chloride co-transporters encoded by the Slc12a one to nine genes [for review see (Gamba, 2005)]. Four KCC co-transporters have been identified so far (KCC1–4). KCC2 is the only KCC isoform exclusively expressed in central neurons (Payne et al., 1996; Williams et al., 1999; Karadsheh and Delpire, 2001; Uvarov et al., 2005). The lack of KCC2 mRNA expression in non-neuronal cells relies on binding of the neuron-restrictive silencer factor (NRSF) to the neuron-restrictive silencer element (NRSE) in KCC2 gene promoter (Uvarov et al., 2005). Two transcription factors up- or down-regulate KCC2 mRNA levels (Uvarov et al., 2006). The transcription factor early growth response 4 (Egr4 or NGFI-C) is a neuron-specific immediate early gene that enhances KCC2 expression (Uvarov et al., 2006). In contrast, the REST transcriptional repressor complex inhibits KCC2 mRNA level by binding to two repressor elements (RE-1) in the KCC2 gene (Yeo et al., 2009). The KCC2 mRNA is expressed with a gradual temporal increase from spinal cord and brainstem to higher brain structures (Li et al., 2002; Stein et al., 2004). Alternative splicing of the KCC2 gene (Slc12a5) gives rise to two KCC2 isoforms, KCC2a and KCC2b (Uvarov et al., 2007). The KCC2a transcript is expressed at low-level between E14-P60 (Uvarov et al., 2007). In contrast, the KCC2b mRNA is up-regulated by 10- and 35-fold in hippocampus and neocortex between E17 and P14 (Uvarov et al., 2007). Similarly to the transcripts, KCC2a protein shows only moderate changes during postnatal development, whereas KCC2b expression is strongly up-regulated (Uvarov et al., 2009). Therefore, KCC2a prevails in neonatal brain whereas KCC2b predominates in adult (Uvarov et al., 2009). KCC2b differs from KCC2a by an extra 40 amino acid residue in its N-terminal part that carries a putative binding site for the Ste20-related proline alanine-rich (SPAK) and oxidative stress response-1 (OSR1) kinases (Uvarov et al., 2007).
The KCC2 protein is a glycoprotein with a predicted topology of 12 membrane spanning segments flanked by two cytoplasmic carboxy- and amino-terminal domains of long and short size, respectively (Payne et al., 1996). The intracellular regions represent half of the total size of the molecule and are known so far to be the targets of several kinases and one phosphatase that regulate the function of the co-transporter (Figure 1). KCC2 carries putative phosphorylation sites for the Src-family tyrosine kinase [residues Tyr903, Tyr1087, (Lee et al., 2010)], the serine/threonine With no lysine kinase (Wnk) family Wnk3, (Kahle et al., 2005); Wnk1 at residues Thr906, Thr1006, (Rinehart et al., 2009); Wnk2, (Rinehart et al., 2011), and the Ca2+/phospholipid-dependent protein kinase C (PKC) residues Thr34, Ser728, Thr787, Ser940, Ser1034, (Payne et al., 1996; Lee et al., 2007). Among the five putative PKC-dependent phosphorylation sites, the Ser940 residue is the major site of PKC phosphorylation in neurons (Lee et al., 2007). This site is specific of the KCC2 transporter (Payne et al., 1996) and can be dephosphorylated by the protein phosphatase 1 (PP1) (Lee et al., 2011).
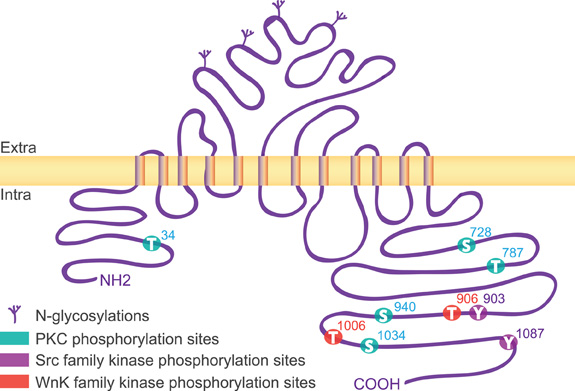
Figure 1. Structure of KCC2. The rat KCC2 co-transporter is a large size (∼140 kDa) protein with a predicted topology of 12 membrane spanning segments, a N-linked glycosylated extracellular domain between transmembrane domains 5 and 6, and is flanked by two cytoplasmic carboxy- and amino-terminal domains of 104 and 481 amino acids, respectively. As indicated, the intracellular regions are the targets of several kinases that regulate the function of the co-transporter.
KCC2 can exist as monomers (∼140 kDa), dimers (∼270 kDa), trimers (∼400 kDa), or tetramers (∼500 kDa) (Blaesse et al., 2006; Uvarov et al., 2009). Immunoblots and co-immunoprecipitation studies from brain extracts reported various oligomeric states of KCC2: KCC2a-KCC2a and KCC2b-KCC2b homo-dimers, KCC2a-KCC2b, and KCC2-KCC4 hetero-dimers (Blaesse et al., 2006; Simard et al., 2007; Uvarov et al., 2009). Although KCC oligomerization domains have not been identified so far, the carboxy-terminal domain appears to be required for oligomerization (Casula et al., 2001; Simard et al., 2004). Thus, truncation of KCC1 in its carboxy-terminus (up to residue 805) fails to oligomerize (Casula et al., 2001). KCC2 oligomers also associate through di-sulfide bounds (Uvarov et al., 2009), as described for other CCCs (Moore-Hoon and Turner, 2000; Blaesse et al., 2006).
Cellular and Subcellular Distribution in Neurons
Regional Expression
KCC2a and KCC2b transcripts are expressed in mature neurons throughout the CNS including all layers of the cortex, neurons of the brainstem, thalamus, olfactory bulb, and spinal cord, CA1–CA4 pyramidal neurons of the hippocampus, granular layer, and cerebellar Purkinje neurons (Payne et al., 1996; Uvarov et al., 2007). Most published immunochemical data on KCC2 have been obtained with antibodies against a carboxy-terminal region of KCC2 common to both KCC2a and KCC2b isoforms (residues 932–1043, commercialized by Sigma, Millipore, AbCam, Life Span…). KCC2a and KCC2b isotype specific antibodies were recently generated: KCC2a, amino-terminal residues DPESRRHSVADPRRLPREDVK, (Uvarov et al., 2009); KCC2b, amino-terminal residues CEDGDGGANPGDGN, (Hubner et al., 2001). Finally, an antibody against the second extracellular loop of KCC2 (IFKAEDASGEAAAML residues) was obtained and characterized in Gagnon et al. (2007) but failed to yield specific staining in our hands. Since KCC2b prevails in adults, immunofluorescence (IF) data from adult tissue predominantly reveals the distribution of this isoform. KCC2 is detected in neurons of the cerebellum (Williams et al., 1999; Takayama and Inoue, 2006), all relay nuclei of the thalamus (Bartho et al., 2004), retina (Vardi et al., 2000; Vu et al., 2000; Gavrikov et al., 2006; Zhang et al., 2006; Li et al., 2008a), lateral superior olive (LSO) of the brainstem (Blaesse et al., 2006), neocortex (DeFazio et al., 2000; Szabadics et al., 2006), somatosensory cortex (Takayama and Inoue, 2006, 2010), hippocampus (Rivera et al., 1999; Gulyas et al., 2001), spinal cord (Hubner et al., 2001; Stil et al., 2009), cochlear nucleus (Vale et al., 2005; Yang et al., 2008), and hypothalamic suprachiasmatic nucleus (SCN) (Belenky et al., 2008).
Cellular Compartmentalization
KCC2 is restricted to the somato-dendritic compartment and excluded from the axon (Hubner et al., 2001; Szabadics et al., 2006; Bartho et al., 2009) including the initial segment (AIS) (Gulyas et al., 2001; Baldi et al., 2010). KCC2 immunoreactivity is usually evenly distributed along the somato-dendritic axis of neurons of the thalamus (Bartho et al., 2009), the granular layer of the cerebellum (Takayama and Inoue, 2006) and dentate gyrus granule cells (Baldi et al., 2010). However, in OFF bipolar cells and starburst cells of the retina, KCC2 is confined in distal dendrites (Vardi et al., 2000; Gavrikov et al., 2006). Similarly, in hippocampal CA1 pyramidal neurons, KCC2 may preferentially accumulate at GABAergic synapses formed onto distal rather than proximal dendrites (Baldi et al., 2010).
Intracellular vs. Membrane Distribution
Intracellular KCC2 labeling is found at a higher level in immature than in mature neurons. KCC2 decorates the membrane of transport vesicles in dendrites of P2 hippocampal neurons (Gulyas et al., 2001). In contrast, only little cytoplasmic KCC2 is detected in mature hippocampal neurons (Gulyas et al., 2001) as well as neurons of the cochlear nucleus (Vale et al., 2005) and isocortex (Szabadics et al., 2006). In the adult brain, organelles including Golgi apparatus and endoplasmic reticulum do not show KCC2 labeling (Takayama and Inoue, 2006) except in neurons of the SCN (Belenky et al., 2008). Instead, most KCC2 protein is found associated with the plasma membrane both in somatic and dendritic compartments (Williams et al., 1999; Gulyas et al., 2001; Hubner et al., 2001; Vale et al., 2005; Blaesse et al., 2006; Szabadics et al., 2006; Takayama and Inoue, 2006; Belenky et al., 2008; Bartho et al., 2009; Baldi et al., 2010). KCC2 exhibits a high turnover rate. It was shown that the membrane pool of KCC2 is partially (Rivera et al., 2004) or totally (Lee et al., 2007) replaced within 10 min under basal conditions.
Synaptic Localization
At the synaptic level, KCC2 is not found within presynaptic boutons except in terminals of the retinal ON bipolar cells (Vardi et al., 2000) and developing photoreceptor cells (Zhang et al., 2006). KCC2-coupled immunogold particles decorate the perisynaptic area of postsynaptic neuronal cell (Gulyas et al., 2001; Bartho et al., 2004; Blaesse et al., 2006; Takayama and Inoue, 2006). Consistent with a major role of the transporter in chloride homeostasis and GABA signaling (see below), KCC2 is found in the postsynaptic membrane near symmetrical inhibitory synapses in brainstem (Blaesse et al., 2006), SCN (Belenky et al., 2008), and spinal cord (Hubner et al., 2001). However, KCC2 also accumulates at or near excitatory synapses formed by cerebellar mossy fiber terminals onto granule cells (Takayama and Inoue, 2006), at terminals formed between the cortico-geniculate fibers and thalamic relay nuclei neurons (Bartho et al., 2004), at synapses in the brainstem (Blaesse et al., 2006) and at glutamatergic synapses in spine heads of principal cells of the hippocampus (Gulyas et al., 2001; Gauvain et al., 2011).
We have studied the subcellular distribution of KCC2 in cultured hippocampal neurons. In line with previous observations (Lee et al., 2007; Watanabe et al., 2009; Gauvain et al., 2011), KCC2 membrane distribution was usually punctate. Using confocal fluorescence microscopy, we found that KCC2 clusters decorate the cell body (Figures 2A,B), dendritic shafts (Figure 2C) and spines (Figures 2A–D2) of mature neurons (29 days in vitro). Optical sectioning through a dendrite highlighted the preferential membrane localization of the transporter (Figures 2D2,D3). On average, KCC2 IF was about 50% higher near cell surface compared with cytoplasm, reflecting a preferential membrane or sub-membrane localization of the transporter. The fluorescence intensity of KCC2 aggregates in spines was about threefold higher than in cytoplasm and twofold higher than on dendritic shaft, demonstrating KCC2 enrichment in spines. KCC2 clustering was also dependent on spine maturation and was maximal in mushroom-type spines, intermediate in stubby spines and absent in filopodia-like structures. KCC2 expression in relation with excitatory and inhibitory synapses was evaluated in hippocampal neurons co-immunolabeled with the glutamate and GABA receptor anchoring proteins PostSynaptic Density protein 95 kD (PSD95) and gephyrin (Figures 2E1–G). KCC2 clusters were found both near inhibitory (Figures 2E1–E3,G) and excitatory (Figure 2F1–F3,G) synapses formed on dendrites as well as on the extrasynaptic membrane. However, although concentrated in spines, the co-transporter did not preferentially accumulate within the postsynaptic differentiation (PSD). Instead, it was often localized at the periphery of the PSD (Figure 2G). Quantification also revealed the absence of specific KCC2 accumulation at inhibitory synapses (Figure 2G). Thus, KCC2 appears to aggregate in close vicinity rather than within synapses, suggesting the existence of specific molecular anchoring mechanisms acting to influence KCC2 clustering.
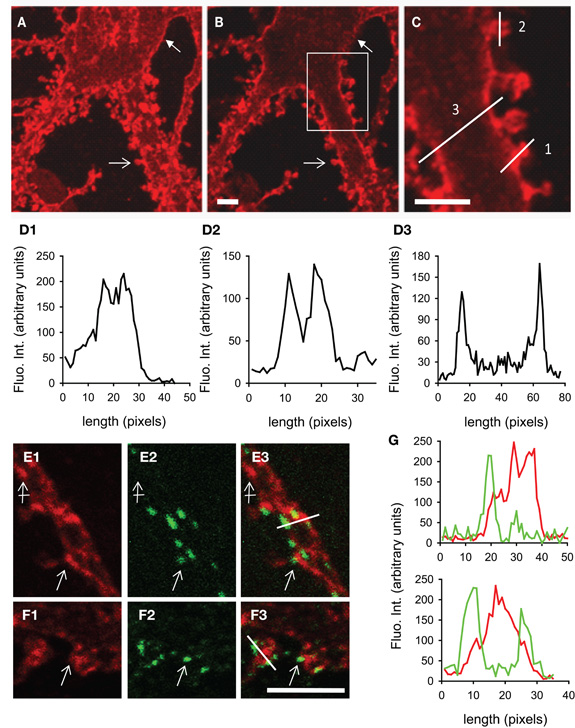
Figure 2. KCC2 clustering in rat hippocampal neurons at 29DIV. (A) Maximum intensity projection of confocal optical sections showing KCC2 at the periphery of somata (filled arrow) and in spines (empty arrow). (B,C) optical sectioning from the same neuron as in (A). Boxed region in (B) is shown enlarged in (C). Adapted from Gauvain et al. (2011) with permission. Scale bars, 5μm. (D) Fluorescence intensity in arbitrary units per pixel along the lines drawn in (C) showing enrichment of KCC2 in dendritic spines (D1), preferential plasma membrane localization of KCC2 in dendritic spines (D2) and shafts (D3) as compared with the cytoplasm. (E1–F3) Dual labeling of KCC2 (red in E1, E3, F1, F3) and the GABA and glutamate receptor anchoring proteins gephyrin (green in E2, E3) or PSD-95 (green in F2, F3). Scale bars, 5μm. KCC2 forms many clusters on dendritic shafts and spines (empty arrows). Clusters are found at distance from synapses (crossed arrow) as well as near gephyrin-labeled inhibitory synapses (empty arrow in E1–3) and PSD95-stained excitatory PSD (empty arrow in F1–3). (G) Fluorescence intensity in arbitrary units per pixel along the lines drawn in (E3) and (F3) showing juxtaposition but no colocalization of KCC2 (red) with gephyrin (green, top) or PSD95 (green, bottom).
Basic Properties of the KCC2 Co-Transporter
Ion Co-Transport
KCC2 is a secondary active transporter, i.e., it uses the energy of the electrochemical K+ gradient to transport Cl−. The electrochemical plasma membrane K+ gradient is generated by the primary active transporter Na-K ATPase (Figure 3). KCC2 colocalizes and structurally interacts with the α2 subunit of the Na-K ATPase (Ikeda et al., 2004). Therefore, an alteration of KCC2 membrane expression may impact the Na-K ATPase distribution and/or activity and vice versa. KCC2 functions near thermodynamic equilibrium since the equilibrium potential of K+ (EK) is close to the resting membrane potential (RMP) (Vm) (Payne, 1997; Payne et al., 2003). KCC2 can function in either direction depending on the external concentration of K+ [K+]o. Even a moderate increase in [K+]o may thus reverse Cl− transport through KCC2. This action may allow K+ buffering when its extracellular concentration is increased upon sustained neuronal activity (Thompson and Gahwiler, 1989b; Payne, 1997).
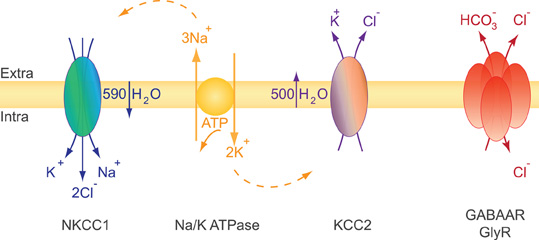
Figure 3. Ions and water transport properties of the co-transporters NKCC1 and KCC2. The secondary active transporters NKCC1 and KCC2 use the energy of the electrochemical Na+ and K+ gradients generated by the primary active transporter Na-K ATPase to trigger a net inflow of 1K+, 1Na+, 2Cl−, and a net outflow of 1K+, 1Cl− ions, respectively. The activity of each transporter is associated with water influx (NKCC1) or efflux (KCC2). NKCC1 and KCC2 are reciprocally expressed and regulated during development. NKCC1 predominates in early development while KCC2 is the main Cl− extruder in mature neurons. This expression profile impacts both cell volume regulation and the efficacy and polarity of GlyR and GABAAR mediated synaptic transmission (see text).
Water Transport and the Control of Cell Volume
Glutamate and GABA/glycine synaptic signaling usually lead to a net influx of Na+ and/or Ca2+ cations and Cl− anions, respectively. These movements of ions are accompanied by water inflow acting to maintain intracellular osmolarity. Massive water influx may consequently lead to neuronal swelling (Collingridge and Lester, 1989) and, if not compensated, to cell death. Since neurons lack aquaporins (Amiry-Moghaddam and Ottersen, 2003; Andrew et al., 2007) they may use an alternative mechanism to face osmotic challenges. CCCs have been extensively studied in this context of osmotic and volume regulation first in erythrocytes and epithelial cells, two non-neuronal cell types facing large osmotic challenges (Zeuthen and MacAulay, 2002; Hoffmann et al., 2009). It is well established that CCCs function is directly or indirectly coupled to water transport (Zeuthen and MacAulay, 2002; Zeuthen, 2010). Water flows in the same direction as ions to counteract osmotic changes due to CCC activity. The electroneutral Na+–K+–2Cl− NKCC1 co-transporter accumulates intracellular chloride using the electrochemical gradient for Na+ and K+ produced by the Na-K ATPase (Gamba, 2005; Blaesse et al., 2009). Both NKCC1 and KCC2 co-transport ions and water and cooperate to maintain ionic and water homeostasis in neurons. It has been estimated that NKCC1 activity triggers a net inflow of 590 water molecules for 1K+, 1Na+, 2Cl− transported ions. Conversely, KCC2 activity results in the extrusion of 500 water molecules per of 1K+ and 1Cl− transported ions [reviewed in (MacAulay and Zeuthen, 2010)]. Consistent with the role of KCC2 in water transport in neurons, experiments using digital holographic microscopy recently revealed that N-methyl-D-aspartate (NMDA) receptor activation in neurons triggers water influx through both NMDAR and NKCC1 while delayed activation of KCC2 compensates this influx by mediating a net outflow of water (Jourdain et al., 2011).
The functional properties of NKCC1 and KCC2 co-transporters are reciprocally regulated by serine/threonine phosphorylation. The Wnk kinase family acts as an osmosensor which can lead to reciprocal regulation of NKCC1 and KCC2 in the central nervous system (Kahle et al., 2010). Upon hyper-osmotic challenge or in conditions of low [Cl−]i, activation of Wnk1/3 leads to phosphorylation and activation of NKCC1 through OSR1/SPAK Ste20-type kinases and direct inhibition of KCC2 through phosphorylation of KCC2 Thr906 and Thr1006 (Gagnon et al., 2006; Rinehart et al., 2009). This results in a net water influx which compensates for cell shrinking due to the hyperosmotic challenge. This process is known as a Regulatory Volume Increase (RVI). Conversely, under hypo-osmotic conditions, activation of KCC2 and inhibition of NKCC1 leads to a Regulatory Volume Decrease (RVD) acting to compensate cell swelling.
Interactions with the Actin Cytoskeleton
Apart from their ion transport function, several ion channels, pumps, transporters, and exchangers appear to act as cytoskeletal anchors of the plasma membrane [reviewed in (Denker and Barber, 2002)]. Together with other integral membrane proteins, several ion transport proteins anchor actin filaments to the plasma membrane via interactions with the linker proteins of the ankyrin and 4.1 families. A particularly well studied example of such interaction has been described in erythrocytes, where the anion exchanger 1 (AE1) plays a critical role in docking actin cytoskeleton to plasma membrane via ankyrin and 4.1R, thereby influencing cell shape (Jons and Drenckhahn, 1992). KCC2 immunoprecipitation assays recently revealed that the carboxy-terminal domain of KCC2 (KCC2 CTD) directly interacts with 4.1N, a neuronal member of the 4.1 protein family (Li et al., 2007). 4.1N, like other 4.1 family members, possesses a 4.1 Ezrin Radixin Moesin (FERM) domain that binds a variety of transmembrane proteins and a spectrin/actin interaction domain (Baines et al., 2009). Therefore, KCC2 is expected to interact with the submembrane actin cytoskeleton. Such interaction may have multiple implications including (1) to regulate KCC2 clustering and thereby its membrane stability and/or activity, (2) to constrain KCC2 localization to specific membrane microdomains, (3) to control the local organization of actin filaments with downstream effects on subcellular morphology (Mohandas et al., 1992; Cancedda et al., 2007; Li et al., 2007; Gauvain et al., 2011), (4) to regulate cell migration (Wei et al., 2011) and (5) to affect the osmoregulatory response since actin is part of the osmotic sensor of the cell (Mills et al., 1994). These major functions of the KCC2 co-transporter are likely at play in all neuronal compartments where it is expressed. Nevertheless, they may be of particular significance at synapses where they appear to influence synaptic structure and maturation as well as efficacy.
Functional Impact of KCC2 on Synaptic Signaling
Function in Synaptogenesis
Numerous studies have revealed a critical role of KCC2 in synaptogenesis. Early overexpression of recombinant KCC2 in immature hippocampal neurons increases the density of GABAergic terminals, the number of postsynaptic GABAAR clusters as well as the frequency and amplitude of GABAAR-mediated mIPSCs (Chudotvorova et al., 2005). Conversely, the frequency of mIPSCs is reduced in hypomorphic KCC2 mice (Tornberg et al., 2005), suggesting KCC2 is required for the functional maturation of GABAergic synapses. However, the role of KCC2 in the formation of glutamatergic synapses is more controversial. The controversy may result from differences in preparations and experimental methodologies. For instance, early overexpression of KCC2 in Xenopus laevis embryos reduced the amplitude and frequency of mEPSCs in tectal neurons (Akerman and Cline, 2006) suggesting elevated [Cl−]i may be required for functional maturation of excitatory synapses. Instead, overexpression of KCC2 had no effect on the density of vesicular glutamate transporter isoform 1 (VGlut1)-immunopositive terminals or mEPSC amplitude or frequency in cultured hippocampal neurons (Chudotvorova et al., 2005). This observation, however, contrasts with the effects of the genetic ablation of KCC2 which leads to a reduced number of functional excitatory synapses in immature hippocampal neurons (Li et al., 2007). Finally, in striking contrast with these data, Khalilov et al. (2011) reported a sixfold increase in the density of synaptophysin immunoreactive terminals and increased frequency of spontaneous IPSCs and EPSCs as well as enhanced network activity in CA3 hippocampal neurons from KCC2−/− E18.5 mouse embryos. These discrepancies may result from the timing of both KCC2 manipulations and functional observations and suggest KCC2 differentially modulates synaptogenesis in a very specific time window.
KCC2 may influence synaptogenesis through an ion-transport-independent mechanism (Li et al., 2007; Khalilov et al., 2011). However, the effects of KCC2 on the development of retinotectal circuits rely on a modulation of GABA signaling through shifting transmembrane chloride gradients (Akerman and Cline, 2006). Thus, depolarizing GABA signals may cooperate with NMDAR-mediated transmission to promote the maturation of glutamatergic synapses and the establishment of the balance of excitation and inhibition in developing circuits [for review see (Ben-Ari et al., 2007)].
Functionnal Impact on GABA and Glycine Signaling
Here, we will present a synthetic view of the well-known impact of KCC2 on inhibitory synaptic transmission and will refer to recent and complete reviews (Ben-Ari, 2002; Ben-Ari et al., 2007; Blaesse et al., 2009). The KCC2-mediated K-Cl co-transport critically determines the electrochemical gradient of chloride ions in neurons. Therefore, a major impact of KCC2 function is on the efficacy or even the polarity of synaptic GABAergic and glycinergic transmissions which both rely on chloride fluxes.
Both GABAARs and GlyRs are primarily permeable to chloride and, to a lesser extent, bicarbonate ions (Bormann et al., 1987). Although these signals are classically considered as ‘inhibitory’, their polarity and functional impact are dependent on (1) the transmembrane gradients in chloride and bicarbonate ions and (2) the local RMP. Thus, GABAAR-mediated currents are hyperpolarizing only when EGABA (the reversal potential of GABAAR currents, which depends on both ECl and EHCO3) is hyperpolarized to RMP. Since, under physiological conditions, EHCO3 is depolarized as compared to RMP [around −12 mV, (Staley et al., 1995)], a rise in [Cl]i may be sufficient to depolarize EGABA above RMP, leading to depolarizing actions of GABAAR-mediated currents. This may occur, for instance, during sustained GABAergic activity leading to intraneuronal chloride accumulation (Thompson and Gahwiler, 1989a). It should be noted however, that depolarizing glycine or GABAAR-mediated currents may still be functionally inhibitory due to the electrical shunt of the membrane input resistance generated by the opening of these receptors (Staley and Mody, 1992).
Although measuring [Cl−]i in neurons remains a technical challenge potentially subject to many pitfalls (Bregestovski et al., 2009), several studies converge to suggest it may range relatively high values during early postnatal development [25–40 mM; refs in (Blaesse et al., 2009)]. This likely reflects the expression and activity of the NKCC1 transporter which acts to accumulate chloride in neurons and the expression of which precedes that of KCC2 (Plotkin et al., 1997; Hubner et al., 2001). Elevated internal chloride concentration depolarize EGABA above RMP and predicts depolarizing actions of GABA acting on GABAARs in immature neurons. Although this view has been taken for granted for many years (Ben-Ari, 2002), it was recently challenged by experiments suggesting both RMP and EGABA may be strongly dependent on the energy supply to immature neurons, which may be inappropriate in experimental conditions used to measure these variables (Rheims et al., 2009; Ruusuvuori et al., 2010; Tyzio et al., 2011). Nevertheless, several actions of GABA have been associated with membrane depolarization due to low KCC2 function and high intracellular chloride in immature neurons. Those include the generation of population activities such as giant depolarizing potentials (GDPs) in vitro (see Ben-Ari, 2002), the maturation of retinotectal circuits (Akerman and Cline, 2006) and cortical interneuron migration (Bortone and Polleux, 2009) in vivo.
It is generally agreed that an increase in KCC2 function correlates with a progressive hyperpolarization of EGABA reflecting a reduction of [Cl−]i in mature neurons to about 5 mM (Khirug et al., 2008; Tyzio et al., 2008). This developmental switch in chloride electrochemical gradient occurs in almost all brain structures and organisms analyzed, but with variation in the spatio-temporal developmental course (Ben-Ari, 2002). It should be noted however that steady-state [Cl−]i is an average and shows intercellular and intracellular variability. GABAAR-mediated currents impinging onto neighboring neurons or even onto different subcellular compartments onto a given neurons may, therefore, be very different. In particular, [Cl−]i may differ up to two to threefold between the somatic and dendritic compartments (Duebel et al., 2006) or even from one dendritic branch to the other (Waseem et al., 2010). Similarly, an axo-somatic gradient of EGABA has been reported which may range up to 15–20 mV (Szabadics et al., 2006; Khirug et al., 2008). Thus, GABAAR-mediated currents are usually depolarizing on axon terminals [e.g., (Ruiz et al., 2003); see (Kullmann et al., 2005)] or AISs [(Szabadics et al., 2006; Khirug et al., 2008); but see (Glickfeld et al., 2009)] and hyperpolarizing on somato-dendritic compartments [(Banke and McBain, 2006; Tyzio et al., 2006; Khirug et al., 2008); but see (Tyzio et al., 2008)]. This axo-somatic gradient likely reflects the differential subcellular distribution of KCC2 and NKCC1 (Khirug et al., 2008). In addition, local, activity-dependent alteration of [Cl−]i along a single dendrite is correlated with a transient and local shift in EGABA (Dallwig et al., 1999; Staley and Proctor, 1999; Kuner and Augustine, 2000; Isomura et al., 2003; Jedlicka et al., 2011), the dynamics of which likely depends on KCC2 function [see (Doyon et al., 2011)]. Thus, KCC2 dynamically regulates the efficacy of GABA signaling through a local control over [Cl−]i. Therefore, modulation of KCC2 function may not only impact the steady-state efficacy of GABA signaling through GABAARs but also its dynamic operation. This may occur for instance upon repetitive neuronal activity (Fiumelli and Woodin, 2007) as well as in several pathological conditions which have been shown to both suppress KCC2 expression and affect the efficacy of GABA signaling. These include epilepsy (Cohen et al., 2002; Huberfeld et al., 2007), spinal cord injury (Boulenguez et al., 2010), neuropathic pain (Coull et al., 2003) and others (see Kahle et al., 2008).
Function at Excitatory Synapses
Although KCC2 primarily influences GABAergic transmission in cortical neurons, its accumulation in dendritic spines near excitatory synapses (Gulyas et al., 2001) suggests a possible interaction with glutamatergic signaling. Most excitatory synapses onto cortical neurons are formed onto dendritic spines which appear concomitantly with the up-regulation of KCC2 during early postnatal development (Rivera et al., 1999; Yuste and Bonhoeffer, 2004). In the absence of KCC2 expression in knock-out animals, spine morphogenesis is largely compromised in immature neurons, leading to anomalously long, filopodia-like protrusions (Li et al., 2007). This defect in spine maturation is associated with a reduced number of functional excitatory synapses as detected by a decreased density of synaptic clusters of the glutamate receptor anchoring proteins PSD95 and Homer and reduced mEPSC frequency. Importantly, this effect can be rescued by expression of a non-functional, N-terminal deficient KCC2 and mimicked by expression of the KCC2 CTD (Li et al., 2007). This observation suggests that KCC2 influences spine morphogenesis and functional maturation through a mechanism independent of its ion transport function.
KCC2 CTD directly interacts with the neuronal FERM-domain actin-binding protein 4.1N (Li et al., 2007). F-actin constitutes the main cytoskeleton of dendritic spines and its dynamic organization is known to play a critical role in spine morphogenesis (Tada and Sheng, 2006). In particular, submembrane actin dynamics have been shown to be essential during spine head formation and the transition from filopodium to dendritic spine (Hotulainen et al., 2009). Thus, KCC2–4.1N interaction may contribute to organize submembrane actin cytoskeleton in developing dendritic spines. Consistent with this hypothesis, overexpression of 4.1N FERM domain to prevent KCC2–4.1N interaction is sufficient to prevent spine morphogenesis in hippocampal neurons (Li et al., 2007).
Remodeling of the actin cytoskeleton not only impacts spinogenesis but also affects excitatory synaptic function (Hotulainen and Hoogenraad, 2010). Actin-depolymerizing factors, in particular, induce changes in synaptic function that may reflect changes in glutamate receptor membrane diffusion (Rust et al., 2010). We recently reported that chronic suppression of KCC2 after spine formation does not compromise spine maintenance in mature (>24 DIV) hippocampal neurons (Gauvain et al., 2011). Instead, it is associated with a reduction in the efficacy of excitatory synapses and aggregation of the GluA1 subunit in dendritic spines. Again, this effect is independent of a loss of KCC2 function since it is mimicked by overexpressing the KCC2 CTD but not by application of a selective KCC2 antagonist. Using single particle tracking (SPT) techniques, we showed that suppression of KCC2 expression induced an increase in the lateral diffusion of rapidly moving, likely extrasynaptic AMPA receptors (Tardin et al., 2003; Petrini et al., 2009) in dendritic spines but not on dendritic shafts. Immobile (likely postsynaptic) receptors were also unaffected. Thus, KCC2 interaction with intracellular partners is essential to spine morphogenesis but not maintenance, and contributes to hinder lateral diffusion of AMPA receptors within dendritic spines, thereby influencing synaptic efficacy (Figure 4). A loss of KCC2 clusters in dendritic spines induced by sustained excitatory synaptic activity or under pathological conditions may then induce a rapid homeostatic adjustment of synaptic receptor content and efficacy by acting on the lateral diffusion of AMPA receptors.
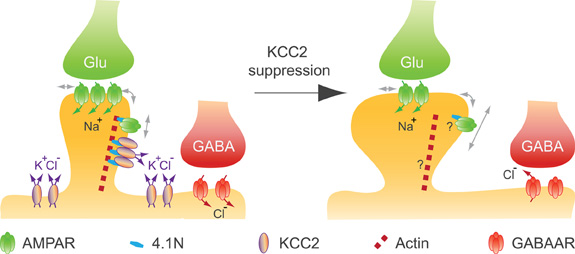
Figure 4. KCC2 controls spine volume and efficacy of glutamatergic AMPAR-mediated synaptic transmission in mature neurons. KCC2 suppression in mature neurons indicates that KCC2 is not required for spine maintenance, while it contributes to both spine head volume regulation and the efficacy of excitatory synapses by physically constraining AMPAR GluA1 subunit in spine head, likely through its interaction with submembrane actin cytoskeleton [adapted from Gauvain et al. (2011)]. A loss of KCC2 clusters induced by sustained excitatory synaptic activity or under pathological conditions may induce a rapid homeostatic adjustment of the efficacy of AMPAR mediated synaptic transmission as well as a reduction in the efficacy of GABAAR-dependent synaptic transmission.
Finally, although KCC2 is not necessary for dendritic spine maintenance in mature neurons, its suppression leads to increased spine head volume, an effect that is mimicked by chronic application of a KCC2 antagonist (Gauvain et al., 2011). As described above, KCC function is essential to volume regulation in many cell types (Gamba, 2005). Since KCC2 is the only CCC leading to solute and water extrusion under isotonic conditions (MacAulay et al., 2004; Jourdain et al., 2011), it likely contributes to osmotic regulation in dendritic spines, in particular to counteract cation influx through postsynaptic receptors. These observations predict that changes in KCC2 aggregation or function by synaptic activity may induce activity-dependent modulation of spine volume through local regulation of transmembrane solute and water efflux.
Regulation of KCC2 Expression and Function
The expression level and/or transport activity of KCC2 are up-regulated during development (Kahle et al., 2005; Vale et al., 2005; Rinehart et al., 2009; Ludwig et al., 2011a,b). In mature neurons, the activity-dependent regulation of KCC2 contributes to the plasticity of inhibitory synapses under both physiological and pathological conditions. In some conditions, the activity-dependent up-regulation of KCC2 may counteract synaptic excitation by strengthening GABA signaling (Banke and Gegelashvili, 2008; Chorin et al., 2011) whereas in some pathological conditions, KCC2 down-regulation may promote excitotoxicity (Hershfinkel et al., 2009) or seizure (Reid et al., 2001; Rivera et al., 2002, 2004; Huberfeld et al., 2007; Pathak et al., 2007; Wake et al., 2007; Li et al., 2008b; Shimizu-Okabe et al., 2011). Thus, activity-dependent regulation of KCC2 participates in the adjustment of the excitatory-inhibitory balance in neuronal networks.
Developmental Up-Regulation of KCC2
In developing hippocampal neurons, KCC2 expression is increased by the trophic factors neurturin (Ludwig et al., 2011a,b) and BDNF (Ludwig et al., 2011b). BDNF and Neurturin activate similar intracellular cascades that include the src homology two domain containing transforming protein/FGF receptor substrate 2 (Shc/FRS-2). This in turn triggers the extracellular signal-regulated kinase 1/2 (ERK1/2) and the mitogen-activated protein kinase (MAPK) pathway. This effect induces enhanced Egr4 expression and Egr4-dependent activation of KCC2b promoter (Ludwig et al., 2011b). Functional activation of KCC2 also requires oligomerization and/or phosphorylation/dephosphorylation of the co-transporter. Blaesse et al. (2006) showed that membrane expression and mature glycosylation pattern were not sufficient to ensure KCC2-mediated Cl− extrusion in immature rat LSO neurons. At P3, KCC2 is mainly expressed as a monomeric form in plasma membrane, whereas in more mature neurons (P30) in which Cl− extrusion was effective, KCC2 was found as oligomers (dimers, trimers, and tetramers). A direct link between oligomerization and ion transport by KCCs is supported by the observation that truncation of KCC1 carboxy-terminal domain prevents both oligomerization and chloride transport (Casula et al., 2001). Phosphorylation and dephosphorylation also play a critical role in KCC2 activation during development. Khirug et al. (2005) observed that the broad-spectrum kinase inhibitor staurosporine, leads to a rapid (5 min) negative shift in EGABA in immature hippocampal cultured neurons. Although this work did not identify the kinase(s) involved or their target(s) (i.e., KCC2 itself and/or molecular partners), KCC2 can be activated by dephosphorylation of Threonine residues. Residues Thr906 and Thr1006 in the CTD of KCC2 are phosphorylated by Wnk1 at birth and dephosphorylated in adult neurons (Rinehart et al., 2009). These phosphorylated residues appear critical for KCC2 function since Wnk1 phosphorylation of KCC2 in mature neurons causes a loss of transport function (Kahle et al., 2005; Rinehart et al., 2009). Although phosphorylation of Ser940 has not been analyzed during development, this residue is phosphorylated in mature hippocampal neurons (Lee et al., 2007), and PKC activation enhances KCC2 function (Banke and Gegelashvili, 2008) through direct phosphorylation of Ser940 (Lee et al., 2007). Therefore, PKC-dependent phosphorylation of Ser940 may also contribute to the developmental or activity-dependent activation of KCC2. Finally, the developmental activation of KCC2 function requires tyrosine kinase activity. Thus, inhibitors of tyrosine phosphorylation such as genistein or lavendustin A impair KCC2 activity in mature hippocampal neurons (Kelsch et al., 2001). KCC2 appears to be a direct target of tyrosine kinases as tyrosine phosphorylation shows a marked increase during postnatal development in cochlear nucleus neurons (Vale et al., 2005). Consistent with this observation, dephosphorylation of Tyr1087 abolishes KCC2 activity in mature neurons (Wake et al., 2007; Watanabe et al., 2009). In conclusion, functional activation of KCC2 during postnatal development appears to rely on dephosphorylation of Thr906 and Thr1006, PKC-dependent phosphorylation of Ser940, and tyrosine kinase phosphorylation of Tyr1087 (Table 1).
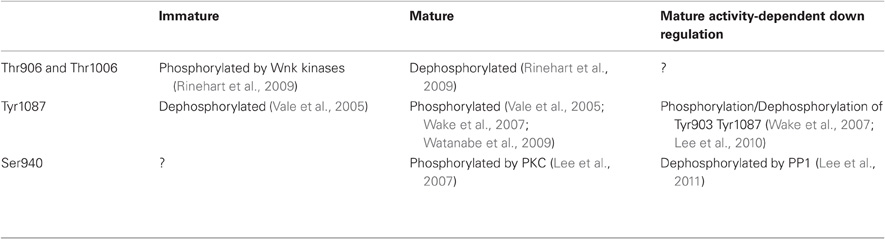
Table 1. Phosphorylation/dephosphorylation states of KCC2 at immature or mature stages of development and in conditions of increased activity..
Activity-dependent regulation
Neuronal activity dynamically up- or down-regulates KCC2 activity. Rapid up-regulation of KCC2 activity is induced upon activation of group I metabotropic GluRs (mGluRs) and Zn2+ extracellular signaling. Tonic activation of both mGluR1 and mGluR5 has been shown to enhance synaptic strength at GABAergic inputs onto CA3 pyramidal neurons through hyperpolarization of EGABA (Banke and Gegelashvili, 2008). This effect may involve synaptic co-release of Zn2+ from mossy fibers and subsequent activation of the metabotropic zinc-sensing receptor G-protein coupled receptor 39 (mZnR/GPR39) which in turn causes a hyperpolarizing shift in EGABA by enhancing KCC2 activity and surface expression in CA3 neurons in hippocampal slices (Chorin et al., 2011). mZnR activation triggers Ca2+ release from intracellular stores via group I mGluRs, activation of the ERK1/2 pathway and PKC phosphorylation of KCC2 (Banke and Gegelashvili, 2008; Besser et al., 2009; Sindreu et al., 2011). In this context, it is remarkable that synaptic accumulation of Zn2+ in mossy fiber terminals and KCC2 activity in pyramidal neurons follow a similar developmental time course (Rivera et al., 1999; Nitzan et al., 2002; Lee et al., 2005; Liguz-Lecznar et al., 2005), suggesting Zn2+ signaling may participate in the developmental up-regulation of KCC2 (Chorin et al., 2011). Interestingly however, Zn2+ appears to operate a bi-directional control over KCC2 activity. In contrast to extracellular Zn2+, a rise in intracellular Zn2+ under pathological conditions such as oxygen-glucose deprivation inhibits KCC2 function resulting in a depolarizing shift in EGABA (Hershfinkel et al., 2009).
KCC2 activity is down-regulated in a variety of physiological and pathological conditions, usually associated with enhanced neuronal activity. These include long term potentiation (Wang et al., 2006b), repetitive pairing of pre- and post-synaptic activities (Woodin et al., 2003), repetitive postsynaptic spiking (Fiumelli et al., 2005), rebound burst activity (Wang et al., 2006a), tetanic stimulation (Kaila et al., 1997), NMDA receptor activation (Kitamura et al., 2008), and epileptic activity (Reid et al., 2001; Rivera et al., 2002, 2004; Huberfeld et al., 2007; Pathak et al., 2007; Wake et al., 2007; Li et al., 2008b; Shimizu-Okabe et al., 2011). All these conditions cause a depolarizing shift in EGABA through reduced KCC2 function and/or expression.
The activity-dependent down-regulation of KCC2 mRNA level involves the BDNF-TrkB signaling and the combination of the Shc/FRS-2 and the phospholipase Cγ (PLCγ) signaling cascades (Rivera et al., 2004). This in turn activates the transcription factor cAMP response element-binding protein (CREB) through phosphorylation (Rivera et al., 2004). At the post-translational level, down-regulation of KCC2 usually relies on Ca2+ signaling and involve either Ca2+ influx through NMDA receptors (Kitamura et al., 2008; Lee et al., 2011) or L type Ca2+ channel (Fiumelli et al., 2005) or Ca2+-induced-Ca2− release from intracellular stores (Fiumelli et al., 2005). Although Ca2+ was initially suggested to activate a PKC-dependent phosphorylation of either KCC2 itself or some associated molecules that will in turn inactivate the transporter (Fiumelli et al., 2005), this seems somewhat unlikely since PKC-dependent phosphorylation of KCC2 on Ser940 enhances rather than reduces its activity (Lee et al., 2007; Banke and Gegelashvili, 2008). Instead, activity-dependent reduction of KCC2 activity may involve dephosphorylation of Ser940 (Lee et al., 2011) and perhaps phosphorylation of Tyr903/1087 residues (Wake et al., 2007; Lee et al., 2010). NMDAR-dependent Ca2+ influx causes PP1 dephosphorylation of KCC2 residue Ser940 (Lee et al., 2011). Thus, PKC-dependent phosphorylation of Ser940 enhances (Lee et al., 2007) while the PP1-mediated dephosphorylation of Ser940 inhibits KCC2 activity (Lee et al., 2011). Lee et al. (2011), therefore, proposed that KCC2 function is controlled by the balance between PKC and PP1 activities. The involvement of residues Tyr903/1087 in the down-regulation of KCC2 by activity is supported by the observation that pilocarpine-induced status epilepticus increases the phosphorylation of KCC2 residues Tyr903/1087 (Lee et al., 2010). However, hyperexcitability induced by BDNF or low external Mg2+ as well as oxidative stress all lead to tyrosine dephosphorylation of KCC2 (Wake et al., 2007). Interestingly, in these experiments, tyrosine phosphorylation of KCC2 could only be unmasked once phosphatase activity was blocked. Thus, neuronal activity appears to recruit both kinases and phosphatases, the relative activity of which determines KCC2 phosphorylation state. It is remarkable that developmental up-regulation and activity-dependent down-regulation of KCC2 involve a reciprocal regulation of its phosphorylation (Table 1).
Regulation of KCC2 by Oligomerization, Clustering and Lateral Diffusion
Membrane expression and glycosylation of KCC2 precede the onset of KCC2 function and the hyperpolarizing shift in EGABA during postnatal development (Kelsch et al., 2001; Khirug et al., 2005; Vale et al., 2005; Blaesse et al., 2006). In addition, enhanced neuronal activity affects KCC2 at the mRNA and protein expression levels within 1–6 h (Rivera et al., 2002, 2004; Wang et al., 2006c; Wake et al., 2007; Ludwig et al., 2011b) whereas activity-dependent regulation of KCC2 function occurs within minutes (Woodin et al., 2003; Wang et al., 2006b; Fiumelli and Woodin, 2007; Wake et al., 2007; Kitamura et al., 2008; Chorin et al., 2011; Lee et al., 2011). Therefore, regulation of KCC2 function in neurons appears to occur at several, partly interdependent levels and the rapid, activity-dependent modulation of the transporter likely occurs predominantly through post-translational modifications. Activity-induced dephosphorylation of Ser940 and phosphorylation of Tyr903/1087 leads to reduced membrane stability of KCC2 through increased endocytosis and targeting to lysosomal degradation (Lee et al., 2010, 2011). However, the loss of KCC2 activity as detected by increased [Cl−]i precedes the removal of KCC2 from cell surface (Wake et al., 2007; Watanabe et al., 2009). Conversely, Zn2+-dependent increase in KCC2 function is observed before the increase in membrane expression (Chorin et al., 2011). These observations suggest that other intermediate mechanisms may be at play to affect KCC2 function independent of its membrane expression level.
As mentioned above, KCC2 oligomerizes and forms clusters at plasma membrane. It is, therefore, tempting to hypothesize that oligomerization and clustering could represent the molecular substrate for such rapid alterations of KCC2 function. So far, only one study has investigated the impact of oligomerization and clustering on KCC2 activity (Watanabe et al., 2009). This study shows that a perturbation of KCC2 oligomerization and clustering by tyrosine kinase inhibitors correlates with a loss of function but no change in membrane expression in hippocampal neurons. Besides, heterologous GT1–7 cells expressing a recombinant KCC2 with either Tyr1087 residue mutated to aspartate (Y1087D) or a deletion of the 28 amino acids distal to the tyrosine phosphorylation site fails to oligomerize and form clusters as wild type KCC2 does (Watanabe et al., 2009). These results support a role for oligomerization and clustering in the regulation of KCC2 function. They also indicate that phosphorylation of Tyr1087 and the carboxyl-terminus of KCC2 play a critical role in oligomerization and clustering.
The mechanisms of KCC2 clustering remain elusive and may involve cholesterol-enriched lipid rafts (Watanabe et al., 2009). Indirect binding of KCC2 to actin through 4.1N interaction (Li et al., 2007) suggests clustering may also involve scaffolding molecules as described for postsynaptic neurotransmitter receptors. Identification of these molecular partners will be of particular importance since the availability of the submembrane scaffold that anchors and stabilizes transmembrane proteins can be rapidly up- or down-regulated by activity (Bruneau et al., 2009) and could be at play to locally control KCC2 aggregation and function.
Lateral diffusion in and outside synapses plays a key role in the activity-dependent regulation of receptor number at synapses (Triller and Choquet, 2008). This raises the question of the impact of lateral diffusion in the control of KCC2 clustering. The mobility of recombinant Flag-tagged KCC2 was analyzed using SPT (Dahan et al., 2003; Bannai et al., 2006). The surface recombinant Flag-tagged KCC2 transporter were labeled with an antibody raised against Flag and subsequently labeled with an intermediate biotinylated Fab fragment and streptavidin-coated quantum dot (QD). QD trajectories were overlaid with fluorescent images of recombinant homer1c-GFP and gephyrin-mRFP clusters in order to identify excitatory and inhibitory synapses, respectively. Trajectories were at/near synapses when overlapping ±2 pixels (440 nm) with homer1c-GFP and gephyrin-mRFP clusters or extrasynaptic for spots further away (Dahan et al., 2003). As shown in Figure 5, KCC2 exploratory behavior (Figure 5A) and mobility (Figure 5B) is reduced and its confinement (Figure 5C) increased in close vicinity of excitatory and inhibitory synapses.
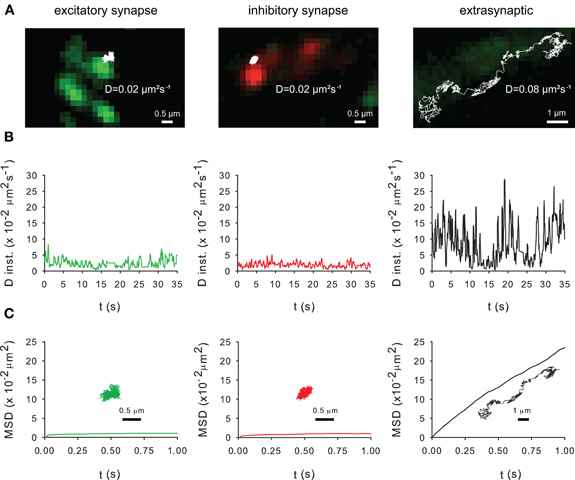
Figure 5. Membrane dynamics of the KCC2 transporter studied with Single Particle Tracking.(A) Representative trajectories of Quantum Dot-bound Flag tagged recombinant KCC2 reconstructed from 35 s recording sequences (Δ t = 0.03 s). QD trajectories were overlaid with fluorescent images of recombinant homer1c-GFP (green) and gephyrin-mRFP (red) clusters in order to identify excitatory and inhibitory synapses, respectively. Note that extrasynaptic QD-bound KCC2 explored larger area of membrane than synaptic/perisynaptic ones. Scale bars for synaptic/perisynaptic and extrasynaptic trajectories, 0.5 μm and 1 μm, respectively. (B) Instantaneous diffusion coefficients of the trajectories shown in (A). Note the reduction in diffusion coefficient values and fluctuations for synaptic/perisynaptic QDs as compared with the extrasynaptic QD. (C) Time-averaged MSD functions of the trajectories shown in (A). Extrasynaptic and synaptic QDs display linear and negatively bent MSD curves, characteristic of random walk and confined movement, respectively. In all graphs: green and red curves, trajectories at/near excitatory and inhibitory synapses, respectively; black, extrasynaptic trajectory.
As shown with confocal microscopy (Figure 2) and with electron microscopy (Gulyas et al., 2001; Bartho et al., 2004; Blaesse et al., 2006; Takayama and Inoue, 2006), KCC2 is localized at the periphery of the postsynaptic density. Thus, the QDs located in close vicinity of synapses (Figure 5A) may correspond to perisynaptic QDs. However, several technical limitations do not permit to strictly separate QDs in the core of the synapse from QDs in the near vicinity of the postsynaptic differentiation (i.e., perisynaptic QDs). Indeed, the optical resolution of the wide field microscope does not permit to precisely segregate the two compartments. Second, due to their large size (20–30 nm), QDs have difficulties to enter the core of the synapse (Groc et al., 2007). Anyway, the extrasynaptic vs. synaptic/perisynaptic diffusion behavior of KCC2 is reminiscent of what was found for neurotransmitter receptors with confined movement of the protein at/near synapses and free diffusion in extrasynaptic area [ref in (Triller and Choquet, 2008)]. More work is now required to examine whether KCC2 membrane dynamics and clustering are modulated by normal and pathological neuronal activities. A regulation of KCC2 diffusion/clustering might locally affect the net function of KCC2-mediated chloride extrusion. This will be particularly relevant for the regulation of GABA signaling which may be modulated at the subcellular level (Foldy et al., 2010).
Conclusions and Perspectives
The neuronal KCC2 transporter has long been considered only with respect to its function of chloride transport and its subsequent influence on GABA/glycine signaling. It is now becoming clear that KCC2 function in neurons extends beyond the mere transport of ions across the plasma membrane. In particular, KCC2 turns out to play a major role in the maturation and functional maintenance of excitatory synapses where it regulates the density of AMPAR in spines, most likely through actin binding property via 4.1N protein. Interestingly, 4.1N is required for activity-induced exocytosis of GluA1 subunit of AMPAR during long term potentiation (Lin et al., 2009). More work is, therefore, needed to fully characterize the contribution of KCC2/4.1N complexes in AMPAR traffic and long term synaptic plasticity. Another emerging field is that of KCC2 membrane traffic and its regulation by activity. Where does KCC2 exocytosis take place and is it regulated by neuronal activity? If so, this may represent a way to rapidly adjust KCC2 membrane expression and function. We propose that lateral diffusion and clustering of the transporter participate in the rapid regulation of its function. More work is needed to identify the scaffolding molecules that anchor KCC2 to the submembrane cytoskeleton. Whether KCC2 membrane diffusion undergoes a “diffusion trap” mechanism similar to postsynaptic neurotransmitter receptors and whether such mechanism may contribute to the activity-dependent modulation of KCC2 also remain to be established. In this context, it is noteworthy that similar cellular (cell surface stability, clustering, and perhaps lateral diffusion) and molecular (phosphorylation/dephosphorylation) mechanisms are at play to regulate KCC2 and GABAAR at the cell surface [for GABAAR regulation, see (Luscher et al., 2011)]. This raises questions about potential interactions of KCC2 and GABAAR and of their role in the coordinated regulation of GABA/glycine synaptic signaling.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguado, F., Carmona, M. A., Pozas, E., Aguilo, A., Martinez-Guijarro, F. J., Alcantara, S., Borrell, V., Yuste, R., Ibanez, C. F., and Soriano, E. (2003). BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development 130, 1267–1280.
Akerman, C. J., and Cline, H. T. (2006). Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J. Neurosci. 26, 5117–5130.
Amiry-Moghaddam, M., and Ottersen, O. P. (2003). The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 4, 991–1001.
Andrew, R. D., Labron, M. W., Boehnke, S. E., Carnduff, L., and Kirov, S. A. (2007). Physiological evidence that pyramidal neurons lack functional water channels. Cereb. Cortex 17, 787–802.
Baines, A. J., Bennett, P. M., Carter, E. W., and Terracciano, C. (2009). Protein 4.1 and the control of ion channels. Blood Cells Mol. Dis. 42, 211–215.
Baldi, R., Varga, C., and Tamas, G. (2010). Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur. J. Neurosci. 32, 1319–1325.
Banke, T. G., and Gegelashvili, G. (2008). Tonic activation of group I mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J. Physiol. 586, 4925–4934.
Banke, T. G., and McBain, C. J. (2006). GABAergic input onto CA3 hippocampal interneurons remains shunting throughout development. J. Neurosci. 26, 11720–11725.
Bannai, H., Lévi, S., Schweizer, C., Dahan, M., and Triller, A. (2006). Imaging the lateral diffusion of membrane molecules with quantum dots. Nat. Protoc. 1, 2628–2634.
Bartho, P., Curto, C., Luczak, A., Marguet, S. L., and Harris, K. D. (2009). Population coding of tone stimuli in auditory cortex: dynamic rate vector analysis. Eur. J. Neurosci. 30, 1767–1778.
Bartho, P., Payne, J. A., Freund, T. F., and Acsady, L. (2004). Differential distribution of the KCl cotransporter KCC2 in thalamic relay and reticular nuclei. Eur. J. Neurosci. 20, 965–975.
Belenky, M. A., Yarom, Y., and Pickard, G. E. (2008). Heterogeneous expression of gamma-aminobutyric acid and gamma-aminobutyric acid-associated receptors and transporters in the rat suprachiasmatic nucleus. J. Comp. Neurol. 506, 708–732.
Ben-Ari, Y. (2002). Excitatory actions of gaba during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739.
Ben-Ari, Y., Gaiarsa, J. L., Tyzio, R., and Khazipov, R. (2007). GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol. Rev. 87, 1215–1284.
Besser, L., Chorin, E., Sekler, I., Silverman, W. F., Atkin, S., Russell, J. T., and Hershfinkel, M. (2009). Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J. Neurosci. 29, 2890–2901.
Blaesse, P., Airaksinen, M. S., Rivera, C., and Kaila, K. (2009). Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838.
Blaesse, P., Guillemin, I., Schindler, J., Schweizer, M., Delpire, E., Khiroug, L., Friauf, E., and Nothwang, H. G. (2006). Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J. Neurosci. 26, 10407–10419.
Bormann, J., Hamill, O. P., and Sakmann, B. (1987). Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J. Physiol. 385, 243–286.
Bortone, D., and Polleux, F. (2009). KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron 62, 53–71.
Boulenguez, P., Liabeuf, S., Bos, R., Bras, H., Jean-Xavier, C., Brocard, C., Stil, A., Darbon, P., Cattaert, D., Delpire, E., Marsala, M., and Vinay, L. (2010). Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307.
Bregestovski, P., Waseem, T., and Mukhtarov, M. (2009). Genetically encoded optical sensors for monitoring of intracellular chloride and chloride-selective channel activity. Front. Mol. Neurosci. 2, 15. doi: 10.3389/neuro.02.015.2009
Bruneau, E. G., Esteban, J. A., and Akaaboune, M. (2009). Receptor-associated proteins and synaptic plasticity. FASEB J. 23, 679–688.
Cancedda, L., Fiumelli, H., Chen, K., and Poo, M. M. (2007). Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235.
Casula, S., Shmukler, B. E., Wilhelm, S., Stuart-Tilley, A. K., Su, W., Chernova, M. N., Brugnara, C., and Alper, S. L. (2001). A dominant negative mutant of the KCC1 K-Cl cotransporter: both N− and C− terminal cytoplasmic domains are required for K-Cl cotransport activity. J. Biol. Chem. 276, 41870–41878.
Cherubini, E., Griguoli, M., Safiulina, V., and Lagostena, L. (2011). The depolarizing action of GABA controls early network activity in the developing hippocampus. Mol. Neurobiol. 43, 97–106.
Chorin, E., Vinograd, O., Fleidervish, I., Gilad, D., Herrmann, S., Sekler, I., Aizenman, E., and Hershfinkel, M. (2011). Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 31, 12916–12926.
Chudotvorova, I., Ivanov, A., Rama, S., Hubner, C. A., Pellegrino, C., Ben-Ari, Y., and Medina, I. (2005). Early expression of KCC2 in rat hippocampal cultures augments expression of functional GABA synapses. J. Physiol. 566, 671–679.
Cohen, I., Navarro, V., Clemenceau, S., Baulac, M., and Miles, R. (2002). On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298, 1418–1421.
Collingridge, G. L., and Lester, R. A. (1989). Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol. Rev. 41, 143–210.
Coull, J. A., Boudreau, D., Bachand, K., Prescott, S. A., Nault, F., Sik, A., De Koninck, P., and De Koninck, Y. (2003). Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942.
Dahan, M., Lévi, S., Luccardini, C., Rostaing, P., Riveau, B., and Triller, A. (2003). Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science 302, 442–445.
Dallwig, R., Deitmer, J. W., and Backus, K. H. (1999). On the mechanism of GABA-induced currents in cultured rat cortical neurons. Pflugers Arch. 437, 289–297.
DeFazio, R. A., Keros, S., Quick, M. W., and Hablitz, J. J. (2000). Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J. Neurosci. 20, 8069–8076.
Denker, S. P., and Barber, D. L. (2002). Ion transport proteins anchor and regulate the cytoskeleton. Curr. Opin. Cell Biol. 14, 214–220.
Doyon, N., Prescott, S. A., Castonguay, A., Godin, A. G., Kroger, H., and De Koninck, Y. (2011). Efficacy of synaptic inhibition depends on multiple, dynamically interacting mechanisms implicated in chloride homeostasis. PLoS Comput. Biol. 7, e1002149. doi: 10.1371/journal.pcbi.1002149
Duebel, J., Haverkamp, S., Schleich, W., Feng, G., Augustine, G. J., Kuner, T., and Euler, T. (2006). Two-photon imaging reveals somatodendritic chloride gradient in retinal ON-type bipolar cells expressing the biosensor Clomeleon. Neuron 49, 81–94.
Fiumelli, H., Cancedda, L., and Poo, M. M. (2005). Modulation of GABAergic transmission by activity via postsynaptic Ca2+-dependent regulation of KCC2 function. Neuron 48, 773–786.
Fiumelli, H., and Woodin, M. A. (2007). Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr. Opin. Neurobiol. 17, 81–86.
Foldy, C., Lee, S. H., Morgan, R. J., and Soltesz, I. (2010). Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat. Neurosci. 13, 1047–1049.
Gagnon, K. B., England, R., and Delpire, E. (2006). Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am. J. Physiol. Cell Physiol. 290, C134–C142.
Gagnon, K. B., Fyffe, R. E., Adragna, N. C., and Lauf, P. K. (2007). Characterization of an extracellular epitope antibody to the neuronal K-Cl cotransporter, KCC2. Clin. Exp. Pharmacol. Physiol. 34, 566–573.
Gamba, G. (2005). Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493.
Gauvain, G., Chamma, I., Chevy, Q., Cabezas, C., Irinopoulou, T., Bodrug, N., Carnaud, M., Levi, S., and Poncer, J. C. (2011). The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc. Natl. Acad. Sci. U.S.A. 108, 15474–15479.
Gavrikov, K. E., Nilson, J. E., Dmitriev, A. V., Zucker, C. L., and Mangel, S. C. (2006). Dendritic compartmentalization of chloride cotransporters underlies directional responses of starburst amacrine cells in retina. Proc. Natl. Acad. Sci. U.S.A. 103, 18793–18798.
Glickfeld, L. L., Roberts, J. D., Somogyi, P., and Scanziani, M. (2009). Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat. Neurosci. 12, 21–23.
Groc, L., Lafourcade, M., Heine, M., Renner, M., Racine, V., Sibarita, J. B., Lounis, B., Choquet, D., and Cognet, L. (2007). Surface trafficking of neurotransmitter receptor: comparison between single-molecule/quantum dot strategies. J. Neurosci. 27, 12433–12437.
Gulyas, A. I., Sik, A., Payne, J. A., Kaila, K., and Freund, T. F. (2001). The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur. J. Neurosci. 13, 2205–2217.
Hershfinkel, M., Kandler, K., Knoch, M. E., Dagan-Rabin, M., Aras, M. A., Abramovitch-Dahan, C., Sekler, I., and Aizenman, E. (2009). Intracellular zinc inhibits KCC2 transporter activity. Nat. Neurosci. 12, 725–727.
Hoffmann, E. K., Lambert, I. H., and Pedersen, S. F. (2009). Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89, 193–277.
Hotulainen, P., and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629.
Hotulainen, P., Llano, O., Smirnov, S., Tanhuanpaa, K., Faix, J., Rivera, C., and Lappalainen, P. (2009). Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol. 185, 323–339.
Huberfeld, G., Wittner, L., Clemenceau, S., Baulac, M., Kaila, K., Miles, R., and Rivera, C. (2007). Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873.
Hubner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K., and Jentsch, T. J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524.
Ikeda, K., Onimaru, H., Yamada, J., Inoue, K., Ueno, S., Onaka, T., Toyoda, H., Arata, A., Ishikawa, T. O., Taketo, M. M., Fukuda, A., and Kawakami, K. (2004). Malfunction of respiratory-related neuronal activity in Na+, K+-ATPase alpha2 subunit-deficient mice is attributable to abnormal Cl− homeostasis in brainstem neurons. J. Neurosci. 24, 10693–10701.
Isomura, Y., Sugimoto, M., Fujiwara-Tsukamoto, Y., Yamamoto-Muraki, S., Yamada, J., and Fukuda, A. (2003). Synaptically activated Cl− accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J. Neurophysiol. 90, 2752–2756.
Jedlicka, P., Deller, T., Gutkin, B. S., and Backus, K. H. (2011). Activity-dependent intracellular chloride accumulation and diffusion controls GABA(A) receptor-mediated synaptic transmission. Hippocampus 21, 885–898.
Jons, T., and Drenckhahn, D. (1992). Identification of the binding interface involved in linkage of cytoskeletal protein 4.1 to the erythrocyte anion exchanger. EMBO J. 11, 2863–2867.
Jourdain, P., Pavillon, N., Moratal, C., Boss, D., Rappaz, B., Depeursinge, C., Marquet, P., and Magistretti, P. J. (2011). Determination of transmembrane water fluxes in neurons elicited by glutamate ionotropic receptors and by the cotransporters KCC2 and NKCC1: a digital holographic microscopy study. J. Neurosci. 31, 11846–11854.
Kahle, K. T., Rinehart, J., De Los Heros, P., Louvi, A., Meade, P., Vazquez, N., Hebert, S. C., Gamba, G., Gimenez, I., and Lifton, R. P. (2005). WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788.
Kahle, K. T., Rinehart, J., and Lifton, R. P. (2010). Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta. 1802, 1150–1158.
Kahle, K. T., Staley, K. J., Nahed, B. V., Gamba, G., Hebert, S. C., Lifton, R. P., and Mount, D. B. (2008). Roles of the cation-chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 4, 490–503.
Kaila, K., Lamsa, K., Smirnov, S., Taira, T., and Voipio, J. (1997). Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J. Neurosci. 17, 7662–7672.
Karadsheh, M. F., and Delpire, E. (2001). Neuronal restrictive silencing element is found in the KCC2 gene: molecular basis for KCC2-specific expression in neurons. J. Neurophysiol. 85, 995–997.
Kelsch, W., Hormuzdi, S., Straube, E., Lewen, A., Monyer, H., and Misgeld, U. (2001). Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J. Neurosci. 21, 8339–8347.
Khalilov, I., Chazal, G., Chudotvorova, I., Pellegrino, C., Corby, S., Ferrand, N., Gubkina, O., Nardou, R., Tyzio, R., Yamamoto, S., Jentsch, T. J., Hubner, C. A., Gaiarsa, J. L., Ben-Ari, Y., and Medina, I. (2011). Enhanced synaptic activity and epileptiform events in the embryonic KCC2 deficient hippocampus. Front. Cell. Neurosci. 5, 23. doi: 10.3389/fncel.2011.00023
Khirug, S., Huttu, K., Ludwig, A., Smirnov, S., Voipio, J., Rivera, C., Kaila, K., and Khiroug, L. (2005). Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur. J. Neurosci. 21, 899–904.
Khirug, S., Yamada, J., Afzalov, R., Voipio, J., Khiroug, L., and Kaila, K. (2008). GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J. Neurosci. 28, 4635–4639.
Kitamura, A., Ishibashi, H., Watanabe, M., Takatsuru, Y., Brodwick, M., and Nabekura, J. (2008). Sustained depolarizing shift of the GABA reversal potential by glutamate receptor activation in hippocampal neurons. Neurosci. Res. 62, 270–277.
Kullmann, D. M., Ruiz, A., Rusakov, D. M., Scott, R., Semyanov, A., and Walker, M. C. (2005). Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog. Biophys. Mol. Biol. 87, 33–46.
Kuner, T., and Augustine, G. J. (2000). A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron 27, 447–459.
Lee, H., Chen, C. X., Liu, Y. J., Aizenman, E., and Kandler, K. (2005). KCC2 expression in immature rat cortical neurons is sufficient to switch the polarity of GABA responses. Eur. J. Neurosci. 21, 2593–2599.
Lee, H. H., Deeb, T. Z., Walker, J. A., Davies, P. A., and Moss, S. J. (2011). NMDA receptor activity downregulates KCC2 resulting in depolarizing GABA(A) receptor-mediated currents. Nat. Neurosci. 14, 736–743.
Lee, H. H., Jurd, R., and Moss, S. J. (2010). Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179.
Lee, H. H., Walker, J. A., Williams, J. R., Goodier, R. J., Payne, J. A., and Moss, S. J. (2007). Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 282, 29777–29784.
Li, B., Mckernan, K., and Shen, W. (2008a). Spatial and temporal distribution patterns of Na-K-2Cl cotransporter in adult and developing mouse retinas. Vis. Neurosci. 25, 109–123.
Li, H., Khirug, S., Cai, C., Ludwig, A., Blaesse, P., Kolikova, J., Afzalov, R., Coleman, S. K., Lauri, S., Airaksinen, M. S., Keinanen, K., Khiroug, L., Saarma, M., Kaila, K., and Rivera, C. (2007). KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033.
Li, H., Tornberg, J., Kaila, K., Airaksinen, M. S., and Rivera, C. (2002). Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 16, 2358–2370.
Li, X., Zhou, J., Chen, Z., Chen, S., Zhu, F., and Zhou, L. (2008b). Long-term expressional changes of Na+ –K+–Cl− co-transporter 1 (NKCC1) and K+–Cl− co-transporter 2 (KCC2) in CA1 region of hippocampus following lithium-pilocarpine induced status epilepticus (PISE). Brain Res. 1221, 141–146.
Liguz-Lecznar, M., Nowicka, D., Czupryn, A., and Skangiel-Kramska, J. (2005). Dissociation of synaptic zinc level and zinc transporter 3 expression during postnatal development and after sensory deprivation in the barrel cortex of mice. Brain Res. Bull. 66, 106–113.
Lin, D. T., Makino, Y., Sharma, K., Hayashi, T., Neve, R., Takamiya, K., and Huganir, R. L. (2009). Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat. Neurosci. 12, 879–887.
Ludwig, A., Uvarov, P., Pellegrino, C., Thomas-Crusells, J., Schuchmann, S., Saarma, M., Airaksinen, M. S., and Rivera, C. (2011a). Neurturin evokes MAPK-dependent upregulation of Egr4 and KCC2 in developing neurons. Neural Plast. 2011, 1–8.
Ludwig, A., Uvarov, P., Soni, S., Thomas-Crusells, J., Airaksinen, M. S., and Rivera, C. (2011b). Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 31, 644–649.
Luscher, B., Fuchs, T., and Kilpatrick, C. L. (2011). GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409.
MacAulay, N., Hamann, S., and Zeuthen, T. (2004). Water transport in the brain: role of cotransporters. Neuroscience 129, 1031–1044.
MacAulay, N., and Zeuthen, T. (2010). Water transport between CNS compartments: contributions of aquaporins and cotransporters. Neuroscience 168, 941–956.
Mills, J. W., Schwiebert, E. M., and Stanton, B. A. (1994). Evidence for the role of actin filaments in regulating cell swelling. J. Exp. Zool. 268, 111–120.
Mohandas, N., Winardi, R., Knowles, D., Leung, A., Parra, M., George, E., Conboy, J., and Chasis, J. (1992). Molecular basis for membrane rigidity of hereditary ovalocytosis. A novel mechanism involving the cytoplasmic domain of band 3. J. Clin. Invest. 89, 686–692.
Moore-Hoon, M. L., and Turner, R. J. (2000). The structural unit of the secretory Na+–K+–2Cl− cotransporter (NKCC1) is a homodimer. Biochemistry 39, 3718–3724.
Nitzan, Y. B., Sekler, I., Hershfinkel, M., Moran, A., and Silverman, W. F. (2002). Postnatal regulation of ZnT-1 expression in the mouse brain. Brain Res. Dev. Brain Res. 137, 149–157.
Pathak, H. R., Weissinger, F., Terunuma, M., Carlson, G. C., Hsu, F. C., Moss, S. J., and Coulter, D. A. (2007). Disrupted dentate granule cell chloride regulation enhances synaptic excitability during development of temporal lobe epilepsy. J. Neurosci. 27, 14012–14022.
Payne, J. A. (1997). Functional characterization of the neuronal-specific K-Cl cotransporter: implications for K+o regulation. Am. J. Physiol. 273, C1516–C1525.
Payne, J. A., Rivera, C., Voipio, J., and Kaila, K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206.
Payne, J. A., Stevenson, T. J., and Donaldson, L. F. (1996). Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 271, 16245–16252.
Petrini, E. M., Lu, J., Cognet, L., Lounis, B., Ehlers, M. D., and Choquet, D. (2009). Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron 63, 92–105.
Plotkin, M. D., Snyder, E. Y., Hebert, S. C., and Delpire, E. (1997). Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J. Neurobiol. 33, 781–795.
Reid, K. H., Li, G. Y., Payne, R. S., Schurr, A., and Cooper, N. G. (2001). The mRNA level of the potassium-chloride cotransporter KCC2 covaries with seizure susceptibility in inferior colliculus of the post-ischemic audiogenic seizure-prone rat. Neurosci. Lett. 308, 29–32.
Rheims, S., Holmgren, C. D., Chazal, G., Mulder, J., Harkany, T., Zilberter, T., and Zilberter, Y. (2009). GABA action in immature neocortical neurons directly depends on the availability of ketone bodies. J. Neurochem. 110, 1330–1338.
Rinehart, J., Maksimova, Y. D., Tanis, J. E., Stone, K. L., Hodson, C. A., Zhang, J., Risinger, M., Pan, W., Wu, D., Colangelo, C. M., Forbush, B., Joiner, C. H., Gulcicek, E. E., Gallagher, P. G., and Lifton, R. P. (2009). Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536.
Rinehart, J., Vazquez, N., Kahle, K. T., Hodson, C. A., Ring, A. M., Gulcicek, E. E., Louvi, A., Bobadilla, N. A., Gamba, G., and Lifton, R. P. (2011). WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J. Biol. Chem. 286, 30171–30180.
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., Kokaia, Z., Airaksinen, M. S., Voipio, J., Kaila, K., and Saarma, M. (2002). BDNF-induced TrkB activation down-regulates the K+–Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 159, 747–752.
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., Pirvola, U., Saarma, M., and Kaila, K. (1999). The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255.
Rivera, C., Voipio, J., Thomas-Crusells, J., Li, H., Emri, Z., Sipila, S., Payne, J. A., Minichiello, L., Saarma, M., and Kaila, K. (2004). Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691.
Ruiz, A., Fabian-Fine, R., Scott, R., Walker, M. C., Rusakov, D. A., and Kullmann, D. M. (2003). GABAA receptors at hippocampal mossy fibers. Neuron 39, 961–973.
Rust, M. B., Gurniak, C. B., Renner, M., Vara, H., Morando, L., Gorlich, A., Sassoe-Pognetto, M., Banchaabouchi, M. A., Giustetto, M., Triller, A., Choquet, D., and Witke, W. (2010). Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 29, 1889–1902.
Ruusuvuori, E., Kirilkin, I., Pandya, N., and Kaila, K. (2010). Spontaneous network events driven by depolarizing GABA action in neonatal hippocampal slices are not attributable to deficient mitochondrial energy metabolism. J. Neurosci. 30, 15638–15642.
Shimizu-Okabe, C., Tanaka, M., Matsuda, K., Mihara, T., Okabe, A., Sato, K., Inoue, Y., Fujiwara, T., Yagi, K., and Fukuda, A. (2011). KCC2 was downregulated in small neurons localized in epileptogenic human focal cortical dysplasia. Epilepsy Res. 93, 177–184.
Simard, C. F., Bergeron, M. J., Frenette-Cotton, R., Carpentier, G. A., Pelchat, M. E., Caron, L., and Isenring, P. (2007). Homooligomeric and heterooligomeric associations between K+–Cl− cotransporter isoforms and between K+–Cl− and Na+–K+–Cl− cotransporters. J. Biol. Chem. 282, 18083–18093.
Simard, C. F., Brunet, G. M., Daigle, N. D., Montminy, V., Caron, L., and Isenring, P. (2004). Self-interacting domains in the C terminus of a cation-Cl− cotransporter described for the first time. J. Biol. Chem. 279, 40769–40777.
Sindreu, C., Palmiter, R. D., and Storm, D. R. (2011). Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc. Natl. Acad. Sci. U.S.A. 108, 3366–3370.
Staley, K. J., and Mody, I. (1992). Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J. Neurophysiol. 68, 197–212.
Staley, K. J., and Proctor, W. R. (1999). Modulation of mammalian dendritic GABA(A) receptor function by the kinetics of Cl− and HCO3− transport. J. Physiol. 519 Pt 3, 693–712.
Staley, K. J., Soldo, B. L., and Proctor, W. R. (1995). Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269, 977–981.
Stein, V., Hermans-Borgmeyer, I., Jentsch, T. J., and Hubner, C. A. (2004). Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J. Comp. Neurol. 468, 57–64.
Stil, A., Liabeuf, S., Jean-Xavier, C., Brocard, C., Viemari, J. C., and Vinay, L. (2009). Developmental up-regulation of the potassium-chloride cotransporter type 2 in the rat lumbar spinal cord. Neuroscience 164, 809–821.
Szabadics, J., Varga, C., Molnar, G., Olah, S., Barzo, P., and Tamas, G. (2006). Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science 311, 233–235.
Tada, T., and Sheng, M. (2006). Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 16, 95–101.
Takayama, C., and Inoue, Y. (2006). Developmental localization of potassium chloride co-transporter 2 in granule cells of the early postnatal mouse cerebellum with special reference to the synapse formation. Neuroscience 143, 757–767.
Takayama, C., and Inoue, Y. (2010). Developmental localization of potassium chloride co-transporter 2 (KCC2), GABA and vesicular GABA transporter (VGAT) in the postnatal mouse somatosensory cortex. Neurosci. Res. 67, 137–148.
Tardin, C., Cognet, L., Bats, C., Lounis, B., and Choquet, D. (2003). Direct imaging of lateral movements of AMPA receptors inside synapses. EMBO J. 22, 4656–4665.
Thompson, S. M., and Gahwiler, B. H. (1989a). Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J. Neurophysiol. 61, 501–511.
Thompson, S. M., and Gahwiler, B. H. (1989b). Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl− in hippocampal CA3 neurons. J. Neurophysiol. 61, 512–523.
Tornberg, J., Voikar, V., Savilahti, H., Rauvala, H., and Airaksinen, M. S. (2005). Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur. J. Neurosci. 21, 1327–1337.
Triller, A., and Choquet, D. (2008). New concepts in synaptic biology derived from single-molecule imaging. Neuron 59, 359–374.
Tyzio, R., Allene, C., Nardou, R., Picardo, M. A., Yamamoto, S., Sivakumaran, S., Caiati, M. D., Rheims, S., Minlebaev, M., Milh, M., Ferre, P., Khazipov, R., Romette, J. L., Lorquin, J., Cossart, R., Khalilov, I., Nehlig, A., Cherubini, E., and Ben-Ari, Y. (2011). Depolarizing actions of GABA in immature neurons depend neither on ketone bodies nor on pyruvate. J. Neurosci. 31, 34–45.
Tyzio, R., Cossart, R., Khalilov, I., Minlebaev, M., Hubner, C. A., Represa, A., Ben-Ari, Y., and Khazipov, R. (2006). Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792.
Tyzio, R., Minlebaev, M., Rheims, S., Ivanov, A., Jorquera, I., Holmes, G. L., Zilberter, Y., Ben-Ari, Y., and Khazipov, R. (2008). Postnatal changes in somatic gamma-aminobutyric acid signalling in the rat hippocampus. Eur. J. Neurosci. 27, 2515–2528.
Uvarov, P., Ludwig, A., Markkanen, M., Pruunsild, P., Kaila, K., Delpire, E., Timmusk, T., Rivera, C., and Airaksinen, M. S. (2007). A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 282, 30570–30576.
Uvarov, P., Ludwig, A., Markkanen, M., Rivera, C., and Airaksinen, M. S. (2006). Upregulation of the neuron-specific K+/Cl− cotransporter expression by transcription factor early growth response 4. J. Neurosci. 26, 13463–13473.
Uvarov, P., Ludwig, A., Markkanen, M., Soni, S., Hubner, C. A., Rivera, C., and Airaksinen, M. S. (2009). Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J. Biol. Chem. 284, 13696–13704.
Uvarov, P., Pruunsild, P., Timmusk, T., and Airaksinen, M. S. (2005). Neuronal K+/Cl− co-transporter (KCC2) transgenes lacking neurone restrictive silencer element recapitulate CNS neurone-specific expression and developmental up-regulation of endogenous KCC2 gene. J. Neurochem. 95, 1144–1155.
Vale, C., Caminos, E., Martinez-Galan, J. R., and Juiz, J. M. (2005). Expression and developmental regulation of the K+–Cl− cotransporter KCC2 in the cochlear nucleus. Hear. Res. 206, 107–115.
Vanhatalo, S., Palva, J. M., Andersson, S., Rivera, C., Voipio, J., and Kaila, K. (2005). Slow endogenous activity transients and developmental expression of K+–Cl− cotransporter 2 in the immature human cortex. Eur. J. Neurosci. 22, 2799–2804.
Vardi, N., Zhang, L. L., Payne, J. A., and Sterling, P. (2000). Evidence that different cation chloride cotransporters in retinal neurons allow opposite responses to GABA. J. Neurosci. 20, 7657–7663.
Vinay, L., and Jean-Xavier, C. (2008). Plasticity of spinal cord locomotor networks and contribution of cation-chloride cotransporters. Brain Res. Rev. 57, 103–110.
Vu, T. Q., Payne, J. A., and Copenhagen, D. R. (2000). Localization and developmental expression patterns of the neuronal K-Cl cotransporter (KCC2) in the rat retina. J. Neurosci. 20, 1414–1423.
Wake, H., Watanabe, M., Moorhouse, A. J., Kanematsu, T., Horibe, S., Matsukawa, N., Asai, K., Ojika, K., Hirata, M., and Nabekura, J. (2007). Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 27, 1642–1650.
Wang, L., Kitai, S. T., and Xiang, Z. (2006a). Activity-dependent bidirectional modification of inhibitory synaptic transmission in rat subthalamic neurons. J. Neurosci. 26, 7321–7327.
Wang, W., Gong, N., and Xu, T. L. (2006b). Downregulation of KCC2 following LTP contributes to EPSP-spike potentiation in rat hippocampus. Biochem. Biophys. Res. Commun. 343, 1209–1215.
Wang, W., Wang, H., Gong, N., and Xu, T. L. (2006c). Changes of K+–Cl− cotransporter 2 (KCC2) and circuit activity in propofol-induced impairment of long-term potentiation in rat hippocampal slices. Brain Res. Bull. 70, 444–449.
Waseem, T., Mukhtarov, M., Buldakova, S., Medina, I., and Bregestovski, P. (2010). Genetically encoded Cl−Sensor as a tool for monitoring of Cl-dependent processes in small neuronal compartments. J. Neurosci. Methods 193, 14–23.
Watanabe, M., Wake, H., Moorhouse, A. J., and Nabekura, J. (2009). Clustering of neuronal K+–Cl− cotransporters in lipid rafts by tyrosine phosphorylation. J. Biol. Chem. 284, 27980–27988.
Wei, W. C., Akerman, C. J., Newey, S. E., Pan, J., Clinch, N. W., Jacob, Y., Shen, M. R., Wilkins, R. J., and Ellory, J. C. (2011). The potassium chloride cotransporter 2 (KCC2) promotes cervical cancer cell migration and invasion by an ion transport-independent mechanism. J. Physiol. 589, 5349–5359.
Williams, J. R., Sharp, J. W., Kumari, V. G., Wilson, M., and Payne, J. A. (1999). The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J. Biol. Chem. 274, 12656–12664.
Woodin, M. A., Ganguly, K., and Poo, M. M. (2003). Coincident pre- and post-synaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron 39, 807–820.
Yang, K., Huang, Z. W., Huang, J., Zhang, X. J., and Xiao, B. K. (2008). Expression of the neuron-specific potassium chloride cotransporter KCC2 in adult rat cochlear. Neurosci. Lett. 441, 205–209.
Yeo, M., Berglund, K., Augustine, G., and Liedtke, W. (2009). Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 29, 14652–14662.
Yuste, R., and Bonhoeffer, T. (2004). Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. Neurosci. 5, 24–34.
Zeuthen, T., and MacAulay, N. (2002). Cotransporters as molecular water pumps. Int. Rev. Cytol. 215, 259–284.
Keywords: KCC2, neuronal activity, excitatory and inhibitory synapses, post-translational regulation
Citation: Chamma I, Chevy Q, Poncer JC and Lévi S (2012) Role of the neuronal K-Cl co-transporter KCC2 in inhibitory and excitatory neurotransmission. Front. Cell. Neurosci. 6:5. doi: 10.3389/fncel.2012.00005
Received: 02 December 2011; Accepted: 30 January 2012;
Published online: 21 February 2012.
Edited by:
Enrico Cherubini, International School for Advanced Studies, ItalyReviewed by:
Melanie A. Woodin, University of Toronto, CanadaEnrica M. Petrini, Italian Institute of Technology, Italy
Copyright: © 2012 Chamma, Chevy, Poncer and Lévi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Sabine Lévi and Jean Christophe Poncer, INSERM UMR-839/UPMC, Institut du Fer à Moulin, Plasticity in Cortical Networks and Epilepsy, 17 rue du Fer à Moulin, 75005 Paris, France. e-mail:c2FiaW5lLmxldmlAaW5zZXJtLmZy;amVhbi1jaHJpc3RvcGhlLnBvbmNlckBpbnNlcm0uZnI=