 Benjamin H. Levy1
Benjamin H. Levy1 Jeffrey G. Tasker1,2*
Jeffrey G. Tasker1,2*
- 1 Neuroscience Program, Tulane University, New Orleans, LA, USA
- 2 Department of Cell and Molecular Biology, Tulane University, New Orleans, LA, USA
Dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis has been implicated in a range of affective and stress-related disorders. The regulatory systems that control HPA activity are subject to modulation by environmental influences, and stressful life events or circumstances can promote subsequent HPA dysregulation. The brain is a major regulator of the HPA axis, and stress-induced plasticity of the neural circuitry involved in HPA regulation might constitute an etiological link between stress and the development of HPA dysregulation. This review focuses on the synaptic regulation of neuroendocrine corticotropin-releasing hormone (CRH) neurons of the hypothalamic paraventricular nucleus, which are the cells through which the brain predominantly exerts its influence on the HPA axis. CRH neuronal activity is largely orchestrated by three neurotransmitters: GABA, glutamate, and norepinephrine. We discuss our current understanding of the neural circuitry through which these neurotransmitters regulate CRH cell activity, as well as the plastic changes in this circuitry induced by acute and chronic stress and the resultant changes in HPA function.
Introduction
Glucocorticoids are released in response to physical, emotional, and/or metabolic stress, and many of the effects of glucocorticoids are thought to serve as adaptive responses to stressful events or circumstances. Physiological levels of glucocorticoids are regulated by the hypothalamic–pituitary–adrenal (HPA) axis, which is responsible for a cascade of hormone signals that begins in the brain and ends with glucocorticoid secretion from the adrenal cortex. An additional component of the HPA axis is a glucocorticoid-mediated negative feedback mechanism, wherein glucocorticoids act on the anterior pituitary and in the brain to suppress HPA activity. Given the association between glucocorticoids and stress, it is not surprising that abnormal regulation of the HPA axis is commonly associated with a range of affective and stress-related disorders. Research into these phenomena has dealt mostly with hypersecretion of glucocorticoid in patients with depressive illness (Gillespie and Nemeroff, 2005), although there has also been interest in the hyposecretion of glucocorticoids found in some cases of post-traumatic stress disorder (Yehuda, 2006) and depression (Morphy et al., 1985; Penninx et al., 2007; Ahrens et al., 2008). Dysregulation of the HPA axis has also been associated with panic disorder (Abelson et al., 2007), chronic fatigue syndrome (Van Houdenhove et al., 2009), obsessive-compulsive disorder (Gustafsson et al., 2008), fibromyalgia (Tanriverdi et al., 2007), generalized anxiety disorder (Lenze et al., 2011), and bipolar disorder (Daban et al., 2005). Importantly, stressful life events have been implicated in the onset of each of these disorders (Kendler et al., 1995; Hatcher and House, 2003; Gupta and Silman, 2004; Brawman-Mintzer et al., 2005; Garno et al., 2005; Goodwin et al., 2005; Sarkhel et al., 2011), and dysregulation of the HPA axis may be an etiological link between stress and the subsequent development of pathology.
The physiological origins of stress-induced variation in HPA regulation have remained largely unknown, although rodent models have shed some light on the matter, largely by providing insight into the ways that the brain regulates HPA activity. In fact, the neural circuitry that regulates the HPA axis has been found to be highly plastic, and that plasticity is induced by both stress and changes in glucocorticoid levels. It is possible that exposure to stress and/or the resultant fluctuations in circulating glucocorticoids results in pathology by evoking long-lasting alterations in the circuitry that regulates the HPA axis. Here, we will review what is known about the synaptic regulation of the HPA axis and the plasticity of the circuitry regulating the HPA axis induced under stress conditions. We will focus on the neuroendocrine corticotropin-releasing hormone (CRH) cells, which are located in the paraventricular nucleus (PVN) of the hypothalamus and are situated at the apex of the HPA axis. The CRH cells are of particular interest because it is principally through the regulation of CRH release into the portal circulation that the brain exerts its influence on the HPA axis. As with all neurons, CRH cells are stimulated by depolarization of their cell membrane, which stimulates action potentials and triggers the release of peptide from their axon terminals. However, unlike with classical neurons, these terminals do not form synapses with postsynaptic neurons; but rather, they are incorporated into a region at the base of the brain, the median eminence, where released neuropeptide accesses the pituitary portal circulation via fenestrated portal arterioles. In addition to CRH, the CRH cells can synthesize and release vasopressin (VP). CRH and VP in the portal circulation bind to CRH and VP receptors on a subset of cells, the corticotropes, of the anterior pituitary to stimulate the secretion of adrenocorticotropic hormone (ACTH) into the systemic circulation. The ACTH then stimulates the synthesis and systemic secretion of glucocorticoids from the adrenal cortex. Hypothalamic CRH neuronal activity is largely orchestrated by three neurotransmitters: GABA, glutamate, and norepinephrine. In this review we discuss our current understanding of how these neurotransmitter systems regulate CRH neuron activity (and consequently HPA activity), and the modulation of these neurotransmitter systems by glucocorticoids and stress. One aim of this review is to provide a framework to help guide future investigations of the synaptic regulation of CRH cells that have the potential to advance our understanding of the neurological bases for HPA dysfunction. It is also an aim of this paper to inform those who study stress-induced abnormalities in HPA activity, but who may not be familiar with studies of synaptic transmission, in the hope of making these studies more accessible for the purpose of enhancing communication and promoting interdisciplinary investigations.
Methods of Identifying CRH Cells
Inasmuch as CRH cells play such a central role here, before discussing the synaptic regulation of the CRH neurons, we will briefly address the challenges in identifying the CRH cells in the course of physiological experiments. The PVN can be divided into three major cell types: the magnocellular neuroendocrine cells, which project their axons into the posterior lobe of the pituitary, the parvocellular neuroendocrine cells, which send their axons to the median eminence, and the parvocellular preautonomic neurons, which project to the brain stem and spinal cord. The CRH neurons fall into the parvocellular neuroendocrine cell type. The three cell types can be distinguished with reasonable reliability on the basis of their respective somatic sizes and shapes, dendritic morphologies, and positions in the PVN. Magnocellular neuroendocrine cells tend to have larger, more rounded somata and fewer dendritic branches, often bipolar in morphology, compared to the parvocellular neuroendocrine cells, which tend to be smaller, fusiform, and with multipolar dendritic arbors. The magnocellular neuroendocrine cells are concentrated in the lateral portion of the PVN, while the parvocellular neuroendocrine cells are located mainly in the medial region of the PVN. The dorsal-most and ventral-most regions of the PVN tend to be occupied by the parvocellular preautonomic neurons, which are intermediate in somatic size between the parvocellular neuroendocrine cells and the magnocellular neuroendocrine cells, and have multipolar dendritic arbors. While this morphological and topographical organization of the PVN can be used to identify enriched populations of the different subtypes of PVN neurons, there is fairly extensive overlap of the anatomical characteristics of the cell types within the PVN (Simmons and Swanson, 2009), such that it does not provide for a strict distillation of the three neuron subpopulations.
Distinct electrical properties have also been characterized in the three PVN cell types and have been used to assign electrical fingerprints for the reliable identification of individual cells in in vitro electrophysiological studies (Figure 1). Magnocellular neuroendocrine cells generate a robust A-type voltage-dependent K+ current, which causes a prominent transient outward rectification that delays action potential generation (Tasker and Dudek, 1991). Most parvocellular preautonomic neurons generate a T-type Ca2+ current, which causes a small low-threshold spike and clustering of Na+-dependent action potentials (Stern, 2001; Luther et al., 2002). Parvocellular neuroendocrine cells can be distinguished from both magnocellular neuroendocrine cells and parvocellular preautonomic cells by the absence of both the transient outward rectification and the low-threshold Ca2+ spike (Luther and Tasker, 2000; Luther et al., 2002; Figure 1).
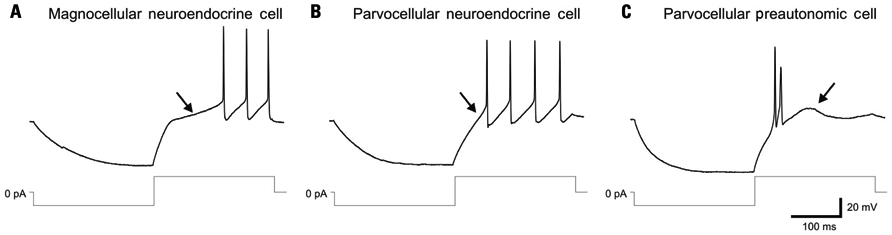
FIGURE 1. Distinct electrophysiological fingerprints of PVN magnocellular and parvocellular neurons. (A) Magnocellular neuroendocrine cells recorded in current-clamp mode generate an A-type K+ current-mediated transient outward rectification (arrow) that is generated by depolarization from a hyperpolarized holding membrane potential and delays the onset of spiking. Lower traces: current injection protocol. (B) Parvocellular neuroendocrine cells fail to generate a transient outward rectification and delay to spiking (arrow) in response to a similar current injection protocol. (C) Parvocellular preautonomic cells do not display a transient outward rectification, but generate a low-threshold spike (arrow), mediated by a T-type Ca2+ current, and clustered action potentials in response to a similar current injection protocol. Modified from Luther and Tasker (2000).
While useful for narrowing down the field of the three cell subtypes, these anatomical and electrophysiological approaches do not allow investigators to specifically distinguish CRH neurons from the other PVN parvocellular neuroendocrine cells, such as thyrotropin releasing hormone and somatostatin cells. Electron and confocal microscopy immunohistochemical studies using selective antibodies for CRH and for neurotransmitters or vesicular neurotransmitter transporters have been useful for characterizing the anatomical innervation of CRH neurons and its plasticity in response to stress and adrenalectomy (Flak et al., 2009). Because basal CRH peptide expression is not robust, attempts to identify CRH neurons during electrophysiological recordings with a combination of intracellular dye injection and post hoc immunostaining for CRH have been largely unsuccessful. One method that has been useful for specifically targeting CRH cells for synaptic analysis is intracellular recording combined with single-cell, reverse transcription–PCR analysis (Di et al., 2003). This technique allows the correlation of molecular expression profiles, including CRH mRNA expression, with electrophysiological properties. Transgenic mice that express green fluorescent protein under the transcriptional control of the CRH promoter have recently become available (Alon et al., 2009) and offer the greatest promise for contributing significantly to the study of the synaptic regulation of identified CRH neurons and the synaptic plasticity of CRH neurons under conditions of stress.
GABA Inputs
Origins of GABA inputs to the PVN have been identified through the use of tract tracing in conjunction with immunohistochemical staining, as well as through in vitro electrophysiological analyses. A retrograde tracing-immunohistochemistry study revealed four discrete origins of significant GABAergic innervation of the PVN, which are: the area surrounding the supraoptic nucleus, the anterior perifornical region, the anterior hypothalamic area, and the anterior one-third of the PVN itself (Roland and Sawchenko, 1993). There is also evidence from tract tracing studies for a prominent GABAergic projection from the bed nucleus of the stria terminalis (BNST) to the PVN that relays inhibitory signaling to CRH cells from the prefrontal cortex (Radley et al., 2009) and the hippocampus (Radley and Sawchenko, 2011). Studies employing in vivo ibotenic acid lesions of subnuclei of the BNST have implicated specifically the posterior medial region of the bed nucleus as an inhibitory relay from limbic structures to the PVN (Choi et al., 2007,2008). An in vitro brain slice electrophysiological study identified robust local GABAergic innervation of both parvocellular and magnocellular neurons of the PVN that originates in the perinuclear area surrounding the PVN (Boudaba et al., 1996). The perinuclear PVN area has also been postulated to serve as an inhibitory relay into the PVN from upstream limbic structures, such as the hippocampus and septum (Ziegler and Herman, 2002).
The inhibitory effect of GABA on CRH cells is mediated primarily by GABAA receptors. In situ hybridization assays detected the expression of the GABAA receptor α2, β1, and β3 subunits in nearly all the CRH cells of the PVN, and expression of α1 and β2 in about half of the CRH cells (Cullinan, 2000). Interestingly, microinjection of a GABAA receptor antagonist into the PVN is sufficient to activate CRH cells and elicit a surge in glucocorticoid secretion (Cole and Sawchenko, 2002), revealing a tonic inhibitory GABAergic input in the PVN that constrains CRH neuronal activity under basal conditions. The tonic inhibition of CRH neurons is preserved in organotypic hypothalamic slice cultures containing the PVN (Bali and Kovacs, 2003), which suggests that local GABA neuronal circuitry (i.e., GABA circuitry retained through the slicing procedure) plays a central role in this mechanism. The source of local GABAergic innervation of the PVN CRH neurons is predominantly extranuclear (Figure 2), including a robust input from the peri-PVN region (Boudaba et al., 1996).
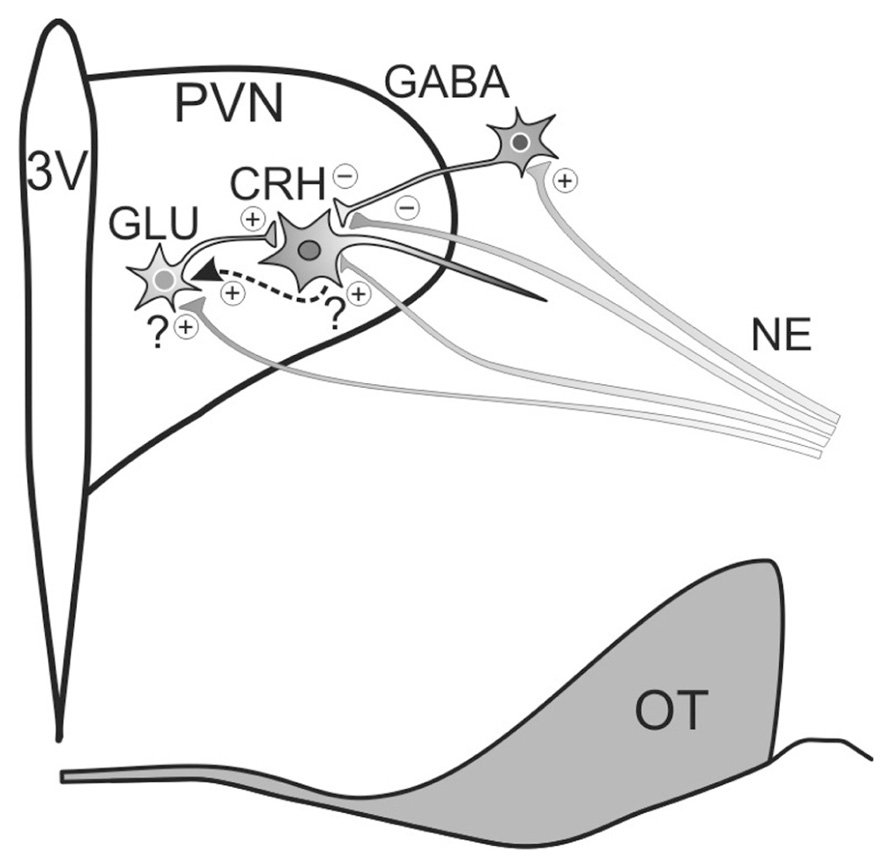
FIGURE 2. Model of noradrenergic regulation of PVN CRH neuron activity. Noradrenergic inputs (NE) originate in the brainstem and regulate CRH neuronal activity by modulating glutamate and GABA release. Norepinephrine both suppresses and enhances GABAergic inhibition of CRH neuron activity, suppressing GABA release via α2-adrenoceptor activation at presynaptic terminals and promoting GABA release by activating α1-adrenoceptors on upstream GABAergic somata/dendrites. Noradrenergic facilitation of glutamate release onto CRH neurons is spike-dependent and is mediated by α1-adrenoceptor activation. Preliminary evidence suggests that the facilitatory effect on glutamate release may be mediated by the release of a retrograde messenger that stimulates upstream local glutamatergic circuits (dashed arrow). 3V, thrid ventricle; OT, optic tract.
Stress Plasticity of GABA Inputs
Acute Stress Plasticity of GABA Inputs
The GABAergic synaptic innervation of PVN neurons undergoes significant plastic changes in response to acute stress, which alters the excitability of the parvocellular neurons and activation of the HPA axis. A 30–60 min acute restraint stress is enough to cause a significant shift in the Cl- gradient across the PVN parvocellular neuron membrane via downregulation of the membrane K+–Cl- co-transporter KCC2. This shift in the Cl- gradient causes a postsynaptic attenuation of the inhibitory GABAergic inputs to these cells and leads to the disinhibition of the HPA axis (Hewitt et al., 2009). It is not known whether glucocorticoids modulate KCC2 expression, although KCC2 expression has been found to be subject to steroidal modulation by sex hormones in substantia nigra neurons wherein, at postnatal day 15, GABAA receptors are hyperpolarizing in females and depolarizing in males (Galanopoulou and Moshe, 2003). Interestingly, a similar phenomenon has been discovered in the magnocellular neurons of the PVN, in which the GABAA reversal potential shifts positive and the GABA signals are transformed into excitatory signals in oxytocinergic magnocellular neurons following dehydration, a chronic physiological stress (Kim et al., 2011); GABA was shown to be excitatory in VP cells, but not in oxytocin cells, under normal conditions (Haam et al., 2012).
Acute stress also appears to induce a long-term presynaptic plasticity in the GABAergic inhibitory innervation of PVN parvocellular neurons via a transient elevation in circulating glucocorticoids. Thus, a 30-min acute restraint stress was found to cause a persistent reduction in inhibitory synaptic inputs to putative parvocellular neurons in recordings performed in brain slices in vitro up to 5 h later, and this effect was mimicked by a previous in vivo subcutaneous injection of glucocorticoid (Verkuyl et al., 2005). These changes in inhibitory inputs were not spike-dependent, suggestive of plasticity in the GABA release probability, which was corroborated with paired-pulse analysis. An acute 20-min glucocorticoid application directly to brain slices had no rapid effect (<20 min) on GABAergic synaptic currents (Di et al., 2003), but resulted in a similar presynaptic suppression of GABA inputs to PVN parvocellular neurons 1–5 h later (Verkuyl et al., 2005), suggesting that these presynaptic glucocorticoid actions are transcriptional and occur directly in the hypothalamus.
GABAergic synaptic plasticity may also play a role in the HPA response to hypoglycemic stress via signaling through neuropeptide Y (NPY). There is a high density of NPY projections that terminate in the medial parvocellular PVN (Cowley et al., 1999), many of which originate in the arcuate nucleus, a hypothalamic region that provides access to the brain of circulating nutritional signals (Wang et al., 2004). Both insulin-induced hypoglycemia (Tuchelt et al., 2000) and intracerebroventricular injection of NPY (Dimitrov et al., 2007) activate the HPA axis and cause an increase in the circulating glucocorticoid level. In vitro electrophysiological studies reported that NPY caused a decrease in the probability of GABA release onto putative parvocellular PVN neurons via actions at multiple presynaptic NPY receptors (Cowley et al., 1999). The role of HPA axis activation in response to hypoglycemia may be related to the fact that glucocorticoids increase glucose bioavailability by shifting metabolic processes towards catabolism.
Chronic Stress Plasticity of GABA Inputs
Chronic stress and sustained changes in systemic glucocorticoid levels also lead to long-term shifts in the GABAergic synaptic innervation of the PVN parvocellular neurons. Adrenalectomy, for example, which eliminates the main endogenous source of circulating glucocorticoids, leads to an increase in the number of GABAergic synapses and in the density of GABA receptors on CRH neurons, suggesting that the loss of circulating glucocorticoids induces a proliferation of afferent GABAergic synaptic inputs to CRH neurons. Thus, quantitative electron microscopic analysis of immunocytochemically labeled GABAergic synaptic profiles revealed a ~55% increase in the number of GABAergic synapses on CRH cells in the PVN of adrenalectomized rats (Miklos and Kovacs, 2002). An in vitro electrophysiological study provided a physiological corroboration with the finding that putative parvocellular PVN neurons undergo a significant increase in inhibitory synaptic inputs in brain slices from adrenalectomized rats, and paired-pulse analysis suggested that this effect of adrenalectomy was due to an increase in the number of GABAergic synapses (Verkuyl and Joels, 2003). Glucocorticoid replacement in this study confirmed that the effect of adrenalectomy was due to the loss of endogenous glucocorticoids. Finally, binding of radiolabeled muscimol, a selective GABAA receptor agonist, was found to increase in the hypothalamus following adrenalectomy, suggesting an increase in postsynaptic expression or membrane localization of GABAA receptors following adrenalectomy (Majewska et al., 1985).
Whereas the loss of endogenous glucocorticoids by adrenalectomy was shown to increase the number of GABAergic synapses on CRH cells, chronic stress and chronically high glucocorticoid levels were reported to suppress GABAergic inhibitory synaptic inputs to PVN parvocellular neurons. In vitro electrophysiological recordings from putative parvocellular neurons in PVN brain slices taken after 3 weeks of a chronic variable stress treatment, which leads to sustained elevated glucocorticoids (Herman et al., 1995), revealed a decrease in GABAergic inhibitory synaptic inputs to parvocellular neurons without an accompanying reduction in the probability of release of GABA, suggesting a reduced number of GABAergic synapses (Joels et al., 2004; Verkuyl et al., 2004). A confocal immunofluorescence study, however, failed to detect a reduction in the density of GABAergic synaptic boutons on CRH neurons in the medial parvocellular PVN, although these measurements were taken following a shorter exposure (1 week) to the chronic variable stress treatment (Flak et al., 2009). The acute glucocorticoid-induced reduction in GABA release probability and the chronic glucocorticoid-induced decrease in GABA synapses, in conjunction with the adrenalectomy-induced increase in GABAergic synapses, suggest that there is an inverse causal relationship between glucocorticoid levels and the efficacy of GABAergic synaptic inhibition of CRH cell activity. Both acute and chronic stress, therefore, appear to downregulate GABAergic synaptic transmission in PVN parvocellular neurons, albeit in different ways. The progression from acute stress to chronic stress plasticity in the functional inhibitory synaptic regulation of parvocellular PVN neurons appears generally to involve a transition from a modulation of GABAergic synaptic strength to a structural modification of afferent GABA inputs.
While, to our knowledge, an analysis of the changes in the density of GABAA receptors in the PVN following chronic variable stress is still lacking, molecular studies on chronic stress-induced changes in GABAA receptor expression in the PVN have been performed, and have produced somewhat contradictory findings. An in situ hybridization study reported a decrease in the expression of the GABAA receptor β1 and β2 subunits (Cullinan and Wolfe, 2000). Another study using a single-cell RNA amplification approach reported no change in the β receptor subunit expression, but an elevated expression of the α5 subunit and decreased expression of the δ subunit in putative parvocellular PVN neurons following chronic variable stress (Verkuyl et al., 2004). However, the absence of a change in β-subunit expression in the latter study may have been a consequence of the small sample size, as the investigators themselves concede. While the regulation of the α5 and δ GABAA receptor subunits may suggest a stress-induced change in extrasynaptic GABAA receptor signaling, the decrease in β1 and β2 subunits expression should lead to the downregulation of functional synaptic GABAA receptors and/or a change in synaptic GABAA receptor efficacy. Corresponding changes in postsynaptic GABAA receptor sensitivity have not been reported to date.
Glutamate Inputs
Double immunohistochemical labeling for the vesicular glutamate transporter VGlut2 and for CRH has shown that PVN CRH neurons are densely innervated by glutamatergic fibers (Wittmann et al., 2005). Electrophysiological experiments have determined that functional local circuit glutamatergic innervation of putative parvocellular neurons originates in areas adjacent to the PVN, largely overlapping sources of peri-PVN GABAergic input, but at a much lower density (Boudaba et al., 1997; Tasker et al., 1998). There is also evidence that glutamatergic input to PVN neurons originates within the PVN itself (Figure 2). First, CRH-, oxytocin-, and VP-expressing neurons in the PVN have all been shown to co-express VGlut2 mRNA (Hrabovszky and Liposits, 2008). In vitro brain slice electrophysiological recordings from putative magnocellular neurons of the PVN revealed a norepinephrine-evoked stimulation of local glutamate circuits within the PVN that was spike-dependent, suggesting the presence of glutamatergic neurons in the PVN that mediate intra-PVN signaling (Daftary et al., 1998). Furthermore, intra-PVN glutamatergic circuits were also revealed in an autoradiographic study in which sources of glutamatergic input to the PVN were identified using the radiolabeled retrograde marker [3H]D-aspartate, which selectively labels glutamate neurons (Csaki et al., 2000). In addition to the peri-PVN and intra-PVN sources of glutamatergic inputs to the PVN, significant glutamatergic afferents to the PVN have also been identified emanating from the ventromedial hypothalamic nucleus, the posterior hypothalamic nucleus, the dorsomedial hypothalamic nucleus, the anterior hypothalamic nucleus, the lateral hypothalamic nucleus, the paraventricular thalamic nucleus, and the medial nucleus of the amygdala (Csaki et al., 2000; Ulrich-Lai et al., 2011).
Microinjection of glutamate into the PVN caused a depletion of CRH in the median eminence and activation of the HPA axis (Feldman and Weidenfeld, 1997), and microinjection of an ionotropic glutamate receptor antagonist into the PVN attenuated the HPA hormonal response to acute restraint stress (Ziegler and Herman, 2000). The inconsistent activation of CRH neurons and the HPA axis by direct PVN glutamate microinjection reported in another study (Cole and Sawchenko, 2002) is likely due to the desensitization of the ionotropic glutamate receptors on the PVN neurons and/or stimulation of opposing inhibitory inputs via the activation of local circuit neurons upstream from the PVN neurons. Double-label immunohistochemical assays, for example, have shown that glutamate microinjection into the PVN increases Fos induction in peri-PVN GABAergic neurons, which may project to the CRH cells and oppose the direct excitatory actions of glutamate.
Molecular studies have reported the expression of multiple subtypes of ionotropic and metabotropic glutamate receptors (mGluRs) in the PVN. One in situ hybridization study that used riboprobes reported that mRNAs for the NMDA receptor subunits GluN1, GluN2A, and GluN2B are abundantly expressed in the medial parvocellular PVN, whereas there is significantly less expression of the GluN2C and GluN2D subunits (Herman et al., 2000). This study also reported substantial expression in the medial parvocellular PVN of the kainate receptor subunits GluK2 and GluK5 and of the AMPA receptor GluA1 subunit. Another in situ hybridization study that used oligonucleotide probes to test for the colocalization of ionotropic glutamate receptor subunits with CRH found mRNAs for the AMPA receptor subunits GluA1 and GluA2 to be present in 46 and 21% of CRH cells, respectively, and mRNA for the kainate receptor subunit GluK2 to be present in 31% of CRH cells (Aubry et al., 1996). This study also found that 70% of CRH cells expressed the NMDA receptor subunit GluN1 mRNA, but no colocalization with either GluN2A or GluN2B subunits was detected. The difference in GluN2 expression between these two studies may be due to the higher sensitivity of riboprobes relative to oligonucleotide probes. Thus, it appears that kainate, AMPA, and NMDA receptors all play a role in mediating glutamatergic input to CRH cells, with the AMPA receptor subunit GluA1 predominating over the GluA2 subunit, and the NMDA receptor subunits GluN2A and GluN2B predominating over the GluN2C and GluN2D subunits.
In addition to ionotropic glutamate receptors, mGluRs play a role in the regulation of CRH cell activity (Durand et al., 2008). Double-label immunocytochemistry revealed the presence of mGluR1a in a significant number of CRH cells (Kocsis et al., 1998). Intracerebroventricular injections of selective group I and group III mGluR agonists were found to elicit an increase in circulating glucocorticoids (Lang and Ajmal, 1995; Johnson et al., 2001). Unexpectedly, intracerebroventricular injections of antagonists of the group I mGluRs were also found to trigger the activation of the HPA axis (Bradbury et al., 2003). Based on previous findings of a pre- and postsynaptic modulation of magnocellular neuroendocrine cells by group I and III mGluRs (Schrader and Tasker, 1997; Tasker et al., 1998), a similar model for the regulation of CRH cell activity by group I and group III mGluRs was proposed (Johnson et al., 2001). According to this model, group III mGluRs disinhibit CRH cell activity by suppressing the release of GABA onto CRH cells, and group I mGluRs inhibit CRH cells through stimulation of afferent GABAergic neurons, while postsynaptic group I mGluRs stimulate CRH cells directly.
Stress Plasticity of Glutamate Inputs
Acute Stress Plasticity of Glutamate Inputs
Glutamatergic synaptic inputs to CRH cells are suppressed by rapid glucocorticoid actions that appear to be involved in the glucocorticoid-mediated fast negative feedback of the HPA axis. Thus, corticosterone and dexamethasone cause a rapid (within 3–5 min) decrease in the frequency of miniature excitatory postsynaptic currents (mEPSCs) in each of the major neuroendocrine cell types of the PVN, including in CRH cells, recorded in hypothalamic slices (Di et al., 2003). This rapid glucocorticoid effect is mediated by a non-genomic mechanism via the activation of a membrane-associated receptor. Importantly, this glucocorticoid effect was blocked by antagonists and mimicked and occluded by agonists of the cannabinoid type 1 receptor (CB1R), suggesting the involvement of the endocannabinoid system. We recently corroborated this with the finding that the rapid glucocorticoid effect is absent in CB1R knockout mice (Nahar et al., submitted). This was supported by the finding that glucocorticoid rapidly (<10 min) triggers the synthesis of the major endocannabinoids anandamide (AEA) and 2-arachidonoylglycerol (2-AG) in slices of the PVN and immediate surround (Malcher-Lopes et al., 2006). Endocannabinoids signal in a retrograde fashion (i.e., from postsynaptic membrane to presynaptic membrane) and cause the inhibition of presynaptic neurotransmitter release (Freund et al., 2003). Consistent with the endocannabinoid acting as a retrograde messenger to inhibit glutamate release at glutamate synapses, the glucocorticoid effect was prevented by blockade of G protein and protein kinase activity and Ca2+ signaling specifically in the postsynaptic cell (Malcher-Lopes et al., 2006; Harris and Tasker, 2011).
Interestingly, the glucocorticoid-induced rapid suppression of glutamate release in the PVN in vitro is not reversible in brain slice electrophysiological studies for over an hour after dexamethasone administration (Di et al., 2003), which indicates that glucocorticoids trigger a form of endocannabinoid-mediated long-term depression of synaptic excitation; long-term depression is recognized as a widespread form of synaptic plasticity in the brain that reduces the efficacy of synaptic interactions (Heifets and Castillo, 2009). The physiological significance of the extended duration of the glucocorticoid-induced suppression of excitatory inputs to CRH cells remains unknown; however, we have preliminary evidence for the desensitization to the rapid glucocorticoid-induced suppression of glutamate release in brain slices from animals that had been subjected to an acute 30-min restraint stress prior to sacrifice (Jiang and Tasker, unpublished observation). This effect is likely due to the long-term depression of glutamatergic synaptic inputs by a tonic activation of presynaptic CB1 receptors via glucocorticoid-induced retrograde endocannabinoid release, since a CB1 receptor-mediated inhibitory tone was observed in glutamate inputs to parvocellular neurons from acutely stressed rats, but not from unstressed rats. A downregulation of rapid glucocorticoid-induced endocannabinoid suppression of synaptic excitation was also reported in putative parvocellular neuroendocrine cells of juvenile rats subjected to a repeated immobilization stress, although the mechanism proposed was a chronic stress-induced desensitization of the cannabinoid signaling system (Wamsteeker et al., 2010), rather than the increased endocannabinoid inhibitory tone that we find after single exposure to an acute stress or stress level of glucocorticoid. It remains to be determined how long the glucocorticoid-induced long-term depression of synaptic excitation lasts, but it may be in place to provide the time necessary for the peptidergic stores in the somata and axon terminals to replenish after a rapid and robust secretion of CRH and VP into the portal circulation. Also, for those mechanisms that are sensitive to rates-of-change in glucocorticoid concentration (Jacobson and Sapolsky, 1993), long-term depression of the synaptic excitation of CRH neurons may reduce CRH release to allow for clearance of circulating glucocorticoids in order to ensure the sensitivity of glucocorticoid targets to the secretion of a subsequent bolus.
N-methyl-D-aspartate receptor expression in the medial parvocellular PVN has also been reported to be modulated by a single exposure to a stress stimulus. An in situ hybridization study found that acute immobilization stress caused a ~35% increase in the expression of mRNA for the GluN1 subunit of the NMDA receptor in several brain regions, including in the medial parvocellular PVN (Bartanusz et al., 1995). The increased GluN1 expression suggests the possibility that acute stress may cause an overall increase in NMDA receptor function in the medial parvocellular PVN, since functional NMDA receptors are comprised of a requisite GluN1 subunit (Cull-Candy et al., 2001). However, a brain slice electrophysiological study demonstrated depressed NMDA receptor function in individual PVN parvocellular neurons following a 30-min acute immobilization stress (Kuzmiski et al., 2010). Paradoxically, the depression of NMDA receptor currents in the parvocellular neurons following acute stress placed the glutamate synapses in a permissive state that allowed them to undergo activity-dependent short-term potentiation in response to high frequency stimulation. Thus, the NMDA receptor mRNA expression and the functionality of the receptor do not appear to be regulated in parallel by stress, which suggests that there may be changes in the receptor protein expression or trafficking that may account for this discrepancy.
Chronic Stress Plasticity of Glutamate Inputs
In addition to inducing an apparent functional downregulation in the inhibitory synaptic inputs to PVN parvocellular neuroendocrine cells (Verkuyl et al., 2004), the chronic variable stress model of stress plasticity also gives rise to significant changes in the excitatory synaptic innervation of PVN parvocellular neurons. A double-labeling immunohistochemical study found that chronic variable stress significantly increased the density of synaptophysin-expressing synaptic boutons and VGlut2-expressing glutamatergic fibers in the medial parvocellular PVN, and increased the number of glutamatergic boutons directly contacting CRH-immunoreactive somata and dendrites (Flak et al., 2009). This suggests that chronic stress causes an increase in the glutamatergic synaptic innervation of the CRH neurons, which, along with the reduced GABAergic innervation of medial parvocellular neurons (Verkuyl et al., 2004), should lead to an increase in the excitability of these neurons following chronic stress exposure. Preliminary electrophysiological findings from our laboratory suggest that the increased density of immunolabeled glutamate synapses on the CRH neurons gives rise to an increase in the functional excitatory synaptic inputs to these cells (Franco et al., 2007).
Glutamate receptor expression in the PVN has also been reported to be modified by chronic stress exposure. An in situ hybridization study found that 2 weeks of chronic variable stress caused a decrease in the mRNA expression of the NMDA receptor subunit GluN2B in the medial parvocellular PVN, but found no changes in GluN1 or GluN2A subunit expression (Ziegler et al., 2005). This effect differed from the glutamate receptor plasticity induced by acute immobilization stress, which caused an increase in the expression of GluN1 reported in the aforementioned study (Bartanusz et al., 1995). The fact that the decrease in GluN2B induction caused by chronic stress was not accompanied by a change in GluN1 expression suggests that the downregulated GluN2B does not reflect a decrease in the number of functional NMDA receptors, since the GluN1 subunit is an essential component of functional NMDA receptors (Cull-Candy et al., 2001). The decrease in NMDA receptor mRNA expression with chronic stress is surprising given the observation of an increase in the density of glutamate synapses with the same chronic stress paradigm (Flak et al., 2009). This suggests either that NMDA receptor mRNA and protein expression are differentially modulated, or that there is less of a contribution of NMDA receptors relative to AMPA receptors at glutamate synapses on CRH neurons following chronic variable stress. Follow-up electrophysiological studies will be necessary to distinguish between these possibilities. Interestingly, the stress-induced downregulation of GluN2B mRNA is apparently not mediated by glucocorticoids, as neither adrenalectomy nor high-dose glucocorticoid administration produced any change in GluN2B mRNA expression in the medial parvocellular PVN (Ziegler et al., 2005). Thus, this plasticity of NMDA receptor expression following chronic variable stress may result from repeated activation of glutamatergic receptors with the chronic activation of stress circuits.
Interestingly, glutamatergic synaptic plasticity resulting from the neonatal environment might be involved in lifelong changes in HPA responsivity. Pups that are handled daily during the first 10 days of life have been found to exhibit a suppressed HPA response to a novel stressor as adults (Liu et al., 1997). It is possible that this effect of handling is mediated by an increase in maternal attention, as handled pups receive more frequent bouts of maternal care in the form of licking, grooming, and arched-back nursing. One study using an early-life handling paradigm found that handled pups have lower VGlut2 protein content in the PVN, fewer VGlut2-immunoreactive synaptic boutons contacting CRH cells, and fewer asymmetrical, putative excitatory, synapses on CRH cells (Korosi et al., 2010). Electrophysiological recordings in putative parvocellular PVN neurons from these animals found that the frequency of miniature EPSCs also was decreased, suggesting a reduced excitatory synaptic input to the CRH cells. The reduced glutamatergic input in this study was found exclusively in the pups and not following maturation to adulthood, but the handled animals exhibited life-long reductions in PVN CRH expression, which suggests that reductions in glutamatergic input early in life may be involved in a cascade of events that leads to a life-long reduction in CRH expression and HPA stress responsivity.
Norepinephrine Inputs
Noradrenergic input to the medial parvocellular PVN originates in the brainstem, particularly in the nucleus of the solitary tract (NTS; Herman et al., 2003). Norepinephrine has long been known to play a prominent role in the regulation of the HPA axis and has been shown to be generally excitatory with regard to modulation of CRH neuron activity (Plotsky et al., 1987). Accordingly, microinjection of norepinephrine directly into the PVN increases CRH mRNA expression and elevates circulating ACTH in a dose-dependent manner (Itoi et al., 1994). There has been debate, however, regarding the role of the brainstem in driving HPA responses to stress. It has been proposed that the brainstem drives HPA responses to systemic stress, whereas HPA responses to psychological stress are driven by regions in the forebrain (Li et al., 1996). In support of this division, the glucocorticoid response to immune stress caused by intraperitoneal injection of interleukin-1α was found to be attenuated by depletion of noradrenergic inputs to the PVN, whereas no such effect of the depleted inputs was observed on the HPA responses to restraint stress (Chuluyan et al., 1992) or footshock stress (Li et al., 1996). However, later findings would suggest that ascending noradrenergic signaling plays a role in driving HPA responses to both systemic and psychological stress (Pacak et al., 1998; Dayas et al., 2001). More recently, Herman et al. (2003) presented an alternative model regarding the role of the brainstem in HPA responses to stress. According to this model, the extent to which the NTS promotes HPA activity results from an interaction between ascending sensory inputs to the NTS and descending inputs to the NTS from the forebrain. It was proposed that the ascending input to the NTS transmits information regarding the current homeostatic state, whereas descending input to the NTS transmits information regarding an anticipated homeostatic state. As a result of this convergence of signaling in the NTS, the HPA response to an anticipated homeostatic state would depend on the current homeostatic state, so that inhibitory signaling from the forebrain can be overridden by current somatic, visceral, or humoral disturbances; alternatively, excitatory signaling from the periphery to the medial parvocellular PVN could be modulated by descending contextual information (Herman et al., 2003).
Electrophysiological analyses have revealed mixed effects of norepinephrine on putative parvocellular neuroendocrine cells of the PVN; some cells are excited and other cells are inhibited by norepinephrine (Yang et al., 2007). The excitatory actions of norepinephrine on CRH cells appear largely to be mediated by modulation of glutamatergic synaptic inputs. Thus, the surge in circulating ACTH and glucocorticoids resulting from electrical stimulation of the ventral noradrenergic bundle was found to be inhibited by intra-PVN microinjection of ionotropic glutamate receptor antagonists in a dose-dependent manner (Feldman and Weidenfeld, 2004). An electrophysiological study found that about 36% of putative parvocellular neurons responded to norepinephrine with an increase in glutamatergic input, while another 14% responded with a hyperpolarization (Daftary et al., 2000). Post-recording biocytin labeling revealed that the cells that had exhibited a norepinephrine-induced hyperpolarization tended to be located close to the third ventricle in a region known to contain dopamine and/or somatostatin parvocellular neurons, so it is possible that none of the hyperpolarized neurons were CRH cells. The norepinephrine-induced increase in glutamatergic activity was mediated by α1-adrenoceptors and was spike-dependent. Perhaps the most surprising result from our electrophysiological studies is the absence of putative parvocellular neurons that are excited by norepinephrine directly (Daftary et al., 2000). Electron microscopy has revealed the presence of adrenergic boutons forming synaptic specializations with CRH neurons (Liposits et al., 1986) and dual in situ hybridization has demonstrated that the α1b-adrenoceptor is expressed by nearly all the CRH neurons of the PVN (Day et al., 1999). However, we have found no evidence, and there have been no electrophysiological reports, of norepinephrine directly activating an inward current or depolarization in parvocellular neuroendocrine cells. As noradrenergic receptors are metabotropic, noradrenergic receptors on CRH cells may initiate signal transduction pathways without altering membrane conductance. On the other hand, postsynaptic noradrenergic receptors may stimulate the synthesis of a retrograde messenger that could trigger the activation of upstream local glutamatergic circuits. We have preliminary evidence for just such a retrograde mechanism, in which a messenger is released from CRH neurons in the PVN and stimulates a spike-dependent increase in glutamatergic inputs to the CRH neurons (Jiang and Tasker, unpublished observation; Figure 2).
In addition to regulating glutamate release, norepinephrine modulates GABA release onto parvocellular neurons in the PVN (Figure 2). An electrophysiological study found that about 58% of putative parvocellular neurons responded to norepinephrine with an increase in the frequency of spontaneous inhibitory postsynaptic currents (IPSCs) that was mediated by α1-adrenoceptors and was spike-dependent, suggestive of a locus of action upstream of the GABAergic axon (Han et al., 2002). In another 33% of the neurons tested, norepinephrine reduced the IPSC frequency, and this reduction was mediated by α2-adrenoceptors and was spike-independent, suggesting a presynaptic terminal locus of action (Han et al., 2002). In another study that specifically targeted parvocellular neuroendocrine cells with electrophysiological markers and retrograde staining from the median eminence, norepinephrine suppressed GABAergic input in 46% of the cells tested, and this was increased to 100% following blockade of either α1-adrenoceptors or spike generation (Yang et al., 2008). Consistent with α2-adrenoceptors located on GABAergic boutons inhibiting GABA release onto CRH cells, binding assays have reported substantial binding of radiolabeled clonidine, an α2-adrenoceptor agonist, in the CRH cell region of the medial parvocellular PVN (Cummings and Seybold, 1988). Moreover, electrophysiological analyses indicate that the α2-adrenoceptor-mediated decrease in GABA input is reflective of a reduced probability of GABA release (Yang et al., 2008). In keeping with its suppressive effect on neurotransmitter release, the α2-adrenoceptor is commonly found to suppress norepinephrine release as an autoreceptor (Raiteri et al., 1992). In support of a stimulatory effect of the α2-adrenoceptor on the HPA axis, clonidine has been reported to enhance CRH secretion from organotypic hypothalamic cultures (Calogero et al., 1988), and intracerebroventricular (Szafarczyk et al., 1990) and intraperitoneal (Shimizu, 1984) injections of clonidine in vivo induce a surge in ACTH release.
Plasticity of Norepinephrine Inputs
Soon after the discovery that norepinephrine regulates HPA activity, it was found that ascending noradrenergic signaling plays a role in the glucocorticoid-induced suppression of the HPA axis, since depletion of noradrenergic inputs via knife cuts to the medulla attenuated the increases in hypothalamic CRH expression and CRH release caused by adrenalectomy (Sawchenko, 1988). Adrenalectomy was also found to reduce binding of radiolabeled clonidine in the CRH-expressing region of the medial parvocellular PVN (Cummings and Seybold, 1988), while α1B-adrenoceptor expression in the PVN was upregulated following adrenalectomy (Day et al., 1999). Microdialysis analyses revealed that adrenalectomy increases the synthesis, release, and turnover of norepinephrine in the PVN (Pacak et al., 1993), whereas 1 week of hypercortisolemia, induced by administration of exogenous glucocorticoid, reduced the synthesis, release, and turnover of norepinephrine in the PVN (Pacak et al., 1995). This last finding is surprising inasmuch as 1 week of chronic variable stress has been found to increase both the density of noradrenergic synaptic boutons in the PVN and the number of noradrenergic boutons abutting CRH-immunoreactive somata and dendrites (Flak et al., 2009). Additionally, 1 week of repeated intermittent cold stress was reported to enhance HPA responsivity to an acute immobilization stress via an increase in α1-adrenoceptor signaling within the PVN (Ma and Morilak, 2005).
In addition to the effects of adrenalectomy on norepinephrine release and adrenoceptor expression in the PVN, electrophysiological recordings revealed that adrenalectomy results in an increased fraction of putative parvocellular neuroendocrine cells that are excited by norepinephrine and a decreased fraction inhibited by norepinephrine (Yang et al., 2007). Additionally, it was found that the fraction of cells in which norepinephrine reduced GABA input was doubled following adrenalectomy, whereas there was an 80% decrease in the fraction of cells in which norepinephrine enhanced GABA input (Yang et al., 2008).
Although norepinephrine is thought to be generally excitatory with regard to CRH release, there is a body of evidence that suggests that norepinephrine suppresses CRH neuron activity in the absence of glucocorticoids; thus noradrenergic regulation of CRH cells may be more complex than previously thought. While norepinephrine normally enhances CRH secretion from organotypic hypothalamic cultures, it was found to reduce basal secretion of CRH from cultures maintained in a glucocorticoid-free medium (Szafarczyk et al., 1995). Also, while the α2-adrenoceptor agonist clonidine normally promotes CRH cell activity (Calogero et al., 1988), it was found to reduce basal CRH release from organotypic hypothalamic cultures maintained in a glucocorticoid-free medium (Feuvrier et al., 1998). If noradrenergic signaling from the brainstem does in fact play a more prominent role in driving HPA responses to systemic stress than to psychological stress, adrenalectomized rats might be expected to respond differently to certain stressors than do intact rats.
Perspectives and Outlook
Plasticity of the neural circuitry regulating the HPA axis may constitute an etiological link between stress exposure and the subsequent development of HPA dysregulation and associated pathologies. Research into dysregulation of the HPA axis has focused mainly on hypersecretion of glucocorticoids, which is a condition that has been associated with depressive illness. Autopsies of depressed patients have reported higher than normal levels of CRH and VP mRNA in the PVN (Raadsheer et al., 1994,1995), which is suggestive of elevated pre-mortem CRH cell activity. It is believed that diminished glucocorticoid-induced suppression of the HPA axis is an underlying factor in the HPA hyperactivity found in depressive illness (Gillespie and Nemeroff, 2005). Consistent with this, depressed patients exhibit an attenuated suppression of glucocorticoid secretion when given the dexamethasone suppression test (i.e., the suppression of HPA activity by dexamethasone administration; Juruena et al., 2006). It has been suggested that this glucocorticoid resistance could result from a reduced expression of the glucocorticoid receptor (GR; Modell et al., 1997; Boyle et al., 2005), an idea supported by reports of reduced GR expression in post-mortem brain tissue of depressed patients (Webster et al., 2002). A similar downregulation of GR is seen in the rat brain following chronic stress (Kitraki et al., 1999). It is possible that a stress-induced downregulation of GR serves as a protective mechanism to limit allostatic load during periods of elevated glucocorticoid levels. Indeed, there is evidence that GR mediates the hippocampal deterioration that results from prolonged glucocorticoid exposure (Packan and Sapolsky, 1990). Stress could then result in hypercortisolemia by downregulating GR expression, thereby diminishing glucocorticoid-induced feedback inhibition of the HPA axis. However, we propose that a modified central drive underlies, at least in part, the link between exposure to stress and the subsequent development of HPA abnormalities. To be clear, we refer to the central drive as it pertains to the neural circuitry, namely the glutamatergic, GABAergic and noradrenergic circuitry that regulates the CRH neuronal activity. There is evidence that enhanced central drive of the HPA axis contributes to the hypercortisolemia associated with depressive illness. Specifically, dexamethasone-resistant depressed patients exhibit an enhanced ACTH response to metyrapone, a glucocorticoid synthesis inhibitor (Fava et al., 1984; Ur et al., 1992). This observation is consistent with a hyperactive drive of the HPA axis that is, in fact, suppressed by glucocorticoids. This glucocorticoid-independent overdrive of the HPA axis may reflect neural circuitry that favors CRH cell hyperactivity. Interestingly, an enhanced ACTH response to metyrapone in depressed patients has been correlated with a reduced efficacy of the selective serotonin re-uptake inhibitor fluoxetine for the relief of depressive symptoms (Young et al., 2004), which suggests that resetting to a normal level of central drive to the CRH neurons may prove an effective therapy in cases of depression that are resistant to serotonin-based antidepressants.
Stress- and glucocorticoid-induced plasticity of neural circuitry regulating the HPA axis may constitute a means through which exposure to stress could play a role in a spectrum of HPA abnormalities, including aberrant HPA circadian rhythms, abnormalities in HPA responsivity to stress, and basal HPA dysregulation. We hypothesize that this synaptic plasticity constitutes an etiological link between exposure to stress and a range of affective and stress-related disorders. We frame this hypothesis on the basis of three premises: (1) exposure to stress increases the likelihood of developing subsequent HPA dysregulation and associated pathologies, (2) rodent models have demonstrated that the synaptic mechanisms that regulate the HPA axis are highly plastic, and that synaptic plasticity is induced by both glucocorticoids and stress, and (3) stress exposure and associated pathologies have, at times, been linked to hypoactivity of the HPA axis, and this phenomenon cannot be explained by diminished glucocorticoid-induced suppression of the HPA axis. With regard to this last premise, fibromyalgia (Tanriverdi et al., 2007) and chronic fatigue syndrome (Van Houdenhove et al., 2009) have both been linked to HPA hypoactivity, and stressful life events have been implicated in the onset of each of these disorders (Hatcher and House, 2003; Gupta and Silman, 2004). There is also evidence that HPA hypoactivity is involved in some cases of post-traumatic stress disorder (PTSD; Yehuda, 2006) and depression (Morphy et al., 1985; Penninx et al., 2007; Ahrens et al., 2008). Sexually abused girls (ages 5–7 years) have exhibited reduced salivary glucocorticoid levels within months following an abusive incident (King et al., 2001). Moreover, adult women who were victims of childhood sexual abuse have exhibited enhanced suppression of glucocorticoid release in response to the dexamethasone suppression test (Stein et al., 1997). Lastly, in rats, stress can result in HPA hypoactivity under certain circumstances. One study using female rats found that exposure to intense footshock, followed by 3 weekly reminders of the stimulus, resulted in lower glucocorticoid levels than those recorded previous to the footshock (Louvart et al., 2005). Reminders of the footshock involved placing the rat in a compartment adjacent to the compartment where the footshock had previously occurred, in what was intended as a rodent model of PTSD. Another study found that rats subjected to chronic variable stress, followed by several days recovery from the stress, exhibit attenuated pituitary–adrenal responses to acute novel environment and restraint stresses (Ostrander et al., 2006). It should be noted that there are some mechanisms through which glucocorticoids could actually enhance HPA activity. For example, glucocorticoids seem to attenuate GABAergic restraint on CRH cells, as described above; thus, in certain cell types, downregulated GR expression in response to stress could promote subsequent HPA hypoactivity. However, there is evidence that stress can, at times, result in upregulated GR. GR number was found to be larger in lymphocytes of combat veterans than in those of non-veterans (Yehuda et al., 1995). Also, increased GR binding capacity has been reported in the rat hippocampus following repeated inescapable footshock stress (van Dijken et al., 1993). Moreover, the enhanced response to the dexamethasone suppression test in adult victims of childhood sexual abuse (Stein et al., 1997) is suggestive of an increased sensitivity to glucocorticoids, at least at the anterior pituitary (Cole et al., 2000). Upregulated GR expression could be a protective response to hypocortisolemia, as some degree of GR activity may be critical for survival (Cole et al., 1995), and the initial hypocortisolemia could result from stress-induced synaptic plasticity that promotes CRH cell suppression. Indeed, GR expression generally increases following adrenalectomy (Olpe and McEwen, 1976; Svec et al., 1989; Isenovic et al., 2006).
With respect to future investigations of neural regulation of the HPA axis, recent advances in circuit tracing technology have seen the development of a Cre-dependent retrogradely transported marker derived from the rabies virus that will allow investigators to trace the precise sources of innervation to CRH cells (Wall et al., 2010). This tool will inevitably improve our understanding of how the brain integrates pertinent information into an HPA response. Also, one investigative team recently reported that perinatal stress, caused by an active construction site adjacent to the rodent vivarium, resulted in hypercortisolemia that persisted even after the construction had ended (O’Regan et al., 2010), which underscores the sensitivity of HPA regulatory mechanisms to environmental influences (at least perinatally), but also serves as a warning to investigators of the importance of documenting the living conditions under which their subjects are kept. Lastly, it is important that assays of morphological plasticity and changes in ionotropic receptor expression are accompanied by electrophysiological recordings, as electrophysiology provides invaluable information regarding functional manifestations of synaptic activity. Dysregulation of the HPA axis may be a causal factor in a range of affective and physiological disorders, and there is increasing evidence that stress can result in persistent changes in HPA regulation. Rodent models of the past several years have provided significant insight into the etiology of stress-induced aberrations in HPA activity, but our understanding is still in its infancy. Studies of synaptic transmission will remain invaluable to this provocative and promising line of research, as we uncover future targets for pharmacological intervention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abelson, J. L., Khan, S., Liberzon, I., and Young, E. A. (2007). HPA axis activity in patients with panic disorder: review and synthesis of four studies. Depress. Anxiety 24, 66–76.
Ahrens, T., Deuschle, M., Krumm, B., Van Der Pompe, G., Den Boer, J. A., and Lederbogen, F. (2008). Pituitary–adrenal and sympathetic nervous system responses to stress in women remitted from recurrent major depression. Psychosom. Med. 70, 461–467.
Alon, T., Zhou, L., Perez, C. A., Garfield, A. S., Friedman, J. M., and Heisler, L. K. (2009). Transgenic mice expressing green fluorescent protein under the control of the corticotropin-releasing hormone promoter. Endocrinology 150, 5626–5632.
Aubry, J. M., Bartanusz, V., Pagliusi, S., Schulz, P., and Kiss, J. Z. (1996). Expression of ionotropic glutamate receptor subunit mRNAs by paraventricular corticotropin-releasing factor (CRF) neurons. Neurosci. Lett. 205, 95–98.
Bali, B., and Kovacs, K. J. (2003). GABAergic control of neuropeptide gene expression in parvocellular neurons of the hypothalamic paraventricular nucleus. Eur. J. Neurosci. 18, 1518–1526.
Bartanusz, V., Aubry, J. M., Pagliusi, S., Jezova, D., Baffi, J., and Kiss, J. Z. (1995). Stress-induced changes in messenger RNA levels of N-methyl-D-aspartate and AMPA receptor subunits in selected regions of the rat hippocampus and hypothalamus. Neuroscience 66, 247–252.
Boudaba, C., Schrader, L. A., and Tasker, J. G. (1997). Physiological evidence for local excitatory synaptic circuits in the rat hypothalamus. J. Neurophysiol. 77, 3396–3400.
Boudaba, C., Szabo, K., and Tasker, J. G. (1996). Physiological mapping of local inhibitory inputs to the hypothalamic paraventricular nucleus. J. Neurosci. 16, 7151–7160.
Boyle, M. P., Brewer, J. A., Funatsu, M., Wozniak, D. F., Tsien, J. Z., Izumi, Y., and Muglia, L. J. (2005). Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc. Natl. Acad. Sci. U.S.A. 102, 473–478.
Bradbury, M. J., Giracello, D. R., Chapman, D. F., Holtz, G., Schaffhauser, H., Rao, S. P., Varney, M. A., and Anderson, J. J. (2003). Metabotropic glutamate receptor 5 antagonist-induced stimulation of hypothalamic–pituitary–adrenal axis activity: interaction with serotonergic systems. Neuropharmacology 44, 562–572.
Brawman-Mintzer, O., Monnier, J., Wolitzky, K. B., and Falsetti, S. A. (2005). Patients with generalized anxiety disorder and a history of trauma: somatic symptom endorsement. J. Psychiatr. Pract. 11, 212–215.
Calogero, A. E., Gallucci, W. T., Chrousos, G. P., and Gold, P. W. (1988). Catecholamine effects upon rat hypothalamic corticotropin-releasing hormone secretion in vitro. J. Clin. Invest. 82, 839–846.
Choi, D. C., Furay, A. R., Evanson, N. K., Ostrander, M. M., Ulrich-Lai, Y. M., and Herman, J. P. (2007). Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic–pituitary–adrenal axis activity: implications for the integration of limbic inputs. J. Neurosci. 27, 2025–2034.
Choi, D. C., Furay, A. R., Evanson, N. K., Ulrich-Lai, Y. M., Nguyen, M. M., Ostrander, M. M., and Herman, J. P. (2008). The role of the posterior medial bed nucleus of the stria terminalis in modulating hypothalamic–pituitary–adrenocortical axis responsiveness to acute and chronic stress. Psychoneuroendocrinology 33, 659–669.
Chuluyan, H. E., Saphier, D., Rohn, W. M., and Dunn, A. J. (1992). Noradrenergic innervation of the hypothalamus participates in adrenocortical responses to interleukin-1. Neuroendocrinology 56, 106–111.
Cole, M. A., Kim, P. J., Kalman, B. A., and Spencer, R. L. (2000). Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology 25, 151–167.
Cole, R. L., and Sawchenko, P. E. (2002). Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J. Neurosci. 22, 959–969.
Cole, T. J., Blendy, J. A., Monaghan, A. P., Krieglstein, K., Schmid, W., Aguzzi, A., Fantuzzi, G., Hummler, E., Unsicker, K., and Schutz, G. (1995). Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 9, 1608–1621.
Cowley, M. A., Pronchuk, N., Fan, W., Dinulescu, D. M., Colmers, W. F., and Cone, R. D. (1999). Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron 24, 155–163.
Csaki, A., Kocsis, K., Halasz, B., and Kiss, J. (2000). Localization of glutamatergic/aspartatergic neurons projecting to the hypothalamic paraventricular nucleus studied by retrograde transport of [3H]D-aspartate autoradiography. Neuroscience 101, 637–655.
Cull-Candy, S., Brickley, S., and Farrant, M. (2001). NMDA receptor subunits: diversity, development and disease. Curr. Opin. Neurobiol. 11, 327–335.
Cullinan, W. E. (2000). GABA(A) receptor subunit expression within hypophysiotropic CRH neurons: a dual hybridization histochemical study. J. Comp. Neurol. 419, 344–351.
Cullinan, W. E., and Wolfe, T. J. (2000). Chronic stress regulates levels of mRNA transcripts encoding beta subunits of the GABA(A) receptor in the rat stress axis. Brain Res. 887, 118–124.
Cummings, S., and Seybold, V. (1988). Relationship of alpha-1- and alpha-2-adrenergic-binding sites to regions of the paraventricular nucleus of the hypothalamus containing corticotropin-releasing factor and vasopressin neurons. Neuroendocrinology 47, 523–532.
Daban, C., Vieta, E., Mackin, P., and Young, A. H. (2005). Hypothalamic–pituitary–adrenal axis and bipolar disorder. Psychiatr. Clin. North Am. 28, 469–480.
Daftary, S. S., Boudaba, C., Szabo, K., and Tasker, J. G. (1998). Noradrenergic excitation of magnocellular neurons in the rat hypothalamic paraventricular nucleus via intranuclear glutamatergic circuits. J. Neurosci. 18, 10619–10628.
Daftary, S. S., Boudaba, C., and Tasker, J. G. (2000). Noradrenergic regulation of parvocellular neurons in the rat hypothalamic paraventricular nucleus. Neuroscience 96, 743–751.
Day, H. E., Campeau, S., Watson, S. J. Jr., and Akil, H. (1999). Expression of alpha(1b) adrenoceptor mRNA in corticotropin-releasing hormone-containing cells of the rat hypothalamus and its regulation by corticosterone. J. Neurosci. 19, 10098–10106.
Dayas, C. V., Buller, K. M., and Day, T. A. (2001). Medullary neurones regulate hypothalamic corticotropin-releasing factor cell responses to an emotional stressor. Neuroscience 105, 707–719.
Di, S., Malcher-Lopes, R., Halmos, K. C., and Tasker, J. G. (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J. Neurosci. 23, 4850–4857.
Dimitrov, E. L., Dejoseph, M. R., Brownfield, M. S., and Urban, J. H. (2007). Involvement of neuropeptide Y Y1 receptors in the regulation of neuroendocrine corticotropin-releasing hormone neuronal activity. Endocrinology 148, 3666–3673.
Durand, D., Pampillo, M., Caruso, C., and Lasaga, M. (2008). Role of metabotropic glutamate receptors in the control of neuroendocrine function. Neuropharmacology 55, 577–583.
Fava, G. A., Carson, S. W., Perini, G. I., Morphy, M. A., Molnar, G., and Jusko, W. J. (1984). The metyrapone test in affective disorders and schizophrenia. J. Affect. Disord. 6, 241–247.
Feldman, S., and Weidenfeld, J. (1997). Hypothalamic mechanisms mediating glutamate effects on the hypothalamo–pituitary–adrenocortical axis. J. Neural Transm. 104, 633–642.
Feldman, S., and Weidenfeld, J. (2004). Involvement of endogeneous glutamate in the stimulatory effect of norepinephrine and serotonin on the hypothalamo–pituitary–adrenocortical axis. Neuroendocrinology 79, 43–53.
Feuvrier, E., Aubert, M., Mausset, A. L., Alonso, G., Gaillet, S., Malaval, F., and Szafarczyk, A. (1998). Glucocorticoids provoke a shift from alpha2- to alpha1-adrenoreceptor activities in cultured hypothalamic slices leading to opposite noradrenaline effect on corticotropin-releasing hormone release. J. Neurochem. 70, 1199–1209.
Flak, J. N., Ostrander, M. M., Tasker, J. G., and Herman, J. P. (2009). Chronic stress-induced neurotransmitter plasticity in the PVN. J. Comp. Neurol. 517, 156–165.
Franco, A. J., Zsombok, A., and Tasker, J. G. (2007). Rapid Glucocorticoid-Induced, Endocannabinoid-Mediated Inhibition of Synaptic Excitation in the Hypothalamus Maintained Following Chronic Variable Stress. Program No. 298.25/TT27. Washington, DC: Neuroscience Meeting Planner, Society for Neuroscience. Available at: http:// www.sfn.org/index.aspx?pagename=abstracts_ampublications
Freund, T. F., Katona, I., and Piomelli, D. (2003). Role of endogenous cannabinoids in synaptic signaling. Physiol. Rev. 83, 1017–1066.
Galanopoulou, A. S., and Moshe, S. L. (2003). Role of sex hormones in the sexually dimorphic expression of KCC2 in rat substantia nigra. Exp. Neurol. 184, 1003–1009.
Garno, J. L., Goldberg, J. F., Ramirez, P. M., and Ritzler, B. A. (2005). Impact of childhood abuse on the clinical course of bipolar disorder. Br. J. Psychiatry 186, 121–125.
Gillespie, C. F., and Nemeroff, C. B. (2005). Hypercortisolemia and depression. Psychosom. Med. 67(Suppl. 1), S26–S28.
Goodwin, R. D., Fergusson, D. M., and Horwood, L. J. (2005). Childhood abuse and familial violence and the risk of panic attacks and panic disorder in young adulthood. Psychol. Med. 35, 881–890.
Gupta, A., and Silman, A. J. (2004). Psychological stress and fibromyalgia: a review of the evidence suggesting a neuroendocrine link. Arthritis Res. Ther. 6, 98–106.
Gustafsson, P. E., Gustafsson, P. A., Ivarsson, T., and Nelson, N. (2008). Diurnal cortisol levels and cortisol response in youths with obsessive-compulsive disorder. Neuropsychobiology 57, 14–21.
Haam, J., Popescu, I. R., Morton, L. A., Halmos, K. C., Teruyama, R., Ueta, U., and Tasker, J. G. (2012). GABA is excitatory in adult vasopressinergic neuroendocrine cells. J. Neurosci. 32, 572–582.
Han, S. K., Chong, W., Li, L. H., Lee, I. S., Murase, K., and Ryu, P. D. (2002). Noradrenaline excites and inhibits GABAergic transmission in parvocellular neurons of rat hypothalamic paraventricular nucleus. J. Neurophysiol. 87, 2287–2296.
Harris, C. C., and Tasker, J. G. (2011). A Calcium Requirement For Rapid Glucocorticoid Dependent Modulation Of Glutamate Release In The Supraoptic Nucleus. Program No. 501.01/VV32. Washington, DC: Neuroscience Meeting Planner, Society for Neuroscience. Available at: http://www.sfn.org/am2011/pdf/prelim/MON_Poster_PM_v2.pdf
Hatcher, S., and House, A. (2003). Life events, difficulties and dilemmas in the onset of chronic fatigue syndrome: a case–control study. Psychol. Med. 33, 1185–1192.
Heifets, B. D., and Castillo, P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 71, 283–306.
Herman, J. P., Adams, D., and Prewitt, C. (1995). Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology 61, 180–190.
Herman, J. P., Eyigor, O., Ziegler, D. R., and Jennes, L. (2000). Expression of ionotropic glutamate receptor subunit mRNAs in the hypothalamic paraventricular nucleus of the rat. J. Comp. Neurol. 422, 352–362.
Herman, J. P., Figueiredo, H., Mueller, N. K., Ulrich-Lai, Y., Ostrander, M. M., Choi, D. C., and Cullinan, W. E. (2003). Central mechanisms of stress integration: hierarchical circuitry controlling hypothalamo–pituitary–adrenocortical responsiveness. Front. Neuroendocrinol. 24, 151–180.
Hewitt, S. A., Wamsteeker, J. I., Kurz, E. U., and Bains, J. S. (2009). Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 12, 438–443.
Hrabovszky, E., and Liposits, Z. (2008). Novel aspects of glutamatergic signalling in the neuroendocrine system. J. Neuroendocrinol. 20, 743–751.
Isenovic, E. R., Radojcic, M., Zakula, Z., Koricanac, G., and Ribarac-Stepic, N. (2006). Effect of acute adrenalectomy on rat liver glucocorticoid receptor. Arch. Biol. Sci. 58, 153–159.
Itoi, K., Suda, T., Tozawa, F., Dobashi, I., Ohmori, N., Sakai, Y., Abe, K., and Demura, H. (1994). Microinjection of norepinephrine into the paraventricular nucleus of the hypothalamus stimulates corticotropin-releasing factor gene expression in conscious rats. Endocrinology 135, 2177–2182.
Jacobson, L., and Sapolsky, R. (1993). Augmented ACTH responses to stress in adrenalectomized rats replaced with constant, physiological levels of corticosterone are partially normalized by acute increases in corticosterone. Neuroendocrinology 58, 420–429.
Joels, M., Karst, H., Alfarez, D., Heine, V. M., Qin, Y., Van Riel, E., Verkuyl, M., Lucassen, P. J., and Krugers, H. J. (2004). Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress 7, 221–231.
Johnson, M. P., Kelly, G., and Chamberlain, M. (2001). Changes in rat serum corticosterone after treatment with metabotropic glutamate receptor agonists or antagonists. J. Neuroendocrinol. 13, 670–677.
Juruena, M. F., Cleare, A. J., Papadopoulos, A. S., Poon, L., Lightman, S., and Pariante, C. M. (2006). Different responses to dexamethasone and prednisolone in the same depressed patients. Psychopharmacology (Berl.) 189, 225–235.
Kendler, K. S., Kessler, R. C., Walters, E. E., Maclean, C., Neale, M. C., Heath, A. C., and Eaves, L. J. (1995). Stressful life events, genetic liability, and onset of an episode of major depression in women. Am. J. Psychiatry 152, 833–842.
Kim, J. S., Kim, W. B., Kim, Y. B., Lee, Y., Kim, Y. S., Shen, F. Y., Lee, S. W., Park, D., Choi, H. J., Hur, J., Park, J. J., Han, H. C., Colwell, C. S., Cho, Y. W., and Kim, Y. I. (2011). Chronic hyperosmotic stress converts GABAergic inhibition into excitation in vasopressin and oxytocin neurons in the rat. J. Neurosci. 31, 13312–13322.
King, J. A., Mandansky, D., King, S., Fletcher, K. E., and Brewer, J. (2001). Early sexual abuse and low cortisol. Psychiatry Clin. Neurosci. 55, 71–74.
Kitraki, E., Karandrea, D., and Kittas, C. (1999). Long-lasting effects of stress on glucocorticoid receptor gene expression in the rat brain. Neuroendocrinology 69, 331–338.
Kocsis, K., Kiss, J., Gorcs, T., and Halasz, B. (1998). Metabotropic glutamate receptor in vasopressin, CRF and VIP hypothalamic neurones. Neuroreport 9, 4029–4033.
Korosi, A., Shanabrough, M., Mcclelland, S., Liu, Z. W., Borok, E., Gao, X. B., Horvath, T. L., and Baram, T. Z. (2010). Early-life experience reduces excitation to stress-responsive hypothalamic neurons and reprograms the expression of corticotropin-releasing hormone. J. Neurosci. 30, 703–713.
Kuzmiski, J. B., Marty, V., Baimoukhametova, D. V., and Bains, J. S. (2010). Stress-induced priming of glutamate synapses unmasks associative short-term plasticity. Nat. Neurosci. 13, 1257–1264.
Lang, C. H., and Ajmal, M. (1995). Metabolic, hormonal, and hemodynamic changes induced by metabotropic excitatory amino acid agonist (1S, 3R)-ACPD. Am. J. Physiol. 268, R1026–R1033.
Lenze, E. J., Mantella, R. C., Shi, P., Goate, A. M., Nowotny, P., Butters, M. A., Andreescu, C., Thompson, P. A., and Rollman, B. L. (2011). Elevated cortisol in older adults with generalized anxiety disorder is reduced by treatment: a placebo-controlled evaluation of escitalopram. Am. J. Geriatr. Psychiatry 19, 482–490.
Li, H. Y., Ericsson, A., and Sawchenko, P. E. (1996). Distinct mechanisms underlie activation of hypothalamic neurosecretory neurons and their medullary catecholaminergic afferents in categorically different stress paradigms. Proc. Natl. Acad. Sci. U.S.A. 93, 2359–2364.
Liposits, Z., Phelix, C., and Paull, W. K. (1986). Adrenergic innervation of corticotropin releasing factor (CRF)-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. A combined light and electron microscopic immunocytochemical study. Histochemistry 84, 201–205.
Liu, D., Diorio, J., Tannenbaum, B., Caldji, C., Francis, D., Freedman, A., Sharma, S., Pearson, D., Plotsky, P. M., and Meaney, M. J. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic–pituitary–adrenal responses to stress. Science 277, 1659–1662.
Louvart, H., Maccari, S., Ducrocq, F., Thomas, P., and Darnaudery, M. (2005). Long-term behavioural alterations in female rats after a single intense footshock followed by situational reminders. Psychoneuroendocrinology 30, 316–324.
Luther, J. A., Daftary, S. S., Boudaba, C., Gould, G. C., Halmos, K. C., and Tasker, J. G. (2002). Neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J. Neuroendocrinol. 14, 929–932.
Luther, J. A., and Tasker, J. G. (2000). Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J. Physiol. 523(Pt 1), 193–209.
Ma, S., and Morilak, D. A. (2005). Chronic intermittent cold stress sensitises the hypothalamic–pituitary–adrenal response to a novel acute stress by enhancing noradrenergic influence in the rat paraventricular nucleus. J. Neuroendocrinol. 17, 761–769.
Majewska, M. D., Bisserbe, J. C., and Eskay, R. L. (1985). Glucocorticoids are modulators of GABAA receptors in brain. Brain Res. 339, 178–182.
Malcher-Lopes, R., Di, S., Marcheselli, V. S., Weng, F. J., Stuart, C. T., Bazan, N. G., and Tasker, J. G. (2006). Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J. Neurosci. 26, 6643–6650.
Miklos, I. H., and Kovacs, K. J. (2002). GABAergic innervation of corticotropin-releasing hormone (CRH)-secreting parvocellular neurons and its plasticity as demonstrated by quantitative immunoelectron microscopy. Neuroscience 113, 581–592.
Modell, S., Yassouridis, A., Huber, J., and Holsboer, F. (1997). Corticosteroid receptor function is decreased in depressed patients. Neuroendocrinology 65, 216–222.
Morphy, M. A., Fava, G. A., Perini, G. I., and Molnar, G. (1985). The metyrapone test in depressed males. Prog. Neuropsychopharmacol. Biol. Psychiatry 9, 187–191.
Nahar, J., Haam, J., Glatzer, N. R., Halmos, K. C., Muglia, L. J., Dohanich, G. P., and Tasker, J. G. (2012). Rapid glucocorticoid actions in hypothalamic neuroendocrine cells are dependent on the glucocorticoid receptor. Endocrinology (submitted).
O’Regan, D., Kenyon, C. J., Seckl, J. R., and Holmes, M. C. (2010). Environmental disturbance confounds prenatal glucocorticoid programming experiments in Wistar rats. Lab. Anim. 44, 199–205.
Olpe, H. R., and McEwen, B. S. (1976). Glucocorticoid binding to receptor-like proteins in rat brain and pituitary: ontogenetic and experimentally induced changes. Brain Res. 105, 121–128.
Ostrander, M. M., Ulrich-Lai, Y. M., Choi, D. C., Richtand, N. M., and Herman, J. P. (2006). Hypoactivity of the hypothalamo–pituitary–adrenocortical axis during recovery from chronic variable stress. Endocrinology 147, 2008–2017.
Pacak, K., Kvetnansky, R., Palkovits, M., Fukuhara, K., Yadid, G., Kopin, I. J., and Goldstein, D. S. (1993). Adrenalectomy augments in vivo release of norepinephrine in the paraventricular nucleus during immobilization stress. Endocrinology 133, 1404–1410.
Pacak, K., Palkovits, M., Kvetnansky, R., Matern, P., Hart, C., Kopin, I. J., and Goldstein, D. S. (1995). Catecholaminergic inhibition by hypercortisolemia in the paraventricular nucleus of conscious rats. Endocrinology 136, 4814–4819.
Pacak, K., Palkovits, M., Yadid, G., Kvetnansky, R., Kopin, I. J., and Goldstein, D. S. (1998). Heterogeneous neurochemical responses to different stressors: a test of Selye’s doctrine of nonspecificity. Am. J. Physiol. 275, R1247–R1255.
Packan, D. R., and Sapolsky, R. M. (1990). Glucocorticoid endangerment of the hippocampus: tissue, steroid and receptor specificity. Neuroendocrinology 51, 613–618.
Penninx, B. W., Beekman, A. T., Bandinelli, S., Corsi, A. M., Bremmer, M., Hoogendijk, W. J., Guralnik, J. M., and Ferrucci, L. (2007). Late-life depressive symptoms are associated with both hyperactivity and hypoactivity of the hypothalamo–pituitary–adrenal axis. Am. J. Geriatr. Psychiatry 15, 522–529.
Plotsky, P. M., Otto, S., and Sutton, S. (1987). Neurotransmitter modulation of corticotropin releasing factor secretion into the hypophysial-portal circulation. Life Sci. 41, 1311–1317.
Raadsheer, F. C., Hoogendijk, W. J., Stam, F. C., Tilders, F. J., and Swaab, D. F. (1994). Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 60, 436–444.
Raadsheer, F. C., Van Heerikhuize, J. J., Lucassen, P. J., Hoogendijk, W. J., Tilders, F. J., and Swaab, D. F. (1995). Corticotropin-releasing hormone mRNA levels in the paraventricular nucleus of patients with Alzheimer’s disease and depression. Am. J. Psychiatry 152, 1372–1376.
Radley, J. J., Gosselink, K. L., and Sawchenko, P. E. (2009). A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J. Neurosci. 29, 7330–7340.
Radley, J. J., and Sawchenko, P. E. (2011). A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J. Neurosci. 31, 9683–9695.
Raiteri, M., Bonanno, G., Maura, G., Pende, M., Andrioli, G. C., and Ruelle, A. (1992). Subclassification of release-regulating alpha 2-autoreceptors in human brain cortex. Br. J. Pharmacol. 107, 1146–1151.
Roland, B. L., and Sawchenko, P. E. (1993). Local origins of some GABAergic projections to the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J. Comp. Neurol. 332, 123–143.
Sarkhel, S., Praharaj, S. K., and Sinha, V. K. (2011). Role of life events in obsessive compulsive disorder. Isr. J. Psychiatry Relat. Sci. 48, 182–185.
Sawchenko, P. E. (1988). Effects of catecholamine-depleting medullary knife cuts on corticotropin-releasing factor and vasopressin immunoreactivity in the hypothalamus of normal and steroid-manipulated rats. Neuroendocrinology 48, 459–470.
Schrader, L. A., and Tasker, J. G. (1997). Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J. Neurophysiol. 77, 527–536.
Shimizu, K. (1984). Effect of alpha 1- and alpha 2-adrenoceptor agonists and antagonists on ACTH secretion in intact and in hypothalamic deafferentated rats. Jpn. J. Pharmacol. 36, 23–33.
Simmons, D. M., and Swanson, L. W. (2009). Comparison of the spatial distribution of seven types of neuroendocrine neurons in the rat paraventricular nucleus: toward a global 3D model. J. Comp. Neurol. 516, 423–441.
Stein, M. B., Yehuda, R., Koverola, C., and Hanna, C. (1997). Enhanced dexamethasone suppression of plasma cortisol in adult women traumatized by childhood sexual abuse. Biol. Psychiatry 42, 680–686.
Stern, J. E. (2001). Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J. Physiol. 537, 161–177.
Svec, F., Gordon, S., and Tate, D. (1989). Glucocorticoid receptor regulation: the effects of adrenalectomy, exogenous glucocorticoid, and stress on hepatic receptor number in male and female mice. Biochem. Med. Metab. Biol. 41, 224–233.
Szafarczyk, A., Feuvrier, E., Siaud, P., Rondouin, G., Lacoste, M., Gaillet, S., Malaval, F., and Assenmacher, I. (1995). Removal of adrenal steroids from the medium reverses the stimulating effect of catecholamines on corticotropin-releasing hormone neurons in organotypic cultures. Neuroendocrinology 61, 517–524.
Szafarczyk, A., Gaillet, S., Barbanel, G., Malaval, F., and Assenmacher, I. (1990). [Implication of alpha-2 adrenergic post-synaptic receptors in the central catecholaminergic stimulation of the corticotropic axis in rats]. C R Acad. Sci. III. 311, 81–88.
Tanriverdi, F., Karaca, Z., Unluhizarci, K., and Kelestimur, F. (2007). The hypothalamo–pituitary–adrenal axis in chronic fatigue syndrome and fibromyalgia syndrome. Stress 10, 13–25.
Tasker, J. G., Boudaba, C., and Schrader, L. A. (1998). Local glutamatergic and GABAergic synaptic circuits and metabotropic glutamate receptors in the hypothalamic paraventricular and supraoptic nuclei. Adv. Exp. Med. Biol. 449, 117–121.
Tasker, J. G., and Dudek, F. E. (1991). Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J. Physiol. 434, 271–293.
Tuchelt, H., Dekker, K., Bahr, V., and Oelkers, W. (2000). Dose-response relationship between plasma ACTH and serum cortisol in the insulin-hypoglycaemia test in 25 healthy subjects and 109 patients with pituitary disease. Clin. Endocrinol. (Oxf.) 53, 301–307.
Ulrich-Lai, Y. M., Jones, K. R., Ziegler, D. R., Cullinan, W. E., and Herman, J. P. (2011). Forebrain origins of glutamatergic innervation to the rat paraventricular nucleus of the hypothalamus: differential inputs to the anterior versus posterior subregions. J. Comp. Neurol. 519, 1301–1319.
Ur, E., Dinan, T. G., O’keane, V., Clare, A. W., Mcloughlin, L., Rees, L. H., Turner, T. H., Grossman, A., and Besser, G. M. (1992). Effect of metyrapone on the pituitary–adrenal axis in depression: relation to dexamethasone suppressor status. Neuroendocrinology 56, 533–538.
van Dijken, H. H., De Goeij, D. C., Sutanto, W., Mos, J., De Kloet, E. R., and Tilders, F. J. (1993). Short inescapable stress produces long-lasting changes in the brain–pituitary–adrenal axis of adult male rats. Neuroendocrinology 58, 57–64.
Van Houdenhove, B., Van Den Eede, F., and Luyten, P. (2009). Does hypothalamic–pituitary–adrenal axis hypofunction in chronic fatigue syndrome reflect a “crash” in the stress system? Med. Hypotheses 72, 701–705.
Verkuyl, J. M., Hemby, S. E., and Joels, M. (2004). Chronic stress attenuates GABAergic inhibition and alters gene expression of parvocellular neurons in rat hypothalamus. Eur. J. Neurosci. 20, 1665–1673.
Verkuyl, J. M., and Joels, M. (2003). Effect of adrenalectomy on miniature inhibitory postsynaptic currents in the paraventricular nucleus of the hypothalamus. J. Neurophysiol. 89, 237–245.
Verkuyl, J. M., Karst, H., and Joels, M. (2005). GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur. J. Neurosci. 21, 113–121.
Wall, N. R., Wickersham, I. R., Cetin, A., De La Parra, M., and Callaway, E. M. (2010). Monosynaptic circuit tracing in vivo through Cre-dependent targeting and complementation of modified rabies virus. Proc. Natl. Acad. Sci. U.S.A. 107, 21848–21853.
Wamsteeker, J. I., Kuzmiski, J. B., and Bains, J. S. (2010). Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J. Neurosci. 30, 11188–11196.
Wang, R., Liu, X., Hentges, S. T., Dunn-Meynell, A. A., Levin, B. E., Wang, W., and Routh, V. H. (2004). The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53, 1959–1965.
Webster, M. J., Knable, M. B., O’grady, J., Orthmann, J., and Weickert, C. S. (2002). Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol. Psychiatry 7, 985–994; 924.
Wittmann, G., Lechan, R. M., Liposits, Z., and Fekete, C. (2005). Glutamatergic innervation of corticotropin-releasing hormone- and thyrotropin-releasing hormone-synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. Brain Res. 1039, 53–62.
Yang, J. H., Li, L. H., Lee, S., Jo, I. H., Lee, S. Y., and Ryu, P. D. (2007). Effects of adrenalectomy on the excitability of neurosecretory parvocellular neurones in the hypothalamic paraventricular nucleus. J. Neuroendocrinol. 19, 293–301.
Yang, J. H., Li, L. H., Shin, S. Y., Lee, S., Lee, S. Y., Han, S. K., and Ryu, P. D. (2008). Adrenalectomy potentiates noradrenergic suppression of GABAergic transmission in parvocellular neurosecretory neurons of hypothalamic paraventricular nucleus. J. Neurophysiol. 99, 514–523.
Yehuda, R. (2006). Advances in understanding neuroendocrine alterations in PTSD and their therapeutic implications. Ann. N.Y. Acad. Sci. 1071, 137–166.
Yehuda, R., Boisoneau, D., Lowy, M. T., and Giller, E. L. Jr. (1995). Dose–response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Arch. Gen. Psychiatry 52, 583–593.
Young, E. A., Altemus, M., Lopez, J. F., Kocsis, J. H., Schatzberg, A. F., Debattista, C., and Zubieta, J. K. (2004). HPA axis activation in major depression and response to fluoxetine: a pilot study. Psychoneuroendocrinology 29, 1198–1204.
Ziegler, D. R., Cullinan, W. E., and Herman, J. P. (2005). Organization and regulation of paraventricular nucleus glutamate signaling systems: N-methyl-D-aspartate receptors. J. Comp. Neurol. 484, 43–56.
Ziegler, D. R., and Herman, J. P. (2000). Local integration of glutamate signaling in the hypothalamic paraventricular region: regulation of glucocorticoid stress responses. Endocrinology 141, 4801–4804.
Keywords: corticosteroid, glutamate, GABA, norepinephrine, neural circuits, depression, synaptic plasticity, paraventricular nucleus
Citation: Levy BH and Tasker JG (2012) Synaptic regulation of the hypothalamic–pituitary–adrenal axis and its modulation by glucocorticoids and stress. Front. Cell. Neurosci. 6:24. doi: 10.3389/fncel.2012.00024
Received: 12 March 2012; Accepted: 26 April 2012;
Published online: 11 May 2012.
Edited by:
Harmen J. Krugers, Universiteit van Amsterdam, NetherlandsReviewed by:
Yang Dan, University of California Berkeley, USACorette Wierenga, Utrecht University, Netherlands
Copyright: © 2012 Levy and Tasker. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jeffrey G. Tasker, Department of Cell and Molecular Biology, Tulane University, 2000 Percival Stern Hall, New Orleans, LA 70118-5221, USA. e-mail:dGFza2VyQHR1bGFuZS5lZHU=