


- 1 Center for Neuroscience, Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, Netherlands
- 2 Department of Neuroscience and Pharmacology, Rudolf Magnus Institute of Neuroscience, University Medical Center Utrecht, Utrecht, Netherlands
One of the core reactions in response to a stressful situation is the activation of the hypothalamus–pituitary–adrenal axis which increases the release of glucocorticoid hormones from the adrenal glands. In concert with other neuro-modulators, such as (nor)adrenaline, these hormones enable and promote cognitive adaptation to stressful events. Recent studies have demonstrated that glucocorticoid hormones and noradrenaline, via their receptors, can both rapidly and persistently regulate the function of excitatory synapses which are critical for storage of information. Here we will review how glucocorticoids and noradrenaline alone and in synergy dynamically tune these synapses in the hippocampus and amygdala, and discuss how these hormones interact to promote behavioral adaptation to stressful situations.
Introduction
Situations which potentially disturb homeostatic processes in body and mind, and which are subjectively perceived as a threat, i.e., stress, initiate the activation of two systems aimed at helping the organism to adapt (de Kloet et al., 2005; Kvetnansky et al., 2009; McEwen and Gianaros, 2010). First, the autonomic nervous system is activated upon arousal, ultimately causing the rapid release of (nor)adrenaline from the adrenal medulla into the circulation which – as a hormone – can rapidly regulate the function of peripheral organs. Via indirect pathways involving the nucleus tractus solitarius as well as more directly via activation of noradrenergic cells in the locus coeruleus, noradrenaline is also abundantly released in the brain, including close to cells in limbic structures like the amygdala and hippocampus (McGaugh, 2004; Valentino and Von Bockstaele, 2008). In these areas, noradrenaline – as a neurotransmitter – regulates neuronal function via α- and β-adrenergic receptors (Gibbs and Summers, 2002; Roozendaal et al., 2009a).
Somewhat later, the hypothalamus–pituitary–adrenal (HPA) axis is activated, which causes the secretion of corticosteroid hormones from the adrenal cortex. The prevailing corticosteroid hormone in most rodents is corticosterone, while in humans cortisol is most prevalent. Corticosteroid hormones are very lipophilic and therefore easily pass the blood–brain barrier, in principle reaching all cells but acting only on those carrying receptors. Two types of corticosteroid receptors are expressed in the brain: (1) mineralocorticoid receptors (MRs), which bind corticosterone, cortisol, and aldosterone with high affinity; and (2) glucocorticoid receptors (GRs) with an approximately 10-fold lower affinity for corticosterone and cortisol (de Kloet et al., 2005). Due to this difference in affinity, MRs are already substantially occupied by the natural ligand under rest, whereas activation of GRs to a large extent only occurs when corticosteroid levels are high, e.g., after exposuretostressful experiences. Pyramidal cells in the hippocampal CA1 area and granule cells in the dentate gyrus abundantly express both MR and GR. In most other limbic areas, including in the basolateral amygdala (BLA), expression of GR is higher than that of MR; the exception is formed by CA3 pyramidal neurons, which highly express MR but have only low levels of GR.
Corticosteroid hormones interact with various neurotransmitter systems, e.g., serotonin, dopamine, and endocannabinoids (for reviews, see Czyrak et al., 2003; Joëls et al., 2007; Hill et al., 2010; Haj-Dahmane and Shen, 2011). In this review, the focus is on the interactive effects of the neurotransmitter noradrenaline and the hormone corticosterone, with respect to synaptic function and behavioral relevance. While most cells in limbic brain regions are exposed to both noradrenaline and corticosteroid hormones after stressful events, the kinetic properties of exposure differ for the two ligands (see Figure 1). In vivo microdialysis studies, e.g., in the amygdala have shown that noradrenaline levels quickly rise after stress, but are normalized within an hour (Quirarte et al., 1998). By contrast, corticosteroid hormone levels in the brain are raised with a delay of approximately 20 min (compared to the rise observed in plasma; Droste et al., 2008) and return to baseline after 1–2 h. Catecholamines such as noradrenaline primarily act through G-protein coupled receptors which, via second messengers, alter the functionality of ion channels. This causes rapid-onset changes in electrical properties of neurons, which generally are also quickly reversible when noradrenaline levels in the synaptic cleft decline. Next to these rapid effects, gene-mediated events may develop, e.g., via CREB, which may lastingly affect synaptic function. This can induce a secondary, delayed response to noradrenaline, starting by the time that rapid effects have subsided, and last for hours.
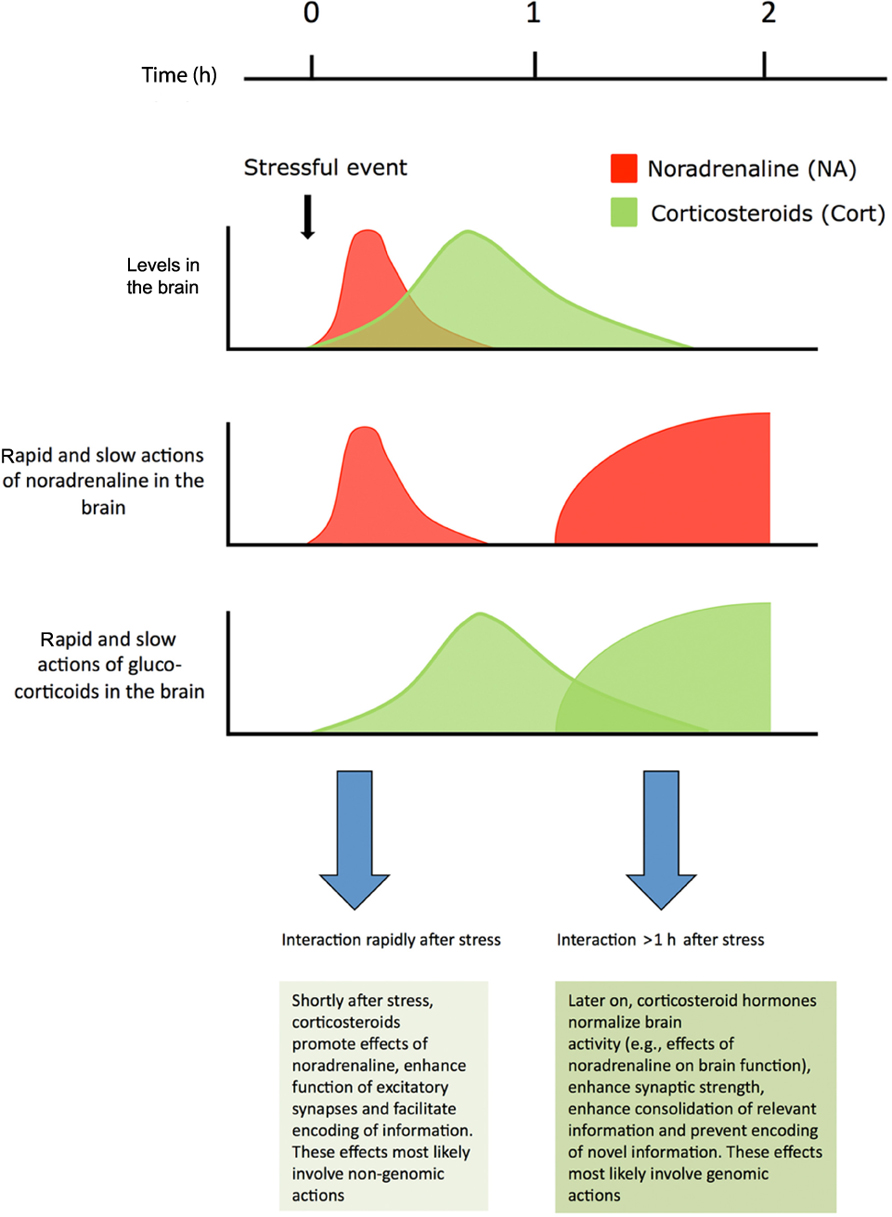
FIGURE 1. Shortly after stress noradrenaline levels in the brain are transiently elevated. Corticosteroids reach the same brain areas later and remain elevated for approximately 2 h. For a restricted period of time neurons are exposed to high levels of both hormones. Noradrenaline primarily works through a rapid G-protein coupled pathway, but long-lasting secondary genomic effects requiring gene transcription may develop. Corticosteroid hormones exert rapid non-genomic actions via membrane receptors, and also slowly, but persistently, regulate neuronal function via nuclear receptors Corticosterone and noradrenaline regulate synaptic transmission and promote memory performance, both alone and in a synergistic fashion. For details, see text. Figure adapted from Joëls et al. (2011).
In contrast to noradrenaline, corticosteroid hormones act through receptors – i.e., MRs and GRs – known to be transcriptional regulators (Duma et al., 2006). Upon binding of the hormone to the intracellular receptor, the latter dissociates from chaperone molecules and translocates to the nucleus, where it binds to response elements in an estimated 1–2% of the genes or, alternatively, changes gene transcription indirectly by interfering with the efficacy of other transcription factors. Both of these pathways were indeed demonstrated to take place in the brain (Morsink et al., 2006). Consequently, corticosteroid effects on neuronal activity are generally slow in onset, developing with a delay of approximately an hour, yet long-lasting. The most prominent of these corticosteroid effects after stress are caused by GR rather than MR activation (for review, see Joëls et al., 2007). Over the past decade, though, it has become increasingly evident that corticosteroid hormones also induce rapid effects on neuronal activity, which involve non-genomic signaling, via activation of MRs and GRs (Orchinik et al., 1991; Venero and Borrell, 1999; Di et al., 2003; Karst et al., 2005, 2010; Groc et al., 2008). Corticosteroid hormones can therefore rapidly and persistently regulate neuronal function, via activation of MRs and GRs.
Despite the different kinetics for adrenergic and steroid signaling, there is a window in time during which cells in limbic brain areas are simultaneously exposed to elevated levels of both catecholamines and corticosteroid hormones, allowing these neuromodulators to affect neuronal processes in concert (Joëls et al., 2011). Although, as mentioned, corticosteroid hormones are known to interact with various neurotransmitter systems, we discuss in this review only how noradrenaline (as a neurotransmitter) and corticosteroid hormones, alone or in interaction, regulate synaptic transmission (particularly via glutamate), synaptic plasticity, and memory formation.
Noradrenaline, Corticosteroid Hormones, and Excitatory Synapses in Limbic Regions
One important function of stress is to induce long-term adaptive responses. Enhanced memory for stressful events is one of these well-known highly adaptive phenomena that help to remember relevant information. The current view of how memories are formed is that neurons are activated during the learning process, thereby changing the strength of synaptic connections between these cells. These changes in synaptic strength are generally believed to underlie storage of information, and learning and memory processes (Whitlock et al., 2006; Neves et al., 2008).
Noradrenergic Effects on AMPA Receptors
There is ample evidence that the dynamic regulation of AMPA-type glutamate receptors (AMPARs) – which mediate most of the fast excitatory synaptic transmission in brain cells – can change synaptic function and regulate storage of information (Kessels and Malinow, 2009). Recent studies have revealed that AMPARs are regulated by noradrenaline and glucocorticoid hormones. Thus, via activation of β-adrenergic receptors, noradrenaline and stress can rapidly – but reversibly – activate PKA and CaMKII, and increase the phosphorylation of GluA1 AMPAR subunits at Ser845 and Ser831 in the hippocampus, a critical step for synaptic insertion of these receptors (Wang et al., 2004; Hu et al., 2007). In addition, activation of β-adrenergic receptors facilitates the induction of hippocampal long-term potentiation (LTP; Thomas et al., 1996; Winder et al., 1999; Hu et al., 2007; Tenorio et al., 2010) and enhances activity-dependent synaptic insertion of AMPARs (Hu et al., 2007). Interestingly, activation of β-adrenergic receptors facilitates LTP in a time-dependent manner, i.e., only when receptors are phosphorylated by β-adrenergic activation, the induction of LTP is enhanced.
Corticosteroid Effects on AMPA Receptors in the Hippocampus and Prefrontal Cortex
Corticosteroid hormones can rapidly and reversibly change hippocampal synaptic transmission. Rapid corticosteroid effects on neuronal activity were first described in detail for parvocellular neurons in the hypothalamic paraventricular nucleus (PVN; reviewed in Tasker et al., 2006). In these cells, corticosterone rapidly decreases the mEPSC frequency, via a retrograde signaling pathway involving the endocannabinoid receptor 1. This potentially suppresses activity of PVN cells. In neurons located in the hippocampal CA1 area (Karst et al., 2005) and dentate gyrus (Pasricha et al., 2011) however, mEPSC frequency is rapidly and reversibly enhanced by corticosterone. Within minutes after application, glucocorticoids increase synaptic transmission in the hippocampus (Karst et al., 2005; Pasricha et al., 2011), via activation of low affinity MRs which are probably located in the cellular membrane. This rapid and reversible increase in synaptic transmission after glucocorticoid exposure most likely results from an increase in the presynaptic release of glutamate (Karst et al., 2005) in which the Erk pathway is critically involved (Olijslagers et al., 2008). Glucocorticoid exposure, via membrane MRs, also rapidly increases lateral diffusion of GluA1 and GluA2 subunits in primary hippocampal cultures, without altering the number of postsynaptic AMPARs (Groc et al., 2008) and promotes the activity-dependent synaptic insertion of GluA2-containing AMPARs (Groc et al., 2008). Furthermore, corticosterone shifts the voltage-dependent activation of a transient K-conductance (IA) to the right, thus reducing its influence during depolarization and thereby its inhibitory action (Olijslagers et al., 2008). All of these actions potentially lead to a transiently raised hippocampal activity shortly after stress. Finally, glucocorticoids facilitate LTP in a time-dependent manner; LTP is only facilitated when elevated corticosteroid levels are present at the moment of high-frequency stimulation (Wiegert et al., 2006). These studies show that both noradrenaline and glucocorticoids can rapidly facilitate hippocampal synaptic plasticity and thereby increase the ability to encode information at the cellular level. The results are not entirely unequivocal, though. Recently, dexamethasone–BSA was reported to rapidly increase the frequency and amplitude of hippocampal spontaneous GABAergic currents within minutes (Hu et al., 2010). GABAergic transmission is also enhanced in the dorsal (but not ventral) hippocampus at a somewhat slower timescale, starting 25 min after treatment onset (Maggio and Segal, 2009). This would potentially decrease the activity of hippocampal cells.
After exposure to a stressful event, plasma corticosteroid levels slowly return to their pre-stress level, a process that requires about 2 h (de Kloet et al., 2005). Still, these hormones exert – via a slow, genomic mode of action – long-lasting effects on excitatory synapses. In hippocampal primary cultures – which contain cells from various hippocampal subregions – elevated glucocorticoid levels increase the membrane expression and synaptic insertion of GluA2-containing AMPARs (Groc et al., 2008; Martin et al., 2009). These effects are mediated via GRs, require more than an hour to develop as well as the synthesis of new proteins, and most likely result from increased lateral diffusion and/or altered ratio of endocytosis/exocytosis of GluA2-containing AMPARs (Groc et al., 2008; Martin et al., 2009). In hippocampal primary neurons as well as identified CA1 pyramidal cells, glucocorticoids slowly increase the amplitude of evoked as well as spontaneous AMPAR-mediated synaptic currents (Karst and Joëls, 2005; Martin et al., 2009), thereby enhancing AMPAR-mediated synaptic transmission in specific synapses. A similar effect has been observed > 1 h after stress in prefrontal neurons, via the induction of serum- and glucocorticoid-inducible kinase and the activation of Rab4 (Yuen et al., 2009, 2011). While LTP is enabled when plasma corticosterone levels are low, elevated plasma hormone levels slowly suppress the ability to induce LTP (Diamond et al., 1992; Kim and Diamond, 2002). Elevated plasma corticosterone levels may hamper synaptic plasticity possibly because these hormones and synaptic plasticity make use of overlapping signaling pathways, which causes occlusion of one by the other (Groc et al., 2008). Corticosteroids facilitate long-term depression (LTD; Coussens et al., 1997; Xu et al., 1997) and increase endocytosis of synaptic AMPARs upon stimuli that weaken synaptic transmission (Martin et al., 2009). A vast amount of studies has seen this pattern of reduced LTP/enhanced LTD first reported for the hippocampus (see review in Kim and Diamond, 2002), but enhanced LT P was reported for the ventral-most part (20%) of the hippocampus (Maggio and Segal, 2007).
Corticosteroid Effects on AMPA Receptors in the Basolateral Amygdala
Activity of neurons in the BLA is mostly enhanced after corticosterone exposure in a slow GR-dependent way (Duvarci and Paré, 2007; Liebmann et al., 2008). BLA neurons respond rapidly to corticosterone in yet another manner. Thus, in slices prepared from animals under rest, having very low circulating corticosterone levels, exposure to corticosterone causes a nongenomic enhancement in mEPSC frequency via MRs, similar to the hippocampus (Karst et al., 2010). However, in BLA cells this enhancement is sustained, a phenomenon that requires not only the presence of MR but also of GR and protein synthesis. This alters the state of BLA neurons such that they respond differently to renewed exposure to corticosterone, now causing a decrease in mEPSC frequency, via a rapid GR-dependent mechanism involving the endocannabinoid receptor 1, reminiscent of the mechanism described in the PVN. BLA cells therefore seem – at least with respect to the nongenomic corticosteroid actions – more sensitive to the recent stress history of the animal than hippocampal CA1 and dentate cells.
Taken together, these studies indicate that exposure of limbic cells to either noradrenaline or corticosteroid hormones can rapidly but also slowly regulate limbic glutamatergic synapses (Krugers et al., 2010; Joëls et al., 2011).
AMPA Receptors, Stress, and Behavior
Behavioral studies indicate that regulation of AMPARs by noradrenaline and corticosterone is relevant for learning and memory. Studies using mice carrying mutations in the GluA1 phosphorylation sites indicate that noradrenaline-regulated phosphorylation of GluA1 facilitates emotional memory in a contextual fear conditioning task (Hu et al., 2007). Moreover, application of pep2m, which blocks trafficking of GluA2-containing AMPA receptors prevents the memory-enhancing effects of stress in the Morris water-maze (Conboy and Sandi, 2010), and cued fearful memories (Migues et al., 2010). Also, stress-induced regulation of Rab4/SGK may underlie stress-effects on AMPA receptor function in the prefrontal cortex and working memory (Yuen et al., 2011).
Crosstalk Between Glucocorticoids and Noradrenaline at the Cellular Level
Noradrenaline and glucocorticoids not only change transmission independent from each other but also interact to regulate excitatory synapses (Joëls et al., 2011). This interaction may take place in two windows of time after exposure to a stressful experience. In the first window, shortly after exposure to stress, rapid non-genomic actions of corticosteroids coincide with initial effects of noradrenaline, so that interactions could take place while both stress mediators are present in substantial amounts within 1 h after stress and act on those cells carrying receptors for both types of modulators. In the second window, presumably hours after exposure to stress, corticosterone might also change noradrenergic actions via its slow genomic pathway, either when given in advance of stress and/or noradrenaline (a pharmacological approach) or in the aftermath of stress. In the following sections we will discuss the current evidence for interactions, at the cellular/circuit level, at the animal behavior level, and from human research.
Hippocampus
Recent data indicates that corticosterone and noradrenaline interact to rapidly regulate AMPA receptor function at the cellular level (Zhou et al., 2011). Thus, application of corticosterone for 15 min to hippocampal slices, at a dose that activates both MRs and GRs (30 nM), did not affect phosphorylation of the AMPAR GluA1 subunit at S845 or S831. Co-application of the β-adrenergic receptor agonist isoproterenol, however, largely increased S845 (but not S831) phosphorylation. Similarly, corticosterone alone did not rapidly change GluA1 and GluA2 surface expression in hippocampal primary cultures. However, combined administration of corticosterone and isoproterenol – which by itself was ineffective – enhanced surface expression. Interestingly, a high dosage of isoproterenol in the absence of corticosterone enhanced GluA1 surface expression, and this effect was decreased by corticosterone. Finally, in hippocampal primary cultures, mEPSC frequency was enhanced by the combination of isoproterenol and corticosterone (ineffective by themselves) while the same combination did not affect the amplitude. Although there are differences in dose-dependency of these various interactive effects of corticosterone and noradrenaline – which might result from differences in preparation –, the data indicates that there is an optimal combination at which noradrenaline and corticosterone interact to regulate AMPAR function and that beyond these concentrations the combined responses decline.
These rapid interactions aimed at glutamatergic transmission may bear relevance to observations at the circuit level in the dentate gyrus (Pu et al., 2007). For instance, perforant path stimulation in a theta-burst pattern – in slices from adult animals – by itself does not induce synaptic potentiation, but application of isoproterenol (1 μM) just prior to and during high-frequency stimulation causes robust synaptic potentiation. The onset of this potentiation is not instantaneous and was found to be accelerated when corticosterone (100 nM) was applied in addition to isoproterenol. One hour after high-frequency stimulation there was no difference between the signals recorded from slices exposed to isoproterenol alone or to the combination of the two hormones, suggesting that there were no interactions in the later time-domain. However, the 1-h delay may have been too short to reveal such interactions. This explanation is supported by the fact that in slices pretreated with a pulse of corticosterone > 1 h before delivery of isoproterenol (and high-frequency stimulation), a significant attenuation of the isoproterenol effect was observed. Given that thus applied corticosterone by itself (i.e., without subsequent isoproterenol administration) did not change synaptic potentiation, these data support interactive rather than additive actions of the two hormones.
These observations in the dentate gyrus are in line with findings reported over two decades ago at the single cell level in the CA1 area (Joëls and de Kloet, 1989). In CA1 pyramidal neurons, noradrenaline (via β-adrenergic receptors) reduces a calcium-dependent K-conductance, causing cells to fire more action potentials during a depolarizing episode. The efficacy to do so was strongly attenuated by pretreatment with corticosterone, via a slow GR-dependent process. Thus, both in the hippocampal CA1 area and dentate gyrus, β-adrenergic facilitation of excitability is markedly attenuated by pretreatment with corticosterone via a slow and presumably gene-mediated pathway.
Basolateral Amygdala
In principal neurons of the BLA, isoproterenol causes a dose-dependent rapid enhancement of AMPAR-mediated synaptic responses, while the NMDAR mediated component is unaffected (Liebmann et al., 2009). This was not altered by simultaneous application of (100 nM) corticosterone. However, if corticosterone was applied > 1 h in advance of isoproterenol, the facilitation of AMPAR-mediated synaptic responses by a moderate dose (0.4 μM) of the β-adrenoceptor agonist was strongly reduced.
This interaction was mirrored in recordings at the circuit level (Pu et al., 2009). Thus, isoproterenol was able to potentiate synaptic (field) responses for at least 60 min after delivery of a mild tetanic stimulation. In contrast to what was seen in the dentate gyrus, no acceleration of this effect by simultaneously applied corticosterone was observed. Instead, corticosterone gradually reversed the effect of isoproterenol; the corticosteroid hormone by itself did not affect synaptic responses after mild tetanic stimulation. The gradually developing attenuation by corticosterone was even more pronounced when the hormone was administered > 1 h in advance of isoproterenol and high-frequency stimulation.
Some Principles About Hormonal Interactions at the Single Cell/Circuit Level
Overall, these data at the single cell and circuit level shows that at the short-term corticosterone may accelerate or enhance the efficacy of noradrenaline to facilitate synaptic transmission and potentiation in limbic cells (Joëls et al., 2011). These effects are relatively mild, though, and not always apparent. There is evidence that these interactions may only occur with intermediate levels of synaptic input and/or moderately high hormone concentrations; when input/hormone levels are too low, interactive effects remain sub-threshold, while too high levels of input/hormone levels seem to cause ceiling or even reversed effects. An important limitation of all studies so far is the fact that in actual life noradrenaline and corticosterone will not reach limbic cells at the exact same moment. None of the studies so far has addressed this issue (further discussed in Section “Conclusion”).
The slow genomic effect by corticosterone seems rather consistent: attenuation of β-adrenergic actions by pretreatment with corticosterone was observed in the hippocampal CA1 area and dentate gyrus as well as the BLA. While an approach in which corticosterone is given > 1 h in advance of noradrenaline is pharmacologically relevant and certainly has helped to delineate the slow corticosteroid actions, there is paucity in studies examining whether corticosterone co-administered with noradrenaline may reverse and normalize noradrenergic actions after approximately 1 h. At this moment we can only infer such effects from the experimental design using corticosteroid pretreatment. The exception is formed by a study on synaptic potentiation in the BLA which provides preliminary evidence that corticosterone can indeed exert such normalizing actions (Pu et al., 2009).
Interactions at the Behavioral Level in Rodents
Noradrenaline and corticosteroid hormones, via their receptors, mediate (at least in part) the memory-enhancing effects of stress and emotion (Roozendaal et al., 2009a; Joëls et al., 2011). Noradrenaline enhances memory formation of emotional events via brain β-adrenergic receptors: application of noradrenaline or β-adrenergic receptor agonists promotes memory consolidation in various aversive memory tasks such as inhibitory avoidance task, fear conditioning and in Morris water-maze learning (Hu et al., 2007; Roozendaal et al., 2009a; but see also Hatfield and McGaugh, 1999; Lee et al., 2001; Bush et al., 2010), and blocking β-adrenergic receptors reduces contextual fear memories (Ji et al., 2003). Activation of α-adrenergic receptors also enhances memory, presumably acting by enhancing β-adrenergic actions (Ferry et al., 1999). Noradrenaline has also been reported to enhance reconsolidation of information (e.g., Debiec and LeDoux, 2004).
Corticosteroid hormones, via MRs have been implicated in the appraisal and response selection during the learning process (Oitzl and de Kloet, 1992; Sandi and Rose, 1994). Recent studies provide evidence that MRs are involved in encoding of information, possibly linked to effects on appraisal and/or response selection. For instance, application of the MR antagonist spironolactone prior to training lastingly suppressed the expression of fear (Zhou et al., 2010). Moreover, genetic deletion of MRs in the forebrain led to various cognitive impairments, including impaired learning in a Morris water-maze task (Berger et al., 2006) and reduced fear learning (Zhou et al., 2010). Via GRs, corticosteroid hormones have been reported to promote long-term consolidation of information (de Kloet et al., 1999; Joëls et al., 2006; Roozendaal et al., 2009a). For instance, a point mutation in the mouse GR was found to impair spatial memory formation (Oitzl et al., 2001), and blocking GRs impairs fear conditioning (Pugh et al., 1997a; Donley et al., 2005). In agreement, in several fearful learning paradigms, such as fear conditioning and inhibitory avoidance learning, post-training application of corticosterone or GR agonists promotes the consolidation of information (Corodimas et al., 1994; Sandi and Rose, 1994; Pugh et al., 1997b; Hui et al., 2004). These studies imply that GRs are involved in consolidation of (fearful) information and that genomic actions are involved. This does not exclude the possibility that other GR-dependent pathways are also involved. For instance, a recent study suggested that membrane-associated GRs too promote long-term memory in an object recognition task via chromatin modification (Roozendaal et al., 2010). Thus, it is possible that both non-genomic as well as genomic actions of corticosteroid hormones, via GRs, promote the storage of relevant information.
In addition to the well-documented effects of stress and glucocorticoids on consolidation processes, these hormones also affect memory retrieval mechanisms (De Quervain et al., 1998). Exposure to stress and elevated corticosteroid levels hamper the retrieval of already stored information (De Quervain et al., 1998). Blocking MRs and GRs also hampers the reconsolidation of context and cue-conditioned fear respectively (Pitman et al., 2011; Zhou et al., 2011). Taken together, there is ample evidence that corticosteroid hormones, via activation of MRs and GRs, have a repertoire of behavioral effects that promote the consolidation and updating of relevant (fearful) information and ultimately favor behavioral adaptation (de Kloet et al., 1999).
Several recent reviews (e.g., Roozendaal et al., 2009a) have highlighted that particularly interactions between noradrenaline and corticosterone affect (emotional) memory formation, a process in which the hippocampus and amygdala play a crucial role. We will here only describe a few examples which nicely illustrate the principles. Thus, the presence of noradrenaline is crucial for facilitation of emotional memory in rodents (Quirarte et al., 1997). Moreover, post-training administration of noradrenaline or β-adrenergic receptor agonists into the BLA produces a dose-dependent enhancement of amygdala-dependent memory formation (Ferry et al., 1999). Corticosterone can modulate noradrenergic effects on memory formation but seems to be unable to enhance memory formation independent of noradrenaline. This is most clearly demonstrated by an experiment in which post-training corticosterone administration enhanced spatial and aversive memory formation, a process blocked by concurrent intra-BLA infusions of a β-adrenoceptor antagonist (Roozendaal et al., 2006). Similarly, corticosterone administered to naïve rats enhanced object recognition, an effect that was again blocked by the β-adrenoceptor antagonist propranolol. Corticosterone was ineffective in rats with reduced training-associated emotional arousal due to prior habituation to the experimental context (Okuda et al., 2004). Conversely, emotional arousal effects were mimicked in well-habituated rats by releasing endogenous noradrenaline via administration of the α2-adrenoceptor antagonist yohimbine (presumably causing higher noradrenaline levels) immediately after object recognition training (Roozendaal et al., 2006).
In contrast to the reduced preparations used for cellular studies, studies with intact animals should consider at least two other aspects of interactions between the two hormones. First, corticosteroids are known to increase the availability of noradrenaline in the BLA (McReynolds et al., 2010). Second, corticosteroid and noradrenergic actions in one region cannot be regarded independent from what happens in associated areas. For instance, interactions between noradrenaline and corticosterone in the BLA time-dependently influence the function of the dentate gyrus (Akirav and Richter-Levin, 1999). Corticosteroids and noradrenaline also interact in the prefrontal or insular cortices to enhance memory consolidation (Miranda et al., 2008; Roozendaal et al., 2009b). Thus, similar to what was described for the BLA, administration of a β-adrenoceptor antagonist into these brain regions prevents the memory enhancement by concurrently administered corticosteroids (Barsegyan et al., 2010). Due to reciprocal connections between the prefrontal cortex and BLA, interactions in one area will almost certainly influence the functionality in the other.
There is substantial evidence from behavioral studies that (of the two types of corticosteroid receptors) at least GRs play a role in the modulation of noradrenergic function (Roozendaal et al., 2009a). The relatively short delay between hormone application and behavioral effects seems to favor a non-genomic mode of action. Rapid and presumably non-genomic effects via MRs, however, are also involved in successful memory formation under arousing conditions (Zhou et al., 2010). To what extent genomic actions of corticosteroids interact with noradrenaline to change memory formation is more difficult to assess. Nevertheless, there is indirect evidence that such interactive effects do play a role. The most straightforward example comes from a study using mice that were genetically modified such that GRs do not homodimerize and thus cannot bind to the DNA (Oitzl et al., 2001). Training of these animals in a water-maze paradigm – which is sufficiently stressful to increase levels of both noradrenaline and corticosterone – resulted in a poor spatial performance compared to the wildtype controls. Calcium currents, and thus calcium-dependent attenuation of firing frequency, were not increased by corticosterone in these mutant mice (Karst et al., 2000). This may allow for more retrograde interference of stress-unrelated information, a possible explanation for the impaired behavioral performance. Interestingly, one behavioral study used a corticosterone-pretreatment paradigm which quite closely resembles that used in cellular investigations. In this study (Borrell et al., 1984), pretreatment with corticosteroids 1 h before adrenaline administration was demonstrated to dramatically reduce the efficacy of the latter to affect amygdala-dependent behavior. Both examples support the view that slow genomic GR effects reduce/normalize noradrenergic actions on behavior, which is in line with the observations at the cellular level.
Interactions in the Human Brain
Most studies in humans indicate that stressful and emotional events are remembered well. This most likely involves endogenous catecholamines like noradrenaline (Cahill et al., 1994; Strange and Dolan, 2004; Onur et al., 2009) but also corticosteroids (Lupien et al., 2002; Marin et al., 2011). Several studies have specifically investigated the interactions between noradrenaline and corticosteroid hormones. Some of these studies are discussed in the following paragraphs.
Smeets et al. (2009) examined if stress exposure prior to encoding of a list of words affected learning and memory performance. The stressor consisted of public speaking about one’s personality in front of an unresponsive panel. The words to be learned were either related to personality or unrelated to personality but of comparable valence. Afterward each subject ranked all words on an arousal-scale, allowing – on an individual basis – a distinction between low-arousing and high-arousing words. It turned out that stress improved learning and long-term memory of high-arousing context-related words, at the cost of memorizing low-arousing context-related words, whereas no effect was seen with regard to the context-unrelated words. The improved memory for context-related high-arousing words was particularly evident in individuals who performed the task immediately after stress exposure, more so than in those who carried out the task 2 h later. If the order was reversed (learning prior to stress exposure), memory performance was unchanged. Interestingly, the memory for high- versus low-arousing context-related words in subjects stressed just prior to learning correlated significantly with a combined index for their salivary alpha-amylase and cortisol levels, which reflect the function of the autonomic and HPA systems respectively, but did not correlate with either of these parameters alone, underlining the potential relevance of interactions between the two systems.
Van Stegeren et al. (2010) used a pharmacological approach, specifically addressing the interactive effect of the two hormones on memory formation. Subjects received yohimbine and hydrocortisone, prior to encoding of arousing and neutral pictures. The timing of hydrocortisone administration (45 min before encoding) was slightly ambiguous, probably allowing the development of non-genomic as well as genomic effects. At the behavioral level, combined drug administration led to the best (surprise) recognition of the pictures, particularly of arousing material. Contrary to the observations in animals, hydrocortisone seemed more effective than yohimbine in improving memory (see also Maheu et al., 2005), but it cannot be excluded that the experimental setting already caused substantial release of endogenous catecholamines, so that exogenous administration of drugs tapping on the same system were less effective. Paradoxically, the very good memory performance in the group receiving both yohimbine and hydrocortisone was linked to reduced activity in the hippocampus, as revealed by simultaneously acquired fMRI data. Interestingly, this reduced hippocampal activity during encoding of later remembered material was also observed when subjects instead of receiving drugs were stressed during encoding (Henckens et al., 2009). At this time one can only speculate about this observation, but one explanation could be that under stressful conditions extensive filtering of the incoming information may take place, causing restricted but highly efficient functioning of the human hippocampus.
A third example illustrating that noradrenaline and corticosteroids interact at the level of the amygdala in the human brain was supplied by Kukolja et al. (2008). In this study subjects received either (i) the noradrenaline-reuptake inhibitor reboxetine, (ii) hydrocortisone, (iii) a combination of these two drugs, or (iv) placebo, 105 min before they entered the scanner. They were exposed to film clips with actors who displayed either a happy, fearful or neutral facial expression. In control subjects happy or fearful (compared to neutral) expressions cause enhanced amygdala activity. Subjects receiving both reboxetine and hydrocortisone showed reduced amygdala activity to happy faces but enhanced activity to fearful facial expressions. This suggests that noradrenaline in interaction with cortisol gives rise to a negative bias of emotional functions.
More recently this was confirmed and extended in a study which specifically targeted the delayed and presumably genomic effects of cortisol administration under conditions that are probably sufficiently arousing to cause substantial release of endogenous noradrenaline (Henckens et al., 2010). Subjects ingested a placebo or a cortisol tablet (10 mg) either 75 or 285 min prior to exposure to a face-morphing task of both happy and fearful facial expressions while they were in a scanner. When given at 75 min prior to the test, cortisol suppressed amygdala activity to both happy and fearful faces. When cortisol was given 285 min in advance, normalized responses to fearful face stimuli but still reduced responses to happy face stimuli were observed. As seen in the study by Kukolja et al. (2008), this suggests a valence-specific slow corticosteroid effect, causing diminished amygdala processing preferentially for happy faces. Interestingly, normalization of the responses to fearful face stimuli after 285 min correlated with increased mPFC-amygdala connectivity, indicating augmented cognitive top-down control (Henckens et al., 2010). This suggests an important influence of delayed corticosteroid actions on executive functions, a field that is currently being explored.
Although the studies reported here represent only a selection of papers on the effect of stress on human cognition, they nicely illustrate the principle that noradrenaline and corticosteroid hormones interact to affect cognitive processing in the human brain.
Open Questions
When an organism experiences a stressful event, its neurons in limbic brain structures are exposed to surges of both noradrenaline and corticosteroids. In addition to these two important stress mediators, there is a myriad of transmitters and hormones that join in the overall central response to a stressful situation, including catecholamines other than noradrenaline such as dopamine and peptides like CRH and vasopressin (Joëls and Baram, 2009). Here we discussed that corticosteroids and noradrenaline regulate the function of excitatory synapses and that this is thought to contribute to the memory-enhancing effects of these hormones (Figure 1). Neuromodulators such as noradrenaline and corticosteroids work in overlapping time-domains and most likely interact. Interactions between other stress mediators, though, have not yet been studied in great detail. There might be some redundancy in the activity of these stress mediators, but the regional distribution and sub-cellular localization of their receptors will confer quite some specificity to their contribution to the overall stress–response (Joëls and Baram, 2009).
In the rapid time-domain (i.e., within 30–60 min after stress, when both hormones are present in high levels in the vicinity of neurons in limbic areas) noradrenaline and corticosterone seem to act synergistically; current evidence supports that noradrenaline is indispensable in the rapid time-domain, whereas corticosterone seems to serve a more permissive role (Roozendaal et al., 2006). MRs are important in this phase, among other things for appraisal of the situation and selection of behavioral strategies (Schwabe et al., 2010). The behavioral consequences of corticosteroids in this time-domain however, particularly in humans, still need to be addressed in detail. Such investigations in human subjects are presently hampered by the fact that (1) there are no (oral) selective ligands available for membrane MRs mediating rapid effects and (2) peripherally administered drugs require some time to reach the brain, which hampers precise timing such as is possible in vitro or with intracerebroventricular administration in rodents. But even in reduced rodent brain preparations, the “natural” order of hormone exposure – i.e., first to noradrenaline and then, with an approximate delay of 20 min, to corticosteroids – has not been examined. This clearly requires dedicated experiments, aligning the experimental designs in the reduced cell preparations, animal behavior and human studies as much as possible.
The cellular studies in rodents and neuroimaging studies in humans regarding delayed effects of corticosteroid hormones on noradrenaline seem to be quite consistent, all finding a suppression of the latter by the former, probably via GRs. The evidence for this view in the human brain, however, is still limited. More importantly, support for this notion from behavioral studies in rodents is near-absent. Dedicated experiments, in which administration of corticosterone is precisely timed relative to mildly arousing learning situations, could resolve this issue. To what extent these experiments with corticosteroid treatment are indicative of what happens several hours after their release during stress also remains to be investigated. If this would be the case, one could postulate that the delayed effects of corticosteroid hormones primarily play a role in response normalization after stress and consolidation of the stress-related information, a notion that is indeed supported by behavioral investigations in humans and experimental animals. Whether the interactive effects of noradrenaline and corticosteroids on excitatory synapses are crucial for the memory-enhancing effects of these neuromodulators needs to be verified.
A final consideration regards the effect of multiple surges of corticosteroid hormones. Recent cellular investigations in the rodent basolateral nucleus of the amygdala suggest that exposure to a single surge of corticosterone changes cellular properties such that these cells respond in the complete opposite way to a second exposure to corticosteroids (Karst et al., 2010); this “flip” in response depends on protein synthesis and activation of GRs. The behavioral relevance of these metaplastic responses needs further investigation, but given the pulsatile release pattern of corticosteroid hormones throughout the day (Lightman and Conway-Campbell, 2010), metaplasticity is likely to change the background excitability of these amygdala cells, even in the absence of stress. How this affects the responsivity of amygdala versus hippocampal cells to stress over the day is one of the challenging questions for the next years.
Conclusion
Shortly after stress, cells in limbic brain areas are exposed to a wave of catecholamines including noradrenaline and, slightly later, to corticosteroid hormones. These two stress mediators regulate synapses and memory performance. They interact in multiple time-domains: (1) up to approximately 1 h after stress via rapid non-genomic actions, i.e., while levels of stress mediators are high; and (2) several hours after stress exposure via genomic effects, i.e., at a time when concentrations of noradrenaline and corticosteroids have returned to pre-stress levels. Cellular studies over the past decade have shown that the two stress mediators act synergistically in the initial time-window, particularly with intermediate concentrations. Animal behavior and human studies indicate that these rapid actions may promote the encoding of stress-context related and relevant information. The latter actions, primarily exerted by corticosteroid hormones, may serve to normalize earlier effects of catecholamines and protect the encoded stress-related information. Corticosteroid hormones given out of the stress context (e.g., 1–4 h in advance of stress) generally suppress noradrenergic facilitation of neuronal activity, as shown in rodents at the cellular level and in the human brain. In the rodent amygdala, corticosterone administration after stress quickly resets neuronal activity. These out-of-context effects of corticosterone could be of relevance for pharmacotreatment of stress-related disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akirav, I., and Richter-Levin, G. (1999). Biphasic modulation of hippocampal plasticity by behavioral stress and basolateral amygdala stimulation in the rat. J. Neurosci. 19, 10530–10535.
Barsegyan, A., Mackenzie, S. M., Kurose, B. D., McGaugh, J. L., and Roozendaal, B. (2010). Glucocorticoids in the prefrontal cortex enhance memory consolidation and impair working memory by a common neural mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 16655–16660.
Berger, S., Wolfer, D. P., Selbach, O., Alter, H., Erdmann, G., Reichardt, H. M., Chepkova, A. N., Welzl, H., Haas, H. L., Lipp, H. P., and Schütz, G. (2006). Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc. Natl. Acad. Sci. U.S.A. 103, 185–200.
Borrell, J., de Kloet, E. R., and Bohus, B. (1984). Corticosterone decreases the efficacy of adrenaline to affect passive avoidance retention of adrenalectomized rats. Life Sci. 34, 99–104.
Bush, D. E., Caparosa, E. M., Gekker, A., and LeDoux, J. (2010). Beta-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front. Behav. Neurosci. 4:154. doi: 10.3389/fnbeh.2010.00154
Cahill, L., Prins, B., Weber, M., and McGaugh, J. L. (1994). Beta-adrenergic activation and memory for emotional events. Nature 371, 702–704.
Conboy, L., and Sandi, C. (2010). Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology 35, 674–685.
Corodimas, K. P., LeDoux, J. E., Gold, P. W., and Schulkin, J. (1994). Corticosterone potentiation of conditioned fear in rats. Ann. N. Y. Acad. Sci. 746, 392–393.
Coussens, C. M., Kerr, D. S., and Abraham, W. C. (1997). Glucocorticoid receptor activation lowers the threshold for NMDA-receptor-dependent homosynaptic long-term depression in the hippocampus through activation of voltage-dependent calcium channels. J. Neurophysiol. 78, 1–9.
Czyrak, A., Mackowiak, M., Chocyk, A., Fijal, K., and Wedzony, K. (2003). Role of glucocorticoids in the regulation of dopaminergic transmission. Pol. J. Pharmacol. 55, 667–674.
Debiec, J., and LeDoux, J. E. (2004). Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 129, 267–272.
de Kloet, E. R., Joëls, M., and Holsboer, F. (2005). Stress and the brain: from adaptation to disease. Nat. Rev. Neurosci. 6, 463–475.
de Kloet, E. R., Oitzl, M. S., and Joels, M. (1999). Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci. 22, 422–426.
De Quervain, D. J., Roozendaal, B., and McGaugh, J. L. (1998). Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394, 787–790.
Di, S., Malcher-Lopes, R., Halmos, K. C., and Tasker, J. G. (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J. Neurosci. 23, 4850–4857.
Diamond, D. M., Bennett, M. C., Fleshner, M., and Rose, G. M. (1992). Inverted-U relationship between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus 2, 421–430.
Donley, M. P., Schulkin, J., and Rosen, J. B. (2005). Glucocorticoid receptor antagonism in the basolateral amygdala and ventral hippocampus interferes with long-term memory of contextual fear. Behav. Brain Res. 164, 197–205.
Droste, S. K., de Groote, L., Atkinson, H. C., Lightman, S. L., Reul, J. M., and Linthorst, A. C. (2008). Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology 149, 3244–3253.
Duma, D., Jewell, C. M., and Cidlowski, J. A. (2006). Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J. Steroid Biochem. Mol. Biol. 102, 11–21.
Duvarci, S., and Paré, D. (2007). Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J. Neurosci. 27, 4482–4491.
Ferry, B., Roozendaal, B., and McGaugh, J. L. (1999). Basolateral amygdala noradrenergic influences on memory storage are mediated by an interaction between β- and α1-adrenoceptors. J. Neurosci. 19, 5119–5123.
Gibbs, M. E., and Summers, R. J. (2002). Role of adrenoceptor subtypes in memory consolidation. Prog. Neurobiol. 67, 345–391.
Groc, L., Choquet, D., and Chaouloff, F. (2008). The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat. Neurosci. 11, 868–870.
Haj-Dahmane, S., and Shen, R. Y. (2011). Modulation of the serotonin system by endocannabinoid signalling. Neuropharmacology 61, 414–420.
Hatfield, T., and McGaugh, J. L. (1999). Norepinephrine infused into the basolateral amygdala posttraining enhances retention in a spatial water maze task. Neurobiol. Learn. Mem. 71, 232–239.
Henckens, M. J., Hermans, E. J., Pu, Z., Joëls, M., and Fernández, G. (2009). Stressed memories: how acute stress affects memory formation in humans. J. Neurosci. 29, 10111–10119.
Henckens, M. J., van Wingen, G. A., Joëls, M., and Fernández, G. (2010). Time-dependent effects of corticosteroids on human amygdala processing. J. Neurosci. 30, 12725–12732.
Hill, M. N., Patel, S., Campolongo, P., Tasker, J. G., Wotjak, C. T., and Bains, J. S. (2010). Functional interactions between stress and the endocannabinoid system: from synaptic signalling to behavioural output. J. Neurosci. 30, 14980–14986.
Hu, H., Real, E., Takamiya, K., Kang, M., LeDoux, J. E., Huganir, R. L., and Malinow, R. (2007). Emotion enhances learning via norepinephrine regulation of AMPA-receptor trafficking. Cell 131, 160–173.
Hu, W., Zhang, M., Czéh, B., Flügge, G., and Zhang, W. (2010). Stress impairs GABAergic network function in the hippocampus by activating nongenomic glucocorticoid receptors and affecting the integrity of the parvalbumin-expressing neuronal network. Neuropsychopharmacology 35, 1693–1707.
Hui, G. K., Figueroa, I. R., Poytress, B. S., Roozendaal, B., McGaugh, J. L., and Weinberger, N. M. (2004). Memory enhancement of classical fear conditioning by post-training injections of corticosterone in rats. Neurobiol. Learn. Mem. 81, 67–74.
Ji, J. Z., Wang, X. M., and Li, B. M. (2003). Deficit in long-term contextual fear memory induced by blockade of beta-adrenoceptors in hippocampal CA1 region. Eur. J. Neurosci. 17, 1947–1952.
Joëls, M., and de Kloet, E. R. (1989). Effects of glucocorticoids and norepinephrine on the excitability in the hippocampus. Science 245, 1502–1505.
Joëls, M., Fernandez, G., and Roozendaal, B. (2011). Stress and emotional memory: a matter of timing. Trends Cogn. Sci. 15, 280–288.
Joëls, M., Karst, H., Krugers, H. J., and Lucassen, P. J. (2007). Chronic stress: implications for neuronal morphology, function and neurogenesis. Front. Neuroendocrinol. 28, 72–96.
Joëls, M., Pu, Z., Wiegert, O., Oitzl, M. S., and Krugers, H. J. (2006). Learning under stress: how does it work? Trends Cogn. Sci. 10, 152–158.
Karst, H., Berger, S., Erdmann, G., Schütz, G., and Joëls, M. (2010). Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc. Natl. Acad. Sci. U.S.A. 107, 14449–14454.
Karst, H., Berger, S., Turiault, M., Tronche, F., Schütz, G., and Joëls, M. (2005). Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. U.S.A. 102, 19204–19207.
Karst, H., and Joëls, M. (2005). Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J. Neurophysiol. 94, 3479–3486.
Karst, H., Karten, Y. J., Reichardt, H. M., de Kloet, E. R., Schütz, G., and Joëls, M. (2000). Corticosteroid actions in hippocampus require DNA binding of glucocorticoid receptor homodimers. Nat. Neurosci. 3, 977–978.
Kessels, H. W., and Malinow, R. (2009). Synaptic AMPA receptor plasticity and behaviour. Neuron 61, 340–350.
Kim, J. J., and Diamond, D. M. (2002). The stressed hippocampus, synaptic plasticity and lost memories. Nat. Rev. Neurosci. 3, 453–462.
Krugers, H. J., Hoogenraad, C. C., and Groc, L. (2010). Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nat. Rev. Neurosci. 11, 675–681.
Kukolja, J., Schläpfer, T. E., Keysers, C., Klingmüller, D., Maier, W., Fink, G. R., and Hurlemann, R. (2008). Modeling a negative response bias in the human amygdala by noradrenergic-glucocorticoid interactions. J. Neurosci. 28, 12868–12876.
Kvetnansky, R., Sabban, E. L., and Palkovits, M. (2009). Catecholaminergic systems in stress: structural and molecular genetic approaches. Physiol. Rev. 89, 535–606.
Lee, H. J., Berger, S. Y., Stiedl, O., Spiess, J., and Kim, J. J. (2001). Post-training injections of catecholaminergic drugs do not modulate fear conditioning in rats and mice. Neurosci. Lett. 303, 123–126.
Liebmann, L., Karst, H., and Joëls, M. (2009). Effects of corticosterone and the beta-agonist isoproterenol on glutamate receptor-mediated synaptic currents in the rat basolateral amygdala. Eur. J. Neurosci. 30, 800–807.
Liebmann, L., Karst, H., Sidiropoulou, K., van Gemert, N., Meijer, O. C., Poirazi, P., and Joëls, M. (2008). Differential effects of corticosterone on the slow afterhyperpolarization in the basolateral amygdala and CA1 region: possible role of calcium channel sub-units. J. Neurophysiol. 99, 958–968.
Lightman, S. L., and Conway-Campbell, B. L. (2010). The crucial role of pulsatile activity of the HPA axis for continuous dynamic equilibration. Nat. Rev. Neurosci. 11, 710–718.
Lupien, S. J., Wilkinson, C. W., Brière, S., Ménard, C., Ng Ying Kin, N. M., and Nair, N. P. (2002). The modulatory effects of corticosteroids on cognition: studies in young human populations. Psychoneuroendocrinology 27, 401–416.
Maggio, N., and Segal, M. (2007). Striking variations in corticosteroid modulation of long-term potentiation along the septotemporal axis of the hippocampus. J. Neurosci. 27, 5757–5765.
Maggio, N., and Segal, M. (2009). Differential corticosteroid modulation of inhibitory synaptic currents in the dorsal and ventral hippocampus. J. Neurosci. 29, 2857–2866.
Maheu, F. S., Joober, R., and Lupien, S. J. (2005). Declarative memory after stress in humans: differential involvement of the beta-adrenergic and corticosteroid systems. J. Clin. Endocrinol. Metab. 90, 1697–1704.
Marin,M. F., Hupbach, A., Maheu, F. S., Nader, K., and Lupien, S. J. (2011). Metyrapone administration reduces the strength of an emotional memory trace in a long-lasting manner. J. Clin. Endocrinol. Metab. 96, E1221–E1227.
Martin, S., Henley, J. M., Holman, D., Zhou, M., Wiegert, O., van Spronsen, M., Joëls, M., Hoogenraad, C. C., and Krugers, H. J. (2009). Corticosterone alters AMPAR mobility and facilitates bidirectional synaptic plasticity. PLoS ONE 4, e4714. doi: 10.1371/journal.pone.0004714
McEwen, B. S., and Gianaros, P. J. (2010). Stress- and allostasis-induced brain plasticity. Annu. Rev. Med. 62, 431–445.
McGaugh, J. L. (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci. 27, 1–28.
McReynolds, J. R., Donowho, K., Abdi, A., McGaugh, J. L., Roozendaal, B., and McIntyre,C. K.(2010). Memory-enhancing corticosterone treatment increases amygdala norepinephrine and Arc protein expression in hippocampal synaptic fractions. Neurobiol. Learn. Mem. 93, 312–321.
Migues, P. V., Hardt, O., Wu, D. C., Gamache, K., Sacktor, T. C., Wang, Y. T., and Nader, K. (2010). PKMzeta maintains memories by regulating GluR2-dependent AMPA receptor trafficking. Nat. Neurosci. 13, 630–634.
Miranda, M. I., Quirarte, G. L., Rodriguez-Garcia, G., McGaugh, J. L., and Roozendaal, B. (2008). Glucocorticoids enhance taste aversion memory via actions in the insular cortex and basolateral amygdala. Learn. Mem. 15, 468–476.
Morsink, M. C., Steenbergen, P. J., Vos, J. B., Karst, H., Joëls, M., de Kloet, E. R., and Datson, N. A. (2006). Acute activation of hippocampal glucocorticoid receptors results in different waves of gene. J. Neuroendocrinol. 18, 239–252.
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat. Rev. Neurosci. 9, 65–75.
Oitzl, M. S., and de Kloet, E. R. (1992). Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav. Neurosci. 106, 62–71.
Oitzl, M. S., Reichardt, H. M., Joëls, M., and de Kloet, E. R. (2001). Point mutation in the mouse glucocorticoid receptor preventing DNA binding impairs spatial memory. Proc. Natl. Acad. Sci. U.S.A. 98, 12790–12795.
Okuda, S., Roozendaal, B., and McGaugh, J. L. (2004). Glucocorticoid effects on object recognition memory require training-associated emotional arousal. Proc. Natl. Acad. Sci. U.S.A. 101, 853–858.
Olijslagers, J. E., de Kloet, E. R., Elgersma, Y., van Woerden, G. M., Joëls, M., and Karst, H. (2008). Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur. J. Neurosci. 27, 2542–2550.
Onur, O. A., Walter, H., Schlaepfer, T. E., Rehme, A. K., Schmidt, C., Keysers, C., Maier, W., and Hurlemann, R. (2009). Noradrenergic enhancement of amygdala responses to fear. Soc. Cogn. Affect. Neurosci. 4, 119–126.
Orchinik, M., Murray, T. F., and Moore, F. L. (1991). A corticosteroid receptor in neuronal membranes. Science 252, 1848–1851.
Pasricha, N., Joels, M., and Karst, H. (2011). Rapid effects of corticosterone in the mouse dentate gyrus via a nongenomic pathway. J. Neuroendocrinol. 23, 143–147.
Pitman, R. K., Milad, M. R., Igoe, S. A., Vangel, M. G., Orr, S. P., Tsareva, A., Gamache, K., and Nader, K. (2011). Systemic mifepristone blocks reconsolidation of cueconditioned fear; propranolol prevents this effect. Behav. Neurosci. 125, 632–638.
Pu, Z., Krugers, H. J., and Joëls, M. (2007). Corticosterone time-dependently modulates beta-adrenergic effects on long-term potentiation in the hippocampal dentate gyrus. Learn. Mem. 14, 359–367.
Pu, Z., Krugers, H. J., and Joëls, M. (2009). Beta-adrenergic facilitation of synaptic plasticity in the rat basolateral amygdala in vitro is gradually reversed by corticosterone. Learn. Mem. 16, 155–160.
Pugh, C. R., Fleshner, M., and Rudy, J. W. (1997a). Type II glucocorticoid receptor antagonists impair contextual but not auditorycue fear conditioning in juvenile rats. Neurobiol. Learn. Mem. 67, 75–79.
Pugh, C. R., Tremblay, D., Fleshner, M., and Rudy, J. W. (1997b). A selective role for corticosterone in contextual-fear conditioning. Behav. Neurosci. 111, 503–511.
Quirarte, G. L., Galvez, R., Roozendaal, B., and McGaugh, J. L. (1998). Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Res. 808, 134–140.
Quirarte, G. L., Roozendaal, B., and McGaugh, J. L. (1997). Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 94, 14048–14053.
Roozendaal, B., Hernandez, A., Cabrera, S. M., Hagewoud, R., Malvaez, M., Stefanko, D. P., Haettig, J., and Wood, M. A. (2010). Membrane-associated glucocorticoid activity is necessary for modulation of long-term memory via chromatin modification. J. Neurosci. 30, 5037–5046.
Roozendaal, B., McEwen, B. S., and Chattarji, S. (2009a). Stress, memory and the amygdala. Nat. Rev. Neurosci. 10, 423–433.
Roozendaal, B., McReynolds, J. R., Van der Zee, E. A., Lee, S., McGaugh, J. L., and McIntyre, C. K. (2009b). Glucocorticoid effects on memory consolidation depend on functional interactions between the medial prefrontal cortex and basolateral amygdala. J. Neurosci. 29, 14299–14308.
Roozendaal, B., Okuda, S., Van der Zee, E. A., and McGaugh, J. L. (2006). Glucocorticoid enhancement of memory requires arousal-induced noradrenergic activation in the basolateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 103, 6741–6746.
Sandi, C., and Rose, S. P. (1994). Corticosterone enhances long-term retention in one-day-old chicks trained in a weak passive avoidance learning paradigm. Brain Res. 647, 106–112.
Schwabe, L., Schächinger, H., de Kloet, E. R., and Oitzl, M. S. (2010). Corticosteroids operate as a switch between memory systems. J. Cogn. Neurosci. 22, 1362–1372.
Smeets, T., Wolf, O. T., Giesbrecht, T., Sijstermans, K., Telgen, S., and Joëls, M. (2009). Stress selectively and lastingly promotes learning of context-related high arousing information. Psychoneuroendocrinology 34, 1152–1161.
Strange, B. A., and Dolan, R. J. (2004). Beta-adrenergic modulation of emotional memory-evoked human amygdala and hippocampal responses. Proc. Natl. Acad. Sci. U.S.A. 101, 11454–11458.
Tasker, J., Di, S., and Malcher-Lopes, R. (2006). Minireview: rapid glucocorticoid signaling via membrane-associated receptors. Endocrinology 147, 5549–5556.
Tenorio, G., Connor, S. A., Guévremont, D., Abraham, W. C., Williams, J., O’Dell, T. J., and Nguyen, P. V. (2010). ’Silent’ priming of translation-dependent LTP by β-adrenergic receptors involves phosphorylation and recruitment of AMPA receptors. Learn. Mem. 23, 627–638.
Thomas, M. J., Moody, T. D., Makhinson, M., and O’Dell, T. J. (1996). Activity-dependent beta-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron 17, 475–482.
Valentino, R. J., and Von Bockstaele, E. (2008). Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur. J. Pharmacol. 583, 194–203.
Van Stegeren, A. H., Roozendaal, B., Kindt, M., Wolf, O. T., and Joëls, M. (2010). Interacting noradrenergic and corticosteroid systems shift human brain activation patterns during encoding. Neurobiol. Learn. Mem. 93, 56–65.
Venero, C., and Borrell, J. (1999). Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus: a microdialysis study in freely moving rats. Eur. J. Neurosci. 11, 2465–2473.
Wang, W., Zhu, W., Wang, S., Yang, D., Crow, M. T., Xiao, R. P., and Cheng, H. (2004). Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/ calmodulin kinase signaling pathway. Circ. Res. 95, 798–806.
Whitlock, J. R., Heynen, A. J., Shuler, M. G., and Bear, M. F. (2006). Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097.
Wiegert, O., Joëls, M., and Krugers, H. (2006). Timing is essential for rapid effects of corticosterone on synaptic potentiation in the mouse hippocampus. Learn. Mem. 13, 110–113.
Winder, D. G., Martin, K. C., Muzzio, I. A., Rohrer, D., Chruscinski, A., Kobilka, B., and Kandel, E. R. (1999). ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron24, 715–726.
Xu, L., Anwyl, R., and Rowan, M. J. (1997). Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387, 497–500.
Yuen, E. Y., Liu, W., Karatsoreos, I. N., Feng, J., McEwen, B. S., and Yan, Z. (2009). Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc. Natl. Acad. Sci. U.S.A. 106, 14075–14079.
Yuen, E. Y., Liu, W., Karatsoreos, I. N., Ren, Y., Feng, J., McEwen, B. S., and Yan, Z. (2011). Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol. Psychiatry 16, 156–170.
Zhou, M., Bakker, E. H., Velzing, E. H., Berger, S., Oitzl, M., Joëls, M., and Krugers, H. J. (2010). Both mineralocorticoid and glucocorticoid receptors regulate emotional memory in mice. Neurobiol. Learn. Mem. 94, 530–537.
Keywords: hippocampus, amygdala, mouse, electrophysiology, glutamate
Citation: Krugers HJ, Karst H and Joels M (2012) Interactions between noradrenaline and corticosteroids in the brain: from electrical activity to cognitive performance. Front. Cell. Neurosci. 6:15. doi: 10.3389/fncel.2012.00015
Received: 02 February 2012; Paper pending published: 20 February 2012;
Accepted: 20 March 2012; Published online: 09 April 2012.
Edited by:
Menahem Segal, Weizmann Institute of Science, IsraelReviewed by:
Menahem Segal, Weizmann Institute of Science, IsraelOliver Von Bohlen Und Halbach, Universitätsklinikum Greifswald der Ernst-Moritz-Arndt-Universität, Germany
Copyright: © 2012 Krugers, Karst and Joels. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Harm J. Krugers, Center for Neuroscience, Swammerdam Institute for Life Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, Netherlands. e-mail:aC5rcnVnZXJzQHV2YS5ubA==