

- 1Departments of Pediatrics, Anatomy/Neurobiology, and Neurology, University of California-Irvine, Irvine, CA, USA
- 2Department of Structural Neurobiology, Center for Molecular Neurobiology, Hamburg, University of Hamburg, Hamburg, Germany
To enhance survival, an organism needs to remember—and learn from—threatening or stressful events. This fact necessitates the presence of mechanisms by which stress can influence synaptic transmission in brain regions, such as hippocampus, that subserve learning and memory. A major focus of this series of monographs is on the role and actions of adrenal-derived hormones, corticosteroids, and of brain-derived neurotransmitters, on synaptic function in the stressed hippocampus. Here we focus on the contribution of hippocampus-intrinsic, stress-activated CRH-CRH receptor signaling to the function and structure of hippocampal synapses. Corticotropin-releasing hormone (CRH) is expressed in interneurons of adult hippocampus, and is released from axon terminals during stress. The peptide exerts time- and dose-dependent effects on learning and memory via modulation of synaptic function and plasticity. Whereas physiological levels of CRH, acting over seconds to minutes, augment memory processes, exposure to presumed severe-stress levels of the peptide results in spine retraction and loss of synapses over more protracted time-frames. Loss of dendritic spines (and hence of synapses) takes place through actin cytoskeleton collapse downstream of CRHR1 receptors that reside within excitatory synapses on spine heads. Chronic exposure to stress levels of CRH may promote dying-back (atrophy) of spine-carrying dendrites. Thus, the acute effects of CRH may contribute to stress-induced adaptive mechanisms, whereas chronic or excessive exposure to the peptide may promote learning problems and premature cognitive decline.
Learning and Memory must be Influenced by Stress, Necessitating means to Influence Synaptic Transmission: What do we know and what are some of the Remaining Gaps?
Stress is generally defined as a signal conveying threat or potential threat (López et al., 1999; Kim and Diamond, 2002; McEwen, 2004; de Kloet et al., 2005; Joëls and Baram, 2009; Lupien et al., 2009), and, operationally as a signal that activates a specific brain system (Pacák and Palkovits, 2001). Stress of different types is common and pervasive. In addition, there is a strong evolutionary advantage to remembering and learning from threatening situations (McEwen, 1999). In contrast, the continuation of normal life requires forgetting severely stressful events, because such haunting memories might interfere with emotional health and with carrying out life's daily tasks, as is found in post-traumatic stress disorder (Wingo et al., 2010; Yehuda et al., 2010). Therefore, it is not surprising that stress has been found to be a powerful modulator of synaptic plasticity and memory (Kim and Diamond, 2002; de Kloet et al., 2005; Joëls and Baram, 2009; Lupien et al., 2009; Sandi, 2011). In the context of this series, the overall focus is on the mechanisms by which stress affects the hippocampus in a time- and severity-dependent manner, with consequences that contribute to a cognitive and emotional health and disease. Thus, whereas acute stress (lasting seconds to minutes) may augment memory and related cellular processes, longer stress tends to impair hippocampus-dependent learning and memory (Kim and Diamond, 2002; de Kloet et al., 2005; Diamond et al., 2006; Joëls and Baram, 2009).
A remarkable body of work that focused on the basis of these effects of stress has centered on the roles of adrenal-derived corticoid stress hormones and on their signaling via glucocorticoid receptors (GRs) and, more recently, mineralocorticoid receptors (MRs) (Kim and Diamond, 2002; de Kloet et al., 2005; Lupien et al., 2009; Joëls and Baram, 2009; Krugers et al., 2010; Segal et al., 2010; Sandi, 2011; Yuen et al., 2011). In view of the fact that MR activation generally increases synaptic plasticity (Joëls and Baram, 2009; Krugers et al., 2010), and the relatively limited distribution of GR on hippocampal CA3 pyramidal cells that are highly vulnerable to stress (Magarinos and McEwen, 1995; Sanchez et al., 2000; de Kloet, 2004; Joëls and Baram, 2009), it is reasonable to consider potential additional factors that may contribute to the actions of stress on the cognitive functions taking place within the hippocampus.
Among the many factors that influence the effects of stress (e.g., type of stress, age, and gender of the involved brain), Time and Space are key. As mentioned, seconds-long stress improves learning whereas chronic, weeks-long stress perturbs both hippocampal function and structure. When and how does the transition occur? Similarly, emerging evidence indicates that stress may affect the dorsal and the ventral hippocampus differentially (Maggio and Segal, 2009; Segal, 2010). In addition, stress may destroy dendritic spine within minutes and hours in hippocampus, yet increase complexity of dendrites in amygdala (Vyas et al., 2002). How is the spatial specificity take place? A large body of work, cited elsewhere in this monograph, has tackled these temporal and spatial issues. In the temporal domain, elegant studies have demonstrated rapid, non-genomic effects of MR and GR activation, followed by slower (hours to weeks) genomic actions. Rapid effects of neurotransmitters may translate to longer actions through influence on enzyme activity (reviewed in Joëls and Baram, 2009). Here we focus on an additional temporal solution: stress-provoked release of a neuropeptide within hippocampus. Neuropeptides classically function in the time-frame of seconds to a few hours depending, among other factors, on their degradation and reuptake (Koch et al., 1974). Similarly, peptides offer an attractive solution to the spatial conundrum of the actions of stress: unlike neurotransmitters, they are exuded into the neuropil that may bathe hundreds and thousands of synapses, providing a means to influence synaptic transmission of defined neuronal populations within a defined spatial domain (Fuxe et al., 1990; Agnati et al., 1995; Landgraf and Neumann, 2004; Nässel, 2009). In the current monograph, we describe the hippocampal corticotropin-releasing hormone (CRH) system and the action of this peptide on hippocampal structure and function. A key remaining challenge is to discover how the actions of neurotransmitters, corticosteroids and peptides interact to influence learning and memory in the stressed hippocampus.
The CRH System of Adult Hippocampus: Cells, Receptors, and MIS-Matched Synapses
Potential Sources of CRH Effects in the Hippocampus
CRH is expressed within adult hippocampus and is released locally during stress (Figure 1, and see below). However, the peptide is also released within the amygdala (Roozendaal et al., 2002), locus ceruleus (Valentino and Wehby, 1988; Snyder et al., 2012) and other brain regions. Because this peptide can travel long distances within the brain (Bittencourt and Sawchenko, 2000), an extra-hippocampal source and transport of the peptide from distal brain regions to act on hippocampal CRFR1 receptors cannot be excluded. However, organotypic cultures of the hippocampus have helped clarify the source of endogenous CRH influencing hippocampal neuronal structure. Growing these cultures (where other brain regions are not included) in the presence of selective blockers of CRH receptor type 1 (the receptor most highly expressed in the hippocampal formation), has resulted in abnormal dendritic growth. Dendritic branching is exuberant and total dendritic length is increased under these conditions, suggesting a role for endogenous hippocampal CRH in selective pruning or sculpting of the dendritic tree of hippocampal pyramidal cells. Because dendrites may grow or die-back (atrophy), as a function of activity of excitatory synapses located on dendritic spines, a potential mechanism of the effects of CRH on dendritic structure is via influencing the integrity of such spines (see below). In addition, the structure of dendritic trees of mice lacking the CRFR1 receptor is abnormal, with similar exuberant branching (Chen et al., 2004a, 2008). Interestingly, this is not found in mice lacking CRFR1 only in principal forebrain neurons (Wang et al., 2011a), though the dendritic trees of these mice seem to be resistant to chronic stress induced atrophy. Taken together, available data largely support the idea that the main source of the CRH that activates CRFR1 within hippocampus is the hippocampus itself.
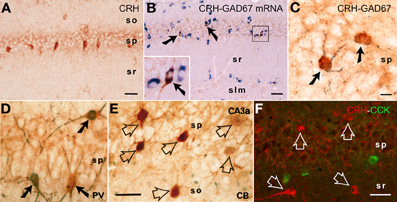
Figure 1. CRH is expressed in interneurons within the hippocampal pyramidal cell layer. (A) CRH-immunoreactive (ir) neuronal somata in the pyramidal cell layer of area CA1. (B,C) All CRH-ir neurons in the pyramidal cell layer in CA1 area are GABAergic interneurons: they co-express the GABA synthesizing enzyme glutamic acid decarboxylase (GAD)-67 at both mRNA and protein levels, using immunocytochemistry coupled with in situ hybridization (GAD67, blue), and dual-labeled immunocytochemistry (CRH, brown; GAD67, black blue), respectively. Arrows indicate the dual-labeled neurons. (D–F) Many CRH-ir neurons in the hippocampus co-express parvalbumin (PV); none co-express calbindin D-28k (CB) or cholecystokinin (CCK). Solid arrows indicate the dual-labeled neurons, and empty arrows denote single-labeled CRH neurons. Abbreviations: so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare. Scale bars = 75 μm (A,B,F), 25 μm (C), and 50 μm (D,E). Reproduced, with permission, from Chen et al. (2004b) (B) and Yan et al. (1998) (D,E).
Who are the Hippocampal CRH-Expressing Cells, and what type of Synapses do they Form?
CRH is produced in several populations of cells in the developing hippocampus (Yan et al., 1998; Chen et al., 2001), including Cajal-Retzius cells (Chen et al., 2001). In adult rodent hippocampus, the large majority of peptide is synthesized and contained within interneurons residing in the pyramidal cell layers of areas CA1 and CA3 (Sakanaka et al., 1987; Yan et al., 1998; Chen et al., 2001; Ivy et al., 2010) (Figure 1). CRH-expressing cells universally express the GABA synthetic enzyme GAD (Figures 1B,C), and include parvalbumin co-expressing basket cells (Figure 1D). Interestingly, there is no co-localization of CRH with calbindin D-28k (Figure 1E) or with cholecystokinin (Figure 1F).
The release site and mode of travel of CRH to target receptors are not fully understood. Light microscopy demonstrated a typical network of CRH-containing axon terminals surrounding the cell bodies of pyramidal cells (Figure 2A). In addition, both light and electron microscopy revealed that CRH is stored in axon terminals (Figures 2B–F) and released from axon terminal-vesicles surrounding the cell bodies and axon initial segments of pyramidal cells (Yan et al., 1998; Chen et al., 2001). These perisomatic release sites are >100 μm away from the location of the CRFR1 receptors on dendritic spines in stratum radiatum (Figures 3A,B). The possibility that CRH is released from interneuronal dendrites closer to the receptors is not supported by electron microscopy studies, which show no evidence for vesicular localization of the peptide in dendrites (Figure 2G). Instead, these data support the idea that CRH released from interneurons in the pyramidal cell layer diffuses locally (via “volume transmission”) (Agnati et al., 1995) to target receptors on dendritic spines (Figure 3C). Remarkably, these data indicate that the hippocampal CRH synapse is “mis-matched”: the release site (pre-synaptic element) is an axon terminal of an interneuron (classically an element of inhibitory synapses), whereas the post-synaptic element resides on dendritic spines, and consists of dense post-synaptic elements, typical of excitatory synapses (Figure 3C).
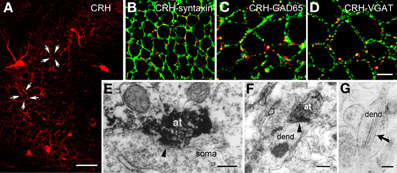
Figure 2. CRH is located in GABAergic axonal terminals. (A) CRH-ir axon terminals form dense perisomatic baskets (arrows) around pyramidal cells in hippocampal area CA3. (B) CRH immunoreactivity localizes to pre-synaptic terminals, demonstrated by dual-labeled immunocytochemistry for CRH and for the pre-synaptic marker syntaxin. (C,D) CRH-containing terminals are GABAergic boutons as indicated by overlapping immunoreactivities of CRH and GAD65 (C) and of CRH and the GABA synaptic vesicular transporter VGAT (D) in axon terminals. (E) CRH-ir axon terminals (at) form axosomatic symmetric synapses (arrowhead) with pyramidal cell body (soma). (F) CRH-ir axon terminals form axodendritic asymmetric synapses (arrowhead) with dendrites (dend). Empty arrow indicates a bouton containing CRH-negative vesicles. (G) Electron micrograph demonstrating CRH-ir gold particles (arrow) within the dendrite, but not within vesicles. Scale bars = 50 μm (A), 10 μm in (D) (25 μm for B and 12.5 μm for C), and 0.2 μm (E–G). Reproduced, with permission, from Chen et al. (2004b) (B–D) and Yan et al. (1998) (E).
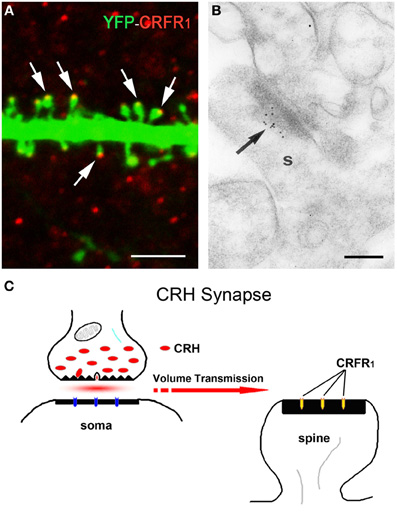
Figure 3. The post-synaptic location of the CRH receptor CRFR1 on dendritic spines. (A) The CRH receptor CRFR1 (red) is located at the dendritic spine heads, as shown by confocal microscopy of CA3 pyramidal neuronal dendrite (green) from a YFP-expressing mouse. Arrows denote spine heads expressing the receptor. (B) Electron micrograph showing CRFR1-immunogold particles (arrow) concentrated at the post-synaptic density (PSD) of an asymmetric (excitatory) synapse on a dendritic spine (s) in stratum oriens of hippocampal area CA3. Very few gold grains were observed elsewhere. (C) A cartoon depicting the concept of the mis-matched CRH synapse: pre-synaptic elements (axon terminal and boutons) are GABAergic (Figure 1), whereas post-synaptic elements on dendritic spines are consistent with excitatory synapses (Figure 3A). CRH that is released from inhibitory pre-synaptic elements may migrate via volume transmission and act on the CRFR1 receptor at relatively distant excitatory post-synaptic sites. Scale bars = 3 μm (A), 0.1 μm (B). Reproduced, with permission, from Chen et al. (2010) (A), (2004b) (B).
CRH-CRFR1 Signaling Contributes to the Effects of Stress on Hippocampal Synaptic Structure and Function
CRH may signal through two identified G-protein coupled receptor family members: CRH receptor type 1 (CRFR1) and type 2 (CRFR2) (Perrin and Vale, 1999). The distribution of CRFR1 and CRFR2 in the brain is different (Chalmers et al., 1995; Chen et al., 2000; Van Pett et al., 2000). In general, CRFR1 is primarily responsible for mediating the synaptic actions of CRH on hippocampal principal cells (Schierloh et al., 2007; Refojo et al., 2011; Stern et al., 2011). In accord, CRFR1 is amply expressed in hippocampal pyramidal cells (Chen et al., 2000, 2004b; Van Pett et al., 2000; Refojo et al., 2011), whereas little CRFR2 expression is observed (Van Pett et al., 2000). Notably, in addition to the cell body, CRFR1 is found on dendrites and within dendritic spines, the location of post-synaptic portions of excitatory synapses (Chen et al., 2004b, 2010). Indeed, a short stress that combining physiological and psychological components activates CRFR1-containing pyramidal cells, indicated by increases in immediate early gene expression. This activation requires CRH-receptor signaling, because selective local blockade of CRFR1 prior to the stress prevents this activation (Chen et al., 2004b, 2006). Of note, CRFR1 signaling may be required for hippocampal plasticity even in the absence of stress: synaptic potentiation is abnormal in hippocampal slices from mice lacking CRFR1 (Schierloh et al., 2007), and these mice have learning deficits (Contarino et al., 1999).
Acute (Seconds to Minutes) Effects of CRH on Synaptic Transmission and Memory
In line with the activating and memory-promoting effects of acute stress, the actions of CRH in the hippocampus are generally excitatory (Baram and Hatalski, 1998). Application of CRH to hippocampal slices in vitro increases the firing rates of pyramidal cells by suppressing the after-hyperpolarization (Aldenhoff et al., 1983), and in the presence of an excitatory stimulus, CRH augments this input (Aldenhoff et al., 1983; Hollrigel et al., 1998). In a physiological context, brief application of CRH in vitro primes and augments LTP (Blank et al., 2002; Refojo et al., 2011) through CRFR1 signaling. In vivo, a short treatment with CRH directly into the brain enhances memory (Wang et al., 1998, 2000; Blank et al., 2002; Joëls and Baram, 2009; Refojo et al., 2011). A significant additional body of work (e.g., Chen et al., 2004b, 2006, 2010) now demonstrates that stress induces rapid release of endogenous, hippocampal-origin CRH into the hippocampal intercellular space (Figure 4), as found also within the amygdala (Roozendaal et al., 2002), locus ceruleus (Van Bockstaele et al., 1996) and cortex (Behan et al., 1995). Taken together these facts suggest that CRH, rapidly released upon the onset of stress, excites synapses, and augments synaptic plasticity. This adaptive mechanism promotes learning and remembering during threatening situations, which might be teleologically advantageous.
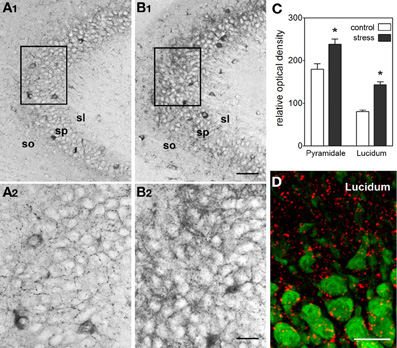
Figure 4. Stress induces rapid release of endogenous CRH into the extracellular space within hippocampus. (A,B) Visualization of stress-induced release of endogenous CRH into the extracellular space (neuropil) in area CA3. Under control conditions (A1,A2), there is little CRH-immunoreactivity outside of the cell bodies and terminals. In contrast, after a 30-min stress (B1,B2), immunoreactive peptide is visible within the neuropil of the pyramidal cell layer and adjacent regions. See Chen et al. (2004b) for experimental details. Frames in (A1,B1) denote areas magnified in (A2,B2), respectively, demonstrating the presence of CRH-immunoreactivity in the extracellular space. (C) Semi-quantitative analysis of CRH-immunoreactivity in presumed extracellular spaces of area CA3 (n = 5 animals, *P < 0.01). (D) Stress-evoked release of endogenous CRH (red) into the extracellular space. NeuN (green) indicates CA3 pyramidal cells. Confocal image from a P18 rat sacrificed after a 30-min stress. Abbreviations: so, stratum oriens; sp, stratum pyramidale; sl, stratum lucidum. Scale bars = 60 μm in (B1,A1), 20 μm in (B2,A2, and D). Reproduced, with permission, from Chen et al. (2004b) (D).
Subacute, Hours-Long Effects of Stress Levels of CRH on Synaptic Function and Structure
The effects of stress on synaptic function vary with the duration of the stress (among other parameters, including the severity, type and context of the stress, and variables intrinsic to the age and gender of the hippocampus itself). Given that CRH is released during stress, it is not surprising that the consequences of stress levels of CRH on hippocampal synapse structure and function vary with the duration of exposure. From the electrophysiological perspective, application of the hormone onto adult hippocampal slices has major effects on synaptic physiology: field EPSPs begin to decline ∼75 min into the infusion period, and short- and long-term synaptic plasticity is obliterated (Chen et al., 2012).
The neuroanatomical basis for this loss of synaptic function involves a loss of synapses. Specifically, CRH at presumed stress levels (Tringali et al., 2009) leads to retraction of dendritic spines that harbor post-synaptic elements of hippocampal excitatory synapses (Chen et al., 2008). This hippocampal effect of CRH supports prior findings in the amygdala (Matys et al., 2004; Bennur et al., 2007). Interestingly, although only a minority of dendritic spines are lost upon hours-long CRH (or stress), their loss results in profound memory impairment and loss of LTP. The magnitude of the functional deficits derives from the fact that spine loss is fairly selective to the subpopulation of thin dendritic spines (Chen et al., 2012). Among the diverse populations of dendritic spines, thin spines are called “learning spines” (Bourne and Harris, 2008; Holtmaat and Svoboda, 2009). They are most influenced by patterned neuronal activity that promotes learning. As these thin spines begin to express more glutamate receptors of the AMPA-GluR1 type, they are converted to mushroom type spines associated with memory storage. Hence, a loss of thin spines will disproportionately hampers the potential for the spine-plasticity process associated with learning and memory (Bourne and Harris, 2008; Maras and Baram, 2012).
The molecular mechanisms by which CRH provokes spine retraction are not fully understood. Whereas CRH-CRFR1 interaction activates several signaling cascades (Swinny and Valentino, 2006; Stern et al., 2011), the activation of an actin-regulating Rho-GTPAse, a specifically RhoA seems to underlie CRH-induced spine loss: blocking RhoA-mediated function rescued dendritic spines from CRH-provoked loss (Chen et al., 2012).
In summary, whereas a short exposure to CRH promotes synaptic function and plasticity, longer exposure, especially to presumed stress-levels of the peptide produces effects on hippocampal neurons that would contribute to stress-related learning and memory impairments. The precise transition from positive to harmful effects of the peptide, and the interaction between length of exposure and CRH levels, as well as the interaction of CRH with glucocorticoid and adrenergic mediators require future study.
Chronic Effects of CRH on Hippocampal Structure and Function
The effects of chronic stress on the integrity and neurotransmission of synapses have been extensively studied. Glucocorticoids, which are released peripherally in response to stress, can have broad impacts on brain function (de Kloet, 2004; McEwen, 2004, 2011; Joëls, 2008; Lupien et al., 2009; Ulrich-Lai and Herman, 2009), whereas the local release of neurotransmitters and neuropeptides within the hippocampus itself provides for more spatially restricted modulation of specific synaptic populations (Joëls and Baram, 2009). The relative roles of glucocorticoids and CRH can be clarified in organotypic slice cultures or acute isolated hippocampal slices, which are not exposed to steroid hormones. Growing organotypic slice cultures chronically in the presence of exogenous CRH stunted dendritic growth (Chen et al., 2004a). These findings support a role for CRH in stress-related modulation of dendritic arborization and pruning.
A second approach to distinguish the requirement for CRH receptor signaling in effects of chronic stress on hippocampal synapses is via the use of transgenic mice, where the receptor is deleted in forebrain or hippocampus only. Adult mice lacking CRFR1 in forebrain were relatively resistant to the deleterious effects of chronic social defeat stress (Wang et al., 2011a). Interestingly, the local deletion of this CRH receptor also protected adult mice from the adverse effects of chronic early life stress on learning and memory in adulthood (Wang et al., 2011b). Chronic early life stress, imposed by creating “simulated poverty” in the cage, results in cognitive problems and dendritic atrophy with loss of dendritic spines and synapses (Brunson et al., 2005). Infusion of CRFR1 blocker immediately following this early life stress prevented the learning and memory defects, rescued LTP and restored the integrity of dendritic structure (Ivy et al., 2010). These findings provide direct evidence for a need for CRH-CRFR1 signaling in the persistent effects of chronic early life stress on hippocampal synapses.
Summary
The body of work reviewed above suggests that, in addition to canonical stress hormones, hippocampal CRH tunes synaptic transmission during stress, influencing memory. In addition, stress is associated with high levels of CRH at hippocampal synapses, and long exposures to these levels result in neuroanatomical and functional defects of hippocampal function. Teleologically, rapid local release of the neuropeptide is adaptive, promoting excitation, and enhanced synaptic function during acute stress. The consequences of protracted elevation of CRH levels during chronic stress are likely maladaptive, providing a potentially treatable cause of stress-related cognitive problems. Challenges in the field include a better understanding of the effects of CRH during relatively short stresses that are common in modern life: namely, those lasting for several hours, with combined physiological and psychological components. A second topic requiring study is the degree and nature of the interaction among concurrently acting stress hormones: glucocorticoids, neurotransmitters, and CRH. How these mediators function, and how they interact in molecular and cellular terms, is a key enigma that impedes our full understanding of the effects of stress on synaptic neurotransmission within the hippocampus.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by NIH grants NS28912, MH73136, NS45260. We thank Barbara Cartwright for editorial help.
References
Agnati, L. F., Bjelke, B., and Fuxe, K. (1995). Volume versus wiring transmission in the brain: a new theoretical frame for neuropsychopharmacology. Med. Res. Rev. 15, 33–45.
Aldenhoff, J. B., Gruol, D. L., Rivier, J., Vale, W., and Siggins, G. R. (1983). Corticotropin releasing factor decreases postburst hyperpolarizations and excites hippocampal neurons. Science 221, 875–877.
Baram, T. Z., and Hatalski, C. G. (1998). Neuropeptide-mediated excitability: a key triggering mechanism for seizure generation in the developing brain. Trends Neurosci. 21, 471–476.
Behan, D. P., Heinrichs, S. C., Troncoso, J. C., Liu, X. J., Kawas, C. H., Ling, N., and De Souza, E. B. (1995). Displacement of corticotropin releasing factor from its binding protein as a possible treatment for Alzheimer's disease. Nature 378, 284–287.
Bennur, S., Shankaranarayana Rao, B. S., Pawlak, R., Strickland, S., McEwen, B. S., and Chattarji, S. (2007). Stress-induced spine loss in the medial amygdala is mediated by tissue-plasminogen activator. Neuroscience 144, 8–16.
Bittencourt, J. C., and Sawchenko, P. E. (2000). Do centrally administered neuropeptides access cognate receptors? An analysis in the central corticotropin-releasing factor system. J. Neurosci. 20, 1142–1156.
Blank, T., Nijholt, I., Eckart, K., and Spiess, J. (2002). Priming of long-term potentiation in mouse hippocampus by corticotropin-releasing factor and acute stress: implications for hippocampus-dependent learning. J. Neurosci. 22, 3788–3794.
Brunson, K. L., Kramár, E., Lin, B., Chen, Y., Colgin, L. L., Yanagihara, T. K., Lynch, G., and Baram, T. Z. (2005). Mechanisms of late-onset cognitive decline after early-life stress. J. Neurosci. 25, 9328–9338.
Bourne, J. N., and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67.
Chalmers, D. T., Lovenberg, T. W., and De Souza, E. B. (1995). Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J. Neurosci. 15, 6340–6350.
Chen, Y., Bender, R. A., Frotscher, M., and Baram, T. Z. (2001). Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J. Neurosci. 21, 7171–7181.
Chen, Y., Bender, R. A., Brunson, K. L., Pomper, J. K., Grigoriadis, D. E., Wurst, W., and Baram, T. Z. (2004a). Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc. Natl. Acad. Sci. U.S.A. 101, 15782–15787.
Chen, Y., Brunson, K. L., Adelmann, G., Bender, R. A., Frotscher, M., and Baram, T. Z. (2004b). Hippocampal corticotropin releasing hormone: pre- and postsynaptic location and release by stress. Neuroscience 126, 533–540.
Chen, Y., Brunson, K. L., Müller, M. B., Cariaga, W., and Baram, T. Z. (2000). Immunocytochemical distribution of corticotropin-releasing hormone receptor type-1 (CRF(1))-like immunoreactivity in the mouse brain: light microscopy analysis using an antibody directed against the C-terminus. J. Comp. Neurol. 420, 305–323.
Chen, Y., Dubé, C. M., Rice, C. J., and Baram, T. Z. (2008). Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J. Neurosci. 28, 2903–2911.
Chen, Y., Fenoglio, K. A., Dubé, C. M., Grigoriadis, D. E., and Baram, T. Z. (2006). Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol. Psychiatry 11, 992–1002.
Chen, Y., Kramár, E. A., Chen, L. Y., Babayan, A. H., Andres, A. L., Gall, C. M., Lynch, G., and Baram, T. Z. (2012). Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol. Psychiatry. doi: 10.1038/mp.2012.17. [Epub ahead of print].
Chen, Y., Rex, C. S., Rice, C. J., Dubé, C. M., Gall, C. M., Lynch, G., and Baram, T. Z. (2010). Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc. Natl. Acad. Sci. U.S.A. 107, 13123–13128.
Contarino, A., Dellu, F., Koob, G. F., Smith, G. W., Lee, K. F., Vale, W., and Gold, L. H. (1999). Reduced anxiety-like and cognitive performance in mice lacking the corticotropin-releasing factor receptor 1. Brain Res. 835, 1–9.
de Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475.
Diamond, D. M., Campbell, A. M., Park, C. R., Woodson, J. C., Conrad, C. D., Bachstetter, A. D., and Mervis, R. F. (2006). Influence of predator stress on the consolidation versus retrieval of long-term spatial memory and hippocampal spinogenesis. Hippocampus 16, 571–576.
Fuxe, K., Agnati, L. F., Härfstrand, A., Zoli, M., von Euler, G., Grimaldi, R., Merlo Pich, E., Bjelke, B., Eneroth, P., Benfenati, F., Cintra, A., Zini, I., and Martire, M. (1990). On the role of neuropeptide Y in information handling in the central nervous system in normal and physiopathological states. Focus on volume transmission and neuropeptide Y/alpha 2 receptor interactions. Ann. N.Y. Acad. Sci. 579, 28–67.
Hollrigel, G. S., Chen, K., Baram, T. Z., and Soltesz, I. (1998). The pro-convulsant actions of corticotropin-releasing hormone in the hippocampus of infant rats. Neuroscience 84, 71–79.
Holtmaat, A., and Svoboda, K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658.
Ivy, A. S., Rex, C. S., Chen, Y., Dubé, C., Maras, P. M., Grigoriadis, D. E., Gall, C. M., Lynch, G., and Baram, T. Z. (2010). Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J. Neurosci. 30, 13005–13015.
Joëls, M. (2008). Functional actions of corticosteroids in the hippocampus. Eur. J. Pharmacol. 583, 312–321.
Kim, J. J., and Diamond, D. M. (2002). The stressed hippocampus, synaptic plasticity and lost memories. Nat. Rev. Neurosci. 3, 453–462.
Koch, Y., Baram, T., Chobsieng, P., and Fridkin, M. (1974). Enzymic degradation of luteinizing hormone-releasing hormone (LH-RH) by hypothalamic tissue. Biochem. Biophys. Res. Commun. 61, 95–103.
Krugers, H. J., Hoogenraad, C. C., and Groc, L. (2010). Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nat. Rev. Neurosci. 11, 675–681.
Landgraf, R., and Neumann, I. D. (2004). Vasopressin and oxytocin release within the brain: adynamic concept of multiple and variable modes of neuropeptide communication. Front. Neuroendocrinol. 25: 150–176. doi: 10.1016/j.yfrne.2004.05.001
López, J. F., Akil, H., and Watson, S. J. (1999). Neural circuits mediating stress. Biol. Psychiatry 46, 1461–1471.
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445.
Magarinos, A. M., and McEwen, B. S. (1995). Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience 69, 83–88.
Maggio, N., and Segal, M. (2009). Differential modulation of long-term depression by acute stress in the rat dorsal and ventral hippocampus. J. Neurosci. 29, 8633–8638.
Maras, P. M., and Baram, T. Z. (2012). Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. PMID: 22386641. [Epub ahead of print].
Matys, T., Pawlak, R., Matys, E., Pavlides, C., McEwen, B. S., Strickland, S., and Matys, T. (2004). Tissue plasminogen activator promotes the effects of corticotropin-releasing factor on the amygdala and anxiety-like behavior. Proc. Natl. Acad. Sci. U.S.A. 101, 16345–16350.
McEwen, B. S. (2004). Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann. N.Y. Acad. Sci. 1032, 1–7.
McEwen, B. S. (2011). The ever-changing brain: cellular and molecular mechanisms for the effects of stressful experiences. Dev. Neurobiol. doi: 10.1002/dneu.20968. [Epub ahead of print].
Nässel, D. R. (2009). Neuropeptide signaling near and far: how localized and timed is the action of neuropeptides in brain circuits? Invert. Neurosci. 9, 57–75.
Pacák, K., and Palkovits, M. (2001). Stressor specificity of central neuroendocrine responses: implications for stress-related disorders. Endocr. Rev. 22, 502–548.
Perrin, M. H., and Vale, W. W. (1999). Corticotropin releasing factor receptors and their ligand family. Ann. N.Y. Acad. Sci. 885, 312–328.
Refojo, D., Schweizer, M., Kuehne, C., Ehrenberg, S., Thoeringer, C., Vogl, A. M., Dedic, N., Schumacher, M., von Wolff, G., Avrabos, C., Touma, C., Engblom, D., Schütz, G., Nave, K. A., Eder, M., Wotjak, C. T., Sillaber, I., Holsboer, F., Wurst, W., and Deussing, J. M. (2011). Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science 333, 1903–1907.
Roozendaal, B., Brunson, K. L., Holloway, B. L., McGaugh, J. L., and Baram, T. Z. (2002). Involvement of stress-released corticotropin-releasing hormone in the basolateral amygdala in regulating memory consolidation. Proc. Natl. Acad. Sci. U.S.A. 99, 13908–13913.
Sakanaka, M., Shibasaki, T., and Lederis, K. (1987). Corticotropin releasing factor-like immunoreactivity in the rat brain as revealed by a modified cobalt-glucose oxidase-diaminobenzidine method. J. Comp. Neurol. 260, 256–298.
Sanchez, M. M., Young, L. J., and Plotsky, P. M. (2000). Distribution of corticosteroid receptors in the rhesus brain: relative absence of glucocorticoid receptors in the hippocampal formation. J. Neurosci. 20, 4657–4668.
Sandi, C. (2011). Glucocorticoids act on glutamatergic pathways to affect memory processes. Trends Neurosci. 34, 165–176.
Schierloh, A., Deussing, J., Wurst, W., Zieglgänsberger, W., and Rammes, G. (2007). Corticotropin-releasing factor (CRF) receptor type 1-dependent modulation of synaptic plasticity. Neurosci. Lett. 416, 82–86.
Segal, M. (2010). Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur. J. Neurosci. 31, 2178–2184.
Segal, M., Richter-Levin, G., and Maggio, N. (2010). Stress-induced dynamic routing of hippocampal connectivity: a hypothesis. Hippocampus 12, 1332–1338.
Snyder, K., Wang, W. W., Han, R., McFadden, K., and Valentino, R. J. (2012). Corticotropin-releasing factor in the norepinephrine nucleus, locus coeruleus, facilitates behavioral flexibility. Neuropsychopharmacology 37, 520–530.
Stern, C. M., Meitzen, J., and Mermelstein, P. G. (2011). Corticotropin-releasing factor and urocortin I activate CREB through functionally selective Gβγ signaling in hippocampal pyramidal neurons. Eur. J. Neurosci. 34, 671–681.
Swinny, J. D., and Valentino, R. J. (2006). Corticotropin-releasing factor promotes growth of brain norepinephrine neuronal processes through Rho GTPase regulators of the actin cytoskeleton in rat. Eur. J. Neurosci. 24, 2481–2490.
Tringali, G., Lisi, L., De Simone, M. L., Aubry, J. M., Preziosi, P., Pozzoli, G., and Navarra, P. (2009). Effects of olanzapine and quetiapine on corticotropin-releasing hormone release in the rat brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 33, 1017–1021.
Ulrich-Lai, Y. M., and Herman, J. P. (2009). Neural regulation of endocrine and autonomic stress responses. Nat. Rev. Neurosci. 10, 397–409.
Valentino, R. J., and Wehby, R. G. (1988). Corticotropin-releasing factor: evidence for a neurotransmitter role in the locus coeruleus during hemodynamic stress. Neuroendocrinology 48, 674–677.
Van Bockstaele, E. J., Colago, E. E., and Valentino, R. J. (1996). Corticotropin-releasing factor-containing axon terminals synapse onto catecholamine dendrites and may presynaptically modulate other afferents in the rostral pole of the nucleus locus coeruleus in the rat brain. J. Comp. Neurol. 364, 523–534.
Van Pett, K., Viau, V., Bittencourt, J. C., Chan, R. K., Li, H. Y., Arias, C., Prins, G. S., Perrin, M., Vale, W., and Sawchenko, P. E. (2000). Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J. Comp. Neurol. 428, 191–212.
Vyas, A., Mitra, R., Shankaranarayana Rao, B. S., and Chattarji, S. J. (2002). Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. Neuroscience 22, 6810–6818.
Wang, H. L., Tsai, L. Y., and Lee, E. H. (2000). Corticotropin-releasing factor produces a protein synthesis–dependent long-lasting potentiation in dentate gyrus neurons. J. Neurophysiol. 83, 343–349.
Wang, H. L., Wayner, M. J., Chai, C. Y., and Lee, E. H. (1998). Corticotrophin-releasing factor produces a long-lasting enhancement of synaptic efficacy in the hippocampus. Eur. J. Neurosci. 10, 3428–3437.
Wang, X. D., Chen, Y., Wolf, M., Wagner, K. V., Liebl, C., Scharf, S. H., Harbich, D., Mayer, B., Wurst, W., Holsboer, F., Deussing, J. M., Baram, T. Z., Müller, M. B., and Schmidt, M. V. (2011a). Forebrain CRHR1 deficiency attenuates chronic stress-induced cognitive deficits and dendritic remodeling. Neurobiol. Dis. 42, 300–310.
Wang, X. D., Rammes, G., Kraev, I., Wolf, M., Liebl, C., Scharf, S. H., Rice, C. J., Wurst, W., Holsboer, F., Deussing, J. M., Baram, T. Z., Stewart, M. G., Müller, M. B., and Schmidt, M. V. (2011b). Forebrain CRF1 modulates early-life stress-programmed cognitive deficits. J. Neurosci. 31, 13625–13634.
Wingo, A. P., Fani, N., Bradley, B., and Ressler, K. J. (2010). Psychological resilience and neurocognitive performance in a traumatized community sample. Depress. Anxiety 27, 768–774.
Yan, X. X., Toth, Z., Schultz, L., Ribak, C. E., and Baram, T. Z. (1998). Corticotropin-releasing hormone (CRH)-containing neurons in the immature rat hippocampal formation: light and electron microscopic features and colocalization with glutamate decarboxylase and parvalbumin. Hippocampus 8, 231–243.
Yehuda, R., Joëls, M., and Morris, R. G. (2010). The memory paradox. Nat. Rev. Neurosci. 11, 837–839.
Keywords: hippocampus, neurotransmission, corticotropin-releasing factor, long-term potentiation, volume transmission, CRF, CRH receptor, CRFR1
Citation: Chen Y, Andres AL, Frotscher M and Baram TZ (2012) Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front. Cell. Neurosci. 6:13. doi: 10.3389/fncel.2012.00013
Received: 25 February 2012; Accepted: 14 March 2012;
Published online: 03 April 2012.
Edited by:
Nicola Maggio, The Chaim Sheba Medical Center, IsraelReviewed by:
Corette Wierenga, Utrecht University, NetherlandsNicola Maggio, The Chaim Sheba Medical Center, Israel
Copyright: © 2012 Chen, Andres, Frotscher and Baram. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Tallie Z. Baram, Departments of Pediatrics, Anatomy/Neurobiology, and Neurology, University of California-Irvine, Medical Sciences I, ZOT: 4475, Irvine, CA 92697-4475, USA. e-mail:dGFsbGllQHVjaS5lZHU=